

Abstract
Manganese exposure produces Parkinson's-like neurologic symptoms, suggesting a selective dysregulation of dopamine transmission. It is unknown, however, how manganese accumulates in dopaminergic brain regions or how it regulates the activity of dopamine neurons. Our in vivo studies in male C57BLJ mice suggest that manganese accumulates in dopamine neurons of the VTA and substantia nigra via nifedipine-sensitive Ca2+ channels. Manganese produces a Ca2+ channel-mediated current, which increases neurotransmitter release and rhythmic firing activity of dopamine neurons. These increases are prevented by blockade of Ca2+ channels and depend on downstream recruitment of Ca2+-activated potassium channels to the plasma membrane. These findings demonstrate the mechanism of manganese-induced dysfunction of dopamine neurons, and reveal a potential therapeutic target to attenuate manganese-induced impairment of dopamine transmission.
SIGNIFICANCE STATEMENT Manganese is a trace element critical to many physiological processes. Overexposure to manganese is an environmental risk factor for neurologic disorders, such as a Parkinson's disease-like syndrome known as manganism. We found that manganese concentration-dependently increased the excitability of dopamine neurons, decreased the amplitude of action potentials, and narrowed action potential width. Blockade of Ca2+ channels prevented these effects as well as manganese accumulation in the mouse midbrain in vivo. Our data provide a potential mechanism for manganese regulation of dopaminergic neurons.
Keywords: dopamine neurons, L-type calcium channel, manganese, Parkinson's disease, synaptic transmission
Introduction
Manganese is a trace element critical to many physiological and developmental processes, including the regulation of macronutrient metabolism, blood glucose, cellular energy, reproduction, digestion, and bone growth (Greene and Madgwick, 1988; Erikson et al., 2005). Manganese is a cofactor for several enzymatic processes and a constituent of metalloenzymes, including arginase, pyruvate carboxylase, and manganese-containing superoxide dismutase (Aschner and Aschner, 2005; Aschner et al., 2007; Guilarte, 2010). Except in children on long-term parenteral nutrition or individuals with mutations in the metal transporter SLC39A8 gene, manganese deficiencies are seldom reported (Greene and Madgwick, 1988; Zogzas and Mukhopadhyay, 2017). In contrast, the toxic effects of manganese can be caused by occupational exposures (Bowler et al., 2006; Park et al, 2009), total parenteral nutrition (Hardy, 2009), drinking water with high manganese concentration (Bouchard et al., 2007; Katsoyiannis and Katsoyiannis, 2006), and exposure to air containing manganese concentrations >5 μg/m3 (Yin et al., 2010). Therefore, manganese exposure can occur under many conditions and throughout an individual's lifespan. Excess manganese accumulation in the brain following environmental exposure is implicated in abnormalities related to the dopaminergic system, including Parkinson-like motor dysfunction (Jankovic, 2005), ataxia (Soriano et al., 2016), and hallucinations (Verhoeven et al., 2011). Animal models of manganism have shown that a single large exposure or prolonged moderate exposure to excess manganese is detrimental to the basal ganglia function (Olanow, 2004; Michalke and Fernsebner, 2014), albeit with less understood mechanisms.
Manganese can enter the CNS through the cerebral spinal fluid or by crossing cerebral capillary endothelial membranes (Aschner et al., 2007). Physiologic concentrations of manganese in the human brain range from 20 to 53 μm (Bowman and Aschner, 2014) but can increase severalfold on overexposure both in humans (Kessler et al., 2003; Crossgrove and Zheng, 2004) as well as rodents (Liu et al., 2006). Existing studies have used a wide range (60-150 μm) of extracellular manganese to investigate manganese-associated neurotoxicity (Tuschl et al., 2013; Bowman and Aschner, 2014). Studies on manganese transport in mammalian systems have largely focused on influx mechanisms (Au et al., 2008). Manganese is transported into neurons, and possibly other CNS cell types, through a number of transporters, including divalent metal transporters (Gunshin et al., 1997), the transferrin receptor (Gunter et al., 2013), store-operated Ca2+ channels (Crossgrove and Yokel, 2005), the choline transporter (Lockman et al., 2001), the magnesium transporter (Goytain et al., 2008), and the NMDA receptor (Itoh et al., 2008). In addition, ZIP14 is shown to uptake manganese in human neuroblastoma cells (Fujishiro et al., 2014). The clinical effects of manganese toxicity are primarily Parkinson-like in nature (Jankovic, 2005). This includes movement disorders characterized by tremor, rigidity, dystonia, and/or ataxia; psychiatric disturbances, including irritability, impulsiveness, agitation, obsessive-compulsive behavior, and hallucinations; and cognitive deficits, such as memory impairment, reduced learning capacity, decreased mental flexibility, and cognitive slowing (Josephs et al., 2005). Neuronal degeneration and altered neurotransmitter release occur in brain regions with abnormally high accumulation of manganese, including the dorsal striatum, internal globus pallidus, and substantia nigra (SN) pars reticulata (Crossgrove and Zheng, 2004; Perl and Olanow, 2007; Uchino et al., 2007; Guilarte, 2010). In addition, neuronal loss and gliosis in the globus pallidus, SN pars reticulata, and striatum are reported with high accumulation of manganese (Olanow, 2004). More recently, mutations in the human metal transporter genes ZNT10 and ZIP14 were shown to cause manganese overload and motor dysfunction (Tuschl et al., 2013; Leyva-Illades et al., 2014). In addition to these clinical findings, previous studies show a correlation between elevated extracellular manganese levels in the brain and dysfunction of dopamine transmission (Madison et al., 2012; Dodd et al., 2013), where manganese reduced dopamine uptake and amphetamine-induced dopamine efflux (Roth et al., 2013). Manganese exposure in developing rats reduces both the levels and activity of striatal D2 receptors (Seth and Chandra, 1984; Rogers et al., 2014), supporting the overarching hypothesis for manganese-mediated dysregulation of the dopaminergic system.
The mechanism by which manganese dysregulates dopamine neurons is poorly understood. One potential mechanism by which manganese can regulate cellular responses is through the modulation of Ca2+ concentrations, which not only regulates membrane potential but also serves as an important signaling molecule (Clapham, 2007). Manganese has been shown to regulate Ca2+ signaling in primary cultures of astrocytes where exposure to manganese results in mitochondrial sequestration of Ca2+, which in turn reduces the available pool of releasable Ca2+ within the endoplasmic reticulum (Tjalkens et al., 2006). This can affect the production of reactive oxygen species, free radicals, and toxic metabolites; alteration of mitochondrial function and ATP production; and depletion of cellular antioxidant defense mechanisms (Puskin and Gunter, 1973; Martinez-Finley et al., 2013). In astrocyte culture, manganese rapidly inhibits ATP-induced Ca2+ waves and Ca2+ transients (Streifel et al., 2013) as well as decreases the influx of extracellular Ca2+ induced by 1-oleoyl-2-acetyl-sn-glycerol, a direct activator of the transient receptor potential channel TRPC3 (Streifel et al., 2013).
The mechanistic relationship between manganese regulation of Ca2+ signaling and excitability of dopamine neurons has remained unclear. Recently, we have shown that changes in Ca2+ homeostasis in the dopamine neurons influence neuronal activity indirectly through Ca2+-activated potassium channels (Lin et al., 2016). In the current study, we report a mechanistic link between manganese regulation of the excitability of dopamine neurons and manganese modulation of Ca2+ channels. Here, we identified a cellular mechanism by which manganese accumulates in the midbrain, enters into dopamine neurons, and regulates the activity of dopamine neurons. Contrary to our initial hypothesis, we found that manganese does not block Ca2+ channels in dopamine neurons but acts as a substrate for Ca2+ channels. Manganese permeability through the nifedipine-sensitive Ca2+ channels was further supported by computational modeling, single-neuron analysis, and in vivo MRI experiments showing that blockade of Cav1.2 channels decreased manganese regulation of dopaminergic neuronal activity and its accumulation in the midbrain. These data address the existing debate in the field regarding manganese regulation of Ca2+ channels in dopamine neurons. The mechanistic results reported here provide a clinically relevant therapeutic target that could attenuate the severity of manganese toxicity in patients exposed to excess manganese.
Materials and Methods
Drugs and reagents
The drugs and reagents used in this study were purchased from Sigma Millipore, unless otherwise stated. Chemical reagents and drugs used for primary neuronal culture are listed in Table 1. Antibodies used for Western blot analysis are listed in Table 2. The catalog number of the reagents and drugs used for electrophysiology and microscopy experiments are listed in Table 3.
Table 1.
Chemical reagents and drugs for primary neuronal culture
| Chemical | Concentration (mm) | Manufacturer, catalog # |
| NaCl | 116 | Millipore Sigma, S7653 |
| KCl | 5.4 | Millipore Sigma, P9541 |
| NaHCO3 | 26 | Millipore Sigma, S5761 |
| D-(+)-Glucose | 25 | Millipore Sigma, G8270 |
| NaH2PO4 | 2 | Millipore Sigma, 71505 |
| MgSO4 | 1 | Millipore Sigma, M7506 |
| Cysteine | 1.3 | Millipore Sigma, C6645 |
| EDTA | 0.5 | Millipore Sigma, E9884 |
| Kynurenate | 0.5 | Millipore Sigma, K3375 |
| Papain | 20 units/ml | Worthington, LS003126 |
| Penicillin/streptomycin | 7.70% | Millipore Sigma, P4333 |
| L-glutamine | 0.90% | Millipore Sigma, 59202C |
| Poly-L-lysine | 100 μg/ml | Millipore Sigma, P1399 |
| Laminin | 5 μg/ml | Millipore Sigma, L2020 |
| Insulin | 0.025 g/ml | Millipore Sigma, I2643 |
| Apotransferrin | 50 ml/ml | Millipore Sigma, T0178 |
| GDNF | 1 ng/ml | Millipore Sigma, SRP3200 |
Table 2.
Antibodies for immunofluorescence staining and Western blot
| Antibody | Host species | Dilution | Manufacturer, catalog # |
|---|---|---|---|
| Cav1.2 antibody | Mouse IgG2bκ | 1:1000 | Millipore, MAB13170 |
| Anti-Ca(v)1.3 alpha1 subunit antibody | Rabbit | 1:500 | Millipore, ABN11 |
Table 3.
Chemical reagents and drugs for electrophysiological and TIRF recordings
| Chemical | Concentration (mm) | Manufacturer, catalog # |
| NaCl | 126 | Millipore Sigma, S7653 |
| KCl | 2.5 | Millipore Sigma, P9541 |
| CaCl2 | 2 | Millipore Sigma, 223506 |
| NaHCO3 | 26 | Millipore Sigma, S5761 |
| NaH2PO4 | 1.25 | Millipore Sigma, 71505 |
| MgSO4 | 2 | Millipore Sigma, M7506 |
| Dextrose | 10 | Millipore Sigma, D9434 |
| Potassium gluconate | 120 | Millipore Sigma, P1847 |
| MgCl2 | 2 | Millipore Sigma, M8266 |
| HEPES | 10 | Millipore Sigma, H3375 |
| EGTA | 0.1 | Millipore Sigma, E3889 |
| ATPNa2 | 2 | Millipore Sigma, A2383 |
| GTPNa | 0.25 | Millipore Sigma, G8877 |
| CsMeSO3 | 110 | Millipore Sigma, C1426 |
| Cesium chloride | 1 | Millipore Sigma, 289329 |
| Tetraethylammonium | 10 | Millipore Sigma, T2265 |
| Tetrodotoxin | 0.0005 | Cayman, 14964 |
| 4-Aminopyridine | 1 | Millipore Sigma, 275875 |
| Manganese chloride | 0.1 | Thermo Fisher Scientific, M87-100 |
| Paxilline | 0.01 | Tocris Bioscience, 2006 |
| Nifedipine | 15 mg/kg | Millipore Sigma, N7634 |
| BAPTA-AM | 10 | Millipore Sigma, A1076 |
| NMDA | 0.1 | Millipore Sigma, M3262 |
| Cadmium chloride | 0.1 | Millipore Sigma, 202908 |
| fura-2 AM | 0.005 | Thermo Fisher Scientific, F1201 |
| Caffeine | 20 | Millipore Sigma, C0750 |
Animals
Midbrain neuronal cultures were obtained from RFP::TH C57BL/6 mice (provided by Douglas McMahon, Vanderbilt University), a transgenic mouse strain where the dopamine neurons are rendered fluorescent by expressing the RFP under the TH promoter (Zhang et al., 2004). Mice of the C57BL/6J genetic background conditionally expressing GCaMP6f in dopaminergic neurons were generated by crossing animals expressing Cre recombinase under control of the Slc6a3 restriction (B6.SJL-Slc6a3tm1.1 (cre)BkmnI J; The Jackson Laboratory, stock #006660) to animals expressing GcaMP6f under control of the LoxP promoter (B6;129s-Gt(ROSA) 26Sortm95.1 (CAG-GCaMP6f HzeI J; The Jackson Laboratory, stock #024105). Mice were housed in the animal care facilities at the University of Florida in accordance with the Institutional Animal Care and Use Committee, under guidelines established by National Institutes of Health. Food and water were available ad libitum in the home cage. Animals were housed under standard conditions at 22°C-24°C, 50%-60% humidity, and a 12 h light/dark cycle.
Manganese concentrations used in this study
The physiological concentration of manganese in the human brain is estimated to be between 5.32 and 14.03 ng manganese/mg protein, equivalent to 20.0-52.8 μm (Bowman and Aschner, 2014). Average cellular manganese content in animal models of chronic manganese exposure is as high as 10.95 μg g−1 (200 μm), which has been shown to decrease the viability of dopamine neurons (Higashi et al., 2004). Using this information, we first performed concentration response experiments (see Figs. 1, 2) and found that 100 μm manganese is a suitable concentration to study manganese regulation of intrinsic firing behavior of dopamine neurons.
Figure 1.
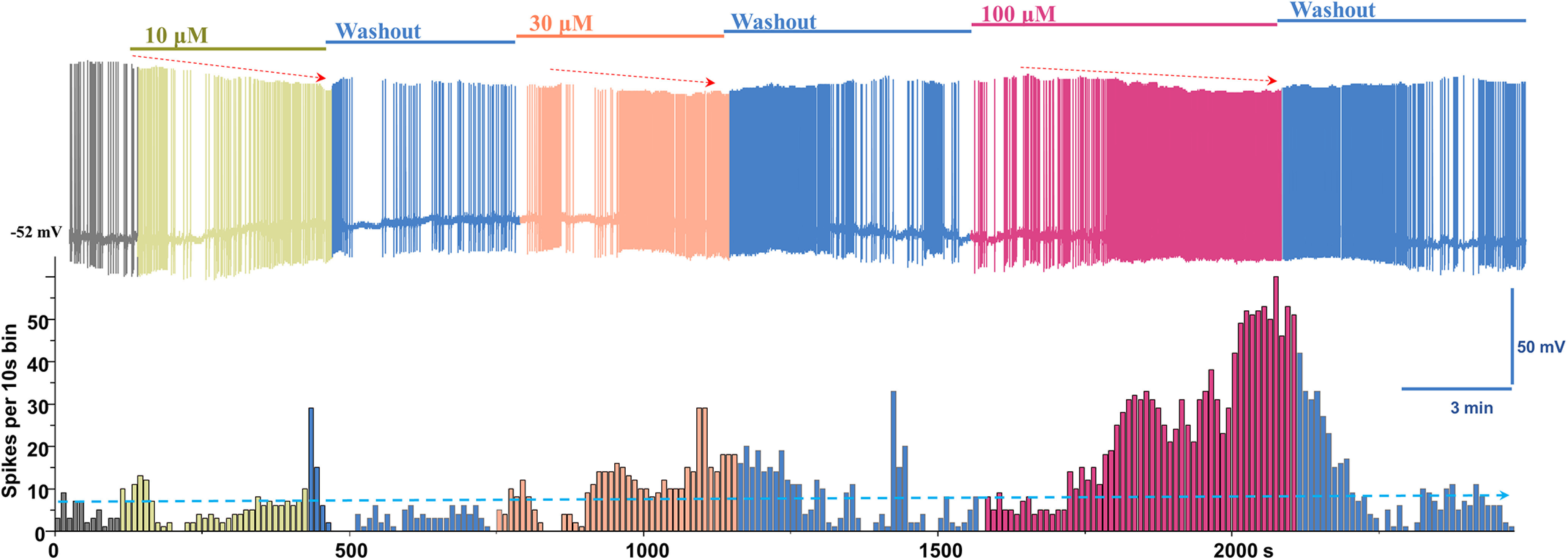
Manganese increases the spontaneous firing activity of dopamine neurons. Top, Representative recording showing the concentration-dependent effect of Mn2+ on the spontaneous firing activity of dopamine neurons and firing rate following washout period. Bottom, Histogram of firing frequency obtained from above trace.
Figure 2.
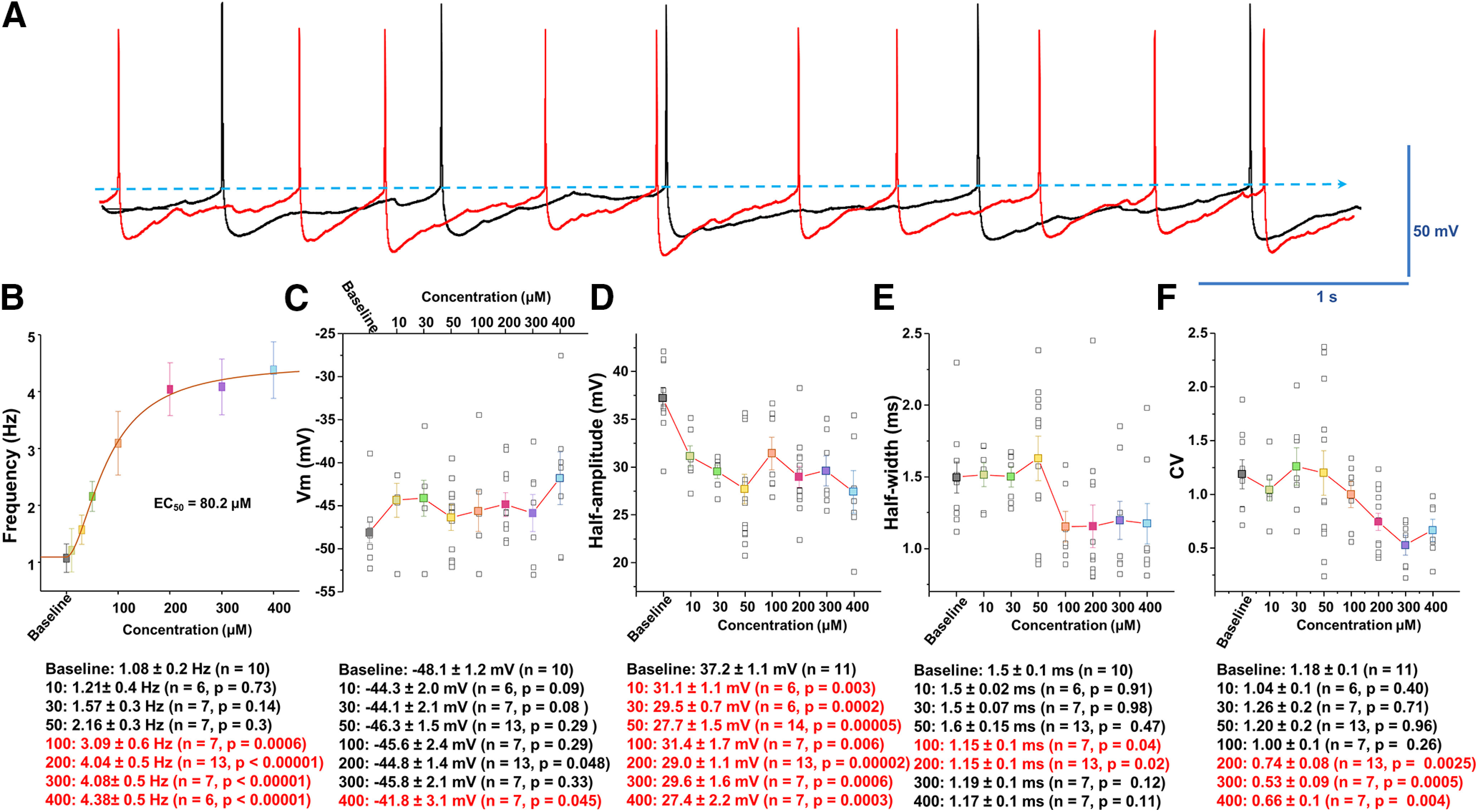
Analysis of spontaneous firing activity and properties of AP following manganese exposure. A, Representative AP trace before (baseline, black trace) and after bath application of 100 μm Mn2+ (red trace). B, Concentration-response relationships of the spontaneous firing frequency of dopamine neurons following Mn2+ exposure. An EC50 of 80.2 μm was obtained by fitting the concentration-dependent curve using a Hill equation. C, Mn2+ did not significantly change membrane potential. D, Mn2+ suppressed the amplitude of AP (as measured as the half-amplitude) at all concentrations examined (baseline: 37.2 ± 1.1 mV; 10 μm manganese: 31.1 ± 1.1 mV; t(15) = 3.6, p = 0.03, two-tailed Student's t test; n = 11 vs 6). E, Mn2+ did not change the half-width of AP at 10-50 μm, but it significantly narrowed the half-width at 100 μm (baseline: 1.5 ± 0.1 ms; 100 μm manganese: 1.15 ± 0.1 ms; t(15) = 2.2, p = 0.04, two-tailed Student's t test; n = 10 vs 7). F, Mn2+ concentration-dependently decreased the CV of interspike intervals (baseline: 1.2 ± 0.1; 200 μm manganese: 0.7 ± 0.1; t(22) = 3.4, p = 0.003, two-tailed Student's t test; n = 11 vs 13).
Mice were injected intraperitoneally with manganese (II) chloride tetrahydrate (70 mg/kg, MnCl2·4H2O) based on a published protocol shown to increase manganese concentrations in the basal ganglia (Dodd et al., 2005).
Neuronal and cells culture
Primary neuronal culture was performed as previously described (Saha et al., 2014; Lin et al., 2016; Sambo et al., 2017). Briefly, mouse midbrain dopamine neurons from 0- to 2-d-old pups of either sex were isolated and incubated in a dissociation medium (in mm) as follows: 116 NaCl, 5.4 KCl, 26 NaHCO3, 25 glucose, 2 NaH2PO4, 1 MgSO4, 1.3 cysteine, 0.5 EDTA, 0.5 kynurenate containing 20 units/ml papain at 34°C-36°C under continuous oxygenation for 2 h. Tissue was triturated with a fire-polished Pasteur pipette in glial medium (in %, v/v): 50 minimum essential media, 38.5 heat-inactivated FBS, 7.7 penicillin/streptomycin, 2.9 (w/v) dextrose (45%), and 0.9 glutamine (200 mm). Dissociated cells were pelleted by centrifugation at 500 × g for 10 min and resuspended in glial medium. Cells were plated on 12 mm round coverslips coated with 100 μg/ml poly-L-lysine and 5 μg/ml laminin in 35 × 10 mm tissue culture Petri dishes. One hour after plating, the medium was changed to neuronal medium: Neurobasal (Invitrogen), 0.9% L-glutamine, 2% B27, and 1 ng/ml GDNF. Neuronal medium was conditioned overnight on cultured glia. The conditioned neuronal medium was supplemented with 1 ng/ml GDNF and 500 μm kynurenate and sterile-filtered before it was added to the neuronal culture.
Plasmids for Cav1.2-GFP and Cav1.3-GFP were generous gifts from Gregory Hockerman (Purdue University). Briefly, cDNAs that encode the cytoplasmic II-III interdomain loop of Cav1.2 (amino acids 758-903) or Cav1.3 (amino acids 752-885) were fused to enhance GFP constructed in the pEGFP-N1 vector (Clontech). Plasmids were transformed into MAX Efficiency DH5α Competent Cells (Invitrogen) and amplified on Luria-Bertani agar plates with an ampicillin concentration of 50 mg/L. Individual colonies were selected and further amplified in 250 ml Luria-Bertani medium with 50 mg/L ampicillin. Plasmid DNAs were purified using the Plasmid Plus Maxi Kit (QIAGEN). The integrity of the clones was confirmed by cDNA sequencing and restriction digest analysis.
HEK 293 cells stably expressing Cav1.2 or 1.3 were a generous gift from Richard B. Silverman (University of Chicago). Cells were maintained in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS (Gemini, catalog #100-106), 1.1 mm sodium pyruvate (Sigma Millipore, catalog #S8636-100ML), 0.1 mg/ml zeocin (InvivoGen, catalog #ant-zn-1), and 0.05 mg/ml hygromycin B (A.G. Scientific, catalog #H-1012-PBS). Additionally, either 0.3 mg/ml G418 (Caisson Labs, catalog #ABL06-20ML) or 0.2 mg/ml blasticidin S hydrochloride (Research Products International, catalog #B12200-0.05) was added for Cav1.2 or Cav1.3, respectively. Cells were passaged using TrypLE Express Enzyme (Invitrogen, catalog #12604-021) at 70%-85% confluency 24-48 h before experimentation.
54Mn uptake
Effectene reagent (QIAGEN) was used for transient transfection of HEK cells with a plasmid vector either empty or containing Cav1.2 or Cav1.3 cDNA. Overexpression of Cav1.2 and Cav1.3 was assessed by Western blotting 48 h after transfection (Sigma Millipore). For 54Mn uptake experiments, following a 48 h transfection, the cells were washed with HBSS and incubated at 37°C in serum-free DMEM containing 40 μm MgCl2 and 54Mn at 0.18 µCi/ml (PerkinElmer). In some experiments, cells were pretreated with 10 μm nifedipine 30 min before 54Mn treatment. After incubation with 54Mn for the times indicated, cells were washed with a chelating buffer (10 mm EDTA, 10 mm HEPES, and 0.9% NaCl) and solubilized in 0.2% SDS 0.2 m NaOH for 1 h. Radioactivity was measured by γ-ray solid scintillation spectrometry. Protein content was measured colorimetrically with BCA reagent (Thermo Fisher Scientific). Results were expressed as counts per minute per mg total protein.
Electrophysiological recordings
Spontaneous firing activity of midbrain dopamine neurons was examined via whole-cell current-clamp recordings as previously described (Saha et al., 2014; Lin et al., 2016; Sambo et al., 2017). The neurons were continuously perfused with aCSF containing the following (in mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 26 NaHCO3, 1.25 NaH2PO4, 2 MgSO4, and 10 dextrose, equilibrated with 95% O2/5% CO2; pH was adjusted to 7.4 at 37°C. Patch electrodes were fabricated from borosilicate glass (1.5 mm outer diameter; World Precision Instruments) with the P-2000 puller (Sutter Instruments). The tip resistance was in the range of 3-5 mΩ. The electrodes were filled with a pipette solution containing the following (in mm): 120 potassium-gluconate, 20 KCl, 2 MgCl2, 10 HEPES, 0.1 EGTA, 2 ATP, and 0.25 GTP, with pH adjusted to 7.25 with KOH. All experiments were performed at 37°C. To standardize action potential (AP) recordings, neurons were held at their resting membrane potential (see below) by DC application through the recording electrode. AP was recorded if the following criteria were met: a resting membrane potential <−35 mV and an AP peak amplitude of >60 mV. AP half-width was measured as the spike width at the half-maximal voltage using Clampfit 10 software (Axon Instruments). Steady-state basal activity was recorded for 2-3 min before bath application of the drug. For experiments involving drug application, each coverslip was used for only one recording. The spontaneous spike activity of midbrain dopamine neurons was obtained by averaging 1 min interval activities at baseline (before manganese) and after 7-10 min of manganese. Mn concentration-response experiments were individually examined for concentration of 10, 30, 50, 100, 200, 300, and 400 μm. Since the maximal spontaneous firing activity at each concentration of Mn applications appeared at ∼3 min, all recordings including baseline, drug application, and washout, were done within 10 min. The relationship between Mn concentration and neuronal firing frequency was fitted according to a Hill equation: E = EmaxMnnH/(EC50 + MnnH) where E is the predicted effect of Mn, Emax is the maximum effect, nH is the slope factor, and the nth root of EC50 gives an estimate of the midpoint of the activation curve.
Recording from HEK cells expressing GFP α subunits
HEK293 cells stably expressing GFP α subunits where generous gift from Robert Brenner (University of Texas Health Science Center at San Antonio). Cell culture was performed as described previously (Goodwin et al., 2009; Wang et al., 2009; Saha et al., 2014). The cells were plated on glass coverslips (Electron Microscopy Sciences). GFP expression was used to identify α subunit-expressing cells. Electrophysiology experiments were performed 3 d after plating the cells. Macropatch recordings were performed using the excised inside-out patch-clamp configuration at 22°C-23°C. Patch pipettes were pulled to a final tip resistance of 1.5-3 mΩ and filled with the internal solution. The electrode solution was composed of the following (in mm): 20 HEPES, 140 KMeSO3, 2 KCl, and 2 MgCl2, pH 7.2. Bath solution was composed of a pH 7.2 solution of the following (in mm): 20 HEPES, 140 KMeSO3, and 2 KCl. Intracellular Ca2+ was buffered with 5 mm HEDTA to give the required 10 μm free [Ca2+] concentration calculated using the MAXCHELATOR program (Winmaxchelator software; Chris Patton, Stanford University). Recordings and data acquisition were described in Single-channel recordings above. Mean current density was plotted against membrane potential (I–V) and was fitted by the Boltzmann equation: I = Imin + (Imax − Imin)/(1 + exp − κ(V − V1/2)), where I is the current, Imax is the maximum current, Imin is the minimum current, κ is a slope factor, and V1/2 is the midpoint potential.
Pharmacological isolation of Ca2+ currents
For whole-cell recordings, we used a cesium methane sulfonate-based intracellular solution composed of the following (in mm): 110 CsMeSO3, 10 TEA-Cl, 10 4-aminopyridine, 10 HEPES, 1 MgCl2, 10 EGTA, 2 ATP, and 0.3 GTP, with pH adjusted to 7.25 with CsOH. Bath solution contained the following (in mm): 110 NaCl, 35 tetraethylammonium (TEA)-Cl, 1 CsCl, 1 MgSO4, 2 CaCl2, 10 HEPES, 11.1 dextrose, and 0.001 TTX, pH 7.4 with CsOH. For the Ca2+-free bath solution, 2 mm CaCl2 was replaced with 3 mm MgCl2, a HEPES solution containing 4 mm Mg2+. Calcium currents were evoked by a series of 200 ms depolarizing steps from −60 to 85 mV in 5 mV increments. To avoid possible interference between responses, the depolarizing voltage steps were delivered every 5 s. Data were obtained in 1 to 3 min intervals while the patches were held at −65 mV. The series resistances were in the range of 5-10 mΩ (typically 5 mΩ) and were compensated 60% online. Membrane potential measurements were not corrected for the liquid junction potential (∼15 mV). Leak currents were subtracted using a standard P/4 protocol. Before seals (5 GΩ) were made on cells, offset potentials were nulled. Capacitance subtraction was used in all recordings. To determine current density (pA/pF), the peak current value of the steady-state current at 180 ms was divided by the membrane capacitance from each recorded cell. Since maximum spontaneous firing appeared ∼3 min after manganese application (see Fig. 1), we have matched this time point for Ca2+ current recording.
Preparation of mouse brain slices
Unless otherwise noted, 30- to 40-d-old C57BL/6 male mice in either WT or mice conditionally expressing GCaMP6f in dopaminergic neurons (described above) were used for slice preparation. Only males heterozygous were selected and used for these experiments. Mice were deeply anesthetized with 4% isoflurane, and the brain was subsequently removed from the cranium. The tissue was glued onto the cutting stage and submersed in ice-cold, oxygenated aCSF (equilibrated with 95% O2/5% CO2). Coronal or horizontal brain slices (200 μm) containing the SN compacta or striatum were cut using a MicroSlicer Zero 1N (Dosaka).
FFN200 loading and multiphoton imaging
Striatal slices of WT mice were incubated in oxygenated aCSF with 10 μm FFN200 (Tocris Bioscience) for 30 min at 22°C-24°C and washed for 45-50 min before imaging. After incubation with FFN200, dorsal striatal slices were transferred into imaging chambers, then continuously perfused with aCSF equilibrated with 95% O2/5% CO2. In vitro multiphoton imaging was performed with a Nikon AR1 MP (Nikon Instruments) equipped with a plan apo LWD 25×, water-immersion (NA = 1.1, WD = 1.43-2.04 mm, DIC N2) objective. Multiphoton excitation was generated with a Spectra Physics 15 W Mai Tai eHP tunable Ti:sapphire femtosecond pulsed IR laser. FFN200 imaging experiments were performed using excitation wavelengths of 830-840 nm and an emission of 430-470 nm. Emission light was directed with a 405 nm dichroic mirror through a 340 nm short pass filter to enhanced hybrid PMTs via fluorophore specific filters. Imaging sites in time lapse experiments were aligned based on the multiphoton imaged dendritic structure and FFN200 puncta. Imaging sites in time lapse experiments were aligned based on the multiphoton-imaged dendritic structure and FFN200 puncta. Images were collected with a 2.4 μs pixel dwell time, with the laser attenuated to 3% of total power, a pixel size of 0.24 μm (XY), and a step size of 0.5 μm (Z) at 1024 × 1024 pixel resolution. After acquiring an initial image (t = 0), perfusion was switched to aCSF containing 100 μm MnCl2 and images acquired every 4s. Control slices labeled with FFN200 were imaged 5-10 min in the absence of MnCl2.
Two-photon images and analysis of fluorescent intensity for FFN200 puncta were performed using NIS Elements Software version 4.5 (Nikon Instruments). ROIs were automatically selected by the program. Background fluorescence intensity was determined as the mean fluorescence intensity of areas of the image that excluded puncta. The mean background-subtracted fluorescent puncta intensity was then calculated by subtracting the mean background intensity from the mean fluorescence intensity measured at the puncta.
fura-2 calcium imaging
For fura-2 calcium imaging, midbrain neuronal cultures were used. Neurons were washed twice in HBSS (Invitrogen) followed by 30 min treatment in 5 μm fura-2 AM (Thermo Fisher Scientific). Cells were then washed twice with HBSS followed by 30 min incubation in HBSS. Cells were then imaged on a Nikon Ti Eclipse inverted microscope. Fluorescence was monitored using dual excitation wavelengths (340/380 nm) and a single emission wavelength (510 nm). fura-2 bound to free calcium is excited at 340 nm, whereas unbound fura-2 is excited at 380 nm. Baseline images were taken for 30 s to 1 min followed by addition of vehicle, 100 μm manganese, or 20 mm caffeine as the positive control group. The concentration of caffeine used in this study was selected based on a previous study examining caffeine regulation of Ca2+ mobilization in the midbrain dopamine neurons (Y. M. Choi et al., 2006). Images were acquired for 4 min after treatment. For analysis, ROIs were drawn along the cell body of each neuron. The method for the selection of ROIs is based on previously published reports (Zipfel et al., 2003; Tian et al., 2009). Ca2+ changes were determined as the fluorescence normalized to the average fluorescence of the first 30 s of baseline imaging for each neuron. The percent fluorescence change over baseline was calculated as follows: %ΔF/F0 = (Fmax – F0)/F0 × 100, where Fmax is the maximal fluorescent value after treatment and F0 is the average fluorescence at baseline. Images were presented as the ratio of 340/380 imaging.
Total internal reflection fluorescence (TIRF) microscopy
For these experiments, HEK293 cells expressing GFP α subunits were plated onto 35 mm glass-bottom dishes to 60%-80% confluence. Ti Eclipse inverted microscope (Nikon) equipped with a multiline solid-state laser source (470, 514, 561 nm) was used for all TIRF microscopy imaging. Lasers were guided through a 60 × 1.4 NA objective. Images were detected digitally using a CCD camera. Image exposure time was coupled with stimulation duration at 100 ms, and laser intensity was maintained at 40%. For quantification of fluorescence intensity at the cell surface, experimenter-defined ROIs were created for each cell to exclude both cell membrane overlap between adjacent cells and measurement of intensity at the peripheral edges of cells. Background fluorescence was subtracted from all images. Mean intensity over time for each ROI was recorded continuously before and after the application of 100 μm manganese to the bath solution. Experiments were performed in the isotonic, isosmotic external solution described above. The baseline fluorescent intensity is defined as the average fluorescent intensity 1 min before drug application. All values were normalized to the baseline fluorescent intensity. Single-count elementary sequential smoothing was applied for image presentation only, and not for analysis. Bleaching at each experimental time point was determined by the amount of decreased fluorescence intensity in untreated cells.
MRI scans
Seventeen adult male mice (35- to 40-d-old male WT C57BL/6 mice) were randomly assigned to two groups either receiving manganese only or manganese plus the L-type Ca2+ channel inhibitor nifedipine (15 mg/kg). The systemic injection of 15 mg/kg nifedipine has been shown to cross the blood–brain barrier (Cain et al., 2002) and inhibits the L-type Ca2+ channels (Jinnah et al., 1999). The treatment doses were based on the previously published protocols (Jinnah et al., 1999; Cain et al., 2002; Dodd et al., 2005). Both experimental groups received manganese (II) chloride tetrahydrate (70 mg/kg) via intraperitoneal injection, but one group received manganese injection after 30 min nifedipine (15 mg/kg, i.p.) injection. Magnetic resonance (MR) scanning was performed 24 h after manganese exposure. On the scanning day, mice were anesthetized using 3%-4% isoflurane delivered in medical grade air (70% nitrogen/30% oxygen; air flow rate 1.5 ml/min). The anesthesia was maintained at 1.0%-1.5% isoflurane during MRI scanning. Core body temperature and spontaneous respiratory rates were continuously recorded during MRI scanning (SA Instruments). Mice were maintained at normal body temperature levels (37°C-38°C) using a warm water recirculation system. The manganese-enhanced MRI (MEMRI) scans were collected in a 4.7T/33 cm horizontal bore magnet (Magnex Scientific) at the Advanced Magnetic Resonance Imaging and Spectroscopy facility in the McKnight Brain Institute of the University of Florida. The MR scanner consisted of a 11.5 cm diameter gradient insert (Resonance Research) controlled by a VnmrJ 3.1 software console (Agilent Technologies). A quadrature transmit/receive radiofrequency coil tuned to 200.6 MHz 1H resonance was used for B1 field excitation and radiofrequency signal detection (airmri). The MEMRI included a multiple repetition time sequence to calculate parametric T1 maps for each group using a fast spin echo sequence with a total of four TRs (0.5, 1.08, 2.33, 5.04 s), and TE = 6.02 ms with the following geometric parameters: 16 × 16 mm2 in plane, 14 slices at 0.8 mm thickness per slice, data matrix = 128 × 128 (125 μm in-plane resolution).
MRI postprocessing
Whole-brain masks were obtained via automatic segmentation with PCNN using high-resolution anatomic scans to remove nonbrain voxels. All cropped data were used to create templates for each cohort using Advanced Normalization Tools (http://stnava.github.io/ANTs/). The templates were then registered to an atlas of the mice brain using the FMRIB Software Library linear registration program flirt (Jenkinson et al., 2002). The atlas was then transformed back to each individual dataset with the registration matrices from Advanced Normalization Tools. To generate parametric T1 maps, multi-TR images were fit to the equation STR = S0(1 – e–TR/T1) using nonlinear regression in QuickVol II for ImageJ (Schmidt et al., 2004). From T1 maps, the T1 relaxation rate (R1 in ms−1) is calculated and exported from ROIs. These methods allowed the measurement of the amount of activity, along with the location of manganese uptake in both the control and drug-exposed groups.
Experimental design and statistical analysis
The data were analyzed under blind condition. We have repeated each experiment from start to finish in multiple animals, multiple sets of neuronal cultures, and multiple set of cell cultures. For primary culture experiments, one coverslip represents one technical replicate. On average, 1-3 coverslips were used from one midbrain primary culture preparation. Each coverslip is used once. Each midbrain tissue provides 1 or 2 slices. Each slice is used once (i.e., 1 or 2 technical replicates). Therefore, n = 8 from four biological replicates means we have used 4 animals and a total of 8 slices or coverslips. All statistical analyses performed on data presented in the manuscript are stated in the figure legends and detailed in Materials and Methods and Results. For each experiment, statistical tests were chosen based on the structure of the experiment and dataset. No outliers were removed during statistical analysis. Sample sizes estimates were based on published studies in the field that used similar models and techniques as those used in this study. The electrophysiology data were acquired using the ClampEx 10 software (Molecular Devices). The data were analyzed offline using pClamp 10. For all experiments, the data are presented as mean ± SEM. N denotes the number of neurons or cells for in vitro experiments, or mice for in vivo experiments. Statistical significance was assessed using two-tailed Student's t test or one-way ANOVA. If ANOVA showed statistical significance, all pairwise post hoc analysis was performed using a Tukey's post hoc test. Differences were considered significant at p < 0.05. The coefficient of variation (CV) is a measure of the relative spread of the data. It is computed as the SD divided by the mean times 100%. SigmaPlot 11 was used for all statistical analysis.
Results
We exploited multiple complementary approaches to identify dopamine neurons for the electrophysiological recordings, as described previously (Lin et al., 2016). A total of 149 midbrain dopamine neurons meeting the criteria, as described in Materials and Methods, were recorded and analyzed. The average resting membrane potential was −48.3 ± 1.1 mV; input resistance was 252.5 ± 21.9 mΩ; membrane time constant was 826.9 ± 52.5 μs; and membrane capacitance was 64.1 ± 3.3 pF.
Manganese altered the intrinsic firing behavior of midbrain dopaminergic neurons
Spontaneous firing activity of dopamine neurons was measured before and after manganese application at concentrations of 10, 30, and 100 μm (Fig. 1). Lower concentrations of 10 and 30 μm are within the normal range of intracellular manganese (Bowman and Aschner, 2014), whereas 100 μm falls within the pathologic range (Gandhi et al., 2018). Acute exposure to manganese increased the spontaneous firing frequency and suppressed the amplitude of APs (red arrows) in a concentration-dependent manner. Superimposed representative traces of spontaneous spikes before (black trace) and after 100 μm manganese (red trace) exposure revealed that manganese increased the firing frequency of dopamine neurons and truncated AP amplitudes (Fig. 2A). The EC50 for the manganese-mediated increase in firing frequency of dopamine neurons was determined to be 80.2 μm by fitting a concentration-dependent curve using a Hill equation (Fig. 2B). While manganese induced a modest membrane depolarization at lower concentrations (10 μm: −44.3 ± 2.0 mV vs 50 μm: −46.3 ± 1.5 mV), it significantly depolarized membrane potential at the highest concentration tested in this study (400 μm), which is consistent with the reported manganese concentration for in vitro studies (Bowman and Aschner, 2014) (Fig. 2C; baseline: −48.1 ± 1.2 mV; 400 μm manganese: −41.8 ± 3.1 mV; t(15) = −2.2, p = 0.045, two-tailed Student's t test; n = 7 vs 10). Although manganese markedly suppressed AP amplitude at the lowest concentration of 10 μm (baseline: 37.2 ± 1.1 mV; 10 μm manganese: 31.1 ± 1.1 mV; t(15) = 3.6, p = 0.03, two-tailed Student's t test; n = 11 vs 6), higher concentrations of manganese did not further suppress AP amplitude (Fig. 2D). In contrast, manganese did not influence AP half-width at low concentrations, but it significantly narrowed AP width at 100 μm (Fig. 2E, baseline: 1.5 ± 0.1 ms; 100 μm manganese: 1.15 ± 0.1 ms; t(15) = 2.2, p = 0.04, two-tailed Student's t test; n = 10 vs 7). Manganese progressively decreased the CV, indicating a reduction in firing variation evident by the small interspike interval (Fig. 2F, baseline: 1.2 ± 0.1; 200 μm manganese: 0.7 ± 0.1 ms; t(22) = 3.4, p = 0.003, two-tailed Student's t test; n = 11 vs 13). These results suggest that manganese regulates the intrinsic firing behavior of dopamine neurons at both physiological and pathophysiological concentrations in a concentration-dependent manner. To address the pathologic effects of manganese, 100 μm was used for the rest of the study based on the determined EC50 in Figure 2B.
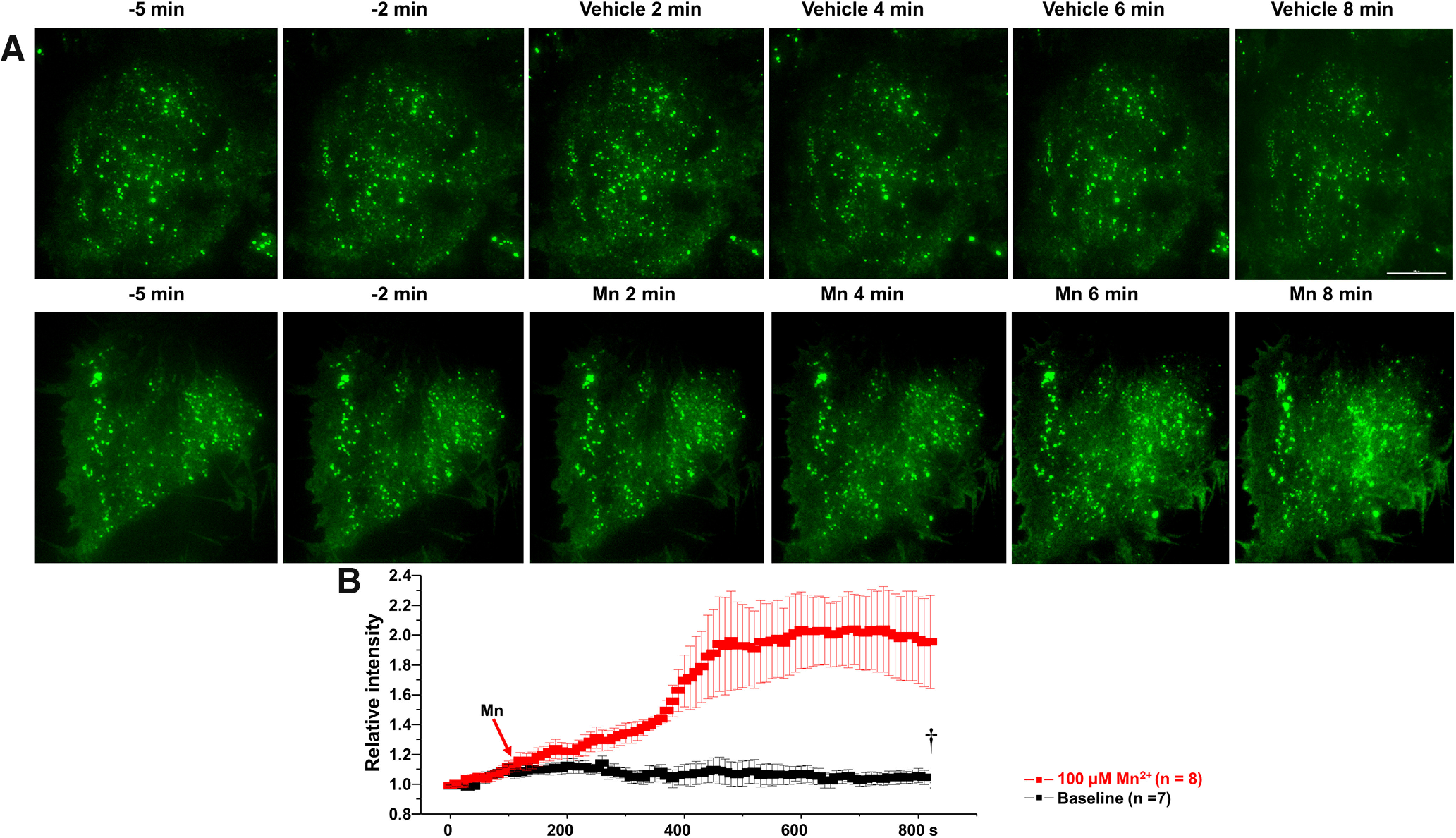
Manganese stimulation of neuronal activity increased the release of fluorescent false neurotransmitter (FFN200)
Next, we asked whether manganese stimulation of firing frequency of dopamine neurons leads to increased neurotransmitter release. To do this, we used FFN200, a fluorescent substrate of VMAT2 that selectively traces monoamine exocytosis in both neuronal culture and striatal slices (Pereira et al., 2016). “Destaining” of FFN200 associated with monoamine vesicular release has been used as a surrogate for neurotransmitter release (Pereira et al., 2016). Consistent with a previous report (Pereira et al., 2016), we found that, following vehicle (aCSF) application, there is a time-dependent and slow destaining of FFN200 at striatal dopamine terminals (Fig. 3), suggesting a direct correlation between spontaneous firing activity of dopamine neurons and neurotransmitter release. Manganese increased FFN200 release as measured by increased rate and magnitude of fluorescence destaining. These data suggest that manganese stimulation of neuronal activity correlates with increased neurotransmitter release (Fig. 3, yellow arrows; mean ± SEM, F(1,148) = 70.2, p < 0.01, one-way ANOVA followed by Tukey's test, n = 4 slices/group). Additional control experiments were performed when the release of FFN200 is inhibited. Since blockade of dopamine transporter (DAT) decreases firing activity of dopamine neurons (Saha et al., 2014; Lin et al., 2016), theoretically, it should decrease the destaining rate or even increase the fluorescent punctate due to accumulation of FFN200-filled synaptic vesicles in the terminal regions. As shown in Figure 3, treatment with DAT inhibitor nomifensine (5 μm) produced a plateau in the fluorescent signal followed by increased fluorescent levels, suggesting inhibition of neurotransmitter release. These data further support the findings that manganese treatment increases the activity of dopaminergic neurons.
Figure 3.
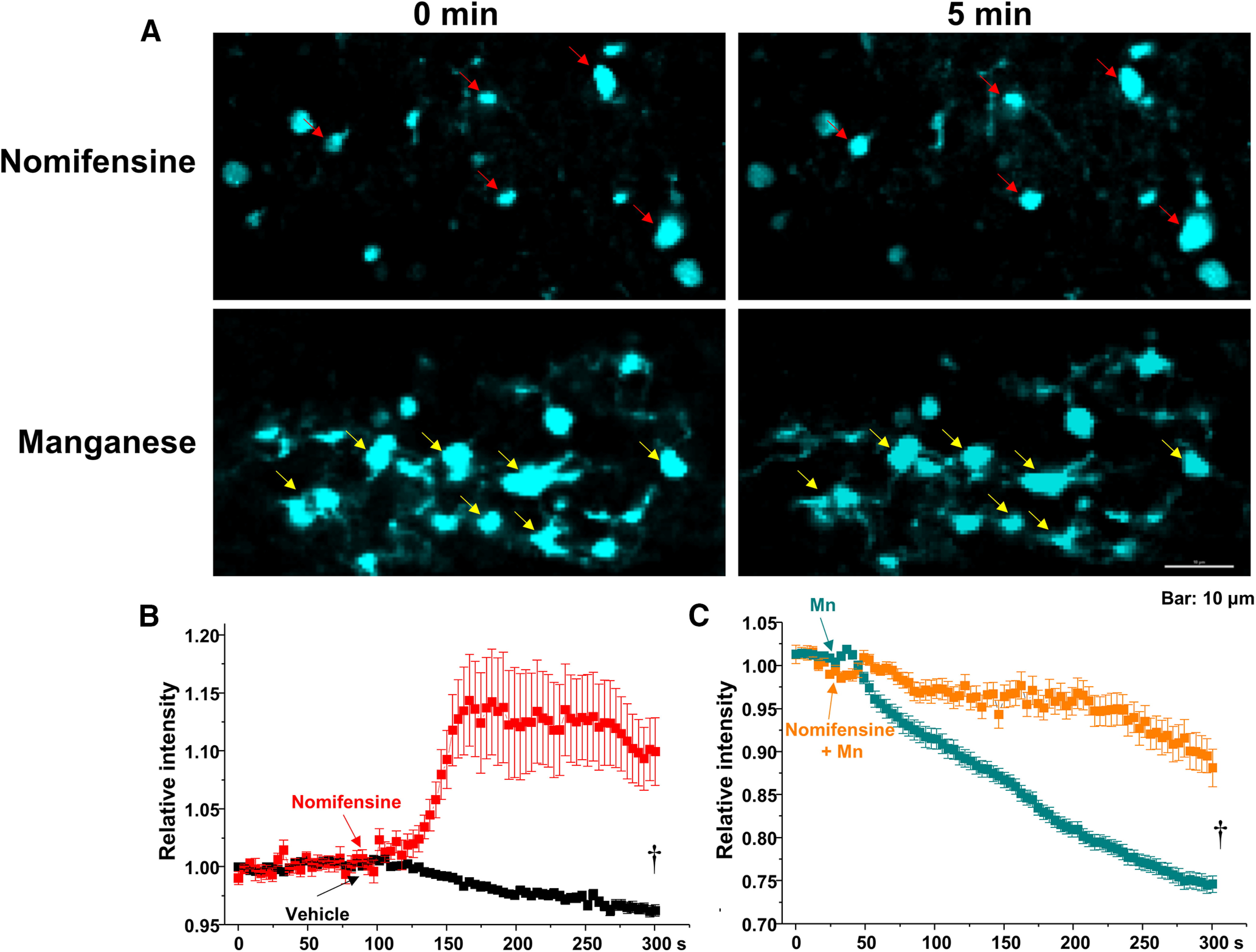
Mn2+ induces release of FFN200 from dorsal striatum. A, Time lapse images of a striatal slice loaded with FFN200 following nomifensine (A, top) or 100 μm Mn2+ treatments (A, bottom). Mn2+ treatment decreased FFN200 puncta intensities (yellow arrows). B, C, Quantification of FFN200 puncta fluorescence normalized to the averaged intensity of first 60 s for each experimental condition. Scale bar, 10 μm. †p < 0.01.
Removal of extracellular Ca2+ did not prevent manganese stimulation of spontaneous spike frequency
Our observed broad APs and slow autonomous pacemaking activities are consistent with known electrophysiological features in dopamine neurons (Bean, 2007). Numerous studies have shown that slow rhythmic spiking is accompanied by large oscillations in intracellular Ca2+ concentrations that are driven by the opening of voltage-dependent Ca2+ channels (Nedergaard et al., 1993; Mercuri et al., 1994; Puopolo et al., 2007; Guzman et al., 2009). Therefore, next we measured manganese regulation of the spontaneous firing activity of dopamine neurons in Ca2+-free extracellular solution. Unexpectedly, removal of extracellular Ca2+ did not prevent manganese stimulation of the firing frequency (Fig. 4A,C; baseline: 1.3 ± 0.2 Hz vs manganese in Ca2+-free: 6.5 ± 1.1 Hz; F(2,18) = 20.9, p = 0.000002, one-way ANOVA followed by Tukey's test; n = 7/group). Ca2+-free solution containing manganese did not change the membrane potential (Fig. 4D), AP half-width (Fig. 4E), or CV (Fig. 4G); however, it significantly truncated the amplitude of AP (Fig. 4F; baseline: 45.4 ± 2.2 mV vs manganese in Ca2+-free: 37.9 ± 0.7 mV; F(2,18) = 16.1, p = 0.0001, one-way ANOVA followed by Tukey's test; n = 7/group). In the absence of manganese, comparison of the firing activity before and after perfusion of a Ca2+-free external solution revealed a reduction in the firing frequency (Fig. 4C; 0.5 ± 0.03 Hz), broadening of AP half-width (Fig. 4E; baseline: 1.6 ± 0.08 ms vs Ca2+-free: 2.9 ± 0.2 ms, F(2,18) = 35.9, p = 0.0001, one-way ANOVA followed by Tukey's test; n = 7/group), truncated AP amplitude (Fig. 4F; 32.5 ± 2.0 mV, n = 7/group), and an increased CV (Fig. 4G; baseline: 1.1 ± 0.2 vs Ca2+-free: 1.99 ± 0.3, F(2,18) = 7.3, p = 0.004, one-way ANOVA followed by Tukey's test; n = 7/group). These results suggest that manganese stimulation of neuronal activity it is not dependent on extracellular Ca2+ entry into the dopamine neuron.
Figure 4.
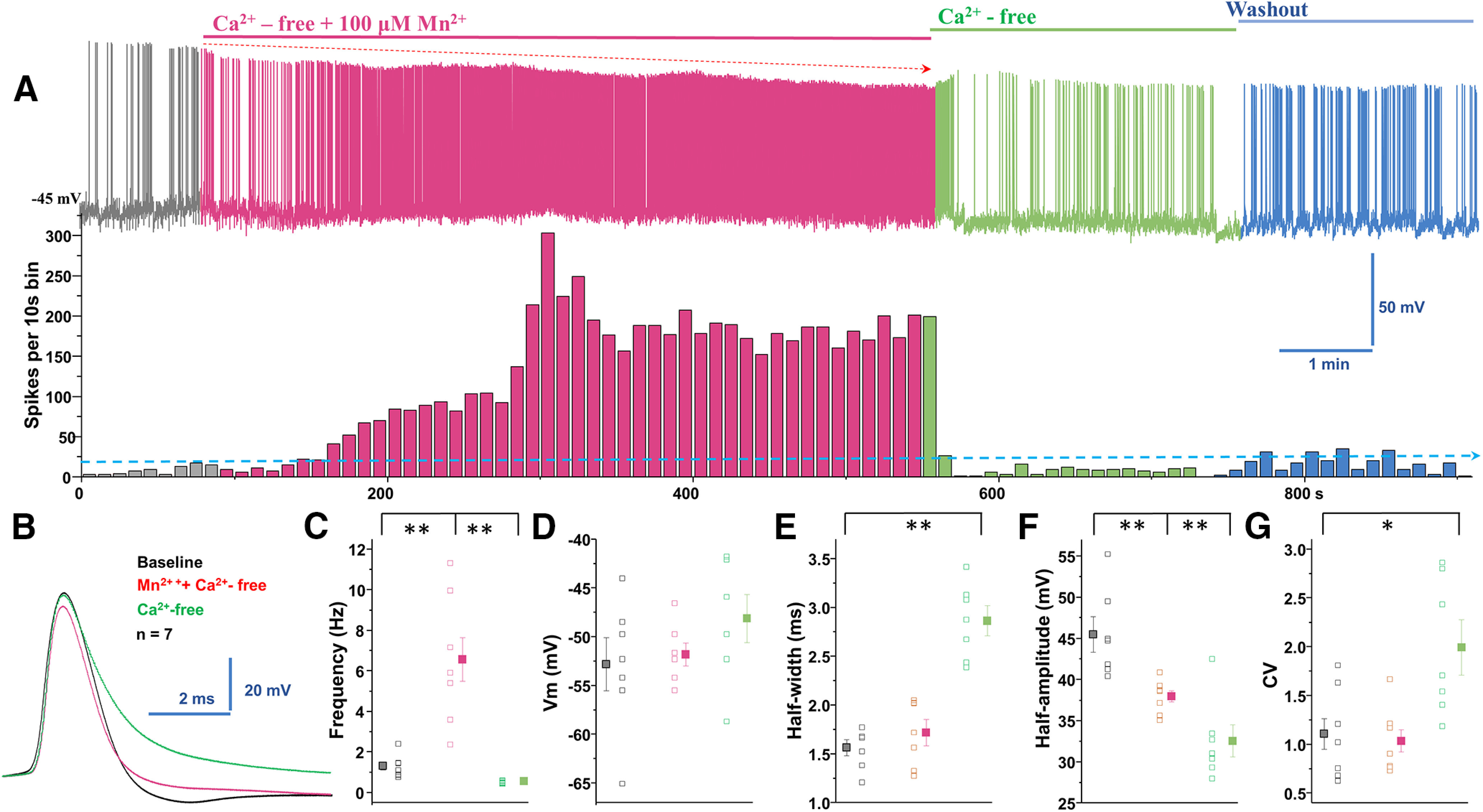
Manganese stimulation of firing activity of dopamine neurons is not dependent on extracellular Ca2+. A, Top, Representative spontaneous spike activities of a neuron exposed to Mn2+ in a Ca2+-free extracellular solution. Middle, Histogram of firing frequency obtained from the above trace showing Mn2+ increased firing frequency. B, Superimposed representative single AP traces shown in A, baseline (black trace), Mn2+ + Ca2+-free solution (red trace), and Ca2+-free solution only (green trace). C, In Ca2+-free solution, Mn2+ increased spontaneous firing rate. Mn2+ washout in Ca2+-free conditions revealed a reduction of firing rate (baseline: 1.3 ± 0.2 Hz vs manganese in Ca2+-free: 6.5 ± 1.1 Hz; F(2,18) = 20.9, p = 0.000002, one-way ANOVA followed by Tukey's test; n = 7/group). D, Mn2+ and Ca2+-free extracellular solution did not change membrane potential. E, Half-width measured at half-maximal voltage of AP was not different between baseline and Mn2+ application, but it was significantly broadened in Ca2+-free condition compared with baseline (baseline: 1.6 ± 0.08 ms vs Ca2+-free: 2.9 ± 0.2 ms, F(2,18) = 35.9, p = 0.0001, one-way ANOVA followed by Tukey's test; n = 7/group). F, Both the Ca2+-free only and Mn2+ + Ca2+-free conditions significantly depressed the amplitude of AP (baseline: 45.4 ± 2.2 mV vs manganese in Ca2+-free: 37.9 ± 0.7 mV; F(2,18) = 16.1, p = 0.0001, one-way ANOVA followed by Tukey's test; n = 7/group). G, The CVs of the interspike intervals were not different between baseline and Mn2+ + Ca2+-free solution (baseline: 1.1 ± 0.2 vs Ca2+-free: 1.99 ± 0.3, F(2,18) = 7.3, p = 0.004, one-way ANOVA followed by Tukey's test; n = 7/group). *p < 0.05; **p < 0.01.
Combined application of Ca2+-free external solution and chelation of intracellular Ca2+ did not prevent manganese stimulation of firing frequency
Since excluding extracellular Ca2+ (Ca2+-free external solution) did not decrease the effect of manganese on the spontaneous firing activity of dopamine neurons, AP half-width, amplitude, and CV (Fig. 4), we asked whether manganese promotes neuronal activation by increasing intracellular Ca2+ mobilization ([Ca2+]i) (Tjalkens et al., 2006). To test this hypothesis, neurons were pretreated with BAPTA-AM (10 mm), a membrane-permeable Ca2+ chelator, to deplete [Ca2+]i followed by application of manganese in the presence of Ca2+-free external solution. Consistent with previous reports (Benedetti et al., 2011; Torkkeli et al., 2012), BAPTA pretreatment before bath application of manganese in Ca2+-free external solution (shown in blue) depolarized the membrane potential (Fig. 5D; baseline: −54.9 ± 1.3 mV vs 100 μm manganese: −49.6 ± 1.9 mV; F(2,27) = 2.5, p = 0.028, one-way ANOVA followed by Tukey's test; n = 9/group) and broadened the AP half-width (Fig. 5B,E; baseline: 1.37 ± 0.2 ms vs 100 μm manganese: 2.50 ± 0.3 ms; F(2,24) = 3.5, p = 0.009, one-way ANOVA followed by Tukey's test; n = 10/group). Interestingly, we found that manganese treatment in Ca2+-free external solution (shown in red) still increased the spontaneous firing activity of the neurons (Fig. 5A,C; baseline: 0.9 ± 0.2 Hz vs 100 μm manganese: 6.9 ± 1.7 Hz, F(2,26) = 13.2, p = 0.003, one-way ANOVA followed by Tukey's test; n = 10/group) and truncated the amplitude of APs (Fig. 5B,F; baseline: 37.5 ± 1.4 mV vs 100 μm manganese: 29.0 ± 1.7 mV; F(2,24) = 6.0, p = 0.001, one-way ANOVA followed by Tukey's test; n = 9/group). Washout of extracellular manganese in neurons pretreated with BAPTA and recorded in Ca2+-free external solution (shown in green) exhibited a significant decrease in the firing frequency (Fig. 5C; 0.4 ± 0.08 Hz), a broadening of AP half-width (Fig. 5B,E; 2.4 ± 0.4 ms, F(2,24) = 4.3, p = 0.029, one-way ANOVA followed by Tukey's test; n = 9/group), and truncated AP amplitude (Fig. 5B,F; 31.7 ± 2.1 mV, F(2,24) = 6.0, p = 0.028, one-way ANOVA followed by Tukey's test; n = 9/group). These results suggest manganese enhancement of firing frequency of dopamine neurons (Figs. 1, 2, 4) is not due to the release of Ca2+ from intracellular stores.
Figure 5.
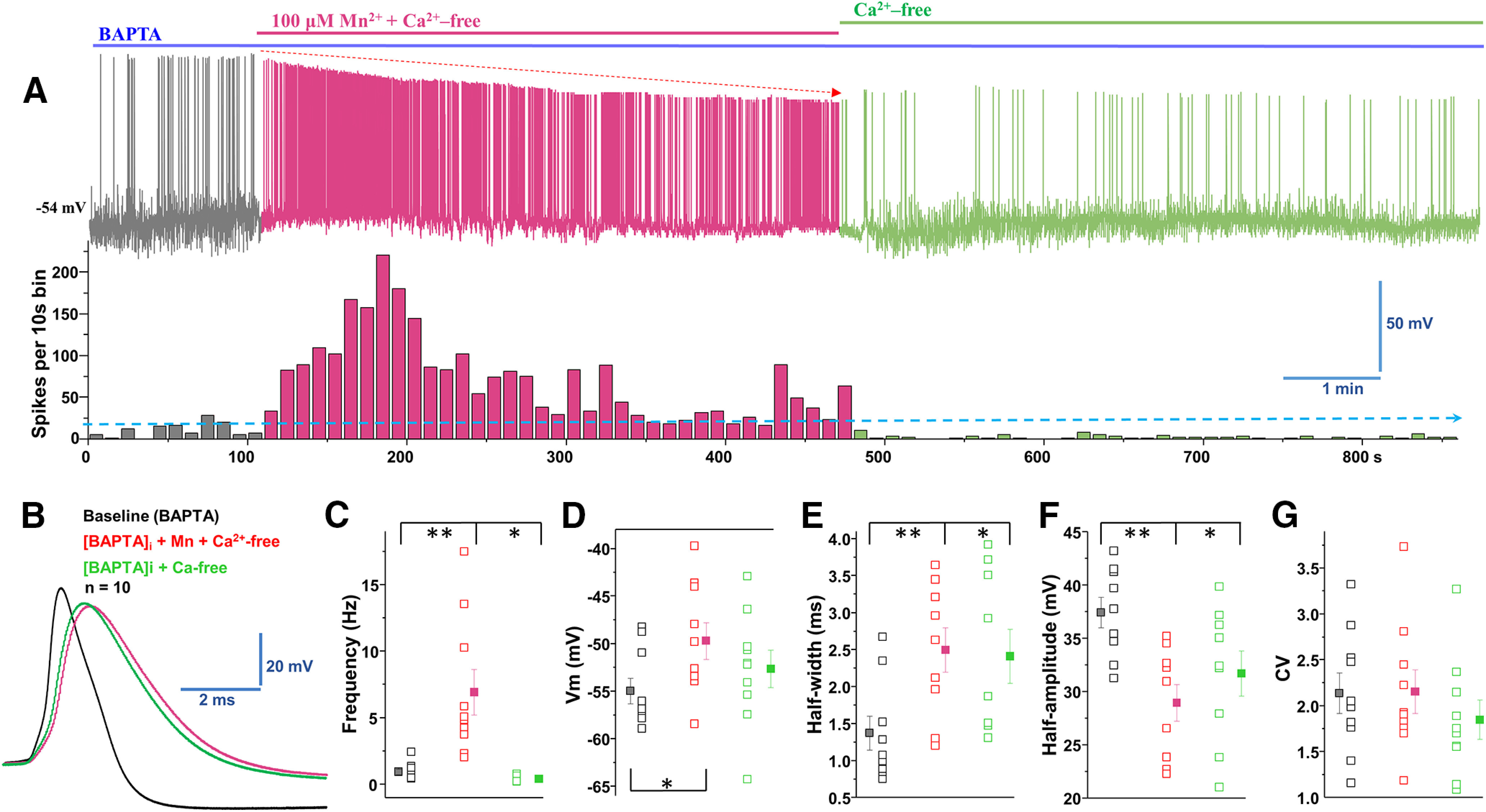
Manganese increases firing rate in the combined absence of extracellular and intracellular Ca2. A, Top, Representative trace of spontaneous spike activity following Mn2+ exposure in a Ca2+-free extracellular solution and an intracellular solution containing the Ca2+ chelator BAPTA (10 mm). Bottom, Histogram of firing frequency obtained from above trace showing Mn2+ increased firing frequency. B, Superimposed representative single AP traces are shown in A: baseline (black trace), 100 μm Mn2+ + Ca2+-free + BAPTA (red trace) and Ca2+-free + BAPTA-AM (green trace). C, Mn2+ increased firing rate, even in the combined absence of extracellular and intracellular Ca2+; after Mn2+ washout, the firing rate decreased under Ca2+-free conditions (baseline: 0.9 ± 0.2 Hz vs 100 μm manganese: 6.9 ± 1.7 Hz, F(2,26) = 13.2, p = 0.003, one-way ANOVA followed by Tukey's test; n = 10/group). D, The combination of extracellular Ca2+-free + Mn2+ and intracellular BAPTA-containing solution depolarized the membrane potential. After Mn2+ washout, the membrane potential returned to the baseline level (Ca2+-free conditions) (baseline: −54.9 ± 1.3 mV vs 100 μm manganese: −49.6 ± 1.9 mV; F(2,27) = 2.5, p = 0.028, one-way ANOVA followed by Tukey's test; n = 9/group). E, The half-width was measured at the half-maximal voltage of APs. The half-width was broadened significantly in the combination of extracellular Ca2+-free + intracellular BAPTA-containing solution in the absence or presence of Mn2+ (baseline: 1.37 ± 0.2 ms vs 100 μm manganese: 2.50 ± 0.3 ms; F(2,24) = 3.5, p = 0.009, one-way ANOVA followed by Tukey's test; n = 10/group). F, Half-amplitude of AP was significantly depressed in extracellular Ca2+-free solution combined with intracellular BAPTA in the absence or presence of Mn2+ (baseline: 37.5 ± 1.4 mV vs 100 μm manganese: 29.0 ± 1.7 mV; F(2,24) = 6.0, p = 0.001, one-way ANOVA followed by Tukey's test; n = 9/group). G, The CVs of the interspike intervals were not significantly different among the experimental groups. *p < 0.05, **p < 0.01. n = 9 or 10 per group.
Manganese does not affect intracellular Ca2+ homeostasis in midbrain dopamine neurons
To directly examine the effect of manganese exposure on intracellular Ca2+ levels [Ca2+]i in dopamine neurons, we measured Ca2+ responses in the midbrain slices from mice expressing GCaMP6f in dopaminergic neurons. Brain slices were continuously perfused with aCSF-containing manganese or NMDA equilibrated with 95% O2/5% CO2. Since NMDA receptors are Ca2+-permeable glutamate receptors (D. W. Choi, 1987; Wild et al., 2014), 100 μm NMDA was used as a positive control. Representative two-photon Ca2+ images are shown in Figure 6, where images for manganese (Fig. 6A) or NMDA (Fig. 6B) were taken for 10 min after 10 min of baseline imaging. We found that manganese exposure (100 μm) did not significantly alter [Ca2+]i compared with vehicle control. In contrast, 100 μm NMDA markedly increased [Ca2+]i in the neurons (positive control group). The relative intensity of two-photon Ca2+ signaling over baseline over time is shown in Figure 6C. The signal intensity was not affected by manganese administration (black) [Ca2+]i, whereas NMDA (red) robustly increased the GCaMP6f response (F(1,592) = 115.1, p < 0.001, one-way ANOVA followed by Tukey's test, n: Mn = 10; NMDA = 11). In complement, ratiometric Ca2+ imaging using fura-2 AM in neuronal culture of dopamine neurons confirmed the findings that manganese does not affect intracellular Ca2+ homeostasis in dopamine neurons (Fig. 7). Collectively, the lack of effect of manganese on Ca2+ homeostasis in dopamine neurons (Figs. 6, 7) is consistent with the notion that manganese stimulation of firing frequency of dopamine neurons is not a result of Ca2+ modulation.
Figure 6.
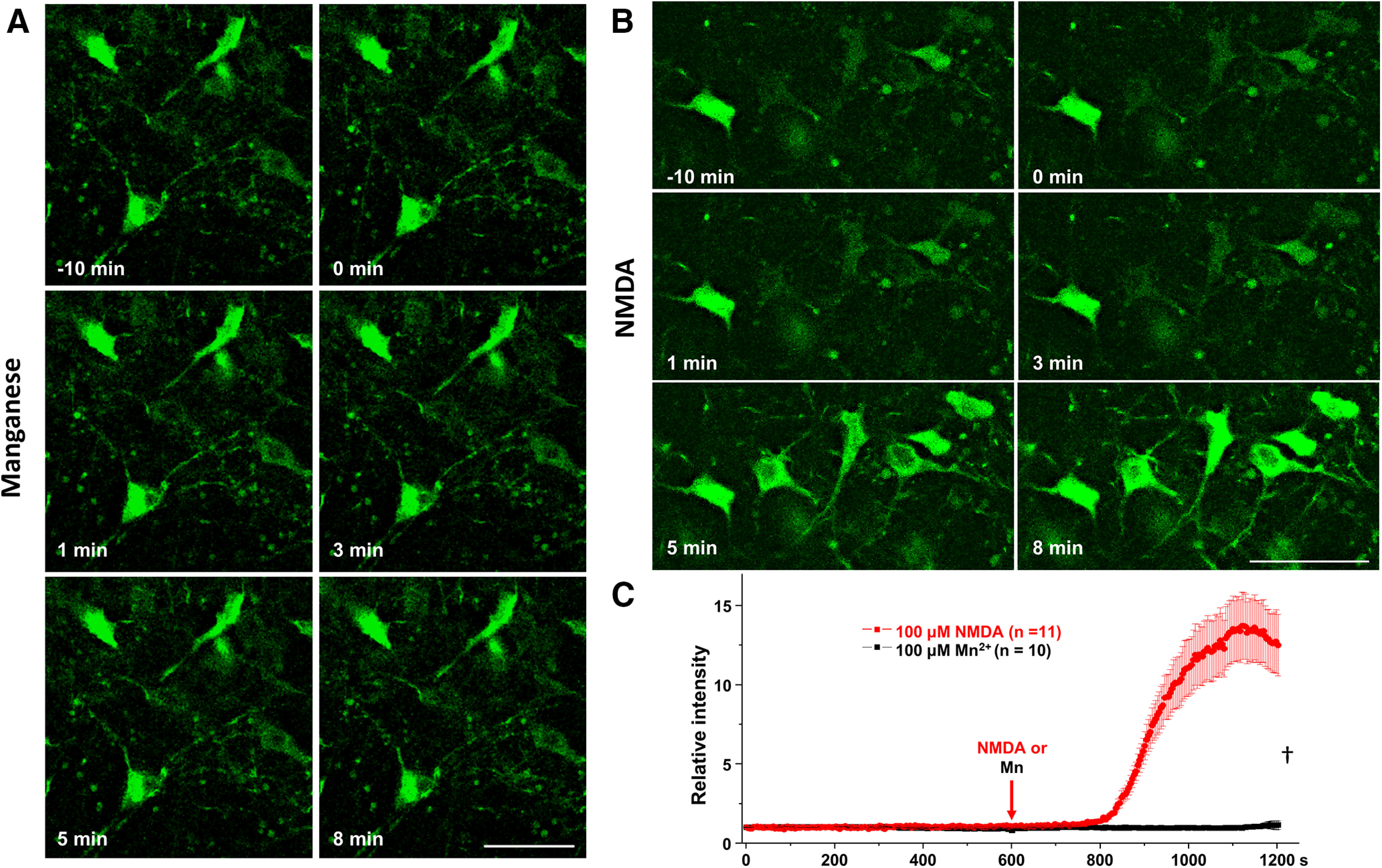
Two-photon imaging of midbrain slices of DATcre-GCaMP6f-expressing dopamine neurons showed that manganese does not alter intracellular Ca2+ homeostasis. A, B, Representative two-photon images of GCaMP6f fluorescent neurons in the following 100 μm Mn2+ or NMDA treatments (positive control group). C, Analyses of relative fluorescence intensities of GCaMP6 neurons following 100 μm Mn2+ or NMDA (F(1,592) = 115.1). †p < 0.001 (one-way ANOVA followed by Tukey's test). n: Mn = 10 frame (71 neurons), NMDA = 11 frame (50 neurons) from three independent experiments. Scale bar, 50 μm. †p < 0.001.
Figure 7.
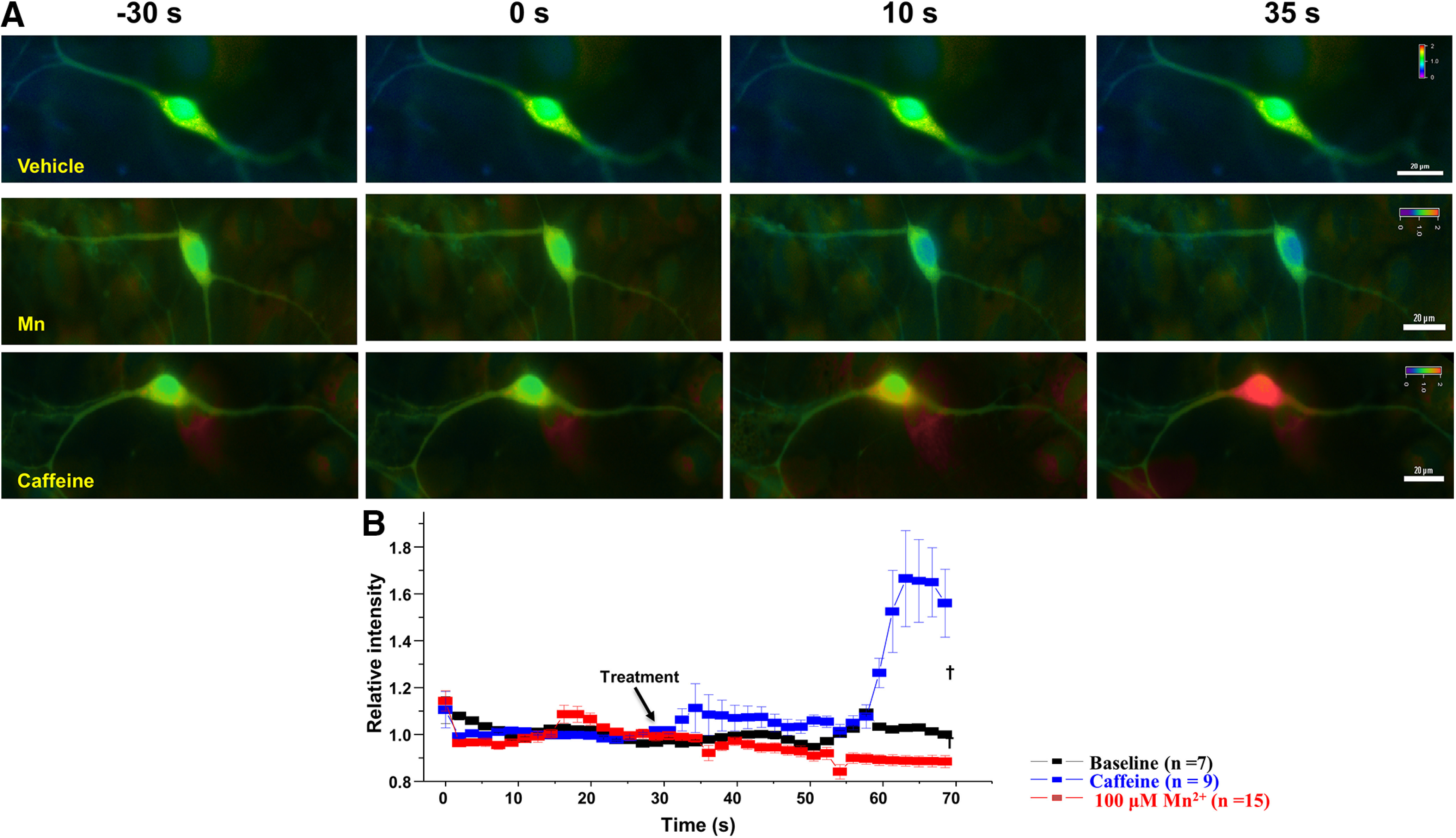
Ratiometric analyses of fura-2-loaded dopamine neurons revealed Mn does not increase intracellular calcium. Ratiometric analyses were performed on fura-2-loaded dopamine neurons. A, Representative pseudo-color images indicating the 340/380 ratio via a visible spectrum heat map. Violet represents minimum Ca2+. Red represents maximum Ca2+. Left, Representative 340/380 ratio images before and after drug or vehicle application. Right, Identical view fields 30 s after vehicle (top), 100 μm Mn2+ (middle), and 20 mm caffeine (bottom). B, Mean fluorescence intensities for each condition (normalized to the initial 30 s intensity). Data are plotted in 1.5 s interval for vehicle (black squares), Mn2+ (red squares), and the positive control group caffeine (blue squares). Scale bar, 20 μm. †p < 0.01.
Cadmium (Cd2+) blockade of voltage-gated Ca2+ channels inhibited manganese stimulation of spontaneous firing activity of dopamine neurons
Thus far, above data suggest that manganese enhances the spontaneous activity of midbrain dopamine neurons, but it is not due to changes in extracellular Ca2+ influx or cytosolic Ca2+ release from intracellular stores. Ca2+ channels are also permeant to other divalent cations, such as barium (Bourinet et al., 1996). Therefore, as a divalent ion with a smaller ionic radius than Ca2+ (see Fig. 14), we hypothesized that manganese can potentially interact with the Ca2+ channels. To examine this possibility, next we tested whether manganese augments the spontaneous activity of dopamine neurons by interacting with Ca2+ channels expressed on dopamine neurons. We examined the effect of a nonselective Ca2+ channel blocker, cadmium (Cd2+, 100 μm) on the intrinsic firing behaviors of dopamine neurons following manganese exposure. As shown in Figure 8, nonselective blockade of Ca2+ channels by bath application of Cd2+ suppressed the spontaneous firing rate of dopamine neurons (Fig. 6A). Unexpectedly, we found that, under this condition, manganese application failed to enhance the firing frequency (Fig. 8A,C; F(2,18) = 8.3, p = 0.002, one-way ANOVA followed by Tukey's test, n = 7 group). Treatment with Cd2+ alone or the coadministration of Cd2+ and manganese did not change the membrane potential (Fig. 8D), AP half-width (Fig. 8B,E), and AP half-amplitude (Fig. 8B,F), but significantly increased the CV (Fig. 8G; F(2,18) = 4.6, p = 0.024, one-way ANOVA followed by Tukey's test, n = 7 group). These findings suggest that, despite observations that manganese-mediated increase in firing activity of dopamine neurons is not dependent on extracellular or intracellular Ca2+ flux, it is dependent on Ca2+ channels expressed on dopamine neurons.
Figure 14.

Superposition of msCav1.1 Ca binding site structure with the msCav1.2 model. The Ca2+ ion (green) as located in the msCav1.1 structure (tan color backbone) (PDB ID 5GJW). The homology model structure for Cav1.2 α chain is shown in the blue color backbone.
Figure 8.
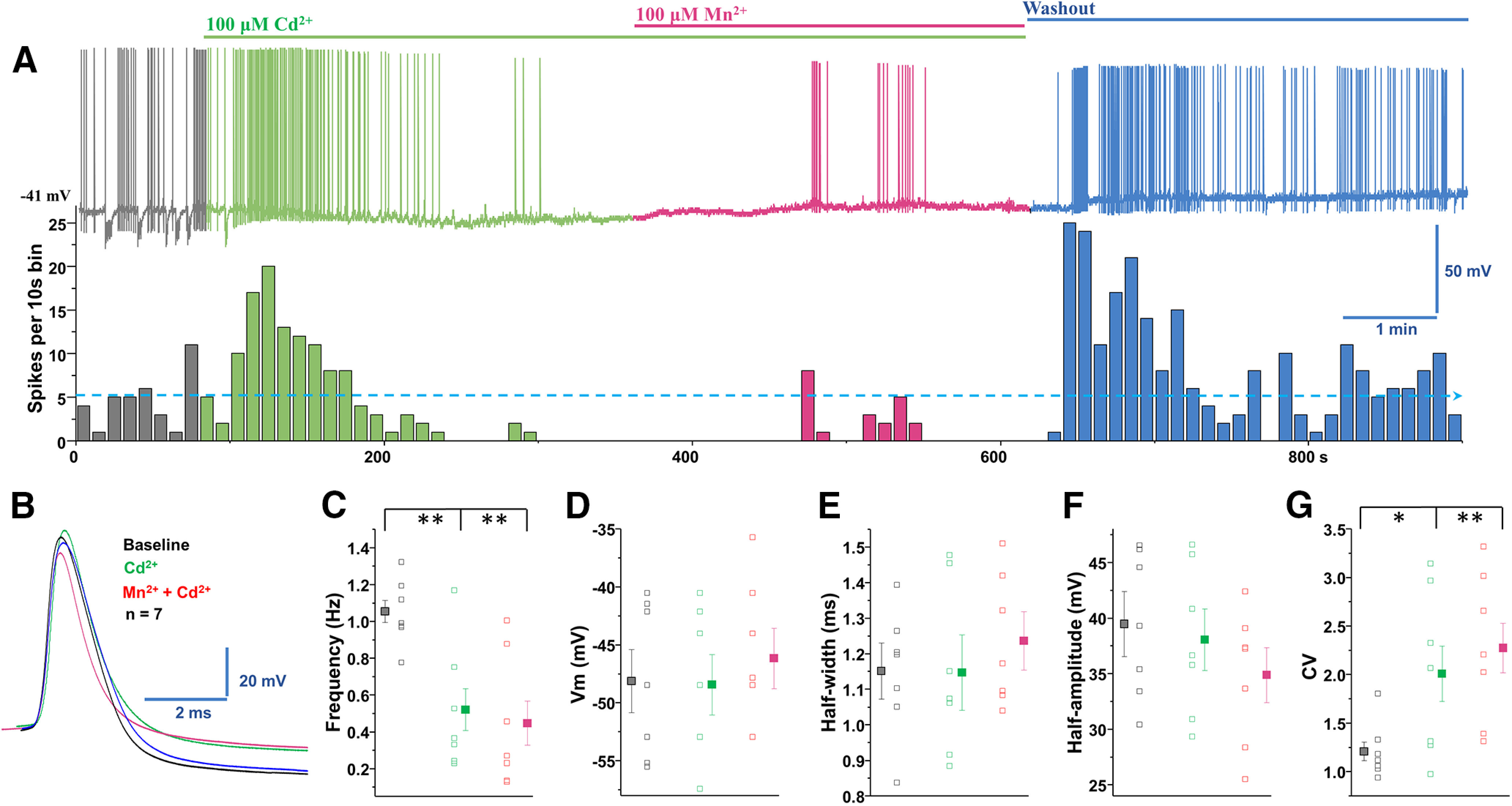
Cd2+ blockade of voltage-gated Ca2+ channels inhibited manganese stimulation of spontaneous firing activity of dopamine neurons. A, Top, Representative spontaneous spike activity of a dopamine neuron before and after exposure to Cd2+ (a nonselective voltage-gated Ca2+ channel blocker) and Cd2+ + Mn2+. Cd2+ blockade of voltage-gated Ca2+ channels inhibited Mn2+ stimulation of spontaneous firing activity. Bottom, Histogram of firing frequency from above trace showing that firing frequencies were lowered in Cd2+ only condition and following or Cd2+ + Mn2+ but return to baseline after washout. B, Superimposed representative single AP traces shown in A: baseline (black trace), 100 μm Cd2+ (green trace), 100 μm Mn2+ + Cd2+ (red trace), and washout (blue trace). C, Spontaneous firing rate significantly decreased following application of Cd2+ or Cd2+ + Mn2+ (F(2,18) = 8.3, p = 0.002, one-way ANOVA followed by Tukey's test, n = 7/group). D, Application of Cd2+ or Cd2+ + Mn2+ did not change the membrane potential. E, Half-width was measured at half-maximal voltage of AP; there was no change in the half-width of the Cd2+ and Cd2+ + Mn2+ experimental groups. F, Cd2+ or Cd2+ + Mn2+ treatments did not change the amplitude of AP. G, Both Cd2+ and Cd2+ + Mn2+ treatments increased the CVs of the interspike interval (F(2,18) = 4.6, p = 0.024, one-way ANOVA followed by Tukey's test, n = 7/group). *p < 0.05, **p < 0.01.
The Ca2+ current involved in pacemaking activity is partly due to L-type currents, which are activated at subthreshold potential and potentially contribute to the tonic firing of the neuron (Putzier et al., 2009). To investigate the possible role of these channels, we asked whether nifedipine blockade of L-type Ca2+ (Cav1) channels inhibited manganese stimulation of spontaneous firing activity of dopamine neurons. As shown in Figure 9, we found that nifedipine inhibited manganese stimulation of spontaneous firing activity. Consistent with the results in Figures 4 and 5, the firing frequencies were increased in the presence of manganese and Ca2+-free condition. Similar to the Cd2+ inhibition of manganese stimulation of firing activity of dopamine neurons, coapplication of nifedipine inhibited the manganese stimulation of firing activity (Fig. 9A,C; F(2,18) = 44.4, p = 0.0006, one-way ANOVA followed by Tukey's test, n = 7 group). Similar to Cd2+, the manganese in Ca2+-free solution condition or the coadministration of manganese plus nifedipine in Ca2+-free solution did not change the membrane potential (Fig. 9D) or AP half-width (Fig. 9B,E), but both manganese in Ca2+-free solution and additional application of nifedipine truncated AP amplitudes (Fig. 9A,F; F(2,18) = 11.80, p =0.001, one-way ANOVA followed by Tukey's test, n = 7 group). The coadministration of manganese in Ca2+-free solution with nifedipine significantly increased the CV (Fig. 9G; F(2,18) = 14.11, p = 0.0016, one-way ANOVA followed by Tukey's test, n = 7 group). These unexpected results support the interpretation that manganese may enter the dopamine neuron through the nifedipine-sensitive voltage-gated Ca2+ channels.
Figure 9.
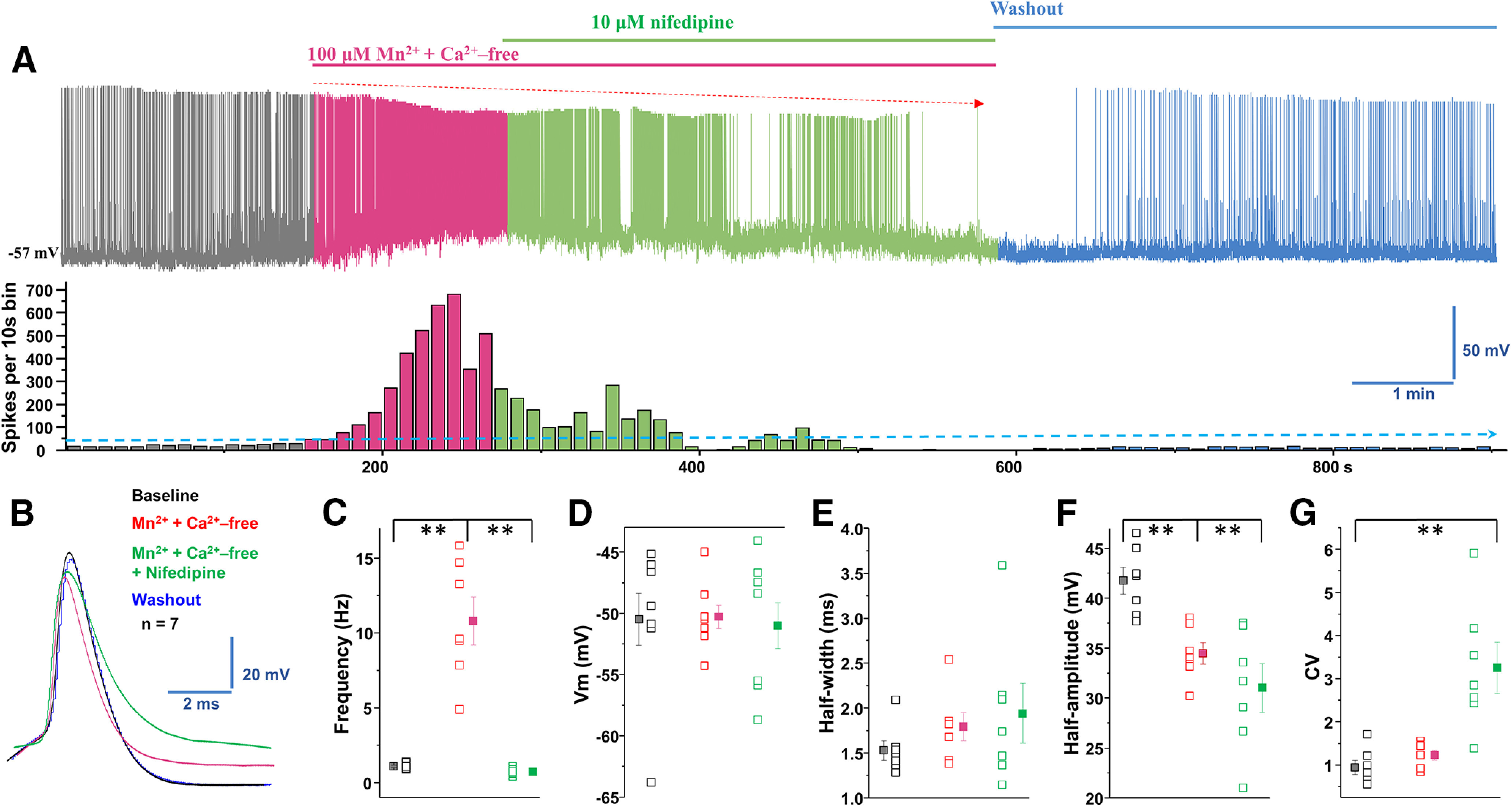
Blockade of L-type Ca2+ channels inhibited manganese stimulation of spontaneous firing activity of dopamine neurons. A, Top, Representative spontaneous spike activity of a dopamine neuron before and after exposure to Mn2+ + Ca2+-free and nifedipine, an L-type Ca2+ channel blocker. Nifedipine blockade of voltage-gated Ca2+ channels inhibited Mn2+ stimulation of spontaneous firing activity. Bottom, Histogram of firing frequency from above trace showing firing frequencies were increased in combination of Mn2+ and Ca2+-free, lowered after nifedipine addition, but returned to baseline after washout. B, Superimposed representative single AP traces shown in A: baseline (black trace), Mn2+ + Ca2+-free (red trace), Mn2+ + Ca2+-free + nifedipine (green trace), and washout (blue trace). C, Mn2+ increased firing rate, even in the combination of Mn2+ and Ca2+-free. Additional application of nifedipine significantly reduced firing rate (F(2,18) = 44.4, p = 0.0006, one-way ANOVA followed by Tukey's test, n = 7/group). D, Application of Mn2+ + Ca2+-free or further administrated nifedipine did not change the membrane potential. E, Half-width was measured at half-maximal voltage of AP; there was no change in the half-width of Mn2+ + Ca2+-free and additional + nifedipine experimental groups. F, Both Mn2+ + Ca2+-free and further added nifedipine condition significantly depressed the amplitude of AP (F(2,18) = 11.80, p = 0.001, one-way ANOVA followed by Tukey's test, n = 7/group). G, Mn2+ + Ca2+-free and additional application of nifedipine increased the CVs of the interspike interval (F(2,18) = 14.11, p = 0.0016, one-way ANOVA followed by Tukey's test, n = 7/group). **p < 0.01.
Single-neuron recording revealed manganese competes with Ca2+ influx and generates Ca2+ channel-like currents
Because the blockade of voltage-gated Ca2+ channels prevented the manganese-mediated increases in the spontaneous firing activity of dopamine neurons independent of the presence of intracellular or extracellular Ca2+, we then examined the hypothesis that voltage-gated Ca2+ channels on dopamine neurons are directly permeable to extracellular manganese. Since maximum spontaneous firing appeared ∼3 min after Mn application (Fig. 1), we have matched this time point for Ca2+ current recording. After replacing external Ca2+ with equimolar manganese, we observed that manganese induced a Ca2+ channel-mediated current, which was evoked by a series of 200 ms depolarizing steps from −60 to 85 mV in 5 mV increments (Fig. 10A1). The currents were composed of a rapidly inactivating transient component and a slowly inactivating persistent component. The current–voltage relationship of the manganese current is shown in Figure 10B. The manganese currents were activated at voltages at ∼20 mV and peaked at ∼30 mV. Next, we asked whether the activity of Ca2+ channels in midbrain dopamine neurons is affected by these manganese-mediated currents. Since there is little information on whether manganese modulates Ca2+ currents in dopamine neurons, we focused on the action of manganese on whole-cell Ca2+ currents. Total Ca2+ currents (Fig. 10A2,C) were activated at voltages of ∼20 mV and peaked at ∼45 mV. Bath application of 100 μm manganese suppressed the peak amplitude of total Ca2+ currents (Fig. 10A3,C; p < 0.05, two-tailed Student's t test; n = 7/group). These data suggest that, in dopamine neurons, manganese might compete with Ca2+ influx and generate Ca2+ channel-gated-like currents that in turn alter the activity of the neurons. To our knowledge, these are the first reported findings that Ca2+ channels may be permeable to manganese and may represent a distinct action of manganese in regulating neuronal activity.
Figure 10.

Manganese reduced voltage-gated Ca2+ currents. A, Representative traces of inward currents in response to voltage steps (5 mV, 250 ms) from −60 mV to 85 mV in a bath solution containing 0.5 μm TTX, 35 mm TEA, and 1 mm CsCl. A1, Inward currents evoked by voltage steps during application of extracellular Ca2+-free + 2 mm Mn2+ solution. A2, Inward currents evoked by voltage steps in the presence of 2 mm Ca2+. A3, Inward currents evoked by voltage steps in the presence of 2 mm Ca2+ + 100 mm Mn2+. B, Plot represents the current–voltage relationship (I–V curves) induced by Mn2+. C, Plot shows current–voltage relationships (I–V curves) of peak voltage-gated Ca2+ current density in dopamine neurons at baseline (black squares) and following Mn2+ exposure (blue squares). *p < 0.05 (two-tailed Student's t test). n = 7 per group.
Manganese-enhanced MRI provides evidence for a Ca2+ channel dependent mechanism in the midbrain in vivo
Next, we asked whether manganese can enter the neuron through the Cav1.2 and Cav1.3 channels and whether nifedipine can block this. To examine this hypothesis in vivo, we performed MEMRI in animals pretreated with saline or manganese. Intraperitoneal injections of 70 mg/kg manganese have previously been shown to significantly increase manganese levels in the basal ganglia (Dodd et al., 2005), and also this dose is used for contrast in MRI (Poole et al., 2017). Manganese frequently serves as a neuronal contrast agent to enhance functional brain mapping at a higher spatial resolution than typical fMRI studies (Lee et al., 2005). In this study, we used MEMRI and T1 mapping to study basal levels of brain activity before and after nifedipine blockade of voltage-gated Ca2+ channels (Figs. 11, 12). A commonly used dose of nifedipine (15 mg/kg) (Giordano et al., 2010; Morellini et al., 2017) was intraperitoneally injected 30 min before manganese exposure. As expected, we found that T1 relaxation (quantified as R1 or the rate of T1 relaxivity in s−1) following manganese treatment exhibited predominantly higher (faster) R1 in VTA and SN compared with other cortical and subcortical regions (Fig. 12; F(8,72) = 2.35, p = 0.026, one-way ANOVA followed by Tukey's test, test, n = 8 or 9/group). This reflects a shortening of VTA and SN T1 relaxation time due to accumulation of the paramagnetic manganese. Importantly, and consistent with single-neuron recordings (Figs. 8–10), we observed that nifedipine blockade of L-type Ca2+ channels reduced T1 shortening effects of manganese in the midbrain region, which is significantly enriched with dopamine neurons (Nair-Roberts et al., 2008) (Fig. 11; VTA: t(15) = −2.2, p = 0.04, two-tailed Student's t test; Sn: t(15) = −2.7, p = 0.015, two-tailed Student's t test; n = 8 or 9/group). This was observed as a slower rate R1 in the nifedipine/manganese group relative to manganese only. Therefore, both in vitro (single-neuron recording) and in vivo studies suggest that nifedipine decreases manganese stimulation of neuronal activity and its accumulation in the multiple brain regions, including the dopamine neuron-enriched midbrain.
Figure 11.
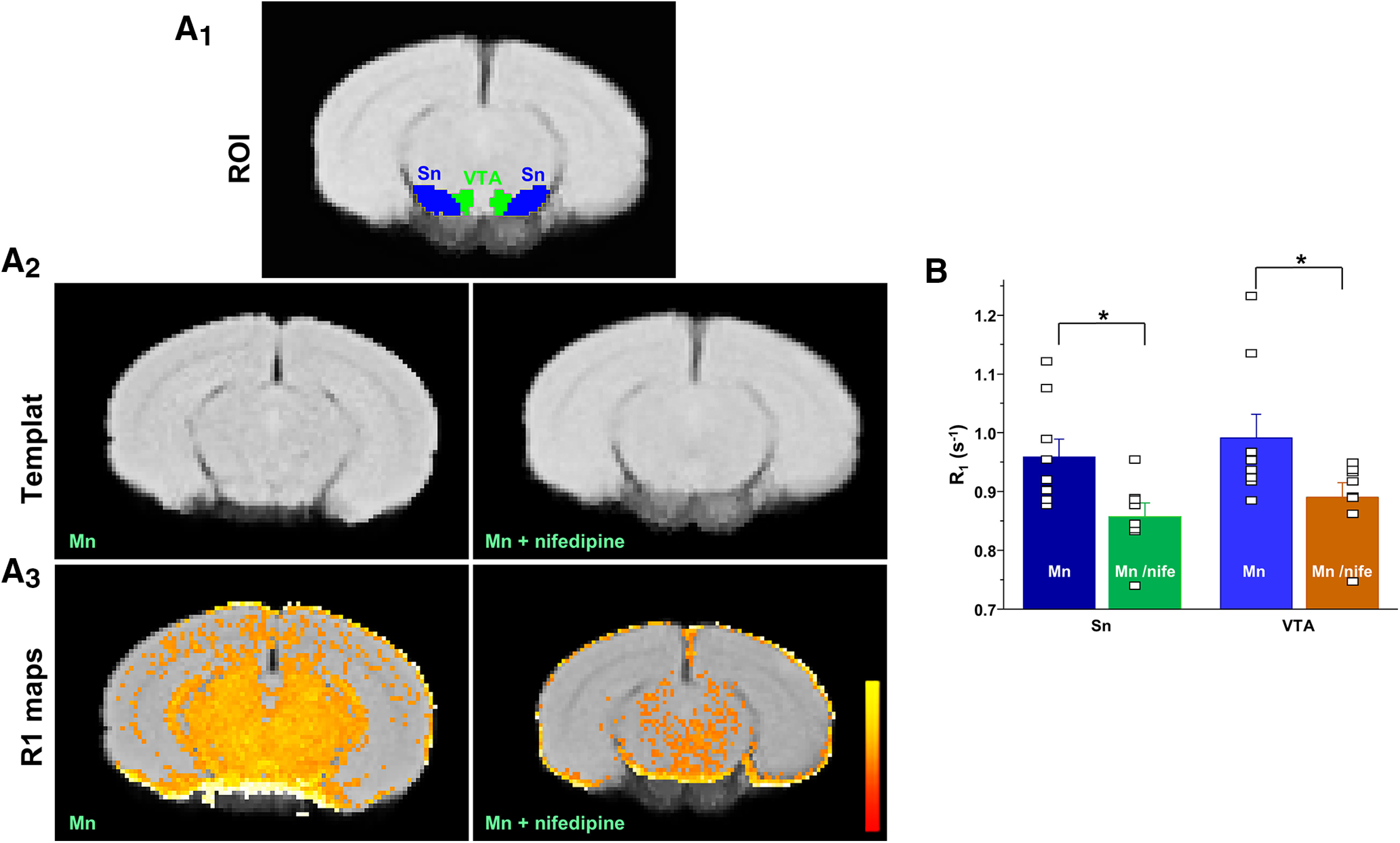
MEMRI provides evidence of an in vivo Ca2+ channel-dependent mechanism involved in midbrain manganese accumulation. R1 (1/T1) maps. Top, The location of the VTA and SN. Middle, A custom template from all controls and experimental groups (nifedipine-administered mice). Bottom, The average R1 map of both cohorts. Maps display values up to 2 s−1. Yellow represents large values (∼2 s^−1). Red represents low values (∼0). The control group shows a small number of voxels of R1 larger within the threshold showing less Mn2+ infiltration in the midbrain. A1, Mouse brain atlas-based segmentation of the VTA and SN. A2, Template brain for Mn alone or with nifedipine treatment that was aligned with mouse brain atlas. A3, Parametric maps of T1 relaxation rate (R1 in ms−1) show that calcium channel blockade with nifedipine treatment reduces R1 in midbrain and surrounding areas. Scale bar indicates intensity of R1. B, Nifedipine reduces T1 relaxation rate (R1) in VTA and SN (VTA: t(15) = −2.2, p = 0.04, two-tailed Student's t test; SN: t(15) = −2.7, p = 0.015, two-tailed Student's t test; n = 8 or 9/group). *p < 0.05.
Figure 12.
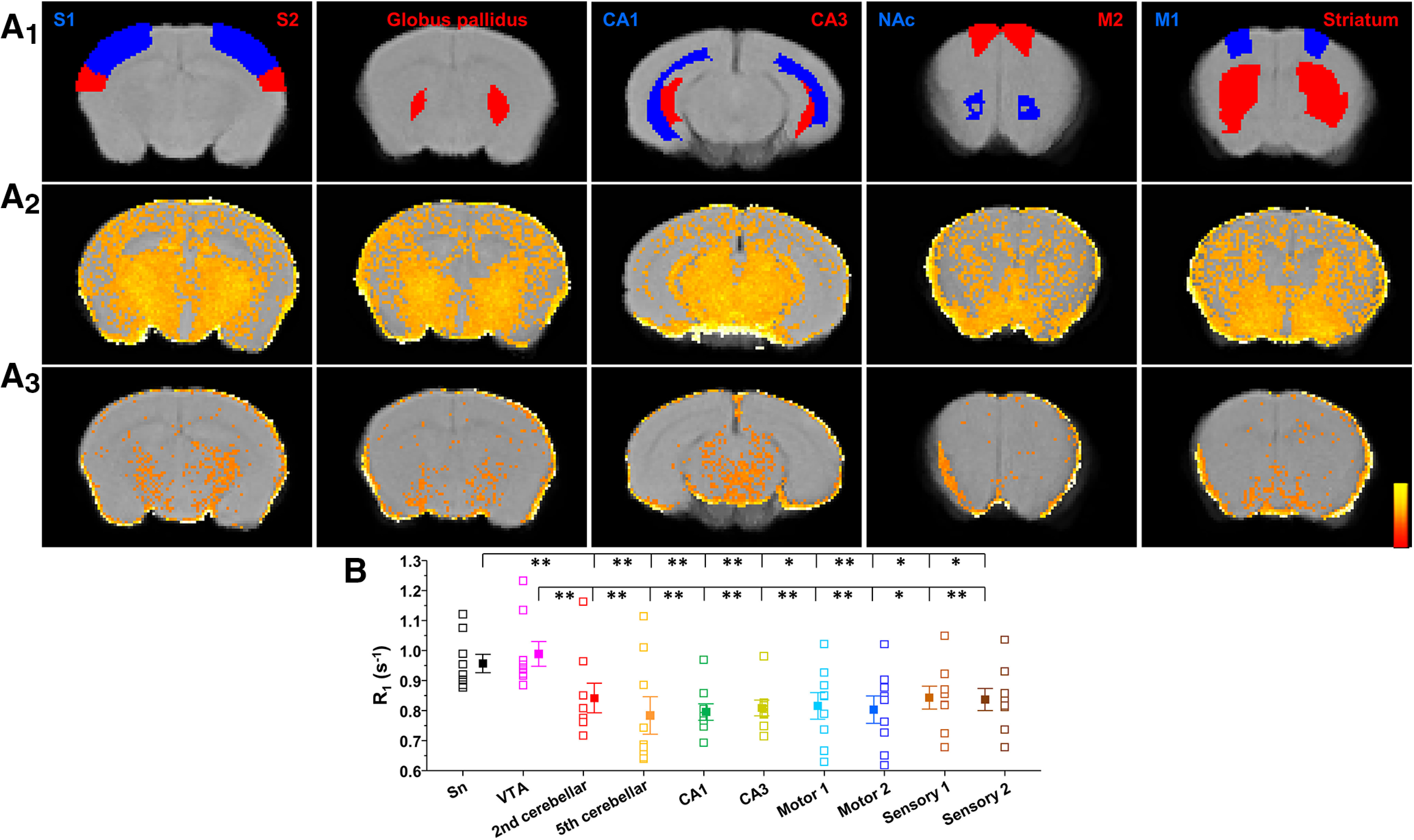
Ca2+ channel blockade using nifedipine reduces the effects of Mn2+ on T1 relaxation in several brain regions. A1, Mouse brain atlas-based segmentation of the primary (S1), secondary (S2) somatosensory cortex, globus pallidus, CA1, CA3, SN compacta (SNc), primary motor cortex (M1), secondary motor cortex (M2), and striatum. A2, Effects of Mn2+ accumulation on increasing brain T1 relaxation rate (R1 in ms−1). A3, Nifedipine treatment reduces the effects of Mn2+ accumulation by reducing its concentration in brain. Thus, maps of mice treated with nifepidine had a slower rate of T1 relaxation, or lower R1 in the case of the shown parametric maps (and lower intensity). Scale bar indicates intensity of R1. B, A greater Mn2+ accumulation produces faster rates of T1 relaxation (R1) in VTA and SN than in other segmented cortical and subcortical nuclei (F(9,72) = 3.96, p < 0.001, one-way ANOVA followed by Tukey's test, n = 8 or 9/group). *p < 0.05, **p < 0.01.
Manganese can enter the cell via a nifedipine-sensitive Cav1.2 channel-mediated mechanism
Our data so far support the interpretation that manganese may enter the cell via nifedipine-sensitive Ca2+ channels. To further support this idea, we measured 54Mn uptake in HEK cells that were transiently transfected with either a control (empty vector) or Cav1.2 or Cav1.3 cDNA. Western blot analysis confirmed the expression of Cav1.2 or Cav1.3 in the transfected cells (Fig. 13A). Overexpression of Cav1.2 increased the uptake of 54Mn at 5 and 15 min after the addition of 54Mn, while there was no difference observed between the Cav1.3-expressing and control cells (Fig. 13B). Uptake of 54Mn by HEK cells overexpressing Cav1.2 was inhibited when the cells were pretreated with nifedipine (Fig. 13C). These findings further support the interpretation that manganese can enter the cell via a nifedipine-sensitive Cav1.2 channel-mediated mechanism.
Figure 13.

54Mn uptake is greater in Cav1.2-overexpressing HEK293 cells. A, Representative Western blot analysis shows the expression of Cav1.2 and Cav1.3 in cells transiently transfected to overexpress Cav1.2 and Cav1.3. B, Greater 54Mn uptake is shown in Cav1.2-overexpressing cells at the indicated time points. C, Nifepidine inhibits 54Mn uptake at 15 min in Cav1.2-overexpressing cells. *p < 0.05 (one-way ANOVA followed by Tukey's test). n = 3 per group.
In silico modeling supports the possibility that manganese can interact with the Ca2+ binding site in the Cav1 channel
Because the crystal structure for Cav1.2 is not available, Cav1.1 was used for homology modeling studies to investigate whether the putative structure of Cav1.2 could inform us about the manganese cation transport. First, the available electron microscopy structure for the homologous Cav1.1 channel was used to build a homology model using the SWISS-MODEL program (Fig. 14) (Waterhouse et al., 2018). The helical regions of the mouse Cav1.1 structure (Wu et al., 2016) and the helical regions for the Cav1.2 model were compared by protein BLAST (Altschul et al., 1990) and found to be 72% identical and 81% similar. Further, when all amino acids within 7 Å of the bound Ca2+ atom found in the Cav1.1 structure were selected and compared with amino acids in the model structure for Cav1.2, we found 100% identity in this region, including the carboxylate ligands for the Ca2+ atom. We conclude the Cav1.2 channel has an extremely similar structure and presumably cation-binding capability compared with Cav1.1. Because the ionic radius of Mn2+ is smaller than for Ca2+ (Persson, 2010), it should be adequately accommodated by the channel. Together, the structural features of the Cav1.2 channel and our experimental findings argue for the possibility of the Cav1.2 channel conducting a manganese ion. While these data collectively suggest that manganese produces a Ca2+ channel-mediated current in dopamine neurons, which increases rhythmic firing activity of dopamine neurons, the connection between manganese-mediated current and increased firing activity of dopamine neurons remains unclear. The next set of experiments were designed to determine the potential mechanism.
Blockade of large-conductance Ca2+-activated potassium (BK) channels decreased the spontaneous firing activity and prevented manganese stimulation of firing frequency
Previously, we have shown that paxilline blockade of BK channels reduces the spontaneous firing activity, broadens the width, and enhances the amplitude of APs in midbrain dopamine neurons (Lin et al., 2016). Since Mn increased the firing frequency and decreased the width and the amplitude of APs (Fig. 1), we postulated that manganese stimulation of the firing frequency, narrowing of AP width, and reduction of AP amplitude might be due to BK channel activation. Consistent with our previous reports, paxilline suppressed the firing frequency of midbrain dopamine neurons (Fig. 15A,B). Pretreatment with paxilline abolished the manganese-induced increase in firing frequency (Fig. 15A,C; baseline: 1.5 ± 0.2 Hz vs Mn + paxilline: 0.51 ± 0.1 Hz; F(2,20) = 11.2, p = 0.0005, one-way ANOVA followed by Tukey's test; n = 8/group) and inhibited manganese's modulation of AP amplitude (Fig. 15A,F). Comparison of the firing frequency after treatment with paxilline alone or paxilline + manganese treatment revealed a further reduction in the firing frequency (Fig. 15C; paxilline: 0.81 ± 0.1 Hz). Consistent with our previous report (Lin et al., 2016), blockade of BK channels depolarized membrane potential (Fig. 15D; baseline: −45.1 ± 1.6 mV vs paxilline: −40.1 ± 1.8 mV; F(2,21) = 3.5, p = 0.04, one-way ANOVA followed by Tukey's test; n = 8/group), broadened AP half-width (Fig. 15B,E; baseline: 1.3 ± 0.1 ms vs paxilline: 1.8 ± 0.2 ms; F(2,21) = 5.9, p = 0.009, one-way ANOVA followed by Tukey's test; n = 8/group), increased AP amplitude (Fig. 15B,F; baseline: 28.1 ± 1.6 mV vs paxilline: 32.6 ± 1.1 mV; F(2,21) = 6.6, p = 0.005, one-way ANOVA followed by Tukey's test; n = 8/group), and increased the CV (Fig. 15G; baseline: 1.8 ± 0.2 vs paxilline: 2.8 ± 0.3; F(2,21) = 2.8, p = 0.015, one-way ANOVA followed by Tukey's test; n = 8/group). After pretreatment with paxilline, we found that manganese did not reverse paxilline-induced membrane depolarization (Fig. 15D), but it did diminish the effects of paxilline on the AP half-width (Fig. 15E), amplitude (Fig. 15F), and CV (Fig. 15G). These results suggest that the manganese stimulation of spontaneous spike activity of dopamine neurons is possibly due to BK channel activation.
Figure 15.
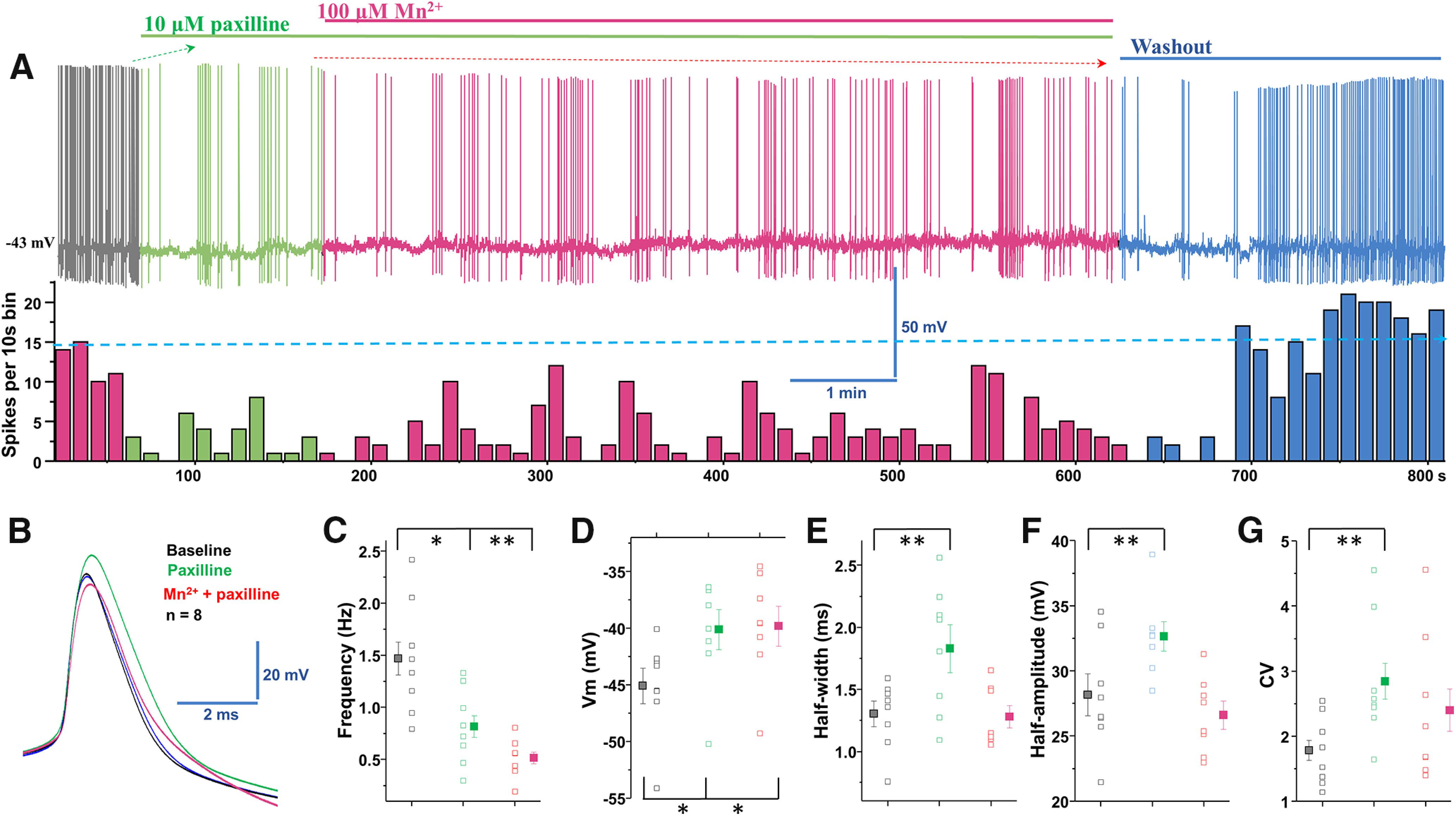
Blockade of BK channels inhibited manganese stimulation of the spontaneous firing activity in dopamine neurons. A, Top, Representative spontaneous firing activities of a dopamine neuron exposed to paxilline followed by Mn2+ treatment. Firing rate returns to baseline after washout with aCSF solution. Bottom, Rate histogram of above trace. B, Superimposed traces of representative single AP shown in A: baseline (black trace), paxilline (green trace), Mn2+ (red trace), and washout (blue trace). C, Paxilline reduced the spontaneous firing rate; Mn2+ treatment did not attenuate the paxilline-induced reduction of firing activity. D, Paxilline and concomitant Mn2+ + paxilline applications significantly depolarized the membrane potential. E, Half-width is measured at half-maximal voltage of AP. The half-width was broadened after paxilline application but returned to baseline after coadministration of Mn2+ and paxillin. F, Paxilline increased the amplitude of AP but diminished the effect of Mn2+ on the AP amplitude. G, Blockade of BK channels exhibited larger CVs of the interspike interval. *p < 0.05, **p < 0.01. n = 8 per group.
Manganese increases membrane expression of BK α subunits
The BK α subunit is the pore-forming unit of the BK channel (Knaus et al., 1994). There is a direct relationship between BK channel regulation of neuronal excitability and the level of BK α subunits at the surface membrane (Shruti et al., 2012; Chen et al., 2013; Cox et al., 2014). Since blockade of BK channels abolished the effect of manganese stimulation of the spontaneous firing frequency of dopamine neurons, we examined the possibility that manganese-mediated enhancement of neuronal excitability is due to increased BK α subunit levels at the plasma membrane. To test this, we used TIRF microscopy to monitor fluorescently tagged BK channel trafficking. The TIRF profile of GFP α subunits was examined in HEK293 cells expressing GFP α subunits in the presence or absence of 100 μm manganese. As shown in Figure 16, the GFP α subunits' fluorescence signal at the membrane remained stable in the vehicle (external solution)-treated control group. The TIRF profile of GFP α subunits was markedly increased following manganese treatment (F(1,164) = 163.8, p < 0.01, one-way ANOVA followed by Tukey's test, n: baseline = 7; Mn = 8). Because TIRF microscopy detects fluorophores at or near the plasma membrane, these results indicate that manganese's stimulation of neuronal excitability is, in part, due to manganese-induced increases in surface BK channel expression and thus activity. To test this possibility, we used excised inside-out patches to record currents in response to test voltage steps. Figure 17 shows representative BK currents in response to test voltage steps in the absence or presence of manganese. The I–V curves of these recordings demonstrate manganese significantly increased peak amplitude of BK currents (p < 0.05, two-tailed Student's t test, n = 6/group). Together, these results suggest manganese enhances BK channel activity that we have previously shown increases firing activity of dopamine neurons (Lin et al., 2016).
Figure 16.
Membrane localization of BK GFP α subunit is increased following manganese exposure. A, Representative TIRF microscopy images of GFP α subunit following vehicle or 100 μm Mn2+ treatments. B, Analyses of relative fluorescence intensities at the surface membrane following vehicle or 100 μm Mn2+. Scale bar, 20 μm. †p < 0.01.
Figure 17.

Manganese increases BK currents. A, Representative traces of outward currents in response to voltage steps (5 mV, 250 ms) from −60 mV to 85 mV in a bath solution containing 0.5 μm TTX, 35 mm TEA, and 1 mm CsCl. A, In cells expressing BK α subunits, families of outward currents were evoked by voltage steps from −60 to 85 mV for 250 ms with 5 mV increments every 5 s from the holding potential of −70 mV, before (A) and after (B) Mn2+ administration. C, Peak current–voltage relationships (I–V curves) before (baseline) and after Mn2+ application.
Discussion
Prolonged exposure to low levels of manganese or single large exposure results in its accumulation in multiple brain regions, leading to dysfunction of CNS and an extrapyramidal motor disorder referred to as manganism (Aschner et al., 2009). In this study, we used multiple complementary approaches to examine the manganese regulation of dopaminergic excitability and to determine the potential mechanisms involved. We found that manganese increased the spontaneous firing activity of dopamine neurons, decreased the amplitude and half-width of APs, and reduced the variation of interspike interval. Unexpectedly, neither the removal of extracellular Ca2+ nor the chelation of intracellular Ca2+ modulated the manganese stimulation of spontaneous firing frequency of dopamine neurons. Live cell two-photon imaging of GCaMP6f-expressing dopamine neurons support the electrophysiology data showing no change in intracellular Ca2+ levels after manganese application. In contrast, we found that manganese regulation of dopamine neurons was blocked by Cd2+, a nonselective Ca2+ channel blocker, or by nifedipine, an L-type Ca2+ channel blocker, suggesting that manganese can potentially enter the neuron through voltage-gated Ca2+ channels. Furthermore, we identified a Ca2+ channel-mediated manganese current that reduced voltage-gated Ca2+ currents, supporting the idea that manganese may compete with Ca2+ influx, leading to activation of BK channels and increased spontaneous firing activity of dopamine neurons.
Manganese competes with Ca2+ entry through voltage-gated Ca2+ channels to enhance excitability of dopamine neurons
Numerous studies demonstrated the neuronal expression of Ca2+ channels, such as Cav1.2 and Cav1.3, during neurodevelopment (Desarmenien and Spitzer, 1991; Gruol et al., 1992; Spitzer et al., 1995; Carey and Matsumoto, 1999; Kamijo et al., 2018). In addition, Ca2+ transients are observed in embryonic mouse neural crest (Carey and Matsumoto, 1999). Maladaptive Ca2+ influx, through mutated Cav1.2, disrupts radial migration of immature neurons (Kamijo et al., 2018). Calcium waves propagate through radial glial cells in the proliferative cortical ventricular zone (Weissman et al., 2004), whereas other reports have shown that calcium signals trigger a variety of developmental events (e.g., Desarmenien and Spitzer, 1991; Gruol et al., 1992; Spitzer et al., 1995). Postnatal dopamine neurons express Cav1.2 and Cav1.3, and they provide a suitable model system to identify manganese regulation of biophysical properties of dopamine neurons. It should be noted that the maturation of primary neuronal culture far exceeds the rate of neuronal maturation in vivo, and the time point of recordings in these cultures (8-10 d) yields neurons that express the markers and electrophysiological properties of mature neurons.
The AP of dopamine neurons is slow and broad, which maximizes Ca2+ entry and promotes slow rhythmic activity (Bean, 2007). The slow, rhythmic activity (2-10 Hz) in these neurons is autonomously generated and accompanied by slow oscillations in intracellular Ca2+ concentration that are triggered by the opening of plasma membrane Cav1 (Cav1.2, Cav1.3) Ca2+ channels and release of Ca2+ from intracellular, endoplasmic reticulum stores (Nedergaard et al., 1993; Puopolo et al., 2007; Guzman et al., 2009; Morikawa and Paladini, 2011). Thus, we tested the hypothesis that manganese increases Ca2+ entry into the neuron and/or releases Ca2+ from intracellular Ca2+ stores. Our data support neither of these possibilities (Figs. 3–9). Unexpectedly, application of manganese in Ca2+-free solution did not significantly alter the membrane potential (Fig. 4D); in contrast, BAPTA pretreatment before bath application of manganese in Ca2+-free external solution depolarized the membrane potential (Fig. 5D). This is consistent with previous reports showing that, under certain conditions, BAPTA induces membrane depolarization (Benedetti et al., 2011; Torkkeli et al., 2012). Consistently, a side-by-side comparison of firing activity of dopamine neurons in Figures 3 and 4 suggests that BAPTA facilitated the effect of manganese on the spontaneous spike activity of dopamine neurons. We that found performing the experiments in either extracellular Ca2+-free condition or chelation of intracellular Ca2+ did not impair manganese stimulation of firing frequency or reduction of AP amplitude. Consistently, live cell two-photon Ca2+ imaging showed that manganese did not alter intracellular Ca2+ concentration in the midbrain dopamine neurons. Thus, although manganese has been previously shown to regulate Ca2+ homeostasis in astrocytes (Xu et al., 2009), this effect is not present in dopamine neurons. Previous studies suggest that manganese can enter cells through a number of transporters (Gunshin et al., 1997; Lockman et al., 2001; Crossgrove and Yokel, 2005; Goytain et al., 2008; Itoh et al., 2008; Gunter et al., 2013). In addition, as a divalent cation, manganese may potentially target Ca2+ channels, which are also permeant to other divalent cations, such as barium (Bourinet et al., 1996). Therefore, if manganese competes with Ca2+ entry at the level of voltage-gated Ca2+ channels, then a nonselective Ca2+ channel blocker, such as Cd2+ or an L-type Ca2+ channel nifedipine, should block the effect of manganese on the firing frequency of dopamine neurons and AP morphology. As shown in Figures 8 and 9, both Cd2+ and nifedipine suppressed manganese stimulation of spontaneous firing activities, which is consistent with recent reports showing that dihydropyridine L-type channel inhibitors slow pacemaker activity of dopamine neurons at submicromolar concentrations (Nedergaard et al., 1993; Puopolo et al., 2007).
Blockade of L-type Ca2+ (Cav1) channels decreases manganese influx
Since the 1980s, manganese has been used as a tool to increase the signal-to-noise ratio in MRI (Lauterbur, 1980). Consistent with previous reports (Aoki et al., 2004; Lee et al., 2005), following systemic manganese administration, manganese is accumulated in multiple brain regions, including dopaminergic neuron-enriched brain regions, such as SN and VTA (Figs. 11, 12). Here we found that nifedipine blockade of L-type Ca2+ channels decreased manganese accumulation in several brain regions as well as manganese uptake in the Cav1.2-expressing HEK cells. Concentration-response experiment for manganese in Ca2+-free buffer revealed a very small current after replacing 2 mm CaCl2 with manganese (Fig. 10A1); therefore, steady-state amplitude of Ca2+ channel-mediated Mn2+ currents at different concentrations was difficult to discriminate. Nevertheless, these data support the interpretation that manganese might be permeable through the Ca2+ channels. Consistent with this idea, the chemical properties of aqueous Ca2+ and Mn2+ ions and the smaller ionic radius of Mn2+ than for Ca2+ also support the hypothesis that manganese might be accommodated by the Cav1 channels. While the crystal structure of Cav1.2 is yet to be determined, in silico analyses of the homology model of the helical regions of the mouse Cav1.1 structure and the helical regions for the Cav1.2 were found to be 72% identical and 81% similar for Cav1.2; therefore, it is possible that Mn2+ enters Cav-expressing cells. Whether manganese is permeable to other Ca2+ channels in dopamine or other neuronal types requires further examination. Furthermore, the possibility that manganese, enters the cell and leads to disruption of mitochondrial function, loss of ATP production, subsequent depolarization due to reduced activity of Na+/K+ ATPase, and increased open probability for high-voltage-activated Ca2+ channels require further investigation.
Manganese permeability through L-type Ca2+ channels was further supported by our MEMRI data. It should be noted that both neuronal and glial cells express Cav1 channels; therefore, the MEMRI and the nifedipine inhibition of this response reported in our studies do not distinguish the cell type. While the effect of manganese on Cav1.2 is consistent across other CNS cell types requires further investigation, our observations of manganese accumulation in the dopamine-enriched brain region and in vitro studies on the effects of manganese on dopaminergic neurons could establish a biological basis for movement disorders associated with manganism, which typically include symptoms related to dysregulation of the dopaminergic system. Furthermore, although the MEMRI data suggest that acute manganese exposure can be detected in multiple brain regions, dopaminergic brains regions, specifically the VTA and SN, displayed a greater enhancement of MRI signal by manganese compared with other brain regions observed in this study. Overall, we found that blockade of Cav1.2 can decrease manganese regulation of dopamine neuron activity and its accumulation in the brain.
Manganese increases membrane expression of BK channel α-subunit leading to enhanced BK channel activity
While it is well established that calcium currents drive the main pacemaking activity of midbrain dopamine neurons (Puopolo et al., 2007; Guzman et al., 2009), sodium currents have also been shown to affect the frequency and pattern of firing of multiple neuronal types (Enomoto et al., 2006; Vervaeke, et al., 2006). In contrast, potassium currents have been shown to exert a major role in AP repolarization (Mitterdorfer and Bean, 2002; Storm, 1987). Under our experimental condition, we found that manganese narrowed the half-width of APs (Fig. 2), suggesting that manganese may regulate potassium channels in dopamine neurons. Furthermore, we and others have shown that activation of BK channels can truncate the amplitude of APs (Van Goor et al., 2001; Petrik et al., 2011; Lin et al., 2016). Consistent with these reports, we found that lower concentrations of manganese truncated the amplitude of APs (Figs. 1, 2), and inhibition of BK channels blocked the manganese modulation of AP amplitude (Fig. 15). Structurally, BK channel complexes contain the pore-forming α subunit (four α subunits form the channel pore) and the regulatory β subunits (Knaus et al., 1994; Brenner et al., 2000; Lu et al., 2006). The intrinsic gating properties of BK channels are dynamically modulated by various kinases (White et al., 1991; Weiger et al., 2002) that regulate BK channel trafficking to the membrane (Chae and Dryer, 2005; Toro et al., 2006). Recently, we have shown that PKC activation decreases membrane expression of GFP α subunits of BK channels (Lin et al., 2016). These data, combined with reports showing manganese activation of protein kinase δ triggers apoptosis in dopaminergic neurons (Kitazawa et al., 2005), led us to ask whether Mn increases GFP α subunit membrane expression. We found that manganese enhanced surface trafficking of BK channels (Fig. 16) and increased BK channel activity (Fig. 17). These data collectively describe a cellular mechanism for the paxilline blockade of Mn-stimulated increases in firing frequency as well as its effect on the AP morphology. Therefore, it is possible that manganese may directly increase cell-surface redistribution of the BK α subunit (Fig. 16) to enhance the activity of dopamine neurons. Future studies will determine the PKC subtype(s) involved in Mn-induced trafficking of GFP α subunit of BK channels and the potential involvement of other BK channels subunits, such as regulatory β subunits. Collectively, these findings reveal the cellular mechanism for manganese regulation of dopamine neurons and reveal unique therapeutic targets to attenuate the untoward consequence of manganese exposure.
Footnotes
The authors declare no competing financial interests.
This work was supported by NIDA grants: NIDA-DA026947, NIDA-DA026947S1 and NIDA-DA043895; NINDA grant: NINDS-NS071122 NIH Office of Director: OD043895. We thank Dr. Gregory Hockerman for providing plasmids for Cav1.2-GFP and Cav1.3-GFP; Dr. Richard B. Silverman for providing HEK 293 cells stably expressing Cav1.2 or 1.3; and Dr. Robert Brenner for providing HEK293 cells stably expressing GFP-BK α subunits.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Aoki I, Naruse S, Tanaka C (2004) Manganese-enhanced magnetic resonance imaging (MEMRI) of brain activity and applications to early detection of brain ischemia. NMR Biomed 17:569–580. 10.1002/nbm.941 [DOI] [PubMed] [Google Scholar]
- Aschner JL, Aschner M (2005) Nutritional aspects of manganese homeostasis. Mol Aspects Med 26:353–362. 10.1016/j.mam.2005.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Guilarte TR, Schneider JS, Zheng W (2007) Manganese: recent advances in understanding its transport and neurotoxicity. Toxicol Appl Pharmacol 221:131–147. 10.1016/j.taap.2007.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Erikson KM, Herrero Hernández E, Hernández EH, Tjalkens R (2009) Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular Med 11:252–266. 10.1007/s12017-009-8083-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au C, Benedetto A, Aschner M (2008) Manganese transport in eukaryotes: the role of DMT1. Neurotoxicology 29:569–576. 10.1016/j.neuro.2008.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. (2007) The action potential in mammalian central neurons. Nat Rev Neurosci 8:451–465. 10.1038/nrn2148 [DOI] [PubMed] [Google Scholar]
- Benedetti B, Matyash V, Kettenmann H (2011) Astrocytes control GABAergic inhibition of neurons in the mouse barrel cortex. J Physiol 589:1159–1172. 10.1113/jphysiol.2010.203224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard M, Laforest F, Vandelac L, Bellinger D, Mergler D (2007) Hair manganese and hyperactive behaviors: pilot study of school-age children exposed through tap water. Environ Health Perspect 115:122–127. 10.1289/ehp.9504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Zamponi GW, Stea A, Soong TW, Lewis BA, Jones LP, Yue DT, Snutch TP (1996) The alpha 1E calcium channel exhibits permeation properties similar to low-voltage-activated calcium channels. J Neurosci 16:4983–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler RM, Gysens S, Diamond E, Nakagawa S, Drezgic M, Roels HA (2006) Manganese exposure: neuropsychological and neurological symptoms and effects in welders. Neurotoxicology 27:315–326. 10.1016/j.neuro.2005.10.007 [DOI] [PubMed] [Google Scholar]
- Bowman AB, Aschner M (2014) Considerations on manganese (manganese) treatments for in vitro studies. Neurotoxicology 41:141–142. 10.1016/j.neuro.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW (2000) Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407:870–876. 10.1038/35038011 [DOI] [PubMed] [Google Scholar]
- Cain CK, Blouin AM, Barad M (2002) L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditional fear in mice. J Neurosci 22:9113–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey MB, Matsumoto SG (1999) Spontaneous calcium transients are required for neuronal differentiation of murine neural crest. Dev Biol 215:298–313. 10.1006/dbio.1999.9433 [DOI] [PubMed] [Google Scholar]
- Chae KS, Dryer SE (2005) The p38 mitogen-activated protein kinase pathway negatively regulates Ca2+-activated K+ channel trafficking in developing parasympathetic neurons. J Neurochem 94:367–379. 10.1111/j.1471-4159.2005.03201.x [DOI] [PubMed] [Google Scholar]
- Chen C, Xu Y, Zhang J, Zhu J, Zhang J, Hu N, Guan H (2013) Altered expression of nNOS/NIDD in the retina of a glaucoma model of DBA/2J mice and the intervention by nNOS inhibition. J Mol Neurosci 51:47–56. 10.1007/s12031-012-9943-x [DOI] [PubMed] [Google Scholar]
- Choi DW. (1987) Ionic dependence of glutamate neurotoxicity. J Neurosci 7:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YM, Kim SH, Chung S, Uhm DY, Park MK (2006) Regional interaction of endoplasmic reticulum Ca2+ signals between soma and dendrites through rapid luminal Ca2+ diffusion. J Neurosci 26:12127–12136. 10.1523/JNEUROSCI.3158-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. (2007) Calcium signaling. Cell 131:1047–1058. 10.1016/j.cell.2007.11.028 [DOI] [PubMed] [Google Scholar]
- Crossgrove J, Zheng W (2004) Manganese toxicity upon overexposure. NMR Biomed 17:544–553. 10.1002/nbm.931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossgrove JS, Yokel RA (2005) Manganese distribution across the blood–brain barrier: IV. Evidence for brain influx through store-operated calcium channels. Neurotoxicology 26:297–307. 10.1016/j.neuro.2004.09.004 [DOI] [PubMed] [Google Scholar]
- Desarmenien MG, Spitzer NC (1991) Role of calcium and protein kinase C in development of the delayed rectifier potassium current in Xenopus spinal neurons. Neuron 7:797–805. 10.1016/0896-6273(91)90282-5 [DOI] [PubMed] [Google Scholar]
- Dodd CA, Ward DL, Klein BG (2005) Basal ganglia accumulation and motor assessment following manganese chloride exposure in the C57BL/6 mouse. Int J Toxicol 24:389–397. 10.1080/10915810500366500 [DOI] [PubMed] [Google Scholar]
- Dodd CA, Bloomquist JR, Klein BG (2013) Consequences of manganese administration for striatal dopamine and motor behavior in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-exposed C57BL/6 mice. Hum Exp Toxicol 32:865–880. 10.1177/0960327112469043 [DOI] [PubMed] [Google Scholar]
- Cox N, Toro B, Pacheco-Otalora LF, Garrido-Sanabria ER, Zarei MM (2014) An endoplasmic reticulum trafficking signal regulates surface expression of β4 subunit of a voltage- and Ca2+-activated K+ channel. Brain Res 1553:12–23. 10.1016/j.brainres.2014.01.028 [DOI] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Wu N, Chandler SH (2006) Participation of sodium currents in burst generation and control of membrane excitability in mesencephalic trigeminal neurons. J Neurosci 26:3412–3422. 10.1523/JNEUROSCI.5274-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erikson KM, Syversen T, Aschner JL, Aschner M (2005) Interactions between excessive manganese exposures and dietary iron-deficiency in neurodegeneration. Environ Toxicol Pharmacol 19:415–421. 10.1016/j.etap.2004.12.053 [DOI] [PubMed] [Google Scholar]
- Fujishiro H, Yoshida M, Nakano Y, Himeno S (2014) Interleukin-6 enhances manganese accumulation in SH-SY5Y cells: implications of the up regulation of ZIP14 and the down regulation of ZnT10. Metallomics 6:944–949. 10.1039/c3mt00362k [DOI] [PubMed] [Google Scholar]
- Gandhi D, Sivanesan S, Kannan K (2018) Manganese-induced neurotoxicity and alterations in gene expression in human neuroblastoma SH-SY5Y cells. Biol Trace Elem Res 183:245–253. 10.1007/s12011-017-1153-5 [DOI] [PubMed] [Google Scholar]
- Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, Kosofsky BE, Rajadhyaksha AM (2010) Molecular switch from L-type Ca v 1.3 to Ca v 1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci 30:17051–17062. 10.1523/JNEUROSCI.2255-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin JS, Larson GA, Swant J, Sen N, Javitch JA, Zahniser NR, De Felice LJ, Khoshbouei H (2009) Amphetamine and methamphetamine differentially affect dopamine transporters in vitro and in vivo. J Biol Chem 284:2978–2989. 10.1074/jbc.M805298200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goytain A, Hines RM, Quamme GA (2008) Huntingtin-interacting proteins, HIP14 and HIP14L, mediate dual functions, palmitoyl acyltransferase and Mg2 + transport. J Biol Chem 283:33365–33374. 10.1074/jbc.M801469200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene AC, Madgwick JC (1988) Heterotrophic manganese-oxidizing bacteria from Groote Eylandt, Australia. Geomicrobiol J 6:119–127. 10.1080/01490458809377829 [DOI] [Google Scholar]
- Gruol DL, Deal CR, Yool AJ (1992) Developmental changes in calcium conductances contribute to the physiological maturation of cerebellar Purkinje neurons in culture. J Neurosci 12:2838–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilarte TR. (2010) Manganese and Parkinson's disease: a critical review and new findings. Environ Health Perspect 118:1071–1080. 10.1289/ehp.0901748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482–488. 10.1038/41343 [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gerstner B, Gunter KK, Malecki J, Gelein R, Valentine WM, Aschner M, Yule DI (2013) Manganese transport via the transferrin mechanism. Neurotoxicology 34:118–127. 10.1016/j.neuro.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sánchez-Padilla J, Chan CS, Surmeier DJ (2009) Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29:11011–11019. 10.1523/JNEUROSCI.2519-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy G. (2009) Manganese in parenteral nutrition: who, when, and why should we supplement? Gastroenterology 137:S29–S35. 10.1053/j.gastro.2009.08.011 [DOI] [PubMed] [Google Scholar]
- Higashi Y, Asanuma M, Miyazaki I, Hattori N, Mizuno Y, Ogawa N (2004) Parkin attenuates manganese-induced dopaminergic cell death. J Neurochem 89:1490–1497. 10.1111/j.1471-4159.2004.02445.x [DOI] [PubMed] [Google Scholar]
- Itoh K, Sakata M, Watanabe M, Aikawa Y, Fujii H (2008) The entry of manganese ions into the brain is accelerated by the activation of N-methyl-D-aspartate receptors. Neuroscience 154:732–740. 10.1016/j.neuroscience.2008.03.080 [DOI] [PubMed] [Google Scholar]
- Jankovic J. (2005) Searching for a relationship between manganese and welding and Parkinson's disease. Neurology 64:2021–2018. 10.1212/01.WNL.0000166916.40902.63 [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Bannister P, Brady M, Smith S (2002) Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage 17:825–841. 10.1016/s1053-8119(02)91132-8 [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Yitta S, Drew T, Kim BS, Visser JE, Rothstein JD (1999) Calcium channel activation and self-biting in mice. Proc Natl Acad Sci USA 96:15228–15232. 10.1073/pnas.96.26.15228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Ahlskog JE, Klos KJ, Kumar N, Fealey RD, Trenerry MR, Cowl CT (2005) Neurologic manifestations in welders with pallidal MRI T1 hyperintensity. Neurology 64:2033–2039. 10.1212/01.WNL.0000167411.93483.A1 [DOI] [PubMed] [Google Scholar]
- Kamijo S, Ishii Y, Horigane SI, Suzuki K, Ohkura M, Nakai J, Fujii H, Takemoto-Kimura S, Bito H (2018) A critical neurodevelopmental role for L-type voltage-gated calcium channels in neurite extension and radial migration. J Neurosci 38:5551–5566. 10.1523/JNEUROSCI.2357-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsoyiannis IA, Katsoyiannis AA (2006) Arsenic and other metal contamination of groundwaters in the industrial area of Thessaloniki, Northern Greece. Environ Monit Assess 123:393–406. 10.1007/s10661-006-9204-y [DOI] [PubMed] [Google Scholar]
- Kessler KR, Wunderlich G, Hefter H, Seitz RJ (2003) Secondary progressive chronic manganism associated with markedly decreased striatal D2 receptor density. Mov Disord 18:217–218. 10.1002/mds.10325 [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Yang Y, Hirata Y, Kanthasamy A, Kanthasamy AG (2005) Activation of protein kinase C delta by proteolytic cleavage contributes to manganese-induced apoptosis in dopaminergic cells: protective role of Bcl-2. Biochem Pharmacol 69:133–146. 10.1016/j.bcp.2004.08.035 [DOI] [PubMed] [Google Scholar]
- Knaus HG, Garcia-Calvo M, Kaczorowski GJ, Garcia ML (1994) Subunit composition of the high conductance calcium-activated potassium channel from smooth muscle, a representative of the mSlo and slowpoke family of potassium channels. J Biol Chem 269:3921–3924. [PubMed] [Google Scholar]
- Lauterbur PC. (1980) Progress in N.M.R. zeugmatography imaging. Philos Trans R Soc Lond B Biol Sci 289:483–487. 10.1098/rstb.1980.0066 [DOI] [PubMed] [Google Scholar]
- Lee JH, Silva AC, Merkle H, Koretsky AP (2005) Manganese-enhanced magnetic resonance imaging of mouse brain after systemic administration of MnCl2: dose-dependent and temporal evolution of T1 contrast. Magn Reson Med 53:640–648. 10.1002/mrm.20368 [DOI] [PubMed] [Google Scholar]
- Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S (2014) SLC30A10 is a cell surface-localized manganese efflux transporter, and Parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J Neurosci 34:14079–14095. 10.1523/JNEUROSCI.2329-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Sambo D, Khoshbouei H (2016) Methamphetamine regulation of firing activity of dopamine neurons. J Neurosci 36:10376–10391. 10.1523/JNEUROSCI.1392-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sullivan KA, Madl JE, Legare M, Tjalkens RB (2006) Manganese-induced neurotoxicity: the role of astroglial-derived nitric oxide in striatal interneuron degeneration. Toxicol Sci 91:521–531. 10.1093/toxsci/kfj150 [DOI] [PubMed] [Google Scholar]
- Lockman PR, Roder KE, Allen DD (2001) Inhibition of the rat blood-brain barrier choline transporter by manganese chloride. J Neurochem 79:588–594. 10.1046/j.1471-4159.2001.00589.x [DOI] [PubMed] [Google Scholar]
- Lu R, Alioua A, Kumar Y, Eghbali M, Stefani E, Toro L (2006) MaxiK channel partners: physiological impact. J Physiol 570:65–72. 10.1113/jphysiol.2005.098913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JL, Wegrzynowicz M, Aschner M, Bowman AB (2012) Disease-toxicant interactions in manganese exposed Huntington disease mice: early changes in striatal neuron morphology and dopamine metabolism. PLoS One 7:e31024. 10.1371/journal.pone.0031024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Finley EJ, Gavin CE, Aschner M, Gunter TE (2013) Manganese neurotoxicity and the role of reactive oxygen species. Free Radic Biol Med 62:65–75. 10.1016/j.freeradbiomed.2013.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Bonci A, Calabresi P, Stratta F, Stefani A, Bernardi G (1994) Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br J Pharmacol 113:831–838. 10.1111/j.1476-5381.1994.tb17068.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalke B, Fernsebner K (2014) New insights into manganese toxicity and speciation. J Trace Elem Med Biol 28:106–116. 10.1016/j.jtemb.2013.08.005 [DOI] [PubMed] [Google Scholar]
- Mitterdorfer J, Bean BP (2002) Potassium currents during the action potential of hippocampal CA3 neurons. J Neurosci 22:10106–10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morellini F, Malyshev A, Volgushev M, Chistiakova M, Papashvili G, Fellini L, Kleene R, Schachner M, Dityatev A (2017) Impaired fear extinction due to a deficit in Ca2+ influx through L-type voltage-gated Ca2+ channels in mice deficient for tenascin-C. Front Integr Neurosci 11:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa H, Paladini CA (2011) Dynamic regulation of midbrain dopamine neuron activity: intrinsic, synaptic, and plasticity mechanisms. Neuroscience 198:95–111. 10.1016/j.neuroscience.2011.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA (2008) Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience 152:1024–1031. 10.1016/j.neuroscience.2008.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard S, Flatman JA, Engberg I (1993) Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol 466:727–747. [PMC free article] [PubMed] [Google Scholar]
- Olanow CW. (2004) Manganese-induced parkinsonism and Parkinson's disease. Ann NY Acad Sci 1012:209–223. 10.1196/annals.1306.018 [DOI] [PubMed] [Google Scholar]
- Park RM, Bowler RM, Roels HA (2009) Exposure-response relationships and risk assessment for cognitive deficits in early welding-induced manganism. J Occup Environ Med 51:1125–1136. [DOI] [PubMed] [Google Scholar]
- Perl DP, Olanow CW (2007) The neuropathology of manganese-induced parkinsonism. J Neuropathol Exp Neurol 66:675–682. 10.1097/nen.0b013e31812503cf [DOI] [PubMed] [Google Scholar]
- Pereira DB, Schmitz Y, Mészáros J, Merchant P, Hu G, Li S, Henke A, Lizardi-Ortiz J, Karpowicz R Jr, Morgenstern TJ, Sonders MS, Kanter E, Rodriguez PC, Mosharov EV, Sames D, Sulzer D (2016) Fluorescent false neurotransmitter reveals functionally silent dopamine vesicle clusters in the striatum. Nat Neurosci 19:578–586. 10.1038/nn.4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson I. (2010) Hydrated metal ions in aqueous solution: how regular are their structures? Pure Appl Chem 82:901–1917. [Google Scholar]
- Petrik D, Wang B, Brenner R (2011) Modulation by the BK accessory β4 subunit of phosphorylation-dependent changes in excitability of dentate gyrus granule neurons. Eur J Neurosci 34:695–704. 10.1111/j.1460-9568.2011.07799.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DS, Doorenweerd N, Plomp JJ, Mahfouz A, Reinders MJ, van der Weerd L (2017) Continuous infusion of manganese improves contrast and reduces side effects in manganese-enhanced magnetic resonance imaging studies. Neuroimage 147:1–9. 10.1016/j.neuroimage.2016.09.030 [DOI] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP (2007) Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27:645–656. 10.1523/JNEUROSCI.4341-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskin JS, Gunter TE (1973) Ion and pH gradients across the transport membrane of mitochondria following Mn++ uptake in the presence of acetate. Biochem Biophys Res Commun 51:797–803. 10.1016/0006-291x(73)91385-5 [DOI] [PubMed] [Google Scholar]
- Putzier I, Kullmann PH, Horn JP, Levitan ES (2009) Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral DA neurons. J Neurosci 29:15414–15419. 10.1523/JNEUROSCI.4742-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers K, Beaubrun I, Catapane E, Carroll M (2014) The toxic effects of manganese on dopamine D2 receptor activation is not due to inactivation of the phospholipase c receptor signal transduction component (1143.7). FASEB J 28:1143. 7. [Google Scholar]
- Roth JA, Li Z, Sridhar S, Khoshbouei H (2013) The effect of manganese on dopamine toxicity and dopamine transporter (DAT) in control and DAT transfected HEK cells. Neurotoxicology 35:121–128. 10.1016/j.neuro.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha K, Sambo D, Richardson BD, Lin LM, Butler B, Villarroel L, Khoshbouei H (2014) Intracellular methamphetamine prevents the dopamine-induced enhancement of neuronal firing. J Biol Chem 289:22246–22257. 10.1074/jbc.M114.563056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambo DO, Lin M, Owens A, Lebowitz JJ, Richardson B, Jagnarine DA, Shetty M, Rodriquez M, Alonge T, Ali M, Katz J, Yan L, Febo M, Henry LK, Bruijnzeel AW, Daws L, Khoshbouei H (2017) The sigma-1 receptor modulates methamphetamine dysregulation of dopamine neurotransmission. Nat Commun 8:2228. 10.1038/s41467-017-02087-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt KF, Ziu M, Schmidt NO, Vaghasia P, Cargioli TG, Doshi S, Albert MS, Black PM, Carroll RS, Sun Y (2004) Volume reconstruction techniques improve the correlation between histological and in vivo tumor volume measurements in mouse models of human gliomas. J Neurooncol 68:207–215. 10.1023/b:neon.0000033364.43142.bf [DOI] [PubMed] [Google Scholar]
- Seth PK, Chandra SV (1984) Neurotransmitters and neurotransmitter receptors in developing and adult rats during manganese poisoning. Neurotoxicology 5:67–76. [PubMed] [Google Scholar]
- Shruti S, Urban-Ciecko J, Fitzpatrick JA, Brenner R, Bruchez MP, Barth AL (2012) The brain-specific beta4 subunit downregulates BK channel cell surface expression. PLoS One 7:e33429. 10.1371/journal.pone.0033429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC, Olson E, Gu X (1995) Spontaneous calcium transients regulate neuronal plasticity in developing neurons. J Neurobiol 26:316–324. 10.1002/neu.480260304 [DOI] [PubMed] [Google Scholar]
- Soriano S, Calap-Quintana P, Llorens JV, Al-Ramahi I, Gutiérrez L, Martínez-Sebastián MJ, Botas J, Moltó MD (2016) Metal homeostasis regulators suppress FRDA phenotypes in a Drosophila model of the disease. PLoS One 11:e0159209. 10.1371/journal.pone.0159209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. (1987) Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol 385:733–759. 10.1113/jphysiol.1987.sp016517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streifel KM, Miller J, Mouneimne R, Tjalkens RB (2013) Manganese inhibits ATP-induced calcium entry through the transient receptor potential channel TRPC3 in astrocytes. Neurotoxicology 34:160–166. 10.1016/j.neuro.2012.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL (2009) Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods 6:875–881. 10.1038/nmeth.1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjalkens RB, Zoran MJ, Mohl B, Barhoumi R (2006) Manganese suppresses ATP-dependent intercellular calcium waves in astrocyte networks through alteration of mitochondrial and endoplasmic reticulum calcium dynamics. Brain Res 1113:210–219. 10.1016/j.brainres.2006.07.053 [DOI] [PubMed] [Google Scholar]
- Torkkeli PH, Meisner S, Pfeiffer K, French AS (2012) GABA and glutamate receptors have different effects on excitability and are differentially regulated by calcium in spider mechanosensory neurons. Eur J Neurosci 36:3602–3614. 10.1111/j.1460-9568.2012.08275.x [DOI] [PubMed] [Google Scholar]
- Toro B, Cox N, Wilson RJ, Garrido-Sanabria E, Stefani E, Toro L, Zarei MM (2006) KCNMB1 regulates surface expression of a voltage and Ca2+-activated K+ channel via endocytic trafficking signals. Neuroscience 142:661–669. 10.1016/j.neuroscience.2006.06.061 [DOI] [PubMed] [Google Scholar]
- Tuschl K, Mills PB, Clayton PT (2013) Manganese and the brain. Int Rev Neurobiol 110:277–312. 10.1016/B978-0-12-410502-7.00013-2 [DOI] [PubMed] [Google Scholar]
- Uchino A, Noguchi T, Nomiyama K, Takase Y, Nakazono T, Nojiri J, Kudo S (2007) Manganese accumulation in the brain: MR imaging. Neuroradiology 49:715–720. 10.1007/s00234-007-0243-z [DOI] [PubMed] [Google Scholar]
- Van Goor F, Li YX, Stojilkovic SS (2001) Paradoxical role of large-conductance calcium-activated K+ (BK) channels in controlling action potential-driven Ca2+ entry in anterior pituitary cells. J Neurosci 21:5902–5915. 10.1523/JNEUROSCI.21-16-05902.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven WM, Egger JI, Kuijpers HJ (2011) Manganese and acute paranoid psychosis: a case report. J Med Case Rep 5:146. 10.1186/1752-1947-5-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Hu H, Graham LJ, Storm JF (2006) Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron 49:257–270. 10.1016/j.neuron.2005.12.022 [DOI] [PubMed] [Google Scholar]
- Wang B, Rothberg BS, Brenner R (2009) Mechanism of increased BK channel activation from a channel mutation that causes epilepsy. J Gen Physiol 133:283–294. 10.1085/jgp.200810141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TA, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiger TM, Hermann A, Levitan IB (2002) Modulation of calcium-activated potassium channels. J Comp Physiol A Neuroethol Sens Neural Behav Physiol 188:79–87. 10.1007/s00359-002-0281-2 [DOI] [PubMed] [Google Scholar]
- Weissman TA, Riquelme PA, Ivic L, Flint AC, Kriegstein AR (2004) Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43:647–661. 10.1016/j.neuron.2004.08.015 [DOI] [PubMed] [Google Scholar]
- White RE, Schonbrunn A, Armstrong DL (1991) Somatostatin stimulates Ca2+-activated K+ channels through protein dephosphorylation. Nature 351:570–573. 10.1038/351570a0 [DOI] [PubMed] [Google Scholar]
- Wild AR, Jones S, Gibb AJ (2014) Activity-dependent regulation of NMDA receptors in substantia nigra dopaminergic neurones. J Physiol 592:653–668. 10.1113/jphysiol.2013.267310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JP, Yan Z, Li ZQ, Qian XY, Lu S, Dong MQ, Zhou Q, Yan N (2016) Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 Å resolution. Nature 537:191–196. 10.1038/nature19321 [DOI] [PubMed] [Google Scholar]
- Xu B, Xu ZF, Deng Y (2009) Effect of manganese exposure on intracellular Ca2+ homeostasis and expression of NMDA receptor subunits in primary cultured neurons. Neurotoxicology 30:941–949. 10.1016/j.neuro.2009.07.011 [DOI] [PubMed] [Google Scholar]
- Yin Z, Jiang H, Lee ES, Ni M, Erikson KM, Milatovic D, Bowman AB, Aschner M (2010) Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J Neurochem 112:1190–1198. 10.1111/j.1471-4159.2009.06534.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DQ, Stone JF, Zhou T, Ohta H, McMahon DG (2004) Characterization of genetically labeled catecholamine neurons in the mouse retina. Neuroreport 15:1761–1765. 10.1097/01.wnr.0000135699.75775.41 [DOI] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Webb WW (2003) Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol 21:1369–1377. 10.1038/nbt899 [DOI] [PubMed] [Google Scholar]
- Zogzas CE, Mukhopadhyay S (2017) Inherited disorders of manganese metabolism. Adv Neurobiol 18:35–49. 10.1007/978-3-319-60189-2_3 [DOI] [PubMed] [Google Scholar]