

Abstract
The assembly of the transcription machinery is a key step in gene activation, but even basic details of this process remain unclear. Here we discuss the apparent discrepancy between the classic sequential assembly model based mostly on biochemistry and an emerging dynamic assembly model based mostly on fluorescence microscopy. The former model favors a stable transcription complex with subunits that cooperatively assemble in order, whereas the latter model favors an unstable complex with subunits that may assemble more randomly. To confront this apparent discrepancy, we review the merits and drawbacks of the different experimental approaches and list potential biasing factors that could be responsible for the different interpretations of assembly. We then discuss how these biases might be overcome in the future with improved experiments or new techniques. Finally, we discuss how kinetic models for assembly may help resolve the ordered and stable vs. random and dynamic assembly debate.
Introduction
The transcription machinery performs one of the cell’s most fundamental tasks: transcribing DNA into RNA. To do this, it must self-assemble at specific chromatin sites on demand. This requires the coordinated movement of many large subunits, including RNA polymerase, the basal transcription factors, as well as necessary cofactors and chromatin remodelers. How assembly is regulated in the nucleus remains a key question for the postgenomic era. Although we now have a catalog of the main subunits (Roeder 1996; Conaway and Conaway 1993) and highly resolved structures depicting them fully assembled and even in action (Kornberg 2007; Hahn 2004), it remains a great mystery how they were put together in the first place.
Traditionally the assembly of the transcription machinery has been thought of as an ordered process guided by cooperative interactions. The elucidation of this process has taken many researchers many years (Agalioti et al. 2000; Buratowski 1994; Lemon and Tjian 2000). In the classic biochemical model, subunits of the transcription machinery are thought to arrive in sequence to form a stable, functional end product. This straightforward picture is complicated, however, by the fact that assembly occurs on a dynamic chromatin template, requires a large number of cofactors, and is subject to regulation (D’Alessio et al. 2009; Sikorski and Buratowski 2009). Deconstructing the assembly of the transcription machinery is thus a formidable task.
Recently, a new model for the assembly of the transcription machinery has taken root that challenges the classic notion of stable, sequential assembly. In the new model, the transcription machinery is thought to be far more dynamic than originally anticipated, with components that rapidly assemble, perhaps repeatedly throughout a single transcription cycle and perhaps not always in a predefined order (Hager et al. 2009; Darzacq et al. 2009). Support for this new model has come mainly from fluorescence microscopy experiments in single live cells, but some more traditional biochemical studies have also indicated a more dynamic transcription complex. Reconciling this highly dynamic model with the classic sequential assembly paradigm—where one might expect larger complexes to become ever more stable—is a problem that demands closer attention.
In this review we focus on this problem by reevaluating the new fluorescence microscopy experiments that have popularized the dynamic picture of assembly of the transcription machinery. Although these experiments have the advantage of being performed within the natural cellular environment, they suffer from several key limitations that could potentially bias results towards a more dynamic picture. We will discuss these biasing factors in detail and contrast them with limitations of some biochemical assays that may have biased results towards a more static picture. We conclude by discussing how these biases might be overcome in the future with improved experiments, new techniques, and better mathematical modeling.
Sequential assembly of a stable complex: the classic biochemical paradigm
Before 1980 little was known about how the transcription machinery assembles. The pivotal advance came when a transcriptionally active form of RNA polymerase II was first reconstituted in vitro (Manley et al. 1980; Weil et al. 1979). This groundbreaking work enabled researchers to begin dissecting the assembly of the transcription machinery in a controlled environment (Buratowski 1994, 2000; Lemon and Tjian 2000).
With a reliable in vitro transcription system in place, various subunits of the transcription machinery were purified and subsequent addition and subtraction experiments ultimately led to a list of the necessary and sufficient components. From this minimal set, a sequential and cooperative map for the assembly of a stable preinitiation complex (PIC) emerged (Buratowski 1994; Cook 2001; Lemon and Tjian 2000; Roeder 1996; Conaway and Conaway 1993).
The minimal set includes RNA polymerase II—which itself is made up of 12 protein subunits—together with six basal (Sikorski and Buratowski 2009) transcription factors, TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH (all but TFIIB are multisubunit complexes). As this minimal complex cannot transcribe chromatin, chromatin remodelers and modifiers may also be included in the minimal set. The basic picture is as follows: chromatin remodelers initiate large-scale decondensation of chromatin, followed by secondary remodeling to locally rearrange nucleosomes, and expose the promoter (Lemon and Tjian 2000). Once exposed, TFIID—which includes the TATA-binding protein TBP—can be recruited, perhaps with the help of TFIIA, followed by cooperative recruitment of TFIIB, TFIIF/RNA polymerase II (preassembled), and then TFIIE and TFIIH (Buratowski 2000; Cook 2001; Roeder 1996).
Once the PIC is assembled, it remains stable for minutes or even hours in vitro (see Table 1) and may persist through multiple transcription cycles. For example, studies using yeast and human nuclear extracts suggest RNA polymerase remains associated with TFIID (and perhaps a few other basal transcription factors) as a scaffold for facilitated reinitiation of multiple rounds of transcription (Yudkovsky et al. 2000; Zawel et al. 1995).
Table 1.
Comparing biochemical measurements of transcription machinery binding times with live-cell microscopy measurements
| Complex | Estimated binding times (min) | |||
|---|---|---|---|---|
| Biochemistry | Live cell microscopy | |||
| TFIIA:DNA | 1–21 | [toff, TIRFM-array, yeast] (Bonham et al, 2009) | ||
| TFIIB:DNA | 4–260 | [toff TIRFM-array, yeast] (Bonham et al, 2009) | < 0.02 | [t1/2, FRAP, yeast] (Sprouse et al, 2008) |
| TBP:DNA | 65–100 | [t1/2, TATA box, band shift, yeast] (Hoopes et al, 1992) | < 0.02 | [t1/2, FRAP, yeast] (Sprouse et al, 2008) |
| 7–327 | [toff TIRFM-array, yeast] (Bonham et al, 2009) | ~1 | [t1/2, FRAP, human] (Chen et al, 2002) | |
| ~37 / ~63 / ~80 | [ton+off, Pol II / III / I DNA, ChIP, yeast] (van Werven et al, 2009) | 1.7 – 2.8 | [toff, FRAP, human] (de Graaf et al, 2010) | |
| NF-KB:DNA | 3–7 | [toff, SPM, human] (Linnell et al, 2004) | 0.02–0.03 | [t1/2, FRAP, mouse] (Sung et al, 2009) |
| < 0.3 | [toff, p65, FLIP, human] (Bosisio et al, 2006) | |||
| GR:DNA | ~1.5 / ~77 | [toff monomer / dimer, band shift, rat] (Lieberman & Nordeen, 1997) | 0.01 – 0.03 | [toff, FRAP/FCS, mouse] (Stasevich et al, 2010) |
| ~ 151 | [toff, DNAse footprint, rat] (Perlmann et al, 1990) | 0.05 | [toff, FRAP, mouse] (Mueller et al, 2008) | |
| SWI/SNF : chromatin | > 30 | [toff, comp. exp., yeast] (Hassan et al, 2002) | 0.17 | [t1/2, FRAP, mouse] (Johnson et al, 2008) |
| Gal4:DNA | ~ 250 | [toff, SPM, yeast] (Shumaker-Parry et al, 2004) | ||
| ~ 15 | [ton+off, ChIP, yeast] (Nalley et al, 2009) | |||
| TFIIE:Pol II | ~ 4.8 | [toff, SPM, yeast] (Bushnell et al, 1996) | ||
| TFIIB:Pol II | ~ 44 | [toff, SPM, yeast] (Bushnell et al, 1996) | ||
| TFIIF:Pol II | ~ 3.7 | [toff, SPM, yeast] (Bushnell et al, 1996) | ||
The colored columns provide study details, including the binding measurement variable (toff = binding residence time, ton+off = binding turnover time, t1/2=half time for competitive exchange, FRAP recovery, or FLIP loss), details of the binding interaction (only listed if specific rather than mixed or nonspecific), the binding assay, the origin of the complex examined, and the relevant citation
More recently, in vivo cross-linking experiments (ChIP) combined with microarray or deep sequencing technology have largely confirmed the sequential assembly of the PIC, although the number of PIC subunits is ever expanding (Sikorski and Buratowski 2009). This growing list reflects the wide range of promoter signatures. While TBP recognizes the TATA box found in many promoters, many other promoters lack this element and instead rely on other elements and/or their combinations (Juven-Gershon et al. 2008; Sikorski and Buratowski 2009). The question naturally arises: how can a single polymerase identify all these different signatures? The answer seems to be through a host of coregulators and coactivators which can attach to and modify the polymerase DNA-binding interface (Sikorski and Buratowski 2009; D’Alessio et al. 2009; Muller and Tora 2004).
Besides the transcription machinery, biochemical studies of the assembly of many other important biological complexes have generally led to similar models: sequential addition of subunits through cooperative interactions that ultimately result in the formation of stable complexes (see Fig. 1a). The assembly of the ribosome—described by the Nomura assembly map—is the prototypical example (Mizushima and Nomura 1970), but many other important biological complexes appear to assemble sequentially into stable structures in the test tube. Notable examples related to the transcription machinery are the DNA replication (Evrin et al. 2009; Seki and Diffley 2000) and repair machineries (Mu et al. 1996).
Fig. 1.
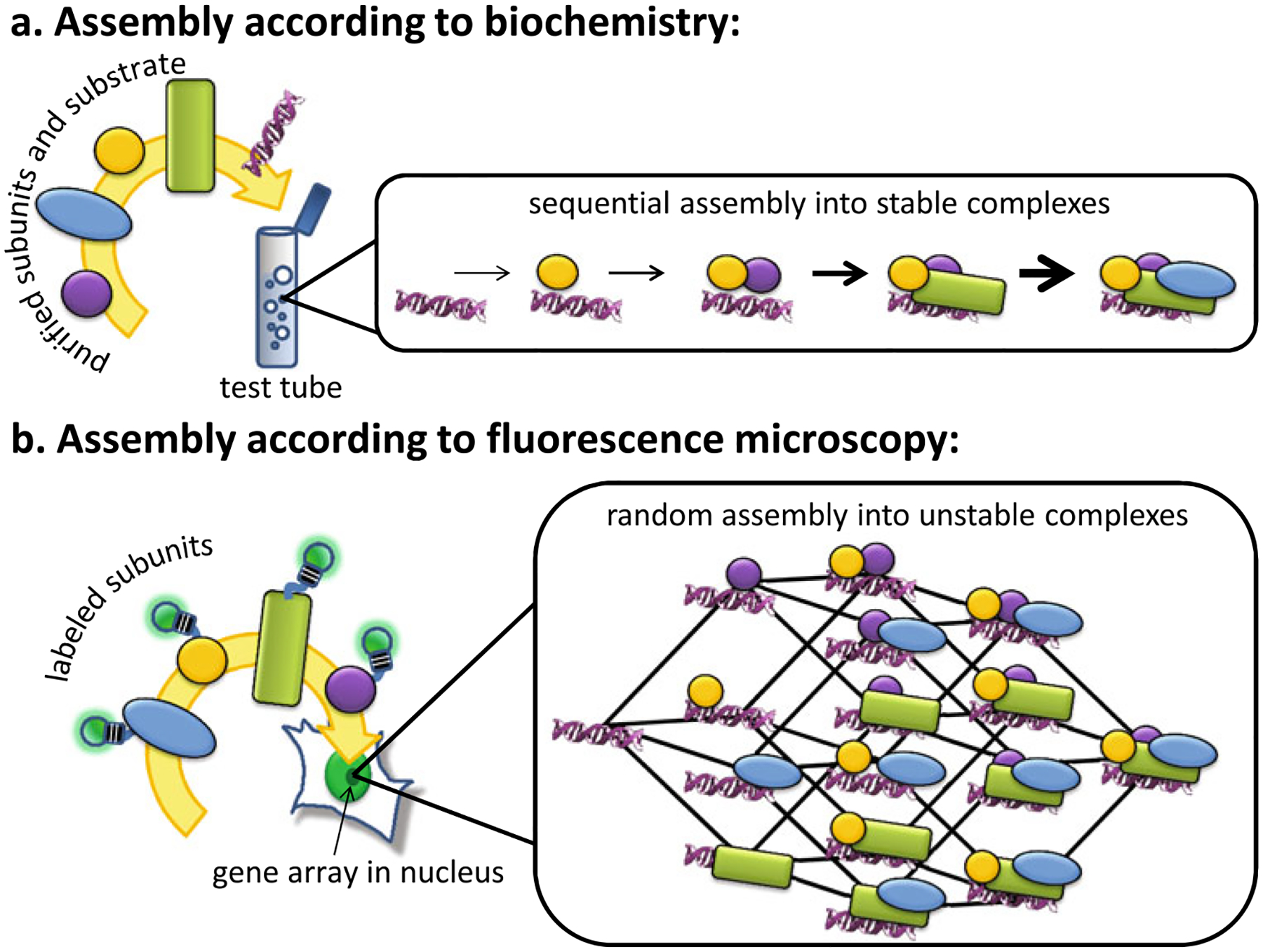
Two models for assembly: a According to biochemistry, macromolecular complexes such as the transcription machinery are stable and assemble in a sequential fashion. b According to live-cell microscopy, these complexes are unstable and assemble in a more random fashion using multiple assembly pathways. The glowing light bulb shape represents a fluorescent label such as GFP. The helix shape represents the DNA-binding substrate
While most biochemical studies are consistent with sequential and stable assembly models, a subset of these studies on the transcription machinery have provided evidence for more dynamic assembly, at least in some species and at some promoters. In particular, a natural TATA-less mouse promoter (DHFR) does not exhibit facilitated reinitiation (Yean and Gralla 1997), and the RNA polymerase II complex from Drosophila embryo extracts seems to fully assemble and disassemble after each round of transcription (Kadonaga 1990). Precisely when and how disassembly occurs is therefore debatable. In theory, cooperative, sequential binding of subunits should result in a “first-on/last-off” assembly/disassembly model, but this may not be universally true; TFIIIC, for example, binds early on, but is apparently displaced once RNA polymerase III binds (Roberts et al. 2003). Biochemical and genetic evidence from several different systems also indicates that some subunits of the transcription machinery can be evicted, replaced, and/or disassembled by cellular regulatory factors such as the proteasome (Collins and Tansey 2006; Daulny et al. 2008), chromatin remodelers (Nagaich et al. 2004), and molecular chaperones (Freeman and Yamamoto 2002). Thus, some number of biochemical studies have suggested that the transcription machinery is more dynamic than the classic sequential assembly model would predict.
Random assembly of a dynamic complex: the fluorescence microscopy paradigm
The notion of a highly dynamic transcription complex has been strongly reinforced by live-cell fluorescence microscopy experiments. Until recently it was extremely difficult to measure protein movement in live cells, much less dissect the assembly of macromolecular complexes. Fortunately this all changed with the green fluorescent protein (GFP) revolution and the availability of high-end commercial fluorescence microscopes. Now such measurements are fairly common. Central to this development has been the use of fluorescence recovery after photobleaching (FRAP). In these experiments, a region of a cell expressing a fluorescently labeled protein is photobleached via a strong and brief laser pulse. The recovery of fluorescence within the photobleached region is then quantified as a function of time. From these measurements it is possible to determine the mobility of proteins in their native environment, in particular their diffusion coefficients and binding times (Mueller et al. 2010).
Early on it became clear that the results of these new experiments did not agree with the classic paradigm for assembly. For example, if the transcription machinery is stable, then each subunit should have a fairly slow FRAP recovery lasting for minutes or more. In contrast, most transcription factors exhibit rapid FRAP recoveries lasting just a few seconds in live cells (Hager et al. 2009). Likewise, depending on the cell and/or tissue type, key components of the PIC, such as TFIIB (Chen et al. 2002), TFIIH (Hoogstraten et al. 2002), TFIID TAFs (de Graaf et al. 2010), and TBP (Sprouse et al. 2008), recover in a matter of seconds, as can certain other preinitiation factors (Dundr et al. 2002). Even the core subunits of the polymerase display biphasic FRAP recoveries (Kimura et al. 2002; Dundr et al. 2002; Darzacq et al. 2007; Yao et al. 2007), indicating a slow binding state attributed to elongation/pausing as well as a fast state that is regulated throughout the cell cycle (Gorski et al. 2008) and attributed to initiation binding kinetics (Kimura et al. 2002; Dundr et al. 2002; Darzacq et al. 2007). Considering the now large body of FRAP studies of transcription associated molecules, rapid binding appears common, even though most factors were predicted to be slow (see Table 1).
In spite of these rapid dynamics, the new data could still be consistent with the biochemical notion of a stable preformed complex or “holoenzyme” (Kimura et al. 1999) containing RNA polymerase and other factors that transiently bind chromatin as a whole. In this case, subunits of the preformed complex should at least exhibit the same FRAP recovery rates. In contrast to this simple model, however, different subunits appear to recover with different kinetics. For example, in mammalian cells, TFIIH (Hoogstraten et al. 2002) and TFIIB (Chen et al. 2002) recover much more quickly than the core subunits of RNA polymerase II (Kimura et al. 2002), and even the core subunits of RNA polymerase I in humans (Dundr et al. 2002) and RNA polymerase II in yeast (Sprouse et al. 2008) can recover with different kinetics from one another.
Thus, the in vivo fluorescence microscopy data argue against stable complexes, but could assembly of these unstable complexes still be sequential as predicted by the classic paradigm? In this case, one would expect the subunits to bind in a specific order, albeit quickly. Since most factors recover with variable kinetics, the consensus view so far has been that different subunits of the transcription machinery arrive at different times and only occasionally, via random “hit and run” collisions, do they form a functional end product (Hager et al. 2009). In this model, assembly is stochastic and the order of assembly variable and random (see Fig. 1b).
This argument for random assembly may be an oversimplified interpretation of the basic observation that different components of the transcription complex show different FRAP recovery rates, since the observation of variable FRAP curves by itself does not directly implicate either ordered or random assembly. However, conclusions about assembly order might in principle be drawn by fitting multiple FRAP recovery curves to estimate the steady-state binding on rate and off rate for each subunit in a complex. Dundr et al. (2002) used a combination of fitted on and off rates from FRAP data to rank the incorporation efficiency of different pol I subunits into an active form. They assumed that subunits showing a higher efficiency of assembly were probably assembled later, and this led to an ordering of several factors consistent with the sequential assembly predicted from in vitro analyses (Dundr et al. 2002). However, it is not clear if this approach is the correct one for determining assembly order. An alternative would be to assume that subunits with faster binding on rates as measured by FRAP should assemble earlier than those with slower binding on rates. At the moment, these are all simply intuitive arguments, underscoring the need for a more rigorous theoretical analysis of how on and off rates of different subunits may relate to their order of assembly.
To more directly test assembly models, inducible gene arrays (Karpova et al. 2008; Rafalska-Metcalf et al. 2010; Yao et al. 2007) have recently been employed. With this sort of system, multicolor time lapse can be used in live cells to find out whether one subunit consistently is recruited to a gene array before another subunit. Unlike FRAP, recruitment experiments do not require complicated kinetic models to interpret assembly order: if binding at a gene array is sequential, then recruitment to the array should be sequential; if binding is random, then recruitment to the array should be simultaneous (due to variation in assembly order from one promoter to another in the array). In two recent studies of this type, results are mixed. Upon heat shock in Drosophila polytene chromatin, heat-shock factor is recruited to the Hsp70 gene loci around 80 s before RNA polymerase II, whereas three different elongation factors arrive 10–20 s later (Zobeck et al. 2010). Thus, these data argue for rapid sequential assembly. In human U2OS cells, on the other hand, three of four factors examined (RNA polymerase II, histone acetyl transferase GCN5, and acetyl-lysine binding protein Brd4) accumulated at an engineered gene array simultaneously with the VP16 activator (Rafalska-Metcalf et al. 2010). Simultaneous appearance of these factors could either mean that their assembly is random or that the temporal resolution is not high enough to detect their sequential assembly. Consistent with the latter possibility is the observation that mRNA also appeared simultaneously with the VP16 activator. However, this could also reflect enhanced sensitivity for the detection of mRNA, which unlike the other factors, was labeled by multiple fluorophores, so even a single transcript could be detected. Importantly, one factor, Brd2, clearly arrived later than all the other factors, suggesting that at least one part of the assembly process is sequential, although this ordering is unexpected since the chromatin remodeler actually comes after elongation has already begun.
Thus, in terms of assembly order, the data from fluorescence microscopy are very limited and somewhat mixed favoring sequential in two cases and perhaps random in one other case. Clearly many more analyses of in vivo assembly order are needed before sound conclusions can be reached. The data from fluorescence microscopy are however much clearer with respect to the dynamics of assembly. Here results strongly suggest that the transcription machinery is unstable, with subunits that rapidly assemble and disassemble on the order of seconds, rather than minutes or hours. This transient binding extends beyond the transcription components to many other factors in the nucleus. As determined by FRAP, most nuclear proteins are highly mobile. Examples include chromatin structural proteins (Catez et al. 2006), components of the DNA replication (McNairn et al. 2005; Xouri et al. 2007) and repair machineries (Dinant et al. 2009; Luijsterburg et al. 2010), as well as components of larger subnuclear structures, such as Cajal (Kaiser et al. 2008) and PML bodies (Weidtkamp-Peters et al. 2008). It should be noted that this dynamic behavior of nuclear proteins is not absolute, as a handful of nuclear proteins do form stable structures (Hemmerich et al. 2010).
Reevaluating measurements of assembly dynamics
As discussed above, a highly dynamic assembly model for the transcription machinery is challenging the classic model of stable, sequential assembly. The dynamic model differs from the classic one in three key ways: (1) binding of individual subunits to chromatin/DNA is transient rather than stable; (2) stable holocomplexes do not exist (or are rare); and (3) assembly order may not be sequential. In what follows, we attempt to identify the source of these differences by reevaluating some of the experiments that have helped produce both models. Since live-cell fluorescence microscopy has significantly bolstered the dynamic assembly model, we begin there by taking a closer look at some intrinsic experimental limitations that could potentially bias results towards a more dynamic viewpoint. We follow up with a counterpoint discussion of some biochemical limitations that may have favored the more static classic viewpoint. In both cases, potential biasing factors could shift one viewpoint into the other (see Fig. 2), leaving the assembly debate largely unsettled in our opinion.
Fig. 2.
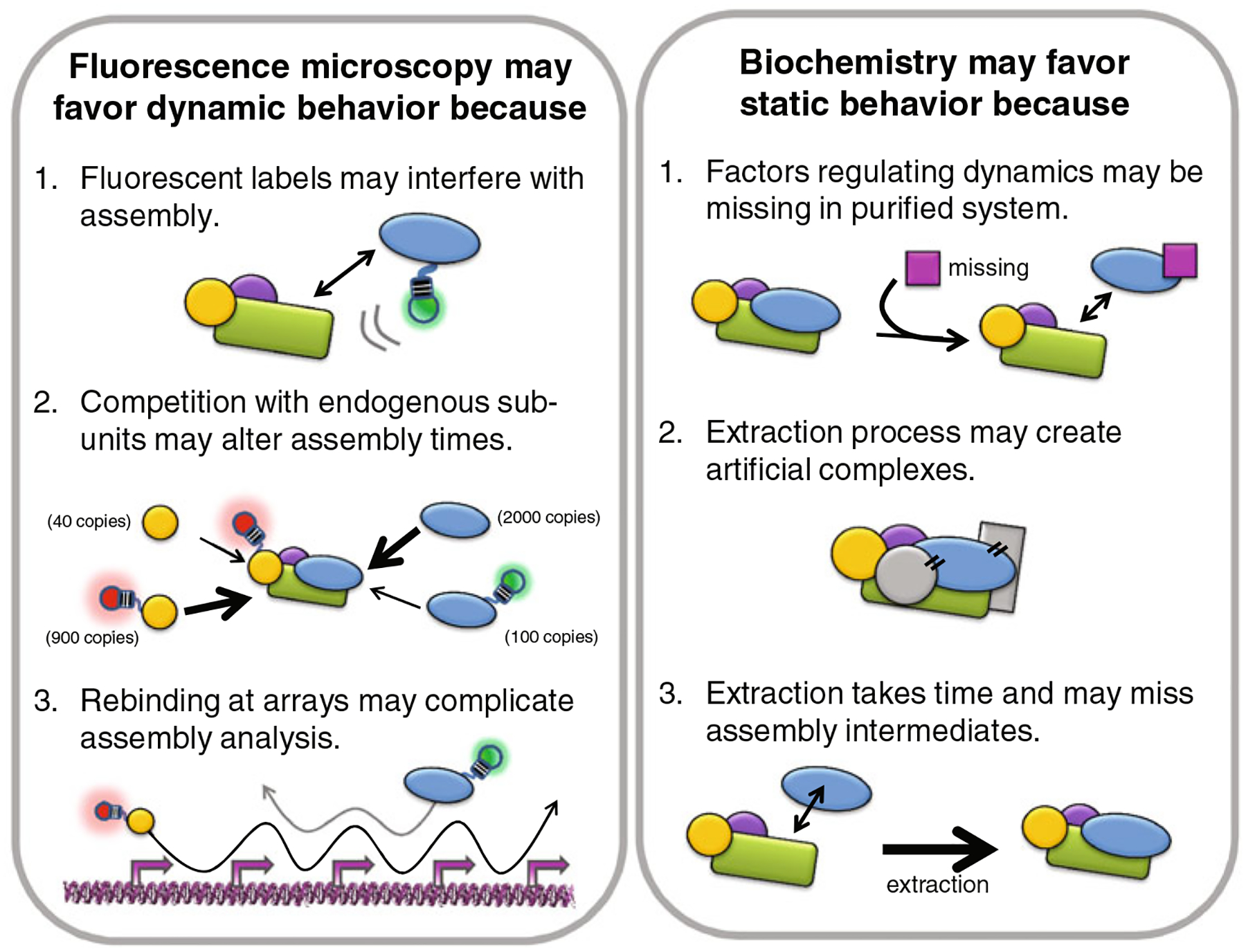
Potential biases of biochemistry and fluorescence microscopy. For the most part, fluorescence microscopy has supported the concept of unstable complexes with subunits that may not assemble in a predefined order, while biochemistry has supported the concept of stable complexes with subunits that assemble sequentially. This discrepancy may reflect biases in the studies. A few potential biasing factors are listed for each type of study
Limitations of fluorescence microscopy
Fluorescent tags may enhance measured assembly kinetics
An obvious criticism of the in vivo experiments is their complete reliance on fluorescent labels. Ideally a fluorescent label would be relatively small and inert. GFP—by far the most common fluorescent label—is fairly inert, but not small. At 27 kD, it often contributes substantially to the size of the fusion protein. It is therefore easy to imagine that such a large label gets in the way of things and generally destabilizes complex formation. Labeled subunits might therefore bind more transiently than endogenous subunits, so any holocomplex would appear smaller than it really is and the apparent order that subunits bind could be skewed.
Besides the physical size of fluorescent labels, their photophysical properties must also be carefully considered when using them to dissect assembly kinetics. For example, fluorophores within a complex might be masked by other subunits, so the fluorescent signal could be biased towards dynamic subunits that are not within a complex. Unaccounted photobleaching and/or blinking can also make things appear more dynamic than they really are (Mueller et al. 2010). In FRAP, for example, a small fraction of bleached GFP molecules may refluoresce in a few seconds and this can enhance the initial part of a recovery curve (Sinnecker et al. 2005). Photobleaching is especially problematic when dissecting long binding events via FRAP variants such as inverse FRAP, fluorescence loss in photo-bleaching (FLIP), or photoactivation/conversion. Unlike standard FRAP, in these variants, the loss of fluorescence from a subcellular region is recorded through time. Presumably the decay represents the unbinding of fluorescent molecules and their subsequent diffusion away from the measurement zone, but photobleaching will also contribute to the decay. Typically, photobleaching is corrected by measuring the decay rate in a neighboring cell contained in the same field of view, but this rate may still underestimate the photobleaching of bound molecules since freely diffusing ones can escape the imaging region or focal plane and thus tend to photobleach more slowly than bound ones. It therefore becomes difficult to distinguish true unbinding events from photobleaching, leading to underestimates of binding times.
Rebinding at arrays may complicate the measurement of assembly
Arrays have played a major role in the study of in vivo assembly kinetics so far. Without arrays, it is difficult to know where a specific DNA-binding site is because fluorescence signal to noise is insufficient. Because arrays contain many copies of the same or similar binding sites, some subunits of a complex could potentially get “caught” inside and undergo multiple binding events before leaving while others may not. This complicates FRAP dynamics because if a subunit of a complex rebinds inside an array, the apparent binding time will be longer than it really is. For example, if a subunit undergoes 10 binding events before exiting an array, its apparent binding time according to FRAP will be 10 times too long (Mueller et al. 2010). Rebinding of some subunits at an array may also complicate assembly and recruitment analyses because rebinding increases the local concentration of these subunits, artificially enhancing their binding kinetics at the array compared to more natural single binding sites. This could alter their assembly order. So far these various possibilities have been neglected in the FRAP and other assembly analyses done at arrays.
Competition with endogenous proteins may cause assembly to appear random
In nearly all of the microscopy studies of transcription so far, the fluorescently labeled proteins must compete with endogenous counterparts. Because the degree of competition could vary from one tagged subunit to another, it is not possible to predict the order that each subunit binds. For example, if the binding initiator does not compete particularly well with its endogenous counterpart, then it may appear to arrive much later than the other labeled subunits, even though its endogenous counterpart arrived first. In this case the predicted binding order would be wrong. Furthermore, since competition depends on relative stoichiometry/expression levels (among other things), cell-to-cell or dayto-day variability could make competition appear variable as well, which in turn would cause the predicted binding order to appear variable and thus random.
Rapid dynamics may depend on the promoter and cell type
Although the majority of nuclear proteins examined so far have had surprisingly fast FRAP recoveries, there is a growing list of exceptions (Hemmerich et al. 2010). Not all transcription factors, for example, exhibit rapid binding. In Drosophila, the FRAP recovery of heat-shock factors on their target genes on polytene chromatin lasts for >5 min rather than just a few seconds (Yao et al. 2006). Similarly, in human cells, TBP FRAP recoveries last minutes (Chen et al. 2002; de Graaf et al. 2010) rather than seconds as in yeast (Sprouse et al. 2008). Moreover, in some knock-in GFP tissue samples, the FRAP recovery time of TFIIH is far longer than it is in cultured cells (Giglia-Mari et al. 2009). Perhaps the rapid dynamics observed in cultured cells is the exception rather than the norm. It should be kept in mind, however, that slow FRAP recoveries do not necessarily imply that binding is stable and slow, since repeated rebinding events could be occurring.
Limitations of biochemistry
Dynamic components may be missing in reconstituted systems
The test tube is a controlled and simplified environment compared to the cell. This makes the analysis of assembly in vitro clean compared to the in vivo microscopy studies discussed above. However, key components could be missing that significantly enhance assembly dynamics in live cells. As noted above, there is growing evidence that cellular regulatory factors can alter the stability of the transcription machinery. In particular, TBP binding is mitigated by TBP associated factor Mot1 in live yeast cells (Sprouse et al. 2008), so assembly in Mot1-free systems appears less dynamic than it would otherwise (Auble et al. 1994). A similar situation holds in live human cells where the Mot1 ortholog BTAF1 destabilizes TBP residence times on chromatin (de Graaf et al. 2010). In these cells, a more complex regulatory network may be involved as a second factor, NC-2, has been found to stabilize TBP binding (de Graaf et al. 2010). Transcription regulatory complexes can also be destabilized by molecular chaperones (Freeman and Yamamoto 2002; Elbi et al. 2004; Dezwaan and Freeman 2008), although stabilization has also been observed (Stavreva et al. 2004). Finally, transcription factors can be actively evicted from target DNA by chromatin remodelers (Karpova et al. 2004; Fletcher et al. 2002; Nagaich et al. 2004) and even core subunits of active RNA polymerase can be ejected by the proteasome (Collins and Tansey 2006; Daulny et al. 2008). As more and more cofactors and coregulators are identified (Lemon and Tjian 2000; Sikorski and Buratowski 2009), the discovery of additional destabilizing (as well as stabilizing) elements seems likely. It therefore remains difficult to predict how much these elements will collectively alter the assembly dynamics of the transcription machinery in vitro, casting doubt on the sequential assembly observed so far.
Artificial aggregation may enlarge and stabilize endogenous complexes
Biochemical studies of the transcription machinery have in large part relied on the isolation of complexes from purified systems and/or cell extracts. During this procedure, the size, composition, and apparent stability of complexes may be altered. For example, hypotonic buffers (Kimura et al. 1999) or low temperatures (McNairn et al. 2005) might cause artificial aggregation and/or reduced mobility. Indeed, TFIIB can stably assemble with TBP alone in vitro, but does not appear to in vivo (Deng and Roberts 2007). Likewise, although large, soluble holoenzymes have been obtained from cellular extracts when more physiological buffers were used, only individual components remained for the most part (Kimura et al. 1999).
Artificial aggregation may also result from cross-linking when performing ChIP. These experiments only yield the sum occupancy of binding sites in a population, so two proteins that ChIP together are not necessarily at the same site at the same time. This may explain why transcription inhibitors have recently been found at some of the most active promoters (Sikorski and Buratowski 2009). In addition, cross-linking can stabilize subunits and thus push the assembly equilibrium closer to a fully assembled complex. This produces larger than average complexes and makes short-lived assembly intermediates even more transient and harder to catch via extraction which can take a relatively long time. Finally, cross-linking is not specific, so nearby but unrelated proteins could become unintentionally fixed together. These potential artifacts raise questions about binding stability and the existence of holocomplexes and generally confound the dissection of assembly order.
Bridging the gap
The preceding two sections show that there are potential problems with both the in vivo fluorescence microscopy and the in vitro biochemical assays used for measuring assembly kinetics. In our view, therefore, the question of random and dynamic or sequential and stable assembly remains unanswered, as does the more basic question of whether or not a “complete” transcription machine ever exists at the promoter at all. For example, it might be that only subcomplexes are bound at the promoter at any given time and that these subcomplexes leave behind marks for future subcomplexes to interpret (Hager et al. 2006). Since the assembly of the transcription machinery and gene regulation are likely to be tightly coupled, unraveling this deepening mystery is critical.
Resolving these issues will require improved experiments and/or new techniques. On the in vivo fluorescence microscopy side, smaller, less invasive labels will be helpful. For example, FlAsH (Martin et al. 2005) or PRIME (Uttamapinant et al. 2010) labels are only ~10 amino acids in length and can be applied posttranslationally for greater temporal control. More generally, the functionality of labeled subunits should always be rigorously tested. For one, labeled subunits should be expressed at appropriate levels. If not, depending on the availability of interacting partners, binding can be artificially weakened [like excess Rpb1 in the cytoplasm (Kimura et al. 2002)] or strengthened [like excess TBP in the nucleus (de Graaf et al. 2010)]. As well, labeled subunits should coimmunoprecipitate with endogenous subunits (Dundr et al. 2002) and should also rescue partial removal (Kuipers et al. 2011), disablement (Darzacq et al. 2007; Kimura et al. 2002), or complete removal (Sprouse et al. 2008) of endogenous counterparts. Whenever possible the latter is ideal because it proves that the labeled subunits are functional and, furthermore, completely avoids artifacts due to competition. Finally, labeled subunits might be avoided altogether by using fluorescent antibody fragments (Fabs) to track the movement of endogenous proteins and/or their modification states in live cells (Hayashi-Takanaka et al. 2009). Since Fabs only bind endogenous targets transiently, dynamics can be tracked with minimal interference to function.
So far, FRAP and array recruitment studies have been the main approaches used to dissect assembly in living cells. Additional cross-validation by complementary techniques is therefore needed to better understand and ultimately resolve the discrepancies with some of the biochemical data. Fluorescence correlation spectroscopy (FCS) and its variants can be used to cross-validate FRAP measured diffusion and binding times in live cells (Stasevich et al. 2010). FCS could also be applied deep within a gene array to check whether or not rapid rebinding is occurring. For dissecting assembly order, fluorescence cross correlation spectroscopy (FCCS) will be useful since two labeled subunits that bind sequentially should produce an asymmetry in the cross-correlation of their fluorescence signals (Sisan et al. 2010). To avoid the complications of gene arrays, single molecule tracking (SMT) (Siebrasse and Kubitscheck 2009; Tokunaga et al. 2008) may make it possible to quantify the recruitment of subunits at a single gene. Finally, experiments should be repeated on a wider range of cell and tissue types and ultimately within intact organs to better assess the generality of results.
On the biochemistry side, new extraction-free methods for accurately quantifying the size and makeup of complexes and assembly intermediates will be helpful, for example quantitative mass spectrometry (Talkington et al. 2005) or paramagnetic nuclear magnetic resonance (Iwahara and Clore 2006). In addition, competition experiments can provide an independent test of the stability of complexes in live cells. This approach was used by the Nomura lab to demonstrate a lack of exchange of core RNA polymerase I subunits in yeast (Schneider and Nomura 2004), in apparent conflict with the FRAP data in mammalian cells (Dundr et al. 2002). The only drawback to competition experiments is their relatively poor temporal resolution. In competition ChIP, for example, the time required to induce the competitor limits the temporal resolution to minutes rather than seconds (Nalley et al. 2006). This means that the very fast exchange rates of seconds detected by some FRAP experiments will be invisible to competition ChIP.
More complex biochemical systems may also be required to faithfully recapitulate assembly dynamics in the cell. For example, chromatin templates have been combined with key remodeling complexes in vitro to increase the in vitro turnover rate of the glucocorticoid receptor by more than an order of magnitude, bringing it closer to that observed in live cells (Nagaich et al. 2004). Along these lines, improved RNA polymerase II systems have recently been developed that contain multiple components of the basal transcription machinery, general coactivators, complex chromatin templates, and chromatin-modifying proteins (Santoso and Kadonaga 2006; Li et al. 2010; Guermah et al. 2009). These kinds of more complex in vitro systems are critical for reconciling the discrepant predictions between some of the biochemistry and fluorescence microscopy experiments.
Finally, a mixture of biochemistry and fluorescence microscopy may prove to be the most efficient way of bridging conflicting results. Such an approach was recently used to dissect the sequential assembly of single spliceosomes in real time (Hoskins et al. 2011). Briefly, yeast whole-cell extracts containing fluorescently labeled subunits of the spliceosome were plated on glass with surface-tethered pre-mRNA. Using multicolor total internal reflection fluorescence microscopy (TIRFM), the authors monitored the ordered recruitment of single spliceosome subunits to the pre-mRNA template. This unique blend of biochemistry and high-resolution fluorescence microscopy can be generalized to other macromolecular complexes. As such, it should be a useful platform for studying the transcription machinery and further testing assembly models.
Towards a quantitative roadmap of assembly kinetics
Besides improved measurements of assembly, improved models for assembly are also essential for proper interpretation. Unfortunately, models for assembly quickly become complex. For example, a general model for just three subunits assembling on a common substrate involves eight assembly states and 12 binding association constants. As the number of subunits increases, the number of assembly states and association constants grows exponentially: N subunits can assemble in 2N states via N2N−1 association constants. Thus, a complex of 20 subunits could have 220 or 1,048,576 states with 10,485,760 association constants.
Within this vast and complex assembly landscape, the purely random and sequential assembly strategies are just two extremes on the full spectrum of possibilities. In all likelihood, assembly falls somewhere in between these two extremes. Nevertheless, it is useful to consider the extreme cases as a starting point.
First, are there any obvious advantages to sequential assembly compared to random assembly or vice versa? To answer this kind of question, a unified model incorporating both sequential and random assembly strategies is required. One simple unified model was recently introduced in the receptor–ligand literature (D’Orsogna and Chou 2005). The model uses a “kinetic chain” to describe random, sequential, and cooperative binding strategies of N identical ligands to a receptor, but it can be extended to nonidentical subunits binding a substrate when assembly is purely random or sequential (see Fig. 3). A key result of the original study (D’Orsogna and Chou 2005) was to show how sequential and random assembly strategies optimize different features. The sequential assembly strategy usually results in higher steady-state levels of fully assembled complex since there are fewer partially assembled possibilities. In contrast, the random assembly strategy usually produces the first fully assembled complex since assembly can progress in more than one way. The only exception to this rule is when binding is noncooperative and on and off rates are about equal. In this case, the random binding strategy can become trapped within the relatively large number of intermediate partially assembled states (D’Orsogna and Chou 2005). Nonetheless, in most cases, random assembly is good for a fast, dynamic response, while sequential assembly is good for a more robust, long-lived response.
Fig. 3.
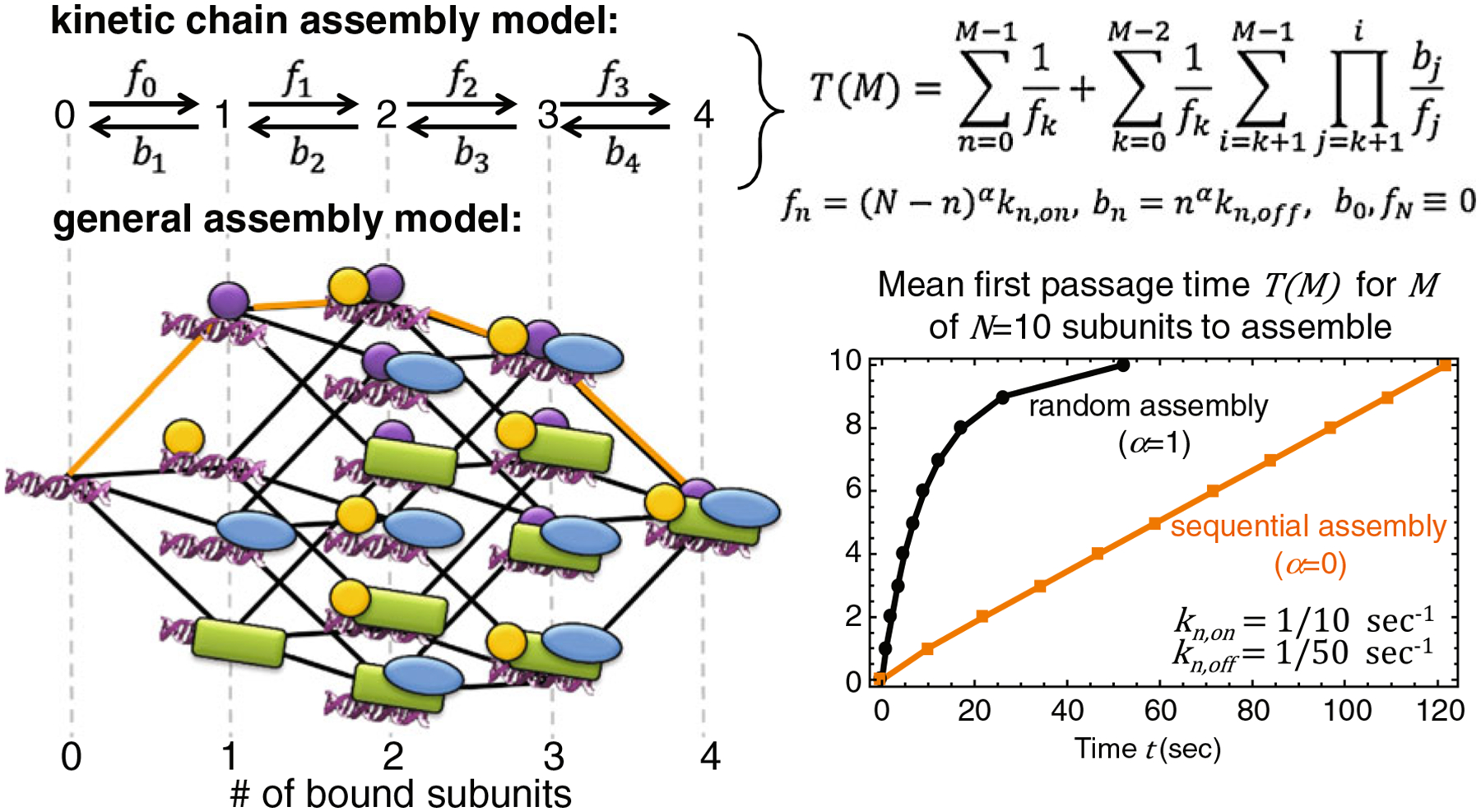
Kinetic chain model for assembly. In the most general assembly model involving N components (either individual subunits or preformed subcomplexes), there are 2N assembly states and N2N−1 association constants. In the kinetic chain model of assembly, this is reduced to just 2N parameters representing forward (fn) and backward (bn) transitions between complexes with n bound subunits. In this simplified model, the mean time T(M) to first form a complex with M of N subunits assembled, referred to as the MFPT, can be explicitly written in terms of fn and bn, which themselves depend on the subunit binding on and off rates, kn,on and kn,off, respectively, with n ranging from 0 to N. In this model, when α=1 assembly is random and when α=0 assembly is sequential. The figure on the left illustrates the simplified kinetic chain model (top of panel) for the general assembly model (bottom of panel) for N=4 subunits. All paths are used in a random assembly strategy, but only one path (thick orange) is used in a sequential assembly strategy. The graph on the right compares the random (α=1) and sequential (α=0) MFPTs for a complex with N=10 subunits with kn, on=1/10 s−1 and kn, off=1/50 s
Besides providing insight into assembly strategies, the kinetic chain model also provides a quantitative connection between FRAP studies and recruitment studies (which so far have only been analyzed qualitatively). Specifically, the average time to first make a partial or complete complex, i.e., the “mean first passage time” (MFPT), can be analytically calculated (Murthy and Kehr 1989, 1990) in terms of binding on and off rates (D’Orsogna and Chou 2005) that are measurable by FRAP. The calculated MFPTs times can then be compared to the recruitment times of each subunit to distinguish random from sequential assembly. To illustrate, suppose 10 subunits bind together to form a complex and their binding on rates are all 1/10 s−1 and their binding off rates are all 1/50 s−1. In this case, all subunits will have the same FRAP recovery curves, but the kinetic chain model predicts a complete complex will first be assembled in ~52 s if assembly is random, whereas it will take ~122 s if assembly is sequential (see Fig. 3). For the 10th subunit this translates into two very distinct recruitment curves: in the random model, the recruitment is rapid and levels off by 50 s, but in the sequential model the recruitment has yet to begin since it takes on average ~109 s for the first nine subunits to preassemble before the 10th subunit can bind.
In all likelihood, as we noted above, assembly will be some mixture of random and sequential. Then the recruitment time of the 10th subunit will fall in between the purely random and purely sequential extremes. Depending on how close it falls to the extremes, it should be possible to quantify a “degree” of assembly randomness. This would be a practical quantity to compare assembly strategies in different cell types and at different promoters. It would also help identify good candidate models from the huge pool of possible models.
One assembly model that mixes random and sequential strategies has recently gained traction in the DNA repair field (Luijsterburg et al. 2010). The model is based on the concept of kinetic proofreading (Hopfield 1974) and explains the apparently conflicting slow accumulation yet rapid binding of subunits of the DNA repair machinery to sites of DNA damage (Luijsterburg et al. 2010; Dinant et al. 2009). The model includes six sequential irreversible steps: DNA damage recognition, partial and complete DNA unwinding, incision, resynthesis, and rechromatinization. Each of these steps is carried out via the random assembly of different sets of subunits. The entire process is highly specific since each step requires the simultaneous binding of many dynamic factors at a single site. The probability for this to occur is only significant at bona fide DNA damage sites, where specific binding sites exist for all of the factors involved. This is the essence of kinetic proofreading. Once the right set accumulates at the right time, the repair process is ratcheted one step closer to completion via an irreversible enzymatic reaction. Since this mixed model involves a sequence of miniassembly events that are each purely random, the entire process can be thought of as a series of linked kinetic chains that could be quantified using MFPTs.
It remains to be seen whether or not the kinetic proofreading model underlies the assembly of the transcription machinery. The transcription machinery shares subunits with the repair machinery, so it is certainly possible that a similar assembly strategy is employed by both. In the case of the transcription machinery, the irreversible steps might be (1) promoter recognition, (2) chromatin remodeling, (3) PIC assembly, (4) initiation, and (5) active elongation. The timing of these transcription signposts in live cells will be interesting to measure. According to the kinetic proofreading model, each of these steps requires some time since each occurs through the chance encounter of multiple subunits. The fact that mRNA can appear within seconds of chromatin remodelers and/or gene activators at some strong promoters (Rafalska-Metcalf et al. 2010) places severe constraints on the kinetic proofreading model, however, since this would imply that up to four distinct complexes were formed in sequence through chance encounters in just a few seconds. Of course, this speed could be a promoter-specific effect. Recent evidence for a surprisingly wide range of promoter signatures (Muller and Tora 2004; Juven-Gershon et al. 2008; Sikorski and Buratowski 2009) coupled with the intriguing prospect that even core subunits of the PIC vary depending on cell and tissue type (D’Alessio et al. 2009) adds yet another dimension to the ongoing search for a unifying assembly model.
Outlook
Although much progress has been made in the study of transcription, new experiments have brought into sharper focus the question of how the transcription machinery assembles. Biochemistry originally produced the classic ordered and stable assembly paradigm, while fluorescence microscopy has popularized a more dynamic assembly paradigm and raised questions about whether assembly is always ordered. These contradictory views may now be inching towards each other. The microscopy studies have stimulated new biochemical studies and also highlighted selected older ones, all of which are consistent with a more dynamic view of assembly. At the same time, some microscopy evidence has emerged for stable behavior. However, it remains completely uncertain where on the range between stability and dynamism a middle ground might be found. Of continuing concern is the relatively strong correlation between the experimental technique and the observed outcome, suggesting that there could be some intrinsic biases in the biochemical or microscopy approaches. Here, we have discussed a few biasing limitations of both techniques, but what is now needed is a concerted effort to bridge the gap. One way this will be achieved is by fitting data to unified models of assembly that include both sequential and random assembly strategies. We have discussed a simple candidate model, although others are certainly possible. In addition, more direct measurements, both in the test tube and in live cells, will be required to better distinguish assembly strategies. In our view the assembly debate is only gaining steam. We anticipate that its resolution will lead to novel techniques and important insights into not only transcription but many other cellular assembly processes.
Acknowledgments
We would like to thank Hiroshi Kimura, David Auble, Akhilesh Nagaich, and Avin Lalmansingh for the critical reading of the manuscript and helpful comments. This research was supported in part by the intramural program of the National Institutes of Health, National Cancer Institute, and the Center for Cancer Research. TJS was also supported by a Japan Society for the Promotion of Science Postdoctoral Fellowship for Foreign Researchers.
Abbreviations
- PIC
Preinitiation complex
- ChIP
Chromatin immunoprecipitation
- GFP
Green fluorescent protein
- FRAP
Fluorescence recovery after photobleaching
- FLIP
Fluorescence loss in photobleaching
- FCS
Fluorescence correlation spectroscopy
- SMT
Single-molecule tracking
- Fabs
Antigen binding fragments
- FCCS
Fluorescence cross-correlation spectroscopy
- MFPT
Mean first passage time
- SPM
Surface plasmon resonance
- TIRFM
Total internal reflection fluorescence microscopy
Contributor Information
Timothy J. Stasevich, Graduate School of Frontier Biosciences, Osaka University, Osaka 565-0871, Japan
James G. McNally, National Cancer Institute, US National Institutes of Health, Bethesda, MD 20892, USA
References
- Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell 103:667–678 [DOI] [PubMed] [Google Scholar]
- Auble DT, Hansen KE, Mueller CG, Lane WS, Thorner J, Hahn S (1994) Mot1, a global repressor of RNA polymerase II transcription, inhibits TBP binding to DNA by an ATP-dependent mechanism. Genes Dev 8:1920–1934 [DOI] [PubMed] [Google Scholar]
- Bonham AJ, Neumann T, Tirrell M, Reich NO (2009) Tracking transcription factor complexes on DNA using total internal reflectance fluorescence protein binding microarrays. Nucleic Acids Res 37:e94–e94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosisio D, Marazzi I, Agresti A, Shimizu N, Bianchi ME, Natoli G (2006) A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-κB-dependent gene activity. EMBO J 25:798–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S (1994) The basics of basal transcription by RNA polymerase II. Cell 77:1–3 [DOI] [PubMed] [Google Scholar]
- Buratowski S (2000) Snapshots of RNA polymerase II transcription initiation. Curr Opin Cell Biol 12:320–325 [DOI] [PubMed] [Google Scholar]
- Bushnell DA, Bamdad C, Kornberg RD (1996) A minimal set of RNA polymerase II transcription protein interactions. J Biol Chem 271:20170–20174 [DOI] [PubMed] [Google Scholar]
- Catez F, Ueda T, Bustin M (2006) Determinants of histone H1 mobility and chromatin binding in living cells. Nat Struct Mol Biol 13:305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Hinkley CS, Henry RW, Huang S (2002) TBP dynamics in living human cells: constitutive association of TBP with mitotic chromosomes. Mol Biol Cell 13:276–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GA, Tansey WP (2006) The proteasome: a utility tool for transcription? Curr Opin Genet Dev 16:197–202 [DOI] [PubMed] [Google Scholar]
- Conaway RC, Conaway JW (1993) General initiation factors for RNA polymerase II. Annu Rev Biochem 62:161–190 [DOI] [PubMed] [Google Scholar]
- Cook PR (2001) Principles of nuclear structure and function, 1st edn Wiley-Liss, New York [Google Scholar]
- D’Alessio JA, Wright KJ, Tjian R (2009) Shifting players and paradigms in cell-specific transcription. Mol Cell 36:924–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH (2007) In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14:796–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Yao J, Larson DR, Causse SZ, Bosanac L, de Turris V, Ruda VM, Lionnet T, Zenklusen D, Guglielmi B, Tjian R, Singer RH (2009) Imaging transcription in living cells. Annu Rev Biophys 38:173–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daulny A, Geng F, Muratani M, Geisinger JM, Salghetti SE, Tansey WP (2008) Modulation of RNA polymerase II subunit composition by ubiquitylation. Proc Natl Acad Sci USA 105:19649–19654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf P, Mousson F, Geverts B, Scheer E, Tora L, Houtsmuller AB, Timmers HTM (2010) Chromatin interaction of TATA-binding protein is dynamically regulated in human cells. J Cell Sci 123:2663–2671 [DOI] [PubMed] [Google Scholar]
- Deng W, Roberts SGE (2007) TFIIB and the regulation of transcription by RNA polymerase II. Chromosoma 116:417–429 [DOI] [PubMed] [Google Scholar]
- Dezwaan DC, Freeman BC (2008) HSP90: the Rosetta stone for cellular protein dynamics? Cell Cycle 7:1006–1012 [DOI] [PubMed] [Google Scholar]
- Dinant C, Luijsterburg MS, Höfer T, von Bornstaedt G, Vermeulen W, Houtsmuller AB, van Driel R (2009) Assembly of multiprotein complexes that control genome function. J Cell Biol 185:21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orsogna MR, Chou T (2005) First passage and cooperativity of queuing kinetics. Phys Rev Lett 95:170603. [DOI] [PubMed] [Google Scholar]
- Dundr M, Hoffmann-Rohrer U, Hu Q, Grummt I, Rothblum LI, Phair RD, Misteli T (2002) A kinetic framework for a mammalian RNA polymerase in vivo. Science 298:1623–1626 [DOI] [PubMed] [Google Scholar]
- Elbi C, Walker DA, Romero G, Sullivan WP, Toft DO, Hager GL, DeFranco DB (2004) Molecular chaperones function as steroid receptor nuclear mobility factors. PNAS 101:2876–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C (2009) A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci USA 106:20240–20245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher TM, Xiao N, Mautino G, Baumann CT, Wolford R, Warren BS, Hager GL (2002) ATP-dependent mobilization of the glucocorticoid receptor during chromatin remodeling. Mol Cell Biol 22:3255–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman BC, Yamamoto KR (2002) Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296:2232–2235 [DOI] [PubMed] [Google Scholar]
- Giglia-Mari G, Theil AF, Mari P-O, Mourgues S, Nonnekens J, Andrieux LO, de Wit J, Miquel C, Wijgers N, Maas A, Fousteri M, Hoeijmakers JHJ, Vermeulen W (2009) Differentiation driven changes in the dynamic organization of basal transcription initiation. PLoS Biol 7:e1000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski SA, Snyder SK, John S, Grummt I, Misteli T (2008) Modulation of RNA polymerase assembly dynamics in transcriptional regulation. Mol Cell 30:486–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermah M, Kim J, Roeder RG (2009) Transcription of in vitro assembled chromatin templates in a highly purified RNA polymerase II system. Methods 48:353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager GL, Elbi C, Johnson TA, Voss T, Nagaich AK, Schiltz RL, Qiu Y, John S (2006) Chromatin dynamics and the evolution of alternate promoter states. Chromosome Res 14:107–116 [DOI] [PubMed] [Google Scholar]
- Hager GL, McNally JG, Misteli T (2009) Transcription dynamics. Mol Cell 35:741–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S (2004) Structure and mechanism of the RNA polymerase II transcription machinery. Nat Struct Mol Biol 11:394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL (2002) Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell 111:369–379 [DOI] [PubMed] [Google Scholar]
- Hayashi-Takanaka Y, Yamagata K, Nozaki N, Kimura H (2009) Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. J Cell Biol 187:781–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerich P, Schmiedeberg L, Diekmann S (2010) Dynamic as well as stable protein interactions contribute to genome function and maintenance. Chromosome Res 19:131–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogstraten D, Nigg AL, Heath H, Mullenders LHF, van Driel R, Hoeijmakers JHJ, Vermeulen W, Houtsmuller AB (2002) Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol Cell 10:1163–1174 [DOI] [PubMed] [Google Scholar]
- Hoopes BC, LeBlanc JF, Hawley DK (1992) Kinetic analysis of yeast TFIID-TATA box complex formation suggests a multi-step pathway. J Biol Chem 267:11539–11547 [PubMed] [Google Scholar]
- Hopfield JJ (1974) Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci USA 71:4135–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ (2011) Ordered and dynamic assembly of single spliceosomes. Science 331:1289–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara J, Clore GM (2006) Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature 440:1227–1230 [DOI] [PubMed] [Google Scholar]
- Johnson TA, Elbi C, Parekh BS, Hager GL, John S (2008) Chromatin remodeling complexes interact dynamically with a glucocorticoid receptor-regulated promoter. Mol Biol Cell 19:3308–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juven-Gershon T, Hsu J-Y, Theisen JW, Kadonaga JT (2008) The RNA polymerase II core promoter—the gateway to transcription. Curr Opin Cell Biol 20:253–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadonaga JT (1990) Assembly and disassembly of the Drosophila RNA polymerase II complex during transcription. J Biol Chem 265:2624–2631 [PubMed] [Google Scholar]
- Kaiser TE, Intine RV, Dundr M (2008) De novo formation of a subnuclear body. Science 322:1713–1717 [DOI] [PubMed] [Google Scholar]
- Karpova TS, Chen TY, Sprague BL, McNally JG (2004) Dynamic interactions of a transcription factor with DNA are accelerated by a chromatin remodeller. EMBO Rep 5:1064–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova TS, Kim MJ, Spriet C, Nalley K, Stasevich TJ, Kherrouche Z, Heliot L, McNally JG (2008) Concurrent fast and slow cycling of a transcriptional activator at an endogenous promoter. Science 319:466–469 [DOI] [PubMed] [Google Scholar]
- Kimura H, Tao Y, Roeder RG, Cook PR (1999) Quantitation of RNA polymerase II and its transcription factors in an HeLa cell: little soluble holoenzyme but significant amounts of polymerases attached to the nuclear substructure. Mol Cell Biol 19:5383–5392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Sugaya K, Cook PR (2002) The transcription cycle of RNA polymerase II in living cells. J Cell Biol 159:777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD (2007) The molecular basis of eukaryotic transcription. Proc Natl Acad Sci USA 104:12955–12961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers MA, Stasevich TJ, Sasaki T, Wilson KA, Hazelwood KL, McNally JG, Davidson MW, Gilbert DM (2011) Highly stable loading of Mcm proteins onto chromatin in living cells requires replication to unload. J Cell Biol 192:29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon B, Tjian R (2000) Orchestrated response: a symphony of transcription factors for gene control. Genes Dev 14:2551–2569 [DOI] [PubMed] [Google Scholar]
- Li G, Margueron R, Hu G, Stokes D, Wang Y-H, Reinberg D (2010) Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo. Mol Cell 38:41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman BA, Nordeen SK (1997) DNA intersegment transfer, how steroid receptors search for a target site. J Biol Chem 272:1061–1068 [DOI] [PubMed] [Google Scholar]
- Linnell J, Mott R, Field S, Kwiatkowski DP, Ragoussis J, Udalova IA (2004) Quantitative high-throughput analysis of transcription factor binding specificities. Nucleic Acids Res 32:e44–e44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, von Bornstaedt G, Gourdin AM, Politi AZ, Moné MJ, Warmerdam DO, Goedhart J, Vermeulen W, van Driel R, Höfer T (2010) Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J Cell Biol 189:445–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley JL, Fire A, Cano A, Sharp PA, Gefter ML (1980) DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc Natl Acad Sci USA 77:3855–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Giepmans BNG, Adams SR, Tsien RY (2005) Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat Biotechnol 23:1308–1314 [DOI] [PubMed] [Google Scholar]
- McNairn AJ, Okuno Y, Misteli T, Gilbert DM (2005) Chinese hamster ORC subunits dynamically associate with chromatin throughout the cell-cycle. Exp Cell Res 308:345–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima S, Nomura M (1970) Assembly mapping of 30S ribosomal proteins from E. coli. Nature 226:1214. [DOI] [PubMed] [Google Scholar]
- Mu D, Hsu DS, Sancar A (1996) Reaction mechanism of human DNA repair excision nuclease. J Biol Chem 271:8285–8294 [DOI] [PubMed] [Google Scholar]
- Mueller F, Wach P, McNally JG (2008) Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys J 94:3323–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller F, Mazza D, Stasevich TJ, McNally JG (2010) FRAP and kinetic modeling in the analysis of nuclear protein dynamics: what do we really know? Curr Opin Cell Biol 22:403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F, Tora L (2004) The multicoloured world of promoter recognition complexes. EMBO J 23:2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy KPN, Kehr KW (1989) Mean first-passage time of random walks on a random lattice. Phys Rev A 40:2082. [DOI] [PubMed] [Google Scholar]
- Murthy KPN, Kehr KW (1990) Erratum: mean first-passage time of random walks on a random lattice. Phys Rev A 41:1160. [DOI] [PubMed] [Google Scholar]
- Nagaich AK, Walker DA, Wolford R, Hager GL (2004) Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell 14:163–174 [DOI] [PubMed] [Google Scholar]
- Nalley K, Johnston SA, Kodadek T (2006) Proteolytic turnover of the Gal4 transcription factor is not required for function in vivo. Nature 442:1054–1057 [DOI] [PubMed] [Google Scholar]
- Nalley K, Johnston SA, Kodadek T (2009) Nalley et al. reply. Nature 461:E8 [Google Scholar]
- Perlmann T, Eriksson P, Wrange O (1990) Quantitative analysis of the glucocorticoid receptor-DNA interaction at the mouse mammary tumor virus glucocorticoid response element. J Biol Chem 265:17222–17229 [PubMed] [Google Scholar]
- Rafalska-Metcalf IU, Powers SL, Joo LM, LeRoy G, Janicki SM (2010) Single cell analysis of transcriptional activation dynamics. PLoS One 5:e10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DN, Stewart AJ, Huff JT, Cairns BR (2003) The RNA polymerase III transcriptome revealed by genome-wide localization and activity–occupancy relationships. Proc Natl Acad Sci USA 100:14695–14700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder RG (1996) The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci 21:327–335 [PubMed] [Google Scholar]
- Santoso B, Kadonaga JT (2006) Reconstitution of chromatin transcription with purified components reveals a chromatin-specific repressive activity of p300. Nat Struct Mol Biol 13:131–139 [DOI] [PubMed] [Google Scholar]
- Schneider DA, Nomura M (2004) RNA polymerase I remains intact without subunit exchange through multiple rounds of transcription in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 101:15112–15117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Diffley JFX (2000) Stepwise assembly of initiation proteins at budding yeast replication origins in vitro. Proc Natl Acad Sci USA 97:14115–14120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker-Parry JS, Aebersold R, Campbell CT (2004) Parallel, quantitative measurement of protein binding to a 120-element double-stranded DNA array in real time using surface plasmon resonance microscopy. Anal Chem 76:2071–2082 [DOI] [PubMed] [Google Scholar]
- Siebrasse JP, Kubitscheck U (2009) Single molecule tracking for studying nucleocytoplasmic transport and intranuclear dynamics. Methods Mol Biol 464:343–361 [DOI] [PubMed] [Google Scholar]
- Sikorski TW, Buratowski S (2009) The basal initiation machinery: beyond the general transcription factors. Curr Opin Cell Biol 21:344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnecker D, Voigt P, Hellwig N, Schaefer M (2005) Reversible photobleaching of enhanced green fluorescent proteins. Biochemistry 44:7085–7094 [DOI] [PubMed] [Google Scholar]
- Sisan DR, Yarar D, Waterman CM, Urbach JS (2010) Event ordering in live-cell imaging determined from temporal cross-correlation asymmetry. Biophys J 98:2432–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprouse RO, Karpova TS, Mueller F, Dasgupta A, McNally JG, Auble DT (2008) Regulation of TATA-binding protein dynamics in living yeast cells. Proc Natl Acad Sci USA 105:13304–13308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasevich TJ, Mueller F, Michelman-Ribeiro A, Rosales T, Knutson JR, McNally JG (2010) Cross-validating FRAP and FCS to quantify the impact of photobleaching on in vivo binding estimates. Biophys J 99:3093–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavreva DA, Muller WG, Hager GL, Smith CL, McNally JG (2004) Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol Cell Biol 24:2682–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung M-H, Salvatore L, De Lorenzi R, Indrawan A, Pasparakis M, Hager GL, Bianchi ME, Agresti A (2009) Sustained oscillations of NF-κB produce distinct genome scanning and gene expression profiles. PLoS One 4:e7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkington MWT, Siuzdak G, Williamson JR (2005) An assembly landscape for the 30S ribosomal subunit. Nature 438:628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, Sakata-Sogawa K (2008) Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 5:159–161 [DOI] [PubMed] [Google Scholar]
- Uttamapinant C, White KA, Baruah H, Thompson S, Fernández-Suárez M, Puthenveetil S, Ting AY (2010) A fluorophore ligase for site-specific protein labeling inside living cells. Proc Natl Acad Sci USA 107:10914–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Werven FJ, van Teeffelen HAAM, Holstege FCP, Timmers HTM (2009) Distinct promoter dynamics of the basal transcription factor TBP across the yeast genome. Nat Struct Mol Biol 16:1043–1048 [DOI] [PubMed] [Google Scholar]
- Weidtkamp-Peters S, Lenser T, Negorev D, Gerstner N, Hofmann TG, Schwanitz G, Hoischen C, Maul G, Dittrich P, Hemmerich P (2008) Dynamics of component exchange at PML nuclear bodies. J Cell Sci 121:2731–2743 [DOI] [PubMed] [Google Scholar]
- Weil PA, Luse DS, Segall J, Roeder RG (1979) Selective and accurate initiation of transcription at the Ad2 major late promotor in a soluble system dependent on purified RNA polymerase II and DNA. Cell 18:469–484 [DOI] [PubMed] [Google Scholar]
- Xouri G, Squire A, Dimaki M, Geverts B, Verveer PJ, Taraviras S, Nishitani H, Houtsmuller AB, Bastiaens PIH, Lygerou Z (2007) Cdt1 associates dynamically with chromatin throughout G1 and recruits Geminin onto chromatin. EMBO J 26:1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Munson KM, Webb WW, Lis JT (2006) Dynamics of heat shock factor association with native gene loci in living cells. Nature 442:1050–1053 [DOI] [PubMed] [Google Scholar]
- Yao J, Ardehali MB, Fecko CJ, Webb WW, Lis JT (2007) Intranuclear distribution and local dynamics of RNA polymerase II during transcription activation. Mol Cell 28:978–990 [DOI] [PubMed] [Google Scholar]
- Yean D, Gralla J (1997) Transcription reinitiation rate: a special role for the TATA box. Mol Cell Biol 17:3809–3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkovsky N, Ranish JA, Hahn S (2000) A transcription reinitiation intermediate that is stabilized by activator. Nature 408:225–229 [DOI] [PubMed] [Google Scholar]
- Zawel L, Kumar KP, Reinberg D (1995) Recycling of the general transcription factors during RNA polymerase II transcription. Genes Dev 9:1479–1490 [DOI] [PubMed] [Google Scholar]
- Zobeck KL, Buckley MS, Zipfel WR, Lis JT (2010) Recruitment timing and dynamics of transcription factors at the Hsp70 loci in living cells. Mol Cell 40:965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]