

Summary
A major unresolved challenge in cell-based regenerative medicine is the absence of non-invasive technologies for tracking cell fate in deep tissue and with high spatial resolution over an extended interval. MRI is highly suited for this task, but current methods fail to provide longitudinal monitoring or high sensitivity, or both. In this study, we fill this technological gap with the first discovery and demonstration of in vivo cellular production of endogenous bright contrast via an MRI genetic reporter system that forms manganese-ferritin nanoparticles. We demonstrate this technology in human embryonic kidney cells genetically modified to stably overexpress ferritin and show that, in the presence of manganese, these cells produce far greater contrast than conventional ferritin overexpression with iron or manganese-permeable cells. In living mice, diffusely implanted bright-ferritin cells produce the highest and most sustained contrast in skeletal muscle. The bright-ferritin platform has potential for on-demand, longitudinal, and sensitive cell tracking in vivo.
Subject Areas: Medical Imaging, Technical Aspects of Cell Biology, Nanomaterials
Graphical Abstract
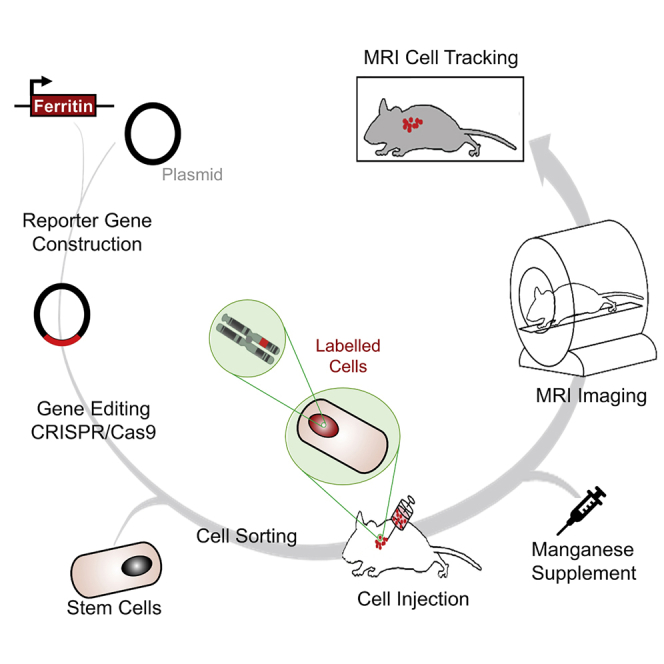
Highlights
-
•
First bright-ferritin MRI gene reporter platform for longitudinal cell tracking
-
•
In vivo assembly of manganese nanoparticles for bright MRI contrast
-
•
On-demand, sensitive, non-invasive in vivo deep imaging of proliferating cells
Medical Imaging; Technical Aspects of Cell Biology; Nanomaterials
Introduction
In vivo cell tracking is valuable across a multitude of applications ranging from stem cell therapy to studies of cancer metastasis. To visualize and distinguish the cells of interest, we must impart to them a differential contrast against background tissue. The simplest approach is to label the cells directly, prior to injection or implantation, with an image modality-specific contrast agent, such as iron oxides for magnetic resonance imaging (MRI) (Li et al., 2013) or 18F-FDG for nuclear medicine imaging (Lang et al., 2013). This exogenous labeling approach, however, works well only for short-term studies and cannot attain the desired capability for monitoring over the long term. Multiple factors underlie this shortcoming, foremost of which are label dilution upon cell division, leakage of contrast agent from cells (Venter et al., 2018), and non-specific labeling of macrophages that take up contrast agents released from dying cells (Ma et al., 2015). Longitudinal cell tracking requires a method that provides sustained contrast specific to the viable cells of interest. To date, the most promising solution to longitudinal cell tracking in vivo is via reporter genes.
A variety of reporter genes have been proposed over the years for use with different imaging modalities. Of note are firefly luciferase for bioluminescence imaging (Bernau et al., 2014), herpes simplex virus 1 thymidine kinase for nuclear medicine (Koehne et al., 2003), and ferritin for MRI (Naumova et al., 2010). Among the modalities suitable for cell tracking, MRI is particularly attractive, because it uniquely affords flexible background tissue contrast, unlimited tissue penetration depth, absence of radiation, and superior spatial resolution compared with nuclear medicine and bioluminescence imaging (Pan et al., 2010). Ferritin, a polymeric iron storage protein, is the most widely used among MR reporters (Cohen et al., 2005; Iordanova and Ahrens, 2012), as other MR gene reporter systems are less accessible owing to very low sensitivity or the requirement for specialized coils tuned to different nuclei (Chen et al., 2011; Patrick et al., 2015). Despite the success achieved with ferritin for cell tracking, however, there remain a number of technical challenges. The change in MR relaxation time is frequently small and the resulting signal drop modest (Naumova et al., 2014; Vande Velde et al., 2011); very high levels of ferritin and/or iron are required to achieve the requisite detection sensitivity (Deans et al., 2006; Genove et al., 2005), and the onset of signal change is slow as iron requires days to accumulate (Iordanova et al., 2010). Sensitive and longitudinal cell tracking remains an unmet need.
In this work, we describe a “bright-ferritin” mechanism for sensitive, longitudinal cell tracking in vivo. This approach uses the cell's ferritin machinery to self-assemble manganese (Mn) nanoparticles, which confer a positive contrast on MRI. While previous work in the 1990s had shown the in vitro nucleation and growth of Mn particles in the cavity of extracted ferritin protein under harsh chemical conditions (Mackle et al., 1993; Meldrum et al., 1991, 1995), we report herein, for the first time, the in vivo self-assembly of endogenous Mn nanostructures. The bright contrast gleaned from Mn-ferritin nanoparticles can overcome many limitations associated with conventional negative contrast from ferritin overexpression. The main advantages are: (1) higher specificity, as negative contrast cannot be clearly distinguished from intrinsically dark sources (e.g., tissue/air interface, microbleeds); (2) higher sensitivity, especially in intrinsically dark background tissues (e.g., skeletal muscle); (3) accurate delineation of cell distribution (i.e., no “blooming” artifact); and (4) the potential for quantitation. Our comparison of bright-ferritin against conventional “dark-ferritin” cell imaging both in vitro and in vivo confirmed a substantially greater sensitivity of cell detection for the former. Bright-ferritin is also shown to rival the sensitivity derived from another positive-contrast reporter gene, the divalent metal transporter-1 (DMT-1), a membrane channel protein whose overexpression leads to increased transmembrane transport of free Mn ions (Bartelle et al., 2013). Our exploitation of the cell's machinery for endogenous production of bright-contrast Mn-ferritin nanoparticles presents a paradigm shift in the utilization of ferritin for on-demand, longitudinal, and sensitive in vivo cell tracking in cell-based therapeutics.
Results
In Situ Manganese Encapsulation by Ferritin-Overexpressing Cells
To demonstrate the bright-ferritin technology, we chose to overexpress the human ferritin protein using a non-viral CRISPR-Cas9 system for targeted integration into the AAVS1 “safe-harbor” locus in human embryonic kidney (HEK293) cells (Figure 1A). Monoclonal cell lines selected for enhanced levels of ferritin expression demonstrated stable gene and protein levels relative to wild-type cells (Figure S1).
Figure 1.

Engineering of Mammalian Bright-Ferritin Reporter Gene System
(A) Plasmid vector diagram for CRISPR-Cas9 gene editing for insertion of the human ferritin transgene at the AAVS1 locus.
(B–D) (B) Schematic of stable cell line generation and assay for intracellular nanoparticle formation. Representative TEM of (C) ferritin nanoparticle subcellular localization and (D) purified electron-dense ferritin nanoparticles (red arrowheads) extracted from ferritin-overexpressing cells supplemented with 0.2 mM MnCl2 for 24 h.
(E) Cellular manganese content from purified ferritin nanoparticles in wild-type (WT) and ferritin-overexpressing (FrT) cells with or without Mn supplementation.
(F) Relative ferritin protein level normalized to α-tubulin in WT and FrT cells. Mn-incubated WT and FrT cells have different ferritin protein expression levels (∗p < 0.05). Data in subfigures (E) and (F) are represented as mean ± SEM.
See also Figures S1 and S2.
The bright-ferritin effect, as will be shown shortly, results from endogenous self-assembly of Mn-ferritin nanoparticles. To confirm this mechanism of intracellular particle formation, stable HEK cell lines overexpressing ferritin were supplemented with free Mn in culture prior to intracellular imaging with transmission electron microscopy (TEM) (Figure 1B). Whole-cell TEM sections of mutant cells revealed visibly distinct aggregates of nanoparticles accumulating in vesicles, which themselves reside in lysosome-like structures (Figure 1C), an observation consistent with previous reports of subcellular localization of ferritin nanoparticles (Sibille et al., 1989). In contrast, wild-type cells contained minimal nanoparticles with no visible aggregates (Figure S2A). Furthermore, cell lysates were collected and immuno-precipitated with ferritin monoclonal antibody to extract and purify the intracellular ferritin. TEM of the ferritin protein purified from mutant cells post-Mn incubation revealed electron-dense metallic particles with discrete mineral cores averaging 5.4 ± 0.3 nm in diameter (Figure 1D), similar in size to endogenous ferritin-iron nanocages measured by TEM (Farrant, 1954; Watabe and Hoshino, 1976). Ferritin extracts purified prior to Mn incubation, however, contained no electron-dense metallic nanoparticles (Figure S2B). To confirm the identity of the metallic core, elemental analysis for Mn was conducted on the purified ferritin particles via inductively coupled plasma atomic emission spectroscopy (ICP-AES). Ferritin-overexpressing cells had over twice the Mn-ferritin content (Figure 1E) and twice the ferritin protein expression levels (Figure 1F) relative to wild-type cells.
Cellular Expression of Ferritin Provides Efficient Bright Contrast on MRI
In vitro MRI reveals substantial contrast enhancement from stable ferritin-overexpressing cells that assemble Mn-ferritin nanoparticles intracellularly. Figure 2 illustrates live-cell imaging on a clinical 3T MR scanner. Ferritin-overexpressing and wild-type cells, both with and without Mn supplementation, were imaged in glass tubes using conventional T1-weighted imaging to visualize bright contrast and T1 mapping to quantify contrast-induced longitudinal relaxation effects (Figure 2A). At baseline, ferritin-overexpressing and wild-type cells displayed similar contrast levels. Upon Mn supplementation, ferritin-overexpressing cells exhibited a T1 that was ∼2.5-fold lower than that of wild-type cells (Figure 2B), thus rendering a higher signal on T1-weighted scans. For this reason, we dub this MRI reporter gene complex bright-ferritin, a nomenclature we shall use hereon to describe the turning “on” of bright contrast via the cell's ferritin machinery.
Figure 2.

Bright-Contrast Efficiency In Vitro on MRI
(A) T1-weighted spin echo image and T1 map of wild-type (WT) and ferritin-overexpressing (FrT) cells incubated with 0.2 mM MnCl2 for 24 h. Arrow indicates the bright-contrast cell pellet.
(B–E) (B) Tabulated mean T1 value ± SD. The contrast efficiencies of all MR gene reporter systems are compared by measuring relaxation rate versus metal concentration; data are represented as mean ± SD (C–E).
We next assessed the sensitivity of the bright-ferritin system relative to that achieved via other MR reporter gene systems (we used CRISPR-Cas9 targeting for all reporters; see Figure S1). The contrast efficiency of bright-ferritin (Figure 2C) was compared with that of conventional ferritin with iron supplementation (Figure 2D) and against free Mn transport via DMT-1 overexpression (Figure 2E). Illustrated in these graphs is the relationship between the change in relaxation rate (R1 = 1/T1 or R2 = 1/T2, depending on the MR reporter) and Mn or iron concentration. A steeper slope indicates a greater relaxivity associated with higher contrast efficiency. The bright-ferritin system had a high relaxivity of 17.7 mM−1s−1, exceeding those of engineered exogenous chemical agents and MR reporter gene gold standards (Alvares et al., 2017; Bartelle et al., 2013; Iordanova and Ahrens, 2012; Kalman et al., 2010; Sana et al., 2012). By comparison, ferritin overexpression with iron supplementation, the most widely used MR reporter system to date (Iordanova and Ahrens, 2012), had a very low relaxivity of 2.15 mM−1s−1. Even when a high dose of 0.9 mM iron was used to supplement ferritin-overexpressing cells, a modest 1.2-fold change in R2 relative to wild-type was achieved. This low efficiency is the result of a lower ferritin protein overexpression from targeted CRISPR-Cas9 transfection, an approach we opted for over common non-targeted methods that entail undesired multiple insertions. To explore differences in signal generated from encapsulated Mn versus free Mn ions, we compared bright-ferritin against DMT-1. The DMT-1 system had a modest relaxivity of 7.1 mM−1s−1, most likely due to the challenge of cellular storage of free Mn. Clearly, bright-ferritin confers the highest contrast efficiency required for sensitive in vivo cell tracking.
Bright-Ferritin Is a Non-toxic MR Reporter Gene Complex
Absence of cellular toxicity is also very important in the setting of cell therapy. Not only must the cell-tracking technology be sensitive, but any modifications made to the therapeutic cells also cannot alter the cell's intended function to grow and replace tissue in the long-term. In our bright-ferritin platform, we introduced two modifications: gene editing for ferritin overexpression and Mn supplementation. To assess potential cytotoxicity associated with these modifications, we measured cell proliferation and metabolic activity for bright-ferritin and the other MR reporter systems at the optimal Mn or iron dose required for visible MR contrast. Cell viability and growth were assessed using a live/dead assay after dosing and expansion (Figure 3A). Cells were stained with a cell permeable/enzymatically activatable “green” fluorescent dye (Calcein AM) and a cell impermeable/DNA-binding “red” dye (EthD-1) to identify live and dead cells, respectively. Cell viability and growth were visually consistent at different time intervals between ferritin-overexpressing and wild-type cells. Minimal cell death and similar cell densities were observed for all reporter systems and treatment groups (Figure 3B). Cell metabolism assessed with WST-1 proliferation assay (Figure 3C), which quantitatively measures enzymatic activity in metabolically viable cells, showed no difference among the different reporter systems and treatment groups, indicating the absence of genomic stress or cytotoxicity from Mn supplementation.
Figure 3.

Biocompatibility of Genetic Reporter Systems
(A) Live (green)/dead (red) fluorescent assay at 0 and 48 h post Mn supplementation revealed minimal cell death and minimal impact on cell growth from ferritin overexpression and supplementation with 0.2 mM Mn for 24 h. Scale bar, 400 μm.
(B) Cell proliferation measured by cell density at 72 h after Mn supplementation (0.2 mM Mn for 24 h) or iron supplementation (0.9 mM Fe for 72 h).
(C) Metabolic (WST-1) assay for the same conditions as (B). Positive controls were treated with 5% dimethyl sulfoxide (DMSO). Negative controls were cultured in standard growth media with no additional supplementation. Data are represented as mean ± SEM.
Bright-Ferritin Is a Superior MR Reporter Gene for In Vivo Cell Tracking
Having validated the superior contrast efficiency of the bright-ferritin platform in vitro, our next challenge was to investigate its performance in vivo, where injected cells are likely to be diffuse and Mn exposure is poorly controlled at the site of cell injection owing to physiological variations. The performance of bright-ferritin, conventional dark-ferritin, and DMT-1 in NOD/SCID mice was evaluated longitudinally on a 3T MR scanner using quantitative MR relaxometry. Cells carrying the genetic reporters were injected into the muscle of one leg, and wild-type cells were injected in the contralateral leg. Histology confirmed the presence of viable cells distributed throughout the leg muscle (Figure S3). For the bright-ferritin and DMT-1 systems, free Mn was administered subcutaneously (s.c.) and allowed to accumulate for 24 h. For the conventional dark-ferritin system, in accordance with previous reports, no supplementation was given for the first 48 h to allow for endogenous iron buildup, after which animals were given daily oral doses of iron for five consecutive days.
Prior to contrast administration (day 1), none of the MR reporter systems presented differential contrast between the two legs (Figure 4A, top row). After Mn injection (day 2), bright contrast appeared in the leg containing bright-ferritin and DMT-1 cells relative to the contralateral leg containing wild-type cells. Notably, the signal enhancement produced by bright-ferritin was sustained visibly for up to 5 days, whereas the signal produced by DMT-1 had diminished by this time. To restore bright signal in the DMT-1 cells, a re-injection of Mn was required. In contrast to bright-ferritin and DMT-1, conventional dark-ferritin imaging produced no visible contrast relative to the contralateral leg. This was true during the first few days without iron supplementation (days 1 and 2) and even at later times with oral iron supplementation (days 5 and 7).
Figure 4.

In Vivo MRI of HEK Cell Injections in Mice
(A) MRI of NOD/SCID mice injected with ferritin or DMT-1-overexpressing cells in the left leg and wild-type cells in the contralateral leg (site of cell injection indicated by yellow arrow). Subcutaneous MnCl2 supplementation (administered subcutaneously at superior-inferior aspect indicated by white arrow) produced large signal enhancement in the leg containing bright-ferritin- (top row) and DMT-1- (bottom row) overexpressing cells. To recover signal loss in DMT-1 cells after 4 days, MnCl2 was re-applied to turn “on” signal. Dark-ferritin cells (middle row) showed no contrast change, both without (day 1 and 2) and with iron supplement (day 5 and 7); oral iron supplementation was given daily after day 2. Quantitative relaxometry revealed (B) significant changes in R1 in the bright-ferritin and DMT-1 legs relative to wild-type but (C) minimal difference in R2∗ on conventional dark-ferritin imaging. Difference in R1 between bright-ferritin and DMT-1 is significant at all times (∗p < 0.05); difference in R1 between DMT-1 and wild-type is significant only at day 2 (#p < 0.05). Data are represented as mean ± SD.
Quantitative MR relaxometry supported these visual findings. Both bright-ferritin and DMT-1 demonstrated a significant increase in R1 after a single dose of Mn (Figure 4B). A maximum R1 increase of 3.5-fold, 2.3-fold, and 1.5-fold relative to pre-contrast levels was achieved with the bright-ferritin, DMT-1, and WT systems, respectively. Enhanced relaxation rates were higher and maintained longer in the bright-ferritin system than in DMT-1. Unsurprisingly, the dark-ferritin system exhibited no temporal changes in R2∗ (Figure 4C). Note that R2∗ is used here instead of R1, because in the dark-ferritin system, contrast arises from susceptibility effects.
In addition to considering temporal contrast changes relative to pre-contrast levels, we also assessed changes in contrast ratios (i.e., ferritin: wild-type or DMT-1: wild-type), as this metric represents the “sensitivity” of the different MR reporter platforms in distinguishing the cells of interest against background tissue (Figure 5A). Bright-ferritin provided the greatest change in relative contrast, with a maximum change in relative R1 of ∼2.0. By comparison, DMT-1 had a maximum change in relative R1 of 1.3 and conventional dark-ferritin had negligible changes in relative R2∗. Based on the literature and observed results, we suggest in the following a potential mechanism for the bright-ferritin and DMT-1 systems (Figure 5B). In the DMT-1 system, free Mn is transported into the cell via DMT-1 and other endogenous pathways (Au et al., 2008). DMT-1 is an active proton-coupled metal ion symporter, which means it catalyzes the co-transport of H+ protons and divalent metals (e.g., Mn2+). At neutral pH, it symports one proton for every divalent metal cation and its import capabilities is enhanced by increasing the extracellular proton concentration (Gunshin et al., 1997; Moos and Morgan, 2000). As more Mn2+ and H+ enter a cell, it is reasonable to expect the intracellular pH to decrease, thus reducing the proton gradient force that drives Mn uptake. The result is a plateau in Mn-induced signal, which we observed between days 2 and 4. As the extracellular concentration of Mn continues to drop, the rate of Mn uptake falls below that of Mn elimination, thus reducing intracellular Mn and bright contrast. It is important to emphasize that this postulation is difficult to validate completely, given our current understanding of the mechanisms underlying proton-coupled transporters is incomplete (Ehrnstorfer et al., 2017). It is also important to note that unlike most previous reports utilizing intravenous and intraperitoneal administration of Mn (Bartelle et al., 2013), we take advantage of s.c. injection for its slow release kinetics to achieve higher contrast and longer signal retention.
Figure 5.

In Vivo Relative Contrast for Different MR Reporter Gene Systems
(A) Normalization of relaxation rates of the three gene reporter systems against wild-type HEK cells in vivo. Difference in relative R1 between bright-ferritin and DMT-1 is significant from day 2 to 5 (∗p < 0.01); difference in relative R1 between DMT-1 and dark-ferritin is significant at day 2 (#p < 0.05). Data are represented as mean ± SD.
(B) Proposed mechanisms of contrast generation in the bright-ferritin and DMT-1 systems. Manganese transport and storage vary with time. Red dots represent Mn ions. Green organelles represent lysosome-like bodies.
In the bright-ferritin system, Mn again enters the cell via endogenous mechanisms (Au et al., 2008). Once inside a cell, Mn does not remain in its ionic form but is sequestered by excess ferritin protein inside a mineral core. The sequestration slows the transition toward equilibrium (of free Mn), thus maintaining a gradient that drives Mn uptake into the cell over a prolonged interval. We postulate that Mn-ferritin particles do not remain indefinitely in the cytosol; rather, they are eventually engulfed in autophagosomes and the ferritin is ultimately degraded by lysosomal proteases. This hypothesis would be consistent with our observations on TEM, where we saw numerous dark puncta within a smaller vesicle found inside a lysosome-like structure. As the extracellular concentration of Mn continues to decrease, a point is reached where the rate of Mn-ferritin synthesis falls below the rate of ferritin turnover and Mn excretion. The result is a decline of intracellular Mn content and bright contrast.
Discussion
We report here the first utilization of a cell's own machinery for in vivo cellular synthesis of endogenous Mn-ferritin nanoparticles to enable sensitive and non-invasive bright-contrast cell tracking over long intervals. This MRI platform, which we call bright-ferritin, seeks to attain the long-sought goal of longitudinal cell tracking in regenerative medicine, and it does so via the use of reporter genes for endogenous contrast generation in viable cells. To compete with the sensitivity provided by short-lived exogenous cell labeling methods, the bright-ferritin system must break the low sensitivity ceiling that had hindered MR reporter gene systems. In this work, we proved in vivo and in vitro the superior sensitivity and contrast efficiency of bright-ferritin relative to other MR reporters, including conventional dark-contrast imaging using iron-ferritin particles and bright-contrast cell imaging using DMT-1 overexpression. Importantly, we achieved high detection sensitivity without introducing genomic stress and using low, non-toxic levels of exposure to Mn supplementation. The new bright-ferritin platform meets the key requirements for a practical cell-tracking technology in regenerative medicine: longitudinal monitoring capability, no ionizing radiation or radioactive tracers, unlimited depth penetration, high spatial resolution, high sensitivity to the viable cells of interest, high specificity to exclude non-targeted cells, and absence of cytotoxicity.
An important consideration with genetic modification is the potential for unintended off-target mutations and alteration of vital cell functions. We must limit not only the number of insertions but also the locations of insertions. It is for this reason that we adopted CRISPR-Cas9 to perform a single, targeted insertion at the “safe-harbor” AAVS1 locus. Although our approach minimizes the risk of deleterious effects, it also reduces the protein expression level, and, therefore, the amount of available contrast, compared with common non-targeted insertions that achieve very high overexpression (up to 60-fold) of MR genetic reporters (Bartelle et al., 2013; Genove et al., 2005; Iordanova et al., 2010; Thomas and Smart, 2005; Tiffen et al., 2010). This difference—single, targeted versus multiple, non-targeted insertions—may explain why we obtained negligible contrast with conventional dark-ferritin when many have reported modest contrast changes. The fact that our bright-ferritin system was able to furnish large contrast changes at low ferritin levels is exceedingly beneficial, as high protein overexpression commonly employed can create cellular stress and non-targeted systems are very unlikely to enter the clinical domain. These “safety” attributes of our bright-ferritin platform are essential attributes in any system intended for integration in the body for tissue regeneration.
The attainable sensitivity of a cell-tracking system can be determined only in an in vivo setting, where the diffusion and migration of injected cells against a background of non-uniform tissue, which are absent in a controlled in vitro setting, together with limited control over Mn distribution to the cell injection site, can substantially diminish the contrast achievable. Despite these in vivo challenges, our bright-ferritin system could readily identify and track diffuse cell populations over days in the mouse leg and with a high degree of detection sensitivity. It is equally important to emphasize that the capability for targeted imaging afforded by our system overcomes a longstanding limitation of exogenous nanoparticles and traditional cellular imaging methods (Tietjen et al., 2018). Our system now lays the foundation for enhanced non-invasive cell monitoring, with the ability to assess cell death in the early days post injection, cell migration throughout tissue over time, and continued cell growth from the original injected population. Furthermore, our use of bright contrast (as opposed to dark contrast with traditional ferritin) opens the door to cell tracking in inherently low-signal tissues such as skeletal and cardiac muscle (Naumova et al., 2010). Although not investigated fully in this work, the possibility exists with bright-contrast mechanisms for quantification of cell numbers. On this point, it is interesting to note the consistency we observed in ferritin protein overexpression (2-fold increase), cellular Mn-ferritin content (2.2-fold increase), in vitro change in R1 (2.3-fold increase), and in vivo change in R1 (2-fold increase). This high degree of correlation between ferritin overexpression, Mn content, and MR contrast changes strongly indicates a robust and controllable platform with quantitative cell-tracking capabilities.
One seemingly peculiar phenomenon is why, at low concentrations, Mn is able to provide a large change in contrast whereas iron cannot. The answer lies in the inherent difference by which Mn and iron nanoparticles alter MR relaxation rates underlying contrast changes. Iron nanoparticles, when aggregated at high concentrations, can effectively distort the local magnetic field, thus creating a change in the transverse relaxation rate R2∗ that produces dark signal. However, to effect a significant distortion of the local field, there must be an abundance of iron nanoparticles. With our low level of ferritin overexpression, we did not reach the particle concentration required for a measurable R2∗ effect. On the other hand, Mn nanoparticles produce contrast changes by altering the R1 relaxation rate of the water protons with which the particles interact. High concentrations of Mn particles are not required for a large pool of water molecules to interact with the Mn ion, because water exchanges very rapidly at a rate of approximately one million times per second.
Another consideration on the topic of contrast efficiency is the impact of the environment on the effective relaxivity of Mn. We measured a longitudinal relaxivity of 17.7 mM−1s−1 per Mn ion in our in vitro cell studies, which is over two times higher than an average of 6.2 mM−1s−1 reported for Mn-loaded ferritin in solution without cells (Kalman et al., 2010). This difference is partly due to the rotational diffusion of the complex, which is expected to be slower inside a cell than it is in solution, thus accounting for slower tumbling and increased relaxivity. Furthermore, our measured relaxivity is representative of the entire cellular system, which includes not only ferritin-bound Mn but also free Mn in the cytosol and Mn ions bound to organelle membranes. This latter portion, which is ascribed to non-ferritin Mn, can be approximated by the effect measured in wild-type cells (Figure 2B). However, the precise proportion of bound versus free Mn cannot be derived from T1 changes alone, because relaxation effects differ between free and bound Mn, as the free ion pool has a smaller effect from rotational diffusion but exchanges very rapidly with water.
There is also the question of what bright-contrast mechanism is best for a particular application. In this work, we characterized the performance of both bright-ferritin and DMT-1. Bright-ferritin produces contrast via the endogenous synthesis of Mn particles, whereas DMT-1 produces contrast via enhancing cellular uptake of free Mn ions. The choice of one over the other ultimately depends on whether a differential toxicity threshold exists between the two. For some cells, there may be no difference. For other cell types, there may be lower compatibility with one mechanism versus the other, because Mn in its ionic form affects cells differently compared with Mn stored in a particle. Neural cells, for example, which are highly sensitive to free ion concentrations, are unlikely to work well with DMT-1, given the vast literature evidence on Mn-related neural toxicity (Wennberg, 1994). The utility of bright-ferritin across a wide array of cell types remains to be explored.
The immediate next step is further pre-clinical investigation of the bright-ferritin technology in different cell therapy platforms. The relatively low cost of requisite materials means the method is easily and repeatedly accessible. Conceivably, any desired cell type can be tracked and against any background tissue, including low-signal tissues that previously failed to provide contrast differences relative to injected cells (Bernau et al., 2016; Pereira et al., 2015; Vande Velde et al., 2012; Vande Velde et al., 2011). More detailed longitudinal studies are needed in animal models to characterize the mechanism of particle elimination, test and retest the turning on of bright signal from the target cells, and uncover differences (if any) in tissue regeneration relative to wild-type cells. At a cellular level, mechanistic studies that provide precise characterization of relaxation effects will be valuable. Building on analogous studies in iron-ferritin systems (Herynek et al., 2000; Vymazal et al., 1996), these future investigations would involve cell systems in which the ferritin-bound Mn fraction is separated out; Mn binding location and loading factor within the nanocage would also be determined. Additionally, the influence of Mn in other cellular compartments such as the cytosol and organelles, whether bound or free, and their influence on relaxation, would be elucidated to provide system insight. Translation into the clinical domain, however, would require further technical and safety characterization and optimization. Rigorous genomic testing is required to ensure the cells are completely safe for injection. We may wish to tailor the mode of administering Mn supplement, such as localized delivery to the injection site or oral supplementation, thus providing an opportunity for further dose reduction. Although these questions remain to be answered, we do have insight on the answers to some questions. For example, our Mn-ferritin nanoparticles had standard core diameters. Particle size is an important parameter, because it dictates a particle's function, permeability, and uptake/degradation (Hoshyar et al., 2016; Theil et al., 2008). By maintaining the size of native ferritin, the particle has a high likelihood of behaving naturally like endogenous ferritin and undergoing similar routes of formation and degradation. Also, there is evidence from in vitro studies performed in solution that Mn can initially bind to ferritin with a stoichiometry of eight ions per molecule (Wardeska et al., 1986), and with specific binding to the ferroxidase center (Ardini et al., 2018); following this, a Mn oxyhydroxide (β-MnOOH) core is formed with various loading amounts from 500 to 4,000 Mn atoms per protein (Meldrum et al., 1991, 1995).
In conclusion, we report the first in vivo MRI cell tracking system that exploits ferritin in combination with manganese supplementation for the endogenous production of highly efficient bright contrast with greater sensitivity and retention than current MR reporters. This bright-ferritin system opens the door for accurate longitudinal monitoring of cell fate, available on demand, across a broad spectrum of applications in regenerative medicine.
Limitations of the Study
The generalized value of the bright-ferritin reporter system has not been investigated across a wide variety of cell types. Further studies are required to establish both sensitivity and safety across different cells and systems. Very long-term monitoring over months and years also was not included in this work and will be considered in future trials that are applications driven. Finally, detailed investigations into potential impact on many other important aspects of cell function need to be undertaken.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Prof. Hai-Ling Cheng (hailing.cheng@utoronto.ca).
Materials Availability
There are restrictions to the availability of the reporter gene constructs and the genetically modified cell lines owing to a patent filed on the invention and materials pertaining to the work presented herein. The cells generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
There are restrictions to the availability of dataset owing to a patent filed on the invention, materials, and data pertaining to the work presented herein.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Michael Laflamme for sharing his expertise on CRISPR-Cas9 gene targeting. Prof. Hai-Ling Cheng is funded by Canada First Research Excellence Fund, Medicine by Design Cycle 1 Team Project; Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant; and Canada Foundation for Innovation/Ontario Research Fund. Prof. Hai-Ying Cheng is funded by Canadian Institutes of Health Research Project Grant and Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant.
Author Contributions
H.-L.M.C. contributed to (1) overall direction, conceptualization, and design of study; (2) design of in vitro and in vivo MRI experiments; (3) design and implementation of MRI protocols; (4) development of software for analyzing quantitative MRI data; (5) in vitro and in vivo MRI; and (6) quantitative MRI analysis. H.-Y.M.C. contributed to (1) design and generation of targeting constructs and (2) PCR analysis. D.A.S. contributed to (1) conceptualization and design of study, (2) generation of stable cell lines, (3) western blot and densitometric analysis, (4) cellular Mn uptake studies, (5) design and execution of protein extraction and purification, (6) electron microscopy sample preparation and imaging, (7) analytical manganese content analysis, (8) toxicity assays and histology, (9) in vitro and in vivo MRI, and (10) statistical analysis of MRI data. X.A.L. contributed to (1) generation of stable cell lines, (2) western blot analysis, and (3) protein purification. All authors contributed to manuscript drafting and approved the final version.
Declaration of Interests
A provisional patent on the technology described herein was filed on April 2, 2020.
Cheng HLM, Szulc DA, Cheng HYM, “Ferritin for cellular tracking utilizing T1-weighted MRI,” United Kingdom. 2004892.2. Filed 2020-04-02.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101350.
Supplemental Information
References
- Alvares R.D.A., Szulc D.A., Cheng H.M. A scale to measure MRI contrast agent sensitivity. Sci. Rep. 2017;7:15493. doi: 10.1038/s41598-017-15732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardini M., Howes B.D., Fiorillo A., Falvo E., Sottini S., Rovai D., Lantieri M., Ilari A., Gatteschi D., Spina G. Study of manganese binding to the ferroxidase centre of human H-type ferritin. J. Inorg. Biochem. 2018;182:103–112. doi: 10.1016/j.jinorgbio.2018.02.003. [DOI] [PubMed] [Google Scholar]
- Au C., Benedetto A., Aschner M. Manganese transport in eukaryotes: the role of DMT1. Neurotoxicology. 2008;29:569–576. doi: 10.1016/j.neuro.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelle B.B., Szulc K.U., Suero-Abreu G.A., Rodriguez J.J., Turnbull D.H. Divalent metal transporter, DMT1: a novel MRI reporter protein. Magn. Reson. Med. 2013;70:842–850. doi: 10.1002/mrm.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernau K., Lewis C.M., Petelinsek A.M., Benink H.A., Zimprich C.A., Meyerand M.E., Suzuki M., Svendsen C.N. In vivo tracking of human neural progenitor cells in the rat brain using bioluminescence imaging. J. Neurosci. Methods. 2014;228:67–78. doi: 10.1016/j.jneumeth.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernau K., Lewis C.M., Petelinsek A.M., Reagan M.S., Niles D.J., Mattis V.B., Meyerand M.E., Suzuki M., Svendsen C.N. In vivo tracking of human neural progenitor cells in the rat brain using magnetic resonance imaging is not enhanced by ferritin expression. Cell Transpl. 2016;25:575–592. doi: 10.3727/096368915X688614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A.P., Hurd R.E., Gu Y.P., Wilson D.M., Cunningham C.H. (13)C MR reporter probe system using dynamic nuclear polarization. NMR Biomed. 2011;24:514–520. doi: 10.1002/nbm.1618. [DOI] [PubMed] [Google Scholar]
- Cohen B., Dafni H., Meir G., Harmelin A., Neeman M. Ferritin as an endogenous MRI reporter for noninvasive imaging of gene expression in C6 glioma tumors. Neoplasia. 2005;7:109–117. doi: 10.1593/neo.04436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans A.E., Wadghiri Y.Z., Bernas L.M., Yu X., Rutt R.K., Turnbull D.H. Cellular MRI contrast via coexpression of transferrin receptor and ferritin. Magn. Reson. Med. 2006;56:51–59. doi: 10.1002/mrm.20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnstorfer I.A., Manatschal C., Arnold F.M., Laederach J., Dutzler R. Structural and mechanistic basis of proton-coupled metal ion transport in the SLC11/NRAMP family. Nat. Commun. 2017;8:14033. doi: 10.1038/ncomms14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant J.L. An electron microscopic study of ferritin. Biochim. Biophys. Acta. 1954;13:569–576. doi: 10.1016/0006-3002(54)90376-5. [DOI] [PubMed] [Google Scholar]
- Genove G., DeMarco U., Xu H., Goins W.F., Ahrens E.T. A new transgene reporter for in vivo magnetic resonance imaging. Nat. Med. 2005;11:450–454. doi: 10.1038/nm1208. [DOI] [PubMed] [Google Scholar]
- Gunshin H., Mackenzie B., Berger U.V., Gunshin Y., Romero M.F., Boron W.F., Nussberger S., Gollan J.L., Hediger M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- Herynek V., Bulte J.W., Douglas T., Brooks R.A. Dynamic relaxometry: application to iron uptake by ferritin. J. Biol. Inorg. Chem. 2000;5:51–56. doi: 10.1007/s007750050007. [DOI] [PubMed] [Google Scholar]
- Hoshyar N., Gray S., Han H., Bao G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine (Lond) 2016;11:673–692. doi: 10.2217/nnm.16.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanova B., Ahrens E.T. In vivo magnetic resonance imaging of ferritin-based reporter visualizes native neuroblast migration. Neuroimage. 2012;59:1004–1012. doi: 10.1016/j.neuroimage.2011.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanova B., Robison C.S., Ahrens E.T. Design and characterization of a chimeric ferritin with enhanced iron loading and transverse nmr relaxation rate. J. Biol. Inorg. Chem. 2010;15:957–965. doi: 10.1007/s00775-010-0657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman F.K., Geninatti-Crich S., Aime S. Reduction/dissolution of a beta-MnOOH nanophase in the ferritin cavity to yield a highly sensitive, biologically compatible magnetic resonance imaging agent. Angew. Chem. Int. Ed. 2010;49:612–615. doi: 10.1002/anie.200904731. [DOI] [PubMed] [Google Scholar]
- Koehne G., Doubrovin M., Doubrovina E., Zanzonico P., Gallardo H.F., Ivanova A., Balatoni J., Teruya-Feldstein J., Heller G., May C. Serial in vivo imaging of the targeted migration of human HSV-TK-transduced antigen-specific lymphocytes. Nat. Biotechnol. 2003;21:405–413. doi: 10.1038/nbt805. [DOI] [PubMed] [Google Scholar]
- Lang C., Lehner S., Todica A., Boening G., Franz W.M., Bartenstein P., Hacker M., David R. Positron emission tomography based in-vivo imaging of early phase stem cell retention after intramyocardial delivery in the mouse model. Eur. J. Nucl. Med. Mol. Imaging. 2013;40:1730–1738. doi: 10.1007/s00259-013-2480-1. [DOI] [PubMed] [Google Scholar]
- Li L., Jiang W., Luo K., Song H., Lan F., Wu Y., Gu Z. Superparamagnetic iron oxide nanoparticles as MRI contrast agents for non-invasive stem cell labeling and tracking. Theranostics. 2013;3:595–615. doi: 10.7150/thno.5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma N., Cheng H., Lu M., Liu Q., Chen X., Yin G., Zhu H., Zhang L., Meng X., Tang Y. Magnetic resonance imaging with superparamagnetic iron oxide fails to track the long-term fate of mesenchymal stem cells transplanted into heart. Sci. Rep. 2015;5:9058. doi: 10.1038/srep09058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackle P., Charnock J.M., Garner C.D., Meldrum F.C., Mann S. Characterization of the manganese core of reconstituted ferritin by x-ray absorption spectroscopy. J. Am. Chem. Soc. 1993;115:8471–8472. [Google Scholar]
- Meldrum F.C., Douglas T., Levi S., Arosio P., Mann S. Reconstitution of manganese oxide cores in horse spleen and recombinant ferritins. J. Inorg. Biochem. 1995;58:59–68. doi: 10.1016/0162-0134(94)00037-b. [DOI] [PubMed] [Google Scholar]
- Meldrum F.C., Wade V.J., Nimmo D.L., Heywood B.R., Mann S. Synthesis of inorganic nanophase materials in supramolecular protein cages. Nature. 1991;349:684–687. [Google Scholar]
- Moos T., Morgan E.H. Transferrin and transferrin receptor function in brain barrier systems. Cell Mol. Neurobiol. 2000;20:77–95. doi: 10.1023/A:1006948027674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova A.V., Balu N., Yarnykh V.L., Reinecke H., Murry C.E., Yuan C. Magnetic resonance imaging tracking of graft survival in the infarcted heart: iron oxide particles versus ferritin overexpression approach. J. Cardiovasc. Pharmacol. Ther. 2014;19:358–367. doi: 10.1177/1074248414525999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova A.V., Reinecke H., Yarnykh V., Deem J., Yuan C., Murry C.E. Ferritin overexpression for noninvasive magnetic resonance imaging-based tracking of stem cells transplanted into the heart. Mol. Imaging. 2010;9:201–210. [PMC free article] [PubMed] [Google Scholar]
- Pan D., Caruthers S.D., Chen J., Winter P.M., SenPan A., Schmieder A.H., Wickline S.A., Lanza G.M. Nanomedicine strategies for molecular targets with MRI and optical imaging. Future Med. Chem. 2010;2:471–490. doi: 10.4155/fmc.10.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick P.S., Kettunen M.I., Tee S.S., Rodrigues T.B., Serrao E., Timm K.N., McGuire S., Brindle K.M. Detection of transgene expression using hyperpolarized 13C urea and diffusion-weighted magnetic resonance spectroscopy. Magn. Reson. Med. 2015;73:1401–1406. doi: 10.1002/mrm.25254. [DOI] [PubMed] [Google Scholar]
- Pereira S.M., Moss D., Williams S.R., Murray P., Taylor A. Overexpression of the MRI reporter genes ferritin and transferrin receptor affect iron homeostasis and produce limited contrast in mesenchymal stem cells. Int. J. Mol. Sci. 2015;16:15481–15496. doi: 10.3390/ijms160715481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sana B., Poh C.L., Lim S. A manganese-ferritin nanocomposite as an ultrasensitive T2 contrast agent. Chem. Commun. 2012;48:862–864. doi: 10.1039/c1cc15189d. [DOI] [PubMed] [Google Scholar]
- Sibille J.C., Ciriolo M., Kondo H., Crichton R.R., Aisen P. Subcellular localization of ferritin and iron taken up by rat hepatocytes. Biochem. J. 1989;262:685–688. doi: 10.1042/bj2620685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theil E.C., Liu X.S., Tosha T. Gated pores in the ferritin protein nanocage. Inorg. Chim Acta. 2008;361:868–874. doi: 10.1016/j.ica.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P., Smart T.G. HEK293 cell line: a vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods. 2005;51:187–200. doi: 10.1016/j.vascn.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Tietjen G.T., Bracaglia L.G., Saltzman W.M., Pober J.S. Focus on fundamental: achieving effective nanoparticle targeting. Trends Mol. Med. 2018;24:P598–P606. doi: 10.1016/j.molmed.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffen J.C., Bailey C.G., Ng C., Rasko J.E., Holst J. Luciferase expression and bioluminescence does not affect tumor cell growth in vitro or in vivo. Mol. Cancer. 2010;9:299. doi: 10.1186/1476-4598-9-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde G., Raman Rangarajan J., Vreys R., Guglielmetti C., Dresselaers T., Verhoye M., Van der Linden A., Debyser Z., Baekelandt V., Maes F. Quantitative evaluation of MRI-based tracking of ferritin-labeled endogenous neural stem cell progeny in rodent brain. Neuroimage. 2012;62:367–380. doi: 10.1016/j.neuroimage.2012.04.040. [DOI] [PubMed] [Google Scholar]
- Vande Velde G., Rangarajan J.R., Toelen J., Dresselaers T., Ibrahimi A., Krylychkina O., Vreys R., Van der Linden A., Maes F., Debyser Z. Evaluation of the specificity and sensitivity of ferritin as an MRI reporter gene in the mouse brain using lentiviral and adeno-associated viral vectors. Gene Ther. 2011;18:594–605. doi: 10.1038/gt.2011.2. [DOI] [PubMed] [Google Scholar]
- Venter A., Szulc D.A., Loai S., Ganesh T., Haedicke I.E., Cheng H.L. A manganese porphyrin-based T1 contrast agent for cellular MR imaging of human embryonic stem cells. Sci. Rep. 2018;8:12129. doi: 10.1038/s41598-018-30661-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vymazal J., Zak O., Bulte J.W., Aisen P., Brooks R.A. T1 and T2 of ferritin solutions: effect of loading factor. Magn. Reson. Med. 1996;36:61–65. doi: 10.1002/mrm.1910360111. [DOI] [PubMed] [Google Scholar]
- Wardeska J.G., Viglione B., Chasteen N.D. Metal ion complexes of apoferritin. Evidence for initial binding in the hydrophilic channels. J. Biol. Chem. 1986;261:6677–6683. [PubMed] [Google Scholar]
- Watabe T., Hoshino T. Observation of individual ferritin particles by means of scanning electron microscope. J. Electron Microsc. (Tokyo) 1976;25:31–33. [Google Scholar]
- Wennberg A. Neurotoxic effects of selected metals. Scand. J. Work Environ. Health. 1994;20:65–71. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are restrictions to the availability of dataset owing to a patent filed on the invention, materials, and data pertaining to the work presented herein.