

Abstract
Despite extensive research into understanding synaptic mechanisms of striatal plasticity, the functional role played by astrocytes in this region remains to be fully elucidated. It was recently demonstrated that high-frequency stimulation (HFS) of cortical inputs induced long-term depression (LTD) mediated by adenosine A1 receptor (A1R) activation at corticostriatal synapses of the direct pathway [cortico-striatal projection neuron (dSPN)] in the dorsolateral striatum (DLS). Because astrocyte-derived adenosine has been shown to regulate synaptic transmission in several brain areas, we investigated whether this form of neuron-astrocyte signaling contributes to synaptic plasticity in the DLS of male and female mice. We found that cortical HFS increases calcium (Ca2+) levels in striatal astrocytes through activation of metabotropic glutamate receptor type 5 (mGluR5) signaling and that this astrocyte-mediated response is necessary for A1R-mediated LTD. Consistent with this, astrocyte activation with Gq designer receptors exclusively activated by designer drugs (DREADDs) induced A1R-mediated synaptic depression at cortico-dSPN synapses. Together, these results indicate that astrocytes are integral elements of striatal A1R-mediated LTD.
SIGNIFICANCE STATEMENT Abnormal striatal circuit function is implicated in several disorders such as Parkinson's disease and Huntington's disease. Thus, there is a need to better understand the mechanisms supporting proper striatal activity. While extensive work has revealed the many important contributions from neurons in striatal function, far less is known about the role of astrocytes in this brain area. We show that long-term depression (LTD) at corticostriatal synapses of the direct pathway is not strictly a neuronal phenomenon; astrocytes respond to corticostriatal stimulation and this astrocyte response is necessary for LTD. This research adds to the accumulating evidence that astrocytes are active and integral players in synaptic communication, and that neuron-astrocyte interactions are key cellular processes involved in brain function.
Keywords: astrocyte calcium signaling, astrocytes, long-term depression, striatal synaptic plasticity
Introduction
Increasing evidence is producing a fundamental shift in our understanding of intercellular communication at synapses. Rather than exclusive neurotransmitter signaling between presynaptic and postsynaptic neurons, astrocytes can respond to synaptically-released neurotransmitters and release gliotransmitters that can influence synaptic transmission (Araque et al., 2014; Papouin et al., 2017). Such bidirectional signaling between neurons and astrocytes has been established in several brain areas (Araque et al., 2014), and recent studies have focused on the role of astrocytes in the striatum (Martín et al., 2015; Jiang et al., 2016; Khakh, 2019). However, the involvement of astrocytes on synaptic plasticity mechanisms in this brain area remains largely incomplete.
The striatum is the main input nucleus of the basal ganglia (Cox and Witten, 2019). The most abundant striatal cell subpopulations are the inhibitory GABAergic striatal projection neurons (SPNs) of the direct (dSPN) and indirect output pathway (iSPN). While dSPNs express the dopamine receptor type 1 (D1R), iSPNs express dopamine receptor type 2 (D2R; Kreitzer and Malenka, 2008). The activity of SPNs strongly depends on the excitatory cortical and thalamic inputs, since SPNs are largely quiescent. Thus, striatal circuit output can be influenced by plastic changes at excitatory synapses, which consequently regulate action selection and initiation (Kravitz et al., 2012; Sippy et al., 2015; O'Hare et al., 2016). Appropriate motor control relies on proper coordination of direct and indirect SPN activity, as well as coordinated regulation of corticostriatal synaptic plasticity at the level of the dorsolateral striatum (DLS; Cui et al., 2013; Tecuapetla et al., 2014; Trusel et al., 2015).
Extensive work has identified key presynaptic mechanisms that cooperate with postsynaptic dopaminergic signaling to enable cell-type-specific forms of high-frequency stimulation-dependent long-term depression (HFS-LTD) at cortico-iSPN and cortico-dSPN synapses (Wang et al., 2006b; Adermark and Lovinger, 2007; Kreitzer and Malenka, 2007; Bagetta et al., 2011; Trusel et al., 2015; Wu et al., 2015; Augustin et al., 2018). We have previously shown that when plasticity is induced by cortical stimulation, HFS-LTD is governed by endocannabinoids (eCBs) at cortico-iSPN synapses, whereas this is done by adenosine at cortico-dSPN synapses through the activation of adenosine A1 receptor (A1R; Trusel et al., 2015; Spigolon et al., 2018). These forms of LTD are concurrently and oppositely regulated by postsynaptic dopaminergic signaling in iSPNs and dSPNs, downstream of D2R and D1R, respectively (Trusel et al., 2015; Wu et al., 2015; Spigolon et al., 2018). The loss of this concurrent and pathway-specific regulation of corticostriatal synaptic plasticity has motor implications in a mouse model of Parkinson's disease (Trusel et al., 2015). eCB-dependent corticostriatal plasticity has been widely studied. Indeed, it is well known that SPNs release eCBs that bind presynaptic cannabinoid receptor type 1 (CB1R) to suppress subsequent glutamate release from corticostriatal terminals (Kreitzer and Malenka, 2007). However, the mechanisms underlying adenosine-dependent LTD are not fully elucidated, in particular the source of adenosine. We hypothesized that astrocytes were the adenosine source, because they can release ATP that, after being hydrolyzed into adenosine, can influence synaptic transmission and plasticity at nearby synapses in other brain regions (Zhang et al., 2003; Pascual et al., 2005; Serrano et al., 2006; Chen et al., 2013; Covelo and Araque, 2018).
In this study, we aimed to elucidate the role of astrocytes in mediating adenosine-dependent-LTD at cortico-dSPN synapses in the DLS (Trusel et al., 2015). We found that DLS astrocytes respond to cortical HFS with Ca2+ increases through activation of metabotropic glutamate receptor type 5 (mGluR5) and inositol triphosphate type 2 receptors (IP3R2s). Blocking astrocyte activation prevented the HFS-induced LTD. Furthermore, directly and selectively activating astrocytes with Gq designer receptors exclusively activated by designer drugs (DREADDs) resulted in a depression of cortico-dSPN synapses in an A1R-dependent manner. Taken together, these results are the first to indicate that astrocytes are an integral component of a form of long-term synaptic plasticity in the striatum. These results add to the accumulating evidence that neuron-astrocyte interactions at the tripartite synapse are necessary for proper brain function.
Materials and Methods
Experimental model and subject details
Experiments were performed on 4- to 12-week-old male and female mice of the following strains: C57BL/6J, Drd2-eGFP bacterial artificial chromosome (BAC) transgenic mice, Drd1-tdTomato BAC transgenic mice, and IP3R2−/− (Li et al., 2005). The animals were housed in a controlled environment, on a 12 h light/dark cycle, with unrestricted access to food and water. All procedures involving animals were conducted in accordance with the Animal Care and Use Committee at the University of Minnesota and the Italian Ministry of Health's directives (D.lgs 26/2014) regulating animal research.
Stereotaxic surgery
Four- to six-week-old mice were anesthetized with a ketamine (100 mg/kg)/xylazine (10 mg/kg) cocktail (0.2 ml/kg body weight) and underwent stereotaxic surgery of different viruses. To measure Ca2+ signaling in astrocytes, mice were injected with an adeno-associated virus containing cytosolic GCaMP6f expression (AAV5 pZac2.1 gfaABC1D-cyto-GCaMP6f from Baljit Khakh, Addgene plasmid #52925; http://n2t.net/addgene:52925; RRID: Addgene_52925, Haustein et al., 2014). For chemogenetic activation of Gq signaling in astrocytes, mice were injected with AAV8-GFAP-hM3Dq-mCherry. For control DREADDs experiments, mice received injections of AAV8-GFAP-mCherry. Stereotaxic coordinates used for DLS injections were (relative to bregma in mm) +1.0 AP, ±2.00 ML, and −3.15 DV. Experiments were performed two to six weeks after surgery. All DREADDs viruses were purchased from the UPenn Vector Core, while the plasmid to generate the AAV8-GFAP-mCherry was purchased from Addgene (Plasmid #58909) and was generated by the University of Minnesota Viral Vector and Cloning Core.
Slice preparation
Mice were anesthetized with isoflurane and decapitated, and their brains were transferred to ice-cold artificial CSF (aCSF) containing 110 mm choline chloride, 2.5 mm KCl, 1.25 mm NaH2PO4, 7 mm MgCl2, 0.5 mm CaCl2, 25 mm NaHCO3, 25 mm D-glucose, and 11.6 mm ascorbic acid, saturated with 95% O2 and 5% CO2 for dissection. Horizontal slices (270 μm thick) were prepared using a Vibratome 1000 slicer (Leica), then transferred to new aCSF containing 115 mm NaCl, 3.5 mm KCl, 1.2 mm NaH2PO4, 1.3 mm MgCl2, 2 mm CaCl2, 25 mm NaHCO3, and 25 mm D-glucose, and aerated with 95% O2 and 5% CO2. After incubating for 20 min at 32°C, slices were kept at 22–24°C. During experiments, slices were continuously superfused with aCSF at a rate of 2 ml/min at 28°C.
Slice electrophysiology
Whole-cell patch-clamp recordings were made on dSPN of the DLS in horizontal slices that preserved corticostriatal connections (Nazzaro et al., 2012; Trusel et al., 2015; Cavaccini et al., 2018; Spigolon et al., 2018). dSPNs in the DLS were visualized under infrared-differential interference contrast and were identified based on their morphologic, electrical properties, and also by the expression of adenosine A2A receptor (A2AR; for iSPN) and substance P (SP; for dSPN; Nazzaro et al., 2012; Trusel et al., 2015; Cavaccini et al., 2018; Spigolon et al., 2018). In some experiments, Drd2-eGFP and Drd1-tdTomato BAC mice were used for electrophysiology experiments, where only GFP-negative and tdTomato-positive cells, respectively, were selected for recording. Patch pipettes (3–4 MΩ) were filled with a solution containing 130 mm KMeSO4, 5 mm KCl, 5 mM NaCl, 10 mm HEPES, 0.1 mm EGTA, 2 mm MgCl2, 0.05 mm CaCl2, 2 mm Na2-ATP, and 0.4 mm Na3-GTP (pH 7.2–7.3; 280–290 mOsm/kg).
Data were acquired using a Multiclamp 700B amplifier controlled by pClamp 10 software (Molecular Device), filtered at 10 kHz and sampled at 20 kHz (Digidata 1322, Molecular Device). Alternatively, recordings were acquired using PC-ONE amplifiers (Dagan Instruments) controlled by pClamp 10 software (Molecular Device) and filtered at 1 kHz (DigiData 1440A). All data are reported without corrections for liquid junction potentials. Data where the input resistance or the access resistance changed >20% have been excluded.
Ca2+ imaging
Ca2+ levels in astrocytes were monitored by two-photon microscopy (Leica DM6000 CFS upright multiphoton microscope with a TCS SP5 MP laser). Astrocyte regions of interest (ROIs) were selected by a custom MATLAB program, which detected ROIs whose Ca2+ signal changed >2 SD from baseline. The ROI Ca2+ values were then fed into a second custom MATLAB program that identified a Ca2+ event when the Ca2+ trace surpassed a threshold that was based on the 25th and 50th percentiles of the entire Ca2+ trace. Our measurement of Ca2+ event probability was calculated as the number of Ca2+ elevations grouped in 10-s bins recorded from the astrocytes in the field of view, and mean values were obtained by averaging each different experiment. The trend toward increased Ca2+ event probability for all treatment groups, which perhaps resulted from non-specific cellular activation by prolonged light excitation, was not corrected. References to “n” in the text correspond to the number of experiments performed for each condition.
Immunohistochemistry
Anesthetized mice were perfused through the left cardiac ventricle with 4% paraformaldehyde in 0.1 m PBS. The brains were removed, postfixed overnight with paraformaldehyde and cut into 100-µm slices using a Leica VT 1000S vibratome. Brain sections were then blocked with PBS containing 10% normal goat serum (NGS) or normal donkey serum (NDS) and 0.5% Triton X-100 and incubated for 48 h with the primary antibody diluted in blocking solution. Appropriate fluorochrome-labeled secondary antibodies (Jackson ImmunoResearch) were then incubated overnight and used for detection. Antibodies against glial fibrillary acidic protein (GFAP; catalog #G9269, 1:500, Sigma-Aldrich or catalog #MAB360, 1:500, Chemicon) were used as a marker for astrocytes. Antibodies against neuron-specific nuclear protein (NeuN, catalog #MABN140, 1:500, Millipore or catalog #MAB377, 1:200, Chemicon) were used as a marker for neurons. Antibodies against NG2 chondroitin sulfate proteoglycan (NG2; catalog #AB5320, 1:500, Millipore) was used to identify NG2 glial cells. Antibodies against ionized calcium binding adaptor molecule 1 (Iba1; catalog #019-19741, 1:500, Wako) were used to localize microglial cells. Antibodies against oligodendrocyte lineage transcription factor 2 (Olig2; catalog #NP1-28667, Novus, 1:500) were used to label oligodendrocytes. An antibody against mCherry was used as a marker for DREADDs (catalog #ab205402, 1:500, Abcam). Fluorescent-stained sections were mounted with Vectashield mounting medium (Vector Laboratories). The stained sections were visualized with an Olympus FluoView FV1000 upright confocal microscope and analyzed for colocalization of DREADDs and other cell markers using ImageJ software (NIH).
Immunofluorescence: identification of dSPNs and iSPNs
To identify SPNs of the direct (dSPN) and indirect (iSPN) pathway, neurons were filled with Neurobiotin (0.5 mg/ml, Vector Laboratories) or biocytin (0.5 mg/ml, Sigma-Aldrich) during recordings and subsequently processed for immunostaining of the A2A receptor (marker of iSPNs) and SP (marker of dSPNs; Nazzaro et al., 2012; Trusel et al., 2015). After recording, slices were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer (0.1 m PB; pH 7.4) overnight at 4°C and then incubated in primary antibodies. Rabbit polyclonal antibody to A2A (1:250, Enzo Life Sciences) and rat monoclonal antibody to SP (1:200, Millipore) were diluted in 0.1 m PB containing 0.3% Triton X-100. Sections were subsequently incubated with Alexa Fluor 647-conjugated or Alexa Fluor 488-conjugated secondary antibodies (1:200) and Alexa Fluor 568-conjugated streptavidin (1:1000, ThermoFisher Scientific), mounted on glass slides, and coverslipped with ProLong Gold antifade reagent (ThermoFisher Scientific). Images were acquired with an inverted Leica TCS SP5 AOBS TANDEM confocal microscope.
Data analysis
Excitatory postsynaptic current (EPSC) and potential (EPSP) amplitudes were normalized to the mean value of 5 min before HFS or Clozapine-N-Oxide (CNO) application, and baseline values were considered 5 min before the stimulus. The occurrence and magnitude of synaptic plasticity was evaluated by comparing the normalized EPSP or EPSC amplitudes recorded before and after HFS delivery (25–30 min post-HFS) or CNO application (last 4 min). Time course plots were generated by averaging the peak amplitude of individual EPSPs or EPSCs in 1-min bins. Presynaptic or postsynaptic changes in EPSPs and EPSCs were assessed from their coefficient of variation (CV), which was calculated by the ratio of the SD and the mean EPSP or EPSC amplitude of the last 7 min before compared with the last 7 min after HFS delivery (Shen et al., 2008), and the parameter CV−2 was used.
Experimental design and statistical analyses
The overall goal of this study was to investigate the role of astrocytes in corticostriatal LTD. To this end, EPSPs or EPSCs were evoked by stimulation of the deep Layer V of the somatosensory cortex in the presence of the GABAA receptor antagonist gabazine (10 μm) or picrotoxin (50 μm) and acquired every 10 s (for more details, see above, Slice preparation and Slice electrophysiology). To induce HFS-LTD, 4 × 1-s long 100-Hz trains were repeated every 10 s. During the HFS protocol, the postsynaptic cell was depolarized from −80 to −50 mV to mimic cortically-induced upstates (Fig. 1A). In addition to measuring the effects of HFS on corticostriatal plasticity, we also monitored Ca2+ activity in striatal astrocytes using a genetically-encoded Ca2+ indicator (for more details, see above, Ca2+ imaging). Several pharmacological agents were used to block various receptor subtypes to assess their effects both on LTD and astrocyte Ca2+. Additionally, Guanosine 5′-[β-thio]diphosphate trilithium salt (GDPβS; 10 μm) was used to block G protein-coupled receptor (GPCR) signaling in neurons or in astrocytes. It was added to the intracellular solution described above for neurons or to an intracellular solution specific for astrocytes containing the following: 100 mm KMeSO4, 50 mm KCl, 10 mm HEPES-K, and 2 mm ATP-Na2 (pH 7.3 adjusted with KOH; osmolality 280–290 mOsm/l). To allow the GDPβS to load into the neuron or into the astrocyte and its syncytium, the patched cells were filled for 20 min (min) before starting the experiment. For patch-loaded astrocytes, the pipette was removed after 20 min and then the experiment on dSPNs was performed. To test the effect of directly activating astrocytes on corticostriatal synaptic plasticity, 10 μm CNO was bath-applied to horizontal brain slices from mice injected with either AAV8-GFAP-hM3Dq-mCherry or AAV8-GFAP-mCherry as control.
Figure 1.
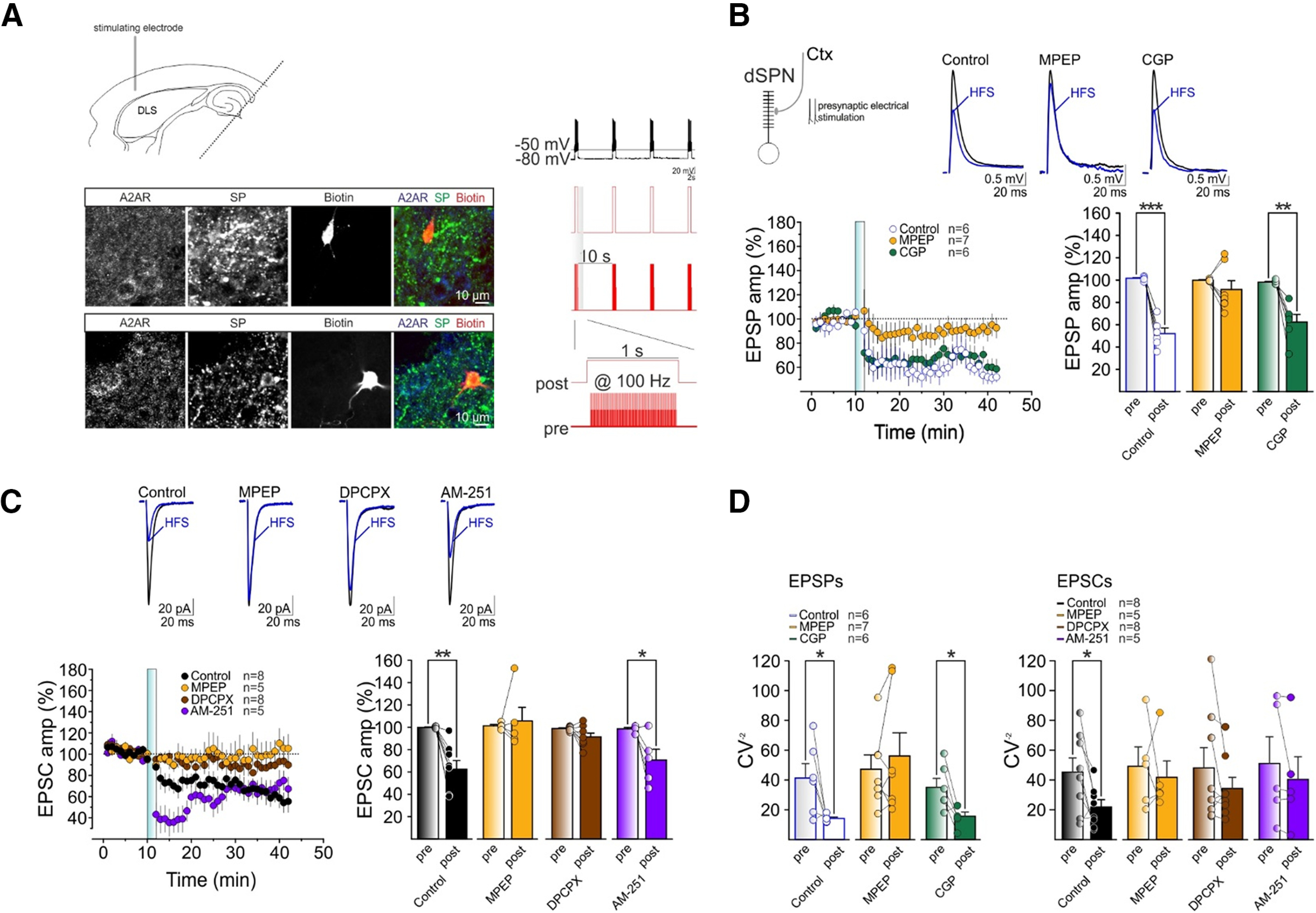
Adenosine and glutamate gate HFS-LTD at cortico-dSPN synapses. A, top left, Experimental configuration in horizontal brain slices of C57BL/6J mice. Bottom left, Confocal laser scanning microscope images of triple immunofluorescence for biotin, A2AR, and SP in patch-recorded neurons. Right, Scheme of HFS protocol. B, top left, Scheme showing glutamatergic cortical inputs to dSPNs, which are recruited following cortical electrical stimulation. Top right, Averaged recordings (10 EPSPs) before (black line) and after (blue line) HFS. Bottom left, Relative EPSP amplitude before and after HFS (vertical blue bar, as in other panels and figures) of cortical connections to dSPN in DLS in control conditions (white, n = 6, 6 mice), in the presence of the mGluR5 antagonist MPEP (yellow, n = 7, 6 mice), and in the presence of the GABABR antagonist, CGP55845 (green, n = 6, 5 mice). Bottom right, Mean normalized EPSP amplitudes before (pre) and 25–30 min after HFS (post; as in other figures) in control (white, n = 6, 6 mice, t = 9.3, df = 5, ***p = 0.0002, paired t test), MPEP (yellow, n = 7, 6 mice, t = 1, df = 6, p = 0.3, paired t test), and CGP55845 (green, n = 6, 5 mice, t = 5, df = 5, **p = 0.0038, paired t test). C, top, Averaged recordings (10 EPSCs) before (black) and after (blue) HFS. Bottom left, Relative EPSP amplitude before and after HFS in control conditions (black, n = 8, 7 mice), in the presence of MPEP (yellow, n = 5, 3 mice), the A1R antagonist DPCPX (brown, n = 8, 7 mice), and the eCB CB1R antagonist AM-251 (purple, n = 5, 3 mice). Bottom right, Mean EPSC amplitudes before and after HFS in control (black, n = 8, 7 mice, t = 4.9, df = 7, **p = 0.0018, paired t test), MPEP (yellow, n = 5, 3 mice, t = 0.4, df = 4, p = 0.7, paired t test), DPCPX (brown, n = 8, 7 mice, t = 2.2, df = 7, p = 0.06, paired t test), or AM-251 (purple, n = 5, 3 mice, t = 2.8, df = 4, *p = 0.049, paired t test). D, left, Mean CV−2 before and after HFS for the EPSP group in control conditions (white, n = 6, 6 mice, t = 2.6, df = 5, *p = 0.048, paired t test) or in the presence of MPEP (yellow, n = 7, 6 mice, W = 10, p = 0.47, Wilcoxon matched-pairs signed-rank test) or CGP (green, n = 6, 5 mice, t = 3.2, df = 5, *p = 0.024, paired t test). Right, Mean CV−2 before and after HFS delivery for the EPSC group in control conditions (black, n = 8, 7 mice, t = 2.9, df = 7, *p = 0.024, paired t test) or in the presence of MPEP (yellow, n = 5, 3 mice, W = −1, p > 0.9999, Wilcoxon matched-pairs signed-rank test), DPCPX (brown, n = 8, 7 mice, t = 1.4, df = 7, p = 0.2, paired t test), or AM-251 (purple, n = 5, 3 mice, t = 12, df = 4, p = 0.3, paired t test). Bar diagrams summarize the ratio of the SD and the mean EPSP or EPSC amplitude of the last 7 min before compared with the last 7 min after HFS delivery (as in other figures). Data are presented as mean ± SEM.
Appropriate parametric statistics were used to test our hypotheses, unless data did not meet the assumptions of the intended parametric test (normality test), in which case, appropriate non-parametric tests were used. Data were analyzed by one-way repeated-measures ANOVA (RM1WA) or a Friedman test for comparisons within a group (GraphPad Prism 6 software). Post hoc analyses (Tukey's and Dunnet multiple comparison test against control) were only performed for ANOVAs with significant main effects. For comparisons of two groups, a paired t test or Wilcoxon non-parametric test was used (GraphPad Prism 6 software). Statistical details of experiments are shown in results, figures, and figure legends. Data are expressed as mean ± SEM.
Data and code availability
Source data that support the findings of this study are available from the corresponding author on reasonable request. Custom MATLAB code for analyses of astrocyte Ca2+ are available from the corresponding author on individual request.
Results
It has been shown that HFS of deep Layer V of somatosensory cortex induces LTD at cortico-dSPN synapses in the DLS that is dependent on A1R activation (Trusel et al., 2015). In this study, we sought to identify the mechanisms mediating this A1R-dependent LTD. Since activated astrocytes have been found in several brain regions to release ATP/adenosine (Zhang et al., 2003; Pascual et al., 2005; Serrano et al., 2006; Chen et al., 2013; Covelo and Araque, 2018), we aimed to test the hypothesis that DLS astrocytes are involved in mediating this A1R-dependent plasticity.
Adenosine and glutamate gate HFS-LTD at cortico-dSPN synapses
We recorded EPSPs or EPSCs evoked by stimulation of deep Layer V of the somatosensory cortex in dSPNs, identified by immunostaining for A2AR (for iSPN) and SP (for dSPN; Fig. 1A). After baseline recordings, we applied an HFS stimulation protocol paired with postsynaptic depolarization to test synaptic plasticity changes at cortico-dSPN synapses (Fig. 1A). Consistent with previous observation (Trusel et al., 2015; Rittiner et al., 2016), HFS resulted in a persistent decrease in amplitude of EPSPs (Fig. 1B) and EPSCs (Fig. 1C). Furthermore, this persistent synaptic depression was associated with a decrease in the CV (CV−2) of EPSPs and EPSCs after HFS (Fig. 1D), which suggests a presynaptic locus of expression of plasticity (Fino et al., 2005; Trusel et al., 2015; Cavaccini et al., 2018). Taken together, these results indicate that HFS of cortico-dSPN synapses induces LTD through a presynaptic mechanism, as previously shown (Trusel et al., 2015).
We then investigated the signaling molecules involved in the HFS-LTD. First, since corticostriatal terminals release glutamate, we tested the involvement of mGluR5. In the presence of the mGluR5 antagonist MPEP (10 μm), HFS was unable to induce LTD at cortico-dSPN synapses (Fig. 1B,C), and no changes in CV−2 occurred (Fig. 1D). Therefore, mGluR5 activation is required for the induction of the HFS-LTD.
Next, since SPNs are GABAergic neurons that, once activated by excitatory inputs, release GABA, we tested whether this neurotransmitter may influence the HFS-LTD. While the contribution of GABAARs could be ruled out because the HFS-LTD was observed in the presence of their antagonists (see Materials and Methods), GABAB receptors might be involved. However, we found that HFS-LTD at cortico-dSPN synapses was unaffected by the GABABR antagonist CGP55845 (10 μm; Fig. 1B), and it was accompanied by a change in CV−2 (Fig. 1D), suggesting that GABABR activation was not involved in HFS-induced LTD.
The HFS-LTD at cortico-dSPN synapses has been reported to be mediated by A1Rs through a presynaptic mechanism (Trusel et al., 2015). Present results agree with previous reports, showing that the HFS-LTD was absent in the presence of the A1R antagonist DPCPX (500 nm; Fig. 1C), and no associated CV−2 changes occurred (Fig. 1D).
Finally, we tested the contribution of CB1Rs. We found that HFS-LTD still occurred in the presence of the CB1R antagonist AM-251 (4 μm; Fig. 1C), suggesting that CB1Rs do not contribute to this form of HFS-LTD, as previously shown (Trusel et al., 2015). Taken together, these results indicate that cortical HFS induces a presynaptic A1R-dependent and mGluR5-dependent LTD at cortico-dSPN synapses.
Cortical HFS enhances Ca2+ transients in DLS astrocytes through mGluR5 activation
Because the HFS-LTD at cortico-dSPN synapses depended on A1Rs (Fig. 1; Trusel et al., 2015; Spigolon et al., 2018) and astrocytes are known to regulate synaptic transmission and plasticity through the release of ATP/adenosine in different brain areas (Araque et al., 2014), we then investigated astrocyte involvement in mediating this form of plasticity. We first monitored astrocyte Ca2+ activity using a viral injection into the DLS of the genetically-encoded cytosolic Ca2+ indicator GCaMP6f under the astrocyte GfaABC1D promoter (AAV5-GfaABC1D-cytoGCaMP6fSV40; Fig. 2A). The astrocyte Ca2+ signal activity was quantified as Ca2+ event probability (measured as the occurrence of Ca2+ events per min). In control conditions (Control), cortical HFS increased Ca2+ event probability during and immediately following the stimulation period compared with baseline (Fig. 2B,D). In a separate control experiment (Control_2), we found that the increase in Ca2+ event probability returned to baseline 12 min after HFS (Post_12; Fig. 2D), indicating that striatal astrocytes sensed the high-frequency activity of corticostriatal inputs.
Figure 2.
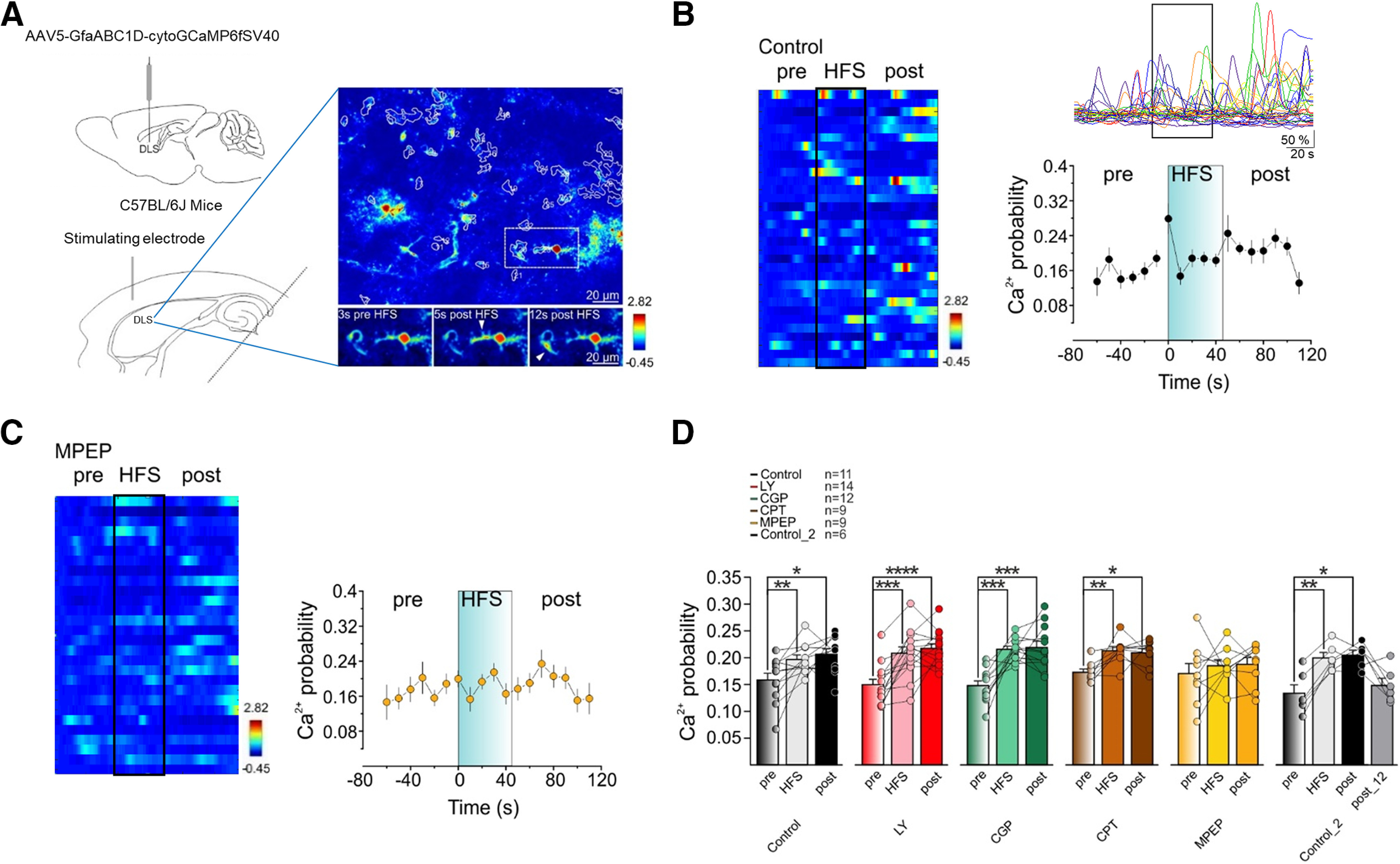
Cortical HFS enhances Ca2+ transients in DLS astrocytes through mGluR5 activation. A, top left, C57Bl/6J mice received bilateral DLS injection of the genetically-encoded cytosolic Ca2+ indicator GCaMP6f under the astrocyte GfaABC1D promoter (AAV5.G, faABC1D.cytoGCaMP6f.SV40). Bottom left, Scheme of experimental configuration for two-photon Ca2+ imaging measurements in DLS astrocytes of horizontal brain slices. Right, Representative image of ROIs automatically generated by CALSEE in control conditions. B, Representative heatmap (left) and traces (top right) of Ca2+ fluorescence from ROIs (y-axis in left) before (pre), during (HFS, black box, as in other panels), and after (post) HFS delivery in control conditions. Bottom right, Time course of Ca2+ event probability averaged in 10-s bins in control conditions before, during, and after HFS delivery. C, left, Representative heatmap of Ca2+ traces from ROIs (y-axis) before, during, and after HFS delivery in the presence of MPEP. Right, Time course of Ca2+ event probability averaged in 10-s bins in MPEP group before, during, and after HFS delivery. D, Mean Ca2+ event probability 60 s before HFS (pre), during HFS (HFS), 70 s after HFS (post), and 12 min after HFS delivery (Post_12) in different pharmacological conditions (black, control, n = 11, 5 mice, pre vs HFS vs post, RM1WA, F(10,20) = 9.082, **p = 0.0019, Dunnett; red, LY, n = 14, 3 mice, RM1WA, F(13,26) = 24.27, ****p < 0.0001, Dunnett; green, CGP, n = 12, 2 mice, RM1WA, F(11,22) = 24.35, ****p < 0.0001, Dunnett; brown, CPT, n = 9, 2 mice, RM1WA, F(8,16) = 8.736, **p = 0.003, Dunnett; yellow, MPEP, n = 9, 2 mice, RM1WA, F(8,16) = 0.6, p = 0.5; black, Control_2, n = 6, 2 mice, RM1WA, F(5,15) = 14.63, **p = 0.0029, Dunnett). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Then, we used pharmacological tools to investigate the signaling mechanisms underlying astrocyte activation. The HFS-induced increase in Ca2+ event probability persisted in thepresence of the mGluR1 antagonist LY367385 (100 μm), the GABABR antagonist CGP54626 (1 mm), and the A1R antagonist CPT (2 μm; Fig. 2D). In contrast, when mGluR5 was blocked with MPEP (50 μm), HFS did not increase Ca2+ event probability (Fig. 2C,D). Therefore, in agreement with numerous previous studies demonstrating that astrocytes express mGluR5 that on activation elevate astrocytic Ca2+ in different brain areas (Porter and McCarthy, 1996; Perea and Araque, 2005; Wang et al., 2006a; Di Castro et al., 2011; Panatier et al., 2011), these results suggest that the DLS astrocytic responses to cortical HFS are mediated by mGluR5.
Blocking astrocyte Ca2+ activity prevents HFS-LTD at dSPN synapses
Given that both HFS-LTD and astrocyte responses to HFS depended on mGluR5 (Figs. 1, 2, respectively), we hypothesized that astrocytes are an integral component mediating the HFS-LTD. Thus, we next asked whether blocking astrocyte activity would influence HFS-LTD. Since mGluR5 are GPCRs, whose activation mobilizes Ca2+ through an IP3-mediated mechanism (Nakahara et al., 1997; D'Ascenzo et al., 2007), we applied two complementary strategies. First, we used IP3R2 knock-out mice (Li et al., 2005), in which IP3R2-mediated Ca2+ signaling in astrocytes is largely absent (Fig. 3B; Durkee et al., 2019). Second, we specifically blocked astrocyte GPCR signaling by loading the astrocytic syncytium with the G-protein inhibitor GDPβS (10 μm; Fig. 3B). As suggested by the spreading of the fluorescent signal across cells on patch-loading an astrocyte with biocytin (Fig. 3B), astrocytes in the DLS are gap-junction coupled to form an astrocyte syncytium. Both the HFS-evoked Ca2+ signaling (Fig. 3A–D) and the HFS-LTD (Fig. 3E–H) were prevented in IP3R2−/− mice and in mice with GDPβS-loaded astrocyte syncytium.
Figure 3.
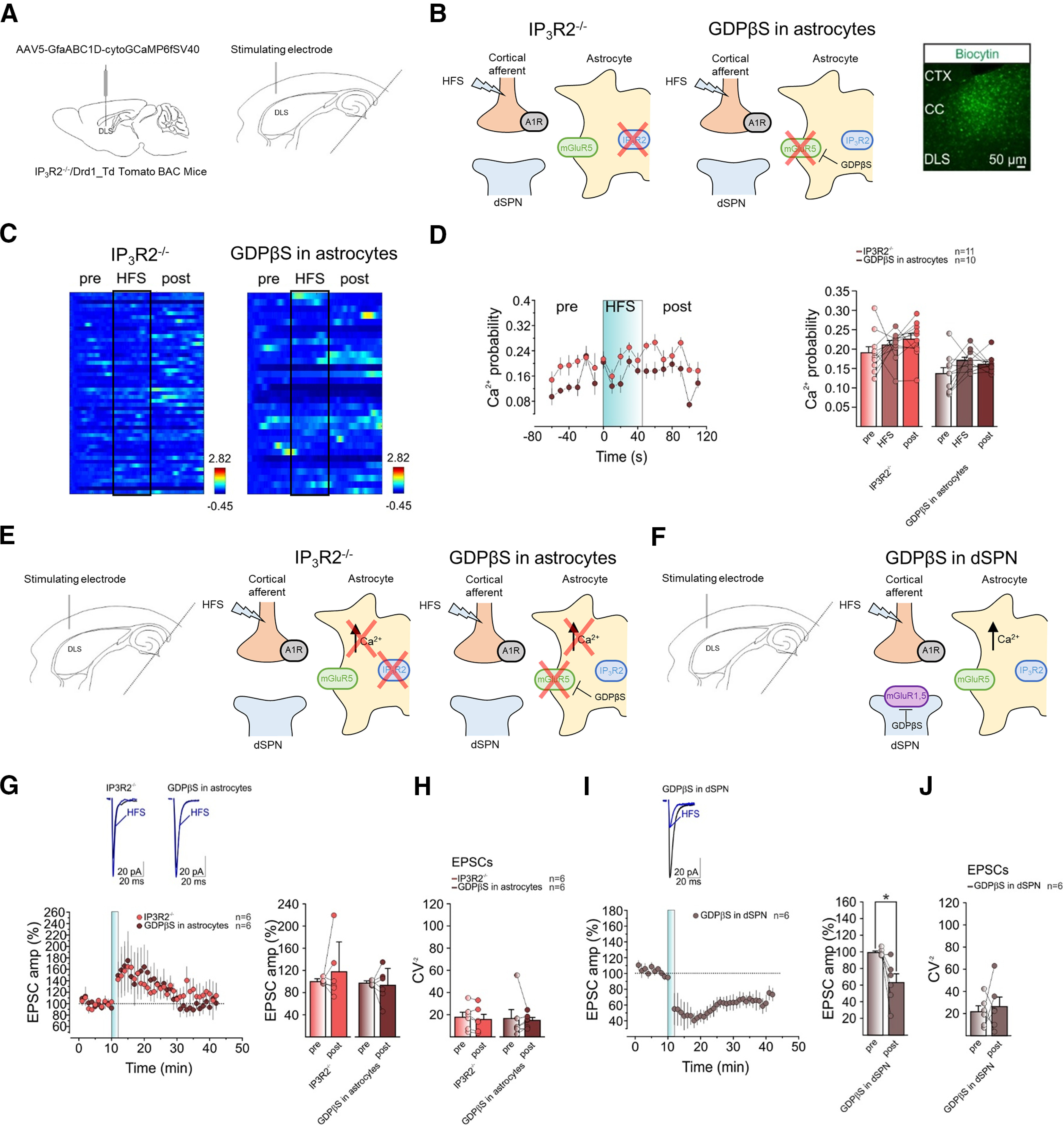
Blocking astrocyte Ca2+ activity prevents HFS-LTD at cortico-dSPN synapses. A, left, IP3R2 knock-out mice (IP3R2−/−) and Drd1-Td Tomato BAC mice received bilateral DLS injection of the genetically-encoded cytosolic Ca2+ indicator GCaMP6f under the astrocyte GfaABC1D promoter (AAV5-GfaABC1D-cytoGCaMP6fSV40). Right, Schematic of experimental configuration for two-photon Ca2+ imaging measurements in DLS astrocytes of horizontal brain slices. B, Scheme of the molecular mechanisms regulating HFS-LTD at tripartite synapses in IP3R2−/− (left) and in Drd1-Td Tomato mice with GDPβS patch-loaded astrocytes (middle). Right, Image of immunofluorescence for biocytin of biocytin-loaded astrocyte syncytium in DLS. C, Representative heatmaps of Ca2+ traces from ROIs before, during, and after HFS delivery in IP3R2−/− (left) and in Drd1-Td Tomato mice with GDPβS patch-loaded astrocyte syncytium (right). D, Left, Time course of Ca2+ event probability before, during, and after HFS in IP3R2−/− (red, n = 11, 2 mice) and in GDPβS patch-loaded astrocyte syncytium (brown, n = 10, 2 mice). Right, Mean Ca2+ event probability before, during, and after HFS in IP3R2−/− (red, n = 11, 2 mice, pre vs HFS vs post, RM1WA, F(10,20) = 1.897, p = 0.2) and in GDPβS patch-loaded astrocyte syncytium (brown, n = 10, 2 mice, pre vs HFS vs post, Friedman test, p = 0.1). E, Scheme of experimental configuration (left) and the molecular mechanisms regulating HFS-LTD at tripartite synapses in IP3R2−/− (middle) and in Drd1-Td Tomato mice with GDPβS patch-loaded astrocytes (right). F, Scheme of experimental configuration (left) and the molecular mechanisms regulating HFS-LTD at tripartite synapses in C57Bl/6J mice with GDPβS patch-loaded dSPN (right). G, top, Averaged recordings (10 EPSCs) before (black) and after (blue) HFS. Bottom left, Relative EPSC amplitudes before and after HFS in IP3R2−/− (red, n = 6, 5 mice) and in Drd1-Td Tomato mice with GDPβS patch-loaded astrocyte syncytium (brown, n = 6, 4 mice). Bottom right, Mean normalized EPSC amplitudes before and after HFS in IP3R2−/− (red, n = 6, 5 mice, t = 0.8, df = 5, p = 0.5, paired t test) and GDPβS in astrocytes (brown, n = 6, 4 mice, t = 0.3, df = 5, p = 0.8, paired t test). H, Mean CV−2 before and after HFS in IP3R2−/− (red, n = 6, 5 mice, t = 1.7, df = 5, p = 0.16, paired t test) and in GDPβS in astrocytes (brown, n = 6, 4 mice, W = 7, p = 0.56, Wilcoxon matched-pairs signed-rank test). I, top, Averaged recordings (10 EPSCs) before (black) and after (blue) HFS. Bottom left, Relative EPSC amplitudes before and after HFS in C57Bl/6J mice with GDPβS patch-loaded dSPN (gray, n = 6, 4 mice). Bottom right, Mean normalized EPSC amplitudes before and after HFS in GDPβS patch-loaded dSPN (gray, n = 6, 4 mice, t = 3.3, df = 5, *p = 0.02, paired t test). J, Mean CV−2 before and after HFS in GDPβS patch-loaded dSPN (gray, n = 6, 4 mice, t = 0.4, df = 5, p = 0.7, paired t test). Data are presented as mean ± SEM.
To test the possibility of indirect effects of chronic global IP3R2 deletion on the potential to induce LTD in IP3R2−/− mice, we assessed basal synaptic transmission properties in IP3R2−/− and wild-type (IP3R2+/+) littermates (Fig. 4). We found that the input-output relationship of corticostriatal stimulation and evoked EPSC amplitude revealed no significant differences between wild-type and IP3R2−/− mice at the stimulation intensities tested (Fig. 4B). Furthermore, both the amplitude and frequency of miniature EPSCs were unaffected in IP3R2−/− mice (Fig. 4C). The unaltered basal synaptic transmission in IP3R2−/− mice suggests that the observed absence of HFS-LTD was not due to changes in synaptic transmission properties, but to the astrocyte signaling impairment. Altogether, these results indicate that IP3R2-mediated and GPCR-mediated astrocyte Ca2+ signaling was required for eliciting the HFS-LTD.
Figure 4.

Basal synaptic transmission is not affected in IP3R2−/− mice. A, Experimental configuration in horizontal brain slices of IP3R2−/− and wild-type (WT) littermates. B, Changes in EPSC amplitude evoked by different stimulus intensities in IP3R2−/− (light red, n = 5, 2 mice) and WT littermates (red, n = 5, 2 mice; F(1,5) = 86.10, RM1WA, p = 0.07). C, left, Representative traces of miniature EPSCs (mEPSCs) in IP3R2−/− (bottom) and WT littermates (top). Right, Mean mEPSC amplitude (pA) in IP3R2−/− (red, n = 6, 2 mice) and WT littermates (light red, n = 6, 2 mice, t = 0.00,019, df = 10, p = 0.99, t test) and mean mEPSC/min in IP3R2−/− (red, n = 6, 2 mice) and WT littermates (light red, n = 6, 2 mice, t = 0.07, df = 10, p = 0.94, t test).
To further test this idea, we blocked the postsynaptic neuronal GPCR signaling by patch-loading dSPN with GDPβS (10 μm) and found that this manipulation had no effect on HFS-LTD (Fig. 3I,J), thus corroborating that astrocytic, not postsynaptic neuronal, GPCR signaling is necessary for HFS-LTD at cortico-dSPN synapses. Taken together, our data suggest that HFS evokes the release of cortical glutamate that activates DLS astrocytes, through mGluR5 activation (see above), resulting in a GPCR-mediated and IP3R2-mediated Ca2+ increase that is necessaryto induce HFS-LTD at cortico-dSPN synapses.
Astrocyte Gq DREADD activation induces A1R-dependent LTD at dSPN synapses
To investigate whether directly activating astrocytes is sufficient for inducing the depression of EPSC amplitude at cortico-dSPN synapses, we used a Gq DREADDs chemogenetic approach (Fig. 5). This approach has previously been shown to reliably elevate astrocyte Ca2+ in a phospholipase C (PLC)-dependent and IP3R2-dependent manner on application of the DREADD ligand CNO (Chai et al., 2017; Martin-Fernandez et al., 2017; Durkee et al., 2019). We injected in the DLS the virus AAV8-GFAP-hM3Dq-mCherry that contains the Gq DREADD hM3Dq, the astroglial promoter GFAP and the reporter mCherry (Fig. 5A). First, we confirmed through immunohistochemistry (Fig. 5B; n = 3 mice) that expression of the virus was largely specific to astrocytes (GFAP+ cells; penetrance: 99.6%, 1010 cells), with negligible penetrance in neurons (3.3% of NeuN+ cells, 1661 cells), microglia (1.2% of Iba+ cells, 81 cells; data not shown), NG2 glia (0% of NG2+ cells, 28 cells; data not shown), and oligodendrocytes (1.8% of Oligo 2+ cells, 281 cells; data not shown).
Figure 5.
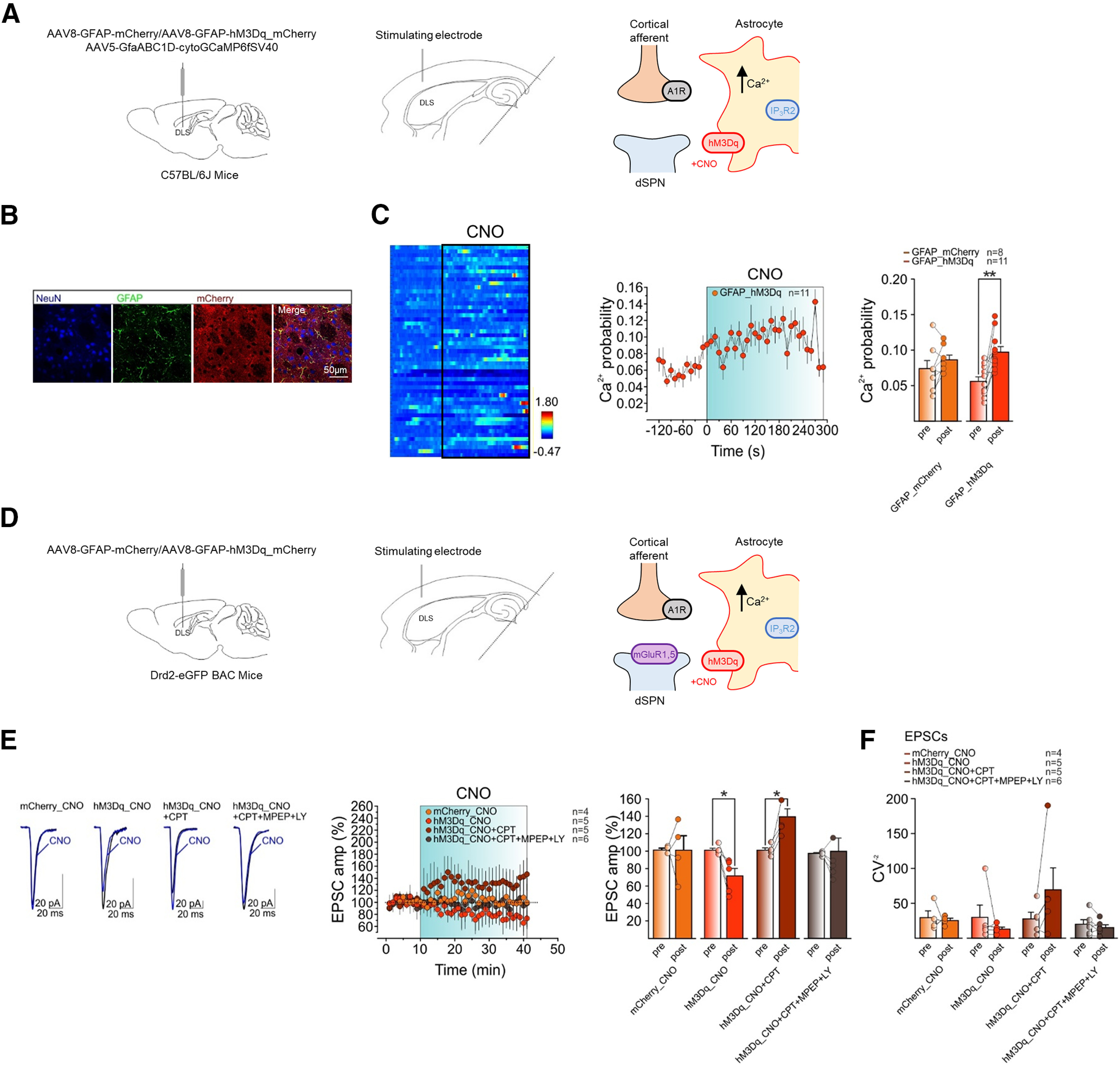
Astrocyte Gq DREADD activation induces A1R-dependent LTD at cortico-dSPN synapses. A, left, C57BL/6J mice received bilateral DLS injection of AAV5-GfaABC1D-cytoGCaMP6fSV40 along with either AAV8-GFAP-mCherry (control condition) or AAV8-GFAP-hM3Dq-mCherry virus. Middle, Schematic of experimental configuration for two photon Ca2+ imaging measurements in DLS astrocytes of horizontal brain slices. Right, Scheme of the molecular mechanisms regulating synaptic plasticity at tripartite synapses in C57Bl/6J mice with astrocyte-specific Gq DREADDs. B, Confocal laser scanning microscope image of immunohistochemistry showing colocalization of hM3Dq-mCherry with the astrocytic marker GFAP, but not with the neuronal marker NeuN. C, left, Representative heatmap of Ca2+ traces from ROIs before and during CNO bath application in mice expressing AAV8-GFAP-hM3Dq-mCherry. Middle, Time course of Ca2+ event probability before and after (last 4 min) CNO bath application in mice expressing AAV8-GFAP-hM3Dq-mCherry. Right, mean Ca2+ event probability before (pre) and after (post) CNO bath application in mice expressing AAV8-GFAP-mCherry (control condition; orange, n = 8, 2 mice, t = 1.5, df = 7, p = 0.2, paired t test) or AAV8-GFAP-hM3Dq-mCherry (red, n = 11, 2 mice, t = 4, df = 10, **p = 0.002, paired t test). D, left, Drd2_eGFP mice received bilateral DLS injection of AAV8-GFAP-mCherry (control condition) or AAV8-GFAP-hM3Dq-mCherry virus. Middle, Scheme of experimental condition. Right, Scheme of the molecular mechanisms regulating synaptic plasticity at tripartite synapses in C57Bl/6J mice with astrocyte-specific Gq DREADDs. E, left, Averaged recordings (10 EPSCs) before (black) and after (blue) CNO bath application. Middle, Relative EPSC amplitudes before and after CNO bath application in mice expressing AAV8-GFAP-mCherry (orange, n = 4, 3 mice), or in mice expressing AAV8-GFAP-hM3Dq-mCherry in control (red, n = 5, 5 mice), in CPT (maroon, n = 5, 4 mice), or in CPT plus MPEP and LY (gray, n = 6, 4 mice). Right, Mean normalized EPSC amplitudes before and after CNO in mice expressing AAV8-GFAP-mCherry (orange, n = 4, 3 mice, W = 0, p > 0.9999, Wilcoxon matched-pairs signed-rank test), or in mice expressing AAV8-GFAP-hM3Dq-mCherry in control (red, n = 5, 5 mice, t = 3.6, df = 4, *p = 0.02, paired t test), in CPT (maroon, n = 5, 4 mice, t = 4.3, df = 4, *p = 0.01, paired t test) or in CPT plus MPEP and LY (gray, n = 6, 4 mice, t = 0.2, df = 5, p = 0.9, paired t test). F, Mean CV−2 before and after CNO in mice expressing AAV8-GFAP-mCherry (orange, n = 4, 3 mice, W = 0, p > 0.9999, Wilcoxon matched-pairs signed-rank test), in mice expressing AAV8-GFAP-hM3Dq-mCherry in control (red, n = 5, 5 mice, W = −7, p = 0.44, Wilcoxon matched-pairs signed-rank test), in CPT (maroon, n = 5, 4 mice, W = 13, p = 0.13, Wilcoxon matched-pairs signed-rank test), or in CPT plus MPEP and LY (gray, n = 6, 4 mice, t = 1.3, df = 5, p = 0.26, paired t test). Data are presented as mean ± SEM.
In a region of DREADD-expressing astrocytes, we observed that CNO application (10 μm) increased astrocyte Ca2+ event probability (Fig. 5C). This effect was absent in control mice injected with the virus AAV8-GFAP-mCherry (i.e., lacking the Gq DREADD; Fig. 5C). Next, we asked whether this CNO-induced increase in astrocyte Ca2+ levels would suppress EPSCs in an adenosine A1R-dependent manner similar to that observed for HFS-evoked LTD. We found that in mice with DREADD-expressing astrocytes, CNO perfusion for 30 min induced depression of EPSC amplitude (Fig. 5D,E), whereas this effect was absent in control mice injected with AAV8-GFAP-mCherry (Fig. 5E). Furthermore, in the presence of the A1R antagonist CPT (2 μm), not only was the CNO-induced LTD prevented in mice with DREADD-expressing astrocytes but the EPSC amplitude was surprisingly potentiated (Fig. 5E). This CNO-induced potentiation occurred with no significant changes in CV−2 and was abolished by antagonizing A1Rs with CPT and Group I mGluRs with MPEP (mGluR5) and LY367385 (mGluR1; Fig. 5E,F).
We then tested whether the CNO-evoked synaptic depression occluded the HFS-LTD (Fig. 6). We first applied CNO, which persistently depressed the EPSC amplitude (Fig. 6B,C; compare with Fig. 5E). Indeed, EPSC amplitudes recorded 25–30 min after CNO application (Post-CNO) were significantly reduced relative to 5 min of baseline (Pre-CNO; Fig. 6B,C). Then, 30 min after applying CNO, we delivered the HFS protocol, which still induced LTD manifested as a further depression of the EPSC amplitude (Fig. 6B,C). Indeed, the amplitude of EPSCs recorded 15–20 min after HFS (Post-CNO_Post-HFS) was significantly depressed from amplitudes recorded 5 min before the HFS (i.e., after CNO; Post-CNO_Pre-HFS; Fig. 6B,C). Since HFS still depressed EPSC amplitude after the initial CNO-evoked EPSC depression, these results indicate that HFS-LTD was not occluded by the previous depression induced by CNO, suggesting that the mechanisms underlying CNO-induced LTD were not saturated or that additional mechanisms triggered by CNO may be present (see Discussion).
Figure 6.
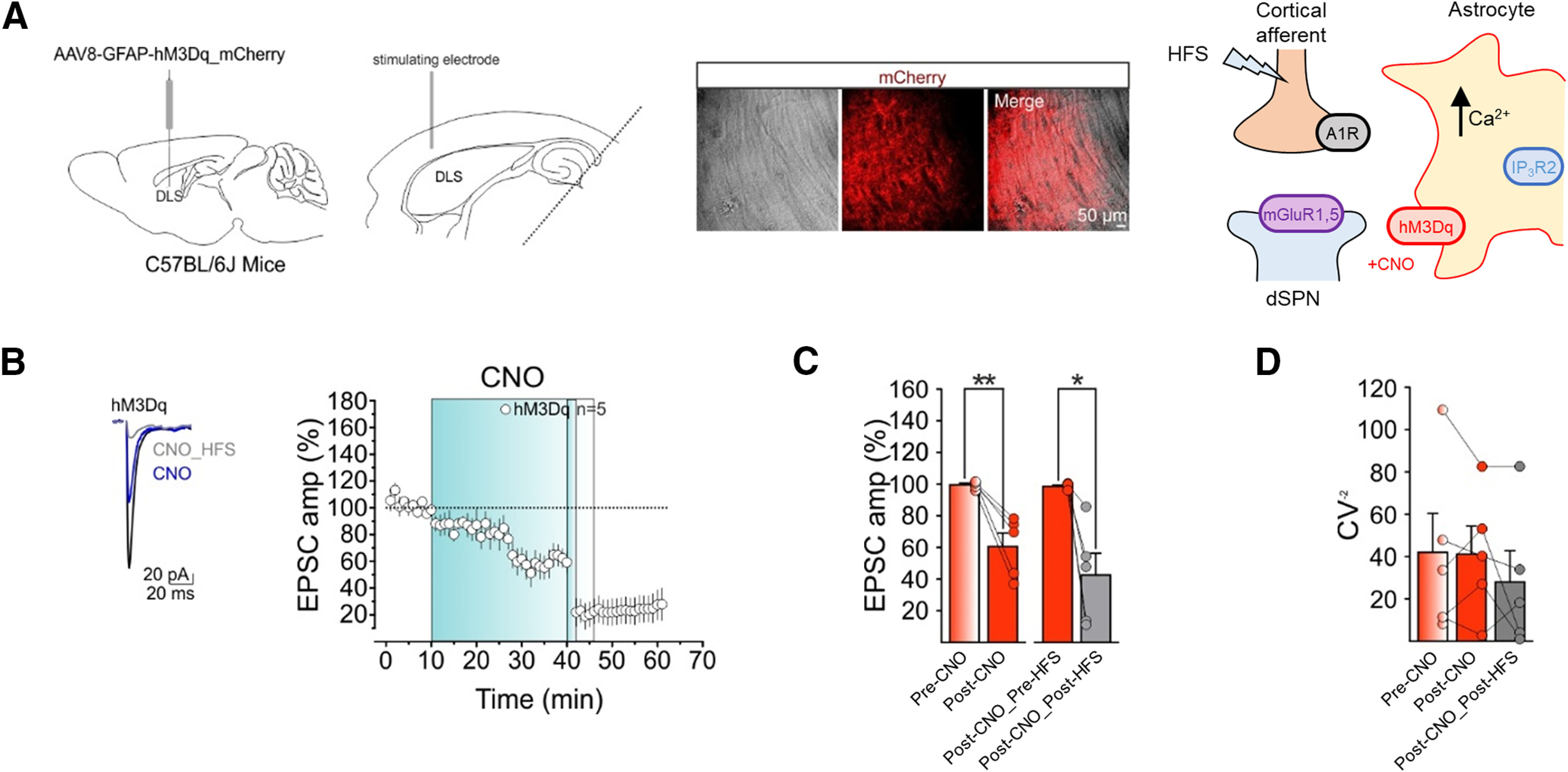
CNO-induced synaptic depression does not occlude HFS-LTD. A, left, C57BL/6J mice received bilateral DLS injection of AAV8-GFAP-hM3Dq-mCherry. Middle left, Experimental configuration in horizontal brain slices. Middle right, Bright field image (left) and confocal laser scanning microscope image (middle) showing the expression of mCherry in the DLS. Right, Scheme of the molecular mechanisms regulating synaptic plasticity at tripartite synapses in C57Bl/6J mice with astrocyte-specific Gq DREADDs. B, Left, Averaged recordings (10 EPSCs) before CNO bath application (black), after CNO bath application (blue), and after CNO+HFS (gray). Right, Relative EPSC amplitudes (relative to Pre-CNO) before and after CNO bath application and subsequent HFS in C57BL/6J mice expressing AAV8-GFAP-hM3Dq-mCherry. C, Relative mean EPSC amplitudes 5 min before (Pre-CNO) and 25–30 min after CNO (Post-CNO; red, n = 5, 3 mice; Pre-CNO vs Post-CNO, t = 4.8, df = 4, **p = 0.009, paired t test) and relative mean EPSCs amplitudes 5 min before (Post-CNO_Pre-HFS) and 15–20 min after HFS (Post-CNO_Post-HFS; silver, n = 5, 3 mice; Post-CNO_Pre-HFS vs Post-CNO_Post-HFS, t = 4.1, df = 4, *p = 0.015, paired t test). D, Mean CV−2 before (orange) and after (red) CNO application and subsequent HFS (silver; n = 5, 3 mice, RM1WA F(4,8) = 0.87, p = 0.45). All data are presented as mean ± SEM.
Discussion
The present results propose that astrocytes are integral to HFS-induced LTD at cortico-dSPN synapses in the DLS. We found that striatal astrocytes respond to HFS with Ca2+ elevations dependent on mGluR5, G-protein signaling, and IP3R2 activation. Blocking signaling from any of those sources prevented HFS-LTD, suggesting that astrocytic mGluR5-mediated GPCR signaling and internal release of Ca2+ via IP3R2 are required for the induction of LTD. Furthermore, activating striatal astrocytes with Gq DREADDs in the absence of cortical HFS mimicked the LTD in an A1R-dependent manner. Since astrocytes are able to release ATP/adenosine in different brain areas (Volterra and Meldolesi, 2005; Araque et al., 2014), present results suggest that astrocytes may be the origin of ATP/adenosine that mediates this form of striatal LTD (Trusel et al., 2015; Spigolon et al., 2018). These findings better elucidate the full complement of astrocyte function on synaptic plasticity in the DLS.
Adenosine and glutamate gate HFS-LTD at direct corticostriatal synapses
We report here that HFS-induced LTD at cortico-dSPN synapses is mediated by A1Rs, which is consistent with recent observation (Trusel et al., 2015). A1R is expressed in many brain regions at glutamatergic terminals and, given that it is Gi/o coupled, its activation results in a decrease in glutamate release (Fredholm et al., 2005). Interestingly, while several studies implicated adenosine/A1R signaling in striatal plasticity (Lovinger and Choi, 1995; Ciruela et al., 2006; Borycz et al., 2007), the adenosine origin remained undefined. In the hippocampus, adenosine signaling was originally shown to mediate heterosynaptic depression via presynaptic A1R-mediated suppression of glutamate release (Manzoni et al., 1994), but the mechanisms upstream of A1R activation were unknown. Later studies demonstrated that hippocampal heterosynaptic depression is mediated by astrocytic-derived adenosine (Zhang et al., 2003; Pascual et al., 2005; Serrano et al., 2006). Present results suggest that astrocytic release of ATP/adenosine might be a general mechanism involved in synaptic depression. Further studies in other brain areas are required to test this exciting and likely possibility.
In addition to requiring A1R, HFS-LTD was also dependent on mGluR5 activation. This finding is consistent with previous reports showing that activation of Group I mGluRs can mediate both LTD and LTP in the striatum (Sung et al., 2001; Gubellini et al., 2003; Lüscher and Huber, 2010). The mechanisms underlying striatal LTD are largely disputed, particularly regarding LTD at cortico-dSPN versus cortico-iSPN synapses, but in general, it is thought that postsynaptic Group I mGluR and L-type Ca2+ channel activation results in the mobilization of eCBs that bind presynaptic CB1Rs to suppress glutamate release (Gerdeman et al., 2002; Kreitzer and Malenka, 2007; Atwood and Lovinger, 2017). Studies have shown that pharmacological activation of Group I mGluRs is sufficient to induce eCB-mediated LTD and that blocking Group I mGluRs prevents LTD induction (Calabresi et al., 1999; Sung et al., 2001; Kreitzer and Malenka, 2007; Shen et al., 2008; Yin et al., 2008; Chepkova et al., 2009; Fino and Venance, 2010; Trusel et al., 2015). In the present study, the mGluR5 antagonist MPEP indeed blocked both the HFS-induced LTD and the striatal astrocyte responses. Interestingly, Trusel and colleagues found that HFS-LTD at cortico-dSPN synapses was not dependent on postsynaptic Ca2+ or on eCB signaling, the latter of which was also found in the present study (Trusel et al., 2015). Furthermore, we also found that blocking dSPN GPCR signaling did not affect HFS-LTD. Together, these data argue against the requirement for postsynaptic neuronal Group I mGluR-mediated Ca2+ and eCB signaling to induce LTD at cortico-dSPN synapses.
Corticostriatal HFS activates astrocytes via mGluR5
In addition to being expressed neuronally, mGluR5 is also expressed on striatal astrocytes and results in gliotransmitter release (Biber et al., 1999; D'Ascenzo et al., 2007; Bradley and Challiss, 2012; Tamura et al., 2014). Despite controversy regarding mGluR5-dependent Ca2+ signaling in adult mice (Sun et al., 2013), a number of subsequent studies have observed such signaling (Tamura et al., 2014; Chai et al., 2017; Durkee et al., 2019). Indeed, we found that blocking mGluR5 prevented both the HFS-induced astrocyte response and LTD. Although we did not specifically interrogate mGluR5 signaling in astrocytes and contribution of neuronal mGluR5 cannot be ruled out, the fact that astrocytic mGluR5 activation elevates astrocytic Ca2+ in different brain areas (Porter and McCarthy, 1996; Perea and Araque, 2005; Wang et al., 2006a; Di Castro et al., 2011; Panatier et al., 2011; Covelo and Araque, 2018) suggests that present results are indicative of astrocytic mGluR5 involvement in HFS-LTD. Antagonizing mGluR1 did not influence the astrocytic response but has previously been found to prevent HFS-LTD (Gubellini et al., 2003), suggesting that neuronal mGluR1 and astrocytic mGluR5 may work in concert to induce HFS-LTD. Of note, GABAB receptors, which have been found to mediate hippocampal heterosynaptic depression (Zhang et al., 2003; Pascual et al., 2005; Serrano et al., 2006; Covelo and Araque, 2018), are not involved in the astrocyte response to HFS and HFS-LTD. Importantly, while A1R antagonism blocked HFS-LTD, we found that it did not block the astrocytic Ca2+ response, indicating that A1R activation is downstream of astrocyte activation. Taken together, these data suggest that A1R-mediated HFS-LTD occurs following mGluR5-mediated astrocyte activation.
Blocking astrocyte GPCR and IP3R2 signaling prevents HFS-LTD
To more directly investigate astrocyte involvement in HFS-LTD, we prevented IP3R2-mediated Ca2+ signaling in astrocytes with IP3R2−/− mice or blocked GPCR signaling via patch-loading GDPβS into the astrocyte syncytium. In these conditions, HFS did not increase astrocyte Ca2+ signaling nor did it produce LTD. These data suggest that blocking the astrocyte Ca2+ response to cortical HFS prevents a form of astrocyte-neuron signaling that is necessary for the observed A1R-mediated suppression of glutamate release. Taken together, these data show that disrupting Ca2+ signaling in astrocytes prevents HFS-LTD at cortico-dSPN synapses, suggesting that LTD is mediated by presynaptic A1R and that the source of adenosine appears to be astrocytic.
Activating astrocytes with Gq DREADDs depresses synaptic strength at cortico-dSPN synapses
We found that Gq DREADD activation in astrocytes persistently depressed EPSC amplitude through A1R activation (Fig. 5E). Furthermore, in the presence of the A1R antagonist CPT, CNO application in Gq DREADD-expressing astrocytes induced a long-lasting synaptic potentiation that was sensitive to Group I mGluR antagonists (Fig. 5E). Interestingly, it has been reported in the DLS that astrocyte Ca2+ elevations induced by eCBs or Ca2+ uncaging stimulate the release of glutamate that increases synaptic transmission at distant synapses through activation of Group I mGluRs (Martín et al., 2015). Thus, astrocytes may potentially release both ATP/adenosine and glutamate with Gq DREADD activation, each of which would oppose the effect of the other on corticostriatal glutamate release. Indeed, it was recently demonstrated in the hippocampus that low levels of stimulation activated single astrocytes to release glutamate and that higher levels of stimulation resulted in corelease of bothglutamate and ATP/adenosine, resulting in a biphasic effect on synaptic transmission: an initial Group I mGluR-mediated potentiation and a secondary A1R-mediated depression (Covelo and Araque, 2018). Accordingly, Gq DREADD activation in the present study may have led to release of both glutamate and ATP/adenosine, whereby the adenosine-mediated depression was attenuated by glutamate-mediated potentiation. In line with this, the A1R antagonist CPT blocked the CNO-induced depression and instead revealed an enhancement of synaptic transmission that was dependent on Group I mGluRs. It is interesting that HFS-evoked astrocyte activation did not appear to induce an initial glutamate-mediated potentiation followed by adenosine-mediated depression. Nor did HFS in the presence of the A1R antagonist result in LTP. Furthermore, present results show that HFS-LTD was not occluded by the previous synaptic depression induced by CNO (Fig. 6B,C), suggesting that the mechanisms underlying LTD were not saturated. Alternatively, CNO activation of Gq DREADD-expressing astrocytes may trigger additional mechanisms involving alternative signaling pathways. Further studies, out of the scope of the present work, are required to determine the potential contribution of astrocytic glutamate, which is released from astrocytes activated by eCBs or Ca2+ uncaging (Martín et al., 2015). As shown in other brain areas, different modalities of astrocyte activation have differential effects on gliotransmission and on its synaptic regulatory effects (Covelo and Araque, 2018; Savtchouk and Volterra, 2018; Durkee and Araque, 2019).
Taken together, these data suggest that chemogenetically-activated DLS astrocytes have the potential to regulate extrasynaptic levels of both glutamate and ATP/adenosine, with sustained chemogenetic activation resulting in the predominance of adenosine-mediated inhibition over glutamate-mediated potentiation. HFS-induced astrocyte activation, however, led only to the A1R-mediated synaptic depression. Whether this is due to a predominance of adenosine-mediated over glutamate-mediated effects or whether glutamate-mediated regulation occurs at relatively distant heterosynapses (Martín et al., 2015), and which were not monitored in the present study, will be a matter of future investigations. Nevertheless, the present results add to the complexity of bidirectional astrocyte-neuron signaling, showing the capability of astrocytes to respond to different stimuli that may trigger different downstream signaling effects on gliotransmitter release and synaptic regulation (Perea and Araque, 2005; Shigetomi et al., 2008).
In summary, present data show that astrocyte Ca2+ signaling is necessary for A1R-mediated LTD at cortico-dSPN synapses in the DLS. First, striatal astrocytes respond to cortical HFS with Ca2+ increases mediated by mGluR5, GPCR, and IP3R2 signaling. Second, blocking astrocyte signaling prevented LTD induction. Lastly, activating astrocytes induced LTD at cortico-dSPN synapses in a A1R-dependent manner. Thus, since astrocytes have been shown to respond in different brain areas to glutamatergic signaling and to release ATP/adenosine (Araque et al., 2014; Gundersen et al., 2015; Illes et al., 2019), we propose that astrocyte mGluR5-mediated Ca2+ signaling and downstream release of ATP/adenosine levels are integral to LTD at cortico-dSPN synapses (Fig. 7). These results reveal a key role of astrocytes in striatal synaptic plasticity.
Figure 7.

Working model of astrocyte-mediated LTD at cortico-dSPN synapses. HFS of deep Layer V of somatosensory cortex (1) induces glutamate release from cortical afferents that activates mGluR5 on astrocytes (2), leading to GPCR-IP3R2-mediated Ca2+ increases (3), and extracellular accumulation of astrocyte-derived adenosine (4). Adenosine activates A1R located on cortical afferents to suppress subsequent glutamate release, resulting in LTD (5).
Footnotes
This work was supported by the Fondazione Istituto Italiano di Tecnologia (R.T.), the Compagnia di San Paolo Grant 2013 0942 (to R.T.), the Fondazione Cariplo Grant 2013 0871 (to R.T.), the National Institutes of Health (NIH)-National Institute of Neurological Disorders and Stroke (NINDS) Grant R01NS097312 (to A.A.), the NIH-National Institute on Drug Abuse Grant R01DA048822 (to A.A.), and the NIH-NINDS Grant F31NS93751 (to C.D.). We thank Justin Lines for the CALSEE program for Ca2+ analysis; Kendrick Kay, Justin Lines, and Mario Martin-Fernandez for MATLAB and CALSEE assistance; Mark Sanders, Guillermo Marques, and Jason Mitchell at the University of Minnesota–University Imaging Centers for assistance using the Leica SP5 Multiphoton Confocal Upright Microscope; Alice Gino and Luca Nava at the Istituto Italiano di Tecnologia for technical assistance during immunohistochemistry and stereotaxic surgery; Ju Chen for generously donating the IP3R2−/− mice; and the University of North Carolina at Chapel Hill Vector Core for providing the DREADDs viruses. Viral vectors used in this study were prepared by the University of Minnesota Viral Vector and Cloning Core.
The authors declare no competing financial interests.
References
- Adermark L, Lovinger DM (2007) Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proc Natl Acad Sci USA 104:20564–20569. 10.1073/pnas.0706873104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A (2014) Gliotransmitters travel in time and space. Neuron 81:728–739. 10.1016/j.neuron.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Lovinger DM (2017) Endocannabinoid-dependent synaptic plasticity in the striatum. In: Endocannabinoids and lipid mediators in brain functions (Melis M, ed), pp 109–153. Cham: Springer International Publishing. [Google Scholar]
- Augustin SM, Chancey JH, Lovinger DM (2018) Dual dopaminergic regulation of corticostriatal plasticity by cholinergic interneurons and indirect pathway medium spiny neurons. Cell Rep 24:2883–2893. 10.1016/j.celrep.2018.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagetta V, Picconi B, Marinucci S, Sgobio C, Pendolino V, Ghiglieri V, Fusco FR, Giampà C, Calabresi P (2011) Dopamine-dependent long-term depression is expressed in striatal spiny neurons of both direct and indirect pathways: implications for Parkinson's disease. J Neurosci 31:12513–12522. 10.1523/JNEUROSCI.2236-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biber K, Laurie DJ, Berthele A, Sommer B, Tölle TR, Gebicke-Härter PJ, van Calker D, Boddeke HW (1999) Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem 72:1671–1680. 10.1046/j.1471-4159.1999.721671.x [DOI] [PubMed] [Google Scholar]
- Borycz J, Pereira MF, Melani A, Rodrigues RJ, Köfalvi A, Panlilio L, Pedata F, Goldberg SR, Cunha RA, Ferré S (2007) Differential glutamate-dependent and glutamate-independent adenosine A1 receptor-mediated modulation of dopamine release in different striatal compartments. J Neurochem 101:355–363. 10.1111/j.1471-4159.2006.04386.x [DOI] [PubMed] [Google Scholar]
- Bradley SJ, Challiss RA (2012) G protein-coupled receptor signalling in astrocytes in health and disease: a focus on metabotropic glutamate receptors. Biochem Pharmacol 84:249–259. 10.1016/j.bcp.2012.04.009 [DOI] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Centonze D, Sancesario G, Morello M, Giorgi M, Pisani A, Bernardi G (1999) A critical role of the nitric oxide/cGMP pathway in corticostriatal long-term depression. J Neurosci 19:2489–2499. 10.1523/JNEUROSCI.19-07-02489.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaccini A, Gritti M, Giorgi A, Locarno A, Heck N, Migliarini S, Bertero A, Mereu M, Margiani G, Trusel M, Catelani T, Marotta R, De Luca MA, Caboche J, Gozzi A, Pasqualetti M, Tonini R (2018) Serotonergic signaling controls input-specific synaptic plasticity at striatal circuits. Neuron 98:801–816.e7. 10.1016/j.neuron.2018.04.008 [DOI] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, Cohn W, Rajendran PS, Vondriska TM, Whitelegge JP, Coppola G, Khakh BS (2017) Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95:531–549.e9. 10.1016/j.neuron.2017.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Tan Z, Zeng L, Zhang X, He Y, Gao W, Wu X, Li Y, Bu B, Wang W, Duan S (2013) Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia 61:178–191. 10.1002/glia.22425 [DOI] [PubMed] [Google Scholar]
- Chepkova AN, Fleischer W, Kazmierczak T, Doreulee N, Haas HL, Sergeeva OA (2009) Developmental alterations of DHPG-induced long-term depression of corticostriatal synaptic transmission: switch from NMDA receptor-dependent towards CB1 receptor-dependent plasticity. Pflugers Arch 459:131–141. 10.1007/s00424-009-0714-7 [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortés A, Canela EI, López-Giménez JF, Milligan G, Lluis C, Cunha RA, Ferré S, Franco R (2006) Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci 26:2080–2087. 10.1523/JNEUROSCI.3574-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covelo A, Araque A (2018) Neuronal activity determines distinct gliotransmitter release from a single astrocyte. Elife 7:e32237 10.7554/eLife.32237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Witten IB (2019) Striatal circuits for reward learning and decision-making. Nat Rev Neurosci 20:482–494. 10.1038/s41583-019-0189-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, Costa RM (2013) Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494:238–242. 10.1038/nature11846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, Moss SJ, Haydon PG (2007) mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci USA 104:1995–2000. 10.1073/pnas.0609408104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, Tiret P, Volterra A (2011) Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci 14:1276–1284. 10.1038/nn.2929 [DOI] [PubMed] [Google Scholar]
- Durkee CA, Araque A (2019) Diversity and specificity of astrocyte-neuron communication. Neuroscience 396:73–78. 10.1016/j.neuroscience.2018.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkee CA, Covelo A, Lines J, Kofuji P, Aguilar J, Araque A (2019) Gi/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia 67:1076–1093. 10.1002/glia.23589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Venance L (2010) Spike-timing dependent plasticity in the striatum. Front Synaptic Neurosci 2:6. 10.3389/fnsyn.2010.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L (2005) Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci 25:11279–11287. 10.1523/JNEUROSCI.4476-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM (2005) Adenosine and brain function. Int Rev Neurobiol 63:191–270. 10.1016/S0074-7742(05)63007-3 [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM (2002) Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5:446–451. 10.1038/nn832 [DOI] [PubMed] [Google Scholar]
- Gubellini P, Saulle E, Centonze D, Costa C, Tropepi D, Bernardi G, Conquet F, Calabresi P (2003) Corticostriatal LTP requires combined mGluR1 and mGluR5 activation. Neuropharmacology 44:8–16. 10.1016/s0028-3908(02)00214-9 [DOI] [PubMed] [Google Scholar]
- Gundersen V, Storm-Mathisen J, Bergersen LH (2015) Neuroglial transmission. Physiol Rev 95:695–726. 10.1152/physrev.00024.2014 [DOI] [PubMed] [Google Scholar]
- Haustein MD, Kracun S, Lu X-H, Shih T, Jackson-Weaver O, Tong X, Xu J, Yang XW, O'Dell TJ, Marvin JS, Ellisman MH, Bushong EA, Looger LL, Khakh BS (2014) Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 82:413–429. 10.1016/j.neuron.2014.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illes P, Burnstock G, Tang Y (2019) Astroglia-derived ATP modulates CNS neuronal circuits. Trends Neurosci 42:885–898. 10.1016/j.tins.2019.09.006 [DOI] [PubMed] [Google Scholar]
- Jiang R, Diaz-Castro B, Looger LL, Khakh BS (2016) Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington's disease model mice. J Neurosci 36:3453–3470. 10.1523/JNEUROSCI.3693-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS. (2019) Astrocyte-neuron interactions in the striatum: insights on identity, form, and function. Trends Neurosci 42:617–630. 10.1016/j.tins.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, Kreitzer AC (2012) Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci 15:816–818. 10.1038/nn.3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC (2007) Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature 445:643–647. 10.1038/nature05506 [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC (2008) Striatal plasticity and basal ganglia circuit function. Neuron 60:543–554. 10.1016/j.neuron.2008.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zima AV, Sheikh F, Blatter LA, Chen J (2005) Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res 96:1274–1281. 10.1161/01.RES.0000172556.05576.4c [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Choi S (1995) Activation of adenosine A1 receptors initiates short-term synaptic depression in rat striatum. Neurosci Lett 199:9–12. 10.1016/0304-3940(95)12024-x [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM (2010) Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65:445–459. 10.1016/j.neuron.2010.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Manabe T, Nicoll RA (1994) Release of adenosine by activation of NMDA receptors in the hippocampus. Science 265:2098–2101. 10.1126/science.7916485 [DOI] [PubMed] [Google Scholar]
- Martín R, Bajo-Graneras R, Moratalla R, Perea G, Araque A (2015) Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 349:730–734. 10.1126/science.aaa7945 [DOI] [PubMed] [Google Scholar]
- Martin-Fernandez M, Jamison S, Robin LM, Zhao Z, Martin ED, Aguilar J, Benneyworth MA, Marsicano G, Araque A (2017) Synapse-specific astrocyte gating of amygdala-related behavior. Nat Neurosci 20:1540–1548. 10.1038/nn.4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara K, Okada M, Nakanishi S (1997) The metabotropic glutamate receptor mGluR5 induces calcium oscillations in cultured astrocytes via protein kinase C phosphorylation. J Neurochem 69:1467–1475. 10.1046/j.1471-4159.1997.69041467.x [DOI] [PubMed] [Google Scholar]
- Nazzaro C, Greco B, Cerovic M, Baxter P, Rubino T, Trusel M, Parolaro D, Tkatch T, Benfenati F, Pedarzani P, Tonini R (2012) SK channel modulation rescues striatal plasticity and control over habit in cannabinoid tolerance. Nat Neurosci 15:284–293. 10.1038/nn.3022 [DOI] [PubMed] [Google Scholar]
- O'Hare JK, Ade KK, Sukharnikova T, Van Hooser SD, Palmeri ML, Yin HH, Calakos N (2016) Pathway-specific striatal substrates for habitual behavior. Neuron 89:472–479. 10.1016/j.neuron.2015.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Vallée J, Haber M, Murai KK, Lacaille JC, Robitaille R (2011) Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146:785–798. 10.1016/j.cell.2011.07.022 [DOI] [PubMed] [Google Scholar]
- Papouin T, Dunphy J, Tolman M, Foley JC, Haydon PG (2017) Astrocytic control of synaptic function. Philos Trans R Soc Lond B Biol Sci 372:20160154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG (2005) Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–116. 10.1126/science.1116916 [DOI] [PubMed] [Google Scholar]
- Perea G, Araque A (2005) Glial calcium signaling and neuron-glia communication. Cell Calcium 38:375–382. 10.1016/j.ceca.2005.06.015 [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD (1996) Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci 16:5073–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittiner JE, Caffall ZF, Hernández-Martinez R, Sanderson SM, Pearson JL, Tsukayama KK, Liu AY, Xiao C, Tracy S, Shipman MK, Hickey P, Johnson J, Scott B, Stacy M, Saunders-Pullman R, Bressman S, Simonyan K, Sharma N, Ozelius LJ, Cirulli ET, et al. (2016) Functional genomic analyses of mendelian and sporadic disease identify impaired eIF2α signaling as a generalizable mechanism for dystonia. Neuron 92:1238–1251. 10.1016/j.neuron.2016.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchouk I, Volterra A (2018) Gliotransmission: beyond black-and-white. J Neurosci 38:14–25. 10.1523/JNEUROSCI.0017-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R (2006) GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci 26:5370–5382. 10.1523/JNEUROSCI.5255-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ (2008) Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321:848–851. 10.1126/science.1160575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS (2008) Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci 28:6659–6663. 10.1523/JNEUROSCI.1717-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippy T, Lapray D, Crochet S, Petersen CC (2015) Cell-type-specific sensorimotor processing in striatal projection neurons during goal-directed behavior. Neuron 88:298–305. 10.1016/j.neuron.2015.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spigolon G, Cavaccini A, Trusel M, Tonini R, Fisone G (2018) cJun N-terminal kinase (JNK) mediates cortico-striatal signaling in a model of Parkinson's disease. Neurobiol Dis 110:37–46. 10.1016/j.nbd.2017.10.015 [DOI] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y, Nedergaard M (2013) Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 339:197–200. 10.1126/science.1226740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM (2001) Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. J Neurophysiol 86:2405–2412. 10.1152/jn.2001.86.5.2405 [DOI] [PubMed] [Google Scholar]
- Tamura A, Yamada N, Yaguchi Y, Machida Y, Mori I, Osanai M (2014) Both neurons and astrocytes exhibited tetrodotoxin-resistant metabotropic glutamate receptor-dependent spontaneous slow Ca2+ oscillations in striatum. PLoS One 9:e85351. 10.1371/journal.pone.0085351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Matias S, Dugue GP, Mainen ZF, Costa RM (2014) Balanced activity in basal ganglia projection pathways is critical for contraversive movements. Nat Commun 5:4315. 10.1038/ncomms5315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusel M, Cavaccini A, Gritti M, Greco B, Saintot PP, Nazzaro C, Cerovic M, Morella I, Brambilla R, Tonini R (2015) Coordinated regulation of synaptic plasticity at striatopallidal and striatonigral neurons orchestrates motor control. Cell Rep 13:1353–1365. 10.1016/j.celrep.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6:626–640. 10.1038/nrn1722 [DOI] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, Kang J, Takano T, Nedergaard M (2006a) Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci 9:816–823. 10.1038/nn1703 [DOI] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ (2006b) Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50:443–452. 10.1016/j.neuron.2006.04.010 [DOI] [PubMed] [Google Scholar]
- Wu YW, Kim JI, Tawfik VL, Lalchandani RR, Scherrer G, Ding JB (2015) Input- and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep 10:75–87. 10.1016/j.celrep.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Adermark L, Lovinger DM (2008) Neurotensin reduces glutamatergic transmission in the dorsolateral striatum via retrograde endocannabinoid signaling. Neuropharmacology 54:79–86. 10.1016/j.neuropharm.2007.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S (2003) ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40:971–982. 10.1016/s0896-6273(03)00717-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Source data that support the findings of this study are available from the corresponding author on reasonable request. Custom MATLAB code for analyses of astrocyte Ca2+ are available from the corresponding author on individual request.