

Abstract
Triple A syndrome (Allgrove syndrome) is characterized by a triad of specific features, namely, alacrimia, adrenal insufficiency, and achalasia cardia. It is a rare autosomal recessive disorder. In the present study, an 18-year-old boy was presented with complaints of decreased tears, darkening of the skin, difficulty in walking and standing up from sitting position, and difficulty in swallowing liquids. Adrenal insufficiency, alacrimia, achalasia, and neurological manifestations were confirmed with relevant laboratory investigations. His condition improved with steroids and artificial teardrops. However, a vigilant eye of the clinician for clinical clues of syndromic manifestation will help in early diagnosis and proper management.
Keywords: Allgrove's syndrome, Alacrimia, Adrenal insufficiency, Achalasia cardia, Triple A syndrome
Introduction
The triple A (Allgrove) syndrome, an autosomal recessive disease with a triad of alacrimia, adrenal insufficiency, and achalasia cardia, was first described in 1978.[1] Skin and neurological manifestations are also commonly present along with the classical triad. Associated other symptoms may include short stature, microcephaly, osteoporosis, and dysmorphic features.[2] The triple A syndrome has a characteristic genetic abnormality on chromosome 12q13 (AAAS gene). This location encodes for a 546 amino acid protein called ALADIN (alacrimia-achalasia-adrenal insufficiency and neurologic disorder).[3] There is a significant gap between initial symptoms and the diagnosis of the triple A syndrome.[4] The clinician should be vigilant with a high index of suspicion. Herein, we diagnosed a case of triple A syndrome presented to us with skin and neurological manifestations.
Case Report
An 18-year-old male referred from district hospital to our department with complaints of decreased tears from eyes since childhood, intermittent history of loose stools for 7 years, darkening of the skin from 10 months, and difficulty in walking and standing up from sitting position for last 10 months. The patients also complained of difficulty in swallowing liquids for the last 4 months. He was born of full-term normal vaginal delivery, from a non-consanguineous marriage. There was no history of any developmental delay during childhood. On general physical examination, the patient was lean built and hyperpigmentation was present on the tongue, buccal mucosa, and skin [Figure 1]. Hypopigmented macules were present over the chest and back. He was having pes cavus deformity on both feet. His pulse was 78/min and blood pressure was 100/60 mmHg.
Figure 1.
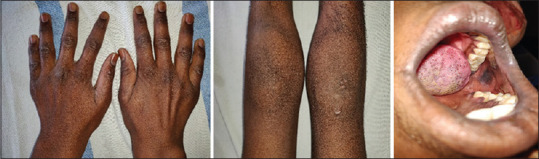
Hyperpigmentation on the dorsum of hands, knee joints, and buccal mucosa
Neurological examination revealed normal mental function and normal cranial nerve involvement. Motor system examination revealed the wasting of the thenar and hypothenar muscles of both hands as well as in the intrinsic muscles of both feet. He had MRC (Medical Research Council) grade 3 weakness at shoulder, elbow, wrist and ankle joints, and MRC grade 4 weakness at hip, ankle and finger grip, and abduction. Superficial reflexes were absent and bilateral plantar reflexes were flexor. All deep tendon reflexes including biceps, triceps, knee, and ankle were brisk with clonus. Sensory examination was unremarkable and no cerebellar and meningeal signs were present. Other system examinations were normal.
He had normal hematological parameters, renal, and liver functions. His serum electrolytes, arterial blood gas, and creatinine kinase (CK NAC) were normal. He had normal fasting plasma glucose (98 mg/dL) and serum vitamin B12 (350 mg/dL). His serum 25 hydroxyvitamin D levels were low (10.20 ng/mL). In view of skin and buccal mucosa hyperpigmentation, he was evaluated for adrenal insufficiency. Serum cortisol, baseline, and post adrenocorticotropic hormone (ACTH) stimulation were less than 0.50 μg/dL with raised baseline serum ACTH level confirming the diagnosis of primary adrenal insufficiency. Other hormonal parameters were within normal limits. Schirmer's test was positive (0 mm in both eyes) suggesting alacrimia. Barium swallow and upper GI endoscopy were suggestive of achalasia [Figure 2]. MRI brain showed bilateral atrophied lacrimal glands [Figure 3] and focal T2 hyperintense signal intensity in the left side of the medulla and inferior cerebellar peduncle showing restriction on diffusion-weighted imaging suggestive of focal demyelination [Figure 4]. The MRI spine was normal. Contrast-enhanced computed tomography (CECT) abdomen showed atrophied adrenal glands [Figure 5], dilatation of esophageal lumen at upper and lower 1/3rd without any wall thickening.
Figure 2.
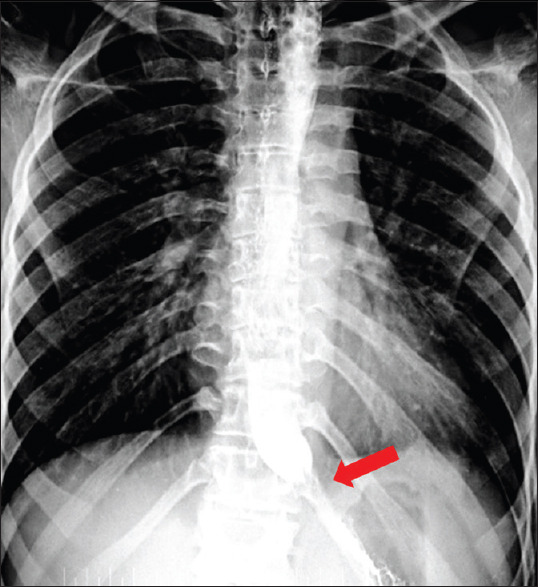
Bird beak appearance in Barium shallow
Figure 3.
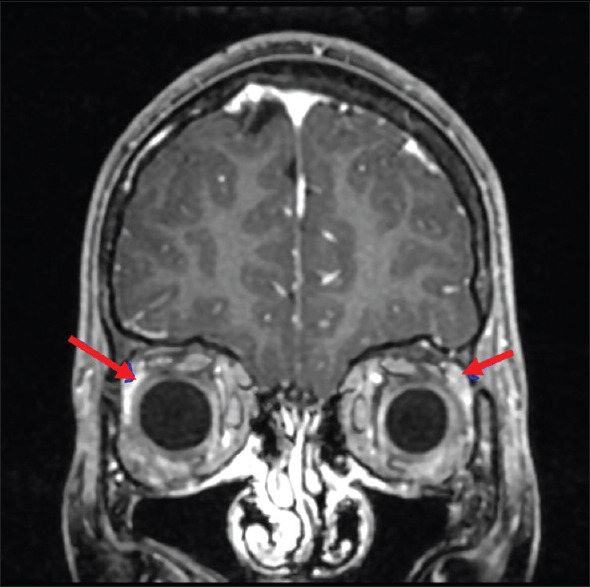
MRI brain shows small and atrophied lacrimal glands
Figure 4.
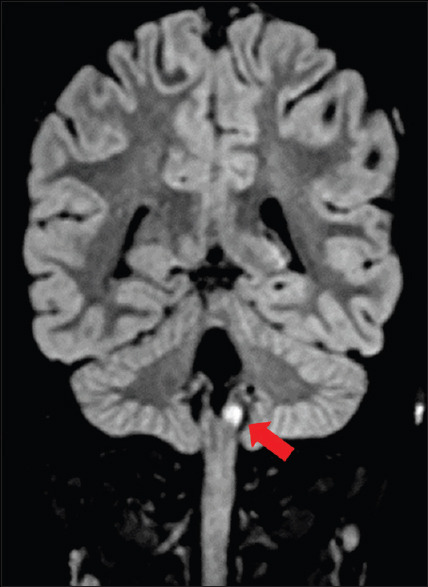
Focal FLAIR hyperintensity in the left inferior cerebellar peduncle
Figure 5.
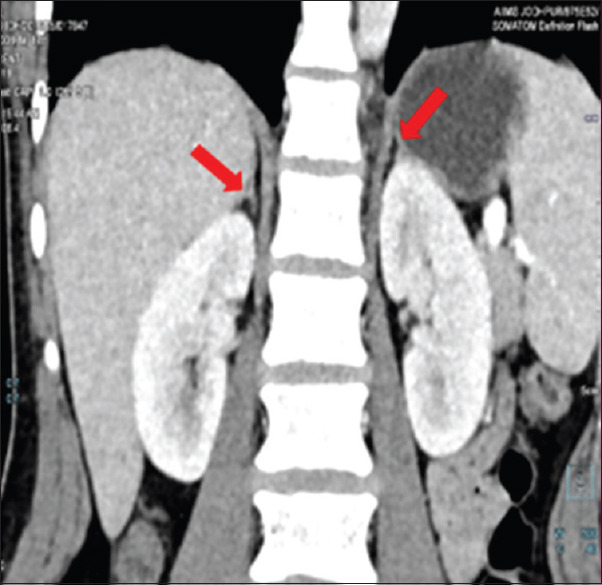
Coronal CECT abdomen and pelvis showing atrophied right adrenal gland with a streak like limbs
As the patient was having a classical triad of adrenal insufficiency, alacrimia, and achalasia, a diagnosis of triple A syndrome (Allgrove syndrome) was made. The patient was started on adrenal replacement therapy for adrenal insufficiency and artificial tears for alacrimia. Since the patient was not having significant dysphagia, regular follow-up was advised. The patient is currently on follow-up and has shown significant improvement.
Discussion
Triple A syndrome (Allgrove syndrome) is a rare autosomal recessive disease and clinically it presents with the classical triad of alacrimia, achalasia, and adrenal insufficiency. The adrenal insufficiency is due to ACTH resistance.[5]
The earliest clinical presentation in our patient, which has also been reported by Misgar et al., was alacrimia which was confirmed by Schirmer's test and MRI.[6] The biopsy of the lacrimal gland may show neuronal degeneration and depletion of secretory granules in the acinar cell.[7]
Our patient developed symptoms of adrenal insufficiency in the second decade of life. In the three cases reported by Prpic et al. there were variable clinical presentations. Two patients had a typical triad of achalasia, alacrimia, and adrenocortical deficiency along with neurologic symptoms. The third patient had only achalasia and neurologic dysfunction but not alacrimia and adrenocortical deficiency.[8] Adrenal insufficiency and achalasia usually manifested during the first decade of life and adrenal insufficiency may cause severe hypoglycemia and sudden death.[9] Mineralocorticoid deficiency is present in around 15% cases and associated with electrolyte abnormalities.[10] In our patient, electrolytes were normal, although we did not measure plasma aldosterone level.
Achalasia causes dysphagia which is more for liquids and, 75% of cases have this symptom.[6] Our patient had dysphagia in the second decade which was more for liquids. The cause of achalasia is fibrosis of the intermuscular plane and absent myenteric ganglia in the lower end of the esophagus.[11]
Neurological manifestations are common in triple A syndrome but these would manifest later than other clinical features. Neurologic features included central, peripheral, and autonomic nervous system abnormalities. The common neurological clinical features include mental retardation, muscle wasting and weakness, cranial nerve involvement, especially lower cranial nerve resulting in dysarthria and nasal speech, hyperreflexia, ataxia, optic atrophy, and less commonly sensory neuropathy.[10] Our patient had proximal and distal muscle weakness in all four limbs and exaggerated deep tendon reflexes with clonus suggesting pyramidal tract involvement. The mobility of the patient was limited due to muscle weakness. The sensory and autonomic nervous system examinations were normal. Early diagnosis of neurological manifestations in the pediatric age group and imperative therapy improves the quality of life in these patients.[12] MRI scan has revealed demyelination of focal demyelination at medulla and inferior cerebellar peduncle. There is no specific MRI brain parenchymal finding mentioned in the literature.
Linkage studies have been conducted to find out the locus for the AAAS gene, in the 12q13 region.[13] The product of this gene is ALADIN, which is nucleoporin at the nuclear pore complex.[14] Nucleoporin is an important transporter between nucleus and cytoplasm and mutant ALADIN results in defective DNA repairs. The presence of this mutation is a diagnostic feature of Allgrove syndrome. Due to resource constraints, we did not opt for mutation analysis. Moreover, the cost of mutation analysis is not affordable by all patients in India, hence our focus was on the syndromic diagnosis of triple A syndrome. Acute adrenal insufficiency is most dreaded complication of of triple A syndrome and early diagnosis and initiation of steroids in these patients avoid to land up in acute adrenal insufficiency.
Conclusion
The presented case of triple A syndrome, with a classical triad of alacrimia, achalasia, and adrenal insufficiency along with neurological manifestations, underscores the importance of keeping a vigil over syndromic manifestations for early diagnosis so that the lag in presentation, diagnosis, and proper management can be minimized. The thoughtful analysis of a clinical clue may open the path for diagnosing a syndrome with its associated manifestations.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Allgrove J, Clayden G, Grant D, Macaulay J. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet. 1978;311:1284–6. doi: 10.1016/s0140-6736(78)91268-0. [DOI] [PubMed] [Google Scholar]
- 2.Huebner A, Elias LL, Clark AJ. ACTH resistance syndromes. J Pediatr Endocrinol Metab. 1999;12:277–93. [PubMed] [Google Scholar]
- 3.Shah SWH, Butt AK, Malik K, Alam A, Shahzad A, Khan AA. AAA syndrome, case report of a rare disease. Pak J Med Sci. 2017;33:1512–6. doi: 10.12669/pjms.336.13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh K, Puri RD, Bhai P, Arya AD, Chawla G, Saxena R, et al. Clinical heterogeneity and molecular profile of triple A syndrome: A study of seven cases. J Pediatr Endocrinol Metab. 2018;31:799–807. doi: 10.1515/jpem-2018-0023. [DOI] [PubMed] [Google Scholar]
- 5.Aftab S, Manzoor J, Talat N, Khan HS, Subhanie M, Khalid NA. Allgrove syndrome: Adrenal insufficiency with hypertensive encephalopathy. J Coll Physicians Surg Pak. 2016;26:790–2. [PubMed] [Google Scholar]
- 6.Misgar RA, Pala NA, Ramzan M, Wani AI, Bashir MI, Laway BA. Allgrove (Triple A) syndrome: A case report from the Kashmir Valley. Endocrinol Metab. 2015;30:604–6. doi: 10.3803/EnM.2015.30.4.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gazarian M, Cowell CT, Bonney M, Rigor WG. The “4A” syndrome: Adrenocortical insufficiency associated with achalasia, alacrima, autonomic and other neurological abnormalities. Eur J Pediatr. 1995;154:18–23. doi: 10.1007/BF01972967. [DOI] [PubMed] [Google Scholar]
- 8.Prpic I, Huebner A, Persic M, Handschug K, Pavletic M. Triple A syndrome: Genotype-phenotype assessment. Clin Genet. 2003;63:415–7. doi: 10.1034/j.1399-0004.2003.00070.x. [DOI] [PubMed] [Google Scholar]
- 9.Yasawy MI. Allgrove's syndrome: Case report and literature review. J Family Community Med. 2009;16:37–40. [PMC free article] [PubMed] [Google Scholar]
- 10.Grant DB, Barnes ND, Dumic M, Ginalska-Malinowska M, Milla PJ, von Petrykowski W, et al. Neurological and adrenal dysfunction in the adrenal insufficiency/alacrima/achalasia (3A) syndrome. Arch Dis Child. 1993;68:779–82. doi: 10.1136/adc.68.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khelif K, De Laet MH, Chaouachi B, Segers V, Vanderwinden JM. Achalasia of the cardia in Allgrove's (triple A) syndrome: Histopathologic study of 10 cases. Am J Surg Pathol. 2003;27:667–72. doi: 10.1097/00000478-200305000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Bouliari A, Lu X, Persky RW, Stratakis CA. Triple A syndrome: Two siblings with a novel mutation in the AAAS gene. Hormones (Athens) 2019;18:109–12. doi: 10.1007/s42000-018-0089-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber A, Wienker TF, Jung M, Easton D, Dean HJ, Heinrichs C, et al. Linkage of the gene for the triple A syndrome to chromosome 12q13 near the type II keratin gene cluster. Hum Mol Genet. 1996;5:2061–6. doi: 10.1093/hmg/5.12.2061. [DOI] [PubMed] [Google Scholar]
- 14.Kurnaz E, Duminuco P, Aycan Z, Savaş-Erdeve Ş, Muratoǧlu Şahin N, Keskin M, et al. Clinical and genetic characterisation of a series of patients with triple A syndrome. Eur J Pediatr. 2018;177:363–9. doi: 10.1007/s00431-017-3068-8. [DOI] [PubMed] [Google Scholar]