

Abstract
Familial Mediterranean fever (FMF) is an autoinflammatory disease caused by homozygous or compound heterozygous gain-of-function mutations in MEFV, encoding pyrin, an inflammasome protein. Heterozygous carrier frequencies for multiple MEFV mutations are high in several Mediterranean populations, suggesting that they confer selective advantage. Among 2,313 Turks, we found extended haplotype homozygosity flanking FMF-associated mutations, indicating evolutionarily recent positive selection of FMF-associated mutations. Two pathogenic pyrin variants independently arose >1,800 years ago. Mutant pyrin interacts less avidly with Yersinia pestis virulence factor YopM than wild type human pyrin, thereby attenuating YopM-induced IL-1β suppression. Relative to healthy controls, leukocytes from FMF patients harboring homozygous or compound heterozygous mutations and from asymptomatic heterozygous carriers released heightened IL-1β specifically in response to Y. pestis. Y. pestis-infected MefvM680I/M680I FMF knock-in mice exhibited IL-1-dependent increased survival relative to wild-type knock-in mice. Thus, FMF mutations that were positively selected in Mediterranean populations confer heightened resistance to Y. pestis.
INTRODUCTION
Familial Mediterranean fever (FMF) is the prototype for a family of disorders of innate immunity termed autoinflammatory diseases. Patients present with self-limited episodes of fever, serositis, arthritis, and rash. Prior to the development of effective therapies, FMF was a common cause of systemic amyloidosis and premature death among eastern Mediterranean populations. The gene responsible for FMF, MEFV, was identified by positional cloning1, 2. MEFV encodes the pyrin protein, which nucleates the macromolecular pyrin inflammasome complex that catalyzes the cleavage and release of the proinflammatory cytokines IL-1β and IL-18, and induces an inflammatory form of cell death termed pyroptosis.
The major FMF-associated mutations, including MEFV_p.M680I, MEFV_p.M694V, and MEFV_p.V726A, are gain-of-function, but the inheritance pattern is usually described as recessive, with well over 50% of patients harboring homozygous or compound heterozygous mutations that reside in the C-terminal domain of the protein3–5. Despite reduced survival of FMF patients, the combined carrier frequency of heterozygous mutations among Jewish, Arab, Armenian, and Turkish populations is approximately 10%6, 7, 8, 9. To place this in perspective, the carrier frequency for cystic fibrosis, the most common lethal recessive disorder among ancestrally European people in North America, is approximately 4%10. Haplotype data obtained at the time MEFV was identified indicated that the major FMF mutations arose on ancient Middle Eastern founder chromosomes1. The selective pressure to generate the observed mutation carrier frequency requires the geographic convergence of the mutations and an agent of selection such as an infection responsible for substantial population mortality, as is the case for hemoglobin S and malaria. Nevertheless, genetic drift remains an alternative explanation for high FMF carrier frequencies.
Inhibition of RhoA, a member of the Ras homology family of small GTPases, induces activation of the pyrin inflammasome11, 12. RhoA activates the serine/threonine protein kinase C-related kinase (PKN, aka PRK), the kinase domain of which binds to pyrin directly and phosphorylates two serine residues (S208 and S242) of human pyrin12. The 14-3-3ε and τ proteins bind phosphorylated pyrin and block inflammasome activation when RhoA is active. Because RhoA activation is essential for leukocyte cytoskeletal assembly, many bacteria utilize RhoA-inactivating toxins to suppress host defense. Pyrin inflammasome activation induced by RhoA inactivation represents an innate immune response that elicits proinflammatory cytokine release and pyroptosis. Unlike other pathogenic bacteria that have RhoA-targeting toxins, pathogenic Yersinia species (Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica) are unique in having an additional virulence factor, YopM, that specifically inhibits the pyrin inflammasome13, 14.
The well-documented series of bubonic plague epidemics over human history raises the possibility that Y. pestis may have played a role in selecting for the high prevalence of FMF mutations in modern-day Mediterranean populations. However, to date there are no population genetic data directly addressing the possibility of natural selection of MEFV mutations, and no data directly examining the effect of human pyrin mutations on its interactions with Y. pestis. Therefore, we evaluated the Turkish population for a genetic imprint of evolutionarily recent natural selection and investigated the host-pathogen interactions involving wild type (WT) or mutant pyrin in human and mouse experimental models. Our results provide strong evidence that FMF-associated pyrin mutations have undergone episodic selection in the Turkish population, that modern-day FMF mutation carriers are descended from common ancestors who likely lived in the Middle East before the known historical plague epidemics, and that FMF-associated pyrin mutations confer resistance to Y. pestis infection biochemically and in ex vivo human systems and in an in vivo mouse model. These findings are consistent with the hypothesis that plague selected for FMF mutations in Middle Eastern-derived populations.
RESULTS
Population genetic analysis suggests evolutionarily recent positive selection of FMF mutations
Despite reduced survival of FMF patients, carrier frequencies of several FMF mutations are high in modern day Mediterranean populations (Supplementary Table 1)7. In 4,626 Turkish haplotypes including 91 with MEFV_p.V726A (rs28940579) and 145 with MEFV_p.M694V (rs61752717), both pathogenic MEFV mutations, we observed extended linkage disequilibrium flanking the mutant alleles (Fig. 1a). We further found extended haplotype homozygosity among haplotypes bearing the derived alleles (i.e., mutations) compared with haplotypes with the ancestral alleles (Fig. 1b) and this extended homozygosity was greater than expected by a haplotype diversity test, iHS15. The scores were below the 1.1st percentile of GWAS markers with similar allele frequency and local recombination rate for both MEFV_p.V726A and MEFV_p.M694V (Fig. 1c). Our sample was underpowered to conduct a similar analysis for MEFV_p.M680I, the third major pathogenic mutation in the Turkish population.
Figure 1. Turkish population haplotypes provide evidence of evolutionarily recent positive selection of FMF-associated pyrin mutations.

a, Extended linkage disequilibrium (LD) surrounding MEFV_p.V726A and MEFV_p.M694V, but not MEFV_p.E148Q, in 2,313 Turkish individuals. (LD depicted by the four-gamete rule with black boxes indicating pairs of markers for which the 4th (recombinant) gamete frequency is less than 0.003, Haploview). b, Extended haplotype homozygosity (EHH) of haplotypes bearing FMF mutations compared with haplotypes with the ancestral alleles determined in 4,626 Turkish haplotypes. c, Histograms of unstandardized iHS statistics of GWAS markers37 with similar frequency and local recombination rate as each FMF mutation. Top panel, 1,570 markers similar to MEFV_p.V726A; middle panel, 2,677 markers similar to MEFV_p.M694V; bottom panel, 6,166 markers similar to MEFV_p.E148Q. The most extreme negative iHS values have the strongest evidence of recent positive selection. d, Representative trajectories from forward-time simulation of episodic selection. The depicted trajectories reflect a single run of the simulated evolutionary process. The plague pandemics are depicted by the hatched rectangles. The red curve depicts a representative trajectory for MEFV_p.M694V from one initial allelic copy 197 generations ago to an allele frequency of 2.1×10−3 at the beginning of the first plague pandemic and to a present-day allele frequency of 0.0313. The blue curve depicts a representative trajectory for MEFV_p.V726A from one initial allelic copy 163 generations ago to 1.6×10−3 at the beginning of the first plague pandemic and to a present-day allele frequency of 0.0197.
These observations suggest that the former two FMF mutations, found on two different haplotypes at high frequency in the Turkish population, have undergone evolutionarily recent positive selection, and the data argue against the possibility that the high frequencies are due to genetic drift. The MEFV_p.E148Q (rs3743930) variant, generally thought to be a functional polymorphism but not to cause clinical FMF, is also found at high frequency in the Turkish population. We identified 232 MEFV_p.E148Q haplotypes among the 4,626 Turkish haplotypes. Despite its high frequency, MEFV_p.E148Q showed no evidence of recent positive selection by the iHS test, with a score in the 69th percentile of the GWAS markers with similar allele frequency and local recombination rate (Fig. 1c). A related haplotype diversity statistic, nSL16, which does not rely upon an estimated genetic map, had even more extreme values for MEFV_p.V726A and MEFV_pM694V, but not for MEFV_p.E148Q (Extended Data Fig. 1a–c).
We used the genetic lengths of haplotypes bearing MEFV_ p.V726A and MEFV_p.M694V and the model-free Gamma method to estimate that the most recent common ancestor of individuals with the MEFV_ p.V726A mutation lived 3,372 (95%CI, 2,585 to 4,356) years ago and the most recent common ancestor of individuals with the MEFV_p.M694V mutation lived 3,541 (95%CI, 2,023 to 6,266) years ago (Supplementary Table 2)17, 18. We also co-estimated the selection coefficients and allele ages for these two mutations using a hidden Markov model of recent positive selection19. The selection coefficients s, 0.068 (95% CI, 0.054 to 0.082) and 0.077 (95% CI, 0.065 to 0.090) for MEFV_p.V726A and MEFV_p.M694V, respectively, were consistent with strong recent positive selection (Extended Data Fig. 1d–f). By comparison, the lactase persistence associated allele rs4988235, a well-accepted example of a variant under strong selection, has s = 0.056 (95%CI, 0.0486 to 0.0654) in Northern Europeans19. The ages of the mutations estimated by this method were 2,192 (95% CI, 1,826 to 2,754) years and 2,108 (95% CI, 1,798 to 2,501) years for MEFV_p.V726A and MEFV_p.M694V, respectively.
We next considered whether lethality due to FMF has played a balancing evolutionary role. Although the clinical presentation of FMF begins with fever episodes during childhood, patients survived into the fourth decade before modern treatment with colchicine4. Thus, lethality due to FMF is unlikely to have had a major impact on reproductive fitness. Consequently, the relative fitness of individuals homozygous for the derived allele is not expected to be low, ruling out balancing selection, and favoring the hypothesis of positive selection. In support of this assumption, we observed that iHH, the integrated haplotype homozygosity values for the ancestral alleles, were not significantly different from the ancestral alleles of other GWAS markers with similar allele frequency and local recombination rate, indicating that evolutionarily recent positive selection was acting only on the derived alleles (Extended Data Fig. 1g–i).
To explain the differences in estimates of mutation age, we investigated three key assumptions of the hidden Markov model of recent positive selection19. The hidden Markov model assumes that allele frequency changes are deterministic (i.e., with no random component), that the selection coefficient is constant over time, and that the derived allele was present in one copy at the beginning of positive selection. We used single-locus forward-time simulation to investigate a more complex episodic model. To account for the durations of plague pandemics, we used historical records to inform a model of episodic selection, with two episodes of positive selection corresponding to the first (541 CE to 767 CE) and second (1346 to 1875 CE in the Middle East) plague pandemics20. In the absence of plague, we modeled allele frequency changes based on random genetic drift. The simulation demonstrates that MEFV_p.M694V is older than MEFV_p.V726A and ends in a higher allele frequency, and that the increase in both variants occurred mostly during the second pandemic (Fig. 1d). The estimated mutational ages are older than those estimated by the hidden Markov model and more consistent with the estimates from the Gamma method (Supplementary Table 2).
In addition, we investigated whole genome sequences from 352 Eurasian archeological samples, including 261 from before the Christian era (Supplementary Table 3), and many from non-Middle Eastern sources. One individual, living in modern day Russia approximately 4,045 to 4,298 years ago, and one in modern day Spain 4,916 to 5,261 years ago, may have carried the MEFV_p.M694V mutation (based on a single read in each case), although neither had surrounding sequence consistent with the modern Turkish haplotype, to the sensitivity of the low-pass DNA sequencing.
In contrast, we determined the mutation haplotypes in ethnically and geographically diverse modern-day carriers of 54 MEFV_p.M694V alleles and 19 MEFV_p.V726A alleles and found a core 48 kb haplotype containing MEFV_p.M694V and a core 77 kb haplotype containing MEFV_p.726A that were identical to the mutation haplotypes identified in the Turkish population (Extended Data Fig. 2a,b). From these data we can infer that these modern-day mutations each occurred on a single ancestral chromosome and that the mutations were subsequently present in diverse populations (Turks, Armenians, Arabs, Sephardi and Ashkenazi Jews, Druze, Italians, and Iraqis) either by shared ancestry or gene flow. We do not have sufficient genomic data from populations other than the Turks to rigorously test the hypothesis of positive selection in these additional ethnically and geographically diverse sources.
The Yersinia virulence factor YopM suppresses the human pyrin inflammasome by inducing pyrin phosphorylation
Among the bacteria known to activate the pyrin inflammasome, Y. pestis is a particularly attractive candidate to have selected for FMF mutations, because of the known history of bubonic plague pandemics and of the presence of a unique virulence factor, YopM, that specifically inhibits the pyrin inflammasome13, 14. Consistent with previous reports, yopM mutant (ΔyopM) Y. pestis induced IL-1β release from lipopolysaccharide (LPS)-primed bone marrow-derived macrophages (BMDMs) by the pyrin inflammasome-dependent pathway, which is also activated by RhoA-inactivating Clostridium botulinum C3 toxin (Fig. 2a). IL-1β release from C3-toxin-treated BMDMs was markedly diminished by infection with WT Y. pestis (Extended Data Fig. 3a). Conversely, the C3-toxin-induced IL-1β release was substantially increased when the BMDMs were infected with the ΔyopM mutant in a manner dependent upon multiplicity of infection (MOI) (Extended Data Fig. 3b). RhoA-targeting toxins, such as C3, activate the human pyrin inflammasome by preventing the phosphorylation of pyrin at residues S208 and S242, and thereby reduce the binding of inhibitory 14-3-3 proteins to pyrin12. However, as shown in human monocytes, Y. pestis increases 14-3-3 binding to pyrin, and counters the effect of C3 toxin to decrease 14-3-3 binding (Fig. 2b). In addition, the constitutive IL-1β release from retroviral-transduced U937 cells expressing phosphorylation-resistant pyrin mutated at the Ser residues (S208A or S242A) was not inhibited by Y. pestis (Fig. 2c, Extended Data Fig. 3c). Of note, there was no significant IL-1β release from the cells expressing wild-type (WT) pyrin or S209A mutant pyrin, which does not affect 14-3-3 binding. In sum, these data suggest that YopM suppresses pyrin by enhancing phosphorylation of Ser 208 and Ser 242 to induce 14-3-3 binding.
Figure 2. The pyrin inflammasome is suppressed by Y. pestis YopM through phosphorylation and subsequent 14-3-3 binding.
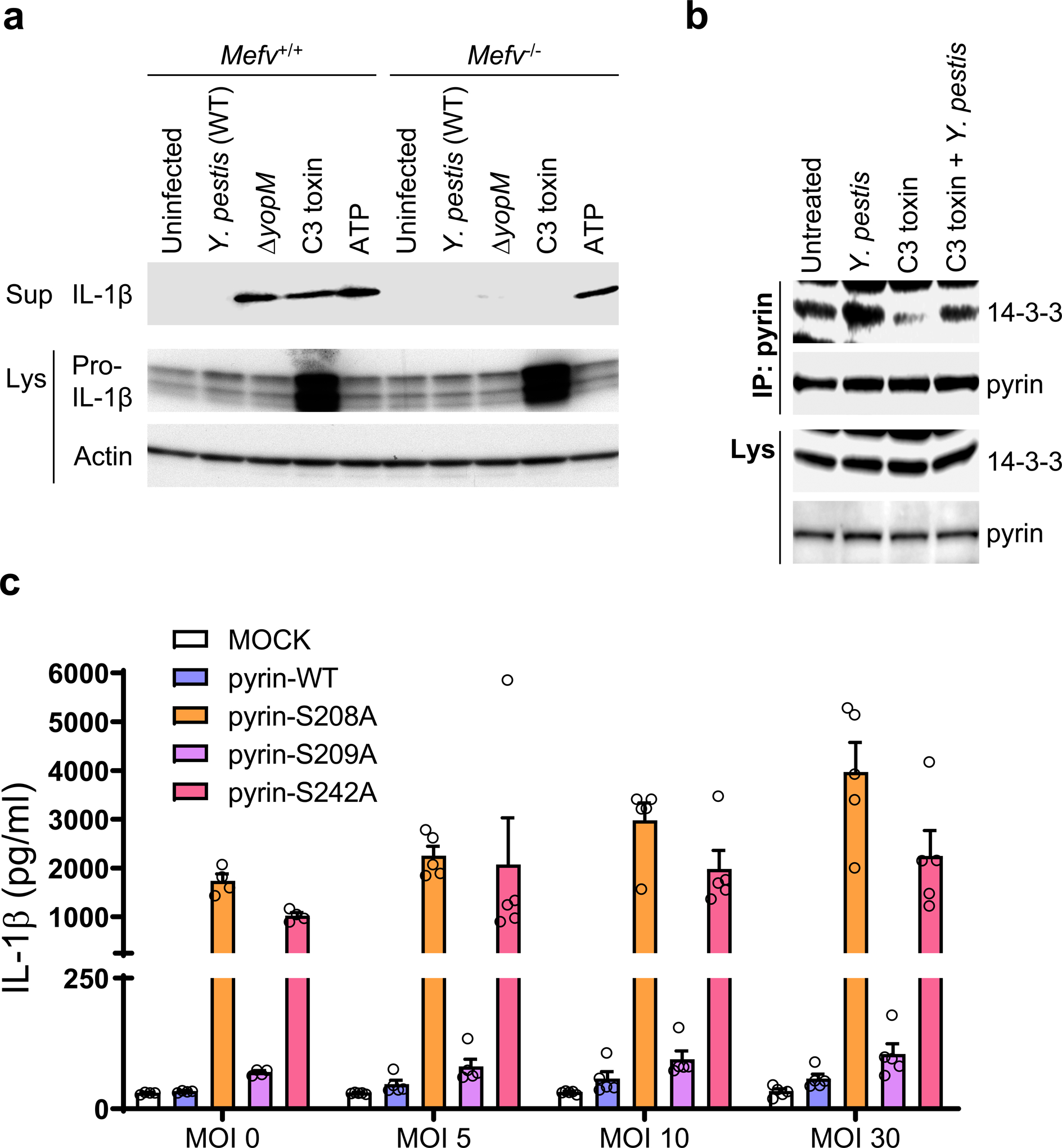
a, Immunoblot analysis of IL-1β in culture supernatants (Sup) and lysates (Lys) of WT (Mefv+/+) or Mefv−/− mouse BMDMs primed with LPS and treated with either C3 toxin or ATP, or infected with WT or ΔyopM Y. pestis strains at MOI 10. b, Immunoblot analysis of 14-3-3 in proteins immunoprecipitated (IP) with antibody to human pyrin from lysates (Lys) of control CD14+ monocytes treated with C3 toxin, Y. pestis, or both C3 toxin and Y. pestis at MOI 10. Data are representative of three independent experiments with similar results (a and b). c, IL-1β measurements of culture supernatants of retroviral transduced U937 cells, expressing WT or indicated mutant pyrin proteins, differentiated with PMA and infected with indicated MOI of WT Y. pestis. Results are presented as mean ± s.e.m., for n=5 independent biological replicates. MOCK, vector control.
Yersinia YopM inhibits the pyrin inflammasome by hijacking ribosome S6 kinase (RSK) proteins
YopM forms a complex with pyrin and with members of two serine-threonine protein kinase families, PKN and ribosomal S6 kinase (RSK)13, 14, 21. We previously observed that PKN-mediated pyrin phosphorylation was enhanced in the presence of YopM in an in vitro kinase assay13, but the role of the RSK-YopM interaction in Yersinia virulence was not examined. We therefore analyzed pyrin phosphorylation by RSK in the presence of YopM. Surprisingly, we found that pyrin is highly phosphorylated by RSK1, but only when YopM is present and there was no further increase of phosphorylation when both kinases were present (Fig. 3a and Extended Data Fig. 4a,b).
Figure 3. YopM recruits host RSK to phosphorylate pyrin and suppress inflammasome activation.
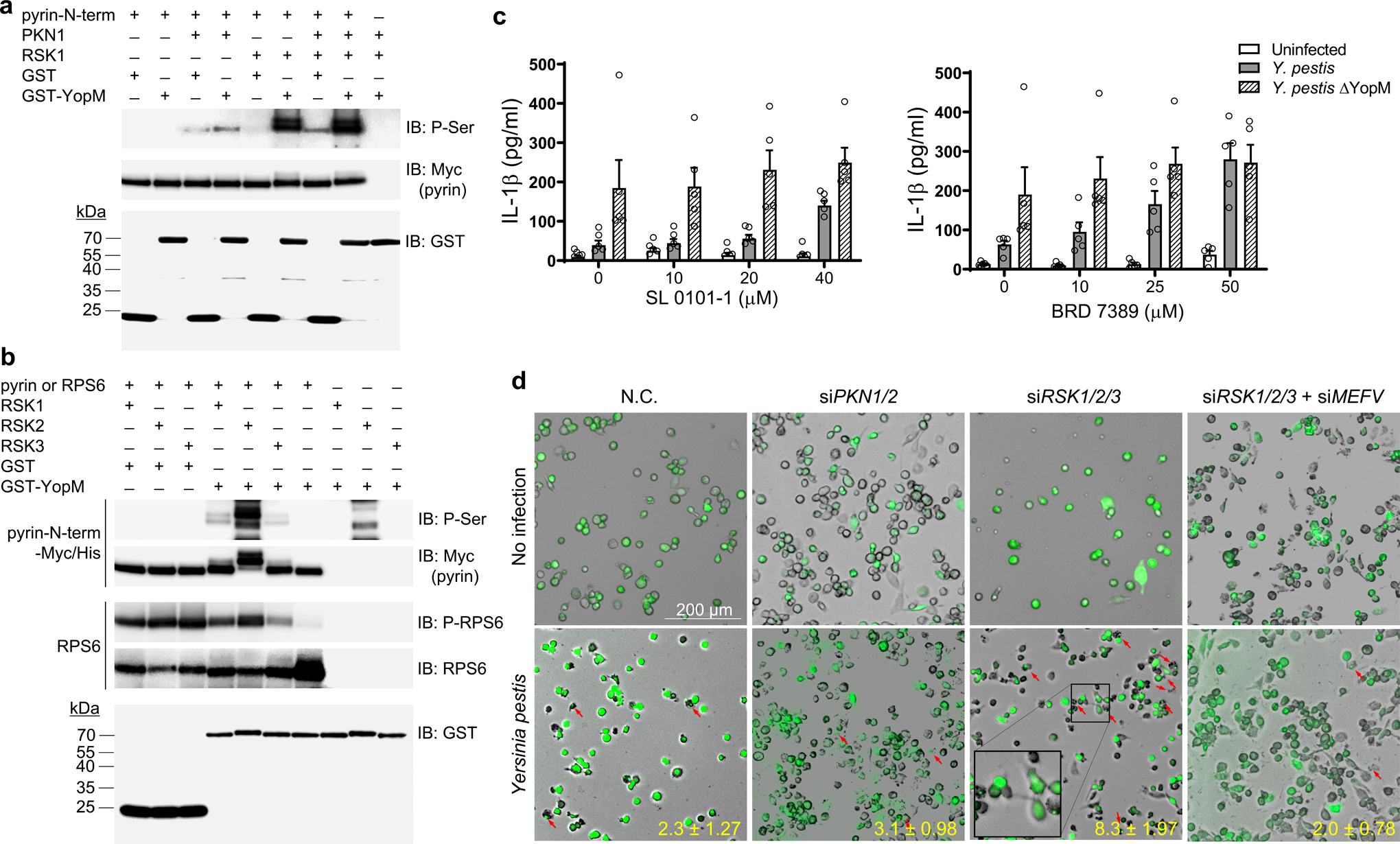
a, b, In vitro kinase assay of purified Myc/His-tagged N-terminal human pyrin (amino acids 1–330) (a, b) or RPS6 (b) incubated with recombinant PKN1 and/or RSK1 (a) or incubated with RSK1, RSK2, or RSK3 (b) in the presence of purified GST or GST-YopM, and analyzed by immunoblot with an antibody specific for phosphorylated serine (a, b) or antibody for phosphorylated RPS6 (b), followed by immunoblot analysis with antibody to Myc or antibody to RPS6. Data are representative of three independent experiments with similar results (a and b). c, IL-1β measurements of culture supernatants of CD14+ monocytes of healthy controls treated with indicated concentrations of SL0101–1 (left) or BRD7389 (right) for 1h and infected with Y. pestis at MOI 10. Results are presented as mean ± s.e.m., for n=5 independent biological replicates. d, ASC speck assay from THP1-ASC-GFP cells transfected with control (negative control, N.C.) or a pool of siRNAs targeting PKN1/2 or RSK1/2/3 with/without siMEFV, and infected with Y. pestis at MOI 10. The cells containing an ASC speck are indicated by red arrows. Data in all panels are representative from three repetitions with similar results.
The RSK family comprises four different isoforms, RSK1–4, and all four isoforms are reported to interact with YopM, which leads to sustained kinase activation by blocking RSK dephosphorylation22. We compared pyrin phosphorylation by each RSK isoform except RSK4 because RSK4 is not expressed in innate immune cells such as human PBMCs or mouse BMDMs (Extended Data Fig. 5a,b). We found that N-terminal pyrin was also phosphorylated by RSK2 and RSK3 when YopM was present, and the pyrin phosphorylation by RSK2 was much stronger than the phosphorylation by the other two isoforms (Fig. 3b). In the absence of pyrin, RSK2 also appeared to phosphorylate GST-YopM at a low level (Fig. 3b). Moreover, when ribosomal protein S6 (RPS6), a known RSK substrate, was tested in the same kinase assay, without YopM, RPS6 phosphorylation by all three RSK isoforms was observed, and the RPS6 phosphorylation by RSKs was not enhanced by addition of YopM (Fig. 3b). These data indicate that YopM suppresses pyrin inflammasome activation by shifting the specificity of RSKs as well as by activating PKN, both of which result in pyrin phosphorylation and subsequent 14-3-3 binding to pyrin. Indeed, the suppression of IL-1β release from human monocytes by Y. pestis infection was substantially attenuated when the cells were pretreated with SL 0101–1 or BRD 7389, specific RSK inhibitors (Fig. 3c). Moreover, in macrophage-like THP-1 cells with siRNA-mediated triple knock-down of RSK1, RSK2, and RSK3, a substantial increase of pyrin-dependent ASC specks or oligomerization, a readout for inflammasome activation11, was observed upon Y. pestis infection (Fig. 3d and Extended Data Fig. 6a,b).
YopM interacts with pyrin and RSK to suppress inflammasome activation
To understand how pyrin is phosphorylated by RSKs, we tested whether pyrin interacts with RSK in the cytosol, similarly to PKN112. We observed no interaction between pyrin and RSK2 in a co-immunoprecipitation assay from lysates of normal human monocytes, but, when the cells were infected with Y. pestis, an interaction of endogenous pyrin with RSK2 was observed (Fig. 4a). Endogenous pyrin also interacted with YopM in the Y. pestis-infected lysates (Fig. 4b). In a GST pull-down assay using purified recombinant GST-tagged YopM and the purified N-terminal half (amino acids 1–330) or C-terminal half (amino acids 331–781) of human pyrin, we found that YopM directly interacted with the N-terminal part of human pyrin (Fig. 4c). When considered with previous reports showing YopM-RSK interaction13, 14, 21, 22, the data suggest that YopM functions as an adaptor connecting RSKs to pyrin.
Figure 4. YopM interacts with pyrin and RSK.
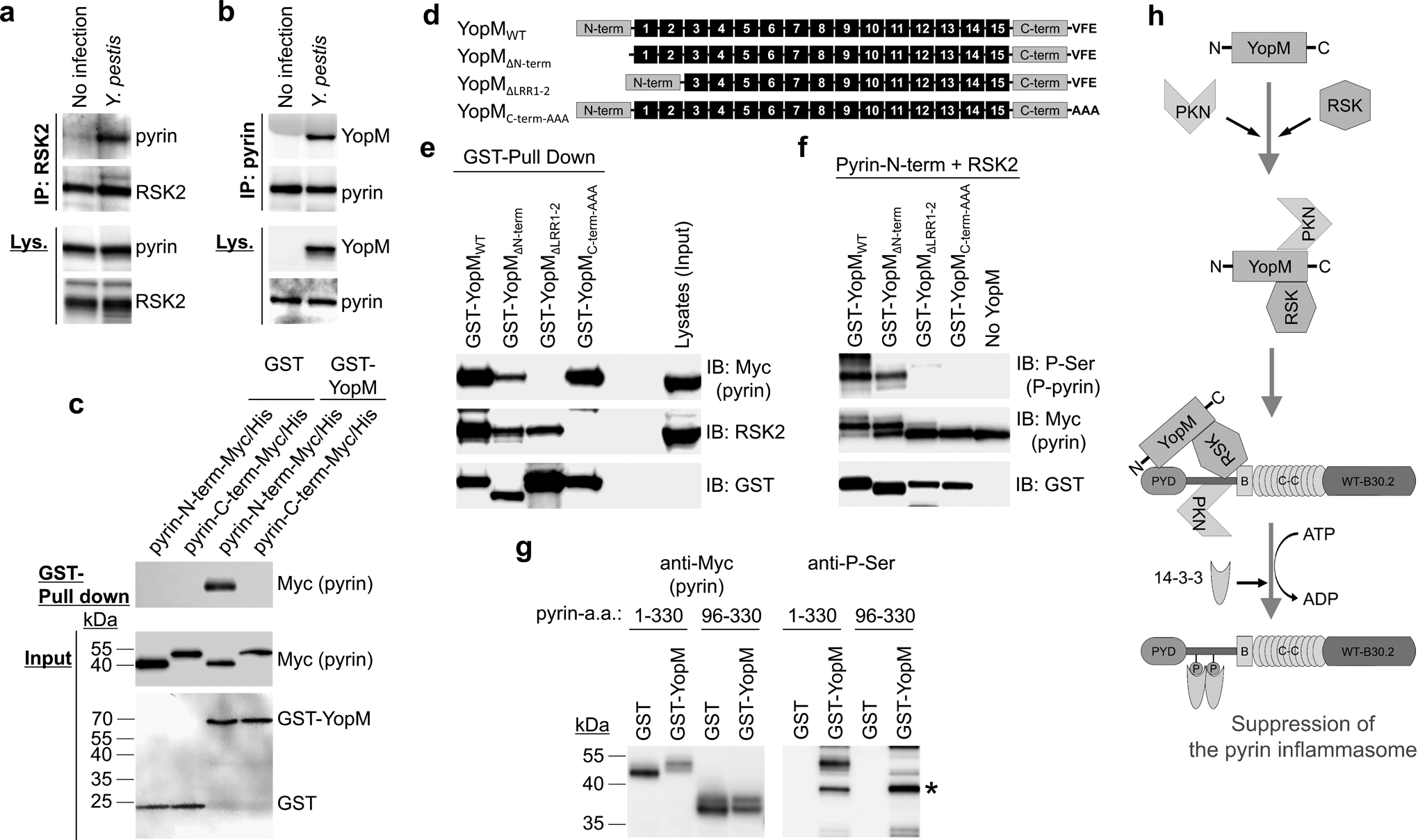
a, b, Immunoblot analysis for pyrin (a) or YopM (b) of endogenous proteins immunoprecipitated (IP) with antibody to human RSK2 (a) or pyrin (b) from lysates (Lys) of control CD14+ monocytes with or without Y. pestis infection. c, GST-pulldown assay of purified Myc/His-tagged N-terminal (amino acids 1–330) or C-terminal (amino acids 331–781) of human pyrin with purified GST or GST-YopM. d, Schematic structure of WT YopM of Y. pestis with various mutant YopM structures used in YopM-pyrin binding and kinase assay. e, GST-pulldown assay of lysates of HEK293T cells, expressing Myc/His-tagged human pyrin, with GST-tagged WT YopM or indicated mutant YopM. f, In vitro kinase assay of purified Myc/His-tagged N-terminal human pyrin with recombinant RSK2 in the presence of GST-tagged WT YopM or indicated mutant YopM. g, In vitro kinase assay of purified Myc/His-tagged N-terminal human pyrin or PYRIN domain-deleted N-terminal pyrin (amino acids 96–330) with recombinant RSK2 in the presence of purified GST or GST-YopM, and analyzed by immunoblot with an antibody specific for phosphorylated serine. Asterisk denotes phosphorylated GST-YopM. h, Proposed model for mechanism of PKN-YopM-RSK-mediated pyrin inflammasome suppression. Data are representative of three independent experiments with similar results (a–c, e–g).
All pathogenic Yersinia species have YopM with similar structure, which consists of an N-terminal domain containing a secretion signal followed by two α-helices, multiple leucine-rich repeat (LRR) domains, and an unstructured C-terminal domain that is highly conserved among all YopM isoforms23. Depending on the Yersinia species, the YopM protein contains 13 to 21 LRRs that make up most of the protein, and an internal portion encompassing LRR6 to LRR15 is required for interaction with PKN24. The last three amino acid residues of the C-terminal tail of YopM in Y. pseudotuberculosis are essential for RSK binding25. A mutant YopM with alanine substitutions of the last three C-terminal amino acids failed to inhibit IL-1β release from LPS-primed BMDMs14, which indicates that the inhibition of the pyrin inflammasome by YopM in vivo requires binding to RSKs.
To investigate the relationship of the pyrin-YopM interaction and pyrin phosphorylation by RSK, we generated various GST-tagged mutant YopM proteins of Y. pestis (Fig. 4d). In a GST pull-down assay, we found that mutant YopM with LRR1–2 deletion (YopMΔLRR1–2) did not interact with pyrin but still interacted with RSK (Fig. 4e). Conversely, mutant YopM with alanine substitutions of the last three C-terminal amino acids (YopMC-term-AAA) bound to pyrin but not to RSK (Fig. 4e). Both mutants failed to induce pyrin phosphorylation by RSK, while both WT or mutant YopM with an N-terminal domain deletion (YopMΔN-term) interacted with pyrin and RSK and induced RSK-mediated pyrin phosphorylation (Fig. 4e,f).
YopM bound to multiple regions of N-terminal pyrin (Extended Data Fig. 7a–c). N-terminal pyrin without the PYRIN domain (pyrin a.a. 96–330) was not phosphorylated by RSK with YopM (Fig. 4g). These data suggest that both the pyrin-YopM and YopM-RSK interactions, as well as the PYRIN domain, are required for suppressing pyrin inflammasome activation (Fig. 4h).
YopM binding to and phosphorylation of FMF mutant human pyrin is substantially reduced relative to WT human pyrin
We next considered the possible effect of FMF-associated mutations on the binding of YopM to pyrin. In Y. pestis-infected human CD14+ monocytes, the binding of YopM to endogenous pyrin (Fig. 5a) and resulting pyrin phosphorylation (Fig. 5b) was diminished in the cells from FMF patients with homozygous or compound heterozygous common B30.2 domain mutations relative to healthy controls. Taken together with the reduced binding of PKN to disease-associated mutant pyrin12, the decreased binding of mutant human pyrin to YopM may provide an additional advantage to defend the host from Y. pestis infection.
Figure 5. YopM binding to and phosphorylation of FMF mutant human pyrin is substantially reduced relative to WT human pyrin.
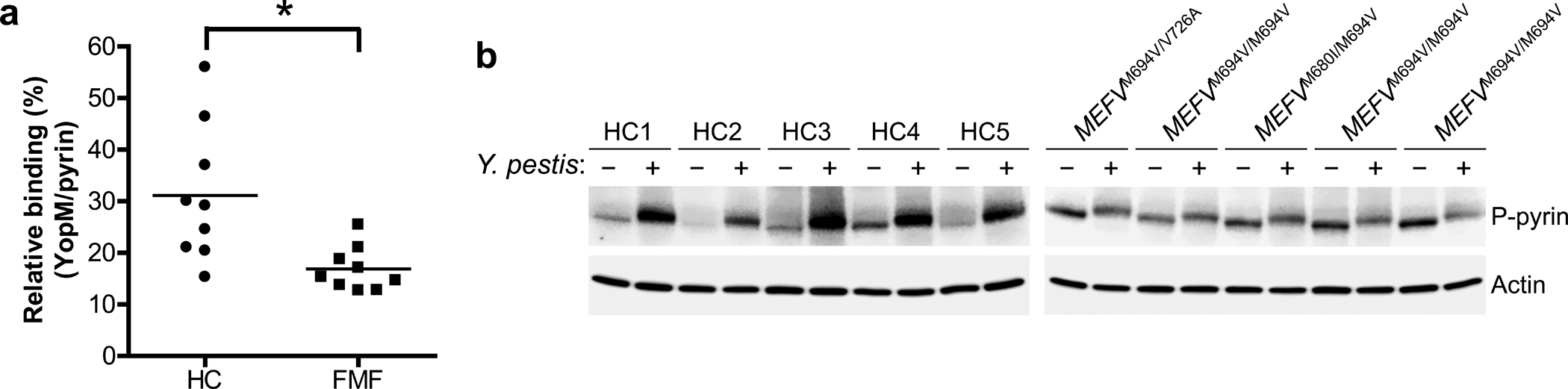
a, YopM-pyrin binding assay with immunoprecipitation from lysates of CD14+ monocytes of PBMCs from healthy controls (n=9) or FMF patients (n=9) with homozygous or compound heterozygous common FMF mutations (MEFV_p.M680I, MEFV_p.M694V, or MEFV_p.V726A) infected with Y. pestis. Proteins immunoprecipitated with antibody to pyrin were analyzed by immunoblotting with an antibody specific to YopM. The densities of YopM or pyrin bands were measured by Fc-Odyssey (Li-cor), and each YopM band in immunoprecipitated proteins was normalized by pyrin band. Each symbol represents an individual person; horizontal lines indicate the mean. *P = 0.0121 (unpaired two-tailed t test). b, Immunoblot analysis of phosphorylated pyrin in lysates of CD14+ monocytes from healthy controls (n=5) or FMF patients (n=5) with indicated mutations infected with Y. pestis, MOI 0 (−) or MOI 30 (+). Shown is an immunoblot from a single biological replicate.
FMF mutations are associated with increased Y. pestis-induced IL-1β release from human PBMCs
Based on the differential binding of mutant pyrin to YopM and decreased YopM-induced pyrin phosphorylation, we hypothesized that FMF patients and carriers would demonstrate heightened responsiveness of the pyrin inflammasome to Y. pestis. To examine the effect of Y. pestis infection on the pyrin inflammasome activated by FMF-associated mutations in humans, we first generated human monocytic U937 cells ectopically expressing WT, MEFV_p.E148Q variant or mutant human pyrin of the three most common FMF mutations (MEFV_p.M680I, MEFV_p.M694V, and MEFV_p.V726A) in the B30.2 domain by retroviral transduction. After phorbol 12-myristate 13-acetate (PMA)-induced differentiation, all cells expressing pyrin proteins released IL-1β, but the levels of IL-1β released from cells expressing FMF-associated mutant pyrin were significantly higher than the cells expressing WT pyrin or MEFV_p.E148Q variant protein, which is not associated with typical FMF (Fig. 6a). When the cells were infected with Y. pestis, the IL-1β release caused by the FMF-associated mutant pyrin was not inhibited by Yersinia, but rather, increased in a dose-dependent manner, while the IL-1β released from the cells expressing WT pyrin was substantially less than the cells expressing mutant pyrin (Fig. 6a). However, a dose-dependent increase of IL-1β release was observed in the cells expressing WT human pyrin when infected with ΔyopM Y. pestis (Fig. 6b). These data suggest that the WT pyrin inflammasome is suppressed by YopM of Y. pestis, but the pyrin inflammasome with FMF mutations is not suppressed by YopM.
Figure 6. FMF mutations are associated with increased Y. pestis-induced IL-1β release from human myeloid cell lines and PBMCs.
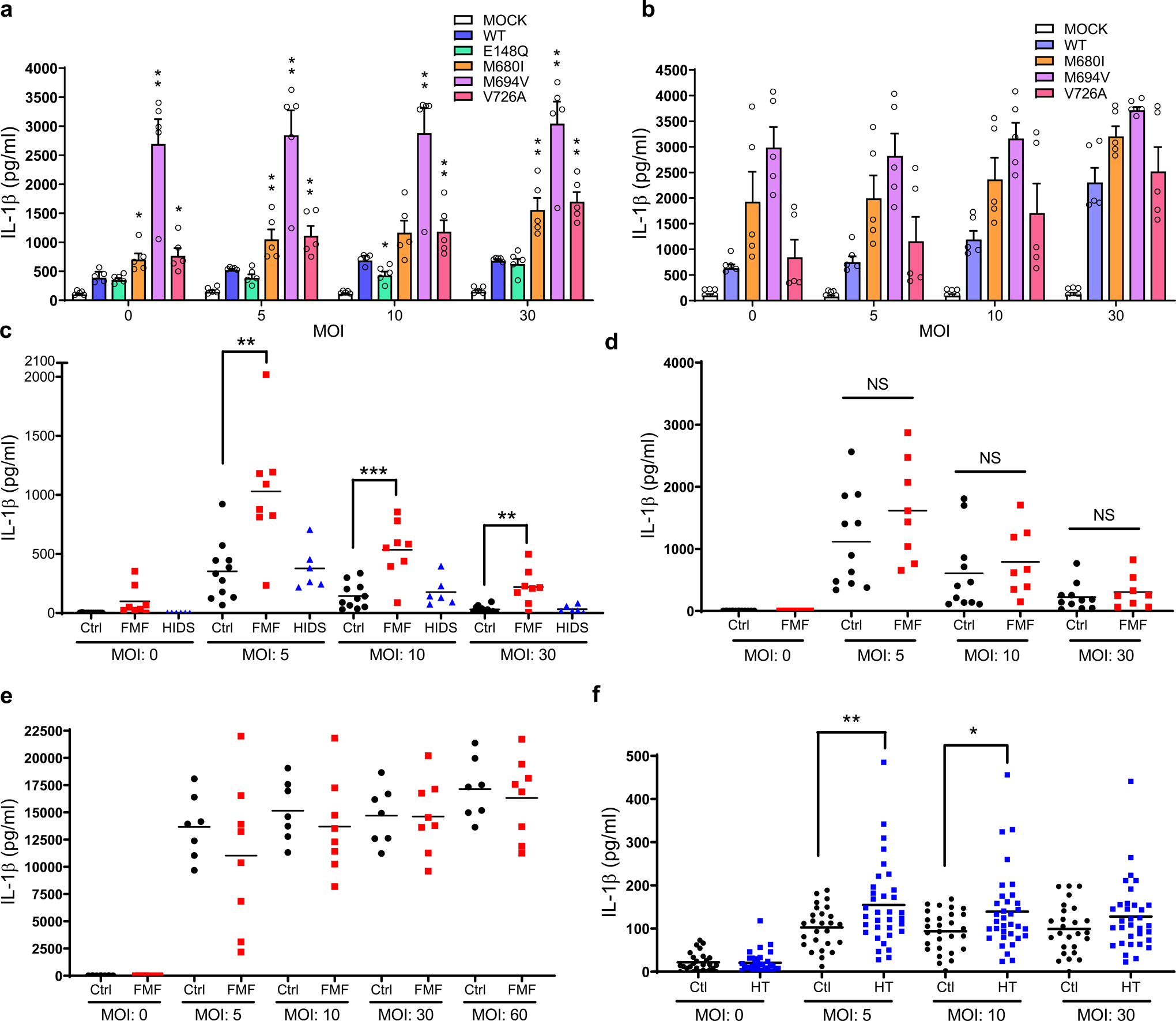
a, b, IL-1β measurements of culture supernatants of retroviral transduced U937 cells, expressing WT or indicated mutant pyrin proteins, differentiated with PMA and infected with indicated MOI of WT (a) or ΔyopM (b) Y. pestis strains. Results are presented as mean ± s.e.m., for n=5 independent biological replicates. *P = 0.0159 and **P = 0.0079 (unpaired two-tailed t test) compared to WT. c, IL-1β measurements of culture supernatants of PBMCs from healthy controls (n=11), FMF patients (n=8) with homozygous or compound heterozygous common FMF mutations (MEFV_p.M680I, MEFV_p.M694V, or MEFV_p.V726A) or HIDS patients (n=6) infected with indicated MOI of Y. pestis. **P = 0.0036 and ***P = 0.0012 (unpaired two-tailed t test) d, IL-1β measurements of culture supernatants of PBMCs from healthy controls (n=11) or FMF patients (n=8) with homozygous or compound heterozygous common FMF mutations (MEFV_p.M680I, MEFV_p.M694V, or MEFV_p.V726A) infected with indicated MOI of ΔyopM Y. pestis. e, IL-1β measurements of culture supernatants of PBMCs from healthy controls (n=7) or FMF patients (n=8) infected with indicated MOI of Burkholderia cenocepacia. f, IL-1β measurements of culture supernatants of CD14+ monocytes from healthy controls (n=28) or asymptomatic FMF carriers (HT, n=34) with one of the FMF mutations (MEFV_p.M680I, MEFV_p.M694V, or MEFV_p.V726A) infected with indicated MOI of Y. pestis. Each symbol represents an individual person; small horizontal lines indicate the mean (c–f). *P = 0.0141 and **P = 0.009 (unpaired two-tailed t test).
Consistent with this hypothesis, PBMCs from FMF patients released significantly more IL-1β than healthy controls when the cells were infected with Y. pestis (Fig. 6c), while IL-1β release from FMF patient PBMCs did not differ from healthy controls when infected with ΔyopM Y. pestis (Fig. 6d). There was no difference in the IL-1β levels subsequent to Y. pestis infection between healthy controls and patients with hyperimmunoglobulinemia D syndrome (HIDS), another IL-1β-mediated autoinflammatory disease caused by defects in prenylation and subsequent pyrin inflammasome activation12, in which there are no pyrin mutations (Fig. 6c). IL-1β release from PBMCs of both FMF patients and healthy controls was uniformly increased when the cells were infected with Burkholderia cenocepacia (Fig. 6e), which induces pyrin inflammasome activation with a RhoA-inactivating toxin, TecA26, but has no pyrin-suppressing effector such as YopM. Consistent with the possible enrichment of FMF carriers during a plague pandemic, CD14+ monocytes from asymptomatic heterozygous carriers of the MEFV_p.M694V, MEFV_p.M680I, and MEFV_p.V726A mutations released more IL-1β than monocytes from healthy controls in response to Y. pestis (Fig. 6f).
A role of the B30.2 domain in regulating the pyrin inflammasome
It is noteworthy that, if human pyrin were linear, the C-terminal B30.2 domain in which FMF mutations are found would be remote from the N-terminal domain where YopM binds pyrin, and from the serine residues where the pyrin inflammasome is regulated by phosphorylation. Nevertheless, mutations in the B30.2 domain appear to play a major role in regulating the activation of the human pyrin inflammasome, by virtue of their impact in FMF and on Y. pestis-induced IL-1β release (Fig. 6).
To study the effect of the B30.2 domain on the pyrin inflammasome activated by Y. pestis infection, we generated human monocytic U937 cells ectopically expressing WT or B30.2 domain-deleted human pyrin (ΔB30.2 pyrin) by retroviral transduction. The ΔB30.2 pyrin exhibited decreased pyrin phosphorylation relative to full-length human pyrin, and the cells expressing the ΔB30.2 pyrin showed constitutive IL-1β release by LPS priming alone (Fig. 7a). When infected with Y. pestis, the ΔB30.2 pyrin-expressing cells released significantly more IL-1β than the cells expressing WT pyrin (Fig. 7b), and the ΔB30.2 pyrin exhibited decreased YopM binding upon infection with Y. pestis (Fig. 7c). In an overexpression system, N-terminal pyrin immunoprecipitated C-terminal pyrin, with mutant C-terminal pyrin interacting less strongly than wild type pyrin (Fig. 7d). Taken together, the data indicate that the C-terminal B30.2 domain of human pyrin plays an important role in the regulation of the pyrin inflammasome and its interactions with YopM.
Figure 7. The human C-terminal pyrin B30.2 domain regulates pyrin inflammasome activation.
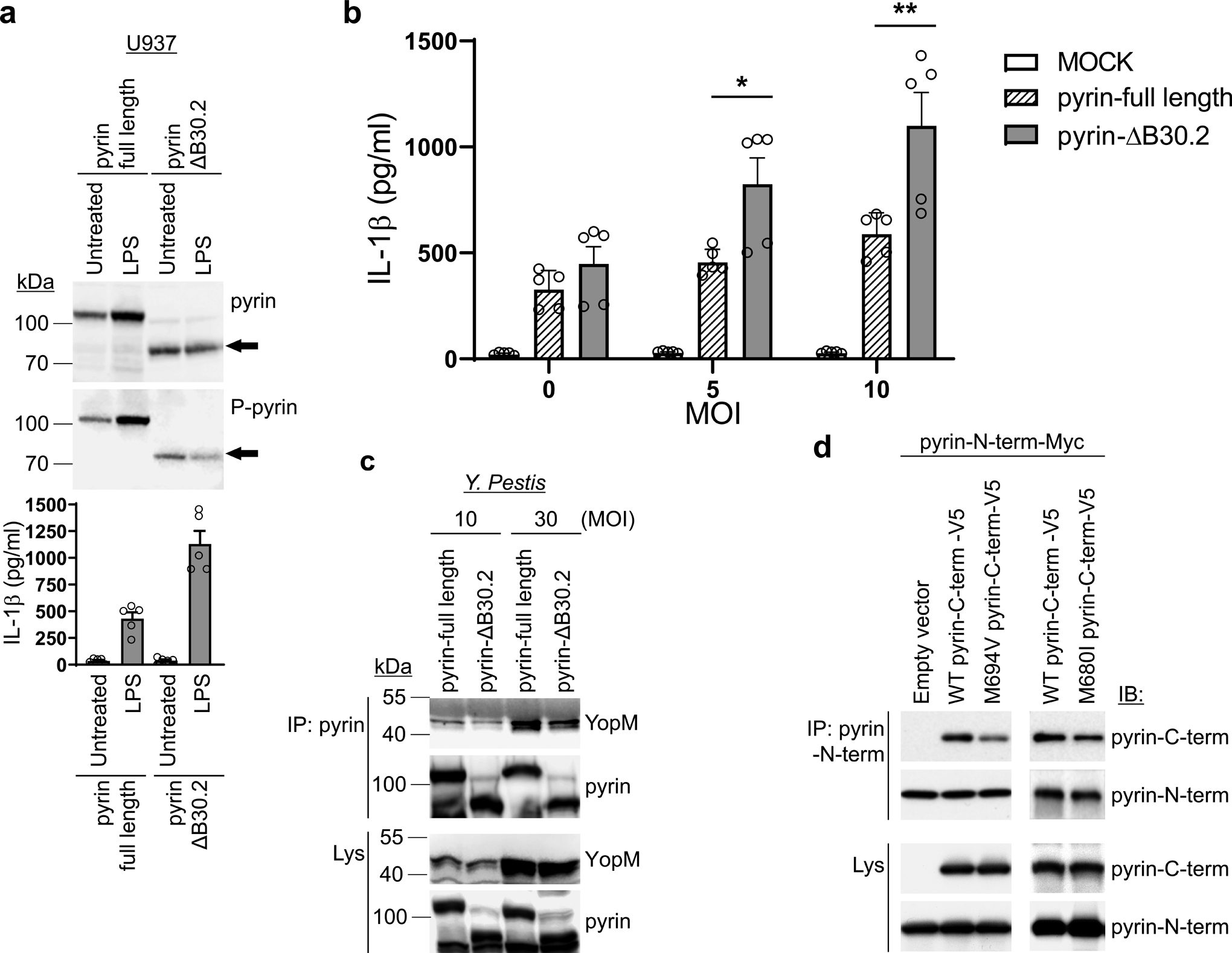
a. Immunoblot analysis of pyrin and phosphorylated pyrin in lysates of retroviral transduced U937 cells, expressing WT or B30.2 domain-deleted (ΔB30.2) pyrin, using antibodies specific for human pyrin and phosphorylated pyrin (top), and IL-1β measurements of culture supernatants with/without LPS treatment (bottom). Results are presented as mean ± s.e.m., for n=5 independent biological replicates. b. IL-1β measurements of culture supernatants from U937 cells expressing WT or B30.2 domain-deleted pyrin infected with indicated MOI of Y. pestis. Results are presented as mean ± s.e.m., for n=5 independent biological replicates. *P = 0.0317 and **P = 0.0079 (unpaired two-tailed t test). c. Immunoblot analysis of YopM in proteins immunoprecipitated with antibody to human pyrin (IP: pyrin) from lysates of U937 cells expressing WT or B30.2 domain-deleted pyrin infected with indicated MOI of Y. pestis. d. Immunoblot analysis of pyrin in proteins immunoprecipitated with antibody specific to N-terminal human pyrin (IP: Pyrin-N-term) from lysates of 293T cells expressing Myc- tagged N-terminal (amino acids 1–330) human pyrin with V5-tagged WT or mutant (p.M694V or p.M680I) C-terminal (amino acids 331–781) human pyrin. Data are representative of three independent experiments with similar results (a, c and d).
MF knock-in mice exhibit increased survival when infected with Y. pestis
While the initial demonstration of the protective effect of HbS for malaria involved both epidemiologic studies and deliberate exposure of human research subjects to infected mosquitoes27, Y. pestis poses an unacceptable risk for direct exposure studies. We therefore considered studying FMF knock-in mice. As the result of a frame-shift mutation, neither mouse nor rat pyrin includes a C-terminal B30.2 domain28, and thus it is important to compare a given FMF knock-in line with knock-ins for the WT human B30.2 domain. Although FMF knock-in mice exhibit an inflammatory phenotype, even the introduction of the wild-type human B30.2 domain into mouse pyrin causes a modest degree of inflammation, relative to WT mice12, and the relative severity of specific FMF knock-in lines does not always parallel the relative severity of the corresponding human mutations3. Thus, the study of FMF knock-ins is of potential value, but, like all animal models, there are limitations in their fidelity to the human condition.
With those caveats in mind, we compared the survival of WT (Mefv+/+) mice lacking the pyrin B30.2 domain with the survival of both MefvB30.2/B30.2 mice with the WT human B30.2 domain and the MefvM680I/M680I FMF knock-in strain. WT mice were highly susceptible to systemic infection with Y. pestis, but both MefvB30.2/B30.2 and MefvM680I/M680I mice were substantially less susceptible to Yersinia-induced lethality (Fig. 8a). Furthermore, there was a statistically significant increase in survival in the MefvM680I/M680I FMF knock-in mice, relative to the WT B30.2 knock-ins. We also crossed MefvM680I/M680I mice with IL-1 receptor-deficient (Il1r1−/−) mice, and found that the MefvM680I/M680IIl1r1−/− mice were highly susceptible to Y. pestis infection, indicating that the survival advantage of the FMF KI mice was mediated by the IL-1β signaling pathway (Fig. 8a). Consistent with an important protective role for IL-1β, Yersinia-stimulated BMDMs from the MefvM680/M680I mice produced increased levels of this cytokine relative to BMDMs from both MefvB30.2/B30.2 and Mefv+/+ mice (Fig. 8b). BMDMs from MefvV726A/V726A mice, which were not subjected to survival studies because of their severe spontaneous autoinflammatory phenotype, produced even higher levels of Y. pestis-induced IL-1β (Fig. 8b). Taken together, these data are consistent with the hypothesis that FMF-associated pyrin mutations confer protection against Y. pestis infection.
Figure 8. FMF mutations confer an IL-1β-dependent survival advantage against Y. pestis infection in mice.
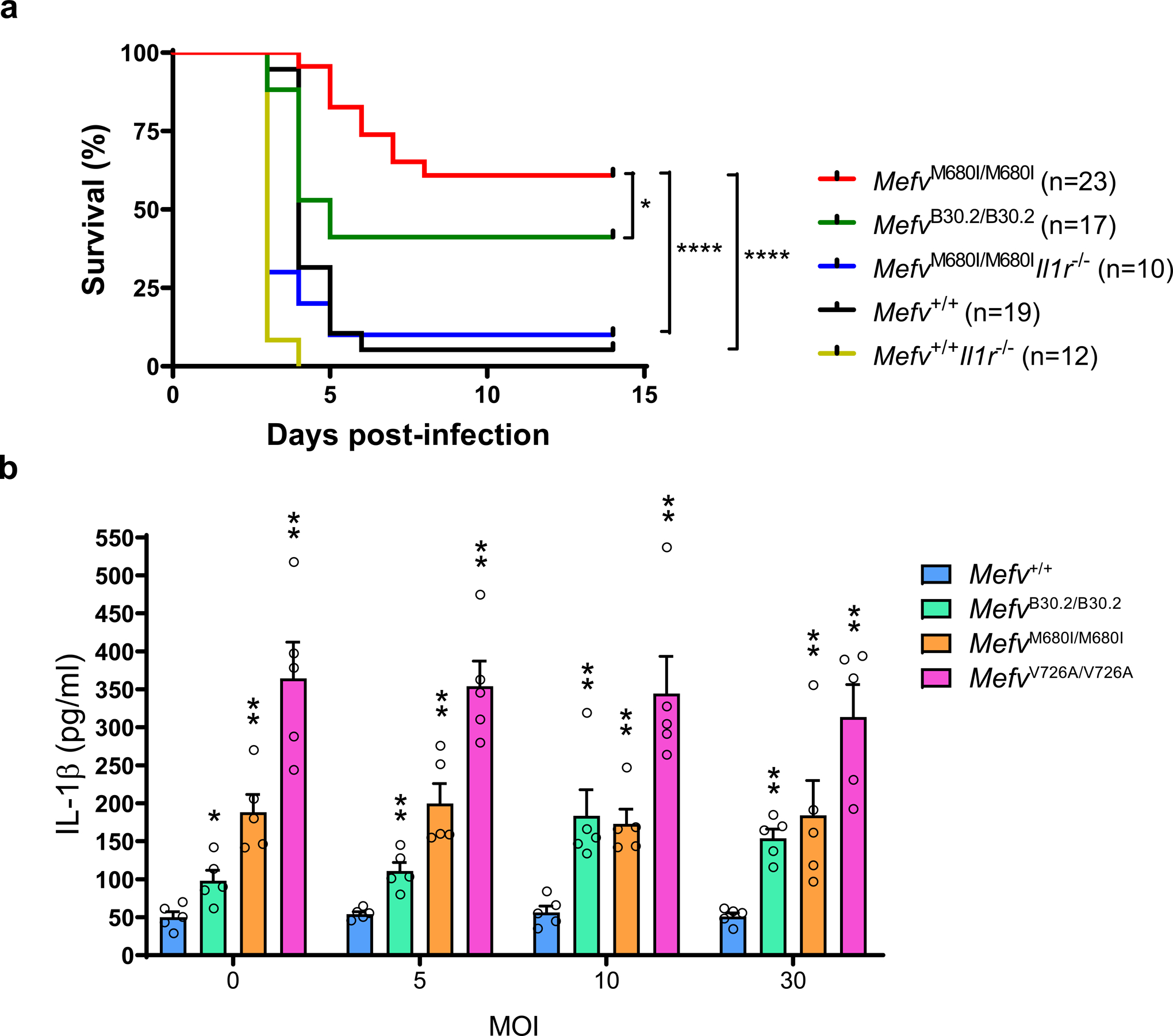
a, Kaplan-Meier survival curves for 12–16 week-old Mefv+/+ (n=19), MefvB30.2/B30.2 (n=17), MefvM680I/M680I (n=10), MefvM680I/M680IIl1r1−/− (n=10) and Mefv+/+Il1r1−/− (n=12) mice infected with approximately 100 or 150 CFU of Y. pestis by tail vein injection. *P = 0.0203 and ****P < 0.0001 (Gehan-Breslow-Wilcoxon test). b, IL-1β measurements of culture supernatants of Mefv+/+ or indicated FMF-KI BMDMs primed with LPS and infected with Y. pestis. Results are presented as mean ± s.e.m., for n=5 independent biological replicates. *P = 0.0159 and **P = 0.0079 (unpaired two-tailed t test) compared to WT.
DISCUSSION
This report provides new insights into the population genetics and biology of FMF, tying a prototypic autoinflammatory disease with a pathogen that has shaped human history. We provide strong evidence of evolutionarily recent positive selection for FMF-associated MEFV mutations, and insight into the origins and dissemination of these mutations. In examining the possibility that Y. pestis might have played an important role in selecting for FMF mutations, we build upon advances in our understanding of Yersinia pathogenesis to provide a molecular account of how FMF variants may have been selected. Finally, we demonstrate that PBMCs from FMF patients and heterozygous carriers do in fact release increased amounts of IL-1β in response to ex vivo Y. pestis infection, and that FMF knock-in mice infected with Yersinia exhibit a survival advantage over knock-ins of the WT human B30.2 domain.
Extended lengths of the present-day Turkish MEFV_p.M694V and MEFV_p.V726A conserved haplotypes compared to haplotypes of other similar genetic variants support the hypothesis of recent positive selection rather than random genetic drift in the Turkish population. The failure to document strong natural selection for the MEFV_p.E148Q polymorphism is noteworthy. Using three different population genetic methods, we estimate that the most recent common ancestors of both MEFV_p.M694V and MEFV_p.V726A lived at least ~1800 years ago, well before the onset of the Justinian plague, 541 CE. The data also support strong selection intensity, and simulations are consistent with observed modern-day allele frequencies, based on the estimated selection intensity and historical data. Consistent with these analyses, the Iraqi Jews, some of whom are descendants of the Babylonian captivity ~2,600 years ago, carry the same MEFV_p.M694V core haplotype as other Jewish Diaspora populations. Similarly, the core Middle Eastern MEFV_p.V726A haplotype is found in the Druze, a religious group that has been ethnically and geographically isolated for over 1,000 years.
Evidence for natural selection for variants of an important immune-related protein such as pyrin raises the question of whether these variants confer protection against an infectious agent. The pyrin inflammasome is an important host defense against a spectrum of bacteria with virulence factors that inactivate RhoA to disable leukocyte cytoskeletal assembly. Y. pestis stands out as having been capable of selective pressure on the frequency of pyrin mutations, based on the highly lethal plague pandemics over recorded human history. Previous work had established the Y. pestis YopM toxin as an important inhibitor of the pyrin inflammasome13, 14, but in the present work we expose YopM as the intracellular saboteur that redirects the specificity of human RSK isoforms to phosphorylate pyrin, and we establish the resistance of FMF mutant human pyrin to this process. These data suggest that, although the C-terminal B30.2 domain of pyrin is remote - in the linear sequence of the protein - from the YopM binding site and phosphorylation sites of pyrin, these sites probably interact in three dimensions. Biochemical and functional studies of ΔB30.2 human pyrin corroborate this view, pending structural confirmation. Although the evidence that FMF mutations render pyrin resistant to YopM strongly implicates Y. pestis infection as an agent for positive selection, we cannot formally rule out the possibility that these mutations protect against other endemic pathogens as well. Moreover, given the severity and geographic scope of plague pandemics, it is likely that plague has selected for additional human genetic variants, as has recently been proposed for a variant in the leukocyte N-formylpeptide receptor29.
The foregoing establishes the basis for the prediction that leukocytes from FMF patients and heterozygous carriers would release increased amounts of IL-1β in response to Y. pestis, a prediction borne out by the data. Arguing against the hypothesis that increased IL-1β release is simply a nonspecific effect of immune activation in FMF patients and carriers, we did not observe increased IL-1β release from FMF patients’ cells in response to Δ yopM Y. pestis, or to B. cenocepacia, which lacks YopM, relative to healthy controls. Although it is difficult to extrapolate these findings to the mouse, which lacks an endogenous pyrin B30.2 domain, we did observe an increase in protection in knock-in mice for the p.M680I mutation, relative to knock-in mice for the wild type human B30.2 domain.
The data are consistent with historical accounts and genomic reconstructions of the two major plague pandemics, the Justinian plague (which began in 541 CE and recurred intermittently for two centuries) and the second pandemic (which began with the Black Death in 1347 CE and recurred intermittently in the Middle East through the end of the nineteenth century)20, 30, 31, 32. The Middle East was a major crossroads in the dissemination of both pandemics, and thus would have been ideally located to select for FMF variants that are protective against Yersinia. In addition, significant outbreaks of plague persisted in the Middle East for a total of at least 700 years20. FMF mutations were not selected globally because they were only present at the appropriate time in a limited geographic distribution, as evidenced by the failure to find modern-day FMF carriers with common non-Middle Eastern haplotypes. The MEFV_p.V726A mutation has a carrier frequency of 8% in the Ashkenazi Jewish population(ref33; gnomAD, https://gnomad.broadinstitute.org/), which has roots in the Middle East but migrated to and expanded markedly in Eastern Europe34, 35. Although there are numerous other microbial pathogens that affect the activity of the pyrin inflammasome36, taking the population genetics, historical record, and biology of pyrin and YopM into account, it is likely that Y. pestis exerted evolutionarily recent selective pressure for FMF-associated pyrin variants in the Middle East and Mediterranean basin.
METHODS
Turkish population genetic data analysis
Genome-wide genotype data37 and MEFV mutation genotypes6 from 2,313 Turkish individuals were used for modern-day population genetic analyses. Linkage disequilibrium was determined with Haploview38. Genome-wide genotypes were phased without a reference using Eagle 2.439. The resultant haplotypes were evaluated for extended haplotype homozygosity40 around each of the FMF mutations using selscan41. The genome-wide haplotypes were also analyzed with selscan to determine the unstandardized haplotype diversity statistic, iHS15 of each genome-wide marker using the selscan default parameters, except with --max-extend 0 (for no limit) and the related unstandardized haplotype diversity statistic, nSL16 using the default parameters except with --max-extend-nsl 300. Integrated haplotype homozygosity values of the ancestral alleles, iHH0, were also determined with selscan. Genetic map positions were interpolated from the combined populations HapMap genetic map hg19 reference (map obtained from http://data.broadinstitute.org/alkesgroup/Eagle/downloads/) using predictGMAP (https://github.com/szpiech/predictGMAP). The unstandardized iHS and nSL haplotype diversity statistics of each mutation were compared to markers with similar frequency (+ or − 0.01) and with similar local recombination rate (+ or − 1.0 cM/Mb), omitting markers from the classical MHC region (hg19 chr6:29,000,000–34,000,000). Markers with extreme values (less than the 2nd percentile) relative to all other similar variants were considered likely under or in LD with markers under positive selection. The mutations were dated by the Gamma method (with correlated genealogy)17 using an R Shiny app (https://shiny.wehi.edu.au/rafehi.h/mutation-dating/). Left and right arm haplotype genetic lengths were determined from the interpolated genetic map positions. The Turkish population chromosome 16 haplotypes and genetic map positions of 800 GWAS markers37 centered on the MEFV mutations were also used to co-estimate the selection intensity and the duration of selection of each mutation with a hidden Markov model method19. Mutation dates were determined from number of generations, assuming 28.1 years per generation18 and assuming the date of the modern-day haplotypes was 2020 AD.
Forward-time simulation of random genetic drift with positive selection
We performed single-locus forward-time simulations that combined random genetic drift with recent positive selection in order to explore a model of episodic selection based on historical records of plague in the Middle East. To do this, we defined five epochs and relative fitnesses for genotypes AA, Aa, and aa, with A being the ancestral allele and a being the derived allele. In epochs 1 (from the unknown time of the mutation to 541 CE), 3 (from 767 CE to 1346 CE), and 5 (from 1875 CE to the present), there is no plague, the selection coefficient s is 0, and the allele frequency of the derived allele a changes from one generation to the next solely by random genetic drift. In epochs 2 (from 541 CE to 767 CE) and 4 (from 1346 CE to 1875 CE), the allele frequency changes according to random genetic drift plus positive selection. Given an allele frequency p for allele a at generation t, the frequency of a in generation t + 1 is . For the tth generation, we simulated random draws from a binomial distribution of 2N trials with the probability of success equal to pt. We estimated the mutational ages of MEFV_p.M694V and MEFV_p.V726A using a two-step procedure. First, given the present-day allele frequencies at the end of epoch 5, we estimated the allele frequencies at the end of the first epoch using 10,000 independent replicates, assuming a uniform prior distribution from 1×10−5 to 5×10−3. Second, given the estimated allele frequencies at the end of the first epoch, we estimated the numbers of generations required for random genetic drift to reach those frequencies starting from one initial allelic copy, using 100,000 independent replicates. To complete the model, we made use of the following four constraints. (1) Under the additive model, the degree of dominance h is 0.5. (2) Under the model of Chen et al.19, the product of the selection coefficient and the number of generations is constant. Given historical evidence of plague in the Middle East for a total of 27 generations, we rescaled the selection coefficient based on rescaling the age estimate. (3) We set the effective population size at 19,602, based on autosomal heterozygosity of the six samples S_Abkhasian-1, S_Armenian-1, S_Druze-1, S_Georgian-1, S_Georgian_2, and S_North_Ossetian-1 from the Simons Genome Diversity Project42 and a mutation rate of 1.17×10−8 per-site per-generation43. The number of trials 2N during the random sampling was therefore 39,204. (4) We assumed a generation time of 28.1 years18.
Mutation haplotypes in geographically diverse mutation carriers
Seventy-three geographically diverse carriers of MEFV_p.M694V or MEFV_p.V726A were genotyped on Illumina Infinium OmniExpressExome-8v1-6_A1 chips. MEFV_p.V726A and MEFV_p.M694V mutations were determined by Sanger sequencing. Chromosome 16 genotypes including mutation genotypes were phased with Eagle 2.4.
Ancient DNA Analyses
We retrieved BAM files from a total of 352 individuals (PMID 22426219, 24762536, 25230663, 26062507, 26351665, 26567969, 26595274, 26748850, 27274049, 27417496, 29743352, and 29743675). These individuals came from sites in Armenia, Czech Republic, Denmark, Estonia, Georgia, Germany, Hungary, Iran, Italy, Kazakhstan, Kyrgyzstan, Lithuania, Luxembourg, Macedonia, Mongolia, Montenegro, Poland, Russia, Slovakia, Spain, Sweden, Switzerland, Turkey, and Turkmenistan. The time periods represented by these individuals extends from ~12,000 calBCE to ~1850 calCE. We called genotypes for MEFV_p.M694V (rs61752717) and MEFV_p.V726A (rs28940579) using bam2mpg (https://github.com/nhansen/bam2mpg), with a quality filter of 20.
Mice
Wild-type C57BL/6/J and IL-1R−/− mice were from The Jackson Laboratory. FMF-KI mice harboring a human B30.2 domain (MefvB30.2/B30.2) or FMF-associated mutant human B30.2 domain (MefvM680I/M680I and MefvV726A/V726A) and Mefv-deficient (Mefv−/−) mice have been described previously3, 12, 44. Mice used in each experiment were randomized by age and sex. The investigators were blinded to allocation during experiments and outcome assessment for experiments shown in Fig. 8a and b. All animal studies were performed according to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of National Human Genome Research Institute or by the Stony Brook University Institutional Animal Care and Use Committee.
Bacterial strains
The Y. pestis KIM5 yopM+ and isogenic ΔyopM mutant strains have been previously described (PMID 26810037) and are excluded from Select Agent guidelines due to the deletion of the chromosomal pgm locus (Δpgm). B. cenocepacia strain was purchased from ATCC (BAA245).
Gene knockdown and ASC speck assay
siRNAs targeting RSK1, RSK2, RSK3, PKN1, PKN2, MEFV and scrambled siRNA were purchased from Invitrogen. The three siRNAs for RSK knockdown were 5’-CACUGAUUCUGAAGGCGAA-3’ (s12273); 5’-GAGAUUUGUUUACACGCUU-3’ (s12279), 5’-GGUUUUCCCUAAGAAGAUG-3’ (1054). The siRNA for MEFV was 5’-CUUUCUGAAACAAACUGAAF-3’ (s502557). The two siRNAs for PKN were 5’-GGACAGUAAGACCAAGAUU-3’ (s11113) and 5’- GACGAGAAGAUGUUAGUAA-3’ (S11116). For siRNA gene knockdown experiments, 50–250 pmol siRNA were transfected into THP1-ASC-GFP cells (thp-ascgfp, Invivogen) by electroporation and plated in a chamber slide (1 X 106 cells per well) or 6 well plate (2 X 106 cells per well). The plated cells were treated with 500 nm PMA for 3h and medium replaced with fresh RPMI medium containing 10% FBS. After 72 h, the transfected cells were infected with Y. pestis (MOI 30). For quantification of ASC specks, ten random fields were selected and cells with an ASC speck were counted from GFP positive cells. For the ASC oligomerization assay, cells were lysed and then insoluble pellets were chemically crosslinked with 2.5 mM disuccinimidyl suberate (DSS) (21658,Thermo Scientific) for 30 min at RT and eluted by SDS sample loading buffer and analyzed by immunoblot with anti-ASC (sc-271054, Santa Cruz Biotechnology) antibody.
Immunoprecipitation, GST pull down and immunoblot
The lysates from mouse BMDMs or human monocytes were immunoprecipitated with anti-RSK2, anti-mouse pyrin44 or anti-human pyrin45 antibodies, followed by Protein A plus Agarose beads (Thermo Scientific, Rockford, IL). After washing with PBS, bound proteins were eluted from the beads by 2X SDS sample buffer and analyzed by immunoblot for RSK2, pyrin, 14-3-3β, 14-3-3ε, or 14-3-3τ. Myc/his-tagged full-length, N-terminal (aa’s 1–330), or C-terminal (aa’s 330–781) human pyrin proteins were transfected in 293T cells. The pyrin proteins were purified using His-SpinTrap™ (28-4013-53, GE Healthcare). Purified full-length, N-terminal, or C-terminal pyrin was incubated with GST or GST-tagged YopM (WT or various mutant forms) for 30 min followed by binding to GST beads. Various deletion constructs of N-terminal pyrin were transfected into 293T cells, and the lysates of the cells were incubated with GST-YopM followed by GST bead binding. After washing with Mammalian Protein Extraction (M-PER) (78505, Thermo Scientific), bound proteins were eluted by 2X SDS sample loading buffer and analyzed by immunoblot with anti-GST or anti-myc antibodies. Immunoblots were prepared with Novex® Tris-Glycine Gel Systems (Invitrogen) and probed overnight at 4°C with anti-mouse IL-1β Ab (AF-401-NA, R&D Systems); anti-14-3-3ε Ab (8C3, sc-23957, Santa Cruz Biotechnology); anti-14-3-3β Ab (60C10, sc-59419, Santa Cruz Biotechnology); anti-RSK1 Ab (A-10, sc-393147, Santa Cruz Biotechnology); anti-RSK2 Ab (E-1, sc-9986, Santa Cruz Biotechnology); anti-RSK3 Ab (3C4C8, sc-517283, Santa Cruz Biotechnology); anti-actin Ab (C-11, sc-1615, Santa Cruz Biotechnology); anti-myc Ab (9E10, sc-40, Santa Cruz Biotechnology); anti-RPS6 Ab (C-8, sc-74459HRP, Santa Cruz Biotechnology); anti-phospho RPS6 Ab (AF3918, R&D Systems); anti-mouse pyrin Ab44; anti-human pyrin Ab45; anti-phospho pyrin (EPR19570, ab200420, Abcam); anti-YopM Ab10 or anti-V5 Ab (R960–25, Invitrogen).
Cell preparation
Mouse BMDMs were obtained by differentiating bone marrow progenitors from the tibia and femur in Iscove’s Modified Dulbecco’s Medium (IMDM) containing 20 ng ml−1 of M-CSF (Peprotech, Rocky Hill, NJ), 10% heat-inactivated fetal bovine serum (FBS, Invitrogen, Carlsbad, CA), 1 mM sodium pyruvate, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin (Invitrogen) for 7 days. BMDMs were then replated in 12-well plates one day before experiments. Blood specimens from healthy controls, FMF patients, and carriers were drawn after obtaining informed consent under a protocol approved by the NIAMS/NIDDK Institutional Review Board or approved by the ethics committee of Hacettepe University Faculty of Medicine, Turkey. Human PBMCs were isolated by LSM-Lymphocyte Separation Medium (50494, MP Biomedicals, Santa Ana, CA) from freshly drawn peripheral venous blood from healthy controls or patients. All FMF patients have common homozygous or compond heterozygous B30.2 domain mutations (MEFV_p.M680I, MEFV_p.M694V, and MEFV_p.V726A).
Macrophage or monocyte infection
At 18h prior to infection, mouse BMDMs (1.0 × 106 cells per well) were plated in 12-well plates in RPMI 1640 (Invitrogen) containing 10% FBS, and antibiotics and then primed with 1 μg ml−1 LPS for 6h. For BMDM infections, Y. pestis strains were grown overnight in Luria broth (LB) at 28°C. The following day, cultures were diluted 1:40 in LB containing 20 mM sodium oxalate and 20 mM MgCl2 and grown at 28°C for 1h, then shifted to 37°C for 2h. LPS-primed BMDMs were left uninfected or infected with Y. pestis or ΔyopM strains cultured at various MOI. Tissue culture plates were centrifuged at 95 × g for 5 min to promote contact of Yersinia with BMDMs and incubated at 37°C. At 90 min post-infection, supernatants were collected and processed for IL-1β measurement, while cell lysates were harvested for western blot analysis. For infection experiments with C3 toxin, C3 toxin was added simultaneously with bacteria to BMDMs at the start of infection. For human monocyte infection, monocytes were purified from PBMCs using CD14 microbeads (130-050-201, Miltenyi Biotec). Monocytes (0.4 × 106 cells per well) were plated in 24-well plates and then infected with Y. pestis or B. cenocepacia. At 90 min post-infection, cell culture media were changed to fresh media and cells were incubated for 3h. For infection experiments with inhibitors, SL0101–1 (2250, Tocris Bioscience) or BRD 7389 (4037, Tocris Bioscience), cells were pretreated with inhibitors for 1h, and infected with Y.pestis after washing twice with PBS.
In vitro kinase assay
Purified N-terminal pyrin and recombinant human ribosomal protein S6 (H00006194-P01, Abnova) that was purchased from Novusbio were used as substrates. Kinase assays were carried out in kinase buffer (50 mM HEPES pH7.5, 0.01% BRIJ-35, 10 mM MgCl2, 1 mM EGTA, 500 μM ATP and 2.5 mM DTT) with 0.5 μg of RSK1 (PV3680, Thermo Scientific), RSK2 (PV3323, Thermo Scientific), RSK3 (A31518, Thermo Scientific) or PKN1 (PR7255B, Thermo Scientific) at 30°C for 30 min. Reactions were stopped by adding SDS loading buffer and separated by SDS/PAGE. Phosphorylation status of pyrin protein was analyzed by immunoblotting with anti-phospho Ser antibody (4E2, 9606, Cell Signaling) or anti-phospho pyrin (EPR19570, ab200420, Abcam) antibody. Immunoblot was visualized on Fc-Odyssey (Li-cor).
Retroviral transduction
WT MEFV, or MEFV with the MEFV_p.S208A, MEFV_p.S209A, MEFV_p.E148Q, MEFV_p.S242A, MEFV_p.M680I, MEFV_p.M694V, or MEFV_p.V726A mutation was cloned into pLNCX2 vector (631503, Takara) and then transfected into RetroPackTM PT67 cells (631510, Takara). After 10h of transfection, media were changed to fresh media and cells were incubated overnight. The cell culture media of the transfected PT67 cells, which contain viral particles, were collected by filtration with low protein binding 0.22 μm syringe filter. The viral particle-containing media were transfered to U937 cells for viral infection in the presence of polybrene (8 μg ml−1). Cells were centrifuged at 1200 × g for 90 min at 32°C and incubated for 8h. Transduced cells were selected with G418 (500 μg ml−1) for 3 weeks. Selected U937 cells were differentiated into macrophages with 500 nM PMA for 24h and then infected with Y.pestis for 1.5h. All cell lines were negative for mycoplasma.
Quantitative RT-PCR
Total RNA was harvested from mouse BMDMs or human PBMCs with Trizol reagent (15596–018, Invitrogen) after treatment with LPS or infection with Y. pestis. From the RNA, cDNA was synthesized using SuperScript First-Strand Synthesis System (11904–018, Invitrogen). PCR was run on ViiA 7 (Applied Biosystems) with 2x TaqMan Fast Universal PCR Master Mix (4352042, Applied Biosystems) using fast protocol. All primers and probes used are from Applied Biosystems: Rps6ka1 (Mm00436395_m1), Rps6ka2 (Mm00441179_m1), Rps6ka3 (Mm00455829_m1), Rps6ka6 (Mm01225184_m1), RPS6KA1 (Hs01546654_m1), RPS6KA2 (Hs00179731_m1), RPS6KA3 (Hs00177936_m1), and RPS6KA6 (Hs00914519_m1).
Mouse infection
Y. pestis was grown overnight in LB at 28°C and harvested by centrifugation at 20,000 X g for 5 min. Y. pestis pellets were resuspended in PBS at approximately 1,000 or 1,500 CFU/ml. A volume of 100 μl was delivered by tail vein injection into C57BL/6 or various FMF-KI mice. Time to death was monitored for 15 days, upon which the remaining mice were euthanized.
Data analysis, statistics and experimental repeats
Statistical analyses were performed with the t-test for two groups or one-way ANOVA for multiple groups using GraphPad Prism (GraphPad Software, San Diego CA). No statistical methods were used to predetermine sample size and the experiments were not randomized. The number of reproduced experimental repeats is described in the relevant figure legends. The investigators were not blinded to allocation during experiments and outcome assessment, except as noted above. The datasets generated during and/or analysed during the current study are available from the corresponding author on request.
Code Availability
Computer code for the forward-time simulations is available at https://github.com/dshriner/forward-time-simulators under the GNU General Public License v3.0.
DATA AVAILABILITY
The datasets generated during and/or analysed during the current study are available from Supplementary Information and Source Data files in the online version of the paper and also available from the corresponding author on request.
Extended Data
Extended Data Fig. 1. Haplotypes from 2,313 Turkish individuals provide evidence of evolutionarily recent positive selection and estimates of selection intensity and duration of selection on FMF-associated pyrin mutations.

a-c, Histograms of unstandardized nSL statistics of GWAS variants37 with similar frequency and local recombination rate as each FMF mutation. a, 1,402 markers similar to MEFV_p.V726A; b, 2,380 markers similar to MEFV_p.M694V; and c, 5,442 markers similar to MEFV_p.E148Q. The most extreme negative nSL values have the strongest evidence of recent positive selection. d-f, Log-likelihood plots and selection co-efficient estimates with 95% confidence intervals and mutation age estimates with 95% confidence intervals of three MEFV mutations, determined with a hidden Markov model method from the multi-locus haplotype structure of 4,626 Turkish chromosomes. d, MEFV_p.V726A; e, MEFV_p.M694V; and f, MEFV_p.E148Q. g-i, Histograms of iHH values for the ancestral alleles of markers with similar allele frequency and local recombination rate as each MEFV mutation, demonstrating that the ancestral alleles have not been under positive selection, thereby providing evidence that the mutations have not been under balancing selection. g, 1,570 markers similar to MEFV_ p.V726, h, 2,677 markers similar to MEFV_p.M694, and i, 6,166 markers similar to MEFV_p.E148.
Extended Data Fig. 2. Geographically diverse FMF mutation carriers exhibit extended conserved haplotypes.
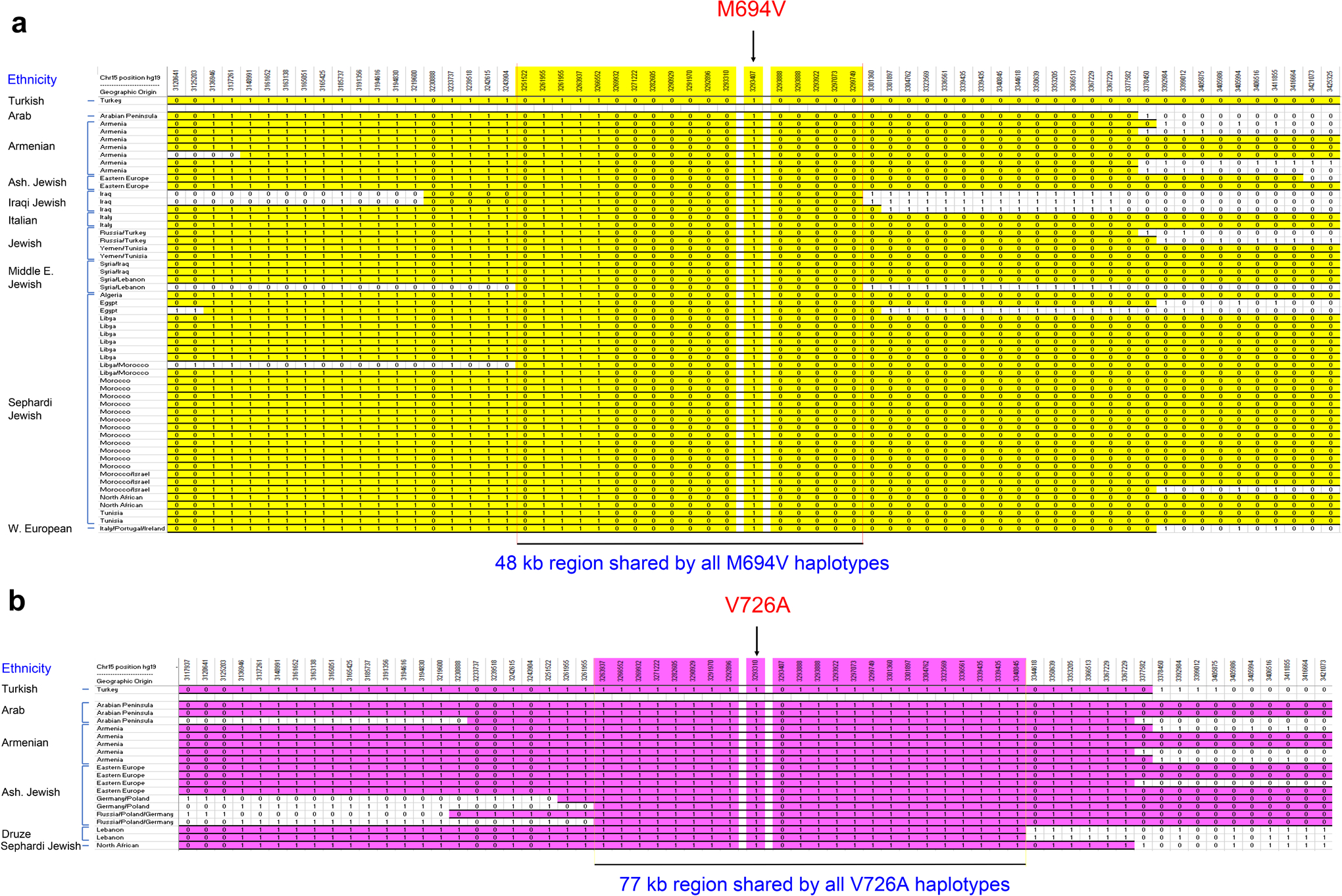
a, MEFV_p.M694V conserved haplotypes (yellow). The length of the MEFV_p.M694V haplotype shared by all the diverse carrier chromosomes = 48 kb and shared by greater than 50% of the diverse carrier chromosomes = 307 kb. b, MEFV_p.V726A conserved haplotypes (pink). The length of the MEFV_p.V726A haplotype shared by all the diverse carrier chromosomes = 77 kb and shared by greater than 50% of the diverse carrier chromosomes = 262 kb. The fully conserved shared haplotypes in the diverse mutation carriers are identical to the respective haplotypes in the Turkish population.
Extended Data Fig. 3. Y. pestis YopM suppresses the pyrin inflammasome.
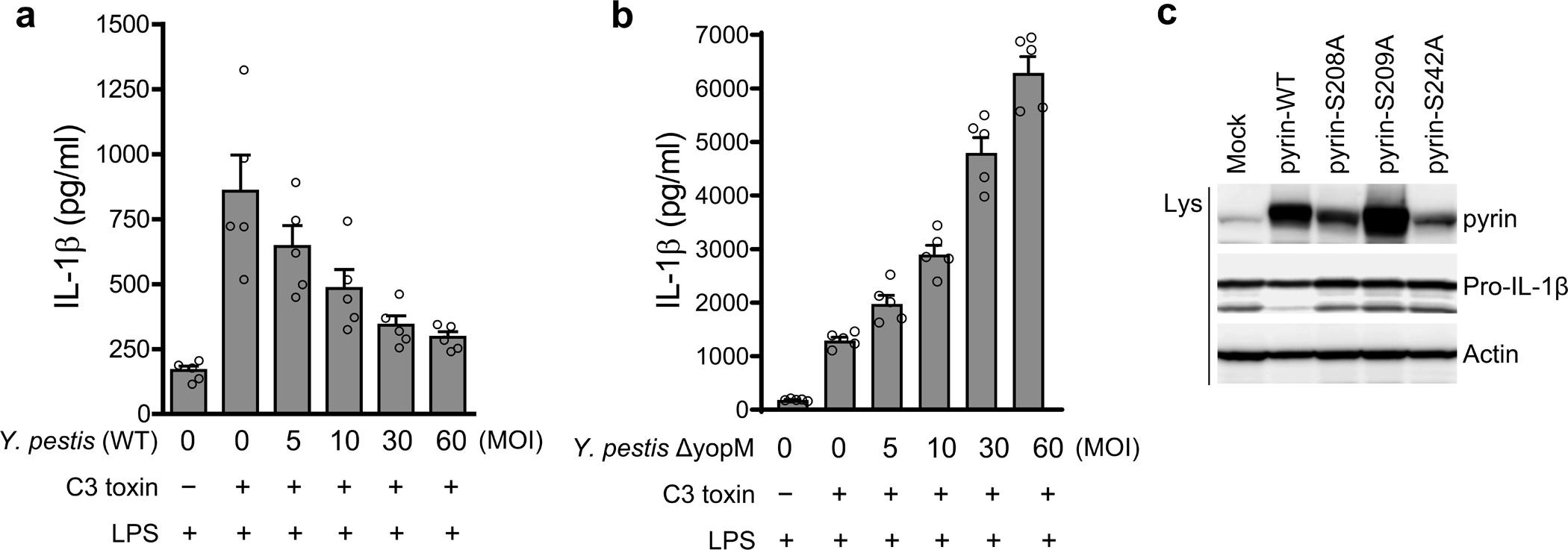
a,b, IL-1β measurements of culture supernatants of Mefv+/+ BMDMs primed with LPS, treated with or without C3 toxin, and infected with indicated MOI of WT (a) or ΔyopM (b) Y. pestis strains. Results are presented as mean ± s.e.m., for n=5 independent biological replicates. c, Immunoblot analysis of pyrin and pro-IL-1β in lysates of retroviral transduced U937 cells, expressing WT or indicated Ser to Ala mutant pyrin proteins. Data are representative of three independent experiments with similar results.
Extended Data Fig. 4. RSK-YopM mediated pyrin phosphorylation is independent of PKN.
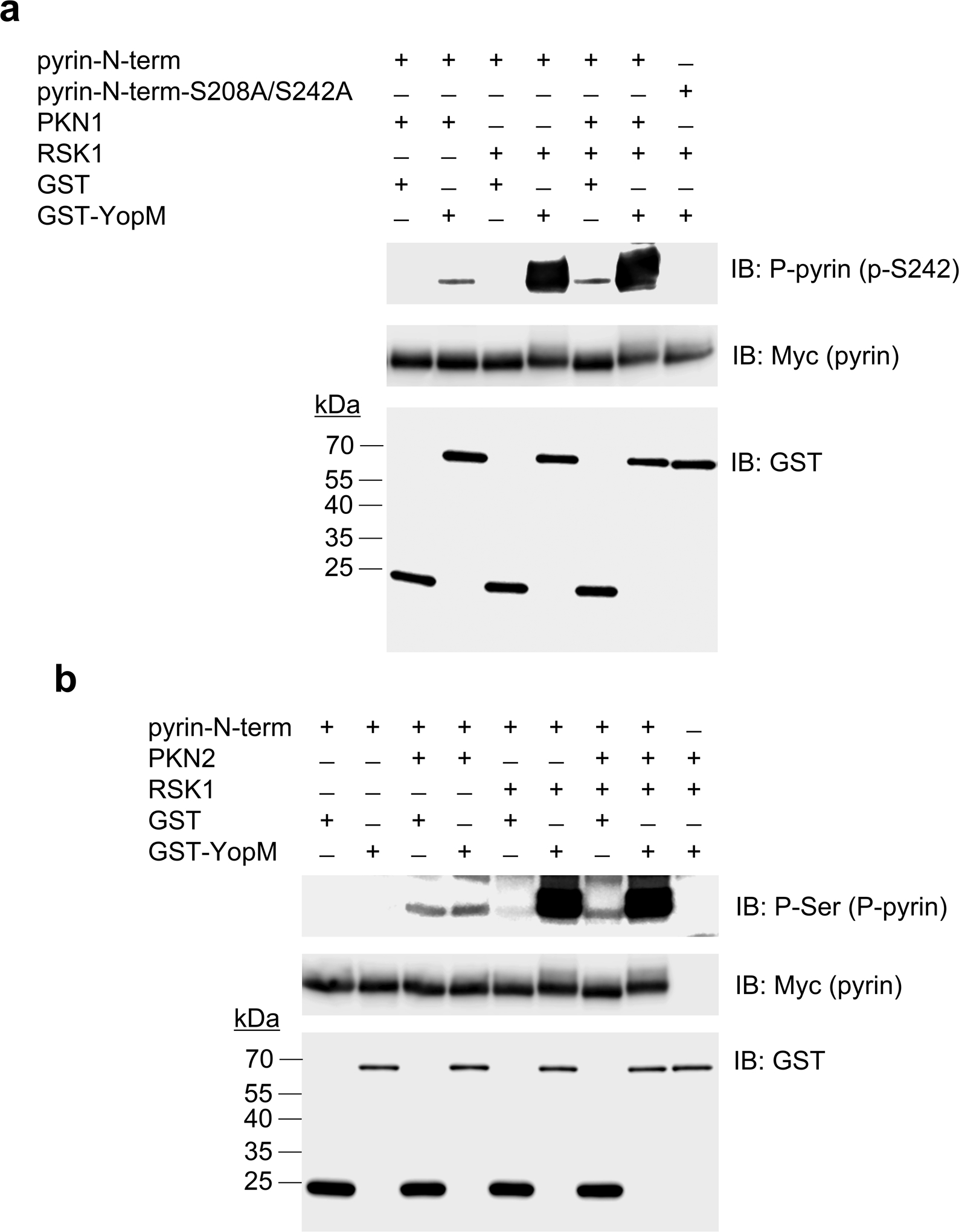
In vitro kinase assay of purified Myc/His-tagged N-terminal human pyrin (amino acids 1–330) incubated with recombinant PKN1 and/or RSK1 (a) or PKN2 and/or RSK1 (b) in the presence of purified GST or GST-YopM, and analyzed by immunoblot with antibody specific for S242 phosphorylated pyrin (a) or phosphorylated serine (b). Data are representative of three independent experiments with similar results.
Extended Data Fig. 5. Human and murine RSK isoforms exhibit a restricted distribution of gene expression in leukocytes.
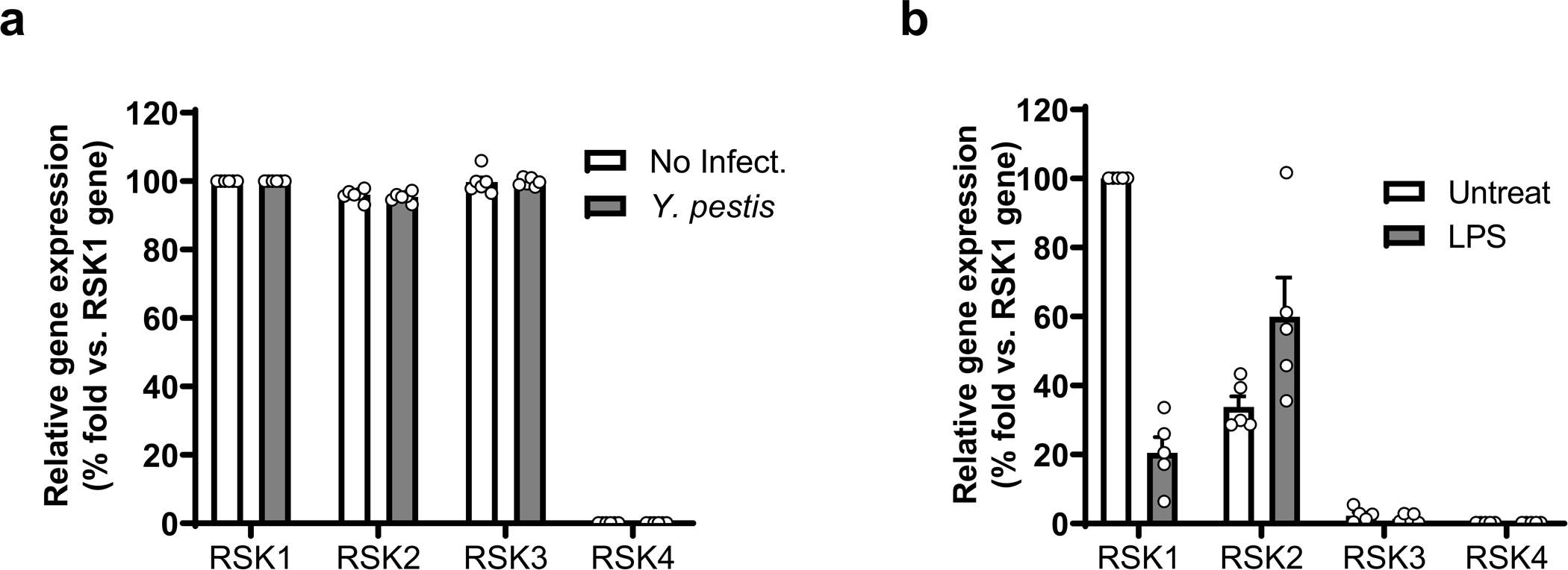
5 a,b, Relative gene expression profile of RSK isoforms in human PBMCs with or without Y. pestis infection (a) and mouse BMDMs with or without LPS priming (b), and assayed by RT-QPCR. Results are presented as mean ± s.e.m., for n=5 independent biological replicates.
Extended Data Fig. 6. Knockdown of RSK1, RSK2 and RSK3 in THP-1 cells induces ASC oligomerization.

a, Immunoblot analysis of lysates of THP-1 cells transiently transfected with negative control siRNA with no substantial sequence similarity to human gene sequences (N.C.) or a mixture of siRNAs targeting RSK1, RSK2 and RSK3. b, ASC oligomerization analysis by immunoblot from THP-1 cells transfected with N.C. or a mixture of siRNAs targeting RSK1, RSK2 and RSK3, and infected with Y. pestis (MOI 30). Cell lysates and the disuccinimidyl suberate (DSS)-treated pellets were analyzed by immunoblot with anti-ASC antibody. Data are representative of three independent experiments with similar results.
Extended Data Fig. 7. YopM binds to multiple regions of N-terminal pyrin.
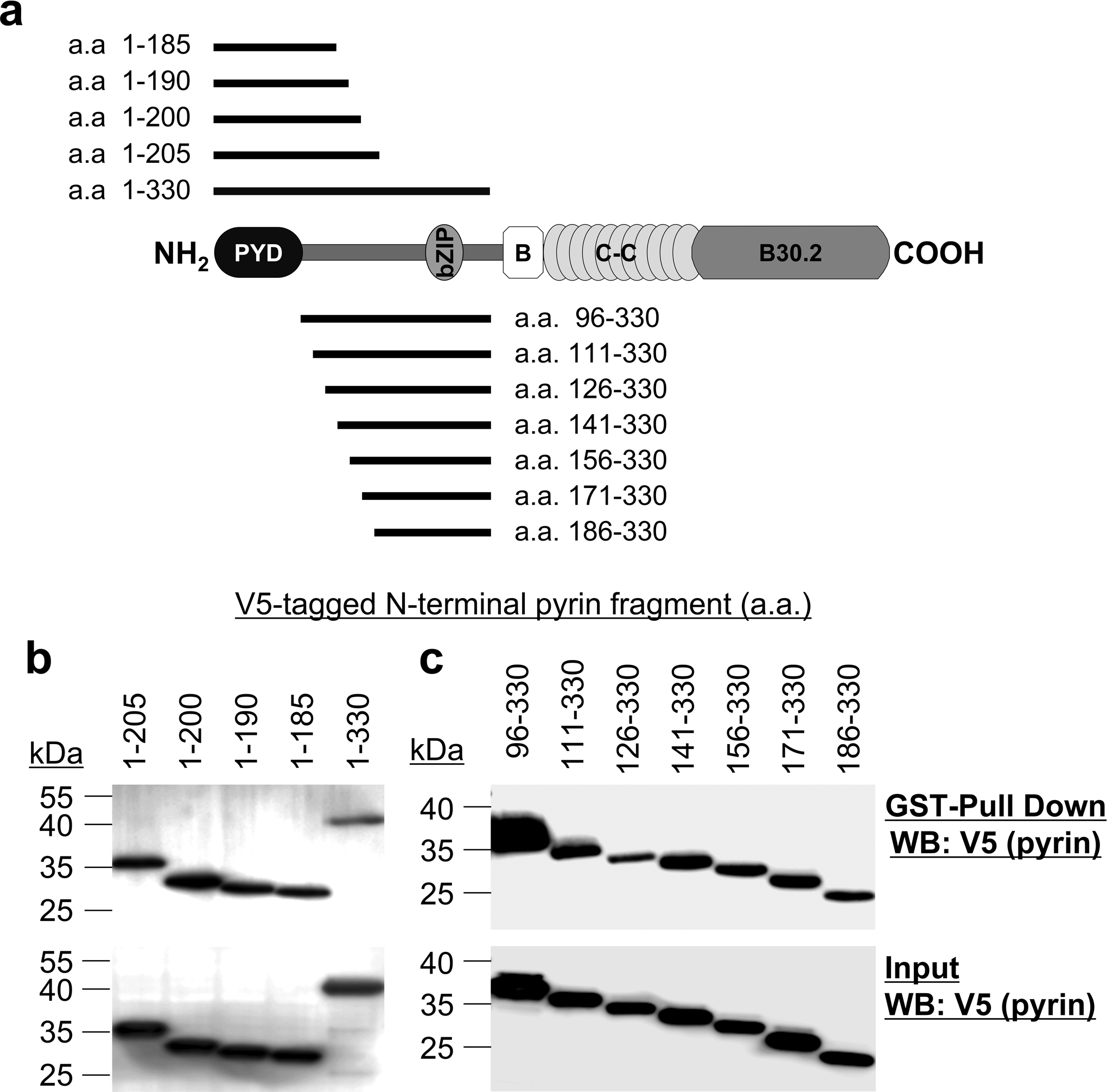
a, Schematic structure of human pyrin with various deletion fragments of N-terminal human pyrin. b,c, GST-pulldown assay of V5-tagged various N-terminal human pyrin fragments with purified GST-YopM. Data are representative of three independent experiments with similar results.
Supplementary Material
ACKNOWLEDGMENTS
We thank the patients enrolled in our clinical protocols for providing research specimens, A. Jones, T. Romeo, L. Poe, and R. Laird for help with caring for patients, and A. Schaffer for thoughtful discussions. This work was supported by the Intramural Research Programs of the National Human Genome Research Institute, the National Institute of Allergy and Infectious Diseases, the National Institite of Arthritis and Musculoskeletal and Skin Diseases, and the Center for Research on Genomics and Global Health. This research was also supported by the National Institute of Allergy and Infectious Diseases award R01AI099222 (to J.B.B.) and utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Footnotes
COMPETING INTERESTS
The authors declare no competing interests
REFERENCES
- 1.International_FMF_Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90, 797–807 (1997). [DOI] [PubMed] [Google Scholar]
- 2.French_FMF_Consortium. A candidate gene for familial Mediterranean fever. Nat. Genet 17, 25–31 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Chae JJ et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity 34, 755–768 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sohar E, Gafni J, Pras M & Heller H Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am. J. Med 43, 227–253 (1967). [DOI] [PubMed] [Google Scholar]
- 5.Touitou I The spectrum of Familial Mediterranean Fever (FMF) mutations. Eur. J. of Hum. Genet 9, 473–483 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Kirino Y et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behcet disease. Proc. Natl. Acad. Sci. USA 110, 8134–8139 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papadopoulos VP, Giaglis S, Mitroulis I & Ritis K The population genetics of familial mediterranean fever: a meta-analysis study. Ann Hum Genet 72, 752–761 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Yilmaz E et al. Mutation frequency of Familial Mediterranean Fever and evidence for a high carrier rate in the Turkish population. Eur. J. Hum. Genet 9, 553–555 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Koshy R, Sivadas A & Scaria V Genetic epidemiology of familial Mediterranean fever through integrative analysis of whole genome and exome sequences from Middle East and North Africa. Clinical Genetics 93, 92–102 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Zvereff VV, Faruki H, Edwards M & Friedman KJ Cystic fibrosis carrier screening in a North American population. Genet Med 16, 539–546 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Xu H et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Park YH, Wood G, Kastner DL & Chae JJ Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol 17, 914–921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung LK et al. The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome. Cell Host Microbe 20, 296–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ratner D et al. The Yersinia pestis Effector YopM Inhibits Pyrin Inflammasome Activation. PLoS Pathog 12, e1006035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voight BF, Kudaravalli S, Wen X & Pritchard JK A map of recent positive selection in the human genome. PLoS Biol 4, e72 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrer-Admetlla A, Liang M, Korneliussen T & Nielsen R On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol. Biol. Evol 31, 1275–1291 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gandolfo LC, Bahlo M & Speed TP Dating rare mutations from small samples with dense marker data. Genetics 197, 1315–1327 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moorjani P et al. A genetic method for dating ancient genomes provides a direct estimate of human generation interval in the last 45,000 years. Proc. Natl. Acad. Sci. USA 113, 5652–5657 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H, Hey J & Slatkin M A hidden Markov model for investigating recent positive selection through haplotype structure. Theor Popul Biol 99, 18–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashemi Shahraki A, Carniel E & Mostafavi E Plague in Iran: its history and current status. Epidemiol Health 38, e2016033 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald C, Vacratsis PO, Bliska JB & Dixon JE The yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. The Journal of biological chemistry 278, 18514–18523 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Hentschke M et al. Yersinia virulence factor YopM induces sustained RSK activation by interfering with dephosphorylation. PLoS One 5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evdokimov AG, Anderson DE, Routzahn KM & Waugh DS Unusual molecular architecture of the Yersinia pestis cytotoxin YopM: a leucine-rich repeat protein with the shortest repeating unit. J. Mol. Biol 312, 807–821 (2001). [DOI] [PubMed] [Google Scholar]
- 24.McPhee JB, Mena P & Bliska JB Delineation of regions of the Yersinia YopM protein required for interaction with the RSK1 and PRK2 host kinases and their requirement for interleukin-10 production and virulence. Infect Immun 78, 3529–3539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCoy MW, Marre ML, Lesser CF & Mecsas J The C-terminal tail of Yersinia pseudotuberculosis YopM is critical for interacting with RSK1 and for virulence. Infect Immun 78, 2584–2598 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aubert DF et al. A Burkholderia Type VI Effector Deamidates Rho GTPases to Activate the Pyrin Inflammasome and Trigger Inflammation. Cell Host Microbe 19, 664–674 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Allison AC Protection afforded by sickle-cell trait against subtertian malarial infection. Br Med J 1, 290–294 (1954). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chae JJ et al. Isolation, genomic organization, and expression analysis of the mouse and rat homologs of MEFV, the gene for familial mediterranean fever. Mamm Genome 11, 428–435 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Osei-Owusu P, Charlton TM, Kim HK, Missiakas D & Schneewind O FPR1 is the plague receptor on host immune cells. Nature 574, 57–62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Achtman M et al. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. USA 96, 14043–14048 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner DM et al. Yersinia pestis and the plague of Justinian 541–543 AD: a genomic analysis. Lancet Infect Dis 14, 319–326 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Achtman M How old are bacterial pathogens? Proc Biol Sci 283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kogan A et al. Common MEFV mutations among Jewish ethnic groups in Israel: high frequency of carrier and phenotype III states and absence of a perceptible biological advantage for the carrier state. Am. J. Med. Genet 102, 272–276 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Ben-Chetrit E & Touitou I Familial mediterranean Fever in the world. Arthritis Rheum 61, 1447–1453 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Ben-Chetrit E & Levy M Familial Mediterranean fever. Lancet 351, 659–664 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y & Shao F Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence. Curr. Opin. Microbiol 29, 37–42 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Remmers EF et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet’s disease. Nat. Genet 42, 698–702 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrett JC, Fry B, Maller J & Daly MJ Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265, doi: 10.1093/bioinformatics/bth457 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Loh PR, Palamara PF & Price AL Fast and accurate long-range phasing in a UK Biobank cohort. Nat. Genet 48, 811–816, doi: 10.1038/ng.3571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabeti PC et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 419, 832–837, doi: 10.1038/nature01140 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Szpiech ZA & Hernandez RD selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol 31, 2824–2827, doi: 10.1093/molbev/msu211 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mallick S et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Genomes Project C et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chae JJ et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell 11, 591–604 (2003) [DOI] [PubMed] [Google Scholar]
- 45.Chae JJ et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc. Natl. Acad. Sci. USA 103, 9982–9987 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from Supplementary Information and Source Data files in the online version of the paper and also available from the corresponding author on request.