

Abstract
Objective:
In this study, we characterized elvitegravir activity in the context of raltegravir resistance mutations.
Design:
Using site-directed mutagenesis, we generated recombinant integrase proteins and viruses harboring raltegravir resistance mutation to assess the biochemical and cellular activity of elvitegravir in the presence of such mutants.
Methods:
Recombinant proteins were used in gel-based assays. Antiviral data were obtained with reporter viruses in a single-round infection using a luciferase-based assay.
Results:
Although main raltegravir resistance pathways involving mutations at integrase position 148 and 155 confer cross-resistance to elvitegravir, elvitegravir remains fully active against the Y143R mutant integrase and virus particles.
Conclusion:
In addition to favorable pharmacokinetics compared to raltegravir, our findings provide the rationale for using elvitegravir in patients failing raltegravir because of the integrase mutation Y143.
Keywords: elvitegravir, HIV-1, integrase inhibitor, integrase resistance, raltegravir
Introduction
HIV-1 integrase is required for viral replication and raltegravir (RAL) is the first approved integrase inhibitor due to its remarkable antiviral efficacy and minimal side-effects [1]. The usefulness of RAL tends to be limited by the rapid occurrence of integrase mutations. These mutations involve three main pathways associated with integrase residues 143, 148, and 155 [2]. The Q148H mutation is generally associated with a secondary mutation at position 140 [e.g. G140S-Q148H (SH)] that rescues the deleterious effects of the primary Q148H mutation on integrase catalytic activity and further increases RAL resistance [2,3].
Elvitegravir (EVG) is the next most advanced integrase inhibitor [4]. Unlike RAL, EVG blood concentration is not limited and can be boosted by co-administration with ritonavir (HIV-1 protease inhibitor) or cobicistat (GS-9350; not active against HIV-1 protease) in a once-daily formulation [5]. Mutations at integrase positions 148 and 155 occurring in patients failing RAL therapy confer cross-resistance to EVG [3,6]. Recently, a prospective study including antiretroviral-experienced patients receiving RAL-based regimens showed that viruses harboring integrase mutations Y143C/H/R exhibited only limited resistance to EVG [7]. However, those viruses isolated from two heavily pretreated patients also showed additional integrase mutations at residues 97 and 72 and 163 [7]. In the present study, we generated the three clinically relevant integrase point mutants at position 143 to assess whether EVG could overcome RAL resistance both at the integrase and replication levels in those mutants.
Materials and methods
Integrase substrates
Oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, Iowa, USA). Oligonucleotides 21t (GTGTGGAAAATCTCTAGCAGT), 19t (GTGTGGAAAATCTCTAGCA), and 21b (ACTGCTAGAGATTTTCCACAC) were used to generate the in-vitro substrates for integrase assays. Single-stranded oligonucleotides 21t and 19t were labeled at the 5′-end using T4 polynucleotide kinase (New England Biolabs, Ipswich, Massachusetts, USA) with [γ−32P] ATP (Perkin-Elmer Life and Analytical Sciences, Boston, Massachusetts, USA) according to the manufacturers’ instructions. Unincorporated isotopes were removed using the Mini Quick Spin Oligo Columns (Roche Diagnostics, Indianapolis, Indiana, USA). The DNA duplexes 21t/21b (3′-processing substrate) and 19t/21b (strand transfer substrate) were annealed by addition of an equal concentration of the complementary strand, heating to 95°C, and slow cooling to room temperature.
Integrase reactions
Integrase reactions were carried out by mixing 20 nmol/l DNA with 400 nmol/l integrase in a buffer containing 50 mmol/l, 3-(N-morpholino)propanesulfonic acid (MOPS) pH 7.2, 7.5 mmol/l MgCl2, 14 mmol/l 2-mercaptoethanol, and drugs [3] or 10% DMSO (dimethyl sulfoxide, the drug solvent). Reactions were performed at 37°C for 120 min and quenched by addition of an equal volume of loading buffer [formamide containing 1% SDS (sodium dodecyl sulfate), 0.25% bromophenol blue, and xylene cyanol]. Reaction products were separated in 16% polyacrylamide denaturing sequencing gels. Dried gels were visualized using a Typhoon 8600 (GE Healthcare, Piscataway, New Jersey, USA). Densitometry analysis was performed using ImageQuant 5.1 software from GE Healthcare.
Antiviral assay
Reporter viruses were obtained using lentiviral vectors only capable of single-round infection. After deletion of the envelope-coding region, a luciferase reporter gene was cloned into the Nef region of a full length HIV-1 genome. Co-transfection of this construct with a vesicular stomatitis virusglycoprotein(VSV-G) encoding plasmid in293Tcells leads after 64 h to the production of VSV-G pseudo-typed virions. Culture supernatants were clarified by centrifugation and stored at −70 °C for further use. To produce mutant integrase-containing particles, a fragment of gagpol was cloned into a pGEM shuttle vector used for the site-directed mutagenesis. The mutated fragment was then exchanged back in the HIV-1Δenv instead of the wild-type gag-pol. To assess antiviral activity of drugs, 293T cells were plated at 12000 cells per well in 96-well culture plates. After 16 h, cells were incubated for 4 h at 37 °C with serial dilutions of drugs before infection. Wild-type and mutant viruses’ replication was determined after 48 h using the Bright-GloTM reagent (Promega, Madison, Wisconsin, USA) with a Victor Wallace luminometer.
Results and discussion
First, we generated recombinant enzymes with each of the clinically relevant mutations: Y143R/C/H (Fig. 1) [8–11]. The biochemical activity of the mutants Y143R and Y143H was near normal for both 3′-processing (3′-P) and strand transfer, whereas the Y143C mutant was impaired for strand transfer (Fig. 1a). In agreement with the clinical resistance data [8,9], the Y143R mutant was highly resistant to RAL and its resistance level was between the N155H and the G140S-Q148H (SH) mutants (Fig. 1b). Surprisingly, the Y143C/H mutants selected in vivo showed limited resistance to RAL (Fig. 1b). Histidine, like tyrosine, may be able to generate a π–π interaction with the oxadiazole ring of RAL, explaining the lack of resistance. Because the mutation Y143R requires two adjacent mutations in the integrase coding sequence, whereas the Y143C and Y143H mutants are coded by each of the single mutants that lead to the Y143R mutation, the existence of the Y143C/H suggests they represent genetic intermediates leading to the emergence of the highly resistant Y143R mutant [11].
Fig. 1. Biochemical and pharmacological properties of clinically relevant mutants.
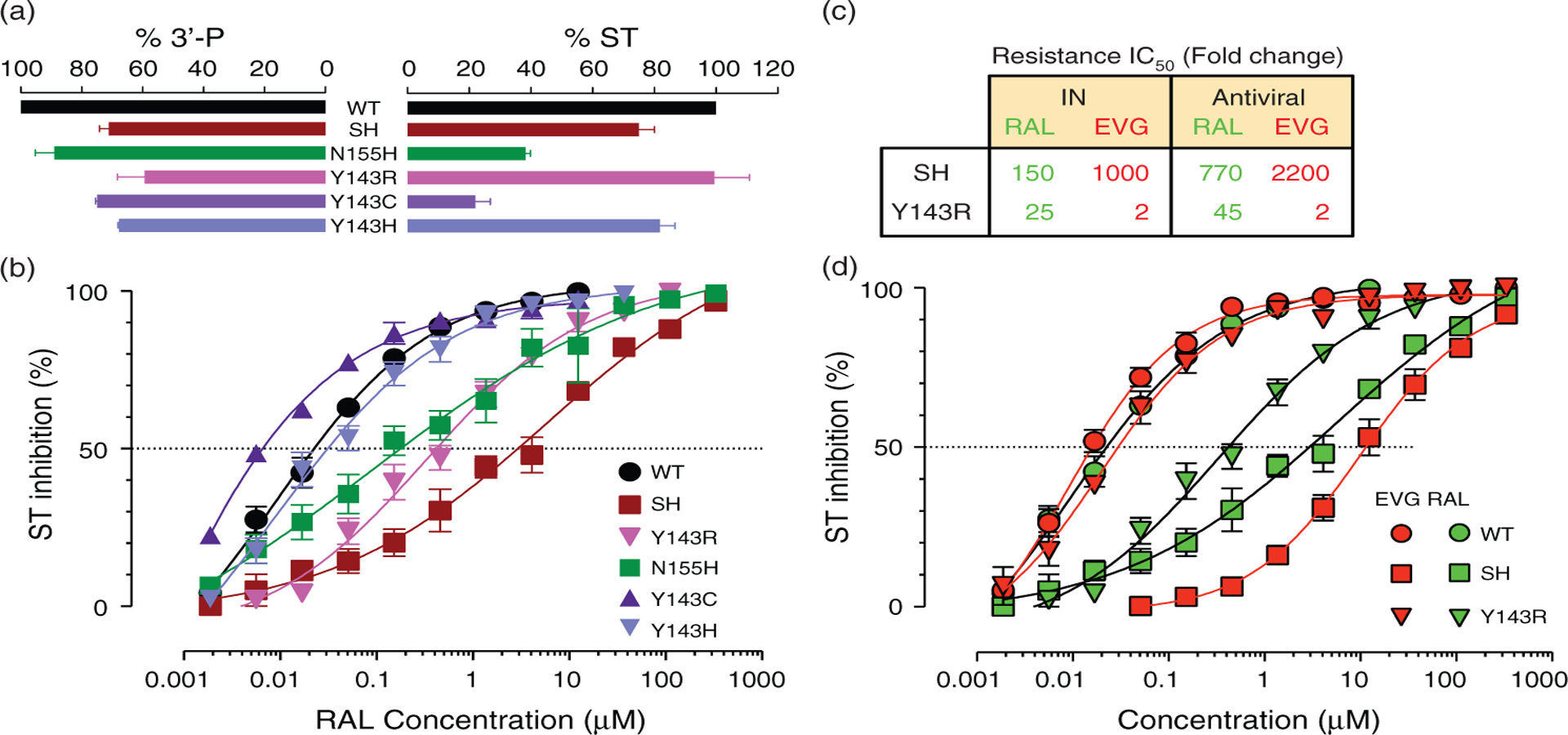
(a) Biochemical activity of the integrase mutants at residues 140, 143, 148, and 155. 3′-processing (3′-P) and strand transfer (ST) activities of the mutants are normalized and represented as percentage of wild-type activity. (b) Pharmacological properties of the integrase mutants Y143R/C/H, SH, and N155H compared to the wild-type enzyme. Strand transfer inhibition of the recombinant integrase enzymes by raltegravir (RAL) was monitored as described [3]. (c) Inhibition of wild-type and mutants (SH and Y143R) recombinant integrase by RAL and elvitegravir (EVG). Mutant enzymes were generated and strand transfer activity was measured as described [3]. Mean and standard deviations are from three to six independent determinations. (d) Fold change IC50 for the SH and Y143R mutants in the presence of RAL and EVG compared to wild-type. Antiviral data were obtained with pseudo-typed viruses in a single round infection using a luciferase-based assay.
When we tested EVG against the integrase mutants, the N155H and SH mutants showed cross-resistance to EVG (Fig. 1c), consistent with a recent report [3]. However, the Y143R mutant showed only minimal resistance to EVG (only a two-fold increase in IC50), whereas the SH mutation was 1000-fold cross-resistant to EVG compared to the wild-type enzyme (Fig. 1c and d)[3].
Next, we generated the two corresponding mutations, the double mutation SH and the mutation Y143R in virus particles to determine whether differential resistance to EVG and RAL was also observed in vivo. Consistent with prior results [7,10,11], both mutant viruses were highly resistant to RAL and the SH mutant was resistant to both drugs (Fig. 1d). However, the Y143R virus remained sensitive to EVG with the same two-fold change in IC50 (Fig. 1d). Our results clearly demonstrate that EVG overcomes RAL resistance induced by the integrase mutation Y143R. They are also consistent with a recent publication showing that viruses isolated from two patients failing RAL therapy and harboring integrase mutations including mutations at position Y143, exhibited only minimal cross-resistance to EVG.
Recently, the complete crystal structure of the prototype foamy virus (PFV) integrase has been resolved in complex with a substrate mimicking the viral DNA end after its 3′-processing (3′-P) by integrase, providing the first three-dimensional structure of an active integrase active site [12]. The active site of the PFV and HIV enzymes are structurally and functionally related, and RAL as well as EVG is able to inhibit PFV integrase in vitro [13]. A recent NMR study using the HIV-1 integrase core domain showed that the HIV-1 and PFV integrase flexible loops (residues 140–149 in HIV and 209–218 in PFV) are highly similar [14] and modelization of the HIV integrase intasome provided further evidence for the similarities between the active sites of the PFV and HIV integrase enzymes [15]. Introducing mutations in PFV integrase corresponding to HIV-1 integrase amino acids 155 and 148 results in comparable RAL-resistance profiles [16]. Notably, the PFV-drug-DNA co-crystal structures reveal that only RAL forms π–π interaction with Y212 of PFV integrase (corresponding to integrase residue Y143, Fig. 2a) [2,3]. The interaction of RAL with Y143 provides a rationale for the resistance of Y143 mutants to RAL. By contrast, EVG, unlike RAL does not interact with this tyrosine (Fig. 2b) [12,15], which provides a molecular explanation for the selective activity of EVG against the Y143R mutant (Fig. 2).
Fig. 2. Differential binding of raltegravir and elvitegravir within the prototype foamy virus integrase active site.
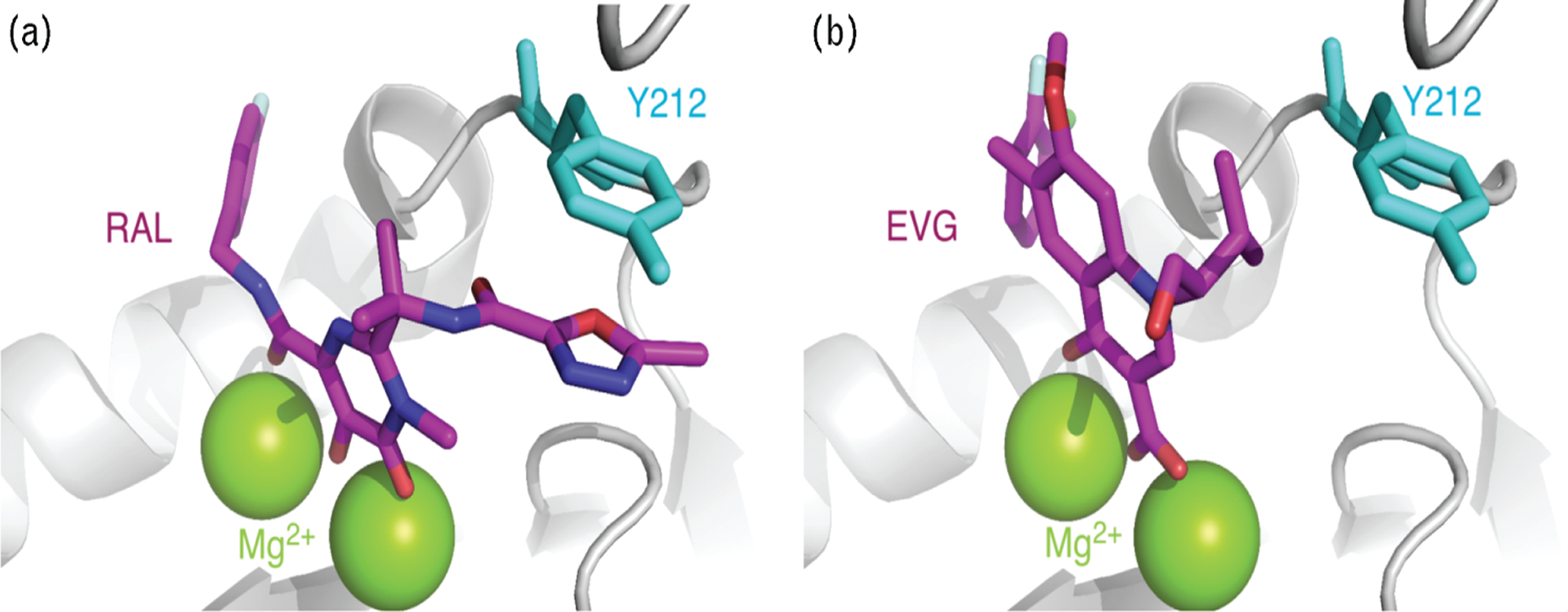
Differential binding of raltegravir (RAL) (a) and elvitegravir (EVG) (b) within the prototype foamy virus (PFV) integrase active site. Representations were generated using the PFV integrase crystal structures [PDB ID code 3L2T for RAL (a) and 3L2U for EVG (b)] [12] and are displayed using the MacPyMol software. Integrase is shaded in light gray except for residue Y212 (light blue) that corresponds to HIV-1 integrase residue Y143 implicated in RAL resistance. The two metal co-factors Mg2+ are in green. RAL and EVG are in magenta with nitrogen atoms in blue, oxygens in red, fluorines in gray, and chlorides in green.
Our study provides a molecular rationale for using EVG in patients failing RAL treatment because of Y143R mutation. It also underlines the value of biochemical assays with recombinant integrase to rationalize the molecular determinants of resistance to integrase inhibitors and to test novel inhibitors to overcome resistance to clinically used integrase inhibitors.
Acknowledgements
M.M., D.R., C.M., and Y.P conceived and designed the experiments. M.M., N.V., X.Z., and A.N performed the experiments. M.M., N.V., and Y.P analyzed the data. K.M. contributed to reagents/materials. M.M., C.M., and Y.P. wrote the paper. Our studies are supported by the Intramural Research Program of the NIH, National Cancer Institute (NCI), Center for Cancer Research (CCR) (Z01BC007333-09LMP), and by a grant from IATAP (Intramural AIDS Target Antiviral Program).
References
- 1.Grinsztejn B, Nguyen BY, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, et al. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet 2007; 369:1261–1269. [DOI] [PubMed] [Google Scholar]
- 2.Metifiot M, Marchand C, Maddali K, Pommier Y. Resistance to integrase inhibitors. Viruses 2010; 2:1347–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metifiot M, Maddali K, Naumova A, Zhang X, Marchand C, Pommier Y. Biochemical and pharmacological analyses of HIV‒1 integrase flexible loop mutants resistant to raltegravir. Biochemistry 2010; 49:3715–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchand C, Maddali K, Metifiot M, Pommier Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr Top Med Chem 2009; 9:1016–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramanathan S, Mathias AA, German P, Kearney BP. Clinical pharmacokinetic and pharmacodynamic profile of the HIV integrase inhibitor elvitegravir. Clin Pharmacokinet 2011; 50:229–244. [DOI] [PubMed] [Google Scholar]
- 6.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry 2008; 47:9345–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.da Silva D, Van Wesenbeeck L, Breilh D, Reigadas S, Anies G, Van Baelen K, et al. HIV-1 resistance patterns to integrase inhibitors in antiretroviral-experienced patients with virological failure on raltegravir-containing regimens. J Antimicrob Chemother 2010; 65:1262–1269. [DOI] [PubMed] [Google Scholar]
- 8.Hu Z, Kuritzkes DR. Effect of raltegravir resistance mutations in HIV-1 integrase on viral fitness. J Acquir Immune Defic Syndr 2010; 55:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reigadas S, Anies G, Masquelier B, Calmels C, Stuyver LJ, Parissi V, et al. The HIV-1 integrase mutations Y143C/R are an alternative pathway for resistance to raltegravir and impact the enzyme functions. PLoS One 2010; 5:e10311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canducci F, Marinozzi MC, Sampaolo M, Boeri E, Spagnuolo V, Gianotti N, et al. Genotypic/phenotypic patterns of HIV-1 integrase resistance to raltegravir. J Antimicrob Chemother 2010; 65:425–433. [DOI] [PubMed] [Google Scholar]
- 11.Delelis O, Thierry S, Subra F, Simon F, Malet I, Alloui C, et al. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob Agents Chemother 2010; 54:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010; 464:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valkov E, Gupta SS, Hare S, Helander A, Roversi P, McClure M, et al. Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic Acids Res 2009; 37:243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitzkee NC, Masse JE, Shen Y, Davies DR, Bax A. Solution conformation and dynamics of the HIV-1 integrase core domain. J Biol Chem 2010; 285:18072–18084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc Natl Acad Sci U S A 2010; 107:15910–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci U S A 2010; 107:20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]