

Abstract
Interfacial inhibitors belong to a broad class of natural products and synthetic drugs that are commonly used to treat cancers as well as bacterial and HIV infections. They bind selectively to interfaces as macromolecular machines assemble and are set in motion. The bound drugs transiently arrest the targeted molecular machines, which can initiate allosteric effects, or desynchronize macromolecular machines that normally function in concert. Here, we review five archetypical examples of interfacial inhibitors: the camptothecins, etoposide, the quinolone antibiotics, the vinca alkaloids and the novel anti-HIV inhibitor raltegravir. We discuss the common and diverging elements between interfacial and allosteric inhibitors and give a perspective for the rationale and methods used to discover novel interfacial inhibitors.
Living cells rely on polymers for cellular architecture (for example, membranes and filaments) and information storage (for example, nucleic acids). Their molecular machines can be extremely complex, consisting of many components — many of them bound to each other non-covalently and reversibly. For instance, the eukaryotic ribosome consists of three RNA chains (5S, 28S and 18S RNAs) and at least 80 polypeptides, the spliceosome and the proteasome consist of approximately 40 different polypeptides each, and the replicative DNA polymerase and RNA polymerase II complexes consist of at least 10 protein subunits each. These molecular machines must also function at high speed, under conditions in which their parts move relative to each other with great precision and robustness. For instance, DNA and RNA polymerases track along their DNA substrates while incorporating nucleobases into new DNA and RNA polymers at average speeds of 30 bases per second1. DNA topoisomerases I and II (TOP1 and TOP2, respectively) function at up to 100 cycles per second and 4 cycles per second, respectively (K. C. Neuman, personal communication). Moreover, to allow the movement of the polymerase complexes relative to their DNA substrate, DNA helicases and topoisomerases must open and recoil the DNA in concert with the polymerases, thereby creating ‘supermolecular machines’. Uncoupling helicases, topoisomerases and polymerases can be highly lethal, as discussed below.
Molecular machines have a large number of fast-moving parts and interdependent velocities; this makes them particularly vulnerable to small molecules that can infiltrate their hinges and interfere with the movements of individual parts relative to each other. It is therefore relatively easy to understand how small-molecule interfacial inhibitors can alter the function of large molecular machines (such as topoisomerases, integrase, polymerases, GTPases, and so on) and the proper organization of large cellular polymers (such as microtubules).
The interfacial inhibitor concept
Molecular pharmacology and structural studies of topoisomerase inhibitors have led to the conceptualization and demonstration of the interfacial inhibitor concept. The first evidence that topoisomerase inhibitors could trap topoisomerase as the enzyme cleaves DNA (that is, that the drugs trap topoisomerase cleavage complexes) was revealed approximately 30 years ago with the discovery that the antibiotics nalidixic acid and oxolinic acid produce gyrase-mediated DNA double-strand breaks2,3. Furthermore, it was shown that anticancer drugs — including doxorubicin (also known as adria mycin), amsacrine, etoposide and camptothecins — target eukaryotic topoisomerases via a comparable mechanism; at the time, it was proposed that the drugs were trapping topoisomerase cleavage complexes by forming ternary complexes with a drug molecule bound at the interface of the enzymes and the cleaved DNA4. It took about 10 years to validate the ternary complex hypothesis, which was achieved when the crystal structure of the TOP1 ternary complex was solved with topotecan (a clinical derivative of camptothecin) intercalated in the cleaved DNA and specifically bound to TOP1 (REF. 5). Soon after, the ternary complex structure was confirmed for the natural alkaloid camptothecin and the indenoisoquinolines, the non-camptothecin inhibitors of TOP1, which are currently in clinical trials6,7.
We proposed the interfacial inhibitor concept because of the common mechanistic principle underlying the mechanism of action of distantly related natural drugs8,9. Camptothecin and brefeldin A served as the landmark examples, each targeting different macromolecular machines: camptothecin targets TOP1–DNA complexes, whereas brefeldin A targets the protein–protein complex formed by ADP-ribosylation factor and guanine nucleotide exchange factor8. The macromolecular structures of natural inhibitors have been extensively analysed in the literature; this has extended the relevance of the interfacial inhibitor concept to a range of drugs and molecular machines, including cyclosporine, tacrolimus (also known as FK506), forskolin, fusicoccin, rapamycin, colchicine, vinca alkaloids, paclitaxel (also known as Taxol) and α-amanitin8. Since then, anti biotics have been shown to act as interfacial inhibitors for bacterial type II topoisomerases10–12. The interfacial inhibition mechanism has recently been extended to purely synthetic compounds such as the anti-AIDS drugs that target the HIV integrase–viral DNA complexes. Stunning co-crystal structures of integrase with its viral substrates have revealed the trapping of the integrase–DNA complex as the drugs bind at its interface13–15. The interfacial hypothesis for the targeting of eukaryotic TOP2 by etoposide has also recently been confirmed16, as discussed below.
Examples of interfacial inhibitors
Several drug classes and targets of interfacial inhibitors are listed in TABLE 1. Below, we describe several examples of interfacial inhibitors, to clarify the concept of interfacial inhibition.
Table 1 |.
Interfacial inhibitors identified/developed to date
| Drugs | Origin | Substrates | Mechanism of action | Medical use* | Refs |
|---|---|---|---|---|---|
| α-amanitin | Black cap mushroom | RNA polymerase II | Binds to Rpb1–Rpb2 interface and prevents Pol II translocation | Not reported | 60,61 |
| Adriamycin (doxorubicin) and anthracyclines | Soil bacteria (Streptomyces peucetius) | TOP2-DNA complex | Trap cleavage complexes by binding at the enzyme–DNA interface | FDA-approved | 28 |
| Anabaseine | Sea worm (Paranemertes peregrina) | α7 nicotinic acetylcholine receptor (homopentamer) | Agonizes α7 nicotinic acetylcholine receptor by binding at the interface between each of the α7 subunit monomers | Not reported | 62 |
| Anaesthetics (desflurane and propofol) | Synthetic | Pentameric ligand-gated ion channels | Act as ion channel blockers | FDA-approved | 63 |
| Benzodiazepines | Synthetic | GABAA receptor | Increase conduction of chloride ion channels | FDA-approved | 64 |
| Brefeldin A | Fungus (Eupenicillium brefeldianum) | ARF•GDP–GEF complex | Stabilizes a kinetic protein–protein intermediate undergoing conformational changes | Not reported | 65,66 |
| Camptothecin | Chinese tree (Camptotheca acuminata) | TOP1–DNA complex | Stabilizes a kinetic intermediate of the enzymatic DNA cleavage reaction | FDA-approved | 5,18 |
| Colchicine | Crocus (Colchicum autumnale) | Tubulin heterodimer | Prevents tubulin polymerization by stabilizing a curved tubulin heterodimer | FDA-approved | 67 |
| Curares | Plant (Strychnos toxifera) | Nicotinic acetylcholine receptor | Act as nicotinic acetylcholine receptor blockers | Not reported | 53 |
| Cyclosporine | Soil fungus (Tolypocladium inflatum) | Cyclophilin–Calcineurin complex | Hinders access to the active site of the protein phosphatase calcineurin by artificially creating a protein–protein interface | FDA-approved | 68,69 |
| Dexrazoxane (ICRF-187) | Synthetic | TOP2–ATP complex | Stabilizes the closed TOP2 dimer | FDA-approved | 41,70 |
| Ecteinascidin 743 | Sea squirt (Ecteinascidia turbinata) | Transcription-coupled nucleotide excision repair | Poisons the repair complex after incision of the DNA by preventing normal repair ligation | Clinically approved | 71 |
| Epothilone A | Soil-dwelling myxobacterium (Sorangium cellulosum) | β-tubulin | Stabilizes microtubules | FDA-approved | 72 |
| Etoposide and teniposide | Synthetic | TOP2–DNA complex | Stabilize a kinetic intermediate of the enzymatic DNA cleavage reaction | FDA-approved | 16 |
| Tacrolimus (FK506) | Soil bacteria Streptomyces tsukubaensis | FKBP–calcineurin complex | Hinders access to the active site of the protein phosphatase calcineurin by artificially creating a protein–protein interface | FDA-approved | 73,74 |
| Forskolin | Indian Coleus plant (Coleus forskohlii) | Heterodimer of cytoplasmic domains of adenylyl cyclase | Activates the enzyme by stabilizing the heterodimer catalytic site in an active conformation | Not reported | 75 |
| Fusicoccin | Plant parasite fungus (Fusicoccum amygdali) | 14-3-3–ATPase complex | Overstabilizes a regulatory complex | Not reported | 76 |
| Fusidic acid | Fungus (Fusidium coccineum) | Bacterial ribosome | Stabilizes the translation elongation factor EF-G | Clinically approved | 77,78 |
| Indenoisoquinolines | Synthetic | TOP1-DNA complex | Stabilizes a kinetic intermediate of the enzymatic DNA cleavage reaction | In Phase I trials | 7,79 |
| Kirromycin | Fungus (Streptomyces collinus) | Antibiotics, bacterial ribosomes | Stabilizes translation elongation factor Tu | Not reported | 77,78 |
| Mitoxantrone | Synthetic | TOP2–DNA complex | Stabilizes a kinetic intermediate of the enzymatic DNA cleavage reaction | FDA-approved | 16‡ |
| Nalidixic acid and quinolone antibiotics | Synthetic | Gyrase–DNA or topoisomerase IV–DNA complexes | Trap cleavage complexes by binding at the interface between DNA gyrase or topoisomerase IV subunits and DNA | FDA-approved | 10–12 |
| Nicotine | Plant (Nicotiana tabacum) | Nicotinic acetylcholine receptor | Increases activity of nicotinic acetylcholine receptors | FDA-approved | 53 |
| Raltegravir, elvitegravir and dolutegravir | Synthetic | HIV-1 integrase–DNA complexes | Chelate metal ions in the catalytic site of the integrase–DNA complex | FDA-approved and in Phase III trials | 13,15,45 |
| Rapamycin (sirolimus) | Soil bacteria (Streptomyces hygroscopicus) | FKBP–mTOR complex | Promotes dimerization of FKBP12 with mTOR and inhibits mTOR | FDA-approved | 80 |
| SAHM1 | Stapled peptide | Notch transcription factor complex | Antagonizes Notch signalling pathway and cell proliferation by binding to the intracellular domain of Notch 1 and the DNA-bound CSL transcription factor | Not reported | 81 |
| Simocyclinone | Bacteria (Streptomyces antibioticus) | DNA gyrase subunit A homotetramer | Poisons DNA gyrase by binding at the DNA gyrase A59 subunit homotetramer interface | Not reported | 82 |
| Paclitaxel and docetaxel | Trees (Taxus baccata, Taxus brevifolia) | β-tubulin | Stabilize microtubules | FDA-approved | 83 |
| Thiostrepton | Bacteria (Streptomyces azureus, Streptomyces laurentii) | Bacterial ribosome | Prevents EF-G binding by altering the interface between ribosomal protein L11 and ribosomal RNA | Veterinary use | 84 |
| Vinblastine | Madagascar periwinkle plant (Catharanthus roseus) | Tubulin heterodimer | Prevents tubulin polymerization by stabilizing a curved tubulin heterodimer | FDA-approved | 44 |
ARF, ADP-ribosylation factor; CSL, DNA-binding protein complex consisting of the mammalian protein CBF1 (also known as RBPJ-κ), the Drosophila melanogaster protein Su(H) and the Caenorhabditis elegans protein LAG1; FDA, US Food and Drug Administration; FKBP, FK506-binding protein; GABAA, γ-aminobutyric acid type A; GEF, guanine nucleotide exchange factor; mTOR, mammalian target of rapamycin; Pol II, DNA polymerase II; Rpb1, RNA polymerase II subunit 1; Rpb2, RNA polymerase II subunit 2; SAHM1, stapled α-helical peptides derived from mastermind like-1 (MAML1); TOP1, topoisomerase I; TOP2, topoisomerase II.
A large fraction (20 out of 30 drug classes listed) of these interfacial inhibitors are in clinical use (approved by the FDA, approved outside the United States, or in clinical trials.
The mechanism of action of mitoxantrone as a TOP2–DNA interfacial inhibitor was reported at the 2011 Topoisomerase conference in Taipei, Taiwan, October 2011, by the group of Nei-Li Chan, National Taiwan University16.
Camptothecins and non-camptothecin anticancer TOP1 inhibitors.
Topoisomerases belong to a large family of enzymes that regulate the topology of DNA without changing its primary sequence. They are ubiquitously expressed in eukaryotes, prokaryotes and archaea, and are also encoded by some viruses. Topoisomerases can be viewed as ‘3 in 1 machines’ because a single enzyme performs three consecutive reactions. First, a topoisomerase cleaves the DNA backbone (endonuclease-equivalent function); second, it allows changes in DNA supercoils (DNA strand passage or untwisting activity); and third, it re-ligates the cleaved DNA (ligase or recombinase-like activity). Topoisomerases are classified as type I or type II, depending on whether they cleave one or both DNA strands in concert, respectively17.
The human genome contains six topoisomerase genes that encode two type IB enzymes (TOP1 and mitochondrial TOP1 (TOP1MT)), two type IA enzymes (TOP3α and TOP3β) and two type IIA enzymes (TOP2α and TOP2β). TOP1MT is only found in vertebrates. It is encoded in the nucleus but only acts on mitochondrial DNA4. The bacterium Escherichia coli encodes four topoisomerases: two type IA enzymes (topoisomerase I and topoisomerase III) and two type IIA enzymes (topoisomerase IV and DNA gyrase).
Topoisomerase inhibitors are important medicinal drugs. They are widely used as antibiotics and anticancer drugs. The quinolone derivatives (nalidixic acid, oxolinic acid, norfloxacin, ciprofloxacin, levofloxacin, moxifloxacin and gemifloxacin) target the bacterial type II enzymes DNA gyrase and topoisomerase IV without affecting the host type II enzymes (TOP2α and TOP2β), which explains their high antibacterial selectivity4. There are 14 anticancer drugs targeting topoisomerases that have been approved by the US Food and Drug Administration (FDA), and several more are in clinical development. Two of these 14 drugs — topo tecan and irinotecan — target TOP1, whereas the other 12 (doxorubicin, daunorubicin, doxil, myocet, idarubicin, epirubicin, aclarubicin, dexrazoxane, mitoxantrone, etoposide, etopophos and teniposide) target TOP2 (both TOP2α and TOP2β). Notably, anticancer drugs targeting topoisomerases are highly selective for either TOP1 or TOP2 but they do not target both enzymes.
FIGURE 1 shows the structure of the ternary complex of camptothecin bound at the interface of TOP1 and its nicked DNA substrate6,7,18. It is crucial to note that camptothecin binds to both DNA and TOP1. The binding of camptothecin to DNA is established by π–π interactions between the heterocyclic ring system of the drug and the nucleobases flanking the cleavage site7, which hinders the rotation of the DNA19. When binding to the TOP1 polypeptide, the drug forms hydrogen bonds between its B, D and E rings and three amino acid residues (Asp533, Arg364 and Asn722)5,6,18. Mutations in any of these single residues render TOP1 highly resistant to camptothecin and its clinical derivatives, despite the fact that camptothecin can still be seen in crystal structures18,20. This apparent contradiction highlights that interfacial inhibitors are primarily kinetic inhibitors and that all the contacts are essential to retain the drugs in the ternary complex (that is, reduce the ‘off-rate’). Off-rates cannot be inferred in crystal structures in which the ligand is used at saturating concentrations.
Figure 1 |. Structure of a topoisomerase I cleavage complex trapped by camptothecin.
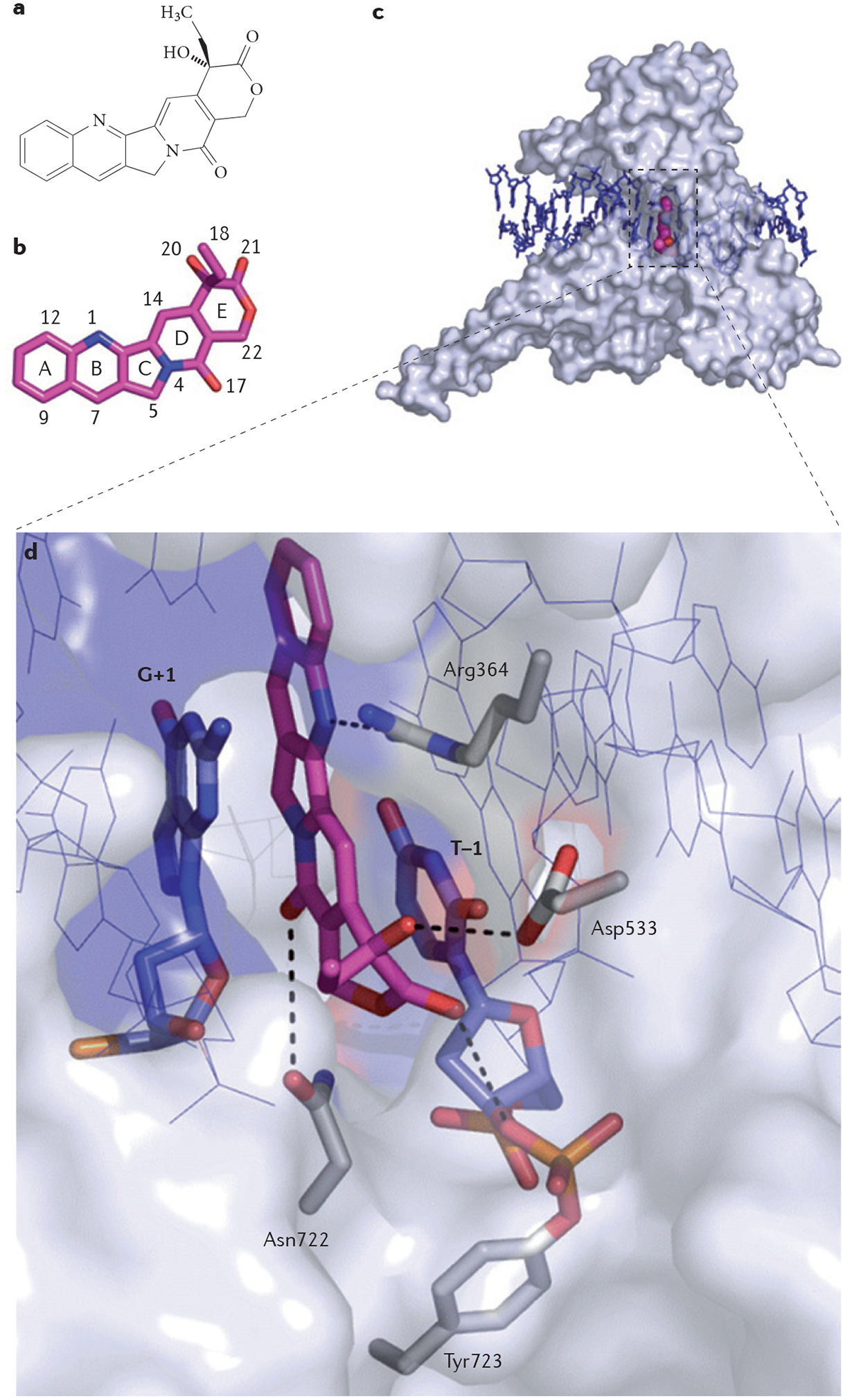
a | Chemical structure of camptothecin (CPT). b | Three-dimensional structure of CPT (stick representation with carbon, nitrogen and oxygen atoms coloured in magenta, blue and red, respectively; Protein Data Bank ID code 1T8I6; numbering according to REF. 26). c | Topoisomerase I (TOP1) is shown in surface representation (grey) to highlight the depth of the CPT binding pocket. CPT (shown in magenta) is bound inside the catalytic core of TOP1, intercalated between the DNA base pairs (shown in blue) flanking the TOP1 cleavage site. This mechanism of action is common to other TOP1 poisons7. d | Interaction network between CPT, DNA and TOP1 in the drug–TOP1–DNA ternary complex. DNA contacts by π-stacking with the flanking DNA base pairs (G+1 and T–1) are indicated in blue. Protein contacts are illustrated, with the following hydrogen bonds demonstrated using dashed lines: hydrogen bonding between the N1 nitrogen atom of CPT and the guanidinium group of Arg364; hydrogen bonding between the 20 hydroxyl group (see panel a) of CPT and the carboxylic functional group of Asp533; and hydrogen bonding between the C17 pyridone ring oxygen and the side chain nitrogen of Asn722.
The stereospecific requirement for the optimum binding of camptothecin21, which is a general characteristic of interfacial inhibitors, is reflected by the fact that only the natural 20S isomer fits inside the TOP1 interfacial binding pocket (FIG. 1); the synthetic 20R isomer, which is inactive against TOP1, cannot fit inside the interfacial binding pocket21.
The exquisite selectivity of camptothecin for TOP1 is also reflected by two genetic observations. First, yeast cells in which the TOP1 gene is inactivated are completely immune to camptothecin22,23. Second, camptothecin-producing plants are immune to the drug because the TOP1 produced by these plants is homogenously mutated at a single asparagine residue next to the catalytic tyrosine residue24. Notably, the corresponding mutation in human TOP1 (Asn722 to Ser722) had previously been identified in camptothecin-resistant human leukaemia cells25.
Understanding how the trapping of TOP1 leads to DNA damage and anticancer activity is crucial for rationalizing the use of TOP1-targeted drugs and their combination with other drugs26,27. As indicated above, the transient stabilization of the TOP1 cleavage complex slows down the TOP1 catalytic cycles, which leads to DNA damage as the fast movements of the replication and transcription complexes produce collisions with the drug-stalled TOP1 cleavage complex.
Etoposide and anticancer TOP2 inhibitors.
Anticancer TOP2 inhibitors were the first drugs that were hypothesized to form ternary complexes by intercalating between the base pairs flanking the cleavage sites generated by a topoisomerase28,29. This hypothesis was derived from the observation that each chemical class of inhibitors selectively traps TOP2 at different sites30,31. Furthermore, sequencing a large collection of drug-induced TOP2 cleavage sites revealed that these differences are defined by the base pairs that flank those sites32. Namely, doxorubicin, daunorubicin, epirubicin and other anthracyclines trap TOP2 cleavage complexes with an adenine at the 3′ end of the break (the A–1 site)28,33, whereas amsacrine preferentially traps the A+1 sites; etoposide, teniposide and mitoxantrone preferentially trap TOP2 cleavage complexes at C–1 sites29,34.
Chan and colleagues16 have successfully co-crystallized etoposide in a ternary complex with TOP2β, based on the preference of etoposide and teniposide for the trapping of TOP2 at the C–1 base32. The crystal structure confirms the prediction29 that the etoposide drug molecule stacks against the C–1 or G+5 base pair and forms a network of hydrogen bonds and van der Waals interactions with TOP2 (FIG. 2). Furthermore, the same group has successfully co-crystallized mitoxantrone in the TOP2 cleavage complex and demonstrated that mitoxantrone has a similar mechanism of action to etoposide (N. L. Chan, personal communication).
Figure 2 |. Structure of a topoisomerase Iiβ cleavage complex trapped by etoposide.
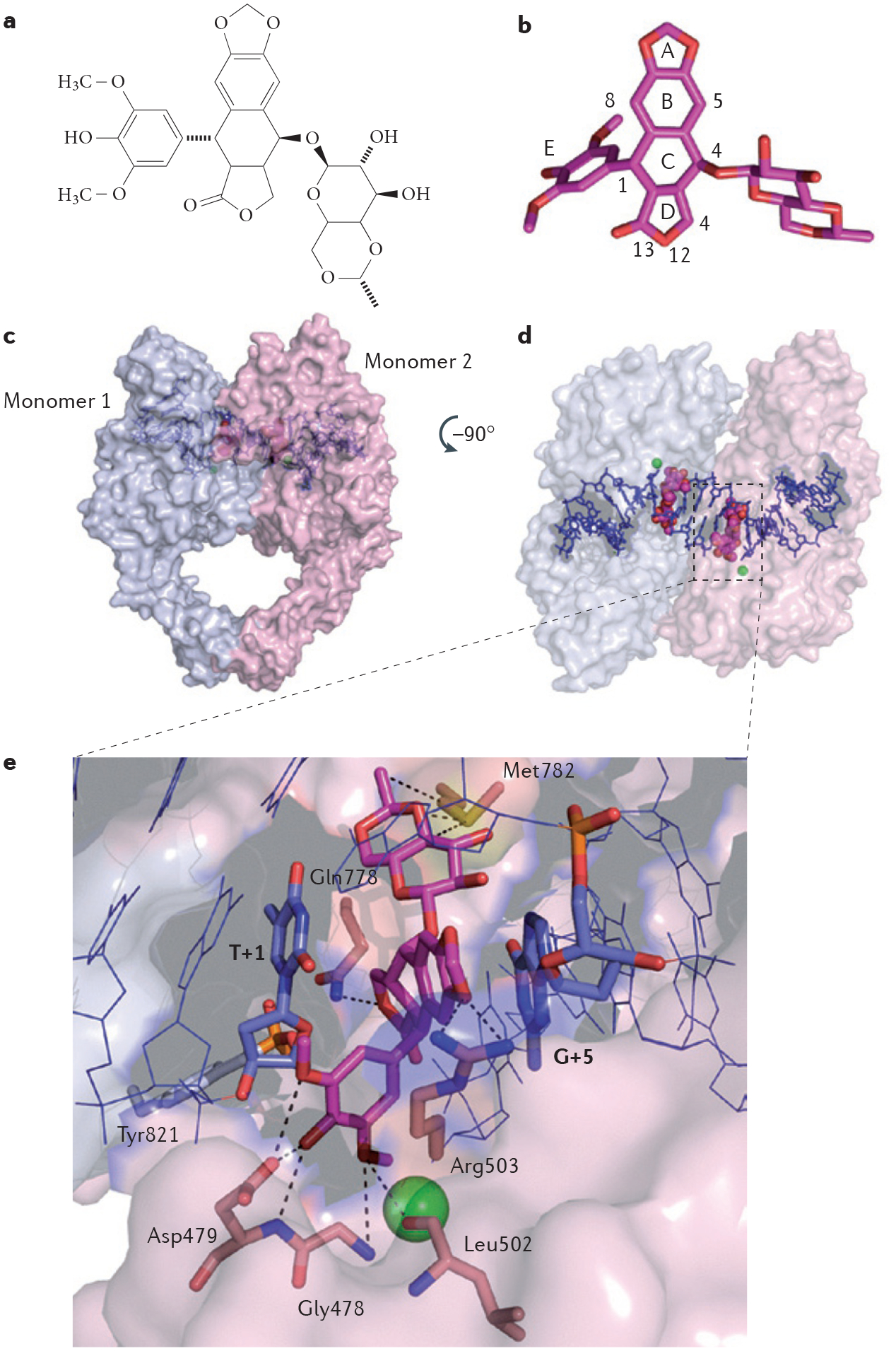
a | Chemical structure of etoposide. b | Three-dimensional structure of etoposide (stick representation with carbon and oxygen coloured in magenta and red, respectively; Protein Data Bank ID code 3QX3 (REF. 16); numbering according to REF. 16). c | Topoisomerase IIβ (TOP2β) is shown in surface representation and functions as a homodimer (represented by the light blue and light pink subunits). Etoposide (shown in magenta) is bound inside the catalytic core of each TOP2β subunit and stabilizes the cleavage complex by intercalating between the DNA base pairs (shown in blue) flanking the TOP2β cleavage sites. d | The image shown is the surface representation of TOP2β (in the left panel) after 90° rotation. e | Interaction network between etoposide in the drug–TOP2β–DNA ternary complex. DNA contacts by π-stacking with the flanking DNA base pairs (T+1 on the cleaved strand and G+5 on the uncleaved strand) are indicated in blue. The active site of TOP2β is assembled in trans with the catalytic Tyr821 residue (shown in grey) from monomer 1 and the magnesium (green sphere)-chelating triad of acidic residues (not shown) from monomer 2. Protein contacts from monomer 2 with etoposide are shown, with the following hydrogen bonds and Van der Waals interactions depicted using dashed lines: hydrogen bonding and Van der Waals interactions between the E ring oxygen atoms of etoposide and Gly478, Asp479 and Leu502 residues; between the A ring oxygen of etoposide and the Arg503 residue; between oxygen 12 on the D ring of etoposide and the Gln778 residue; and between the glycosidic group of etoposide and the Met782 residue.
Quinolone antibiotics: inhibitors of bacterial type II topoisomerases.
Antibiotics have also been successfully co-crystallized with the bacterial type II topoisomerases topoisomerase IV10,11 and DNA gyrase12, and have been shown to act as interfacial inhibitors. Because topoisomerase IV and DNA gyrase are essential for bacterial replication and are sufficiently different from the eukaryotic TOP2α and TOP2β enzymes, which are insensitive to the antibiotics, bacterial type II topoisomerases are prime targets for antibiotics4. Moreover, the same antibacterial topoisomerase inhibitors usually target both DNA gyrase and topoisomerase IV owing to the high structural similarities between the two prokaryotic enzymes4.
Quinolone antibiotics represent the archetype of bacterial type II topoisomerase poisons. They were originally limited to Gram-negative bacteria but the introduction of a fluorine atom in their structure broadened their antibacterial spectrum. Several generations of fluoroquinolone derivatives have led to some of the most potent antibiotics available to date, such as ciprofloxacin and levofloxacin4 (FIG. 3). Similarly to other topoisomerase poisons, fluoroquinolones trap bacterial type II topoisomerases in a stabilized cleavage complex by stacking between the base pairs flanking the cleavage sites at the interface of the parC and parE subunits of topoisomerase IV10,35 (FIG. 3).
Figure 3 |. Structure of a topoisomerase IV cleavage complex trapped by the fluoroquinolone antibiotic levofloxacin.
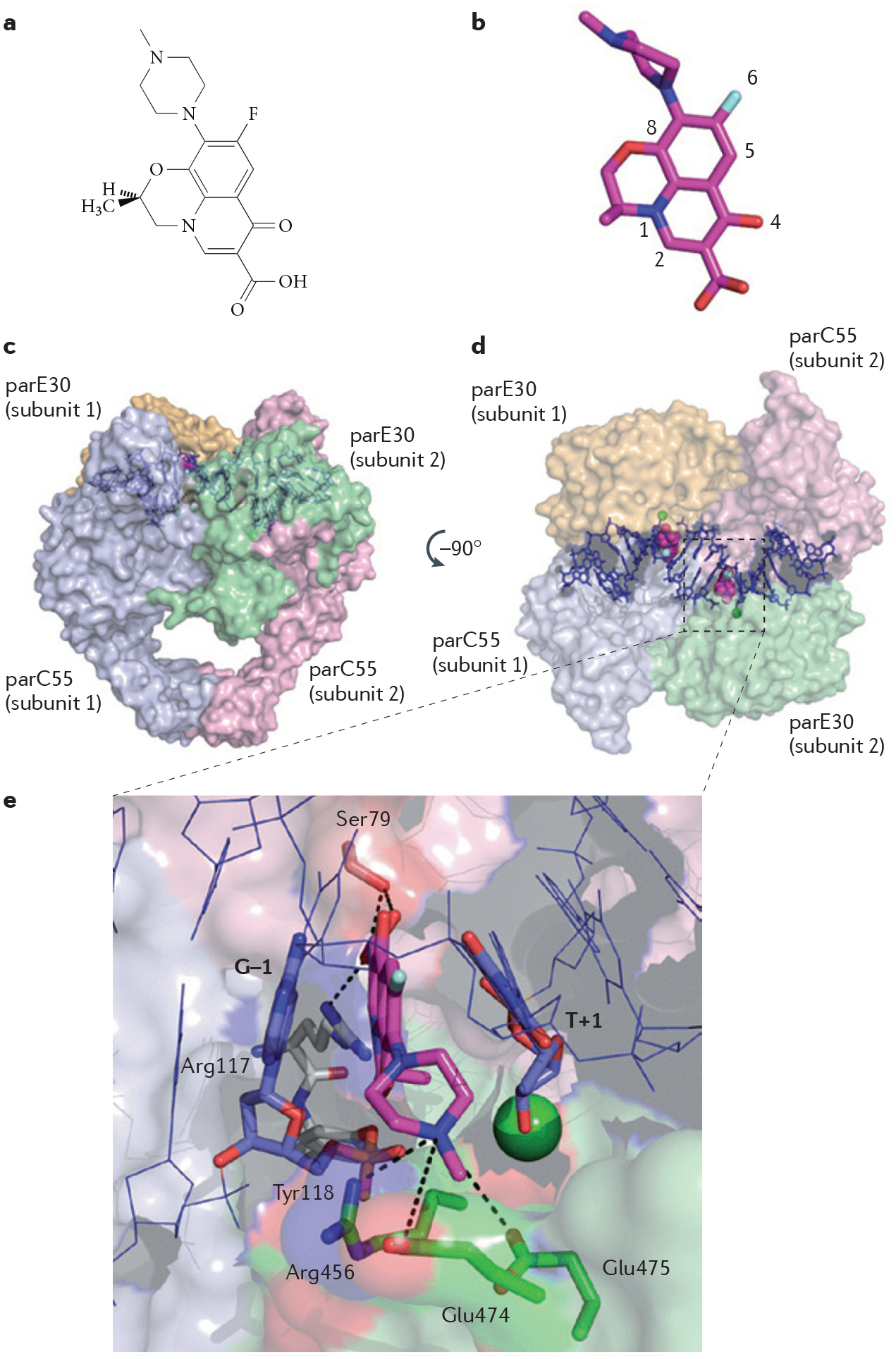
a | Chemical structure of levofloxacin. b | Three-dimensional structure of levofloxacin (stick representation with carbon, nitrogen, oxygen and fluorine atoms coloured in magenta, blue, red and grey, respectively; Protein Data Bank ID code 3K9F10,11; numbering according to REF. 11). c | Bacterial topoisomerase IV functions as a tetramer of two parC55 (light blue and light pink) and two parE30 (light green and yellow) subunits4, shown in surface representation. Levofloxacin (shown in magenta) stabilizes the cleavage complex by intercalating between the DNA base pairs (shown in blue) flanking the topoisomerase IV cleavage sites. d | The image shown is the surface representation of topoisomerase IV (in the left panel) after 90 rotation. e | Interaction network between levofloxacin in the ternary complex formed by the drug, topoisomerase IV and DNA. DNA contacts by π-stacking with the flanking DNA base pairs (G–1 and T+1) are indicated in blue. The active site of topoisomerase IV is assembled in trans with the catalytic Tyr118 residue (shown in grey) from the parC55 subunit 1 and the magnesium (green sphere)-chelating triad of acidic residues (not shown) from the parE30 subunit 2. Protein contacts are shown, with the following hydrogen bonds depicted using dashed lines: hydrogen bonding between the C3 carboxyl group of levofloxacin and the residues Ser79 and Arg117 of the parC55 subunit; and the hydrogen bonds involving the Arg456, Glu474 and Glu475 residues of the parE30 subunit, which hold together the N4 nitrogen atom of the piperazine ring at the other side of the levofloxacin molecule.
In the context of DNA gyrase and interfacial inhibition, it is relevant to mention the ccd operon of the F plasmid of E. coli, which represents an archetype of the toxin–antitoxin modules that are present in many bacteria and archaea, and function as biological inter facial inhibitors36. These plasmid-encoded toxin–antitoxin systems may be used for the development of novel antibiotics37. The ccd operon on the F plasmid encodes two proteins: the toxin CcdB and its antitoxin CcdA. CcdB poisons DNA gyrase by binding as a homodimer to both sub units A of DNA gyrase in the centre of the heterodimer, keeping the gyrase in an open conformation with the cleaved DNA38. CcdA also functions as a homodimer36; it neutralizes and rejuvenates DNA gyrase by binding to the CcdB dimer interface, thereby interfering with the binding of CcdB to DNA gyrase subunit A and forming CcdB–CcdA tetramers that act as repressors of the ccd operon (reviewed in REF. 36). Hence, the toxin–antitoxin ccd operon represents a highly accomplished interfacial inhibitor system that uses DNA gyrase as its endogenous target.
Another type of interfacial inhibition has been described for the catalytic inhibitors of eukaryotic TOP2, represented by the bisdioxopiperazine ICRF-193 and its chemotherapeutic derivative dexrazoxane (ICRF-187)39–41. In this complex TOP2 does not cleave DNA, and the enzyme homodimer is trapped encircling both DNA double helices after re-ligation of the passing strand4. Dexrazoxane binds to a site at the interface of the ATP domains of two TOP2 molecules and consequently stabilizes TOP2 in this trapped intermediate state41.
Tubulin inhibitors.
Interfacial inhibitors can also target the protein–protein interface of structural rather than enzymatic macromolecular complexes such as microtubules. Microtubules have important roles in cellular development, proliferation, trafficking, signalling and migration. They represent key components of the cytoskeleton. Microtubules have been the targets of anticancer therapy for decades, and there are currently 42 microtubule-binding agents that have either been approved or are in clinical trials for the treatment of cancer42. Microtubules are composed of α- and β-tubulin heterodimers arranged in the form of long filamentous, tube-shaped polymers that can be several micrometers long. Microtubule dynamics are crucial for mitosis and protein trafficking. Mitotic spindle microtubules have a very high exchange rate (with half-times as low as 10 seconds) with the tubulin molecules in the soluble pool. Any reduction in microtubule dynamics results in a mitosis block that may lead to apoptosis43.
Microtubule-binding agents are often classified into two main categories — the microtubule-destabilizing agents and the microtubule-stabilizing agents — owing to their differential effects on microtubule mass at high doses. The destabilizing agents either bind to the vinca domain or to the colchicine domain of tubulin. Vinca domain-binding agents include the vinca alkaloids such as vinblastine and vincristine. Colchicine domain-binding agents include colchicine and its analogues, such as podophyllotoxin and combretastatin. The stabilizing agents bind to the taxane-binding site, and include taxanes such as paclitaxel and docetaxel. However, this classification of stabilizing or destabilizing agents has been shown to be oversimplistic, as both categories of microtubule-binding agents exhibit the same powerful suppression of microtubule dynamics at concentrations up to 100-fold lower than needed to observe either the stabilization or destabilization of microtubules42.
The vinca alkaloid vinblastine (FIG. 4a,b) binds at the interface of an α- and β-tubulin heterodimer44 in a site involving residues from both monomers (FIG. 4c,d). The effect of vinblastine binding results in the suppression of spindle microtubule dynamics and stabilizes curved microtubule polymers, which interferes with their proper extension and leads to the slowing or blocking of mitosis and the induction of apoptotic cell death.
Figure 4 |. Structure of vinblastine bound in a ternary complex with tubulin heterodimers.
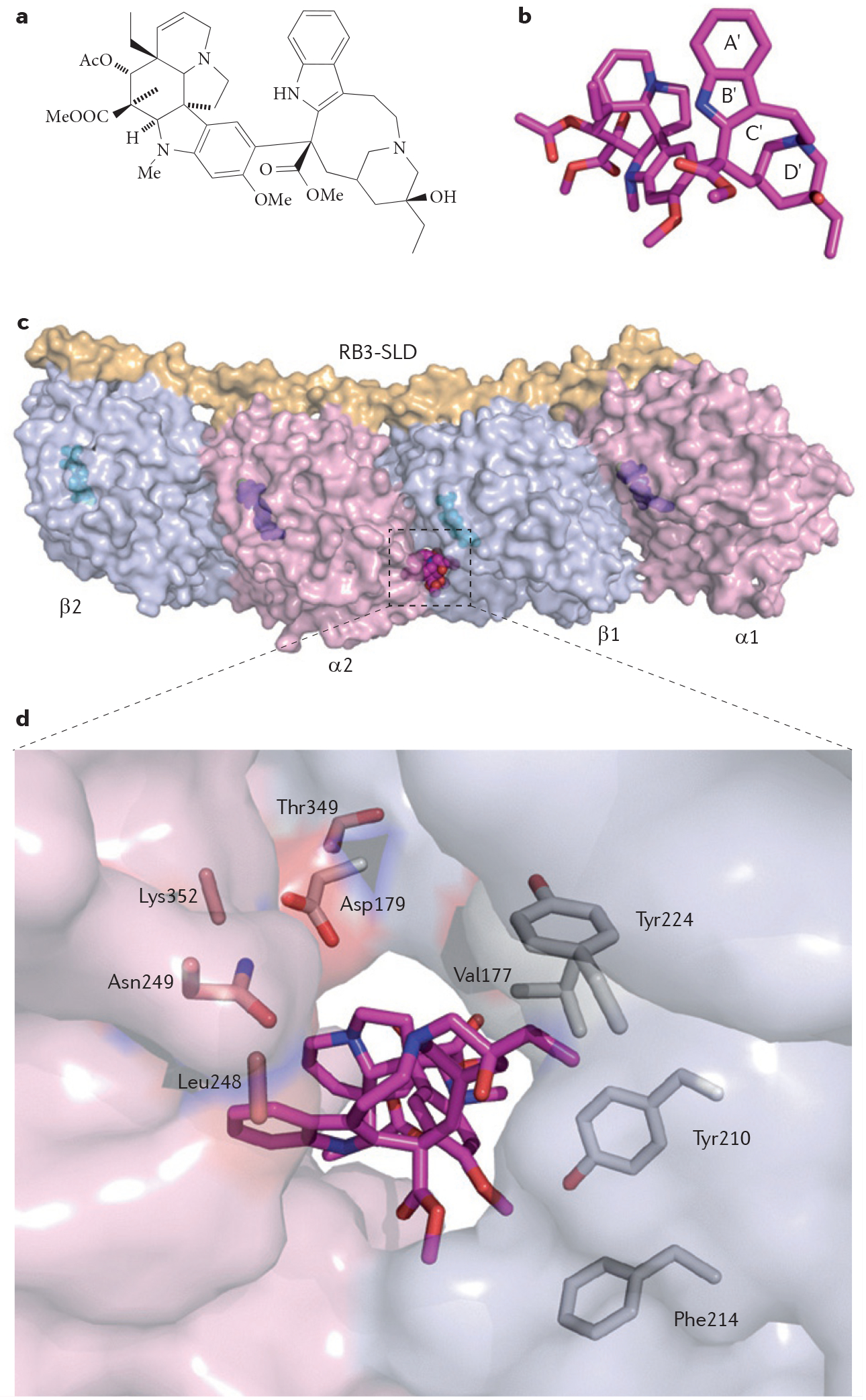
a | Chemical structure of vinblastine. b | Three-dimensional structure of vinblastine (in sphere representation with carbon, nitrogen and oxygen atoms coloured in magenta, blue and red, respectively; Protein Data Bank (PDB) ID code 1Z2B44). Heterocycles labelled A′ to D′ correspond to the catharanthine domain, and the other portion of the molecule corresponds to the vindoline portion of vinblastine44. c | Vinblastine binds between an α-subunit (α2; shown in light pink) and a β-subunit (β1; shown in light blue) of two tubulin α-β heterodimers, in complex with the Rhodopirellula baltica protein 3 (RB3) stathmin-like domain (SLD), as shown in surface representation. GDP and GTP molecules are shown in sphere representation in cyan and magenta, respectively. d | Interaction network between vinblastine and tubulin heterodimers. Vinblastine is buried inside the complex and its orientation allows its catharanthine and vindoline moieties to interact with both tubulin heterodimers. Following vinblastine binding, the β-subunit residue Asp179 and the α-subunit residue Thr349 from each heterodimer move towards the drug and contribute to its binding. Vinblastine is stabilized on one side by the α-subunit residues Leu248, Asn249 and Lys352. On the other side, it is stabilized by the β-subunit residues Val177, Tyr210, Phe214 and Tyr224.
HIV integrase strand transfer inhibitors: synthetic interfacial inhibitors.
As shown in TABLE 1, most of the interfacial inhibitors that were historically identified were natural products. The elucidation of the crystal structure of integrase inhibitors bound to the enzyme–DNA complex13,15,45 demonstrates that synthetic molecules can be successfully developed into archetypical interfacial inhibitors.
HIV integrase is the most recently validated target for the treatment of AIDS46. The FDA approved raltegravir (Isentress; Merck), an integrase inhibitor (FIG. 5), in 2007 based on the remarkable activity and good tolerance of the drug. Several second-generation integrase inhibitors are in development, including elvitegravir (GS-9137; Gilead Sciences) and dolutegravir (GSK-1349572; GlaxoSmithKline & Shionogi-ViiV), which are in Phase III clinical trials with the aim of overcoming the resistance to raltegravir and improving their pharmacokinetics profile45,47,48. HIV integrase is one of the three enzymes encoded by the HIV pol gene, together with pro-tease and reverse transcriptase (reviewed in REF. 49). HIV integrase inserts the viral genome into a host chromosome following reverse transcription. Integration is required for viral replication because only the integrated genome is efficiently transcribed and able to produce both viral RNA genome copies and the viral mRNA that generates the proteins for new virus particles.
Figure 5 |. Structure of the retroviral prototype foamy virus intasome in ternary complex with the strand transfer integrase inhibitor raltegravir.
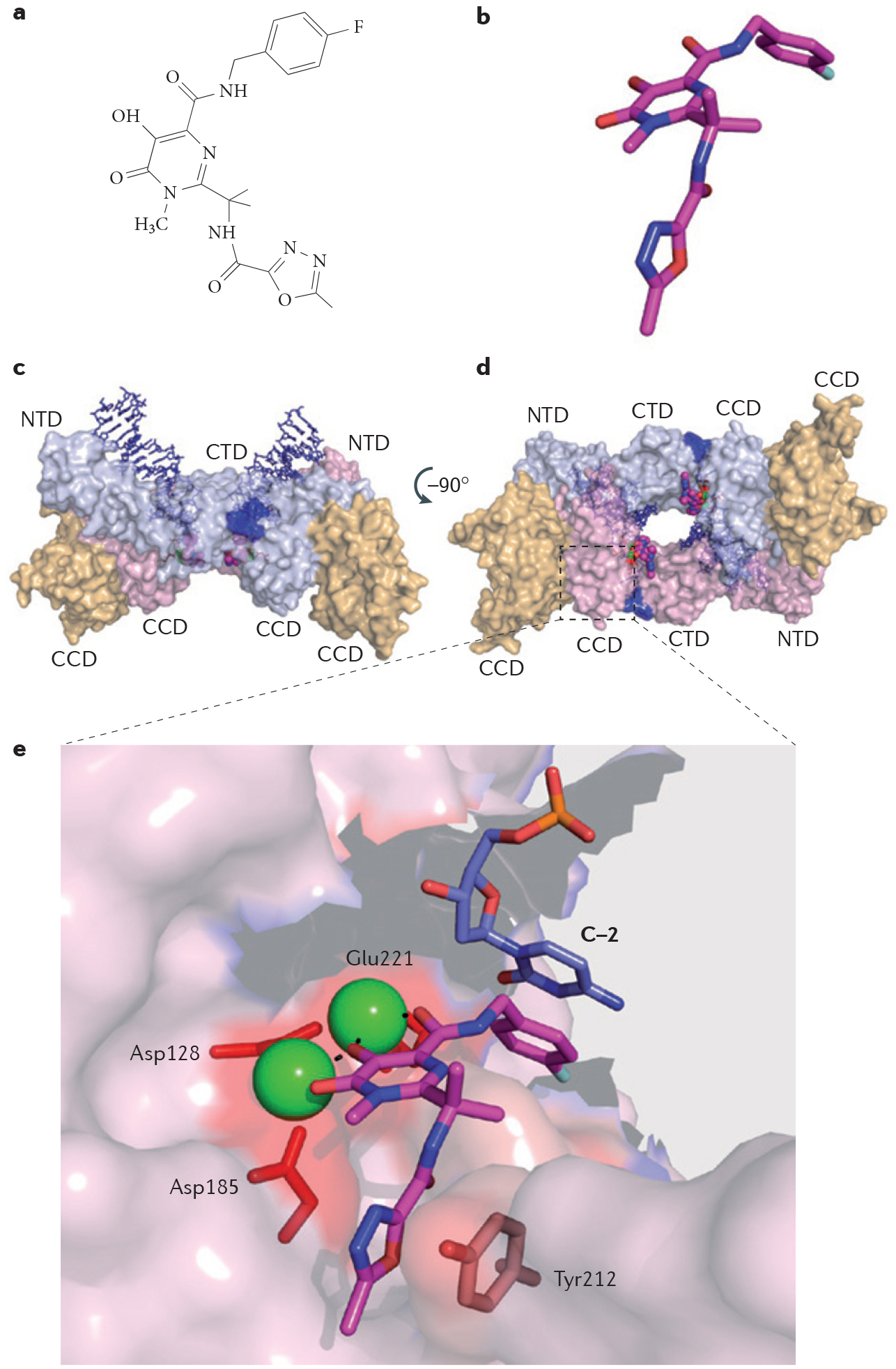
a | Chemical structure of raltegravir. b | Stick representation of raltegravir with carbon, nitrogen and oxygen atoms coloured in magenta, blue and red, respectively; Protein Data Bank (PDB) ID code 3OYA15,59. c | Surface representation of the retroviral intasome formed by a dimer of prototype foamy virus integrase, two viral DNA ends (shown in blue) and two molecules of raltegravir (shown in magenta). Each full-length monomer (shown in light blue and light pink) consists of three domains: the core catalytic domain (CCD), the carboxy-terminal domain (CTD) and the amino-terminal domain (NTD), and is associated in the crystal structure with a structural CCD (shown in yellow). d | Surface representation of the intasome rotated 90 around the horizontal axis to reveal the site of integration into the host DNA, occupied by two molecules of raltegravir. e | Interaction network between raltegravir, viral DNA (stick representation; shown in blue) and integrase residues (shown in red). Raltegravir forms three types of contacts: chelation of the catalytic Mg2+ ions (represented by green spheres) via its carbonyl groups (the three catalytic acidic residues, Asp128, Asp185 and Glu221, that chelate the Mg2+ ion on the other side are represented by red sticks); DNA binding via π-π interactions with the penultimate base of the cleaved viral DNA (C–2; shown in blue); and integrase binding by π-π interactions with the Tyr212 residue.
Integrase acts in two consecutive steps. First, it activates the viral DNA by generating 3′-deoxyribose hydroxyl ends as the enzyme catalyses the removal of a few (generally two) nucleotides from the 3′ends of the viral DNA immediately after the conserved CA dinucleotide sequences that are common to retroviruses. This first reaction, which is referred to as 3′ processing, takes place in the cytoplasm immediately after reverse transcription. The activated viral DNA is then packaged into pre-integration complexes with integrase bound to the viral ends. After 2–4 hours, once they have passed through the nuclear membrane, the pre-integration complexes reach a host chromosome. The second step of the integration reaction is referred to as strand transfer. It is an integrase-mediated electrophilic attack from the 3′-hydroxyl ends of the viral DNA towards the phosphodiester DNA backbones of a host chromosome. This results in the insertion of the viral DNA into the host chromosome49,50.
The integrase-mediated strand transfer reaction relies on the precise positioning of the phosphodiester backbone of the host DNA and the viral 3′ ends of the DNA (the attacking nucleophiles). The host DNA is held in place by two divalent metal cations (Mg2+ or Mn2+; FIG. 5) that also coordinate the three crucial catalytic residues of HIV integrase (Asp64, Asp116 and Glu152 for human HIV integrase) (FIG. 5). Crystal structures of raltegravir bound to the integrase from prototype foamy virus (which has an active site that is highly conserved with HIV integrase) showed that raltegravir binds at the interface of the integrase–DNA–Mg2+ complex13,15. FIGURE 5 summarizes the essential features of the interfacial binding of raltegravir. Raltegravir occupies the active site of integrase and inactivates integrase by blocking the binding of the host DNA as it displaces the terminal adenylyl nucleotide. Raltegravir is held in the active site of integrase by three key contacts: chelation of both Mg2+ ions by the drug carbonyl groups; π-stacking of the halophenyl ring of the drug with the penultimate cytosine base of the viral DNA; and π-stacking of the oxadiazole ring of the drug with the Tyr212 residue of prototype foamy virus integrase (Tyr143 in HIV integrase) (FIG. 5).
In spite of — and possibly owing to — its exclusive targeting of integrase–DNA complexes, raltegravir selects for drug-resistant viruses in some patients, who then become refractory to the drug47. The identification and characterization of the corresponding integrase mutations are being effectively used to generate second-generation integrase inhibitors to overcome such mutations45,47,48,51. Co-crystal structures of elvitegravir and dolutegravir with the prototype foamy virus intasome have recently revealed that both drugs also act as interfacial inhibitors15,45.
Definition of interfacial inhibition
Here, we summarize and further clarify the common properties of interfacial inhibitors based on the five examples described above and the widespread nature of the interfacial inhibition paradigm. It should be noted that targeting a protein interface does not always qualify for our definition of interfacial inhibitors. We also discuss interfacial inhibition in the context of other inhibition mechanisms, and define the common and differential elements between interfacial and allosteric inhibition in more detail.
Common and essential characteristics of interfacial inhibitors that differentiate them from drugs that target unbound (free) macromolecular interfaces.
BOX 1 summarizes the salient features of interfacial inhibitors, and TABLE 1 lists a number of interfacial inhibitors. One of the essential characteristics of interfacial inhibitors is their binding to a site generated at the interface of two (or more) biomolecules that are bound to each other and engaged in a functional complex. As illustrated by the examples in FIGS 1–5 and TABLE 1, these biomolecules can be polypeptides, nucleic acids (DNA or RNA) or even divalent metal cations. If the interface is generated by proteins (such as α-tubulin or β-tubulin; FIG. 4), the polypeptides have to be encoded by separate genes. However, if the interface is generated by different protein domains from a single protein encoded by one gene, we do not refer to this binding as interfacial. Although some biological interfaces could be generated by a small endogenous metabolite or mediator, such as a nucleotide or a hormone bound to a polypeptide, such examples have not yet come to our attention. Hence, it seems most appropriate to use the term ‘biomolecules’ to define the individual components that generate the binding interface.
Box 1 |. Characteristics of interfacial inhibitors.
The drugs target a macromolecular system or machine consisting of two or more components. These components can be proteins or nucleic acid–protein complexes.
The drugs bind at the interface generated by the binding of the macromolecules to each other (for example, protein–DNA interfaces or protein–protein interfaces).
The drug-binding ‘hotspot’ is generated by the movements of the macromolecular machine as it opens sites at the interface of the individual macromolecules (for example, cleavage of DNA or bending of the microtubule filament).
Only one drug stereoisomer tends to be active because of the tight fit between the drug and the interfacial site (most natural products follow this rule).
Interfacial inhibitors generally bind reversibly to the macromolecular interface by hydrogen bonds, π-stacking or metal chelation (see examples described in the article).
In addition to competitive and allosteric inhibition, interfacial inhibitors can act by selectively slowing down the targeted molecular machine (eventually bringing it to a grinding halt), which desynchronizes it from other components of the biological process (for example, slowed reversal of topoisomerase cleavage complexes leads to replication and transcriptional collisions).
It is important to clarify that our definition of an interfacial inhibitor excludes a drug that targets a macromolecular inter face by blocking the binding of another endogenous macromolecule (such as a protein or nucleic acid) to that same surface. For instance, the B cell lymphoma-extra large (BCL-XL) inhibitor ABT-737, which blocks the heterodimerization of the proapoptotic BCL-2-homologous antagonist/killer (BAK) or BCL-2 antagonist of cell death (BAD) to BCL-XL, does not qualify as an interfacial inhibitor; rather, it qualifies as a competitive inhibitor of BAK or BAD to BCL-XL. Similarly, nutlins block the binding of cellular tumour antigen p53 to the oncoprotein HDM2 (also known as MDM2) by binding to the p53-binding pocket of HDM2. However, they do not bind to the p53–HDM2 interface and therefore do not qualify as interfacial inhibitors but instead as competitive inhibitors (BOX 2) (reviewed in REF. 52).
Box 2 |. Drug inhibition mechanisms.
It is important to note that inhibitors can have more than one characteristic; for instance, they can be reversible, allosteric, non-competitive or allosteric interfacial.
We refer to ligands as the substrates or cofactors that are chemically modified by enzymes. Ligands also include molecules that induce signals as they bind to a biomolecule (for instance, hormones or growth factors in the case of receptors). Ligands bind to active sites or receptor sites.
Reversible inhibition can be overcome by increasing the ligand concentration or by dilution (for example, washout or diffusion after drug removal). Reversible inhibitors bind non-covalently via hydrogen bonds, van der Waals interactions, π-stacking, ionic bonds or chelation. Notably, some covalent bonds are reversible, such as Schiff bases or the binding of ecteinascidin 743 (also known as trabectedin) to DNA57.
Competitive inhibitors bind in a reversible way to the macromolecule active site by competing via steric hindrance for ligand binding; this is known as the mass action law. ATP-mimetic protein kinase inhibitors, for instance, are competitive inhibitors. This type of inhibition can be overcome at high ligand concentrations. For enzymatic reactions, the Vmax (velocity of the enzyme-catalysed reaction at infinite concentration of substrate) is not altered by competitive inhibitors but the Km (the Michaelis constant) is increased. Competitive inhibitors are generally isosteric (see below). They can also be allosteric if they hinder a proximal ligand binding site.
Non-competitive inhibitors are also reversible inhibitors. They do not bind to or alter the active sites and their binding is unaffected by ligand binding. They reduce biomolecule activity. Vmax decreases as the drug concentration increases. Km remains unchanged, as non-competitive inhibitors do not affect substrate binding (for example, nifedipine inhibits cytochrome P450 2C9 via a non-competitive inhibitory mechanism).
Uncompetitive inhibitors bind in a reversible way but alter the active site (for instance, by binding to the ligand–active site complex). This type of inhibition causes the Vmax to decrease and the Km to increase.
Mixed inhibitors bind reversibly to both the ligand-bound and ligand-free biomolecules (enzymes or receptors) but their affinities (the inhibition constant Ki) for the ligand-bound and ligand-free forms are different. They interfere with ligand binding (by increasing Km) and hamper catalysis (by decreasing Vmax).
Irreversible inhibitors usually covalently modify the biomolecule. However, in some cases they bind non-covalently with extremely slow off-rates (for example, raltegravir). Their effect cannot be overcome by increasing ligand concentration or by dilution (washout or drug removal); it can only be overcome by the generation of intact biomolecules (enzymes or receptors).
Allosteric inhibitors bind to a site distinct from the active site on an intrinsically flexible macromolecule target. Their long-range effect on the active site influences the binding of the natural ligand (by reducing the Vmax and increasing the Km). The two models of allosteric inhibition are: the concerted two-state model put forth by Monod, Wyman and Changeux (also referred to as the MWC model)56; and the sequential model described by Khoshland, Nemethy and Filmer58. The latter model postulates that the binding of an allosteric inhibitor induces a conformational allosteric transition in the macromolecular complex, whereas the MWC model assumes that the inhibitor binds to a relaxed conformation of the complex that is present before drug binding.
Orthosteric inhibitors bind to the biomolecule target (an enzyme or a receptor) on the active site where endogenous ligands would normally bind.
Interfacial inhibitors (BOX 1) can act as allosteric inhibitors (for example, curare alkaloids or nicotine for the nicotinic acetylcholine receptor) or as orthosteric inhibitors (for example, raltegravir can be viewed as an orthosteric inhibitor as it binds to the active site of HIV integrase, formed by the HIV integrase polypeptide, the viral end of the DNA and the divalent metal cofactor).
Historically, interfacial inhibitors were discovered before their target (or targets) had been identified. These first interfacial inhibitors were generally natural products (TABLE 1), which led us to propose in our first description of the interfacial inhibitor concept that interfacial inhibitors represented “one of Nature’s paradigms” for generating inhibitors or toxins8,9. It is important to keep this principle in mind when dealing with natural products and looking for their molecular targets. This is even more relevant if the natural product contains racemic centres and if the natural isomer is markedly more active than the synthetic stereoisomers. As indicated above, this is the case with camptothecin; only the natural 20S isomer, and not the synthetic 20R isomer, targets TOP1.
In recent years, several examples of synthetic interfacial inhibitors have emerged (TABLE 1). For instance, the indenoisoquinoline drugs targeting TOP1 were developed based on the principles established by studying the camptothecins7. Raltegravir was discovered as a specific HIV integrase inhibitor by searching chemical libraries of compounds with antiviral activity. These two examples of synthetic inhibitors demonstrate the feasibility of synthetic drug development programmes for the discovery of interfacial inhibitors.
Comparison of interfacial and allosteric inhibitors.
Allosteric inhibitors generally act as interfacial inhibitors, as in many cases they bind at the interface of biomolecules. A classical example of an allosteric interfacial inhibitor is the nicotine molecule, which binds to the neuronal acetylcholine receptor53. However, some allosteric inhibitors bind to a single polypeptide with flexible domains, such as allosteric inhibitors of the cyclic AMP-specific 3′,5′-cyclic phosphodiesterase 4D, which bind and stabilize an inactive form of the enzyme with its regulatory domain closed across the active site54. In fact, in the case of allosteric inhibitors that bind and distort a single polypeptide or a single nucleic acid effector, the distinction between allosteric inhibitors and interfacial inhibitors is that allosteric inhibitors alter the movement or the arrangement of flexible protein domains that are encoded by a single gene, whereas in the case of allosteric interfacial inhibitors the domains are represented by separately encoded and non-covalently linked polypeptides.
Allostery has become a prominent biological and pharmacological topic55; however, in some contexts its definition has evolved. For instance, allosteric effects are commonly used to refer to propagated effects that produce conformational changes within a biomolecule. This definition is different from the original one proposed by Changeux, Jacob and Monod55,56 (BOX 2) and cannot be readily related to interfacial interactions.
Concluding remarks and prospects
Targeting complex macromolecular systems is becoming mainstream in drug discovery. As protein surfaces tend to be relatively shallow, it is thermodynamically difficult to identify or synthesize small molecules that compete with large endogenous ligands. Targeting multicomponent molecular machines using interfacial inhibitors that bind at their interfacial hinges overcomes this difficulty because these inhibitors are thermodynamically more efficient and likely to be more selective than competitive inhibitors, which need to cover a large protein–protein or protein–nucleic acid interface. Interfacial inhibitors take advantage of two intrinsic characteristics of macromolecular machines. First, the binding of different molecules tends to generate deep molecular clefts (hinges) at their junction, which can be targeted by small interfacial inhibitors. Second, the macromolecular machines need to be flexible; they must undergo a range of movements around intermolecular hinges to create transient clefts that can be stabilized by the binding of interfacial inhibitors. The ‘jamming’ of the molecular machines, even if reversible, tends to be highly toxic because of the necessary orderly timing and concerted actions of biological systems.
Interestingly, interfacial inhibitors represent a large fraction of the drug market (TABLE 1). Quinolones represent one of the largest fraction (17%) of the antibiotics market, with annual sales above US$7 billion. In the anti-HIV market, the recently approved integrase inhibitor raltegravir is now routinely used both as a first-line therapy and in patients who fail to benefit from the highly active antiretroviral therapy regimen. The market shares for raltegravir amounted to over $1 billion in 2010 and are expected to increase in 2011. The outstanding activity and good tolerance of the two other integrase inhibitors in late-stage clinical trials, elvitegravir and dolutegravir, makes it likely that they will be approved in 2012. In addition, some non-nucleoside reverse transcriptase inhibitors may act not only as allosteric inhibitors but also as interfacial inhibitors. In the anticancer drug market, both the camptothecin and non-camptothecin drugs targeting TOP1 act as interfacial inhibitors, similarly to the anticancer drugs targeting TOP2, tubulin and target of rapamycin (TOR). Drugs that are clinically approved for treating human diseases are listed in TABLE 1.
The objective of this article was to define and elucidate the concept of interfacial inhibition by providing examples and defining its identity with respect to other modes of inhibition. As described above, many allosteric inhibitors are interfacial inhibitors, but the converse is not always true.
There are several obvious implications of the interfacial inhibitor paradigm for drug discovery. First, the assays for discovering interfacial inhibitors need to take into account the stabilization of macromolecular complexes rather than only testing for inhibition of macromolecule binding, which — to our knowledge — is not common practice in drug discovery and high-throughput screening. Second, most interfacial inhibitors act as non-competitive or uncompetitive inhibitors, which emphasizes the importance of developing screening tests that go beyond competitive inhibition. Third, molecular modelling of interfacial inhibitors can be demanding. It requires generating structures of large complexes including at least three components, two or more biomolecules and the drug. Moreover, the dynamic nature of the biological interfaces generated by the movement of the targeted molecular machine can be difficult to model. In any case, we are hopeful that in the near future new drugs will be generated based on interfacial and allosteric principles. Such approaches are logical in the context of complex biological systems that need to utilize large-sized molecular machines with multiple components that move at a high speed relative to each other. Cell surface receptors, G protein-coupled receptors, tyrosine kinases, hormone receptors and transcription factors are all obvious candidates for allosteric and interfacial inhibition.
Acknowledgements
Our studies are supported by the Center for Cancer Research, the Intramural Program of the National Cancer Institute, US National Institutes of Health.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Conti C et al. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol. Biol. Cell 18, 3059–3067 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sugino A, Peebles CL, Kreuzer KN & Cozzarelli NR Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc. Natl Acad. Sci. USA 74, 4767–4771 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gellert M, Mizuuchi K, O’Dea MH, Itoh T & Tomizawa JI Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc. Natl Acad. Sci. USA 74, 4772–4776 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier Y, Leo E, Zhang H & Marchand C DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol 17, 421–433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staker BL et al. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl Acad. Sci. USA 99, 15387–15392 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staker BL et al. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem 48, 2336–2345 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Marchand C et al. A novel norindenoisoquinoline structure reveals a common interfacial inhibitor paradigm for ternary trapping of topoisomerase I-DNA covalent complexes. Mol. Cancer Ther 5, 287–295 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pommier Y & Cherfils J Interfacial protein inhibition: a nature’s paradigm for drug discovery. Trends Pharmacol. Sci 28, 136–145 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Pommier Y & Marchand C Interfacial inhibitors of protein-nucleic acid interactions. Curr. Med. Chem. Anti‑Canc. Agents 5, 421–429 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Laponogov I et al. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nature Struct. Mol. Biol 16, 667–669 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Laponogov I et al. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS ONE 5, 8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bax BD et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Hare S et al. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl Acad. Sci. USA 107, 20057–20062 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan L et al. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc. Natl Acad. Sci. USA 107, 15910–15915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hare S, Gupta S S., Valkov, E., Engelman, A. & Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464, 232–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu CC et al. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333, 459–462 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Forterre P in DNA Topoisomerases and Cancer (ed. Pommier Y) 1–52 (Springer, New York, 2012). [Google Scholar]
- 18.Chrencik JE et al. Mechanisms of camptothecin resistance by human topoisomerase I mutations. J. Mol. Biol 339, 773–784 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Koster DA, Palle K, Bot ESM, Bjornsti MA & Dekker NH Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 448, 213–217 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Pommier Y, Pourquier P, Urasaki Y, Wu J & Laco G Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist. Updat 2, 307–318 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Jaxel C, Kohn KW, Wani MC, Wall ME & Pommier Y Structure–activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: evidence for a specific receptor site and a relation to antitumor activity. Cancer Res. 49, 1465–1469 (1989). [PubMed] [Google Scholar]
- 22.Eng WK, Faucette L, Johnson RK & Sternglanz R Evidence that DNA topoisomerase I is necessary for the cytotoxic effects of camptothecin. Mol. Pharmacol 34, 755–760 (1988). [PubMed] [Google Scholar]
- 23.Nitiss J & Wang JC DNA topoisomerase-targeting antitumor drugs can be studied in yeast. Proc. Natl Acad. Sci. USA 85, 7501–7505 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sirikantaramas S, Yamazaki M & Saito K Mutations in topoisomerase I as a self-resistance mechanism coevolved with the production of the anticancer alkaloid camptothecin in plants. Proc. Natl Acad. Sci. USA 105, 6782–6786 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujimori A, Harker WG, Kohlhagen G, Hoki Y & Pommier Y Mutation at the catalytic site of topoisomerase I in CEM/C2, a human leukemia cell resistant to camptothecin. Cancer Res. 55, 1339–1346 (1995). [PubMed] [Google Scholar]
- 26.Pommier Y Topoisomerase I inhibitors: camptothecins and beyond. Nature Rev. Cancer 6, 789–802 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Pommier Y et al. Repair of topoisomerase I-mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol 81, 179–229 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capranico G, Kohn KW & Pommier Y Local sequence requirements for DNA cleavage by mammalian topoisomerase II in the presence of doxorubicin. Nucleic Acids Res. 18, 6611–6619 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pommier Y, Capranico G, Orr A & Kohn KW Local base sequence preferences for DNA cleavage by mammalian topoisomerase II in the presence of amsacrine or teniposide. Nucleic Acids Res. 19, 5973–5980 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen GL et al. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem 259, 13560–13566 (1984). [PubMed] [Google Scholar]
- 31.Tewey KM, Chen GL, Nelson EM & Liu LF Intercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem 259, 9182–9187 (1984). [PubMed] [Google Scholar]
- 32.Pommier Y, Capranico G, Orr A & Kohn KW Distribution of topoisomerase II cleavage sites in SV40 DNA and the effects of drugs. J. Mol. Biol 222, 909–924 (1991). [DOI] [PubMed] [Google Scholar]
- 33.Capranico G, Zunino F, Kohn KW & Pommier Y Sequence-selective topoisomerase II inhibition by anthracycline derivatives in SV40 DNA: relationship with DNA binding affinity and cytotoxicity. Biochemistry 29, 562–569 (1990). [DOI] [PubMed] [Google Scholar]
- 34.Capranico G, De Isabella P, Tinelli S, Bigioni S & Zunino F Similar sequence specificity of mitoxantrone and VM-26 stimulation of in vitro DNA cleavage by mammalian DNA topoisomerase II. Biochemistry 32, 3032–3048 (1993). [DOI] [PubMed] [Google Scholar]
- 35.Heddle JG, Barnard FM, Wentzell LM & Maxwell A The interaction of drugs with DNA gyrase: a model for the molecular basis of quinolone action. Nucleosides Nucleotides Nucleic Acids 19, 1249–1264 (2000). [DOI] [PubMed] [Google Scholar]
- 36.De Jonge N et al. Rejuvenation of CcdB-poisoned gyrase by an intrinsically disordered protein domain. Mol. Cell 35, 154–163 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Williams JJ & Hergenrother PJ Exposing plasmids as the Achilles’ heel of drug-resistant bacteria. Curr. Opin. Chem. Biol 12, 389–399 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dao-Thi MH et al. Molecular basis of gyrase poisoning by the addiction toxin CcdB. J. Mol. Biol 348, 1091–1102 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Ishida R et al. Inhibition of DNA topoisomerase II by ICRF-193 induces polyploidization by uncoupling chromosome dynamics from other cell cycle events. J. Cell Biol 126, 1341–1351 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andoh T & Ishida R Catalytic inhibitors of DNA topoisomerase II. Biochim. Biophys. Acta 1400, 155–171 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Classen S, Olland S & Berger JM Structure of the topoisomerase II ATPase region and its mechanism of inhibition by the chemotherapeutic agent ICRF-187. Proc. Natl Acad. Sci. USA 100, 10629–10634 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dumontet C & Jordan MA Microtubule-binding agents: a dynamic field of cancer therapeutics. Nature Rev. Drug Discov 9, 790–803 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jordan MA & Wilson L Microtubules as a target for anticancer drugs. Nature Rev. Cancer 4, 253–265 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Gigant B et al. Structural basis for the regulation of tubulin by vinblastine. Nature 435, 519–522 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Hare S et al. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol 80, 565–572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchand C, Maddali K, Metifiot M & Pommier Y HIV-1 IN inhibitors: 2010 update and perspectives. Curr. Top. Med. Chem 9, 1016–1037 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Metifiot M, Marchand C, Maddali K & Pommier Y Resistance to integrase inhibitors. Viruses 2, 1347–1366 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Metifiot M et al. Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS 25, 1175–1178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pommier Y, Johnson A & Marchand C Integrase inhibitors to treat HIV/AIDS. Nature Rev. Drug Discov 4, 236–248 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Li X, Krishnan L, Cherepanov P & Engelman A Structural biology of retroviral DNA integration. Virology 411, 194–205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao XZ et al. Development of tricyclic hydroxy-1H-pyrrolopyridine-trione containing HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett 21, 2986–2990 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wells JA & McClendon CL Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 450, 1001–1009 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Changeux JP & Taly A Nicotinic receptors, allosteric proteins and medicine. Trends Mol. Med 14, 93–102 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Burgin AB et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nature Biotech. 28, 63–70 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Changeux J-P 50th anniversary of the word “allosteric”. Protein Science 20, 1119–1124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monod J, Changeux JP & Jacob F Allosteric proteins and cellular control systems. J. Mol. Biol 6, 306–329 (1963). [DOI] [PubMed] [Google Scholar]
- 57.Pommier Y et al. DNA sequence- and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the carribean tunicate Ecteinascidia Turbinata. Biochemistry 35, 13303–13309 (1996). [DOI] [PubMed] [Google Scholar]
- 58.Koshland DE Jr, Nemethy G & Filmer D Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385 (1966). [DOI] [PubMed] [Google Scholar]
- 59.Maertens GN, Hare S & Cherepanov P The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 468, 326–329 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brueckner F & Cramer P Structural basis of transcription inhibition by α-amanitin and implications for RNA polymerase II translocation. Nature Struct. Mol. Biol 15, 811–818 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Bushnell DA, Cramer P & Kornberg RD Structural basis of transcription: α-amanitin–RNA polymerase II cocrystal at 2.8 Å resolution. Proc. Natl Acad. Sci. USA 99, 1218–1222 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hibbs RE et al. Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal α7 nicotinic acetylcholine receptor. EMBO J. 28, 3040–3051 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nury H et al. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Sigel E & Luscher BP A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr. Top. Med. Chem 11, 241–246 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Renault L, Guibert B & Cherfils J Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 426, 525–530 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Mossessova E, Corpina RA & Goldberg J Crystal structure of ARF1• Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 12, 1403–1411 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Ravelli RBG et al. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 428, 198–202 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Jin L & Harrison SC Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc. Natl Acad. Sci. USA 99, 13522–13526 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huai Q et al. Crystal structure of calcineurin–cyclophilin–cyclosporin shows common but distinct recognition of immunophilin–drug complexes. Proc. Natl Acad. Sci. USA 99, 12037–12042 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roca J, Ishida R, Berger JM, Andoh T & Wang JC Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc. Natl Acad. Sci. USA 91, 1781–1785 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takebayashi Y et al. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nature Med. 7, 961–966 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Nettles JH et al. The binding mode of epothilone A on α,β-tubulin by electron crystallography. Science 305, 866–869 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Kissinger CR et al. Crystal structures of human calcineurin and the human FKBP12–FK506–calcineurin complex. Nature 378, 641–644 (1995). [DOI] [PubMed] [Google Scholar]
- 74.Griffith JP et al. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 82, 507–522 (1995). [DOI] [PubMed] [Google Scholar]
- 75.Tesmer JJ, Sunahara RK, Gilman AG & Sprang SR Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα•GTPγS. Science 278, 1907–1916 (1997). [DOI] [PubMed] [Google Scholar]
- 76.Wurtele M, Jelich-Ottmann C, Wittinghofer A & Oecking C Structural view of a fungal toxin acting on a 14-3-3 regulatory complex. EMBO J. 22, 987–994 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Agrawal RK & Frank J Structural studies of the translational apparatus. Curr. Opin. Struct. Biol 9, 215–221 (1999). [DOI] [PubMed] [Google Scholar]
- 78.Laurberg M et al. Structure of a mutant EF-G reveals domain III and possibly the fusidic acid binding site. J. Mol. Biol 303, 593–603 (2000). [DOI] [PubMed] [Google Scholar]
- 79.Ioanoviciu A et al. Synthesis and mechanism of action studies of a series of norindenoisoquinoline topoi somerase I poisons reveal an inhibitor with a flipped orientation in the ternary DNA–enzyme–inhibitor complex as determined by X-ray crystallo graphic analysis. J. Med. Chem 48, 4803–4814 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Choi J, Chen J, Schreiber SL & Clardy J Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239–242 (1996). [DOI] [PubMed] [Google Scholar]
- 81.Moellering RE et al. Direct inhibition of the NOTCH transcription factor complex. Nature 462, 182–188 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edwards MJ et al. A crystal structure of the bifunctional antibiotic simocyclinone D8, bound to DNA gyrase. Science 326, 1415–1418 (2009). [DOI] [PubMed] [Google Scholar]
- 83.Snyder JP, Nettles JH, Cornett B, Downing KH & Nogales E The binding conformation of taxol in β-tubulin: a model based on electron crystallographic density. Proc. Natl Acad. Sci. USA 98, 5312–5316 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bowen WS, Van Dyke N, Murgola EJ, Lodmell JS & Hill WE Interaction of thiostrepton and elongation factor-G with the ribosomal protein L11-binding domain. J. Biol. Chem 280, 2934–2943 (2004). [DOI] [PubMed] [Google Scholar]