

Abstract
Purpose of review
HIV infection is controlled but not cured by combination antiretroviral therapy. HIV may persist for a number of reasons, including ongoing cycles of HIV infection or viral persistence as latent, or HIV replication in long-lived cells containing HIV proviruses. Therapeutic consequences of these alternative mechanisms are significant and distinct. If ongoing replication remains during current antiretroviral therapy, then improvements in potency will be useful in eradication strategies. Alternatively, long-lived cells with integrated proviruses will not be affected by improvements in therapy directed against active infection, and new strategies will be necessary for HIV eradication. Technologic advances have made it possible to carry out a series of drug intensification protocols in well suppressed patients; these and other analyses for HIV replication have been useful to elucidate the nature of HIV persistence on therapy.
Recent findings
A number of clinical studies intensifying antiretroviral therapy carried out in the last several years have yielded new findings regarding the ability to detect the presence of ongoing replication. Decreases in persistent viremia have not been consistently detected in individuals on potent combination antiretroviral therapy. Evidence for persistent replication has been reported in patients using sensitive assays of cell-associated HIV.
Summary
HIV viremia persists despite combination antiretroviral therapy. Antiretroviral drug intensification does not lower the level of HIV measured in plasma, suggesting current therapy arrests active virus replication. HIV eradication will most likely require therapy in addition to potent antiretroviral therapy.
Keywords: active cycles of replication, drug intensification, HIV infection
Introduction
HIV infection results in a chronic persistent infection in humans that can be suppressed but not eradicated by current combination antiretroviral therapy. The reasons why potent therapy can inhibit but not completely eliminate HIV infection are not well understood, but a number of potential scenarios are possible and testable. One principal mechanism by which HIV may persist is through active complete cycles of HIV replication that continue at low levels despite therapy. By this mechanism, HIV spread continues to uninfected susceptible host cells maintaining persistent active infection. Alternatively, long-lived cells containing HIV proviruses may persist, producing HIV at low levels either continuously or episodically after undergoing reactivation. Therapeutic consequences of these alternative mechanisms are distinct. If ongoing replication remains during current antiretroviral therapy, then improvements in potency or administration will be useful in eradication strategies. Alternatively, long-lived cells with integrated proviruses will not be affected by improvements in therapy directed against active infection, and new strategies will be necessary to cure HIV infection. Several advances have enabled new studies of the nature of HIV replication during antiretroviral suppression. Development of sensitive detection techniques for HIV during therapy has been an essential component of this effort [1,2]. New assays for HIV and for virus replication have enabled detection of HIV viremia at levels below 1 copy/ml plasma, and have enabled quantitation of cell-associated nucleic acid [3]. Cohorts of patients with suppressed viral RNA levels have been followed for prolonged periods. New potent antiretroviral agents, such as, raltegravir, have been developed with mechanisms of action that target steps in replication that are independent from prior agents [4,5]. As a consequence, it has been possible to carry out a series of drug intensification studies and other analyses to investigate the nature of HIV replication in well suppressed patients.
The present review will focus on virological and clinical aspects of combination of antiretroviral therapy in suppressing HIV infection. A number of different terms have been utilized to describe HIV infection. For the purposes of this review, ‘active replication’ will refer to complete HIV replication cycles from initial viral attachment through integration in the host genome and ending with budding of virions from the cell surface. HIV DNA integrated into host genome is referred to as the ‘HIV provirus’. ‘Latent’ infection refers to HIV infection with integrated proviruses that are transcriptionally inactive.
HIV replication
HIV replication is initiated by infection through HIV viral receptor (CD4) and coreceptor (typically CCR5 or CXCR4); a broad spectrum of host cells are susceptible to HIV infection, including short-lived activated T cells, as well as long-lived lymphocytes and cells derived from monocyte/macrophage lineage that may persist in vivo for extended periods (Fig. 1). HIV replication proceeds through a unique mechanism that converts virion RNA into a double-stranded DNA via the viral enzyme reverse transcriptase [6,7]. In the process, terminal repeats pre sent in RNA are expanded into long terminal repeats containing promoter elements at the 5′ end and polyadenylation sequences at the 3′ end. Reverse transcriptase is an error prone RNA directed DNA polymerase; error rates of 3–5 × 10−5 incorrectly incorporated nucleotides/per nucleotide position per cycle of replication [8]. Nascent DNA is integrated into the host genome via viral-encoded integrase executing end processing of long terminal repeats, and transfer of the processed strands into host genome. The half-life of linear unintegrated HIV DNA is thought to be relatively rapid, and integration begins within hours of infection [9,10]. Integration is not reversible and proviruses persist throughout the lifetime of the infected cell. Not all newly reverse transcribed HIV DNA become integrated into host genome in the form of the HIV provirus; alternatively, cellular ligases can convert newly synthesized linear double-stranded HIV into circular DNA in an autointegration dead end pathway (Fig. 1). These circular forms of HIV replication are not in general transcriptionally competent, but are detectable in vivo and have been suggested as recent markers of reverse transcription [11–13] and are central to understanding several current intensification trials. A critical aspect of their utility is their half-life; if 2-LTR circles decay rapidly, they will be useful marker for recent infection; evidence supporting labile [11,12,14–16] and stable [17–21] forms has been reported, and characterization of such forms remains under study.
Figure 1. Key steps in HIV replication.
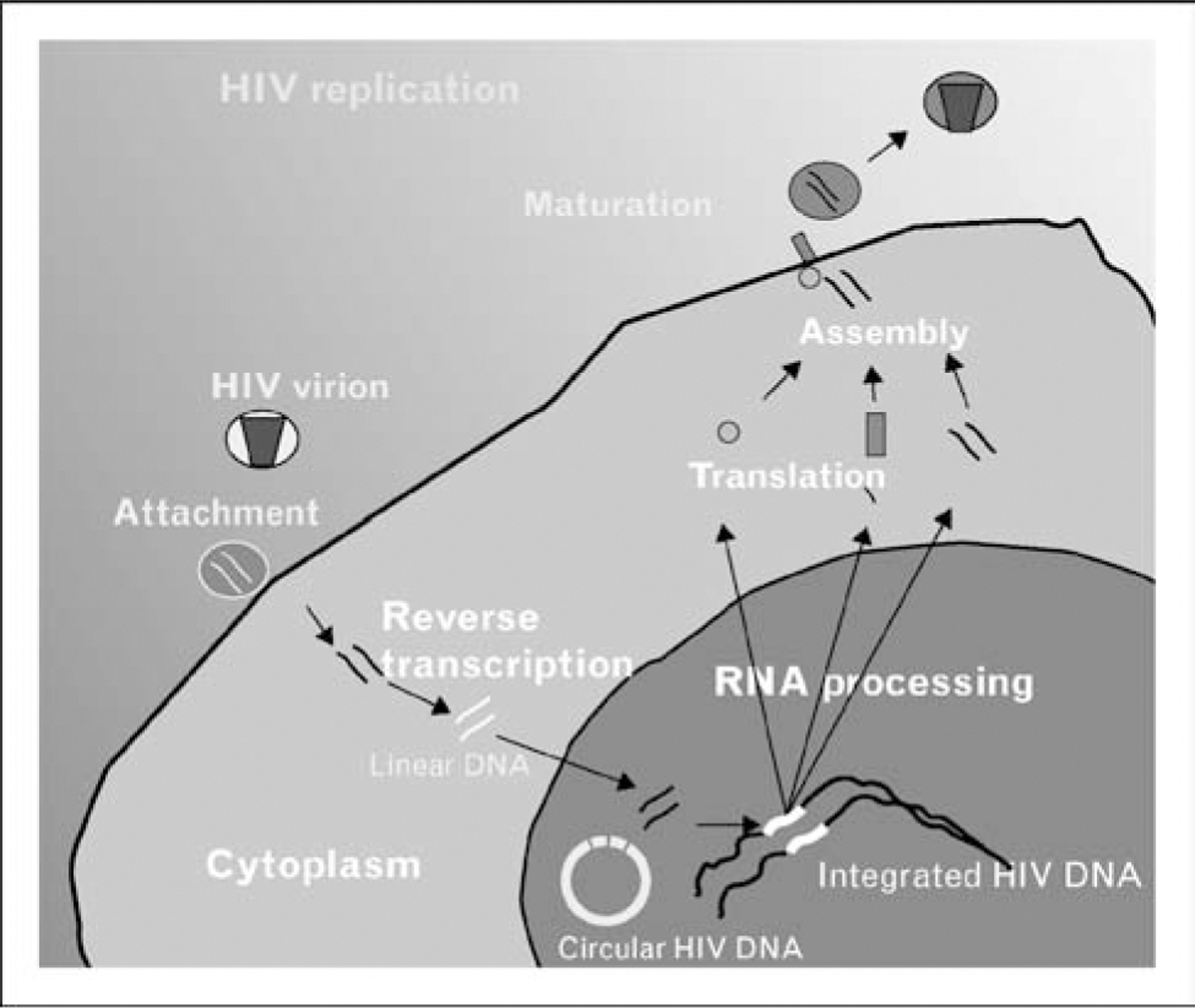
HIV replication is rapid and error prone and includes a number of steps that can facilitate establishing chronic infection, including host cell selection by receptor/coreceptor interactions, reverse transcription, and integration. A number of cell-associated HIV DNA forms, including linear, circular, and integrated are present in infected cells. Linear DNA has a relatively short half-life, but the half-life of circular DNA, and therefore its utility in tracking ongoing replication, remains under study.
Expression of viral RNA from integrated proviruses requires interactions between viral transcription factors and RNA-binding sequences (Tat protein and TAR RNA, respectively) that stimulate assembly of cellular transcription factors resulting in synthesis of full-length HIV RNA [22,23]. Transcription typically continues at high levels until cell death or immune-mediated elimination, but it is possible that virus-producing cells revert to a latent state of expression without detectable HIV RNA production. Latently infected cells are reinducible by appropriate antigen or other stimulation. Following alternative splicing, transport, and translation of HIV RNA, take place via interactions between viral and cellular factors, and virions are assembled by appropriating endosomal sorting complex required for transport (ESCRT) pathways and bud from the cell surface [24,25]. During the final steps of replication, HIV protease is required to complete maturation of the virion particle and confer infectivity on the virion. In the absence of protease activity, virions may be produced but are noninfectious. The entire replication cycle is relatively rapid and complete in 1–2 days. Replication may result in cell death, but chronic HIV production from cells with integrated proviruses for prolonged periods is also possible [26].
Several key observations relevant to understanding HIV replication during antiretroviral therapy derive directly from an understanding of the HIV replication cycle. HIV may persist in vivo by ongoing complete cycles of active infection, which in turn are capable of initiating new cycles of infection before they are eliminated by the host immune system or die by viral-triggered mechanisms. A reproductive ratio of at least one newly infected cell per transmission event ensures steady state persistence [27]. Infection may also persist through infection of cells that are not eliminated, but persist for prolonged periods after infection with persistent or inducible production of virions. Cell-associated HIV DNA may take several forms, including linear, circular, or integrated. Protease is required for production of infectious virus, but not for production of virions per se. In the absence of protease activity, numerical production of virions remains intact, but the virions are not capable of transmitting infection to new cells [28–30].
Detection of HIV during antitretroviral therapy
Limits of HIV detection are approximately 50–75 copies HIV RNA/ml plasma in commercial assays [31,32]. Early amplification techniques were able to detect HIV below this commercial limit. More recent development of sensitive single-copy assays that include controls to assess viral RNA recovery, concentration of plasma virus by ultracentrifugation, and real-time amplification have permitted quantitation of HIV RNA at levels that are essentially limited by the volume of plasma under study [3]. Using 10 ml plasma, the limit of quantitation is in the range of 0.2–1 copy. Key points regarding detection of HIV viremia during suppressive therapy are as follows: at this low level of viremia, variability in detection is a Poisson limited process; in the absence of contribution of factors, such as, assay conditions or biological variation, the variance viral RNA levels is determined by the mean level of viremia. As the clearance of plasma RNA is relatively rapid [33–35], HIV RNA that is detected is likely to have been produced within few hours prior to phlebotomy. Quantitative assay of RNA is limited to determination of levels only, no information is provided regarding infectivity of the measured virus. The level of viremia detected below the commercial limit is not known to be predictive of clinical outcome.
Quantitation of cell-associated HIV nucleic acid is a more detailed process than measuring plasma HIV; in general three additional complexities limit precise detection. Although total cell-associated HIV DNA is readily quantitated, assays to distinguish linear unintegrated, circular, and integrated forms of HIV DNA (see Fig. 1) are necessary to obtain a precise description of replication. Sensitive detection of linear DNA is not well described, but circular forms of DNA are quantifiable with single-copy sensitivity. Precise determination of the level of integrated DNA is difficult but newer assays now have been reported to improve quantitation [36,37]. A second layer of complexity is provided by cell types infected by HIV. As HIV infects subsets of CD4-positive cells, quantitative analysis of cell-associated HIV DNA, or RNA requires normalization to account for potential differences in the level of these CD4 cell subsets in the sample. Finally, because cells with integrated proviruses may persist for prolonged periods, cell-associated DNA is not a dynamic measure of HIV replication. Proviral DNA declines slowly and to a limited degree (generally <10-fold) compared with profound decreases in HIV plasma viremia [38–40], and levels of cell-associated DNA consist of cells that have been recently or remotely infected with HIV. HIV replication is error prone, and many integrated proviruses are incapable of replication. As a consequence, analysis of cell-associated DNA requires sensitive detection to discriminate true differences in HIV DNA above the background of cells harboring a ‘cemetery’ of defective proviruses.
Dynamics of response to antiretroviral therapy
Untreated, HIV viremia is measurable at a relatively stable RNA level for prolonged periods after infection. After introduction of antiretroviral therapy, this set point viremia declines dramatically. Notably, exponential decay is not linear throughout decline. After a period of rapid decline with a t1/2 of 0.8–1 day during which the majority (>90%) of circulating viremia is eliminated, viral decay kinetics slow with t1/2 of 12–14 days [33–35]. Plasma clearance of virions is generally rapid (15 min to 2 h) and shorter than decay rates of viremia described upon initiating antiretroviral therapy. Thus, introduction of antiretroviral therapy with prevention of transmission of infection results in decline in viremia because of the death or elimination of virus-infected cells. Differing rates of decline represent removal of cells with different elimination rates; cells producing the majority of HIV are rapidly eliminated.
Additional information regarding the dynamics of response to therapy has been reported with the incorporation of integrase inhibitors into combination therapy. Prior to integrase inhibitors, antiretroviral therapy prevented newly infected cells at early steps (attachment, fusion, reverse transcription) or late steps (protease-mediated maturation). Upon initiation of combination therapy, infected cells that have completed reverse transcription can still undergo integration and virion production, and contribute to level of viremia. In the presence of integrase inhibitors, however, cells that contain unintegrated linear DNA will be blocked from integration, and will not contribute to overall virion production [41]. Reduction in the size of second-phase compartment [41,42], which can be explained by the drug’s mechanism of action [43].
Source of persistent viremia
Combination therapy has been implemented for over 10 years, permitting investigation of the long-term effects of antiretroviral therapy on viremia in patients suppressed for prolonged periods. A number of studies have demonstrated that HIV persists despite prolonged suppression, suggesting no further decay in viremia. Analysis of patients suppressed for prolonged periods (7 years) revealed that viral RNA levels continue to decline for several years on therapy followed by a relatively stable persistent viremia [44].
The source of HIV viremia on therapy in such patients is uncertain [45]. Viremia may be the result of long-lived cells with integrated proviruses that constitutively produce HIV at low-levels or remain latently infected with HIV and are induced to produce virus as the result of either antigenic or other immune stimulation. A reservoir of persistent cells can be maintained directly by cellular longevity, as many immune cells have long half-lives or indirectly by cellular expansion of infected cells. Alternatively, persistent viremia may be the result of active HIV replication, and residual viremia is the product of low-level spread of HIV to uninfected susceptible host cells. Incomplete drug suppression and/or anatomic sanctuary sites such as central nervous system have been proposed as mechanisms to explain ongoing active cycles of replication in HIV infection.
Current combination antiretroviral therapy suppresses viral RNA levels utilizing different mechanisms. Using sensitive single-copy assays and comparing pretherapy and posttherapy RNA levels in individuals initiating combination therapy nonnucleoside reverse transcriptase inhibitor (NRTI) + protease inhibitor- based therapy, the median suppression of HIV viremia was over 17 000 fold [46]. It remains uncertain, however, whether active replication persists in patients despite profound suppression. In order to investigate whether active replication was present in suppressed patients, we compared relative levels of HIV RNA in patients treated with NNRTI and protease inhibitors containing regimens. Despite varying mechanisms of inhibition and differing regimen potencies, we measured comparable levels of persistent viremia in the range of 3 copies/ml plasma [46]. These data suggested the unexpected result that HIV replication may be completely arrested by combination antiretroviral therapy. The explanation for differences in clinical outcome of regimens with different potency may be related not to ongoing replication, but other factors such as adherence and or preexisting drug resistance. Additional genetic data have also suggested the lack of active replication during therapy [47–49].
In contrast, a number of studies have suggested the presence of ongoing replication during drug suppression, including genetic analysis reporting the accumulation of new genetic diversity during suppressive therapy, and quantitative analysis of the decay in latently infected cells in HIV reservoirs during suppressive therapy [50–54].
Understanding the source of persistent viremia in suppressed patients is critical to designing curative strategies for HIV infection (Fig. 2). If ongoing replication is responsible for persistent viremia, then improvements in antiretroviral therapeutic regimens represent useful approach to eradicate HIV infection. Alternatively, if reservoirs of long-lived cells are responsible for persistent viremia, then new strategies in addition to antiretroviral therapy will be required to cure infection.
Figure 2. Identifying the source of HIV replication during suppressive antiretroviral therapy is essential in designing new strategies to eradicate HIV infection.
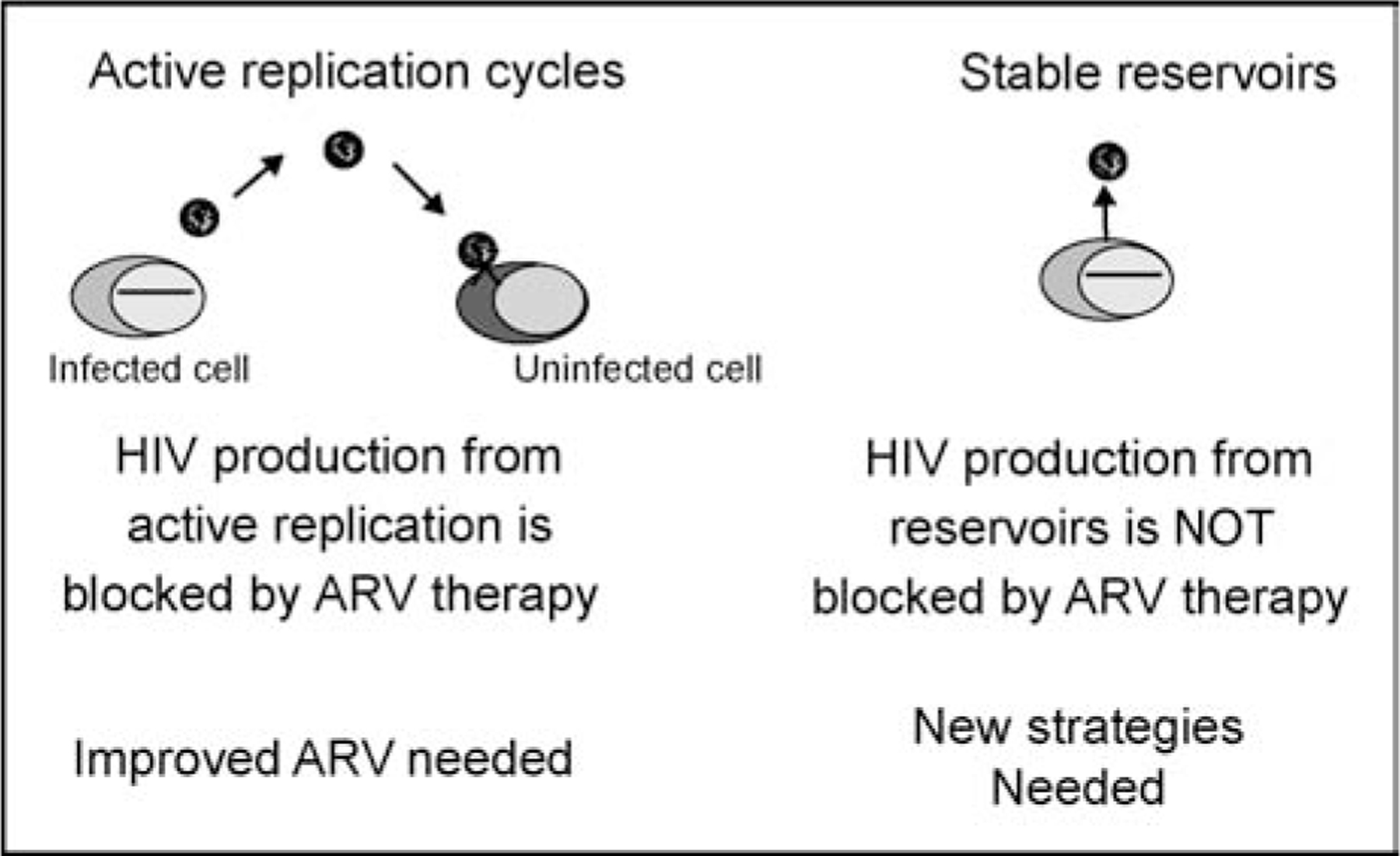
The presence of ongoing replication implies current therapy is inadequate, and new advances in antiviral chemotherapy are necessary. The predominance of reservoirs of long-lived cells with integrated proviruses implies new strategies will be necessary for HIV eradication. ARV, antiretroviral.
Drug intensification studies
In order to investigate the source of residual viremia, interventional studies have been designed to quantitate HIV viremia during suppressive therapy. In principle, such studies investigate whether the inclusion of an additional antiretroviral to a suppressed regimen results in a change in level of detectable HIV. If active cycles of replication are responsible for persistence, drug intensification may result in decreases in viremia but increasing the overall potency of drug therapy, or expanding the penetration of drugs into anatomic sanctuaries of HIV replication.
Havlir et al. [55] performed an early intensification study in individuals with viremia suppressed below commercial limits of detection, but were on dual therapy of indinavir and efavirenz, not standard three drug regimens. Intensification of individuals by the addition of the NRTI abacavir resulted in rapid declines in viremia as measured by sensitive PCR amplification. These data suggested the presence of ongoing replication in patients that was suppressible by drug intensification. Viremia decayed with a half-life of 6–7 days, suggesting that relatively short-lived cells were responsible for persistent viremia. Of note, the level of persistent viremia prior to intensification (c. 3–20 copies) was generally higher than that detected in studies of individuals on three drug regimens.
Additional intensification studies were implemented to investigate whether ongoing replication was taking place in individuals with suppressed viremia undergoing therapy with standard three-drug combination therapy. Dinoso et al. [56••] studied patients with suppression on standard antiretroviral therapy and no prior resistance; individuals undergoing protease inhibitor-based therapy were intensified with efavirenz, and patients on NNRTI-based regimens were intensified with ritonavir-boosted atazanavir or lopinavir. Intensification was carried out for 4–8 weeks and sensitive single copy assays detected no significant changes in viral RNA levels as a result of intensification (Fig. 3). Drug level monitoring demonstrated that therapeutic levels of intensified agent were achieved. These data demonstrated that viremia persisted during intensification without decay suggesting active replication in relatively short-lived cells was not responsible for persistent viremia. Yilmaz et al. [57] performed an intensification study with a similar design but included an investigation of central nervous system replication by comparing viral RNA levels in blood and cerebrospinal fluid (CSF), measuring markers of inflammation in CSF, and sequencing intensification agents that penetrate central nervous system well (e.g., maraviroc) or poorly (enfuvirtide). No decreases in plasma or CSF viremia, and no changes in soluble inflammatory markers neopterin or β2-microglobulin were detected.
Figure 3. HIV RNA levels in nine patients undergoing intensification with atazanavir/ritonavir, efavirenz, or lopinavir/ritonavir.
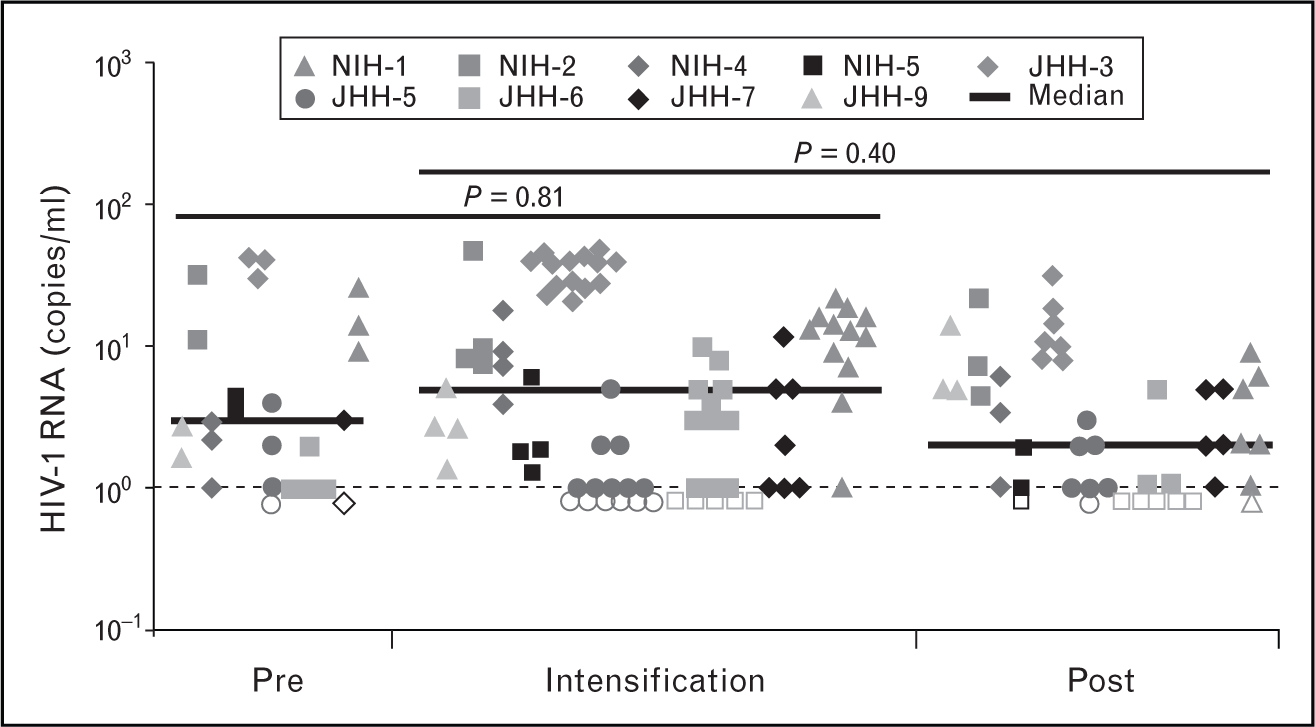
No significant differences in viral RNA levels were detected prior to during or following intensification. Reproduced with permission from [56••].
In these initial studies, reverse transcriptase or protease inhibitors were chosen as intensifying agents. As described above, the persistence of unintegrated linear DNA products that slowly integrate into the host genome may continue as drug therapy with NRTI/protease inhibitor-based therapy in initiatated. Additional studies using the first-in-class integrase inhibitor were carried out to investigate whether raltegravir intensification will result in decreases in persistent HIV viremia by preventing integration of linear DNA. McMahon et al. [58] detected no decrease in viremia after 4-week intensification with raltegravir; therapeutic levels of raltegravir were achieved, and these data suggested no evidence of ongoing replication to persistent viremia. Subsequently, ACTG5244 [59••] investigated a longer (12-week) raltegravir intensification in a randomized crossover design, and detected no change in viral RNA levels as a result of integrase inhibitor intensification, again suggesting active infection of relatively short-lived cells was not responsible for persistent viremia.
Hatano et al. [60•] investigated the presence of ongoing replication in a select group of patients undergoing antiretroviral therapy who had minimal recovery of CD4 cells following initiation of antiretroviral therapy. The cause of lack of immune cell increases in such patients is unknown, but ongoing replication represents an important possibility to evaluate. In a placebo-controlled 24-week trial of raltegravir intensification of these immune nonresponders, no decreases in persistent viremia or improvement in immune parameters was detected, suggesting ongoing replication was not responsible for relatively small number of CD4 cells recovered on therapy. Baseline viral RNA levels in the patients in this study were similar to that measured in other studies of low-level HIV viremia. Thus, the reason for blunted CD4 cells recovery was not elevated persistent viremia or evidence of active replication.
Buzon et al. [61••] carried out a randomized study of 69 participants, 44 of whom received raltegravir intensification for 48 weeks. In addition to measuring persistent viremia, cell-associated HIV DNA was also analyzed. The authors detected transient increases in the levels of 2-LTR circles in a minority (14/44) of patients undergoing intensification. The accumulation of 2-LTR circles suggested ongoing replication in cells in these patients. The transient nature of the increase in circles suggests these newly infected cells decayed over time. It is not known whether these transient changes resulted in changes in plasma viremia; no changes in persistent viremia were detected using single copy assays, but sampling did not coincide with periods when increases in circular forms of DNA. Increases in 2-LTR circles for prolonged periods suggest a relatively large number of cells are undergoing active infection. Following increases, 2-LTR circles declined, but the kinetics of decline appeared more variable than standard first-phase or second-phase decay. The decay in 2-LTR circles did not, however, result in decrease in plasma viremia by the end of the intensification period, and no changes in total or integrated HIV DNA were detected by the end of the study.
Recently, Yukl et al. [62] intensified a series of suppressed patients with raltegravir and carried out an extensive anatomic analysis of HIV infection in gastrointestinal tract; although no decreases in peripheral viremia were detected, this group did identify changes in levels of unspliced RNA in CD4-positive T cells c. 5-fold decreases in unspliced HIV RNA in CD4 cells obtained from the terminal ileum, a site in which CD4 cells have HIV RNA/cell copy numbers c. 10-fold higher compared with other compartments and peripheral blood. These data suggested raltegravir therapy reduced a focus of ongoing replication in a localized compartment. Other findings included substantial increases in HIV DNA levels in duodenum, which are of uncertain cause.
The reasons explaining differences in the findings of intensification studies with respect to ongoing replication are not yet clear. Patient selection, duration of suppression, presence of drug resistance may reconcile some, but not all, results. These studies illustrate the complexities in detecting cell-associated nucleic acid and suggest additional comprehensive analyses of plasma and cell-associated HIV will be useful to further investigate the nature of persistent viremia on therapy. One common observation in all of these studies is the lack of effect of drug intensification in HIV viremia, even in patients who had evidence suggestive of ongoing replication. The finding that, to a substantial degree, residual viremia is not subject to additional suppression by antiretrovirals, strongly suggests additional measures will be necessary to address large reservoirs of HIV-infected cells.
Conclusion
The persistence of plasma virus despite suppressive therapy remains an intriguing observation. As described above, HIV virions have a short half-life in peripheral blood, so the persistent viremia measured during therapy, although relatively low, is likely to reflect substantial production from a large reservoir of cells. The inability of drug intensification to reduce persistent viremia implies that activated short-lived cells are not responsible for the majority of this virus production. New approaches designed to eliminate chronic production from long-lived reservoirs will be necessary as part of strategies to cure HIV infection.
Key points.
HIV replication is both rapid and error prone, infecting a wide variety of cells and generating viral diversity within infected individuals.
Combination antiretroviral therapy suppresses but does not eradicate infection, yielding a remaining reservoir of infected cells. Understanding the nature of replication during therapy will inform strategies to eradicate infection.
A number of drug addition studies have been carried out to investigate whether persistent viremia can be further suppressed by antiretroviral intensification. Intensification with integrase inhibitors does not result in decreases in viremia, suggesting active infection did not take place. Transient increases in 2-LTR circles during raltegravir intensification in some patients in one study of prolonged raltegravir intensification suggested the presence of virus replication.
Viremia persists without decrease despite regimen intensification.
New strategies to address long-lived viral reservoirs will be necessary to eradicate HIV infection.
Acknowledgements
The authors would like to thank S. Palmer, A Wiegand, J. Kovacs, H.C. Lane, H. Masur, J. Mellors for insightful discussions.
The study received funds from National Cancer Institute, National Institutes of Health.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 88).
- 1.Wong JK, Hezareh M, Gunthard HF, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997; 278:1291–1295. [DOI] [PubMed] [Google Scholar]
- 2.Dornadula G, Zhang H, VanUitert B, et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999; 282:1627–1632. [DOI] [PubMed] [Google Scholar]
- 3.Palmer S, Wiegand AP, Maldarelli F, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol 2003; 41:4531–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hazuda D, Iwamoto M, Wenning L. Emerging pharmacology: inhibitors of human immunodeficiency virus integration. Annu Rev Pharmacol Toxicol 2009; 49:377–394. [DOI] [PubMed] [Google Scholar]
- 5.Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis 2009; 48:931–939. [DOI] [PubMed] [Google Scholar]
- 6.Gotte M, Rausch JW, Marchand B, et al. Reverse transcriptase in motion: conformational dynamics of enzyme-substrate interactions. Biochim Biophys Acta 2010; 1804:1202–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarafianos SG, Marchand B, Das K, et al. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol 2009; 385:693–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol 1995; 69:5087–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dimitrov DS, Willey RL, Sato H, et al. Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol 1993; 67:2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato H, Orenstein J, Dimitrov D, Martin M. Cell-to-cell spread of HIV-1 occurs within minutes and may not involve the participation of virus particles. Virology 1992; 186:712–724. [DOI] [PubMed] [Google Scholar]
- 11.Pauza CD, Trivedi P, McKechnie TS, et al. 2-LTR circular viral DNA as a marker for human immunodeficiency virus type 1 infection in vivo. Virology 1994; 205:470–478. [DOI] [PubMed] [Google Scholar]
- 12.Morlese J, Teo IA, Choi JW, et al. Identification of two mutually exclusive groups after long-term monitoring of HIV DNA 2-LTR circle copy number in patients on HAART. AIDS 2003; 17:679–683. [DOI] [PubMed] [Google Scholar]
- 13.McDermott JL, Martini I, Ferrari D, et al. Decay of human immunodeficiency virus type 1 unintegrated DNA containing two long terminal repeats in infected individuals after 3 to 8 years of sustained control of viremia. J Clin Microbiol 2005; 43:5272–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharkey M, Triques K, Kuritzkes DR, Stevenson M. In vivo evidence for instability of episomal human immunodeficiency virus type 1 cDNA. J Virol 2005; 79:5203–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reigadas S, Andreola ML, Wittkop L, et al. Evolution of 2-long terminal repeat (2-LTR) episomal HIV-1 DNA in raltegravir-treated patients and in in vitro infected cells. J Antimicrob Chemother 2010; 65:434–437. [DOI] [PubMed] [Google Scholar]
- 16.Panther LA, Coombs RW, Aung SA, et al. Unintegrated HIV-1 circular 2-LTR proviral DNA as a marker of recently infected cells: relative effect of recombinant CD4, zidovudine, and saquinavir in vitro. J Med Virol 1999; 58:165–173. [DOI] [PubMed] [Google Scholar]
- 17.Pierson TC, Kieffer TL, Ruff CT, et al. Intrinsic stability of episomal circles formed during human immunodeficiency virus type 1 replication. J Virol 2002; 76:4138–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delaugerre C, Charreau I, Braun J, et al. Time course of total HIV-1 DNA and 2-long-terminal repeat circles in patients with controlled plasma viremia switching to a raltegravir-containing regimen. AIDS 2010; 24:2391–2395. [DOI] [PubMed] [Google Scholar]
- 19.Brussel A, Mathez D, Broche-Pierre S, et al. Longitudinal monitoring of 2-long terminal repeat circles in peripheral blood mononuclear cells from patients with chronic HIV-1 infection. AIDS 2003; 17:645–652. [DOI] [PubMed] [Google Scholar]
- 20.Bushman F Measuring covert HIV replication during HAART: the abundance of 2-LTR circles is not a reliable marker. AIDS 2003; 17:749–750. [DOI] [PubMed] [Google Scholar]
- 21.Butler SL, Johnson EP, Bushman FD. Human immunodeficiency virus cDNA metabolism: notable stability of two-long terminal repeat circles. J Virol 2002; 76:3739–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens M, De Clercq E, Balzarini J. The regulation of HIV-1 transcription: molecular targets for chemotherapeutic intervention. Med Res Rev 2006; 26:595–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bannwarth S, Gatignol A. HIV-1 TAR RNA: the target of molecular interactions between the virus and its host. Curr HIV Res 2005; 3:61–71. [DOI] [PubMed] [Google Scholar]
- 24.Fujii K, Hurley JH, Freed EO. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat Rev Microbiol 2007; 5:912–916. [DOI] [PubMed] [Google Scholar]
- 25.Adamson CS, Freed EO. Human immunodeficiency virus type 1 assembly, release, and maturation. Adv Pharmacol 2007; 55:347–387. [DOI] [PubMed] [Google Scholar]
- 26.Venzke S, Keppler OT. Role of macrophages in HIV infection and persistence. Expert Rev Clin Immunol 2006; 2:613–626. [DOI] [PubMed] [Google Scholar]
- 27.Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 1995; 267:483–489. [DOI] [PubMed] [Google Scholar]
- 28.Speck RR, Flexner C, Tian CJ, Yu XF. Comparison of human immunodeficiency virus type 1 Pr55(Gag) and Pr160(Gag-pol) processing intermediates that accumulate in primary and transformed cells treated with peptidic and nonpeptidic protease inhibitors. Antimicrob Agents Chemother 2000; 44:1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humphrey RW, Ohagen A, Davis DA, et al. Removal of human immunodeficiency virus type 1 (HIV-1) protease inhibitors from preparations of immature HIV-1 virions does not result in an increase in infectivity or the appearance of mature morphology. Antimicrob Agents Chemother 1997; 41:1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jardine DK, Tyssen DP, Birch CJ. Effect of protease inhibitors on HIV-1 maturation and infectivity. Antiviral Res 2000; 45:59–68. [DOI] [PubMed] [Google Scholar]
- 31.Pas S, Rossen JW, Schoener D, et al. Performance evaluation of the new Roche Cobas AmpliPrep/Cobas TaqMan HIV-1 test version 2.0 for quantification of human immunodeficiency virus type 1 RNA. J Clin Microbiol 2010; 48:1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elbeik T, Alvord WG, Trichavaroj R, et al. Comparative analysis of HIV-1 viral load assays on subtype quantification: Bayer Versant HIV-1 RNA 3.0 versus Roche Amplicor HIV-1 Monitor version 1.5. J Acquir Immune Defic Syndr 2002; 29:330–339. [DOI] [PubMed] [Google Scholar]
- 33.Ho DD, Neumann AU, Perelson AS, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995; 373:123–126. [DOI] [PubMed] [Google Scholar]
- 34.Wei X, Ghosh SK, Taylor ME, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 1995; 373:117–122. [DOI] [PubMed] [Google Scholar]
- 35.Perelson AS, Neumann AU, Markowitz M, et al. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 1996; 271:1582–1586. [DOI] [PubMed] [Google Scholar]
- 36.Liszewski MK, Yu JJ, O’Doherty U. Detecting HIV-1 integration by repetitive-sampling Alu-gag PCR. Methods 2009; 47:254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu JJ, Wu TL, Liszewski MK, et al. A more precise HIV integration assay designed to detect small differences finds lower levels of integrated DNA in HAART treated patients. Virology 2008; 379:78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torti C, Quiros-Roldan ME, Cologni G, et al. Plasma HIV load and proviral DNA decreases after two standard antiretroviral regimens in HIV-positive patients naive to antiretrovirals. Curr HIV Res 2008; 6:43–48. [DOI] [PubMed] [Google Scholar]
- 39.Koelsch KK, Liu L, Haubrich R, et al. Dynamics of total, linear nonintegrated, and integrated HIV-1 DNA in vivo and in vitro. J Infect Dis 2008; 197:411–419. [DOI] [PubMed] [Google Scholar]
- 40.Ibanez A, Puig T, Elias J, et al. Quantification of integrated and total HIV-1 DNA after long-term highly active antiretroviral therapy in HIV-1-infected patients. AIDS 1999; 13:1045–1049. [DOI] [PubMed] [Google Scholar]
- 41.Murray JM, Emery S, Kelleher AD, et al. Antiretroviral therapy with the integrase inhibitor raltegravir alters decay kinetics of HIV, significantly reducing the second phase. AIDS 2007; 21:2315–2321. [DOI] [PubMed] [Google Scholar]
- 42.Markowitz M, Nguyen BY, Gotuzzo E, et al. Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr 2007; 46:125–133. [DOI] [PubMed] [Google Scholar]
- 43.Sedaghat AR, Dinoso JB, Shen L, et al. Decay dynamics of HIV-1 depend on the inhibited stages of the viral life cycle. Proc Natl Acad Sci U S A 2008; 105:4832–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmer S, Maldarelli F, Wiegand A, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 2008; 105:3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siliciano JD, Siliciano RF. A long-term latent reservoir for HIV-1: discovery and clinical implications. J Antimicrob Chemother 2004; 54:6–9. [DOI] [PubMed] [Google Scholar]
- 46.Maldarelli F, Palmer S, King MS, et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog 2007; 3:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joos B, Fischer M, Kuster H, et al. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci U S A 2008; 105:16725–16730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey JR, Sedaghat AR, Kieffer T, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol 2006; 80:6441–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Persaud D, Pierson T, Ruff C, et al. A stable latent reservoir for HIV-1 in resting CD4(+) T lymphocytes in infected children. J Clin Invest 2000; 105:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chun TW, Justement JS, Moir S, et al. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: implications for eradication of virus. J Infect Dis 2007; 195:1762–1764. [DOI] [PubMed] [Google Scholar]
- 51.Chun TW, Nickle DC, Justement JS, et al. HIV-infected individuals receiving effective antiviral therapy for extended periods of time continually replenish their viral reservoir. J Clin Invest 2005; 115:3250–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shiu C, Cunningham CK, Greenough T, et al. Identification of ongoing human immunodeficiency virus type 1 (HIV-1) replication in residual viremia during recombinant HIV-1 poxvirus immunizations in patients with clinically undetectable viral loads on durable suppressive highly active antiretroviral therapy. J Virol 2009; 83:9731–9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gunthard HF, Frost SD, Leigh-Brown AJ, et al. Evolution of envelope sequences of human immunodeficiency virus type 1 in cellular reservoirs in the setting of potent antiviral therapy. J Virol 1999; 73:9404–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frenkel LM, Wang Y, Learn GH, et al. Multiple viral genetic analyses detect low-level human immunodeficiency virus type 1 replication during effective highly active antiretroviral therapy. J Virol 2003; 77:5721–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Havlir DV, Strain MC, Clerici M, et al. Productive infection maintains a dynamic steady state of residual viremia in human immunodeficiency virus type 1-infected persons treated with suppressive antiretroviral therapy for five years. J Virol 2003; 77:11212–11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56••.Dinoso JB, Kim SY, Wiegand AM, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci U S A 2009; 106:9403–9408. [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration of the lack of decline of HIV RNA during drug intensification with therapeutic levels of intensifying drug.
- 57.Yilmaz A, Verhofstede C, D’Avolio A, et al. Treatment intensification has no effect on the HIV-1 central nervous system infection in patients on suppressive antiretroviral therapy. J Acquir Immune Defic Syndr 2010. [DOI] [PubMed]
- 58.McMahon D, Jones J, Wiegand A, et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clin Infect Dis 2010; 50:912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59••.Gandhi RT, Zheng L, Bosch RJ, et al. The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: a randomized controlled trial. PLoS Med 2010; 7:e1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]; Randomized study of raltegravir intensification demonstrating no effect of the addition of integrase inhibitor on the level of plasma viremia.
- 60•.Hatano HH, Hayes TL, Dahl V, Sinclair E, Lee T-H, Hoh R, Lampris H, Hunt PW, Palmer S, McCune JM, Martin JN, Busch MP, Shacklett BL, Deeks SG. A randomized, controlled trial of raltegravir intensification in antiretroviral-treated, HIV-infected patients with a suboptimal CD4+ T cell response. J Infect Dis (in press). [DOI] [PMC free article] [PubMed]; Raltegravir intensification of immune nonresponders does not result in suppression of viremia or improvements in immune profiles. First demonstration that active replication does not explain a blunted CD4 cells recovery following antiretroviral suppression.
- 61••.Buzon MJ, Massanella M, Llibre JM, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med Apr 2010; 16:460–465. [DOI] [PubMed] [Google Scholar]; Detection of 2-LTR circles in patients during raltegravir intensification suggesting the presence of ongoing replication in a subset of patients.
- 62.Yukl SA, Shergill AK, McQuaid K, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010; 24:2451–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]