

SUMMARY
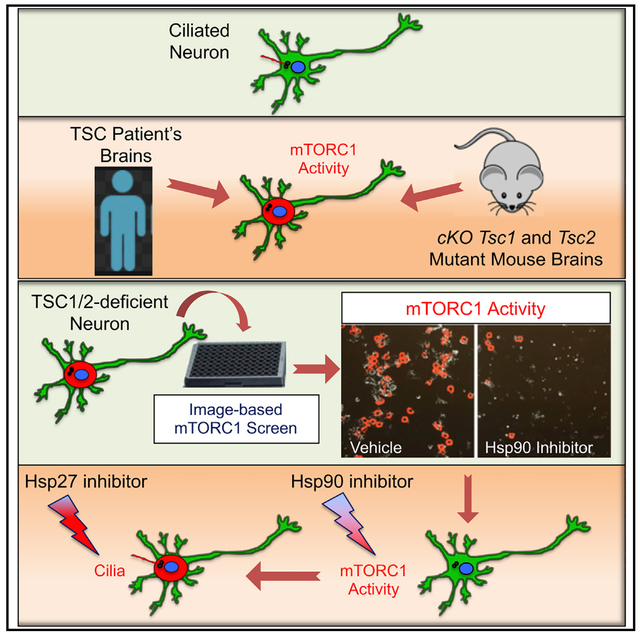
Tuberous sclerosis complex (TSC) is a neurogenetic disorder that leads to elevated mechanistic targeting of rapamycin complex 1 (mTORC1) activity. Cilia can be affected by mTORC1 signaling, and ciliary deficits are associated with neurodevelopmental disorders. Here, we examine whether neuronal cilia are affected in TSC. We show that cortical tubers from TSC patients and mutant mouse brains have fewer cilia. Using high-content image-based assays, we demonstrate that mTORC1 activity inversely correlates with ciliation in TSC1/2-deficientneurons.To investigate the mechanistic relationship between mTORC1 and cilia, we perform a phenotypic screen for mTORC1 inhibitors with TSC1/2-deficient neurons. We identify inhibitors ofthe heat shock protein 90 (Hsp90) that suppress mTORC1 through regulation of phosphatidylinositol 3-kinase (PI3K)/Akt signaling. Pharmacological inhibition of Hsp90 rescues ciliation through downregulation of Hsp27. Our study uncovers the heat-shock machinery as a druggable signaling node to restore mTORC1 activity and cilia due to loss of TSC1/2, and it provides broadly applicable platforms for studying TSC-related neuronal dysfunction.
In Brief
Di Nardo et al. find that cortical tubers from TSC patients and mutant mouse brains have fewer cilia. An image-based screening of mTORC1 activity in TSC1/2-deficient neurons leads to the identification of the heat-shock machinery as a druggable signaling node to restore mTORC1 activity and cilia.
Graphical Abstract
INTRODUCTION
Tuberous sclerosis complex (TSC) is a neurodevelopmental disorder with an incidence of 1 in 6,000 caused by mutations in either the TSC1 or TSC2 genes, which encode proteins that form the TSC1/2 protein complex. TSC is associated with benign tumors called hamartomas in multiple organs as well as central nervous system (CNS) manifestations including epilepsy, intellectual disability, and autism spectrum disorder (ASD;Tsai and Sahin, 2011). The neurological symptoms of TSC have been correlated with brain lesions called cortical tubers, which are characterized by the presence of giant cells and dysmorphic neurons with immature features (Curatolo et al., 2015). The TSC1/2 complex is an inhibitory regulator of the mechanistic target of rapamycin complex 1 (mTORC1), which coordinates key neurodevelopmental processes (Lipton and Sahin, 2014). Disrupted mTORC1 signaling has been clearly implicated in several aspects of the CNS pathogenesis seen in TSC (Lipton and Sahin, 2014). However, the molecular mechanisms downstream of mTORC1 hyperactivation that contribute to the neuronal abnormalities remain unclear.
Primary cilia (hereafter "cilia") are evolutionarily conserved membrane extensions of the cell surface made of microtubules that extend from a centriole-derived structure called the basal body (Lee and Gleeson, 2011). Cilia are often referred to as sensory antennae since they coordinate extracellular ligand-based signaling, playing a critical role in tissue homeostasis (Gerdes et al., 2009). Mutations in genes required for cilia assembly and/or function underlie a broad spectrum of genetic disorders called ciliopathies. In the CNS, ciliopathies are associated with severe neurodevelopmental outcomes including brain malformations, ASD, and intellectual disability (Bettencourt-Dias et al., 2011; Guemez-Gamboa et al., 2014). A recent study showed that patients with focal malformation of cortical developments (FMCDs) caused by somatic mutations in MTOR have a reduction in neuronal cilia (Park et al., 2018). However, elevated mTORC1 activity caused by loss of Tsc1 or Tsc2 genes in mouse embryonic fibroblasts (MEFs) resulted in a rapamycin-insensitive increase of cilia and ciliary length (Hartman et al., 2009). These and other studies indicate a connection between the mTORC1 and cilia with different outcomes dependent on the cellular type and on the etiology of disrupted mTORC1 signaling (DiBella et al., 2009; Rosengren et al., 2018).
Here we investigated the effect of disinhibition of mTORC1 on cilia using in vivo and in vitro models of TSC and specimens from patients. We observed that neuronal ciliation is reduced in brains of Tsc1- and Tsc2-conditional knockout mice, and in cortical tubers resected from TSC patients with refractory epilepsy. To investigate the mechanism by which disinhibition of mTORC1 affects neuronal cilia, we performed a phenotypic screen for mTORC1 inhibitors using Tsc2 gene knockdown (KD) in hippocampal neurons. We identified inhibitors of the molecular chaperone for the heat shock protein 90 (Hsp90), geldanamycin (GA), and 17-allylamino-GA (17-AGG), as compounds that suppress mTORC1 through regulation of phosphatidylinositol 3-kinase (PI3K)/Akt signaling components. Notably, we showed that 17-AGG improved ciliation at doses far below mTORC1 inhibition during a specific developmental window, and further demonstrated that this effect was through reduced expression of HspB1 gene expression, which encodes the small Hsp27. Together, these data indicate that TSC displays features of a ciliopathy and identify the heat-shock response as a regulator at different nodes within the mTORC1 signaling cascade.
RESULTS
Brains of TSC Patients and CNS-Knockout Mouse Models Have Reduced Ciliated Neurons that Are Restored by Rapamycin In Vivo
Previous work demonstrated that altered cilia gene expression is a risk factor for neuropsychiatric disorders (Marley and von Zastrow, 2012; Migliavacca et al., 2015). To determine whether the ciliary gene signature might be altered in TSC patients, we examined the expression of cilia genes from the Syscilia database in a comprehensive set of TSC-associated cortical tubers and healthy controls recently reported in a genomic study (Martin et al., 2017). We found that genes associated with cilia were more likely to be differentially expressed compared to random genes (Figure 1A; Table S1). To investigate whether differential expression of cilia genes reflected changes in ciliation, we examined cilia in brain specimens resected from patients with refractory epilepsy with or without TSC (Table S2). We stained the ciliary membrane-bound adenylyl cyclase III (ACIII; Bishop et al., 2007)toidentify cilia and co-labeled these with the pan-neurofilament marker SMI311 (Talos et al., 2012) to identify the giant cells present in the cortical tubers of TSC patients. We found that compared to the brains of the non-TSC cases, giant cells in the cortical tubers had a significant reduction in ciliation (Figures 1B and 1C).
Figure 1. Brains of TSC Patients and CNS-Knockout Mouse Models Have Reduced Ciliated Neurons that Are Restored by Rapamycin In Vivo.

(A) Heatmap of differential cilia gene expression (red, low; green, high) in healthy control brains (non-TSC, n = 8) and TSC patient brains (TSC/tuber, n = 16; p < 0.005).
(B) Representative confocal images of epileptic brain specimens from non-TSC (a and d) and TSC (b, c, e, and f), patients stained with SMI311 (giant cell, green),ACIII (cilia, red), and Hoechst (nuclei, blue). white arrows indicate cilia.
(C) Quantification of ciliation in the human tissues (non-TSC n = 6; TSC n = 6; Kruskal-Wallis test, with Dunn’s multiple comparison test, *p < 0.05, ns, nonsignificant).Data represent mean ± SEM.
(D) Representative confocal images of hippocampi from vehicle-treated Tsc2 control (n = 5) and Tsc2 mutant (n = 6) and from rapamycin (Rapa)-treated Tsc2 control (n=5) and Tsc2 mutant (n=8) mice. Sections were stained with ACIII (red, a-d; grayscale, e-h) and with NeuN (green, a–d).
(E) Quantification of the percentage of ciliated neurons (400 neurons/mouse; one-way ANOVA with Tukey’s post hoc test, *p < 0.05, ns, non-significant). Data are mean ± SEM. Scale bars are 10 μm.
We also examined neuronal cilia in TSC mouse models with either a conditional deletion of Tsc1 or Tsc2 driven by the Synapsin-1 promoter, which results in loss of TSC1/2 proteins in postmitotic neurons of the cortex and of the hippocampus. The Tsc1/SynCre mice (Tsc1 mutants) have a shorter lifespan (median age post-natal day 35, P35), and they recapitulate many of the neurological manifestations of TSC, including seizures and presence of ectopic giant cells (Meikle et al., 2007). The Tsc2del3/SynCre mice (Tsc2 mutants) are globally heterozygous for the Tsc2 knockoutallelethroughoutthebodyandalsocarryahypomorphic Tsc2 (del3) allele that retains partial function in Synapsin-1 expressing post-mitoticneurons(Pollizzietal.,2009).Duetopartially retained TSC2 expression, these mice develop seizures around eightweeks,andtheysurviveuntilabouttwelveweeksofage.Cilia were assayed in the Tsc2 mutant animals at eight weeks (P56) by staining with ACIII and co-labeling with NeuN to identify neurons. Consistent with the findings in TSC patient brains, we found that Tsc2 mutant mice have decreased cilia in pyramidal neurons of the CA1 region of the hippocampus compared to controls(Figures 1D and 1E). Similarly, ciliation was reduced in the cortex and hippocampus in the Tsc1 mutant mice at P21 (Figures S1A–S1C). Notably, these ciliation defects were prevented by mTORC1 inhibition with early rapamycin treatment starting at P7 in both TSC models (Figures 1D and 1E; Figures S1A–S1C).
Disinhibition of mTORC1 Activity Due to Tsc2 KD in Neurons Leads to Reduced Ciliation
To investigate how mTORC1 dysregulation due to neuronal TSC loss affected ciliation, we developed high-content image-based assays (HCAs) for unbiased quantification of cilia and mTORC1 activity (hereafter ciliaHCA and mTORC1HCA) in primary neurons. As an in vitro model of TSC, we used rat hippocampal neurons transduced with lentiviral vectors (LVs) expressing a short-hairpin RNA (shRNA) directed against either the Tsc2 (Tsc2-sh) or the luciferase gene as a control (ctrl-sh) tagged with GFP. We have previously shown that LV-mediated Tsc2 KD recapitulates several in vivo manifestations observed in mouse models of TSC (Di Nardo et al., 2009, 2014; Ebrahimi-Fakhari et al., 2016; Ercan et al., 2017; Nie et al., 2015, 2010). In vitro phenotypes include robust TSC2 protein downregulation and mTORC1 activation as shown by the time course of increased phosphorylation of ribosomal protein S6 at serine 240/244 (pS6; Figures S2A and S2B). Both the ciliaHCA and mTORC1HCA were optimized as imaged-based assays, where we monitored cilia and mTORC1 activity by immunofluorescent staining of ACIII to identify cilia and pS6 as a proxy of mTORC1 activation (Figures S2C–S2F). Neurons were transduced in culture at days in vitro (DIV) 1, and cilia and mTORC1 activity were assessed in a time-course experiment at DIV2, DIV5, DIV7, DIV13, and DIV20 (Figure 2A). LV-infected neurons with cilia or with phosphorylated S6 (GFP+ACIII+ and GFP+pS6+, respectively) were identified by automated algorithms to detect subcellular structures and their co-localization specifically optimized for each assay. In control neurons, we observed cilia development between DIV2 and DIV5, which remained stable between DIV7 and DIV20 (Figures 2B and 2D). In contrast, the percentage of ciliated Tsc2-deficient neurons progressively decreased between DIV7 and DIV20 (Figures 2B and 2D). Notably, in the Tsc2-deficient neurons, the decrease in ciliation between DIV7 and DIV13 correlated with an increase in mTORC1 activation, unlike in control neurons where mTORC1 activity declined (Figures 2C and 2E; Figures S2A and S2B). Reduced ciliation was validated by manual imaging and quantification of LV-infected neurons co-labeled with ACIII/centrin (Figures S2G and S2H). No changes in size or ACIII intensity were detected in the remaining cilia of the Tsc2-sh neurons (Figures S2I and S2J). These data show that mTORC1 activity is inversely correlated with cilia in neurons, and suggest that mTORC1 disinhibition acts as a brake on ciliation.
Figure 2. Disinhibition of mTORC1 Activity Due to Tsc2 KD in Neurons Leads to Reduced Ciliation.

(A) Outline of time-course experiment to monitor ACIII and pS6 at DIV2, DIV5, DIV7, DIV13, and DIV20 in hippocampal neurons using the ciliaHCA and the mTORC1HCA.
(B) CiliaHCA: Raw images (a and e) and spot detector algorithm identification of ctrl-sh and Tsc2-sh neurons stained with Hoechst (nuclei, b and f), GFP (LV-infected neurons c and g), and ACIII (cilia, d and h) at DIV13. Arrows in yellow indicate cilia. Scale bar is 10 μm.
(C) mTORC1HCA: Raw images (i and n) and spot detector algorithm identification of ctrl-sh and Tsc2-sh neurons stained with Hoechst (nuclei, j and o), GFP (LV-infected neurons, k and p), and ps6 (mTRORC1 avtivity, m and q) at DIV13. Scale bar is 100 μm.
(D and E) Quantification of ciliation (D) and mTORC1 activity (E). Data represent GFP+ACIII+ neurons (n = 5–18 biological replica/experiment; n = 1–6 experiment/condition, D) and FFP+ps6+ neurons (n=7–8 biologicalbreplica/experiment; n=1–4 experiment/ condition, E) as average percentage of GFP+ cells. Data represent mean ± SEM (unpaired Studenťs t test, **p < 0.005, ****p < 0.0001).
See also Figure S2.
A Phenotypic Screen in Tsc2-KD Neurons Identifies Hsp90 as a Drug Target for mTORC1 through Regulation of PI3K/Akt Signaling Components
To explore potential mTORC1-dependent pathways involved in disrupted ciliation in Tsc2-deficient neurons, we performed a high-content screen to identify bioactive compounds that inhibit S6 phosphorylation using the mTORC1HCA as described above. Control and Tsc2-deficient neurons were transduced at DIV1, and the screen was performed at DIV20 since we found optimal assay robustness and reproducibility at that age in culture (Z prime = 0.18, Figure 3A; Figure S3A). The screen was carried out in duplicate using the Biomol collection library, which includes well-characterized bioactive compounds with known mechanisms of action. The neurons were treated with each compound in duplicate, followed by fixation and immunofluorescent staining with GFP, pS6, and Hoechst. Staining for GFP was used to identify the LV-infected neurons, pS6 intensity was used to determine mTORC1 activity, and Hoechst was used to identify the nuclei. Plate normalization was done by converting the percent of GFP+ pS6+ neurons of compound-treated Tsc2-KD wells into Z scores (Figures 3B and 3C; Table S3). We found that rapamycin was the top hit in our screen, indicating robustness of the mTORC1HCA. Other than rapamycin, positive hits included: two inhibitors of the Hsp90, GA, and 17-AGG, the anti-oxidant Edaravone (MCI-186), the calcium channel blocker Nicardipine-HCl, and the kinase inhibitors K252A, Tyrphostin 9, and LY-294002 (2-Morpholin-4-yl-8-phenylchromen-4-one; Figure 3D). We performed confirmatory nine-point dose-response experiments for both GA and 17-AGG because these compounds hit the same molecular target. Using the mTORC1HCA, we found that GA was more potent (IC50 pS6 = 65 nM) than 17-AGG (IC50 pS6 = 346 nM) (Figures 3E and 3F). These IC50 values were in the range of the average pharmacological potencies for GA and 17-AGG against Hsp90 (GA mean IC50 = 50nM and 17-AGG mean IC50 = 220nM) that were previously reported in a large panel of human cancer cell lines (Kelland et al., 1999). Given that these compounds affect the same target and display rank-order potencies that are well aligned with the literature, it suggests that Hsp90 is a potential regulator of mTORC1 activity in neurons.
Figure 3. A Phenotypic Screen with Tsc2-KD Neurons Identifies Hsp90 as Drug Target for mTORC1 through Regulation of PI3K/Akt Signaling Components.

(A) mTORC1 screen workflow. Tsc2-sh neurons were treated with screen compounds for 24 h between DIV19 and DIV20.
(B) Z score correlation between compound Replica 1 and Replica 2. Percent of GFP+pS6+ neurons was converted to Z score. Z score of −1.8 (p < 0.05) was marked by the red lines. Hits were considered compounds with a Z score < −1.8 in the dotted black box. Compounds used for follow-up experiments are rapamycin (in red), GA (in green), and 17-AGG (in blue); all other compounds are labeled in gray.
(C) Zoom of hits in the dotted black box (B).
(D) List of hits, Z scores, and compound concentration in the library.
(E and F) Validation of GA and 17-AGG by dose-response using the mTORC1HCA. Tsc2-sh neurons were treated with vehicle and with a dose-curve of GA (E) or 17-AGG (F) for 24 h between DIV19 and DIV20. Data are GFP+ ps6+ neurons expressed as average percentage of vehicle-treated Tsc2-sh neurons (n = 7–16 biological replica/condition; one-way ANOVA eith Dunnetťs multiple comparison test, **p < 0.01, ***p < 0.005, ****p < 0.0001). Error bars indicate ± SEM.
(G) Western blot of protein lysates from ctrl-sh and Tsc2-sh treated with vehicle or with 17-AGG dose-curve.
(H–J) Quantification of pS6 (H), total IGF-IRb (I), and p-IGF-IRβ (J) expressed as average fold changes of vehicle-treated ctrl-sh neurons. Western blot data were normalized using GAPDH as loading control. Data quantification relative to vehicle-treated Tsc2-sh neurons (n = 3–4 experiment/condition; one-way ANOVA with Dunnett’s multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.0001). Error bars indicate ± SEM.
(K) Hsp90 inhibition results in IGF-IRβ degradation through the proteasome. Wild-type cortical neurons were treated with vehicle and with 4-μM 17-AGG alone or in the presence of 100-nM bortezomib (BTZ, 24 h at DIV20).
(L) Quantification of western blot (n = 5; one-way ANOVA with Tukey’s multiple comparison test, *p < 0.05, **p < 0.005). Western blot data were normalized using GAPDH as loading control. Data are mean ± SEM.
Hsp90 is a molecular chaperone that protects its client proteins from degradation, and many of its substrates are oncogenic proteins (Neckers and Workman, 2012). Among these, insulin-growth factor-1 Receptor β(IGF-lRβ), Akt, and Raptor are components of the PI3K/mTOR pathway that have been identified as Hsp90 substrates in non-neuronal cells (Basso et al., 2002; Ohji et al., 2006). To test whether Hsp90 affects mTORC1 activity through regulation of IGF-IRβ, Akt, or Raptor in neurons, we examined the expression of these potential client proteins in Tsc2-KD neurons treated with vehicle or with a seven-point dose-response curve at a 3-fold dilution of 17-AGG (assay endpoint DIV20). We found that 17-AGG significantly reduced total IGF-lRβ protein level and pS6 phosphorylation at a dose of 4 μM, while there was no effect on Akt or Raptor levels (Figures 3G–3I; Figures S3B, S3D,and S3F). Similarly, 17-AGG reduced S6 phosphorylation and IGF-IRβ levels in ctrl-sh neurons, indicating that the interaction between Hsp90 and IGF-IRβ represents a general mechanism in neurons (Figures S3G–S3I). We then tested GA in theTsc2-sh neurons and found that it also resulted in a similar reduction of IGF-lRβ expression at dosing in the range of inhibition of S6 phosphorylation (Figures S3J–S3L). Therefore, these data demonstrate that Hsp90 inhibition can reduce the activity of mTORC1, as well as decrease the expression of upstream signaling components.
To investigate whether Hsp90 could function as a chaperone of IGF-IRβ in neurons, we treated wild-type neurons with 17-AGG and then inhibited proteasomal degradation using bortezomib (BTZ). As expected, Hsp90 inhibition led to reduced IGF-IRβ levels, and we found that this reduction was prevented with BTZ. Interestingly, the reduction in S6 phosphorylation was also prevented by BTZ in these neurons (Figures 3K and 3L). These data suggest that Hsp90 protects IGF-IRβ against proteasomal degradation, and therefore, we hypothesized that IGF-IRβ protein stability and signaling may represent a potential mechanism by which Hsp90 inhibition reduces mTORC1 activity. To test our hypothesis, we assessed IGF-IRβ activation by autophosphorylation at Tyr1135/1136. Consistent with previous reports of mTOR-dependent negative feedback on the RTK/Akt pathway (Zhang et al., 2006), we found that IGF-IRβ phosphorylation was reduced in the vehicle-treated Tsc2-sh neurons compared to controls (Figures 3G and 3J). Notably, 17-AGG further reduced phosphorylation of IGF-IRβ receptor, and IGF-IRβ phosphorylation was essentially absent in Tsc2-KD neurons at doses that reduced IGF-IRβ levels and inhibited mTORC1, demonstrating that 17-AGG inhibits IGF-IRβ activity in neurons (Figures 3G and 3J). We also examined downstream components of IGF-IRβ signaling and found that phosphorylation of Akt and of its substrate the proline-rich Akt substrate 40 (PRAS40) were also reduced by 17-AGG (Figures S3B, S3C, and S3E). Interestingly, PRAS40 is an inhibitor of mTORC1, and its phosphorylation reduces PRAS40 binding to mTORC1 (Sancak et al., 2007). Taken together, these data suggest that IGF-IRβ and Akt activity may be permissive to ongoing activation of mTORC1, and that Hsp90 inhibition completely suppresses components of the PI3K/Akt signaling pathway, further contributing to mTORC1 inhibition.
Reduced Ciliation in the Tsc2-KD Neurons Is Prevented by mTORC1 and Hsp90 Inhibition during an Age- and Time-Sensitive Window
We then investigated the acute effect of GA, 17-AGG, and rapamycin on ciliation at concentrations that inhibited mTORC1-inhibition in Tsc2-KD neurons. Neither of the Hsp90 inhibitors nor the rapamycin rescued ciliation under these conditions (Figure S3M). Given that reduced ciliation was improved in the brain of Tsc1 and Tsc2 mouse models by 14 and 49 days of rapamycin treatment, respectively (Figures S1A–S1C; Figures 1D and 1E), we tested whether changing the timing or increasing the duration of rapamycin treatment in Tsc2-KD neurons might show an effect. We assayed ciliation at DIV13 and DIV20, and we used both acute (one day) and prolonged (four to eight days) rapamycin treatment (Figures 4A–4D). We observed a significant increase in ciliation of Tsc2-sh neurons treated with rapamycin for four or eight days (between DIV9 and DIV13, and DIV5 and DIV13), while no effect was seen after acute treatment or at later ages (DIV20) in culture (Figures 4C and 4D), suggesting that defective ciliation due to TSC1/2 loss can be prevented by early mTORC1 inhibition but is irreversible at later times. Based on these results, we examined the potency of 17-AGG on ciliation and S6 phosphorylation between DIV9–13. Strikingly, we found that 17-AGG was ~100 times more potent at restoring ciliation than it was at inhibiting mTORC1 and IGF-IRβ/Akt signaling (cilia rescue: EC50 cilia = 15 nM; mTORC1 inhibition: IC50 pS6 = 1.8 μM; Figures 4E–4K; Figures S4A–S4E). Interestingly, 17-AGG did not increase ciliation in ctrl-sh neurons, suggesting that the mechanism of 17-AGG specifically prevents loss of cilia in Tsc2 KD neurons (Figures S4G–S4J). Neither rapamycin nor a four-day 17-AGG treatment regimen was able to rescue the cilia in Tsc2-sh neurons when tested at later ages in culture (Figure S4F). These data show that mTORC1 hyper-activation can be suppressed by Hsp90 inhibition regardless of the age of the culture (assay endpoint, respectively, DIV13 and DIV20) while amelioration of the cilia phenotype only occurs during an early critical period (DIV9–DIV13). In addition, these data show that 17-AGG likely prevents cilia loss via a distinct mechanism that is independent of inhibition of mTORC1.
Figure 4. Reduced Ciliation in the Tsc2-sh Neurons Is Prevented by mTORC1 and Hsp90 Inhibition during an Age- and Time-Sensitive Window.

(A and B) Overview of rapamycin time-course experiment using the ciliaHCA with endpoint at DIV13 (A) and DIV20 (B).
(C and D) Quantification of cilia in ctrl-sh and in Tsc2-sh-vehicle-treated or -rapamycin-treated neurons (20 nM) with assay endpoint at DIV13 (C) and DIV20 (D) using the ciliaHCA. Data were quantified relative to each respective vehicle-treated group (n = 8–18 biological replica/condition; two-way ANOVA with Dunnett’s multiple-comparison test, *p < 0.05, **p < 0.01, ns, non-significant) and are represented as mean ± SEM.
(E) Effect of 17-AGG dose-curve between DIV9 and DIV13 on ciliation using the ciliaHCA. Data represent GFP+ACIII+ in the Tsc2-sh neurons as percent of vehicle-treated control neurons.
(F) Effect of 17-AGG dose-curve between DIV9 and DIV13 on pS6 using the mTORC1HCA. Data represent GFP+pS6+ in the Tsc2-sh neurons as percent of vehicle-treated Tsc2-sh neurons. Quantification relative to vehicle-treated Tsc2-sh neurons (n = 8–16 biological replica/condition, E; n = 15–64 biological replica/condition, F, one-way ANOVA with Dunnett’s multiple-comparison test, *p < 0.05, ****p < 0.0001). Data are mean ± SEM.
(G) Representative images of cilia in vehicle-treated ctrl-sh and in Tsc2-sh neurons vehicle-treated or treated with 50-nM 17-AGG between DIV9–13. Neurons and cilia were identified by GFP and ACIII staining. Scale bar is 25 μm.
(H) Western blot of protein lysates from ctrl-sh and Tsc2-sh neurons treated with vehicle or with 17-AGG dose-curve between DIV9 and DIV13.
(I–K) Quantification of pS6 (I), total IGF-IRβ (J), and p-IGF-IRβ (K). Western blot data were normalized using GAPDH as loading control. Data are average fold changes of vehicle-treated ctrl-sh neurons. Quantification is relative to vehicle-treated Tsc2-sh neurons (n = 4 experiment/condition; one-way ANOVA with Dunnett’s multiple-comparison test, *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.0001). Error bars indicate ± SEM.
See also Figure S4.
Hsp27 Upregulation Due to Neuronal Tsc1/2 Loss Is Reduced by 17-AGG and by Rapamycin in the Dose Range and within the Critical Window that Restores Ciliation
To investigate the mechanism and explore the pharmacological window by which 17-AGG increases ciliation in Tsc2-KD neurons, we examined the heat-shock response in these neurons. The HSPs are divided into six families based on their size, and these proteins function in multi-component complexes that are closely inter-related (Chatterjee and Burns, 2017). In addition, while most of them are constitutively expressed, some are expressed only under stress (Garrido et al., 2006). We assessed expression levels of Hsp90 and other HSP family members at DIV13 and DIV20 following four days of treatment with different concentrations of the Hsp90 inhibitor 17-AGG. We found that in the absence of any compound, Tsc2-KD neurons had increased levels of the small Hsp27 with no change in Hsp90, Hsp70, Hsp60, and Hsp40 (Figures 5A–5L). Remarkably, the aberrant Hsp27 expression was reduced in the Tsc2 KD cultures at DIV13 following four days of treatment with 17-AGG (ranging from 5 to 450 nM) at concentrations that were similar to that required to prevent cilia loss without any effect on mTORC1 activation (Figures 5A, 5B, 4H, and 4I). In addition, we did not observe any change in Hsp27 expression due to treatment with 17-AGG in Tsc2 KD neurons at DIV20, consistent with the lack of effect on cilia at that age (Figures 5G and 5H; Figure S4F). Inhibition of the Hsp90 pathway by 17-AGG was confirmed by induction of Hsp40 and Hsp70, consistent with the reported release of Hsp90-dependent inhibition on HSF1 (Zou et al., 1998; Figures 5A, 5C, 5E, 5G, and 5K). We then evaluated whether Hsp27 upregulation was linked to mTORC1 hyper-activation. We performed acute (one day) and prolonged (four days) rapamycin treatment in Tsc2-sh neurons and measured Hsp27 levels at DIV13. Remarkably, we found that Hsp27 upregulation was reduced by mTORC1 inhibition with rapamycin in the same pharmacological window (between DIV9 and DIV13) and dosing regimen that restored the cilia (Figures 5M–5O). Together, these data indicate that increased expression Hsp27 is mTORC1-dependent and it can be downregulated through either Hsp90 or mTORC1 inhibition using dosing regimens and concentrations that restore ciliation in Tsc2-deficient neurons. These findings suggest that Hsp27 contributes to the mTORC1-dependent mechanism implicated in defective ciliation under loss of neuronal TSC1/2 activity.
Figure 5. Hsp27 Upregulation Due to Neuronal Tsc1/2 Loss Is Reduced by 17-AGG and by Rapamycin in the Dose Range and within the Critical Window that Restores Ciliation.

Western blots of protein lysates and quantification at endpoint DIV13 (A-F) and DIV20 (G-L) from control and Tsc2-sh neurons treated with vehicle or with a four-day dose curve of 17-AGG. The indicated proteins are expressed as average fold changes of vehicle-treated ctrl-sh neurons. Western blot data were normalized using GAPDH as loading control. Data quantification relative to vehicle-treated Tsc2-sh neurons (n = 3–4 experiment/condition; one-way ANOVA with Dunnett’s multiple-comparison test, *p < 0.05, **p < 0.05, ***p < 0.005, ****p < 0.0001). Error bars indicate ± SEM.
(M) Western blots of protein lysates from control and Tsc2-sh neurons treated with vehicle or with 20-nM rapamycin (Rapa) for one day or four days with endpoint at DIV13.
(N and O) Quantification of Hsp27 (N, n = 3) and pS6 (O, n = 7). Western blot data normalized using GAPDH as loading control. Data are average fold changes of vehicle-treated ctrl-sh neurons ± SEM (one-way ANOVA with Dunnett’s multiple-comparison test, *p < 0.05, **p < 0.005, ****p < 0.0001).
17-AGG Improved Ciliation Downstream of mTORC1 Activation through the Downregulation of HspB1 Gene Expression in the Tsc2-KD Neurons
In previous work, we had found that Tsc2 KD in neurons increases the expression of hspB1, which encodes Hsp27 (Nie et al., 2015). Therefore, we examined whether 17-AGG downregulated transcript levels of hspB1, leading to the decrease in Hsp27. Consistent with our prior data, we found that Tsc2-sh neurons have increased hspB1 expression (Figure 6A). We then treated Tsc2-sh neurons with 17-AGG and observed that increased hspB1 expression was reversed by the same dosing scheme that prevented cilia loss (50 nM between DIV9 and DIV13; Figures 6A–6D). To determine whether a similar dysregulation is present in the brains of TSC patients, we examined the gene expression data previously reported from cortical tubers and unaffected brain tissue (Martin et al., 2017). Strikingly, we found that HSPB1 expression was also significantly increased in cortical tubers (Figure 6E). To test whether Hsp27 downregulation may contribute to restoring ciliation in Tsc2-deficient neurons, we KD Hsp27 by hspB1 gene silencing using lentiviral expression of red fluorescent protein (RFP)-tagged Hsp27shRNA (RFP-Hsp27sh) or scrambled shRNA as a control (RFP-C). We confirmed that KD of Hsp27 in the Tsc2-KD cultures significantly reduced Hsp27 expression without affecting mTORC1 activation (Figures 6F–6H). We then examined ciliation in Tsc2-deficient neurons with concomitant Hsp27 KD using the ciliaHCA. Remarkably, we observed that hspB1 KD resulted in a significant increase in ciliation in the Tsc2-deficient neurons (Figures 6I and 6J). These data identify Hsp27 as part of the signaling cascade downstream of mTORC1 affecting cilia. Taken together, our findings identified multiple pharmacological effects of 17-AGG that act at distinct nodes in TSC1/2-deficient neurons, blocking mTORC1 through the disinhibition of Hsp90-regulated degradation of PI3K/Akt signaling components and improving the cilia deficits with a 100-fold greater potency through the transcriptional downregulation of Hsp27 (see model in Figure 6K).
Figure 6. 17-AGG Improved Ciliation Downstream mTORC1 Activation through Downregulation of HspB1 Gene Expression in the Tsc2-KD Neurons.

(A) HspB1 mRNA levels in vehicle-treated ctrl-sh and vehicle- or 50-nM 17-AGG-treated Tsc2-sh neurons (four days, DIV13 endpoint). Data were normalized to GAPDH, and are averages ± SEM (n = 4; one-way ANOVA with Tukey’s multiple-comparison test, ****p < 0.0005).
(B) Western blot of control and Tsc2-sh neurons vehicle-treated or treated with 50-nM 17-AGG (four days, DIV13 endpoint).
(C and D) Quantification of Hsp27 (C, n = 3) and pS6 (D, n = 4). Western blot data were normalized using GAPDH as loading control. Data are average fold changes of ctrl-sh neurons ± SEM (one-way ANOVA with Dunnett’s multiple-comparison test, *p < 0.05, **p < 0.01).
(E) HSPB1 expression in Non-TSC (n = 8) and TSC/tuber (n = 16) brains (unpaired Student’s t test, p** < 0.005). Data are averages ± SEM.
(F) Western blot of control and Tsc2-sh neurons transduced with RFP-tagged scramble control shRNA (RFP-C) or with RFP-tagged hsp27 shRNA (RFP-hsp27sh).
(G and H) Quantification of Hsp27 (G) and pS6 (H). Western blot data were normalized using GAPDH as loading control. Data are average fold changes of ctrl-sh neurons ± SEM (n = 4; one-way ANOVA with Tukey’s multiple-comparison test, ****p < 0.005).
(I) Effect of Hsp27 KD on ciliation using the ciliaHCA assay. Representative images of ctrl-sh and Tsc2-sh neurons transduced with RFP-C or with RFP-hsp27sh. Raw images (a, c, and e) and spot detector algorithm identification (b, d, and f) of ctrl-sh and Tsc2-sh neurons stained with Hoechst (nuclei in the white mask), GFP (ctrl-sh or Tsc2-sh infected neurons, green), ACIII (cilia, pink), and RFP (RFP-C or RFP-hsp27sh transduced neurons, red). Arrows in yellow indicate cilia. Scale bar is 25 μm.
(J) Quantification of ciliation. Data represent GFP+RFP+ neurons with cilia (ACIII+) as a percentage of ciliated GFP+RFP+ controls ± SEM (n = 6–30 biological replica/experiment; n = 2 experiment/condition; one-way ANOVA with Tukey’s multiple comparison test *p < 0.05).
(K) Model for 17-AGG multi-faceted effects in Tsc1/2-deficient neurons. (a) Under Tsc1/2, loss increased mTORC1 activity results in aberrant Hsp27 expression and reduced ciliated neurons. (b) 17-AGGpS6 inhibits mTORC1 (IC50 = 1.8 μM) through Hsp90-dependent regulation of PI3K/Akt signaling components with no significant change in Hsp27. (c) 17-AGGcilia reverses reduced ciliation at 100-fold-greater potency (Effective Concentration 50 [EC50 ] = 15 nM) independently from mTORC1 through suppression of hspB1/Hsp27.
DISCUSSION
TSC is a multisystem genetic disorder with a broad range of clinical symptoms, making the identification of effective treatments particularly challenging. Despite the clear implications of elevated mTORC1 activity as the mechanistic basis of TSC, an important set of unanswered question revolve around the identification of downstream signaling abnormalities due to disrupted mTORC1 signaling in specific cell types that are affected by the disorder. Notably, mTOR inhibitor-based therapies have thus far been unsuccessful in treating the neuropsychiatric features of TSC (Krueger et al., 2017; Overwater et al., 2019). Alternative pathways to restore other aspects of mTORC1 signaling may provide new drug targets and broaden the therapeutic landscape for this disease.
In this study, we show that mTORC1 hyperactivation caused by neuronal loss of Tsc1/2 leads to disruption of cilia, and we identify a potential molecular mechanism by which Hsp90 inhibition can reverse these pathological processes. Functional links observed between cilia and mTORC1 signaling appear to be critically dependent upon cellular context. Previous studies have shown that TS Clos si nkidne yepithelia lcell so fzebrafis han dmic eresults i nlonge rcilia without affecting the number of ciliated cells (Armour et al., 2012; DiBella et al., 2009). In contrast, studies with Tsc1-or Tsc2-deficient MEFs showed a rapamycin-insensitive enhancement of cilia formation (Hartman et al., 2009) or a different cilium phenotype with either longer or shorter cilia depending on the TS Cgen eaffected(Rosengre ne tal.,2018).On possible explanatio for these divergent outcomes could be that ciliation is differenttially regulated under specific cellular metabolic conditions. For instance, mTORC1-inducing stimuli can promote cilia disassembly through progression into mitotic phase in cycling cells (Yeh et al., 2013). By contrast, mTORC1-inhibitory stimuli can promote ciliation through autophagy-dependent (Pampliega et al., 2013; Tang et al., 2013) or autophagy-independent ( Takahashi et al., 2018) mechanisms in dividing cells. Ciliary signaling has an established role in many developmental settings, including cell-cycle progression, -proliferation, and -differentiation (Kirschen and Xiong, 2017). Critical roles for cilia in the development of the CNS include roles in neuronal migration, neurogenesis, plasticity, and maturation. Interestingly, appearance of neuronal cilia coincides with onset of functional glutamatergic synaptic activity, which suggests that the protein machinery required for ciliogenesis might also be involved in synaptogenesis, and that cilia may signal to the synapse (Kumamoto et al., 2012).
We observed an inverse relationship between ciliation and mTORC1 activity in hippocampal cultures at the time when neurons begin to polarize. We hypothesize that this represents a time-sensitive regulatory role for the Tsc1/2 complex that may act as a brake on mTORC1 signaling to promote neuronal maturation through cilia assembly. Rapamycin has been shown to improve several neurological phenotypes in animal models of TSC, including epilepsy, cognition, and social behavior (Meikle et al., 2008; Tsai et al., 2012, 2018). The fact that rapamycin treatment also reversed the ciliary phenotype suggests that defective ciliary signaling might be contributing to these neurological symptoms.
Cortical tubers are a pathological hallmark of TSC characterized by the presence of immature giant cells and dysplastic neurons, and these tubers are associated with disorganized connectivity and astrogliosis (Curatolo et al., 2015). Given the role of cilia in cell fate choice (Kim et al., 2011; Li et al., 2011; Yeh et al., 2013), lack of ciliation could impact neuronal maturation and contribute to the development of the undifferentiated giant cells present in cortical tubers. Disorganized connectivity could also be a TSC-associated manifestation arising from alterations in cilia-dependent functions such as defective neuronal migration, polarization, or cortical lamination (Park et al., 2018; Pruski and Lang, 2019; Sarkisian and Guadiana, 2015). Finally, given the role of cilia in the control of cell cycle and cell proliferation, altered cilia signaling might also have an impact in the transformation of the subependymal nodules into the low-grade subependymal giant cell astrocytomas (SEGAs) seen in the CNS of TSC patients (Álvarez-Satta and Matheu, 2018; Chan et al., 2004; Ess et al., 2004; Sarkisian and Semple-Rowland, 2019). Interestingly, studies have found that altered expression of genes associated with cilia is a risk factor for several neuropsychiatric disorders (Marley and von Zastrow, 2012;Migliavacca et al., 2015). Therefore, our observation that the expression of cilia genes is more likely to be altered in cortical tubers suggests that the ciliary dysfunction present in patients with TSC may contribute to the neuropsychiatric symptoms associated with TSC.
Our phenotypic screen identified compounds that revealed that Hsp90 inhibition is capable of normalizing overactive mTORC1 and was associated with degradation of the receptor tyrosine kinase (RTK), IGF-IRβ. mTORC1 controls RTKs such as IGF-Iβb through negative-feedback, limiting their activation (Zhang et al., 2006). However, RTK signaling can also be limited through ubiquitination, endocytosis, and degradation, and Hsp90 has been previously shown to regulate stability of IGF-IRβ, as well as other oncogenic RTKs (Zsebik et al., 2006). We found that Hsp90 inhibition led to increased IGF-IRβ degradation in Tsc2-KD neurons. This reduction in IGF-IRβ combined with reduced activity due to mTOR hyperactivation leads to full suppression of Akt activity. Together, these data suggest that further inhibition of upstream components of the PI3/Akt signaling pathway, such as the IGF-Iβb, contributes to reduce mTORC1 activity. We found that Tsc2-KD neurons had increased Hsp27 expression at the transcriptional level and that KD of the hspB1 gene prevented the decrease in cilia due to loss of Tsc2, directly implicating Hsp27 as a downstream effector of mTORC1 hyperactivation in the disruption of cilia. Hsp27 is an ATP-independent chaperone expressed at low levels under physiological conditions that is induced by stress (Garrido et al., 2006). Previous studies have shown that misfolded proteins bind to small oligomers of Hsp27 that shift to large oligomers under stress. Therefore, elevated Hsp27 chaperone activity under pathological conditions has proliferative and anti-apoptotic functions. We have previously shown that disinhibition of mTORC1 signaling due to loss of neuronal Tsc1/2 causes oxidative and endoplasmic reticulum stress (Di Nardo et al., 2009). Thus, increased baseline hspB1 gene expression in the Tsc2-KD neurons might represent a compensatory stress-induced response to mTORC1-dependent accumulation of misfolded proteins and reactive oxygen species. In line with this, higher doses of 17-AGG may exacerbate the stress response, leading to persistent Hsp27 expression and lack of efficacy in preventing cilia loss. Interestingly, under stress conditions, dynamic structural changes in Hsp27 oligomerization stabilize actin-filament formation, which is a known inhibitor of ciliogenesis (Drummond et al., 2018). Therefore, Hsp27-dependent modulation of actin cytoskeleton could be part of the mechanism by which exaggerated Hsp27 expression inhibits cilia in the Tsc2-KD cultures. In addition, the fact that altered ciliation was rescued within a critical developmental period by pharmacological inhibition with 17-AGG through suppression of hspB1 suggests the existence of a distinct mechanism involving regulation of ciliogenesis at the transcriptional level (Choksi et al., 2014) that can be prevented by early intervention, but is irreversible at later times.
Several manifestations of TSC can be alleviated by mTOR inhibitors including rapamycin and its analog everolimus (Winden et al., 2018); however, the beneficial effects are lost when the therapy is discontinued. Furthermore, many aspects of TSC, in particular neurocognitive deficits, are not reversed by mTOR inhibitors (Krueger et al., 2017), highlighting the need to identify alternative therapies. The mTORC1HCA here described provided a valuable screening platform as it identified inhibition of Hsp90 with GA and 17-AGG as an alternative strategy to reverse disrupted mTORC1 signaling in TSC. In addition, the fact that the ciliaHCA uncovered 17-AGG as a compound capable of restoring defective ciliation in a time-sensitive window, independent of disrupted mTORC1 inhibition, underscores the translational implication of our work. Given the relevance of ciliary signaling for neuronal function, future work could be done to examine the efficacy of 17-AGG in vivo. Together, the HCAs we developed and optimized for high-throughput quantitation of mTORC1 and cilia with primary neurons represent broadly applicable platforms for compound screening and/or therapeutic testing of drug candidates.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Mustafa Sahin (mustafa.sahin@childrens.harvard.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a complete Material Transfer Agreement.
Data and Code Availability
All software used in this study are listed in the Key Resources Table. The cilia genes expression data and the primary screen data generated in this study are listed in deposited data of the Key Resources Table. These unique datasets have been included and are available as Tables S1 and S3, respectively. Original data have been deposited to Mendeley Data: [https://data.mendeley.com/datasets/p45bhvf2g3/draft?a=420877d4-d4cb-45f1-adb7-91fcefcec901].
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ACIII | Proteintech | Cat# 19492–1-AP; RRID:AB„10638445 |
| Rabbit monoclonal anti-centrin | Millipore | Cat# 04–1624; RRID:AB_10563501 |
| Rabbit polyclonal anti-Hp90 | Proteintech | Cat#13171–1-AP; RRID:AB_2120924 |
| Rabbit monoclonal antl-GAPDH | Ambion | Cat# AM4300; RRID:AB_437392 |
| Rabbit polyclonal anti-pS6 | Cell Signaling Technology | Cat# 5364; RRID:AB_10694233 |
| Mouse monoclonal anti-S6 | Santa Cruz Biotechnology | Cat# sc-74459; RRID:AB_1129205 |
| Rabbit polyclonal anti-phospho Akt (Ser473) | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| Rabbit monoclonal anti-Akt | Cell Signaling Technology | Cat# 4691; RRID:AB_91578 |
| Rabbit polyclonal anti-phospho PRAS40 (Thr246) | Cell Signaling Technology | Cat#13175; RRID:AB_2798140 |
| Rabbit polyclonal anti-PRAS40 | Cell Signaling Technology | Catt# 2691; RRID:AB_2225033 |
| Rabbit polyclonal anti-phospho IGF-IRβ(Tyr1335/6) | Cell Signaling Technology | Cat# 3024S; RRID:AB_331253 |
| Rabbit polyclonal anti-IGF-IRβ | Santa Cruz Biotechnology | Cat# sc-713; RRID:AB_671792 |
| Rabbit polyclonal anti-Tsc2 | Cell Signaling Technology | Cat# 4308; RRID:AB_10547134 |
| Rabbit monoclonal anti-Hsp70 | Santa Cruz Biotechnology | Cat# sc-24; RRID:AB_627760 |
| Rabbit monoclonal anti-Hsp60 | Santa Cruz Biotechnology | Cat# sc-271215; RRID:AB_10607973 |
| Rabbit monoclonal anti-Hsp27 | Santa Cruz Biotechnology | Cat# sc-13132; RRID:AB_627755 |
| Chicken polyclonal anti-GFP | Thermo Fisher Scientific | Cat# A10262; RRID:AB_2534023 |
| Mouse monoclonal anti-RFP | Thermo Fisher Scientific | Cat# MA515257; RRID;AB_10999796 |
| Goat anti-mouse IRDye 800RD | LI-COR Biosciences | Cat#925–32210; RRID:AB_2687825 |
| Goat anti-mouse IRDye 680RD | LI-COR Biosciences | Cat#925–68070; RRID:AB_2651128 |
| Goat anti-rabbit IRDye 800CW | LI-COR Biosciences | Cat#925–32211; RRID:AB_2651127 |
| Goat anti-rabbit IRDye 680RD | LI-COR Biosciences | Cat#925–68071; RRID:AB_2721181 |
| Goat anti-chicken Alexa Fluor-488 | Molecular Probes | Cat#A11039; RRID:AB_.142924 |
| Goat anti-rabbit Alexa Fluor-647 | Molecular Probes | Cat# A21244; RRID:AB_141663 |
| Goat anti-rabbit Alexa Fluor-546 | Molecular Probes | Cat#A11035; RRID:AB_143051 |
| Goat anti-mouse Alexa Fluor-546 | Molecular Probes | Cat#A11030; RRID:AB_144695 |
| Bacterial and Virus Strains | ||
| Stbl3 competent cells | Thermo Fisher Scientific | Cat# C737303 |
| NEB Stable competent cells | New England Biolabs | Cat#C30401 |
| Tsc2-sh-GFP | Previously reported (See text for details) | N/A |
| Hsp27-shRNA | Sigma | Cat#TRCN0000321339 |
| Biological Samples | ||
| Sprague-Dawley rat brain tissue | Charles River Laboratories | N/A |
| TSC patient brain tissue | BCH Repository, Table S2 | N/A |
| Non-TSC patient brain tissue | BCH Repository, Table S2 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BIOMOL ICCB-L Known Bioactive 2012 library | ICCB-Longwood | https://iccb.med.harvard.edu/biomol-iccb-l-known-bioactives-2012 |
| Rapamycin | LC Laboratories | Cat# R-5000 |
| 17-Allylamino- geldanamycin (17-AGG) | Enzo Life Sciences | Cat# BML- EI308–0001 |
| Geldanamycin (GA) | Enzo Life Sciences | Cat# BML- EI280–0001 |
| Poly-D-Lysine Hydrobromide | MP Biomedical | Cat# 102694 |
| Bortezomib | EMD Millipore | Cat# 5.04314.0001 |
| Critical Commercial Assays | ||
| Pure Link Plasmid Kit | Thermo Fisher Scientific | Cat#K21007 |
| Direct-zol™ RNA Miniprep Kit | Zymo-Research | Cat# R2052 |
| iScript™ cDNA Synthesis Kit | BIO-RAD | Cat# 1708891 |
| PowerUP™ SYBR™ Green Master Mix | Thermo Fisher Scientific | Cat# A25742 |
| Deposited Data | ||
| RNA sequencing data | Previously reported (See text for details) | N/A |
| Cilia genes expression data | This study | Table S1 |
| Primary screen data | This study | Table S3 |
| Mendeley Data | This study | https://data.mendeley.com/datasets/p45bhvf2g3/draft?a=420877d4-d4cb-45f1-adb7-91fcefcec901 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216, RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tsc1tm1Djk/J | The Jackson Laboratory | IMSR Cat# JAX:005680, RRID:IMSR_JAX:005680 |
| Mouse: Tsc2tm1Djk/J | The Jackson Laboratory | IMSR Cat# JAX:004686, RRID:IMSR_JAX:004686 |
| Mouse: Tsc2tm2.1Djk/Mmjax | The Jackson Laboratory | IMSR Cat# JAX: 037154,. RRID:MMRRC_037154-JAX |
| Mouse: B6.Cg-Tg(Syn1-cre)671 Jxm/J | The Jackson Laboratory | IMSR Cat# JAX:003966, RRID:IMSR_JAX:003966 |
| Oligonucleotides | ||
| GAPDH forward primer: 5′-TGTGTCCGTCGTGGATCT GA-3′ | This study | N/A |
| GAPDH reverse primer: 5′-CCTGCTTCACCACCTTCTTGA-3′ | This study | N/A |
| hspB1 forward primer: AGACCAAGGAAGGCGTGGTG | This study | N/A |
| hspB1 reverse primer: CACACCTGGAGGGAGCGTGT | This study | N/A |
| Recombinant DNA | ||
| pLKO-RFP-shCntrl | Addgene | RRID:Addgene_69040 |
| Luciferase-ctrl-sh-GFP | Previously reported (See text for details) | N/A |
| psPAX2 | Addgene | RRID:Addgene_12260. |
| pMD2.G | Addgene | RRID:Addgene_12259. |
| Software and Algorithms | ||
| Fiji Software v2.0.0 | Open Source | http://fiji.se/.RRID:SCR_002285 |
| GraphPad PRISM v8.2.1 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/RRID:SCR_002798. |
| HCS Studio™ Cell Analysis Software v6.6.0 | Thermo Fisher Scientific | N/A |
| Image Studio Lite analysis software v5.2.5 | LI-COR | https://www.licor.com |
| Photoshop CS5.1 v12.1 | Adobe | https://www.adobe.com/products/photoshop.html |
| Syscilia. | Open Source | http://www.syscilia.org/goldstandard.shtml. |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
All experimental procedures were done in agreement with animal protocols approved by the Institutional Animal Care and Use Committee at Boston Children’s Hospital. Both female and male mice were used in the experiments. Mice were maintained on a 12-h light/ dark cycle with free access to food and water according to the Animal Research Committee at Boston Children’s Hospital.
Tsc1 mutant mice: The Tsc1 control (Tsc1 w/w SynCre) and Tsc1 mutant (Tsc1 c/c SynCre) mice used in this study were described previously (Ercan et al., 2017). All mice were in mixed background, derived from C57BL/6, CBA, and 129S4/ SvJae, strains. We use c andw to denote the conditional (floxed) and wild-type alleles of Tsc1, respectively; the formal name of the c allele is Tsc1tm1Djk (Meikle et al., 2007). To generate Tsc1 c/c Syn Cre+ mice, first Tsc1 c/w Syn Cre+ females were crossed with Tsc1 c/c Syn Cre- male mice. Rapamycin treatment was performed by injecting 6 mg/kg intra-peritoneally every other day beginning at P7 until sacrifice (P21). These rapamycin treatment timing and dosing were chosen based on previous pharmacokinetics and pharmacodynamics of rapamycin in the brain (Meikle et al., 2008). Brain levels were above the level required to inhibit mTORC1 and effective in reversing the hypomyelination phenotype described previously (Meikle et al., 2008).
Tsc2 mutant mice: the formal name of the c allele is Tsc1tm2.1Djk (Pollizzi et al., 2009). Litters were generated from crosses between mixed background animals containing Tsc2 k/c Syn Cre- and Tsc2 c/c Syn Cre+ in which the k allele was a full knockout of the Tsc2 gene and the c allele was a conditional mutation of the Tsc2 gene that results in ~7% expression of TSC2. All pups in a litter were treated with either vehicle or rapamycin and animals of the; genotype Tsc2 c/c Syn Cre- were used as control mice and Tsc2 k/c Syn Cre+ were used as mutant mice. For rapamycin treatment, animals were dosed every other day beginning at P7 with 3mg/kg rapamycin in vehicle at a volume of 30 μL until P21 and then at a total volume of 100 μL from P21 to P56. Vehicle consisted of 5% PEG 400 and 5% Tween 80; 4% ethanol was added to the vehicle for control treated animals.
Human subjects
Cortical tubers were collected from patients clinically and neuropathologically diagnosed with TSC, at the time of surgery. Tissues were fixed in 4% phosphate-buffered paraformaldehyde pH 7.4 (PFA), subjected to sucrose gradient and stored frozen before further processing. The control samples were prepared in a similar fashion and were processed together. All patients suffered from chronic epilepsy, with a seizure history. See Table S2 for details. The subjects enrolled in this study were recruited through Boston Children’s Hospital, and the protocol was approved by Boston Children’s Hospital IRB (P0008224). IRB protocol number for the Repository Core was CHERP 09–02-0043. Informed consents were obtained from all participants and/or their parents as appropriate.
Neuronal cultures
Hippocampi and cortexes from 18-day-old rat embryos (Charles River CD1) were isolated under the microscope and collected in Hank’s Balanced Salt Solution containing 10mM MgCl2, 1mM kynurenic acid, 10mM HEPES and penicillin/streptomycin. After 5min dissociation at 37° C in 30U/ml of papain, neurons were mechanically triturated and plated in Neurobasal (NB) medium containing B27 supplement, 2mM L-glutamine, penicillin/streptomycin and primocin (NB/B27). Biochemical analysis was performed on cortical cultures plated at 1×106cells/well onto six-well plates. Immunofluorescent analysis was performed on hippocampal cultures plated at 20×103cells/well onto 96-well plates. All plates were coated with 20 μg/ml poly-D-lysine (PDL).
METHOD DETAILS
Lentivirus production and transduction
Viral stocks for lentiviral infection were prepared by co-transfection of the two packaging plasmids psPAX2 and pMD2.G into HEK293T cells with the plasmid to be co-expressed using PEI. Viral particles were collected 48hrs and 72hrs after transfection and filtered through a 0.45 mm membrane. Hippocampal neurons were infected at 1 day in vitro (1DIV) in the presence of polybrene at 0.6 μg/ml. Six h after infection, the virus-containing medium was replaced by fresh NB/B27 medium. GFP-tagged control shRNA construct (here referred as ctrl-sh) was against the luciferase gene was previously described (Flavell et al., 2006). The lentiviral Tsc2 shRNA construct was previously described (Di Nardo et al., 2009). The target sequence for the Tsc2 gene was the following: 50-GGTGAAGAGAGCCGTATCACA-30. RFP-tagged Hsp27-shRNA (referred here as RFP-Hsp27sh) was purchased from Sigma; pLKO-RFP-shControl (referred here as RFP-C) was purchased from Addgene.
RNA preparation and quantitative real-time PCR (qPCR)
Total RNA was prepared with the RNA kit (Zimo Research) following the instructions of the manufacturer and quantified by a spectrophotometer. A total of 1 μg of poly(A) mRNA was used for reverse transcription using the Reverse Transcriptase (BIO-RAD). Realtime PCRs were performed using Power SYBG Green PCR Master Mix (Applied Biosystems). All quantitative PCR (qPCR) reactions were performed in triplicate and normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Analysis was performed using QuantStudio™ 3 Real-Time PCR System (Thermo Fisher Scientific).The qPCR cycle was: 95C for 10min followed by 40 cycles at 95° C for 15sec and 60° C for 1min.
Immunohistochemistry of brain tissue and manual cilia counting
For histological analysis, animals were anesthetized (1ml of 2.5% Avertin) and perfused transcardially with 4% PFA. Brains were dissected and fixed in 4% PFA for another 24 h. Brains were slowly frozen by cooling down in dry-ice cold isopentane and then prepared by cryostat sectioning at a thickness of 25 μm. Sections were washed 4 times with Tris Buffered Saline pH 7.4 (TBS) and blocked for 2 h at room temperature in blocking buffer (5%BSA, 0.1% Triton X-100, 10% goat serum). The incubation with the primary antibody was done in 1% BSA, 0.1% Triton X-100, at 4° C for overnight. The day after, sections were washed three times in TBS buffer before being incubated with the appropriate fluorochrome-coupled secondary antibody. Stained sections were air-dried, dehydrated, and mounted. Imaging of the Tsc1 control and mutant brains was performed by dividing the cortex into 6 layers and by imaging 5 random regions per layer. Imaging of the Tsc2 control and mutant brains was performed by imaging 6–9 random regions in the CA1 of the hippocampus. Finally, the average percentage of neurons with cilia (NeuN+/ACIII+) was calculated in each of the images. For the human tissue, an average of 40–60 images were acquired in random fields of the specimen. The average percentage of SMI311+ or SMI311- with cilia was calculated. The measurement of the cilia length was performed by tracing the ACIII stained cilia using the ImageJ Software freehand tool. A threshold for cilia count was set such that only ACIII positive objects that measured longer than 1 μm were counted as cilia. All the imaging and the quantification were done in a blinded way. Confocal images were acquired with a Nikon Ultraview Vox Spinning Disk Confocal microscope using 63× oil-immersion objective equipped with Hamamatsu camera. All quantifications were performed in a blinded fashion.
Drug treatments
Stocks of drugs were freshly diluted in NB media before performing each experiment. The same amount of vehicle was used as vehicle-only control. For dose response curves serial dilutions were freshly made in NB media by a manual multichannel pipette using compound dilution plates. Proteasomal inhibition experiments were performed by pretreatment with 100nM bortezomib (BTZ) for 6hrs before incubation with 17-AGG at 4 μM for 24hrs. The BIOMOL Collection Library used for the screen was obtained by the ICCB-Longwood Screening Facility. According to Biomol all of the compounds in this collection have known and well-characterized bioactivities and have undergone safety and bioavailability testing. The compounds were carefully selected to maximize chemical and pharmacological diversity.
Western blot
Protein extracts were lysed in 1× SDS (22mM Tris-HCl pH 6.8, 4% Glycerol, 0.8% SDS, 1.6% β-mercaptoethanol, bromphenol blue) sample buffer, heated to 95° C for 5 min and stored frozen. Before being subjected to discontinuous gel electrophoresis, equal amounts of all protein lysates were verified by Coomassie gel staining. For immunoblotting, equal amounts of protein lysates were subjected to SDS–PAGE, transferred to Immobilon-P Millipore and incubated in LI-COR blocking solution at RT for 2hrs. Primary and secondary antibodies were diluted in LI-COR blocking solution to the appropriate concentrations. LI-COR IRDye secondary antibodies were used. All images were acquired using the LI-COR Odyssey Classic imager and associated Image Studio Lite analysis software (version 5.2.5). Quantification of protein expression was performed by densitometry scans of immunoblots using LI-COR Odyssey imaging system.
Neuronal culturing and processing for high-content assays
Rat hippocampal neurons were plated at a density of 20,000/well in 96-well plates (Greiner # 655090) coated with Poly-D-Lysine (PDL) at 20 μgr/ml. Neurons were transduced with lentivirus as previously described and processed for immunofluorescent staining at endpoint assay by fixation with 4% PFA followed by permeabilization in ice cold 100% Methanol. Neurons were then washed 3 times in PBS/0.05% Tween (PBT) containing 50mM glycine and blocked in 2% bovine serum albumin (Sigma) in PBT (PBT/Block) at RT. For consistent and robust identification of the LV-infected neurons, cultures of both the ciliaHCA and mTORC1HCA were stained with GFP antibody and co-labeled with ACIII or pS6 for the identification of respectively cilia and mTORC1 activity. Primary antibodies were incubated in PBT/Block overnight. The following day, neurons were washed in PBT and incubated with secondary antibodies followed by Hoechst staining. After washing with PBT, neurons were kept in PBS buffer. Aside from primary and secondary antibody administration, all the washing for immunofluorescent staining was done using the Agilent bravo automated liquid handler.
Neuronal culturing and processing for manual cilia counting
For manual cilia counting rat hippocampal neurons were plated at a density of 150,000/well onto coverslips coated with Poly-D-Lysine (PDL) at 20 μgr/ml. Neuronal culturing and processed was performed as previously described. Manual imaging and cilia counting were performed in ctrl-sh and Tsc2-sh cultures stained with GFP antibodies to identify the LV-infected neurons, co-labeled with ACIII and centrin antibodies to identify cilia and basal bodies, respectively. Secondary antibodies goat anti-chicken alexa Fluor 488, goat anti-rabbit alexa Fluor 595 and goat anti-mouse alexa Fluor 647 were used. Coverslips were mounted on glass slides in Vectashield with DAPI (Vector Laboratories). Random images of GFP+ cells were obtained using an Eclipse Ti-E (Nikon) microscope with a Plan Apo 100 × 1.49 NA oil objective, an Evolve electron-multiplying charge-coupled device camera (Photometrics), and Meta-Morph software (Molecular Devices). Images were acquired as a z series (0.2-μm z interval) and are presented as maximum-intensity projections. Cilia length was measured in maximum-intensity projections using ImageJ Software (Feret measurement of ROI identifying cilia). The same ROI was used to determine the total intensity of ACIII from the sum projection of the z stacks. ACIII total intensity was then normalized to the corresponding cilium length. All quantifications were performed in a blinded fashion.
High-throughput imaging and quantification of cilia and of mTORC1
Neurons were imaged using the high content analysis platform Cellomics Arrayscan XTI available at the Human Neuron Core of the Translational Neuroscience Center (Boston Children’s Hospital). The Arrayscan XTI is equipped with Zeiss optics, a 7- color solid state LED light engine, and a large format X1 CCD camera (2208×2208). The DAPI channel was used for focal plane acquisition by the software with an intra-well autofocus interval of one (refocus in each subsequent well). When needed, focal planes were adjusted to the best optimal resolution for each channel. Once optimized, Z offsets were kept the same throughout the scans. Imaging of the ciliaHCA was done with a 40x objective on eighty fields of view per well (20% of the well). The nuclei were detected by Hoechst staining at 386nm emission for 30ms, the LV-transduced neurons were detected using GFP staining at 485nm emission for 20ms at a 1.9 μm Z offset above the focal plane of the nuclei, and cilia were detected using ACIII staining at 647 emission for 130ms using the “image projection tool” which allowed the acquisition of a stack of images at three different focal planes for optimal cilia imaging with a step size of 1.9 μm/step for a total of 5.7 μm(Figures S2C and S2D). Object selection for GFP and ACIII spots identification were filtered using area and shape measurements.
Imaging of mTORC1HCA was performed with a 10x objective on the whole well. The nuclei were detected by Hoechst staining at 386nm emission for 70ms, the LV-transduced neurons were detected using GFP staining at 485nm emission for 50ms at a 11.7 μm Z offset above the nuclei focal plane, and mTORC1 activity was detected using pS6 staining at 546 emission for 40ms (Figures S2E and S2F).
Data analysis was performed using an optimized version of Spot Detector algorithm available with the HCS Studio™ Cell Analysis Software which allowed the detection of subcellular structures and their co-localization. Nuclei identification was done using a circular mask and the identified objects functioned as a region of interest (ROI) for the subsequent channels. GFP+ spots were identified using a circular mask of the size of the nuclei, pS6+ spots were identified using a ring mask with a width of 8 μm outside the nuclei ROI, ACIII+ spots were identified using a circular mask with a diameter of 32 μm which included the nuclei ROI. Object selection for GFP and pS6 spots were filtered using area, and intensity measurements. Objects at the border of the well were always excluded.
mTORC1 high-content screen
The screen was performed using the mTORC1HCA. Assay robustness and screening window between positive (ctrl-sh neurons) and negative controls (Tsc2-sh neurons) were assessed by Z-score and Z’-factor calculation (Zhang, 2011). The following formula was used for Z-score calculation: X-Pave/SD (X = % of GFP+ pS6+ neurons in the well; Pave = mean of % GFP+ pS6+ neurons in Positive control wells, SD = Standard Deviation of the values measured in P). The following formula was used for Z-prime calculation: 1-[3*sum (SDn+SDp)/ Yn-Yp]]; Yn = average negative controls, Yp = average positive controls (Figure S3A). For the screen we used the Biomol collection library which includes 480 bioactive compounds with known MOA (ICCB-Longwood screening facility and Human Neuron Core of the Translational Neuroscience Center at BCH). Every compound of the Biomol library was at a concentration optimized to reflect their potency for known target. The library was used a dilution of roughly 30X the potency for each compound for its target. The screen was performed in 96-well plates in duplicate and edge wells were excluded to avoid variability. Neurons were transduced with lentivirus as previously described and treated either with vehicle (0.1% v/v DMSO) or with the compounds at DIV19 for 24hrs. Each plate included three to four vehicle-treated control-sh neurons and three to four vehicle-treated Tsc2-sh neurons. Compounds were administered using the Agilent bravo automated liquid. We calculated a Z-score for each compound in terms of standard deviations from the mean distribution of all the compound-treated Tsc2-knockdown wells assuming that most compounds would not have an effect on S6 phosphorylation (Zhang, 2011).The following formula was used for Z-score calculation of the screen compounds: X-Cave/ SD of Cave (X = % GFP+ pS6+ neurons in Tsc2-sh compound-treated well, Cave = average % of GFP+ pS6+ neurons in all the Compound-treated Tsc2-sh wells, SD of the values measured C). Potential hits were considered compounds that had a z-score less than 1.8 (p < 0.0) in both replicas. Total DAPI+ counts were used as a criterion for toxic compounds exclusion, toxic compound excluded were 5.8%.
Differentially expressed genes
RNA sequencing data from normal brain and cortical tubers were obtained, and they were normalized using trimmed mean of M values summarized using counts per million (Martin et al., 2017). A list of cilia genes was obtained from Syscilia ( http://www.syscilia.org/goldstandard.shtml), and cilia gene expression was quantified within normal brain, and cortical tubers. To determine whether cilia genes were more likely to be dysregulated in the cortical tubers versus normal brain, we used LIMMA, which allowed us to determine the number of genes expressed at different levels in cortical tubers versus normal brain using an uncorrected p value of 0.05. As controls, random groups of genes of the same size as the group of cilia genes were selected and utilized to identify the number of genes dysregulated within these random groups. We then calculated a Z-score by comparing the number of dysregulated genes in the cilia group versus the random groups, and a p value based on this Z-score.
QUANTIFICATION AND STATISTICAL ANALYSIS
Western blot quantifications were performed by protein normalization using GAPDH as loading control. Level of phosphorylated proteins was expressed as the ratio of phosphorylated/total level after GAPDH normalization. Low sample size datasets were tested for normality and when appropriate they were analyzed with non-parametric tests. IC50 values were calculated using the nonlinear regression equation “dose-response curves -Inhibition” of GraphPad PRISM. Data were expressed as percent of vehicle-treated Tsc2-sh neurons which were considered 100% and drug’s concentrations were transformed to logarithmic 10 scale. EC50 values were calculated using the nonlinear regression equation “dose-response curves -Stimulation” of Prism. Data were expressed as percent of vehicle-treated ctrl-sh neurons which were considered 100% and drug’s concentrations were transformed to logarithmic 10 scale.
Supplementary Material
Highlights.
Tubers from TSC patients have a distinct ciliary gene signature and fewer cilia
High-content assays with TSC-deficient neurons can be used as a drug-screening platform
17-AGG can regulate the mTORC1 signaling cascade at multiple levels
Hsp27 is a druggable target of mTORC1-dependent impaired ciliation
ACKNOWLEDGMENTS
We thank Nina Makhortova and Denise McGinnis for helpful comments on the manuscript and Brianna Godlewski for help with specimen collection from patients. This study was supported by grant funding from the National Institutes of Health (NIH) (R01NS113591) and by the Nancy Lurie Marks Family Foundation. K.D.W. was supported by NIH grant R25NS070682 and by the American Academy of Neurology Neuroscience Research Training Scholarship. The Clinical/Translational Core and Cellular Imaging Cores of the Intellectual and Developmental Disabilities Center at Boston Children’s Hospital (BCH IDDRC; U54HD090255), the F.M. Kirby Award sponsored by Boston Children’s Hospital, and the Tuberous Sclerosis (TS) Alliance Research Grant provided support for this study.
Footnotes
DECLARATION OF INTERESTS
M.S. reports grant support from Novartis, Roche, Pfizer, Ipsen, LAM Therapeutics, Astellas, Bridgebio, and Quadrant Biosciences unrelated to this project. He has served on Scientific Advisory Boards for Sage, Roche, Aeovian, Celgene, and Takeda. M.S. and A.D.N. have a patent pending on this work. R.J.K. is a current employee and shareholder of Biogen. The other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107780.
REFERENCES
- Álvarez-Satta M, and Matheu A (2018). Primary cilium and glioblastoma. Ther. Adv. Med. Oncol. 10, 1758835918801169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armour EA, Carson RP, and Ess KC (2012). Cystogenesis and elongated primary cilia in Tsc1-deficient distal convoluted tubules. Am. J. Physiol. Renal Physiol 303, F584–F592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, and Rosen N (2002). Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 277, 39858–39866. [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, and Godinho SA (2011). Centrosomes and cilia in human disease. Trends Genet. 27, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop GA, Berbari NF, Lewis J, and Mykytyn K (2007). Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J. Comp. Neurol. 505, 562–571. [DOI] [PubMed] [Google Scholar]
- Chan JA, Zhang H, Roberts PS, Jozwiak S, Wieslawa G, Lewin-Kowalik J, Kotulska K, and Kwiatkowski DJ (2004). Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J. Neuropathol. Exp. Neurol. 63, 1236–1242. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, and Burns TF (2017). Targeting heat shock proteins in cancer: a promising therapeutic approach. Int. J. Mol. Sci 18, ijms18091978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi SP, Lauter G, Swoboda P, and Roy S (2014). Switching on cilia: transcriptional networks regulating ciliogenesis. Development 141, 1427–1441. [DOI] [PubMed] [Google Scholar]
- Curatolo P, Moavero R, and de Vries PJ (2015). Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 14, 733–745. [DOI] [PubMed] [Google Scholar]
- Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, and Sahin M (2009). Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J. Neurosci. 29, 5926–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo A, Wertz MH, Kwiatkowski E, Tsai PT, Leech JD, Greene-Co-lozzi E, Goto J, Dilsiz P, Talos DM, and Clish CB (2014). Neuronal Tsc1/ 2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum. Mol. Genet. 23, 3865–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBella LM, Park A, and Sun Z (2009). Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum. Mol. Genet. 18, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond ML, Li M, Tarapore E, Nguyen TTL, Barouni BJ, Cruz S, Tan KC, Oro AE, and Atwood SX (2018). Actin polymerization controls cilia-mediated signaling. J. Cell Biol. 217, 3255–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Saffari A, Wahlster L, Di Nardo A, Turner D, Lewis TL Jr., Conrad C, Rothberg JM, Lipton JO, Kölker, S., et al. (2016). Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep. 17, 1053–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan E, Han JM, Di Nardo A, Winden K, Han MJ, Hoyo L, Saffari A, Leask A, Geschwind DH, and Sahin M (2017). Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J. Exp. Med. 214, 681–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ess KC, Uhlmann EJ, Li W, Li H, Declue JE, Crino PB, and Gutmann DH (2004). Expression profiling in tuberous sclerosis complex (TSC) knockout mouse astrocytes to characterize human TSC brain pathology. Glia 46, 28–40. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, and Greenberg ME (2006). Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, and Kroemer G (2006). Heat shock proteins 27 and 70: anti-apoptotic pro;teins with tumorigenic properties. Cell Cycle 5, 2592–2601. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Davis EE, and Katsanis N (2009). The vertebrate primary cilium in development, homeostasis, and disease. Cell 137, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guemez-Gamboa A, Coufal NG, and Gleeson JG (2014). Primary cilia in the developing and mature brain. Neuron 82, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman TR, Liu D, Zilfou JT, Robb V, Morrison T, Watnick T, and Henske EP (2009). The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 18, 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelland LR, Sharp SY, Rogers PM, Myers TG, and Workman P (1999). DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-de-methoxygeldanamycin, an inhibitor of heat shock protein 90. J. Natl. Cancer Inst. 91, 1940–1949. [DOI] [PubMed] [Google Scholar]
- Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, and Tsiokas L (2011). Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat. Cell Biol. 13, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschen GW, and Xiong Q (2017). Primary cilia as a novel horizon between neuron and environment. Neural Regen. Res. 12, 1225–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DA, Sadhwani A, Byars AW, de Vries PJ, Franz DN, Whitte-more VH, Filip-Dhima R, Murray D, Kapur K, and Sahin M (2017). Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann. Clin. Transl. Neurol. 4, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto N, Gu Y, Wang J, Janoschka S, Takemaru K, Levine J, and Ge S (2012). A role for primary cilia in glutamatergic synaptic integration of adult-born neurons. Nature Neurosci. 15, 399–405, S391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, and Gleeson JG (2011). Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr. Opin. Neurol. 24, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Saito M, Chuang JZ, Tseng YY, Dedesma C, Tomizawa K, Kait-suka T, and Sung CH (2011). Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat. Cell Biol. 13, 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JO, and Sahin M (2014). The neurology of mTOR. Neuron 84, 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marley A, and von Zastrow M (2012). A simple cell-based assay reveals that diverse neuropsychiatric risk genes converge on primary cilia. PLoS ONE 7, e46647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KR, Zhou W, Bowman MJ, Shih J, Au KS, Dittenhafer-Reed KE, Sisson KA, Koeman J, Weisenberger DJ, Cottingham SL, et al. (2017). The genomic landscape of tuberous sclerosis complex. Nat. Commun. 8, 15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, and Kwiatkowski DJ (2007). A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci. 27, 5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, and Kwiatkowski DJ (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 28, 5422–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliavacca E, Golzio C, Mä nnik K, Blumenthal I, Oh EC, Harewood L, Kosmicki JA, Loviglio MN, Giannuzzi G, Hippolyte L, et al. ; 16p11.2 European Consortium (2015). A potential contributory role for ciliary dysfunction in the 16p11.2 600 kb BP4-BP5 pathology. Am. J. Hum. Genet. 96, 784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L, and Workman P (2012). Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res. 18, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie D, Di Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, Dabora S, Codeluppi S, Pandolfi PP, Pasquale EB, and Sahin M (2010). Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat. Neurosci. 13, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie D, Chen Z, Ebrahimi-Fakhari D, Di Nardo A, Julich K, Robson VK, Cheng YC, Woolf CJ, Heiman M, and Sahin M (2015). The stress-induced Atf3-gelsolin cascade underlies dendritic spine deficits in neuronal models of tuberous sclerosis complex. J. Neurosci. 35, 10762–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohji G, Hidayat S, Nakashima A, Tokunaga C, Oshiro N, Yoshino K, Yokono K, Kikkawa U, and Yonezawa K (2006). Suppression of the mTOR-raptor signaling pathway by the inhibitor of heat shock protein 90 geldanamycin. J. Biochem. 139, 129–135. [DOI] [PubMed] [Google Scholar]
- Overwater IE, Rietman AB, van Eeghen AM, and de Wit MCY (2019). Everolimus for the treatment of refractory seizures associated with tuberous sclerosis complex (TSC): current perspectives. Ther. Clin. Risk Manag. 15, 951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampliega O, Orhon I, Patel B, Sridhar S, Díaz-Carretero A, Beau I, Co_-dogno P, Satir BH, Satir P, and Cuervo AM (2013). Functional interaction between autophagy and ciliogenesis. Nature 502, 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Lim JS, Ramakrishina S, Kim SH, Kim WK, Lee J, Kang HC, Reiter JF, Kim DS, and Kim HH (2018). Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron 99, 83–97 e87. [DOI] [PubMed] [Google Scholar]
- Pollizzi K, Malinowska-Kolodziej I, Doughty C, Betz C, Ma J, Goto J, and Kwiatkowski DJ (2009). A hypomorphic allele of Tsc2 highlights the role of TSC1/TSC2 in signaling to AKT and models mild human TSC2 alleles. Hum. Mol. Genet. 18, 2378–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruski M, and Lang B (2019). Primary cilia—an underexplored topic in major mental illness. Front. Psychiatry 10, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengren T, Larsen LJ, Pedersen LB, Christensen ST, and Møller LB (2018). TSC1 and TSC2 regulate cilia length and canonical Hedgehog signaling via different mechanisms. Cell. Mol. Life Sci. 75, 2663–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, and Sabatini DM (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915. [DOI] [PubMed] [Google Scholar]
- Sarkisian MR, and Guadiana SM (2015). Influences of primary cilia on cortical morphogenesis and neuronal subtype maturation. Neuroscientist 21, 136–151. [DOI] [PubMed] [Google Scholar]
- Sarkisian MR, and Semple-Rowland SL (2019). Emerging roles of primary cilia in glioma. Front. Cell. Neurosci. 13,55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Nagai T, Chiba S, Nakayama K, and Mizuno K (2018). Glucose deprivation induces primary cilium formation through mTORC1 inactivation. J. Cell Sci. 131, jcs208769. [DOI] [PubMed] [Google Scholar]
- Talos DM, Sun H, Kosaras B, Joseph A, Folkerth RD, Poduri A, Madsen JR, Black PM, and Jensen FE (2012). Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 71, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, and Zhong Q (2013). Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai P, and Sahin M (2011). Mechanisms of neurocognitive dysfunction and therapeutic considerations in tuberous sclerosis complex. Curr. Opin. Neurol. 24, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, and Sahin M (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Rudolph S, Guo C, Ellegood J, Gibson JM, Schaeffer SM, Mogavero J, Lerch JP, Regehr W, and Sahin M (2018). Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep 25, 357–367 e354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winden KD, Ebrahimi-Fakhari D, and Sahin M (2018). Abnormal mTOR activation in autism. Annu. Rev. Neurosci. 41, 1–23. [DOI] [PubMed] [Google Scholar]
- Yeh C, Li A, Chuang JZ, Saito M, Cáceres A, and Sung CH (2013). IGF-1 activates a cilium-localized noncanonical Gbγ signaling pathway that regulates cell-cycle progression. Dev. Cell 26, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD (2011). Illustration of SSMD, z score, SSMD*, z* score, and t statistic for hit selection in RNAi high-throughput screens. J. Biomol. Screen 16, 775–785. [DOI] [PubMed] [Google Scholar]
- Zhang HH, Lipovsky AI, Dibble CC, Sahin M, and Manning BD (2006). S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol. Cell 24, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Guo Y, Guettouche T, Smith DF, and Voellmy R (1998). Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480. [DOI] [PubMed] [Google Scholar]
- Zsebik B, Citri A, Isola J, Yarden Y, Szöllosi J, and Vereb G (2006). Hsp90 inhibitor 17-AAG reduces ErbB2 levels and inhibits proliferation of the trastuzumab resistant breast tumor cell line JIMT-1. Immunol. Lett. 104, 146–155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All software used in this study are listed in the Key Resources Table. The cilia genes expression data and the primary screen data generated in this study are listed in deposited data of the Key Resources Table. These unique datasets have been included and are available as Tables S1 and S3, respectively. Original data have been deposited to Mendeley Data: [https://data.mendeley.com/datasets/p45bhvf2g3/draft?a=420877d4-d4cb-45f1-adb7-91fcefcec901].
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ACIII | Proteintech | Cat# 19492–1-AP; RRID:AB„10638445 |
| Rabbit monoclonal anti-centrin | Millipore | Cat# 04–1624; RRID:AB_10563501 |
| Rabbit polyclonal anti-Hp90 | Proteintech | Cat#13171–1-AP; RRID:AB_2120924 |
| Rabbit monoclonal antl-GAPDH | Ambion | Cat# AM4300; RRID:AB_437392 |
| Rabbit polyclonal anti-pS6 | Cell Signaling Technology | Cat# 5364; RRID:AB_10694233 |
| Mouse monoclonal anti-S6 | Santa Cruz Biotechnology | Cat# sc-74459; RRID:AB_1129205 |
| Rabbit polyclonal anti-phospho Akt (Ser473) | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| Rabbit monoclonal anti-Akt | Cell Signaling Technology | Cat# 4691; RRID:AB_91578 |
| Rabbit polyclonal anti-phospho PRAS40 (Thr246) | Cell Signaling Technology | Cat#13175; RRID:AB_2798140 |
| Rabbit polyclonal anti-PRAS40 | Cell Signaling Technology | Catt# 2691; RRID:AB_2225033 |
| Rabbit polyclonal anti-phospho IGF-IRβ(Tyr1335/6) | Cell Signaling Technology | Cat# 3024S; RRID:AB_331253 |
| Rabbit polyclonal anti-IGF-IRβ | Santa Cruz Biotechnology | Cat# sc-713; RRID:AB_671792 |
| Rabbit polyclonal anti-Tsc2 | Cell Signaling Technology | Cat# 4308; RRID:AB_10547134 |
| Rabbit monoclonal anti-Hsp70 | Santa Cruz Biotechnology | Cat# sc-24; RRID:AB_627760 |
| Rabbit monoclonal anti-Hsp60 | Santa Cruz Biotechnology | Cat# sc-271215; RRID:AB_10607973 |
| Rabbit monoclonal anti-Hsp27 | Santa Cruz Biotechnology | Cat# sc-13132; RRID:AB_627755 |
| Chicken polyclonal anti-GFP | Thermo Fisher Scientific | Cat# A10262; RRID:AB_2534023 |
| Mouse monoclonal anti-RFP | Thermo Fisher Scientific | Cat# MA515257; RRID;AB_10999796 |
| Goat anti-mouse IRDye 800RD | LI-COR Biosciences | Cat#925–32210; RRID:AB_2687825 |
| Goat anti-mouse IRDye 680RD | LI-COR Biosciences | Cat#925–68070; RRID:AB_2651128 |
| Goat anti-rabbit IRDye 800CW | LI-COR Biosciences | Cat#925–32211; RRID:AB_2651127 |
| Goat anti-rabbit IRDye 680RD | LI-COR Biosciences | Cat#925–68071; RRID:AB_2721181 |
| Goat anti-chicken Alexa Fluor-488 | Molecular Probes | Cat#A11039; RRID:AB_.142924 |
| Goat anti-rabbit Alexa Fluor-647 | Molecular Probes | Cat# A21244; RRID:AB_141663 |
| Goat anti-rabbit Alexa Fluor-546 | Molecular Probes | Cat#A11035; RRID:AB_143051 |
| Goat anti-mouse Alexa Fluor-546 | Molecular Probes | Cat#A11030; RRID:AB_144695 |
| Bacterial and Virus Strains | ||
| Stbl3 competent cells | Thermo Fisher Scientific | Cat# C737303 |
| NEB Stable competent cells | New England Biolabs | Cat#C30401 |
| Tsc2-sh-GFP | Previously reported (See text for details) | N/A |
| Hsp27-shRNA | Sigma | Cat#TRCN0000321339 |
| Biological Samples | ||
| Sprague-Dawley rat brain tissue | Charles River Laboratories | N/A |
| TSC patient brain tissue | BCH Repository, Table S2 | N/A |
| Non-TSC patient brain tissue | BCH Repository, Table S2 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BIOMOL ICCB-L Known Bioactive 2012 library | ICCB-Longwood | https://iccb.med.harvard.edu/biomol-iccb-l-known-bioactives-2012 |
| Rapamycin | LC Laboratories | Cat# R-5000 |
| 17-Allylamino- geldanamycin (17-AGG) | Enzo Life Sciences | Cat# BML- EI308–0001 |
| Geldanamycin (GA) | Enzo Life Sciences | Cat# BML- EI280–0001 |
| Poly-D-Lysine Hydrobromide | MP Biomedical | Cat# 102694 |
| Bortezomib | EMD Millipore | Cat# 5.04314.0001 |
| Critical Commercial Assays | ||
| Pure Link Plasmid Kit | Thermo Fisher Scientific | Cat#K21007 |
| Direct-zol™ RNA Miniprep Kit | Zymo-Research | Cat# R2052 |
| iScript™ cDNA Synthesis Kit | BIO-RAD | Cat# 1708891 |
| PowerUP™ SYBR™ Green Master Mix | Thermo Fisher Scientific | Cat# A25742 |
| Deposited Data | ||
| RNA sequencing data | Previously reported (See text for details) | N/A |
| Cilia genes expression data | This study | Table S1 |
| Primary screen data | This study | Table S3 |
| Mendeley Data | This study | https://data.mendeley.com/datasets/p45bhvf2g3/draft?a=420877d4-d4cb-45f1-adb7-91fcefcec901 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216, RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tsc1tm1Djk/J | The Jackson Laboratory | IMSR Cat# JAX:005680, RRID:IMSR_JAX:005680 |
| Mouse: Tsc2tm1Djk/J | The Jackson Laboratory | IMSR Cat# JAX:004686, RRID:IMSR_JAX:004686 |
| Mouse: Tsc2tm2.1Djk/Mmjax | The Jackson Laboratory | IMSR Cat# JAX: 037154,. RRID:MMRRC_037154-JAX |
| Mouse: B6.Cg-Tg(Syn1-cre)671 Jxm/J | The Jackson Laboratory | IMSR Cat# JAX:003966, RRID:IMSR_JAX:003966 |
| Oligonucleotides | ||
| GAPDH forward primer: 5′-TGTGTCCGTCGTGGATCT GA-3′ | This study | N/A |
| GAPDH reverse primer: 5′-CCTGCTTCACCACCTTCTTGA-3′ | This study | N/A |
| hspB1 forward primer: AGACCAAGGAAGGCGTGGTG | This study | N/A |
| hspB1 reverse primer: CACACCTGGAGGGAGCGTGT | This study | N/A |
| Recombinant DNA | ||
| pLKO-RFP-shCntrl | Addgene | RRID:Addgene_69040 |
| Luciferase-ctrl-sh-GFP | Previously reported (See text for details) | N/A |
| psPAX2 | Addgene | RRID:Addgene_12260. |
| pMD2.G | Addgene | RRID:Addgene_12259. |
| Software and Algorithms | ||
| Fiji Software v2.0.0 | Open Source | http://fiji.se/.RRID:SCR_002285 |
| GraphPad PRISM v8.2.1 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/RRID:SCR_002798. |
| HCS Studio™ Cell Analysis Software v6.6.0 | Thermo Fisher Scientific | N/A |
| Image Studio Lite analysis software v5.2.5 | LI-COR | https://www.licor.com |
| Photoshop CS5.1 v12.1 | Adobe | https://www.adobe.com/products/photoshop.html |
| Syscilia. | Open Source | http://www.syscilia.org/goldstandard.shtml. |