

Abstract
After three decades of false hopes and failures, a pipeline of therapeutic drugs that target the actual root cause of Alzheimer's disease (AD) is now available. Challenging the old paradigm that focused on β‐amyloid peptide (Aβ) aggregation in amyloid plaques, these compounds are designed to prevent the neurotoxicity of Aβ oligomers that form Ca2+ permeable pores in the membranes of brain cells. By triggering an intracellular Ca2+ overdose, Aβ oligomers induce a cascade of neurotoxic events including oxidative stress, tau hyperphosphorylation, and neuronal loss. Targeting any post‐Ca2+ entry steps (e.g., tau) will not address the root cause of the disease. Thus, preventing Aβ oligomers formation and/or blocking their toxicity is by essence the best approach to stop any progression of AD. Three categories of anti‐oligomer compounds are already available: antibodies, synthetic peptides, and small drugs. Independent in silico‐based designs of a peptide (AmyP53) and a monoclonal antibody (PMN310) converged to identify a histidine motif (H13/H14) that is critical for oligomer neutralization. This “histidine trick” can be viewed as the Achilles' heel of Aβ in the fight against AD. Moreover, lipid rafts and especially gangliosides play a critical role in the formation and toxicity of Aβ oligomers. Recognizing AD as a membrane disorder and gangliosides as the key anti‐oligomer targets will provide innovative opportunities to find an efficient cure. A “full efficient” solution would also need to be affordable to anyone, as the number of patients has been following an exponential increase, affecting every part of the globe.
Keywords: Alzheimer, amyloid pore, cholesterol, ganglioside, lipid raft, neurodegenerative disease, oligomer, Parkinson
1. THE DISEASE
Alzheimer's disease (AD) is a neurodegenerative disorder currently affecting 50 million people worldwide, with 100 million expected in 2050. AD is characterized by the progressive dysfunction of synapses and neuronal loss leading to dementia. For about three decades, the major paradigm for explaining the root cause of the disease has been the accumulation of misfolded brain proteins resulting in two types of abnormalities, plaques in the extracellular milieu and neurofibrillary tangles in neurons. 1 This paradigm is generally formulated as the amyloid hypothesis, according to which the culprit is the β‐amyloid peptide (Aβ). Aβ is produced by the proteolytic cleavage of a membrane protein called “amyloid precursor protein” (APP), a 770 amino acid protein expressed by neurons. 2 Two mechanisms of APP processing have been characterized: the non‐amyloidogenic pathway leading to the release of the secreted APP α (sAPPα) by α‐secretase and the amyloidogenic pathway generating various Aβ peptides with 38, 40, 42, or 43 amino acids through the successive action of β‐ and γ‐secretase. 3 Only the amyloid pathway (Figure 1) is considered pathogenic, since sAPPα controls and modulates key neural functions including neuronal excitability, synaptic plasticity, neurite outgrowth, synaptogenesis, and cell survival. 6 On its side, Aβ peptides exert important physiological functions that are beneficial for the brain, including protecting the body from infections, repairing leaks in the blood–brain barrier (BBB), promoting recovery from injury, and modulating synaptic plasticity and memory. 7 , 8 , 9 Consistent with the role of Aβ peptides in the regulation of such major brain functions, mutations in the Aβ‐coding region of the APP gene have dramatic consequences, such as being responsible for familial forms of AD. 10 APP mutants include for instance D678N (D7N in Aβ), 6 E693G (E22G in Aβ), 11 V717I, 12 or V717F. 13 These point mutations confirmed the causative link between AD and Aβ and reinforced the notion that aberrant APP processing and/or overexpression of Aβ peptides were the primary cause of AD. The detection of Aβ peptides in amyloid plaques from AD patients suggested that the aggregated peptide was the main neurotoxic species that caused the disease. 14 , 15 , 16 Subsequently, the microtubule‐associated protein tau was identified as the main component of neurofibrillary tangles. 17 Then, it was shown that Aβ peptides could induce the abnormal phosphorylation of the tau protein 18 that resulted in its aggregation and microtubule fragilization. 19 In 1991, Hardy and Allsop summarized the formulation of the amyloid hypothesis in a sentence: “The pathological cascade for the disease process is most likely to be: beta‐amyloid deposition—tau phosphorylation and tangle formation—neuronal death” 20 and in 1992, the “amyloid cascade hypothesis” was published in Science. 21
FIGURE 1.

β‐amyloid peptide (Aβ) oligomers (and not amyloid plaques) as the root cause of Alzheimer's disease. According to the classical amyloid cascade pathway, the amyloid precursor protein (APP) is successively cleaved by two proteases (secretases), resulting in the production of the Aβ in the extracellular milieu. Excess Aβ may self‐aggregate into various assemblies, from small oligomers to large fibers and plaques, or be degraded be degraded through several distinct pathways (microglial/hepatic/renal clearance). The neurotoxicity of Aβ has been matter of speculation for several decades. It is now established that the mere presence of amyloid plaques in the brain is not correlated with Alzheimer's disease, as amyloid plaques are nontoxic structures that can be found in the brain of healthy persons with no cognitive decline. In fact, the paradigm has shifted to small Aβ oligomers, now identified as the real root mechanism of Alzheimer's disease. Once embedded in the membrane, Aβ oligomers induce Ca2+ (•) signal and free radicals that, in combination, trigger toxic downstream events leading to cell death and Alzheimer's disease pathogenesis and symptoms. Ca2+ ions activate GSK3 and CaMII kinases 4 , 5 which catalyze the phosphorylation of tau, a microtubule stabilizing protein. Hyperphosphorylated tau proteins detach from microtubule, self‐aggregate and form neurofibrillary tangles. All these events induce neuronal dysfunction and death. Note that Aβ oligomeric pores can be either preassembled in the extracellular milieu and then inserted in the plasma membrane or formed in the membrane by Aβ monomers under a lipid‐assisted oligomerization process. The dosage of Aβ oligomers in the cerebrospinal fluid (CSF) can be used as an early diagnosis for Alzheimer's disease but also as endpoint for assessing the efficiency of anti‐oligomer therapies. Any approach that could block the formation of Aβ oligomeric pores in the plasma membrane of brain cells could prevent and/or stop the progression of Alzheimer's disease
2. THREE DECADES OF THERAPEUTIC FAILURES
Although the model seemed solid, its interpretation and subsequent extrapolation to therapeutic strategies have not been straightforward. For instance, until very recently, most efforts had been focused on amyloid plaques and Aβ aggregation, ignoring microtubules, tangles, and tau. Moreover, the physiological role of Aβ had been totally neglected so that one popular belief to cure AD had simply been to block or reduce Aβ production with secretase (generally γ‐secretase) inhibitors. Indeed, nearly 50 clinical trials have been conducted using potential γ‐secretase inhibitors for AD, yet all these trials had failed. 22 Different reasons may explain such a general failure, from the risky approach of targeting a physiological process 22 to total lack of expected activity of drugs (e.g., semagacestat which appeared to have no anti‐γ‐secretase activity). 23 Severe side effects triggered by γ‐secretase inhibitors include related proteolytic processes such as those involved in Notch signaling. 24 After secretases, amyloid plaques had retained much attention for developing AD therapies.
Inasmuch as amyloid plaques are considered as the root cause of AD, anti‐plaque therapies seem to be a good idea. The very weak point of this popular belief is that amyloid plaques are in no way specific to AD patients as they are also regularly found in the brain of cognitively normal individuals. 25 , 26 , 27 , 28 , 29 , 30 The proponents of the plaque theory still consider that amyloid plaques are “necessary but not sufficient” to cause AD, or that their presence is indicative of a pre‐AD status. However, this assumption is questionable since amyloid plaques are not found in the brain of patients with an inherited form of AD, the Osaka E693Δ deletion (E22Δ in Aβ). 31 , 32 Overall, all these data and observations converge to clear amyloid plaques as being the root cause of AD, explaining why all clinical strategies targeting amyloid plaques have failed. 33 , 34 , 35 Thus, the old paradigm which incriminated amyloid plaques as the root cause of AD did not provide a cure or even stopped the progression of the disease. The problem is that the old paradigm seems to be as resistant as Captain Haddock's sticking plaster.
There are several reasons for that, explaining key observations—presence of amyloid plaques in healthy people—had been totally ignored. On the one hand, amyloid protein plaques can be easily detected by brain imaging so that they can be widely used as both an early diagnose method for AD and a reference endpoint for clinical trials. 36 On the other hand, amyloid plaques are the hallmark of transgenic animal models of AD on which potential anti‐AD therapeutic molecules are tested before first‐in‐man trials. There is a clear issue on animal models of neurodegenerative diseases, including AD and PD, as they are probably responsible for the systematic failure of all clinical trials so far. 37 As a matter of fact, while many animal models of AD and PD have been created, no single model, either based on toxins or genetic, has been able to recreate all the key features of these neurodegenerative diseases. 38 , 39 As recently stated by an AD researcher “the biggest mistake you can make is to think you can ever have a mouse with AD.” 37 As a matter of fact, the classic scheme (in silico => in vitro => in vivo => clinical trials) has not been successfully applied for the neurodegenerative domain. To summarize, there is a translational gap in AD and PD research, with promising drugs based on work in rodent models failing in clinical trials. 35 , 37 , 38 , 39 , 40 , 41 Thus, despite three decades of intense research efforts, there is still no cure for AD and the only possibility is to try to relieve symptoms, which has a very limited impact on patients. Under these circumstances, innovative strategies with new targets, new molecules, and no side effects are urgently needed. These approaches need to challenge the old paradigm with an open mind, keeping what is sound, eliminating what is wrong. 42
3. AΒ OLIGOMERS, THE ROOT CAUSE OF AD
The issues raised against the amyloid hypothesis have divided the scientific community in two schools of thought: those who consider that AD can be caused independently of Aβ 43 , 44 and those who still consider that Aβ is involved, yet independently of amyloid plaques. 45 This debate is of primary importance because the identification of the real culprit will orient the therapeutic strategies for the next years. After the false accusation against amyloid plaques, which has been misleading for three decades, a new error would still impair the development of efficient anti‐AD drugs. Most experimental studies now converge to suggest that Aβ is indeed the culprit, but in the form of regularly assembled small structures called oligomers instead of amyloid plaques. 25 , 46 , 47
The identification of the actual neurotoxic forms of Aβ came from two distinct fields of research, clinical and biochemical. The new paradigm, initially formulated as “The calcium hypothesis of AD,” 48 did not arise in 1 day. Intracellular Ca2+ concentrations are tightly regulated and are essential for neuronal function and survival, with excess Ca2+ being related to neuronal cell death. 49 Over the years, a solid experimental background has established that the homeostasis of intracellular Ca2+ concentration plays a key role in brain aging. Accordingly, sustained changes in intracellular Ca2+ levels would be responsible for age‐associated brain changes. This led several researchers to postulate that Aβ peptides could alter Ca2+ fluxes in brain cells by several possible mechanisms. 49 , 50 , 51 , 52 Using planar bilayer membranes, Arispe et al. were the first to demonstrate that Aβ peptides can self‐organize into oligomeric Ca2+ channels. 53 These channels were selectively blocked by Zn2+ ions, suggesting the presence of histidine residues lining the pore mouth. 54 , 55 Atomic force microscopy studies of mutant Aβ peptides showed globular structures which were interpreted as oligomeric “amyloid pores.” 56 , 57 Finally, Aβ oligomers were detected in the brain and cerebrospinal fluid of AD patients, 46 , 58 , 59 , 60 and, unlike amyloid plaques, the oligomer burden was found to correlate with cognitive symptoms. 25
The cascade of neurotoxic effects triggered by the Ca2+ overdose through membrane‐embedded Aβ oligomers is summarized in Figure 1 and detailed in Table 1. Downstream effects following Ca2+ entry into brain cells include a broad range of deleterious effects, especially oxidative stress and tau pathology, two classical hallmarks of AD. 47
TABLE 1.
Neurotoxic effects induced by Aβ oligomers in AD
| AD‐related mechanism | References |
|---|---|
| Membrane damage of brain cells |
Williams et al. 61 Sciacca et al. 62 |
| Ca2+ overdose |
Arispe et al. 53 Quist et al. 57 Lee et al. 63 |
| Oxidative stress | Tabner et al. 64 |
| Tau/microtubule disruption |
de Felice et al. 65 Zempel et al. 66 Rudenko et al. 67 |
| Neurotransmitter dysfunctions | Nunes‐Tavares et al. 68 |
| Synapse deterioration |
Lacor et al. 69 Shankar et al. 70 Tu et al. 71 Kawahara et al. 72 |
| Reduced LTP and increased LTD | Hong et al. 73 |
| Increased glutaminyl cyclase expression | de Kimpe et al. 74 |
| Neuronal toxicity and death |
Lambert et al. 75 Kim et al. 76 |
| Neuroinflammation | Forloni and Balducci 77 |
| Plasticity dysfunction | Shankar et al. 46 |
Abbreviations: Aβ, β‐amyloid peptide; AD, Alzheimer's disease; LTD, long‐term depression; LTP, long‐term potentiation.
The scientific literature on Aβ oligomers includes thousands of articles and the number is still growing exponentially. 47 , 58 , 59 , 60 , 78 , 79 Over the years, this huge body of work resulted in an alternative formulation of the neurotoxic cascade according to which Aβ oligomers are the main neurotoxic species at work in AD. 80 , 81 , 82 Hence, the oligomer model has all but supplanted the classical amyloid cascade, laying the foundations for a new understanding of neurodegenerative diseases through strikingly common molecular mechanisms. 83 , 84 , 85 , 86
4. MOLECULAR MECHANISMS OF AΒ OLIGOMER TOXICITY: KEY ROLE OF LIPID RAFTS
Given the prominent role of Ca2+ in AD pathogenesis, understanding the mechanisms controlling the formation Ca2+‐permeable Aβ oligomers in the plasma membrane of brain cells is of crucial importance. All studies in this field converged to identify lipid rafts as privileged platforms of pore formation and/or insertion. 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 Basically, there are two possible (and nonmutually exclusive) mechanisms accounting for the formation of functional Ca2+ permeable pores by Aβ oligomers (Figure 2). In the first mechanism, annular Aβ oligomers which are preassembled in the extracellular milieu bind to the membrane plasma membrane to form ion‐permeable pores. 93 The affinity of these oligomers for the plasma membrane supposes that once assembled, the structure displays an accessible membrane‐binding domain. Then, fine conformational tuning by surrounding lipids may be required for sealing the contacts between the oligomeric pore and the membrane. In the second mechanism, Aβ monomers bind to the membrane, after which they penetrate the outer leaflet where the oligomerization process takes place. 85 Both mechanisms require the assistance of two distinct membrane lipids that are coexpressed by lipid rafts 94 : ganglioside for binding, cholesterol for insertion. 84 , 85 , 86 Both lipids have been shown to be critical for the formation of Ca2+ permeable Aβ oligomers. Indeed, transient depletion of gangliosides and cholesterol (respectively, with metabolic inhibitors and cyclodextrins) prevented the formation of Ca2+ permeable pores from Aβ monomers. 92 , 93 Moreover, preformed neurotoxic oligomers reduced long‐term potentiation and increased long‐term depression in hippocampal slices through a ganglioside‐dependent mechanism. 73
FIGURE 2.

Two molecular mechanisms of β‐amyloid peptide (Aβ) oligomeric pore formation. Aβ oligomers may be assembled in the extracellular space (soluble oligomers) or in the plasma membrane of brain cells (from monomers). In both cases, the formation of neurotoxic oligomeric pores (amyloid pores) requires a ganglioside which acts as a specific membrane receptor. The functional organization of Aβ monomers or preassembled Aβ oligomers into a Ca2+ permeable pore is a finely tuned lipid‐driven process involving brain membrane lipids, including gangliosides and cholesterol
How can Aβ interact with both ganglioside and cholesterol? If we look at the amino acid sequence of Aβ1‐42, we can see that the N‐terminal domain is polar, whereas the C‐terminal domain is apolar. This is not totally surprising if we consider the initial topology of the Aβ part of the APP precursor since the C‐terminal part belongs to the transmembrane domain and the N‐terminal domain is extracellular. 95 Peptide scanning analysis combined with mutational studies 96 , 97 have allowed identifying a ganglioside‐binding domain (GBD) in the N‐terminal domain (aa 5–16) and a cholesterol‐binding domain (CBD) in the C‐terminal domain (aa 22–35) (Figure 3). Interestingly, there are only three amino acid differences between human and rodent Aβ. All these differences are in the GBD and the corresponding amino acid residues (especially Arg‐5 and His‐13) play a critical role in ganglioside recognition. 97 , 98 Therefore, rodent Aβ does not interact with gangliosides and does not form Ca2+ permeable oligomers (Figure 3). The intrinsic inability of rodent Aβ to oligomerize into Ca2+ permeable pores provide a rational explanation for the natural resistance of rodents to AD. 99 Moreover, this observation confirms the crucial role of the GBD of Aβ in the mechanism of Ca2+ permeable oligomeric pore formation. Taken together, these data indicate that membrane lipid composition is an important parameter that controls the vulnerability of brain cells to the neurotoxicity of Aβ oligomers. 89 For this reason, AD is primarily a membrane disorder. 88 , 100 Consequently, therapeutic strategies should consider the plasma membrane and especially lipid raft gangliosides as the key target to cure AD. 84 , 101
FIGURE 3.
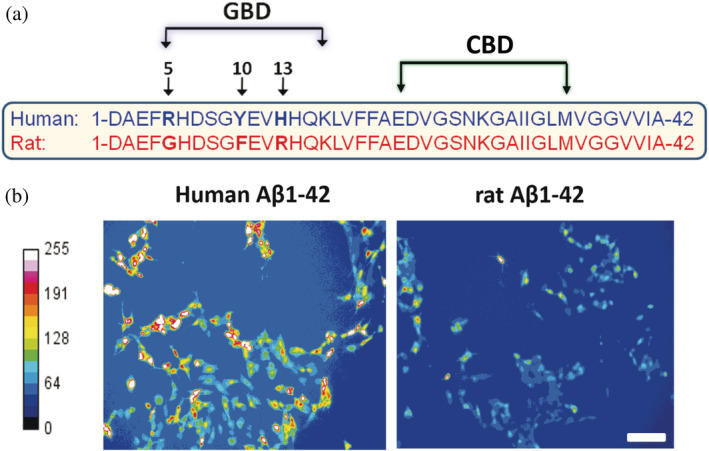
Functional lipid‐binding domains of β‐amyloid peptide (Aβ)1‐42 and their relationship with Alzheimer's disease. (a) Localization of the ganglioside and cholesterol binding domains in human Aβ1‐42. Sequence alignments of rodent and human Aβ1‐42 explain why rodents do not have Alzheimer's disease. (b) Calcium imaging studies demonstrate that rodent Aβ1‐42 do not form Ca2+ permeable oligomeric pores in brain cells (Adapted from Reference 98)
5. NEW PARADIGM, NEW TARGETS
How can we exploit these discoveries to develop efficient therapeutic strategies for AD patients? What can we expect if we prevent the formation of neurotoxic Aβ oligomers? Would it be a preventive therapy, before the appearance of symptoms? Would it cure AD patients, and at which stage? Although it is difficult to answer these questions, the recent demonstration that the deleterious effects of Aβ oligomers on neurons are reversible 102 might be positively interpreted. In this experiment, cultured neurons were exposed to Aβ oligomers for 2 days, a time sufficient to detect neurotoxic insults, including abnormal tau phosphorylation and perturbations of synaptic organization. However, these effects were rapidly reversed upon removal of Aβ oligomers from the cultures. The reversible nature of the neurotoxicity of Aβ oligomers has significant implications for AD therapy. As stated by the authors of this study, any treatment targeting oligomers could be effective in halting or even reversing the progression of the very early pathology of AD.
Therapeutic strategies targeting Aβ oligomers are currently under development, which shows that the paradigm has changed. These therapies can be classified in three structural categories: antibodies, synthetic peptides, and small molecules (Table 2).
TABLE 2.
Pipeline of anti‐oligomer therapeutic compounds for AD
| Compound | Structure | Action | Reference |
|---|---|---|---|
| PMN310 antibody | Monoclonal antibody against a constrained cyclic peptide derived from the 13–16 fragment (HHQK) of Aβ | Binds to an oligomer‐specific Aβ epitope, neutralizes preformed Aβ oligomers | 103 |
| AmyP53 | Synthetic chimeric peptide designed on the basis of the GBDs of α‐synuclein and Aβ: KEGVLYVGHHTK | Binds to membrane gangliosides and prevents Ca2+ permeable oligomeric pore formation (broad neutralization of amyloid pores) | 96 |
| PRI‐002 | Synthetic peptide (d‐amino acids, amidated C‐ter) PTLHTHNRRRRR | Blocks Aβ oligomerization | 104 |
| Alz‐801 |

|
Blocks Aβ oligomerization | 105 |
| Anle138b |
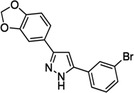
|
Blocks Ca2+ entry through oligomeric pores (broad neutralization of amyloid pores) | 106 |
| PQ912 |
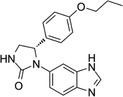
|
Glutaminyl cyclase inhibitor: Selectively blocks the formation of PyrGlu neurotoxic Aβ oligomers | 107 |
Abbreviations: Aβ, β‐amyloid peptide; AD, Alzheimer's disease; GBD, ganglioside‐binding domain.
Immunotherapies for AD patients are not new, and until now, all have failed. 33 The novelty is that these antibodies are no longer directed against amyloid plaques, but against Aβ oligomers. This new strategy includes repositioned antibodies such as Crenezumab or Aducanumab which in fact recognize both amyloid plaques and oligomers. 108 Other antibodies, such as PMN310 have been deliberated designed against oligomer‐specific epitopes so that they do not recognize Aβ monomers or amyloid plaques. 103 The rationale of PMN310 design is worth mentioning. Based on silico prediction of epitope exposure, the authors selected a tetrad of amino acid residues of Aβ spanning the 13–16 region: HHQK. 103 The monoclonal antibody was obtained by immunizing mice with a cyclic peptide displaying the HHQK motif at the tip of a constrained loop (Figure 4a). Indeed, the antibody recognized the constrained cyclic peptide used for immunization, but not its linear counterpart (Figure 4a). These data suggest that the epitope recognized by PMN310 is a constrained loop formed by the 13–16 fragment of Aβ that is fully accessible in the Aβ oligomer. Interestingly, this region was previously identified as a key part of the GBD of Aβ. 96 Of the four residues that constitute the PMN310 epitope, three are directly involved in ganglioside recognition: His‐13, His‐14, and Lys‐16. Most importantly, the pair of histidine residues seems to adopt a very similar “butterfly wings” like topology in both the constrained PMN310 epitope 103 and the Aβ 5–16 peptide bound to ganglioside GM1. 96 This “histidine trick” is one of the key elements of the design of AmyP53 (Figure 4b), a synthetic chimeric peptide built by combining the GBD of Aβ and alpha‐synuclein (Table 2). In this respect, it is important to note that two distinct in silico approaches independently converged to identify the histidine pair His‐13/His‐14 of Aβ as a key target for designing anti‐oligomer compounds. AmyP53 binds to brain gangliosides with a significant higher affinity than the amyloid proteins from which it is derived (Figure 4b). It prevents the formation of neurotoxic Aβ oligomers and all neurotoxic downstream events in neural cell cultures and ex vivo (brain hippocampal slices) and, due to its chimeric design, is also active against α‐synuclein oligomers, including PD associated mutant forms. 85 , 109 In this respect, it should be noted that neurodegenerative disorders share common molecular mechanisms of pathogenesis, including the oligomer‐triggered Ca2+ cascade, that will render possible the use of the same drug for several distinct therapeutic applications. 83 , 86
FIGURE 4.

Molecular mechanism of β‐amyloid peptide (Aβ) and AmyP53 therapeutic peptide binding to gangliosides: relationship to anti‐oligomer therapies. (a) Representative solvent‐exposed conformation of the cyclic HHQK epitope, showing a similarly constrained loop displaying at its tip the histidine pair His‐13/His‐14 of Aβ. This peptide was used to elicit the PMN310 anti‐oligomer antibody. As shown on the right panel, this antibody specifically recognizes the constrained cyclic peptide corresponding to the Aβ oligomer conformation but not the linear unstructured peptide (Reprinted and adapted from Reference 103). (b) Molecular modeling of AmyP53 bound to a ganglioside GM1 dimer. The model is in full agreement with experimental data showing that both His‐13 and His‐14 residues are critical for GM1 binding. Indeed, each histidine interacts with its own GM1, through a complex network of hydrogen (H) and OH–π bonds. Note that the histidine pair of AmyP53 is located at the tip of the constrained loop induced by the ganglioside dimer. The kinetics of AmyP53 interaction with a monolayer of gangliosides GM1, compared with Aβ1‐42 and α‐synuclein (α‐syn) is shown on the right panel (Reprinted and adapted from Reference 96)
Besides AmyP53, several other synthetic peptides are currently considered for AD therapy (Table 2). PRI‐002 (formerly called RD2) is an amidated d‐amino acid peptide with anti‐oligomer properties. 104 Interestingly, this peptide also displays two histidine residues (yet not contiguous) in its amino acid sequence: PTLHTHNRRRRR. Other synthetic d‐amino acid peptides derived from the 16–20 sequence of Aβ (KLVFF) are also considered. 110
Finally, several small organic molecules with experimentally demonstrated anti‐oligomer properties are also under investigation (Table 2). ALZ‐801 is a valine‐conjugated prodrug of homotaurine, a blocker of Aβ oligomerization and aggregation. 105 The diphenyl‐pyrazole derivative anle138b has broad activity against neurotoxic oligomers. 106 PQ912, a polycyclic enzymatic inhibitor of glutaminyl cyclase, an enzyme that catalyzes the formation of a subset of neurotoxic Aβ oligomers. 107 Hopefully, the paradigm shift from amyloid plaques to Aβ oligomers as the root cause of AD will now allow to quickly identify an efficient cure for AD. 45 , 81 , 83 , 84
6. AD THERAPY: PAST, PRESENT, AND FUTURE
If we do not consider symptomatic approaches that do not attack the root cause of the disease (e.g., neurotransmitter and synaptic receptors‐based therapies), 111 there are not many rational therapeutic options for curing AD. Let us briefly review the advantages and drawbacks associated with the means and the targets of possible strategies. Immunotherapies for brain diseases are associated with important side effects. 112 Most anti‐plaque immunotherapies have been abandoned not only because they do not work, but also because they are associated with severe brain inflammatory processes (amyloid‐related imaging abnormalities syndrome). 112 In fact, the percentage of circulating antibodies that can reach the brain is very low (0.1–0.3%) due to the efficacy of the BBB. 113 To overcome this limitation, it has been proposed to disrupt the BBB to gain entry into the brain. 114 This strategy might be at risk, since breaking the BBB, even for short time‐lapses with ultrasounds, could induce severe impairments of brain homeostasis. Maintaining BBB integrity is crucial for tight control of the chemical composition of brain interstitial fluid, which is critical for proper synaptic functioning, information processing, and neuronal connectivity. 115 The intravenous administration of antibodies might also be problematic.
Except for the specific case of d‐enantiomeric peptides, synthetic peptides are made from naturally occurring amino acids which are nontoxic and metabolically tolerable. 116 They are highly specific, active at low doses and they do not trigger undesirable side effects such as those induced by chemical drugs and antibodies. Most therapeutic peptides, such as beta‐sheet breakers 117 are designed to disrupt protein–protein interactions that play a key role in disease pathogenesis. Such therapeutic approaches cannot be implemented with small drugs, because small molecules do not interact with target proteins over a large enough surface area to block the interacting surface. 118 Indeed, the binding pocket size of the small molecules (300–1,000 Å2), does not fit very well with the large area of most larger protein binding interfaces (generally 1,500–3,000 Å2). Another advantage of therapeutic peptides is that they can be safely delivered via the intranasal route, which is far more comfortable and safer than the intravenous pathway. This route of administration is a quite recent, promising alternative pathway to enteral and systemic drug administration for dosing highly potent and efficacious CNS targeted drugs to reach the brain parenchyma. 116 The dosage to be delivered to the olfactory region or to be absorbed neuronally may easily be as low as 0.01–1% of oral dosage. Thus, in contrast with small organic molecules which can induce severe toxic effects, peptides do not accumulate in specific organs (liver, kidney) and this also contributes to minimize their toxicity. Finally, low molecular weight drugs, especially polycyclic compounds, may have specificity, toxicity, and solubility issues 119 , 120 which could require chemical modifications of the initial formula. In any case, as illustrated in Table 2, we have now an arsenal of innovative compounds that, for the first time, deliberately target Aβ oligomers by several angles of attack. Clinical evaluations will identify which one among these therapeutic drug candidates can indeed reach and block the root cause of AD, or whether second generation compounds are needed to cure the disease.
The identification of gangliosides as key targets for blocking the neurotoxicity of Aβ oligomers is an important step that has contributed to transform our vision of AD from a protein misfolding disease to a membrane disorder. If we consider that Aβ belongs to the category of intrinsically disordered proteins which, by essence, fluctuate between numerous equally probable conformations in aqueous solution, 121 the term “misfolding” is not appropriate. In fact, Aβ is a typical α/β discordant peptide 122 which can adopt either α‐rich or β‐rich structures depending on the environment. Membrane lipids, especially gangliosides and cholesterol favor α‐helix structuration, 123 , 124 whereas high concentration in water readily induces β structures and turns. 80 , 125 Thus, which one among the three possible categories of Aβ conformers (unstructured, α, or β) is misfolded? None of them, which indicates that AD can hardly be defined as misfolding disease. Instead, if we consider the Ca2+ overdose triggered by membrane‐embedded Aβ oligomers as the root cause of AD, the conformational status of these oligomers has little importance compared with their location. For this reason, AD should be primarily defined as a membrane disorder due to the toxin‐like activity of Aβ oligomers. Targeting these oligomers, whatever the angle of attack may be, 72 , 101 , 126 , 127 , 128 , 129 , 130 is a new strategy that really addresses the root cause of the disease and could be applied for both preventive and curative approaches against AD. Thus, there is some real hope on the horizon with the arrival of disruptive innovations able to block the amyloid oligomer pathogenic cascade. A “full efficient” solution would also need to be affordable to anyone, as the number of patients has been following an exponential increase, affecting every part of the globe.
CONFLICTS OF INTEREST
N. Y. and J. F. are co‐inventors of the AmyP53 peptide (patent Application EP15709163.8A), currently under development for the treatment of Alzheimer's and Parkinson's diseases by the AmyPore company (France). H. C. is President of the Ethics and Scientific Committee of AmyPore.
AUTHOR CONTRIBUTIONS
Jacques Fantini: Conceptualization; investigation; methodology; supervision; validation; writing‐original draft; writing‐review and editing. Nouara Yahi: Conceptualization; investigation; methodology; supervision; validation; writing‐review and editing. Henri Chahinian: Conceptualization; investigation; methodology; supervision; validation; writing‐review and editing.
Fantini J, Chahinian H, Yahi N. Progress toward Alzheimer's disease treatment: Leveraging the Achilles' heel of Aβ oligomers? Protein Science. 2020;29:1748–1759. 10.1002/pro.3906
REFERENCES
- 1. Maurer K. Historical background of Alzheimer's research done 100 years ago. J Neural Transm. 2006;113:1597–1601. [DOI] [PubMed] [Google Scholar]
- 2. Zheng H, Koo EH. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011;6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dimitrov M, Alattia JR, Lemmin T, et al. Alzheimer's disease mutations in APP but not gamma‐secretase modulators affect epsilon‐cleavage‐dependent AICD production. Nature Commun. 2013;4:2246. [DOI] [PubMed] [Google Scholar]
- 4. Ghosh A, Giese KP. Calcium/calmodulin‐dependent kinase II and Alzheimer's disease. Mol Brain. 2015;8:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Flaherty D, Soria J, Tomasiewicz H, Wood J. Phosphorylation of human tau protein by microtubule‐associated kinases: GSK3b and cdk5 are key participants. J Neurosci Res. 2000;62:463–472. [DOI] [PubMed] [Google Scholar]
- 6. Mattson MP. Cellular actions of beta‐amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. [DOI] [PubMed] [Google Scholar]
- 7. Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L. A physiological role for amyloid‐beta protein: Enhancement of learning and memory. J Alzheimer's Dis. 2010;19:441–449. [DOI] [PubMed] [Google Scholar]
- 8. Puzzo D, Privitera L, Leznik E, et al. Picomolar amyloid‐beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brothers HM, Gosztyla ML, Robinson SR. The physiological roles of amyloid‐β peptide hint at new ways to treat Alzheimer's disease. Front Aging Neurosci. 2018;10:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levy E, Carman MD, Fernandez‐Madrid IJ, et al. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. [DOI] [PubMed] [Google Scholar]
- 11. Nilsberth C, Westlind‐Danielsson A, Eckman CB, et al. The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–893. [DOI] [PubMed] [Google Scholar]
- 12. Yoshioka K, Miki T, Katsuya T, Ogihara T, Sakaki Y. The 717Val–Ile substitution in amyloid precursor protein is associated with familial Alzheimer's disease regardless of ethnic groups. Biochem Biophys Res Commun. 1991;178:1141–1146. [DOI] [PubMed] [Google Scholar]
- 13. Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. [DOI] [PubMed] [Google Scholar]
- 14. Allsop D, Landon M, Kidd M. The isolation and amino acid composition of senile plaque core protein. Brain Res. 1983;259:348–352. [DOI] [PubMed] [Google Scholar]
- 15. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glenner GG, Wong CW. Alzheimer's disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. [DOI] [PubMed] [Google Scholar]
- 17. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule‐associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Busciglio J, Lorenzo A, Yeh J, Yankner BA. Beta‐amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. [DOI] [PubMed] [Google Scholar]
- 19. Alonso AC, Zaidi T, Grundke‐Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:5562–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. [DOI] [PubMed] [Google Scholar]
- 21. Hardy JA, Higgins GA. Alzheimer's disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. [DOI] [PubMed] [Google Scholar]
- 22. Barão S, Moechars D, Lichtenthaler SF, de Strooper B. BACE1 physiological functions may limit its use as therapeutic target for Alzheimer's disease. Trends Neurosci. 2016;39:158–169. [DOI] [PubMed] [Google Scholar]
- 23. Tagami S, Yanagida K, Kodama TS, et al. Semagacestat is a pseudo‐inhibitor of γ‐secretase. Cell Rep. 2017;21:259–273. [DOI] [PubMed] [Google Scholar]
- 24. Geling A, Steiner H, Willem M, Bally‐Cuif L, Haass C. A gamma‐secretase inhibitor blocks Notch signaling in vivo and causes a severe neurogenic phenotype in zebrafish. EMBO Rep. 2002;3:688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Esparza TJ, Zhao H, Cirrito JR, et al. Amyloid‐β oligomerization in Alzheimer dementia versus high‐pathology controls. Ann Neurol. 2013;73:104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Snowdon DA. Aging and Alzheimer's disease: Lessons from the Nun Study. Gerontologist. 1997;37:150–156. [DOI] [PubMed] [Google Scholar]
- 27. Snowdon DA. Nun Study. Healthy aging and dementia: Findings from the Nun Study. Ann Intern Med. 2007;139:450–454. [DOI] [PubMed] [Google Scholar]
- 28. Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rentz DM, Locascio JJ, Becker JA, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol. 2010;67:353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sakr FA, Grothe MJ, Cavedo E, et al. Applicability of in vivo staging of regional amyloid burden in a cognitively normal cohort with subjective memory complaints: The INSIGHT‐preAD study. Alzheimer's Res Ther. 2019;11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomiyama T, Nagata T, Shimada H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer's‐type dementia. Ann Neurol. 2008;63:377–387. [DOI] [PubMed] [Google Scholar]
- 32. Shimada H, Ataka S, Tomiyama T, Takechi H, Mori H, Miki T. Clinical course of patients with familial early‐onset Alzheimer's disease potentially lacking senile plaques bearing the E693Δ mutation in amyloid precursor protein. Dement Geriatr Cogn Disord. 2011;32:45–54. [DOI] [PubMed] [Google Scholar]
- 33. Panza F, Lozupone M, Seripa D, Imbimbo BP. Amyloid‐β immunotherapy for alzheimer disease: Is it now a long shot? Ann Neurol. 2019;85:303–315. [DOI] [PubMed] [Google Scholar]
- 34. Mullane K, Williams M. Alzheimer's disease (AD) therapeutics ‐ 1: Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem Pharmacol. 2018;158:359–375. [DOI] [PubMed] [Google Scholar]
- 35. Mullane K, Williams M. Alzheimer's disease beyond amyloid: Can the repetitive failures of amyloid‐targeted therapeutics inform future approaches to dementia drug discovery? Biochem Pharmacol. 2020;177:113945. [DOI] [PubMed] [Google Scholar]
- 36. Ossenkoppele R, Jansen WJ, Rabinovici GD, et al. Prevalence of amyloid PET positivity in dementia syndromes: A meta‐analysis. JAMA. 2015;313:1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reardon S. Frustrated Alzheimer's researchers seek better lab mice. Nature. 2018;563:611–612. [DOI] [PubMed] [Google Scholar]
- 38. Melrose HL, Lincoln SJ, Tyndall GM, Farrer MJ. Parkinson's disease: A rethink of rodent models. Exp Brain Res. 2006;173:196–204. [DOI] [PubMed] [Google Scholar]
- 39. Mullane K, Williams M. Preclinical models of Alzheimer's disease: Relevance and translational validity. Curr Protoc Pharmacol. 2019;84:e57. [DOI] [PubMed] [Google Scholar]
- 40. King A. The search for better animal models of Alzheimer's disease. Nature. 2018;559:S13–S15. [DOI] [PubMed] [Google Scholar]
- 41. Garner JP. The significance of meaning: Why do over 90% of behavioral neuroscience results fail to translate to humans, and what can we do to fix it? ILAR J. 2014;55:438–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yaari R, Hake A. Alzheimer's disease clinical trials: Past failures and future opportunities. Clin Invest. 2015;5:297–309. [Google Scholar]
- 43. Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer inverted question marks disease. Acta Neuropathol Commun. 2014;2:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chetelat G. Alzheimer disease: Abeta‐independent processes‐rethinking preclinical AD. Nat Rev Neurol. 2013;9:123–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jang H, Connelly L, Arce FT, et al. Alzheimer's disease: Which type of amyloid‐preventing drug agents to employ. Phys Phys Chem Chem Phys. 2013;15:8868–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shankar GM, Li S, Mehta TH, et al. Amyloid‐beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cline EN, Bicca MA, Viola KL, Klein WL. The amyloid‐β oligomer hypothesis: Beginning of the third decade. J Alzheimer's Dis. 2018;64:S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khachaturian ZS. Calcium hypothesis of Alzheimer's disease and brain aging. Ann N Y Acad Sci. 1994;747:1–11. [DOI] [PubMed] [Google Scholar]
- 49. Khachaturian ZS. The role of calcium regulation in brain aging: Reexamination of a hypothesis. Aging. 1989;1:17–34. [DOI] [PubMed] [Google Scholar]
- 50. Berridge MJ. Calcium hypothesis of Alzheimer's disease. Pflugers Archiv Eur J Physiol. 2010;459:441–449. [DOI] [PubMed] [Google Scholar]
- 51. Kim S, Rhim H. Effects of amyloid‐β peptides on voltage‐gated L‐type CaV1.2 and CaV1.3 Ca2+ channels. Mol Cells. 2011;32:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kayed R, Sokolov Y, Edmonds B, et al. Permeabilization of lipid bilayers is a common conformation‐dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–46366. [DOI] [PubMed] [Google Scholar]
- 53. Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A. 1993;90:567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kawahara M, Arispe N, Kuroda Y, Rojas E. Alzheimer's disease amyloid beta‐protein forms Zn(2+)‐sensitive, cation‐selective channels across excised membrane patches from hypothalamic neurons. Biophys J. 1997;73:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rhee SK, Quist AP, Lal R. Amyloid beta protein‐(1‐42) forms calcium‐permeable, Zn2+‐sensitive channel. J Biol Chem. 1998;273:13379–13382. [DOI] [PubMed] [Google Scholar]
- 56. Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature. 2002;418:291. [DOI] [PubMed] [Google Scholar]
- 57. Quist A, Doudevski I, Lin H, et al. Amyloid ion channels: A common structural link for protein‐misfolding disease. Proc Natl Acad Sci U S A. 2005;102:10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Georganopoulou DG, Chang L, Nam JM, et al. Nanoparticle‐based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102:2273–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lesné SE. Breaking the code of amyloid‐β oligomers. Int J Cell Biol. 2013;2013:950783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lasagna‐Reeves CA, Glabe CG, Kayed R. Amyloid‐β annular protofibrils evade fibrillar fate in Alzheimer disease brain. J Biol Chem. 2011;286:22122–22130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Williams TL, Johnson BR, Urbanc B, Jenkins AT, Connell SD, Serpell LC. Aβ42 oligomers, but not fibrils, simultaneously bind to and cause damage to ganglioside‐containing lipid membranes. Biochem J. 2011;439:67–77. [DOI] [PubMed] [Google Scholar]
- 62. Sciacca MF, Kotler SA, Brender JR, Chen J, Lee DK, Ramamoorthy A. Two‐step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys J. 2012;103:702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee J, Kim YH, Arce FT, et al. Amyloid β ion channels in a membrane comprising brain total lipid extracts. ACS Chem Nerosci. 2017;8:1348–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tabner BJ, El‐Agnaf OM, Turnbull S, et al. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem. 2005;280:35789–35792. [DOI] [PubMed] [Google Scholar]
- 65. de Felice FG, Wu D, Lambert MP, et al. Alzheimer's disease‐type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rudenko LK, Wallrabe H, Periasamy A, et al. Intraneuronal Tau misfolding induced by extracellular amyloid‐β oligomers. J Alzheimer's Dis. 2019;71:1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nunes‐Tavares N, Santos LE, Stutz B, et al. Inhibition of choline acetyltransferase as a mechanism for cholinergic dysfunction induced by amyloid‐β peptide oligomers. J Biol Chem. 2012;287:19377–19385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lacor PN, Buniel MC, Furlow PW, et al. Abeta oligomer‐induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid‐beta protein induce reversible synapse loss by modulating an NMDA‐type glutamate receptor‐dependent signaling pathway. J Neurosci. 2007;27:2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Aβ‐induced synaptic dysfunction in Alzheimer's disease. Mol Neurodegener. 2014;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kawahara M, Kato‐Negishi M, Tanaka KI. Amyloids: Regulators of metal homeostasis in the synapse. Molecules. 2020;25:1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hong S, Ostaszewski BL, Yang T, et al. Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron. 2014;82:308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. de Kimpe L, Bennis A, Zwart R, van Haastert ES, Hoozemans JJ, Scheper W. Disturbed Ca2+ homeostasis increases glutaminyl cyclase expression; connecting two early pathogenic events in Alzheimer's disease in vitro. PLoS One. 2012;7:e44674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1‐42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kim HJ, Chae SC, Lee DK, et al. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J. 2003;17:118–120. [DOI] [PubMed] [Google Scholar]
- 77. Forloni G, Balducci C. Alzheimer's disease, oligomers, and inflammation. J Alzheimer's Dis. 2018;62:1261–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Beckman D, Ott S, Donis‐Cox K, et al. Oligomeric Aβ in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proc Natl Acad Sci U S A. 2019;116:26239–26246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aprile FA, Sormanni P, Podpolny M, et al. Rational design of a conformation‐specific antibody for the quantification of Aβ oligomers. Proc Natl Acad Sci U S A. 2020;2020:13509–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jang H, Arce FT, Ramachandran S, Capone R, Lal R, Nussinov R. Beta‐barrel topology of Alzheimer's beta‐amyloid ion channels. J Mol Biol. 2010;404:917–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhao LN, Long H, Mu Y, Chew LY. The toxicity of amyloid beta oligomers. Int J Mol Sci. 2012;13:7303–7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ono K, Yamada M. Low‐n oligomers as therapeutic targets of Alzheimer's disease. J Neurochem. 2011;117:19–28. [DOI] [PubMed] [Google Scholar]
- 83. Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. [DOI] [PubMed] [Google Scholar]
- 84. Fantini J, Yahi N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev Mol Med. 2010;12:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. di Scala C, Yahi N, Boutemeur S, et al. Common molecular mechanism of amyloid pore formation by Alzheimer's β‐amyloid peptide and α‐synuclein. Sci Rep. 2016;6:28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fantini J, Yahi N. Brain lipids in synaptic function and neurological disease: Clues to innovative therapeutic strategies for brain disorders. New York, NY: Elsevier, 2015. [Google Scholar]
- 87. Kim SI, Yi JS, Ko YG. Amyloid beta oligomerization is induced by brain lipid rafts. J Cell Biochem. 2006;99:878–889. [DOI] [PubMed] [Google Scholar]
- 88. Fabiani C, Antollini SS. Alzheimer's disease as a membrane disorder: Spatial cross‐talk among beta‐amyloid peptides, nicotinic acetylcholine receptors and lipid rafts. Front Cell Neurosci. 2019;13:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Evangelisti E, Zampagni M, Cascella R, et al. Plasma membrane injury depends on bilayer lipid composition in Alzheimer's disease. J Alzheimer's Dis. 2014;41:289–300. [DOI] [PubMed] [Google Scholar]
- 90. Rushworth JV, Hooper NM. Lipid rafts: Linking Alzheimer's amyloid‐β production, aggregation, and toxicity at neuronal membranes. Int J Alzheimer's Dis. 2010;2011:603052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Williamson R, Usardi A, Hanger DP, Anderton BH. Membrane‐bound beta‐amyloid oligomers are recruited into lipid rafts by a fyn‐dependent mechanism. FASEB J. 2008;22:1552–1559. [DOI] [PubMed] [Google Scholar]
- 92. Brown AM, Bevan DR. Molecular dynamics simulations of amyloid β‐peptide (1‐42): Tetramer formation and membrane interactions. Biophys J. 2016;111:937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shafrir Y, Durell S, Arispe N, Guy HR. Models of membrane‐bound Alzheimer's Abeta peptide assemblies. Proteins. 2010;78:3473–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Fantini J, Garmy N, Mahfoud R, Yahi N. Lipid rafts: Structure, function and role in HIV, Alzheimer's and prion diseases. Expert Rev Mol Med. 2002;4:1–22. [DOI] [PubMed] [Google Scholar]
- 95. di Scala C, Chahinian H, Yahi N, Garmy N, Fantini J. Interaction of Alzheimer's β‐amyloid peptides with cholesterol: Mechanistic insights into amyloid pore formation. Biochemistry. 2014;53:4489–4502. [DOI] [PubMed] [Google Scholar]
- 96. Yahi N, Fantini J. Deciphering the glycolipid code of Alzheimer's and Parkinson's amyloid proteins allowed the creation of a universal ganglioside‐binding peptide. PLoS One. 2014;9:e104751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Fantini J, Yahi N. Molecular basis for the glycosphingolipid‐binding specificity of α‐synuclein: Key role of tyrosine 39 in membrane insertion. J Mol Biol. 2011;408:654–669. [DOI] [PubMed] [Google Scholar]
- 98. di Scala C, Yahi N, Flores A, et al. Comparison of the amyloid pore forming properties of rat and human Alzheimer's beta‐amyloid peptide 1‐42: Calcium imaging data. Data Brief. 2016;6:640–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lovestone S, Killick R. Is Alzheimer's a disorder of ageing and why don't mice get it? The centrality of insulin signalling to Alzheimer's disease pathology In: Craft S, Christen Y, editors. Diabetes, insulin and Alzheimer's disease. Research and perspectives in Alzheimer's disease. Berlin, Germany: Springer, 2010; p. 129–152. [Google Scholar]
- 100. Lukiw WJ. Alzheimer's disease (AD) as a disorder of the plasma membrane. Front Physiol. 2013;4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Magistretti PJ, Geisler FH, Schneider JS, Li PA, Fiumelli H, Sipione S. Gangliosides: Treatment avenues in neurodegenerative disease. Front Neurol. 2019;10:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tanokashira D, Mamada N, Yamamoto F, et al. The neurotoxicity of amyloid β‐protein oligomers is reversible in a primary neuron model. Mol Brain. 2017;10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gibbs E, Silverman JM, Zhao B, et al. A rationally designed humanized antibody selective for amyloid beta oligomers in Alzheimer's disease. Sci Rep. 2019;9:9870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. An Groen T, Schemmert S, Brener O, et al. The Aβ oligomer eliminating D‐enantiomeric peptide RD2 improves cognition without changing plaque pathology. Sci Rep. 2017;7:16275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hey JA, Kocis P, Hort J, et al. Discovery and identification of an endogenous metabolite of tramiprosate and its prodrug ALZ‐801 that inhibits beta amyloid oligomer formation in the human brain. CNS Drugs. 2018;32:849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Martinez Hernandez A, Urbanke H, Gillman AL, et al. The diphenylpyrazole compound anle138b blocks Aβ channels and rescues disease phenotypes in a mouse model for amyloid pathology. EMBO Mol Med. 2018;10:32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Scheltens P, Hallikainen M, Grimmer T, et al. Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer's disease: Results of a randomized, double‐blind, placebo‐controlled phase 2a study. Alzheimer's Res Ther. 2018;10:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Tolar M, Abushakra S, Sabbagh M. The path forward in Alzheimer's disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimer's Dement. 2019;1–8. [DOI] [PubMed] [Google Scholar]
- 109. di Scala C, Yahi N, Flores A, et al. Broad neutralization of calcium‐permeable amyloid pore channels with a chimeric Alzheimer/Parkinson peptide targeting brain gangliosides. Biochim Biophys Acta. 2016;1862:213–222. [DOI] [PubMed] [Google Scholar]
- 110. Horsley JR, Jovcevski B, Wegener KL, Yu J, Pukala TL, Abell A. Rationally designed peptide‐based inhibitor of Aβ42 fibril formation and toxicity: A potential therapeutic strategy for Alzheimer's disease. Biochem J. 2020;477:2039–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2019. Alzheimer's Dement. 2019;5:272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sperling RA, Jack CR Jr, Black SE, et al. Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: Recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimer's Dement. 2011;7:367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lemere CA. Immunotherapy for Alzheimer's disease: Hoops and hurdles. Mol Neurodegener. 2013;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu HL, Pan CH, Ting CY, Hsiao MJ. Opening of the blood–brain barrier by low‐frequency (28‐kHz) ultrasound: A novel pinhole‐assisted mechanical scanning device. Ultrasound Med Biol. 2010;36:325–335. [DOI] [PubMed] [Google Scholar]
- 115. Daneman R, Prat A. The blood‐brain barrier. Cold Spring Harb Perspect Biol. 2015;7:a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Morimoto BH. Therapeutic peptides for CNS indications: Progress and challenges. Bioorg Med Chem. 2018;26:2859–2862. [DOI] [PubMed] [Google Scholar]
- 117. Liu W, Sun F, Wan M, et al. β‐Sheet breaker peptide‐HPYD for the treatment of Alzheimer's disease: Primary studies on behavioral test and transcriptional profiling. Front Pharmacol. 2018;8:969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Petta I, Lievens S, Libert C, Tavernier J, de Bosscher K. Modulation of protein‐protein interactions for the development of novel therapeutics. Mol Ther. 2016;24:707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Narvekar M, Xue HY, Eoh JY, Wong HL. Nanocarrier for poorly water‐soluble anticancer drugs—Barriers of translation and solutions. AAPS PharmSciTech. 2014;15:822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Harris KL, Banks LD, Mantey JA, Huderson AC, Ramesh A. Bioaccessibility of polycyclic aromatic hydrocarbons: Relevance to toxicity and carcinogenesis. Expert Opin Drug Metab Toxicol. 2013;9:1465–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Uversky VN, Davé V, Iakoucheva LM, et al. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem Rev. 2014;114:6844–6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kallberg Y, Gustafsson M, Persson B, Thyberg J, Johansson J. Prediction of amyloid fibril‐forming proteins. J Biol Chem. 2010;276:12945–12950. [DOI] [PubMed] [Google Scholar]
- 123. Williamson MP, Suzuki Y, Bourne NT, Asakura T. Binding of amyloid beta‐peptide to ganglioside micelles is dependent on histidine‐13. Biochem J. 2006;397:483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ji SR, Wu Y, Sui SF. Cholesterol is an important factor affecting the membrane insertion of beta‐amyloid peptide (a beta 1‐40), which may potentially inhibit the fibril formation. J Biol Chem. 2002;277:6273–6279. [DOI] [PubMed] [Google Scholar]
- 125. Voelker MJ, Barz B, Urbanc B. Fully atomistic Aβ40 and Aβ42 oligomers in water: Observation of porelike conformations. J Chem Theory Comput. 2017;13:4567–4583. [DOI] [PubMed] [Google Scholar]
- 126. Dukhinova M, Veremeyko T, Yung AWY, et al. Fresh evidence for major brain gangliosides as a target for the treatment of Alzheimer's disease. Neurobiol Aging. 2019;77:128–143. [DOI] [PubMed] [Google Scholar]
- 127. Sengupta U, Nilson AN, Kayed R. The role of amyloid‐β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. 2016;6:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Salahuddin P, Fatima MT, Abdelhameed AS, Nusrat S, Khan RH. Structure of amyloid oligomers and their mechanisms of toxicities: Targeting amyloid oligomers using novel therapeutic approaches. Eur J Med Chem. 2016;114:41–58. [DOI] [PubMed] [Google Scholar]
- 129. Galzitskaya OV. Oligomers are promising targets for drug development in the treatment of proteinopathies. Front Mol Neurosci. 2020;12:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shrivastava AN, Aperia A, Melki R, Triller A. Physico‐pathologic mechanisms involved in neurodegeneration: Misfolded protein‐plasma membrane interactions. Neuron. 2017;95:33–50. [DOI] [PubMed] [Google Scholar]