

Abstract
Background
Preclinical data suggest cell cycle checkpoint blockade may induce an immunostimulatory tumor microenvironment. However, it remains elusive whether immunomodulation occurs in the clinical setting. To test this, we used blood and fresh tissue samples collected at baseline and post therapy from a phase II trial of the cell cycle checkpoint 1 inhibitor (CHK1i) prexasertib in recurrent ovarian cancer.
Methods
Paired blood samples and fresh core biopsies, taken before treatment was started at baseline (cycle 1 day 1 (C1D1)) and post second dose on day 15 of cycle 1 (C1D15), were collected. To evaluate changes in the immune responses after treatment, multiparametric flow cytometry for DNA damage markers and immune cell subsets was performed on paired blood samples. RNA sequencing (RNAseq) of paired core biopsies was also analyzed. Archival tissue immune microenvironment was evaluated with immunohistochemistry. All correlative study statistical analyses used two-sided significance with a cut-off of p=0.05.
Results
Flow cytometric analysis showed significantly increased γ-H2AX staining after CHK1i treatment, accompanied by increased monocyte populations, suggestive of an activated innate immune response (median 31.6% vs 45.6%, p=0.005). Increased expressions of immunocompetence marker HLA-DR (Human Leukocyte Antigen DR antigen) on monocytes and of TBK1, a marker of STING (stimulator of interferon genes) pathway activation, in biopsies were associated with improved progression-free survival (PFS) (9.25 vs 3.5 months, p=0.019; 9 vs 3 months, p=0.003, respectively). Computational analysis of RNAseq data indicated increased infiltration of tumor niches by naïve B-cells and resting memory T-cells, suggestive of a possibly activated adaptive immune response, and greater T-reg infiltration after treatment correlated with worse PFS (9.25 vs 3.5 months, p=0.007). An immunosuppressive adaptive immune response, perhaps compensatory, was also observed on flow cytometry, including lymphodepletion of total peripheral CD4+ and CD8+T cells after CHK1i and an increase in the proportion of T-regs among these T-cells. Additionally, there was a trend of improved PFS with greater tumor-infiltrating lymphocytes (TILs) in archival tissues (13.7 months >30% TILs vs 5.5 months ≤30% TILs, p=0.05).
Conclusion
Our study demonstrates that a favorable clinical response in high-grade serous ovarian carcinoma patients treated with CHK1i is possibly associated with enhanced innate and adaptive immunity, requiring further mechanistic studies. It is supportive of current efforts for a clinical development strategy for therapeutic combinations with immunotherapy in ovarian cancer.
Keywords: immunomodulation; immunotherapy; adaptive immunity; immunity, innate
Background
Ovarian carcinoma accounts for the majority of gynecologic cancer deaths in the USA.1 High-grade serous ovarian carcinoma (HGSOC) is the most common subtype of ovarian cancer and typically presents as advanced disease with a high frequency of recurrence, requiring new effective therapeutic strategies. The molecular characteristics of HGSOC include universal TP53 dysfunction that disrupts the G1-S cell cycle checkpoint. This leaves the cells heavily dependent on cell cycle checkpoint-mediated G2-M arrest for DNA repair.2 Cell cycle checkpoint kinase 1 (CHK1), which regulates the G2-M checkpoint, is overexpressed in nearly all HGSOC,3 making it a rational target to induce DNA damage and tumor cell death.
CHK1 is activated by the ataxia telangiectasia and Rad3-related (ATR) and ataxia-telangiectasia mutated (ATM) kinases in response to DNA damage or replication stress.4 On activation, CHK1 phosphorylates and inhibits its substrates, M-phase inducer phosphatases 1 (CDC25A) and 3 (CDC25C), which led to cell cycle arrest at the G2-M checkpoint.4–6 This allows for DNA damage repair and stabilization of stalled replication forks, without which double-stranded DNA breaks and consequent cell death would occur.7 Prexasertib, the second-generation CHK1 inhibitor (CHK1i), has been reported to induce DNA damage and apoptosis in numerous preclinical models including ovarian cancer.8–10
Preclinical data also suggest that the efficacy of CHK1 inhibition may be associated with innate and adaptive immunomodulation, although studies are limited, particularly in ovarian cancer. It has been shown that both CHK1 and ATR activation after DNA damage leads to upregulation of programmed death ligand-1 (PD-L1),11 12 potentially via the STAT/IRF1 pathway.12 In vitro work in osteosarcoma, prostate and lung cancer models demonstrated that PD-L1 expression induced by ionizing radiation-related DNA damage was thereby downregulated by CHK1i.12 ATR inhibition also has been shown to decrease PD-L1 expression, sensitizing tumor cells to T-cell-mediated killing.11 Recently, Sen et al13 reported that DNA repair inhibition, such as with the CHK1i prexasertib, causes increased DNA damage and accumulation of broken DNA fragments in the cytoplasm. Cytosolic DNA fragments can then bind to cyclic GMP-AMP synthase (cGAS), leading to upregulation of the cGAS-stimulator of interferon genes (STING) pathway.13 14 The STING pathway is a potent activator of type I interferons (IFN) and elicits both innate and adaptive anti-tumor immune responses, including increased production of immunostimulatory cytokines and recruitment of T-cells.13–18
However, while CHK1 inhibition enhanced anti-tumor CD8+T cell infiltration, it was also reported to increase endogenous immunosuppressive PD-L1 expression, enabling synergy with anti-PD-L1 therapy.13 These varying results reflect a complex biology as well as different experimental conditions and thus highlight the need for further studies in the context of immunomodulation in the clinical setting. We recently reported promising clinical activity of the CHK1i prexasertib in heavily pre-treated BRCA wild-type (BRCAwt) patients with recurrent ovarian cancer.19 In the present study, we hypothesized that CHK1i may have induced both innate and adaptive immune responses in addition to its effect on the cell cycle regulation, thus contributing to the observed clinical activity in ovarian cancer patients. To test this hypothesis, we investigated changes in the peripheral and tumoral innate and adaptive immune responses in patients treated with CHK1i monotherapy using paired pre-treatment and post-therapy blood and tissue samples. We now report the increases in cellular and molecular markers of innate and adaptive immunity by CHK1i monotherapy that correlated with a favorable clinical response.
Methods
Patients and study design
This single arm proof-of-concept phase II study was approved by the Institutional Review Board of the Center for Cancer Research (CCR), National Cancer Institute (NCI), Bethesda, Maryland, USA, and written informed consent was obtained from participating patients. Comprehensive enrollment criteria have previously been published.19 Briefly, eligible patients had recurrent HGSOC, an Eastern Cooperative Oncology Group (ECOG) Performance Status of 0 to 2 and good end-organ function. The BRCAwt HGSOC cohort completed enrollment in November 2016 for which details were published and data from that cohort are used for this pilot project.19 The BRCA mutation HGSOC cohort is currently enrolling the patients (NCT02203513). There was no limit on number of prior therapies, however, patients had to have at least 4 weeks wash-out period from their last treatment. Clinical study details, including drug administration, safety, adverse events, and tumor response in the BRCAwt HGSOC cohort have been reported19 and the schema for treatment is shown in figure 1A. Paired blood samples, taken before treatment was started at baseline (cycle 1 day 1 (C1D1)) and 6 to 24 hours post second dose on day 15 of cycle 1 (C1D15), were collected from all patients. Pre-treatment and C1D15 percutaneous core biopsies were obtained by interventional radiologists under CT or ultrasound guidance using local anesthesia. All core biopsy samples were processed immediately in real time into optimal cutting temperature compound and stored at −80°C prior to analysis. Paraffin-embedded archival tissue samples from the time of original diagnosis were obtained at trial enrollment for immunohistochemistry as fresh frozen biopsy samples did not yield evaluable immunohistochemistry findings.
Figure 1.
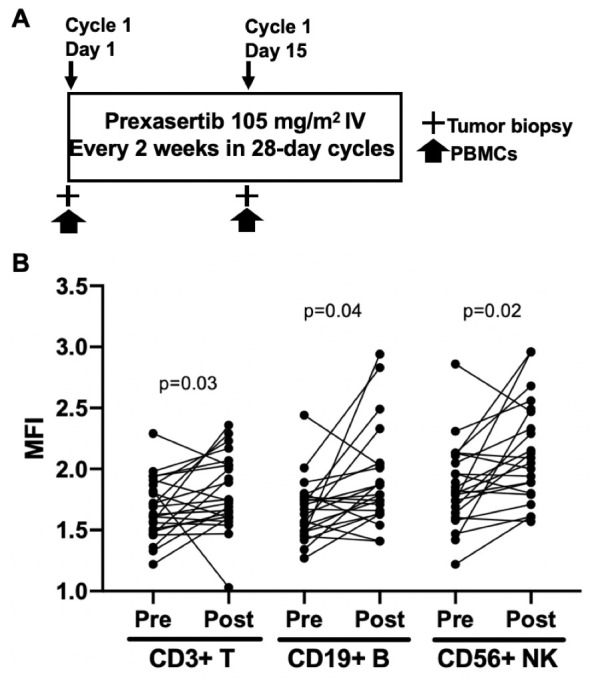
Study schema and increased γ-H2AX, possibly caused by CHK1i. (A) Prexasertib was administered intravenously at 105 mg/m2 every 2 weeks in 28-day cycles until disease progression or unacceptable toxicities. A mandatory baseline tumor biopsy was performed before C1D1 treatment, and an optional second biopsy was performed 6 to 24 hours after treatment on C1D15. PBMCs for flow cytometry were collected at C1D1 and 6 to 24 hours after treatment on C1D15. (B) Flow cytometric analysis showing increased MFI of γH2AX in PBMC isolated from patients before (pre) and after a 2-week treatment with prexasertib (post). Cells were first gated on CD3 (T-cells), CD19 (B cells), or CD56 (NK cells) prior to plotting for γH2AX. C1D1, cycle 1 day 1; C1D15, cycle 1 day 15; MFI, mean fluorescence intensity; NK, natural killer; PBMCs, peripheral blood mononuclear cells.
Flow cytometry
Cells were incubated with Fc receptor blocking agent (Miltenyi Biotec, Bergisch Gladbach, Germany) and stained with monoclonal antibodies for 20 min at 4°C. For intracellular staining for Foxp3 and Ki67 expression, cells were fixed and permeabilized using a Fix/Perm buffer (eBioscience, San Diego, California, USA) according to the manufacturer’s instructions, then stained with anti-Foxp3 or anti-Ki67 antibody. Live cells were discriminated by means of LIVE/DEAD Fixable Aqua Dead Cell Stain (Life Technologies, New York, USA) and dead cells were excluded from the analysis. All analyzes were performed using multiparametric flow cytometry (MACSQuant; Miltenyi Biotec, Bergisch Gladbach, Germany) and data were analyzed using FlowJo software V.10.0.7 (FlowJo, LLC, Ashland, Oregon, USA). Gating was first performed on peripheral blood mononuclear cells (PBMCs) with doublet exclusion and then on viable cells. CD8+ or CD4+T cells were further gated for functional markers PD-1, glucocorticoid-induced TNFR-related protein (GITR), Human Leukocyte Antigen DR antigen (HLA-DR), inducible T-cell costimulatory (ICOS) or Ki-67. Monocytic myeloid-derived suppressor cells (M-MDSCs) were defined as CD11b+CD14+HLA-DR-CD15-, early-stage MDSCs (E-MDSCs) as CD11b+CD3-CD14-CD15-CD19-CD56-HLA-DR-CD33+ and polymorphonuclear MSDCs (PMN-MDSCs) as CD11b+CD14 CD15+. For monocytes, total monocytes were subdivided by CD14 and CD16 expression: classical monocytes (CD14 ++CD16−); non-classical monocytes (CD14 +CD16++); and intermediate monocytes (CD14 ++CD16+). Representative figures describing detailed flow cytometry gating strategies can be found in online supplemental figure 1. The monoclonal antibodies used (all from BioLegend, San Diego, California, USA) are listed in online supplemental table 1.
jitc-2019-000516supp001.pdf (19.9MB, pdf)
RNA sequencing
Total RNA was isolated from pre-treatment and available C1D15 fresh-frozen core biopsies using the RNeasy microkit (Qiagen). RNA quality was evaluated using the Agilent Bioanalyzer 2100 and RNA integrity number (RIN) values were ensured to be >8.0. For total RNA sequencing (RNAseq), each sample (20 to 100 ng) was preprocessed with NEBnext rDNA depletion kit (New England Biolabs, Ipswich, Massachusetts, USA) to remove ribosomal RNA, then barcoded and pooled to ensure at least 100 million reads per sample on a HiSeq3000 sequencing system (Illumina, San Diego, California, USA). The human reference genome Hg38 was used to align reads and gene expression data were generated as counts per million mapped reads (CPM) values. Quality check of sample and sequencing outputs were performed by the CCR sequencing facility and CCR collaborative bioinformatics resource at NCI, Bethesda, Maryland, USA. As previously described,19 RNAseq data of normal ovarian tissues were obtained from the Genotype-Tissue Expression project (GTEx project, NIH; https://www.gtexportal.org/home/, last accessed on January 13, 2019) as Reads Per Kilobase of transcript per Million mapped reads (RPKM) values and then converted to CPM values using the formula RPKM=CPM/L, where L is the exonic length in kilobases. Log2 of CPM values were used throughout. Biopsies determined to be derived from normal tissue were excluded from analysis. HGSOC subtype was determined using a validated single sample Gene Set Enrichment Analysis (ssGSEA) signature as has been previously described.20 21 IFN-γ and expanded immune gene signature scores (online supplemental table 2) were calculated by averaging the normalized log2 expression levels of the included genes, as described by Ayers et al.22 CIBERSORT, a computational algorithm for quantifying immune cell fractions using gene expression profiles, was performed to determine tumorous immune infiltration as previously described.23
Immunohistochemistry
Pre-treatment formalin-fixed paraffin-embedded (FFPE) tissue samples from 24 patients were sent to Johns Hopkins Hospital for pathologic correlative studies; the tissue from one patient (Study ID 40) was inadequate for correlative studies due to tissue preservation. H&E-stained slides of the tissue samples from the remaining 23 patients were evaluated to confirm the pathologic diagnosis of high-grade ovarian carcinoma and to score the presence of tumor-infiltrating lymphocytes (TILs) and tumor-associated macrophages (TAMs). TIL were scored as the percentage (0% to 100%) of tumor stroma area occupied by mononuclear inflammatory cells, as previously described.24 TILs were not scored in tumor specimens lacking stroma, that is, those specimens consisting entirely of detached fragments of carcinoma cells without interface to adjacent stroma. The presence of TAM was recorded as: score 0 (absent), score 1 (focal, largely confined to papillary cores), score 2 (non-focal, involving both papillary cores and tumorous stroma), and score 3 (diffuse, involving papillary cores, tumorous stroma, and carcinoma cell nests). Immunohistochemistry for CD68 (Clone KP1, Dako; Bond RX, Bond Polymer Refine Detection) was performed on all cases to confirm the histologic impression of TAM.25
Immunohistochemistry for PD-L1 was performed manually on unstained slides (PD-L1 clone SP142, 0.096 µg/mL concentration; Spring Bioscience, Pleasanton, California, USA), as described.25 Specifically, the Spring Bioscience PD-L1 clone SP142 was used with a laboratory-developed assay validated against the labeling results of comparable assays.25 Briefly, slides were deparaffinized and rehydrated using standard methods. Antigen retrieval was performed in pH 6.0 CB buffer in a decloaking chamber (Biocare Medical, Pacheco, CA, USA), and slides were treated with peroxidase, protein, avidin, and biotin blocking, and incubated with the primary antibody overnight at 4°C. Slides were then treated with a biotin-labeled anti-rabbit secondary antibody (1 µg/µl concentration). The signal was developed with horseradish peroxidase using the VECTASTAIN Elite ABC Kit (Vector Laboratories, Burlingame, California, USA), amplified using a Tyramide Signal Amplification PLUS Biotin KIT (dilution 1:50; Perkin Elmer, Waltham, Massachusetts, USA), and visualized by 3,3'-Diaminobenzidine (DAB). PD-L1 labeling of carcinoma cells was evaluated in all cases and was scored as percentage (0% to 100%) of malignant cells with membranous labeling, with a cut-off of >1% carcinoma cell labeling considered positive.
Statistical analysis
Comparison of correlative markers were performed using a non-parametric Mann-Whitney test (for unpaired analyses) or a non-parametric Wilcoxon matched-pairs signed-rank test (for paired analyses) using GraphPad Prism, V.6. Immunohistochemistry data analyses were performed using Microsoft Excel (t-test) or GraphPad Online (Fisher’s exact test). The significance of difference between Kaplan-Meier survival curves was compared with a Mantel-Cox log-rank test. All statistical tests used two-sided significance with a cut-off of p=0.05 and are reported without adjustment for multiple comparisons due to the small study cohort and exploratory nature of this analysis.
Study approval
The trial was approved by the Institutional Review Board of the CCR, NCI, Bethesda, Maryland, USA. All patients provided written informed consent before enrollment.
Results
Patients
The treatment schema and correlative studies are shown in figure 1A. Characteristics of the 24 evaluable BRCAwt HGSOC patients enrolled in this phase II trial have previously been described in detail19 and are summarized in table 1. Briefly, in this group of heavily pre-treated patients (median 4.5 prior treatments (1 to 13)), there were eight (33%) partial responses (PR) and six (25%) patients with stable disease (SD) ≥6 months for a clinical benefit rate (CBR; PR +SD ≥6 months) of 58%. For this post hoc exploratory biomarker study, not all patients had available samples from each translational endpoint but 100% of patients had paired pre-treatment and post-treatment blood among which 96% (23/24) were evaluable for flow cytometry, 88% (21/24) had evaluable archival tissue from primary diagnosis, and 38% (9/24) had both paired blood and fresh core biopsy samples from this trial; table 1 also includes the number of patients within each of the substudy elements.
Table 1.
Clinical characteristics and numbers in correlative study elements (n=24)
| Age (years) | |
| Median (IQR) | 64.0 (58.0 to 69.5) |
| Number of previous treatments (n, %) | |
| 1 | 1 (4%) |
| 2 | 8 (33%) |
| ≥3 | 15 (63%) |
| Median (range) | 4.5 (1 to 13) |
| Platinum sensitivity* (n, %) | |
| Platinum-sensitive | 5 (21%) |
| Platinum-resistant | 18 (75%) |
| Platinum-refractory | 1 (4%) |
| Best RECIST response (n, %) | |
| PR | 8 (33%) |
| SD ≥6 months | 6 (25%) |
| Total CBR (PR+SD ≥6 months) | 14 (58%) |
| SD <6 months | 3 (13%) |
| PD | 7 (29%) |
| Correlative studies (n, %) | |
| Tumor biopsies | |
| Pre-treatment | 18 (75%)† |
| Post-treatment | 12 (50%)† |
| Paired | 9 (38%) |
| Flow cytometry | 23 (96%)‡ |
| Immunohistochemistry | 21 (88%)§ |
| CBC | 24 (100%) |
*Platinum-sensitive: recurs 6 or more months after cessation of last platinum-based chemotherapy; platinum-resistant: progression within 6 months of last platinum-based therapy; platinum-refractory: progression while actively on platinum-based therapy.
†All 24 patients had baseline biopsies performed, however six were determined to be non-tumor tissue by RNAseq analysis and were excluded from analysis. Thirteen patients had an optional second biopsy performed, and one was determined to be of non-tumor origin and excluded.
‡C1D15 PBMCs from one patient were missing.
§Archival tissue from one patient was exhausted prior to enrollment in the trial and was unevaluable in two patients due tissue fragmentation and lack of stroma.
CBC, complete blood count; CBR, clinical benefit rate; PBMC, peripheral blood mononuclear cells; PD, progressive disease; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; RNAseq, RNA sequencing; SD, stable disease.
DNA damage in peripheral immune cells
Our findings from paired pre-treatment and post-treatment PBMCs showed an increase in the number of lymphocytes with γ-H2AX staining by flow cytometry, a marker of double-stranded DNA breaks.26 Specifically, we observed increases in median fluorescent intensities (MFIs) of γ-H2AX among CD3+ T (median 1.63 vs 1.74, p=0.03), CD19+ B (median 1.69 vs 1.76, p=0.04), and CD56+Natural Killer (NK) lymphocytes (median 1.82 vs 2.05, p=0.02), suggestive of increased DNA damage in patients treated with CHK1i (figure 1B). Moreover, the observed increase in γ-H2AX corresponded to treatment-induced lymphodepletion as nearly 80% of patients experienced grade 3 or 4 reduced white blood cell counts,19 consistent with previous studies correlating double-stranded DNA damage with peripheral lymphocyte apoptosis.27 28 Change in γ-H2AX was not associated with clinical response or progression-free survival (PFS).
Innate immune activation in recurrent ovarian cancer
To evaluate changes in the peripheral innate immune response, immunostimulatory cell (ie, monocyte) and immunosuppressive cell (ie, myeloid-derived suppressor cell (MDSC)), subpopulations were compared in paired PBMC samples (online supplemental table 3). There was an increase in percent monocytes per total viable cells (median 31.6% (pre) vs 45.6% (post), p=0.005, figure 2A) after CHK1i treatment, although this did not correlate with clinical response or PFS. However, an increase in the immunocompetence functional marker HLA-DR on total monocytes, seen in 12 of 23 patients (MFI fold-increase 1.15 (1.01 to 2.76)), was associated with improved PFS (9.25 vs 3.5 months, p=0.019, figure 2B) suggesting innate immune activation of monocytes may contribute to the clinical efficacy of CHK1i. Similarly, an increase in HLA-DR on classical and non-classical monocytes, seen in 9/23 (39%) and 10/23 (43%) of patients, respectively, correlated with improved PFS (9.5 vs 3.75 months, p=0.04; 11.5 vs 4 months, p=0.002, figure 2C and D). No differences were seen after treatment in MDSC or other immunosuppressive cell subtypes. We further performed differential gene analysis of RNAseq data set from paired tissue samples to identify prexasertib-induced tumorous innate immune activation. We found no significant changes in the RNA expression levels of several cytokines known to play a critical role in the innate immune response, including IL6, IL8, IL12, TNFα and IFNγ.29 We also investigated changes in mRNA expression levels of key STING pathway genes, including cGAS, STING, TANK-binding kinase 1 (TBK1) and IFN regulatory factor 3 (IRF-3),13 30 31 and observed increased expression of TBK1 among six of nine evaluable patients with paired biopsy samples (median fold-increase 1.14 (1.02 to 1.33)). No other genes were significantly changed. TBK1 leads to phosphorylation and dimerization of IRF-3 and ultimately induces transcription of type I IFN genes,31 and increased expression was associated with improved PFS (9 vs 3 months, p=0.003, figure 2E) suggesting STING pathway activation may contribute partly to response to treatment as demonstrated preclinically.13 Downstream expression of IFNβ was undetectable and expression of the chemokines CXCL10 and CCL5, which are direct targets of IRF-3 and have been shown to increase after STING activation,32 were not significantly different after CHK1i treatment.
Figure 2.

Prexasertib-induced changes in innate immunity. (A) An increase from pre-treatment to post-treatment percent monocytes per total viable cells observed with flow cytometric analysis of 23 paired PBMC samples using the Wilcoxon matched-pairs signed-rank test. Lines denote median and 95% CI. (B) Survival curves were compared using the Mantel-Cox log-rank test. An increase in HLA-DR on total monocytes, seen in 12 of 23 patients, is associated with improved PFS (9.25 vs 3.5 months, p=0.019). (C) An increase in HLA-DR on classical monocytes, seen in 9 of 23 patients, is associated with improved PFS (9.5 vs 3.75 months, p=0.04). (D) An increase in HLA-DR on non-classical monocytes, seen in 10 of 23 patients, is associated with improved PFS (11.5 vs 4 months, p=0.002). (E) Increased expression of TBK1, a key protein in the STING pathway, seen in six of nine patients with paired pre-treatment and post-treatment biopsies, is associated with improved PFS (9 vs 3 months, p=0.003). HLA-DR, Human Leukocyte Antigen DR antigen; mono, monocytes, mo, months; PBMC, peripheral blood mononuclear cells; PFS, progression-free survival; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1.
Immunostimulatory and immunosuppressive adaptive immune responses
To characterize tumorous adaptive immune responses to prexasertib treatment, CIBERSORT, an algorithmic tool that estimates proportions of tumor-infiltrating immune cells using RNAseq data,23 was utilized on paired tumor biopsies. We found increased proportions of naïve B-cells (0.5% vs 14%, p=0.003) and resting CD4 memory T-cells (19% vs 25%, p=0.03), as well as decreased resting mast cells after CHK1i (5% vs 0.8%, p=0.03) (figure 3A). However, none of these changes were associated with clinical response or PFS, and there was no significant change in activated T-cells. We also noted greater tumorous infiltration by T-regs (median=2.9%, (range 0% to 6.3%)) was associated with a significantly worse PFS (9.25 vs 3.5 months for above versus below median, p=0.007; figure 3B), suggesting cross-talk between cell cycle checkpoint inhibition and the adaptive immune response influences clinical activity of prexasertib.
Figure 3.

Prexasertib-induced changes in adaptive immunity. Paired pre-treatment and post-treatment PBMCs assessed by flow cytometry compared in 23 patients using a Wilcoxon matched-pairs signed-rank test. Lines in A and C-D show median and 95% CI. (A) CIBERSORT analysis to detect immune cell tumor infiltration among nine paired pre-treatment and post-treatment biopsies show an increase in naïve B-cells and resting memory CD4+T cells, and a decrease in the proportion of infiltrating resting mass was observed. (B) Among 12 available post-treatment core biopsies, high (above median) T-reg tumor infiltration was correlated with a significantly worse PFS (9.25 vs 3.5 months, p=0.007) using the Mantel-Cox log-rank test. (C) Decreased proportion of CD4+ and CD8+T cells per total viable cells observed from pre-treatment to post-treatment. (D) An increase in the proportion of regulatory T-cells and effector regulatory T-cells among total CD4+T-cells was observed post-treatment. eTreg, effector regulatory T-cell; PBMCs, peripheral blood mononuclear cells; Post-tx, post-treatment; PFS, progression-free survival; Treg, regulatory T-cell.
Changes in the peripheral adaptive immune response, including regulatory T-cells (T-regs) and effector T-cells, were compared between pre-treatment and post-treatment PBMCs using flow cytometry (online supplemental table 3). Consistent with the decreased white blood cell count observed in 82% of patients on this trial,19 CHK1i induced peripheral lymphodepletion, including an overall decrease in both CD4+ and CD8+T cells (25% vs 19.5%, p=0.008% and 9.5% vs 7.6%, p=0.005, respectively; figure 3C). There was no significant change in the overall proportion of immunosuppressive T-regs or effector T-regs (eT-regs), defined as active FOXP3highCD45RA− cells,33 among total viable cells. However, a slight increase in the proportion of T-regs and eT-regs was observed among CD4+T cells (4.8% vs 6.8%, p=0.01; 1.5% vs 2.5%, p=0.009, respectively; figure 3D), suggestive of an immunosuppressive environment. No significant changes in Ki67+CD4+ or CD8+effector T-cells were observed after treatment.
Next, using the paired tissue RNAseq data set, we evaluated changes in pre-treatment versus post-treatment expression of an INF-γ and expanded immune gene signature incorporating key genes involved in the adaptive immune response22 (online supplemental table 2). No significant changes were observed. Finally, as CHK1i has been demonstrated to modify PD-L1 expression preclinically,11–13 changes in expression levels of genes involved in immunosuppression, including PD-1, PD-L1, and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), were evaluated using RNAseq from nine paired pre-treatment and post-treatment biopsies. No significant changes were observed.
Investigating tumor-infiltrating lymphocytes in archival tissue samples of study patients
To evaluate whether the tumorous immune environment is prognostic and potentially predictive of response to CHK1i, archival tissue biopsies from the time of initial diagnosis (n=21) or recurrence (n=2) available from 96% (23/24) of patients were examined via H&E and immunohistochemistry for the presence of TILs. TILs were evaluable in 21 patients, with two excluded due to tissue fragmentation and lack of stroma, and contained an average of 20% TIL (range 1% to 90% stromal TIL; online supplemental table 4). In line with reports demonstrating an association between TILs and survival in ovarian cancer patients,34 PFS was significantly improved among patients with >30% TIL (13.7 vs 5.5 months, p=0.05). Moreover, the presence of >30% TIL was seen only in patients who experienced a clinical benefit (25% vs 0%, p=0.22) and the tumors of patients who experienced a clinical benefit from therapy showed a trend towards greater TIL compared with those who lacked clinical benefit (27% vs 11%, p=0.07), suggesting TIL levels may serve as both a prognostic and possibly predictive biomarker, requiring further validation (online supplemental figure 2). Additionally, no association was observed between archival tissue TAMs or PD-L1 carcinoma labeling (positive versus negative) and response to therapy (online supplemental table 4).
Molecular subtypes and correlation with CHK1i response
As the archival tissue immune environment was associated with response to CHK1i, we next assessed whether this was similarly true of pre-treatment tumor biopsies at the time of enrollment. Eighteen available pre-treatment biopsies were classified into four validated transcriptomic molecular ovarian cancer subtypes, which have been shown to carry prognostic and possible therapeutic relevance20 21 35 (figure 4). As with previous studies, individual tumor samples expressed multiple signatures at different levels of activation.35 Thirty-nine percent (7/18) expressed high levels of the immunoreactive subtype gene signature, associated with immune response and characterized by enhanced cytokine expression, T-cell activation, and TILs.20 35 The immunoreactive subtype did not correlate with PFS or response to CHK1i, possibly due to small numbers.
Figure 4.
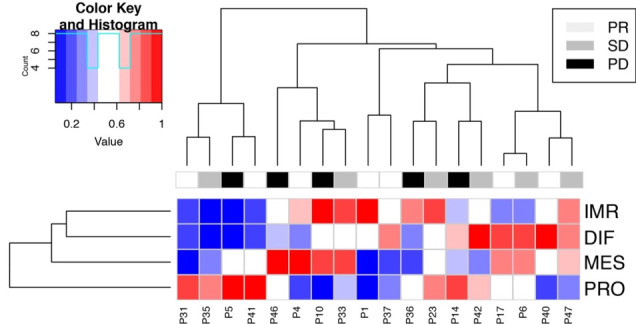
HGSOC molecular subtype. Expression profiles of the four HGSOC molecular subtypes in 18 available pre-treatment biopsies. Thirty-nine percent (7/19) were categorized as highly immunoreactive (IMR; P4, P10, P33, P1, P36, P23, and P47). DIF, differentiated; HGSOC, high-grade serous ovarian cancer; IMR, immunoreactive; MES, mesenchymal; P, patient; PD, progressive disease; PRO, proliferative; PR, partialresponse; SD, stable disease.
In addition, the pre-treatment biopsies of the patients with immunoreactive subtype signatures contained more stromal TILs (mean 26% and median 25%) than those with non-immunoreactive subtype signatures (mean 17% and median 10%; p=0.37). Similarly, a higher proportion of pre-treatment biopsies of the immunoreactive subtype signature contained PD-L1+ carcinoma cells (68%) than those with non-immunoreactive subtype signatures (38%; p=0.39). These features are indicative of an active immune response.
Discussion
Immune responses to the DNA-damaging drugs is a complex process involving multiple elements within the tumor microenvironment. Thus, it is necessary to translate preclinical findings of immune modulation with CHK1i into the clinical setting given the immune microenvironment observed in preclinical models are often not translatable to humans. We hypothesized that CHK1i would induce DNA damage by impairing the DNA damage repair response pathway, leading to increased tumor immunogenicity in ovarian cancer. For this, we used a variety of methods including flow cytometry, immunohistochemistry, and RNAseq to assess the immune landscape of paired clinical trial patient samples before and after treatment. Our exploratory studies indicate increased DNA damage in immune cells of patients treated with prexasertib, enhanced innate and adaptive immune activity, and a potentially compensatory immunosuppressive response.
The role of the innate immune response in malignancy and the tumor immune microenvironment has been well characterized. MDSCs play an immunosuppressive role and are associated with poor prognosis in ovarian cancer.36 On the other hand, anti-tumor effects of activated monocytes have been reported. In the presence of a tumor, monocytes differentiate into pro-inflammatory M1 macrophages which inhibit tumor proliferation, secrete proinflammatory cytokines, and promote NK cell differentiation for enhanced cytotoxicity.37 38 Here, we report that CHK1i treatment led to an increase in the percentage of monocytes among total PBMCs. Moreover, the association between patients with increased HLA-DR positive monocytes and improved PFS suggests that immunocompetent monocytes may contribute to the anti-tumor activity of prexasertib. The STING pathway has been implicated in driving an innate immune response after CHK1i treatment.13 39–41 We therefore evaluated changes in mRNA expression levels of key genes related to the STING pathway and PFS. Patients with increased post-treatment expression of TBK1, a downstream protein critical for production of type I IFNs, had improved PFS, indicating STING-pathway activation and innate immunity may be associated with duration of response to CHK1i.
This highlights the question of whether CHK1i also induces alterations in the adaptive immune response leading to clinical benefit in patients. Several studies have shown an association between the presence of TILs and improved patient survival in ovarian cancer.42 43 In the present study, although we observed increased naïve B-cell and resting memory T-cell infiltration after CHK1i treatment by CIBERSORT,23 neither was associated with PFS. However, a greater proportion of T-reg infiltration after treatment correlated with worse PFS, consistent with previous reports.44 45 These findings further indicate cross-talk between cell cycle checkpoint inhibition and the adaptive immune response and warrant additional investigation.
Recently, Sen et al13 reported that CHK1i treatment enhanced anti-tumor adaptive immunity in a STING-dependent manner but in turn led to upregulation of the immunosuppressive cell surface marker PD-L1. We did not observe increases in PD-L1 expression in our RNAseq analysis. However, it is worth noting the possible immunosuppressive adaptive immune changes after CHK1i treatment, requiring further mechanistic studies. Specifically, flow cytometry analysis of PBMCs revealed peripheral lymphodepletion of CD4+ and CD8+T cells, consistent with the leukopenia observed in patients on treatment19 as well as either selective repopulation with T-regs or selective depletion of effector T-cells in patient samples.
To date, no validated biomarkers have been defined to guide CHK1i therapy. The presence of TILs at diagnosis, but not TAMs, has been associated with improved overall survival in ovarian cancer.34 Our biomarker data also suggest a prognostic association between greater archival tissue TILs, but not TAMs, with longer PFS. This finding should be prospectively tested in a randomized setting for its predictive potential for CHK1i response. Limitations of our study include its small size, the post hoc exploratory nature of this analysis, and all tests reported are without adjustment for multiple comparisons. Therefore, all findings should be interpreted with caution and will need to be validated in a larger, prospective setting. Additionally, while flow cytometry-based detection of γ-H2AX is a validated tool to detect DNA damage and is supported by the observed lymphodepletion after treatment with prexasertib,26 we were unable to confirm CHK1i-induced DNA damage by tissue immunofluorescence due to limited available tissue samples.
The CHK1i prexasertib has marked clinical activity in women with recurrent BRCAwt HGSOC.19 Our exploratory translational studies are the first to report increased DNA damage and depletion of peripheral lymphocytes while apparently activating specific immune subsets that contribute to innate and adaptive immunity in ovarian cancer patients treated with prexasertib. These findings provide insights into the biological effect of CHK1i and present the opportunity to identify potential therapeutic immunotherapy combinations. Further clinical exploration of biomarkers is warranted.
Conclusion
Our findings suggest an immunomodulatory role for CHK1i in BRCAwt HGSOC patients. Changes in molecular and cellular markers after CHK1i treatment suggest enhanced innate and adaptive peripheral and tumorous immunity as well as a potential immunosuppressive compensatory response. This new clinical insight, supported by preclinical evidence, may identify novel predictive biomarkers for response to CHK1i and supports a differentiated clinical development strategy for therapeutic combinations in ovarian cancer. Further investigation is warranted.
Acknowledgments
This research was supported by the Intramural Research Program of the National Cancer Institute (JML, #ZIA BC011525), Center for Cancer Research (CCR). Prexasertib was supplied to the CCR by Eli Lilly and Company under a Collaborative Agreement (Cooperative Research and Development Agreement (CRADA #02915)). This research was made possible through the National Institutes of Health (NIH) Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation (DDCF Grant #2014194), the American Association for Dental Research, the Colgate-Palmolive Company, Genentech, Elsevier and other private donors.
Footnotes
Contributors: JML and EJL developed the study and wrote the manuscript. EJL, ACM, JSL, DA, MJL, AY, JBT, JN and ER performed experiments and analyzed data. All authors discussed the results and implications and revised the manuscript. JML supervised the study.
Funding: This research was supported by the Intramural Research Program of the National Cancer Institute (JML, #ZIA BC011525), CCR. Prexasertib was supplied to the CCR by Eli Lilly and Company under a Collaborative Agreement (Cooperative Research and Development Agreement (CRADA #02915)).
Competing interests: ACM receives research grant funding and consultant support from Bristol-Myers Squibb. JML (Institution) receives research funding from Eli Lilly and AstraZeneca. All other authors: Nothing to declare.
Patient consent for publication: Not required.
Ethics approval: The study was approved by the Institutional Review Board of the Center for Cancer Research, National Cancer Institute, Bethesda, Maryland, USA. All patients provided written informed consent before enrollment.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available upon reasonable request. Unlinked genomic data will be deposited in public genomic databases such as dbGaP after publication or completion of all cohorts of this phase II clinical trial, estimated for approximately December 2020, in compliance with the NIH Genomic Data Sharing Policy (see http://bit.ly/CCR_GDS). All requests for raw and analyzed data and materials will be promptly reviewed by the NCI, NIH for Innovation to verify if the request is subject to any intellectual property or confidentiality obligations. Patient-related data not included in the paper were generated as part of clinical trials and may be subject to patient confidentiality. Any data and materials that can be shared will be released via a Material Transfer Agreement. The trial was approved by the Institutional Review Board of the Center for Cancer Research, National Cancer Institute, Bethesda, Maryland, USA. All patients provided written informed consent before enrollment.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- 2.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, et al. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov 2015;5:1137–54. 10.1158/2159-8290.CD-15-0714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim MK, Min DJ, Wright G, et al. Loss of compensatory pro-survival and anti-apoptotic modulator, IKKε, sensitizes ovarian cancer cells to CHEK1 loss through an increased level of p21. Oncotarget 2014;5:12788–802. 10.18632/oncotarget.2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin AB, McNeely SC, Beckmann RP. Achieving precision death with cell-cycle inhibitors that target DNA replication and repair. Clin Cancer Res 2017;23:3232–40. 10.1158/1078-0432.CCR-16-0083 [DOI] [PubMed] [Google Scholar]
- 5.Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci 2013;70:4009–21. 10.1007/s00018-013-1307-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zannini L, Delia D, Buscemi G. Chk2 kinase in the DNA damage response and beyond. J Mol Cell Biol 2014;6:442–57. 10.1093/jmcb/mju045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kastan MB, Bartek J. Cell-Cycle checkpoints and cancer. Nature 2004;432:316–23. 10.1038/nature03097 [DOI] [PubMed] [Google Scholar]
- 8.Lowery CD, VanWye AB, Dowless M, et al. The checkpoint kinase 1 inhibitor Prexasertib induces regression of preclinical models of human neuroblastoma. Clin Cancer Res 2017;23:4354–63. 10.1158/1078-0432.CCR-16-2876 [DOI] [PubMed] [Google Scholar]
- 9.Brill E, Yokoyama T, Nair J, et al. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017;8:111026–40. 10.18632/oncotarget.22195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King C, Diaz HB, McNeely S, et al. LY2606368 causes replication catastrophe and antitumor effects through Chk1-dependent mechanisms. Mol Cancer Ther 2015;14:2004–13. 10.1158/1535-7163.MCT-14-1037 [DOI] [PubMed] [Google Scholar]
- 11.Sun L-L, Yang R-Y, Li C-W, et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am J Cancer Res 2018;8:1307–16. [PMC free article] [PubMed] [Google Scholar]
- 12.Sato H, Niimi A, Yasuhara T, et al. Dna double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 2017;8:1751. 10.1038/s41467-017-01883-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 2019;9:646–61. 10.1158/2159-8290.CD-18-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 2016;17:1142–9. 10.1038/ni.3558 [DOI] [PubMed] [Google Scholar]
- 15.Chabanon RM, Muirhead G, Krastev DB, et al. Parp inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest 2019;129:1211–28. 10.1172/JCI123319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding L, Kim H-J, Wang Q, et al. Parp inhibition elicits STING-dependent antitumor immunity in BRCA1-deficient ovarian cancer. Cell Rep 2018;25:2972–80. 10.1016/j.celrep.2018.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pantelidou C, Sonzogni O, De Oliveria Taveira M, et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov 2019;9:722–37. 10.1158/2159-8290.CD-18-1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen J, Zhao W, Ju Z, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res 2019;79:311–9. 10.1158/0008-5472.CAN-18-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J-M, Nair J, Zimmer A, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol 2018;19:207–15. 10.1016/S1470-2045(18)30009-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tothill RW, Tinker AV, George J, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 2008;14:5198–208. 10.1158/1078-0432.CCR-08-0196 [DOI] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15. 10.1038/nature10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. 10.1172/JCI91190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen B, Khodadoust MS, Liu CL, et al. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol 2018;1711:243–59. 10.1007/978-1-4939-7493-1_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J-M, Cimino-Mathews A, Peer CJ, et al. Safety and clinical activity of the programmed Death-Ligand 1 inhibitor Durvalumab in combination with poly (ADP-ribose) polymerase inhibitor olaparib or vascular endothelial growth factor receptor 1-3 inhibitor cediranib in women's cancers: a dose-escalation, phase I study. J Clin Oncol 2017;35:2193–202. 10.1200/JCO.2016.72.1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sunshine JC, Nguyen PL, Kaunitz GJ, et al. PD-L1 expression in melanoma: a quantitative immunohistochemical antibody comparison. Clin Cancer Res 2017;23:4938–44. 10.1158/1078-0432.CCR-16-1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johansson P, Fasth A, Ek T, et al. Validation of a flow cytometry-based detection of γ-H2AX, to measure DNA damage for clinical applications. Cytometry B Clin Cytom 2017;92:534–40. 10.1002/cyto.b.21374 [DOI] [PubMed] [Google Scholar]
- 27.Chua MLK, Horn S, Somaiah N, et al. DNA double-strand break repair and induction of apoptosis in ex vivo irradiated blood lymphocytes in relation to late normal tissue reactions following breast radiotherapy. Radiat Environ Biophys 2014;53:355–64. 10.1007/s00411-014-0531-z [DOI] [PubMed] [Google Scholar]
- 28.Korwek Z, Sewastianik T, Bielak-Zmijewska A, et al. Inhibition of ATM blocks the etoposide-induced DNA damage response and apoptosis of resting human T cells. DNA Repair 2012;11:864–73. 10.1016/j.dnarep.2012.08.006 [DOI] [PubMed] [Google Scholar]
- 29.Lacy P, Stow JL. Cytokine release from innate immune cells: association with diverse membrane trafficking pathways. Blood 2011;118:9–18. 10.1182/blood-2010-08-265892 [DOI] [PubMed] [Google Scholar]
- 30.Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 2018;215:1287–99. 10.1084/jem.20180139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger G, Marloye M, Lawler SE. Pharmacological modulation of the sting pathway for cancer immunotherapy. Trends Mol Med 2019;25:412–27. 10.1016/j.molmed.2019.02.007 [DOI] [PubMed] [Google Scholar]
- 32.Parkes EE, Walker SM, Taggart LE, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst 2017;109. 10.1093/jnci/djw199. [Epub ahead of print: 05 10 2016]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol 2014;27:1–7. 10.1016/j.coi.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 34.Morse CB, Toukatly MN, Kilgore MR, et al. Tumor infiltrating lymphocytes and homologous recombination deficiency are independently associated with improved survival in ovarian carcinoma. Gynecol Oncol 2019;153:217–22. 10.1016/j.ygyno.2019.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konecny GE, Wang C, Hamidi H, et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J Natl Cancer Inst 2014;106. 10.1093/jnci/dju249. [Epub ahead of print: 30 09 2014]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui TX, Kryczek I, Zhao L, et al. Myeloid-Derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013;39:611–21. 10.1016/j.immuni.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soderquest K, Powell N, Luci C, et al. Monocytes control natural killer cell differentiation to effector phenotypes. Blood 2011;117:4511–8. 10.1182/blood-2010-10-312264 [DOI] [PubMed] [Google Scholar]
- 38.Green DS, Nunes AT, Annunziata CM, et al. Monocyte and interferon based therapy for the treatment of ovarian cancer. Cytokine Growth Factor Rev 2016;29:109–15. 10.1016/j.cytogfr.2016.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Härtlova A, Erttmann SF, Raffi FA, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015;42:332–43. 10.1016/j.immuni.2015.01.012 [DOI] [PubMed] [Google Scholar]
- 40.Brzostek-Racine S, Gordon C, Van Scoy S, et al. The DNA damage response induces IFN. J Immunol 2011;187:5336–45. 10.4049/jimmunol.1100040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunphy G, Flannery SM, Almine JF, et al. Non-Canonical activation of the DNA sensing adaptor sting by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell 2018;71:745–60. 10.1016/j.molcel.2018.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leffers N, Gooden MJM, de Jong RA, et al. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol Immunother 2009;58:449–59. 10.1007/s00262-008-0583-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005;102:18538–43. 10.1073/pnas.0509182102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams SF, Levine DA, Cadungog MG, et al. Intraepithelial T cells and tumor proliferation: impact on the benefit from surgical cytoreduction in advanced serous ovarian cancer. Cancer 2009;115:2891–902. 10.1002/cncr.24317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004;10:942–9. 10.1038/nm1093 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2019-000516supp001.pdf (19.9MB, pdf)