

Abstract
The medical and psychosocial challenges faced by patients living with Disorders/Differences of Sex Development (DSD) and their families can be alleviated by a rapid and accurate diagnostic process. Clinical diagnosis of DSD is limited by a lack of standardization of anatomical and endocrine phenotyping and genetic testing, as well as poor genotype/phenotype correlation. Historically, DSD genes have been identified through positional cloning of disease-associated variants segregating in families and validation of candidates in animal and in vitro modeling of variant pathogenicity. Owing to the complexity of conditions grouped under DSD, genome-wide scanning methods are better suited for identifying disease causing gene variant(s) and providing a clinical diagnosis. Here, we review a number of established genomic tools (karyotyping, chromosomal microarrays and exome sequencing) used in clinic for DSD diagnosis, as well as emerging genomic technologies such as whole-genome (short-read) sequencing, long-read sequencing, and optical mapping used for novel DSD gene discovery. These, together with gene expression and epigenetic studies can potentiate the clinical diagnosis of DSD diagnostic rates and enhance the outcomes for patients and families.
Keywords: Sexual development, Disorders/differences of sex development, Structural variants, Next-generation sequencing, Whole genome sequencing, Optical genome mapping, Exome sequencing, Long-read sequencing, genetic diagnosis
1. Introduction
Sexual development is one of the most elegant and fascinating mechanisms in developmental biology where spatiotemporally orchestrated molecular signaling events determine the sexual fate of a developing embryo. Sexual development can be broadly divided into two sequential steps: sex determination and sex differentiation (Figure 1). Sex determination in a developing embryo essentially begins with establishment of the sex chromosome complement, inherited from the parental gametes, which in turn triggers a sequence of mutually exclusive molecular signaling events in the presence or absence of the Y chromosome driving the bipotential gonad in the developing embryo towards a male or female fate respectively. Expression of Y-chromosome gene SRY in the somatic cells of the bipotential gonad of an XY embryo kickstarts a cascade of signaling events that lead to testicular organogenesis. In the absence of SRY, female-specific pathways are initiated, triggering ovarian development under the control of genes such as RSPO1, WNT4 and DAX1/NR0B1.
Figure 1: Simplified overview of sex determination and sex differentiation in humans.
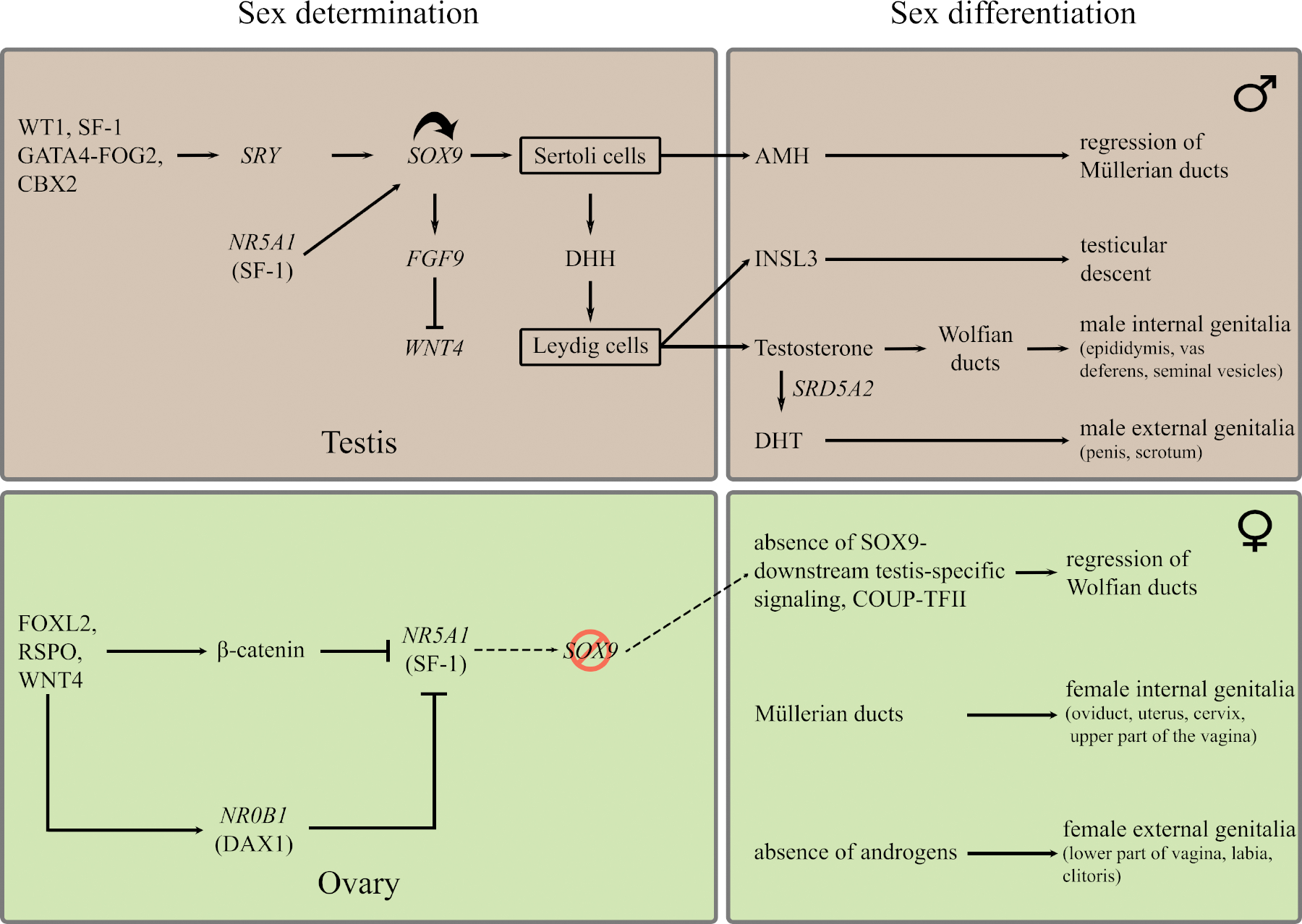
(The figure has been modified from (Arbodela and Vilain, 2009) using Barseghyan et al., 2015; Biason-Lauber, 2012; Croft et al., 2016; Kyriakou et al., 2015; Ohnesorg et al., 2014; Makoto Ono and Harley, 2013; Zhao et al., 2017) and references therein.
The development of a testis or an ovary from a bipotential gonad is followed by secretion of the sex-specific milieu of hormones from those organs, which drives the second step of sexual development – sex differentiation, and results in sex-specific morphogenesis of the reproductive organs. Disruption in any of these developmental processes and the molecular networks driving them, as well as the enzymatic cascades involved in the synthesis of steroid hormones in the adrenal or central control of these processes, can lead to Disorders/Differences of Sex Development (DSD). The molecular mechanisms involved have been expertly reviewed elsewhere (Croft et al., 2016; Délot and Vilain, 2018; Kyriakou et al., 2015; Ohnesorg et al., 2014; Makoto Ono and Harley, 2013).
2. Disorders/Differences of Sexual development, challenges in diagnosis and management
The term DSD was adopted following the 2005 consensus meeting of the Lawson Wilkins Pediatric Endocrine Society (LWPES) and the European Society for Paediatric Endocrinology (ESPE) to replace controversial and potentially confusing terms such as hermaphroditism, pseudohermaphroditism, and intersex. DSD was formally defined as “congenital conditions in which development of chromsomal, gonadal, or anatomical sex is atypical” (Hughes et al., 2006; Lee et al., 2006). As an umbrella term, it encompasses a range of rare and not so rare anatomical phenotypes (e.g. ambiguous genitalia, discordance of appearance external genitalia and chromosomal sex, isolated hypospadias,...); hormonal phenotypes (e.g. androgen insensitivity, over/under secretion of androgens); gonadal phenotypes (e.g. gonadal dysgenesis, ovotestis); and chromosomal complements (e.g. 46,XY, 46,XX, 47,XXY, 45,X). DSD can occur in isolated or syndromic forms (the latter reviewed in (Délot et al., 2017; Hutson et al., 2014)).
Management of DSD is complex as the conditions may be associated with increased infertility, cancer, and gender dysphoria risks, as well as pervasive challenges to psychosocial adaptation for patients and families (Abacl et al., 2015; Arboleda et al., 2014a; Guercio and Rey, 2014; Sandberg et al., 2015; Sandberg D.E., 2014; Słowikowska-Hilczer et al., 2017). Correlation between treatment interventions (surgical, hormonal, psychosocial) and their clinical outcomes is poorly understood. This suggests further research in DSD pathophysiology is needed to provide optimal evidence-based clinical management. Major challenges in DSD management include lack of standardization in descriptions of phenotype and medical/surgical interventions, resulting in variations in diagnostic and treatment practices across medical, surgical and behavioral aspects of health (Sandberg et al., 2015). Substantial unexplained variance in outcomes is revealed by studies examining the relationship between molecular diagnosis, gender development and psychosocial trajectories (Rolston et al., 2017). These challenges complicate the principles of clinical management of DSDs with disagreements among and between healthcare providers, advocacy groups and patient communities regarding the definition of optimal care (Adam and Vilain, 2017). International efforts are underway to standardize practice and diagnosis (e.g. Audí et al., 2018; Délot et al., 2017).
From a diagnostic standpoint too, DSDs are challenging. For example, 46,XY disorders of androgen biosynthesis, which include SRDA2, 17-ßHSD, 3ßHSD, StAR, CYP17A1, and POR deficiencies, are often difficult to distinguish clinically. However, an early genetic diagnosis is critical as these conditions have drastically different natural histories requiring different management (recently reviwed in (Délot and Vilain, 2018)).
3. Genetics of DSDs
The earliest characterized cases of DSDs were cases of sex chromosome aneuploidies such as 45,X (Turner syndrome) and its mosaic variants (e.g. 45,X/46,XY), 47,XXY (Klinefelter syndrome), or 46,XX/46,XY chimerism. These conditions account for about 15% of DSDs (Cox et al., 2014; De Paula et al., 2016; Délot et al., 2017; Demirhan et al., 2015; Heeley et al., 2018). Many of the early identifications of individual DSD genes were achieved by positional cloning and linkage analysis and identifying microscopically discernable structural changes in the karyotype using cytogenetic techniques. A number of genes such as SRY, SOX9, NR0B1/DAX1, DMRT1, WT1, MAP3K1 and RSPO1 were discovered using these approaches. However family-based methods have reduced power for conditions where family size is limited by infertility.
Copy number variants (CNV) affecting coding sequences or regulatory elements of critical dosage-sensitive genes, as well as single nucleotide variants (SNVs) in at least 75 genes involved in gonadal development and/or sex hormone biosynthesis/action are known causes of DSDs (Ahmed and Hughes, 2002; Arboleda A. A.; Vilain, E., 2010; Auchus and Miller, 2012; Buonocore and Achermann, 2016; Croft et al., 2018; Délot et al., 2017; Eggers and Sinclair, 2012; M Ono and Harley, 2013). Genetic etiology has been well established for many sex differentiation conditions. For example, recessive inheritance of variants in CYP21A2 is the most common cause of Congenital Adrenal Hyperplasia (CAH), which can lead to external genital masculinization in 46,XX patients (Nimkarn et al., 2016; Speiser et al., 2010). In 46,XY individuals, sex differentiation anomalies can result from complete or partial androgen insensitivity or defects in biosynthesis of steroid hormones. Androgen insensitivity in its complete form (CAIS) is caused, in >95% of patients, by loss-of-function mutations in AR, the androgen receptor. However, only a minority (~10%) of partial forms of androgen insensitivity (PAIS) are associated with AR variants. For the most difficult to diagnose DSD conditions, such as the non-syndromic forms of 46,XY gonadal dysgenesis, evaluation of historical data suggests that approximately ~15% of are due to SRY defects, ~15% to mutations in SF1/NR5A1, and a few cases due to other rare genetic events (in SOX9, DAX1/NR0B1,...) (Mohnach et al., 2016). More recent data suggest that MAP3K1 variants may explain an additional 10–18% (Baxter et al., 2015; Granados et al., 2017; Pearlman et al., 2010), still leaving nearly half of cases without a genetic diagnosis. In 46,XX individuals, the majority (80–90%) of non-syndromic Testicular DSD are explained by SRY translocations; however, only ~10% of Ovotesticular DSD have detectable Y material (Délot and Vilain, 2015) and etiology remains elusive. In XX individuals, abnormal ovarian development leads to primary ovarian insufficiency, an important medical challenge that affects over 1% of women over the age of 30 (Rossetti et al., 2017). The majority of ovarian dysgenesis cases are linked to abnormal complements of the X chromosome (such as Turner Syndrome). In addition, many genes coding for oocyte-specific transcription factors (e.g. FOXL2), folliculogenesis growth factors (e.g. BMP15), proteins that control ovarian steroidogenesis (e.g. receptor for FSH) or proteins involved in DNA replication or integrity (e.g. STAG3) have now been shown to underlie ovarian dysgenesis or premature ovarian failure, though each explain only a minority of cases and many cases remain unexplained (Aittomaki et al., 1995; Arboleda et al., 2014b; Caburet et al., 2014; Chapman et al., 2015).
The majority of DSD patients don’t receive an accurate diagnosis, which may be due to lack of, or imperfect access to, state-of-the-art genetic testing. We recently showed that 97% of undiagnosed patients in the US-based DSD-TRN registry have not exhausted clinically available diagnostic testing (Délot et al., 2017). In this review, we focus on how the existing and upcoming high throughput genomic technologies have been used to help identify novel DSD-associated genes and how these can be incorporated into genetic diagnostic practice to improve the diagnosis of DSD.
4. Chromosomal Microarrays (CMA)
Karyotyping can typically detect chromosomal rearrangement(s) with 5–10 Mb resolution. Hence, newer genome-wide scanning technologies were developed to achieve a better resolution in identifying gain or loss of genetic material. The two main types of chromosomal microarrays (CMA) used for genetic testing are Comparative Genomic Hybridization (array CGH) and Single-Nucleotide Polymorphism (SNP) arrays. Array CGH involves labelling the patient and control DNA samples with two different fluorophores and hybridizing them to an array containing 50–70 bp long oligonucleotides. The relative fluorescent signal shows regions of gain or loss of genetic material in the patient sample when directly compared with the control sample. It is useful for detecting CNVs, but it is not capable of detecting regions of homozygosity. Single Nucleotide Polymorphism (SNP) arrays, on the other hand, use a single fluorescently labelled (patient) DNA sample and hybridize it with an array containing oligonucleotides representing SNPs and the relative intensity is compared with that of a pool of controls available as online reference. In addition to detecting loss or gain of genetic material, new SNP arrays have incorporated aspects of the array CGH technology to be able to identify homozygosity, heterozygosity, regions of Loss of Homozygosity (LOH) as well as consanguinity (Sund and Rehder, 2014).
CMAs have been instrumental in genetic diagnosis in cases of DSD by identifying genetic changes in regions of the genome housing known DSD gene(s) as well as finding novel variants, potentiating discovery of novel DSD genes (eg. (Aboura et al., 2009)). For example, clinical CMA diagnosed CNV of the X-chromosome dosage-sensitive gene NR0B1/DAX1, whose deletions and duplications cause Adrenal Hypoplasia Congenita and XY gonadal dysgenesis respectively (Bardoni et al., 1994; Ledig et al., 2010; Muscatelli et al., 1994; White et al., 2011). Array CGH analysis for a large cohort of 116 patients with idiopathic DSDs, compared with 8951 controls without urogenital defects, was able to detect clinically relevant CNVs in 21.5% of the patients (Tannour-Louet et al., 2010). A number of chromosomal rearrangements were detected that not only encompassed known DSD genes such as SRY or DMRT1, but also novel ones such as FGFR2, KANK1, ADCY2, or ZEB2. In another study of a cohort of 87 patients, array CGH identified CNVs comprising chromosomal deletions and duplications encompassing known DSD and novel genes for 5 out of 16 cases of syndromic DSD and 21 out of 71 cases of non-syndromic DSD (Ledig et al., 2010).
CMA has also been used to identify novel variants in the non-coding regulatory regions of DSD-associated genes. CMA identified a 250 kb deletion upstream of the NR0B1 locus resulting in its increased expression and consequent sex reversal in an XY woman whose Y chromosome harbored an intact SRY (Smyk et al., 2007). Deletions and duplications upstream of SOX9 have been associated with ovotesticular DSD and 46, XY gonadal dysgenesis (Benko et al., 2011; Cox et al., 2011; Lecointre et al., 2009; Vetro et al., 2011; White et al., 2011). CMA was also used to identify the breakpoint of a 12:17 translocation replacing part of the cis-regulatory region upstream of SOX9 with that of a chromosome 12 pseudogene, causing a gain of SOX9 function and a male phenotype in an XX child (Refai et al., 2010).
Correlation of CMA results from patients with animal model experiments can help validate novel SNVs. For example, a mouse knockout model of Wwox showed abnormal gonadal development (Aqeilan et al., 2009; Ludes-Meyers et al., 2007), but the gene was not associated with human DSD until CMA analysis on an XY DSD patient with abnormal gonadal development revealed an exonic deletion in the WWOX gene (White et al., 2012). SOX3 has a high amino acid sequence similarity (90%) and protein identity (67%) to SRY but is not normally expressed in developing gonads of humans or mice (Bowles et al., 2000). While loss-of-function of SOX3 doesn’t seem to affect sex determination in mice or humans (Weiss et al., 2003), ectopic expression in XX mouse gonads results in a male phenotype (Sutton et al., 2011). Subsequent CMA analysis of human SRY-negative XX male individuals detected SOX3 duplications in several patients (Moalem et al., 2012; Sutton et al., 2011; Vetro et al., 2015), supporting the hypothesis that increased dosage of SOX3 is capable of inducing male sex determination in the absence of SRY.
DSD-relevant CNVs have been recently reviewed (Croft et al., 2018). Many candidates and chromosomal loci with potential roles in sex determination and DSD are awaiting further validation. These include deletions in the 9p23-24 chromosomal region encompassing DMRT1 and KANK1 in cases of XY gonadal dysgenesis (Igarashi et al., 2013; Ledig et al., 2010; Quinonez et al., 2013; Raymond et al., 1999), deletions at 12p13 and 16p11.2 in cases of hypospadias, deletions at 10p14 and Xq28 in cryptorchidism, deletions at 1p36.3, 9p24.3 and 19q12-13.1 in cases of ambiguous genitalia (Tannour-Louet et al., 2010), copy number gains at Xq28 encompassing the VAMP7 gene in cases of newborns with congenital genitourinary tract masculinization disorders (Tannour-Louet et al., 2014).
In addition, clinical CMA typically only reports CNVs above a 25–50 kb cutoff, leaving a diagnostic gap for smaller CNVs. Previous studies in DSD cohorts have identified smaller CNVs, but interpretation remains a challenge (Kon and Fukami, 2015; Mizuno et al., 2014; Norling et al., 2013; Tannour-Louet et al., 2010; White et al., 2011). High-resolution CNV maps (with CNVs as small as 50 bp) are increasingly becoming available and are poised to bridge the gap with the smaller INDELs that are detectable using short-read sequencing. They have been recently used to investigate the potential consequences of small CNVs as well as genomic copy-neutral regions of homozygosity (ROH) in DSD genes (Amarillo et al., 2016). This study identified novel potentially pathogenic mechanisms in well-established DSD genes such as SOX9 or PTPN11, and strengthened the genetic evidence for pathogenicity of recently uncovered DSD genes such as WWOX. It also revealed potential novel recurrent mechanisms of pathogenicity involving imprinted genes, as well as regions that might modify the epigenetic landscape and RNA expression in the developing gonad.
5. Exome sequencing
Endocrine tests, anatomical studies, imaging studies, and the experience of the clinician have historically guided single candidate gene testing. However, in DSD, poor genotype-phenotype correlation and overlapping phenotypes pose a challenge to single-gene testing approaches. In consequence, clinical testing has gradually evolved from single-gene testing (typically using Sanger sequencing) to multi-gene testing allowed by the development and cost decrease of massively parallel next-generation (short-read) sequencing. Exome sequencing (WES) captures and sequences only exons and the immediately adjacent intronic canonical splice sites, covering about 45 million base pairs (or ~1.5 % of the genome). Current options include extended coverage to ~ 60 Mbp to include functional nonprotein coding elements, e.g., micro-RNA, long intergenic noncoding RNA. Alternatively a reduced coverage approach (~35 Mbp, limited to clinically relevant regions) has been used to decrease cost and increase read depth in the regions that are sequenced. WES was originally used to identify single nucleotide variants, but with the advancement of bioinformatic tools, it is now also capable of detecting some CNVs (D’Aurizio et al., 2016).
Exome sequencing involves capture of protein-coding regions, enrichment, sequencing and bioinformatic analysis. The major commercially available exome sequencing platforms differ in the capture and enrichment techniques. Exome capture can be solution-based or array-based. In the solution-based method, the DNA is fragmented; biotinylated oligonucleotide probes selectively hybridizing to a targeted genomic region are immobilized on magnetic streptavidin beads. Hybridized DNA targets are enriched using PCR, at a risk of introducing PCR-associated GC bias, and sequenced. Array-based enrichment is based on the same principles except that it utilizes a library of oligonucleotide probes immobilized on a high-density microarray (Warr et al., 2015). There are several commercial enrichment platforms available for exome sequencing, which differ in target region selection, bait length or density, molecule used for capture, and genomic fragmentation method (Shigemizu et al., 2015; Tobias and McElreavey, 2014).
WES allows for diagnosis of DSDs not only by identifying known DSD pathogenic variants in patients but also the discovery of novel DSD genes. For patients without a diagnosis at a given time, reanalysis of the sequencing data using ever-evolving software and new publications about disease-gene associations can lead to almost a 10% increase in diagnostic yield after a 2–3 years time period (Wenger et al., 2017).
Clinical diagnosis by WES is challenged by factors such as incomplete gene coverage, platform variability, difficulties of variant interpretation, and the well-documented ethical issues linked to incidental findings (e.g. (ACMG Board of Directors, 2014; Allyse and Michie, 2013; Green et al., 2013; Richards et al., 2015; Tobias and McElreavey, 2014)). Since one of the important early steps in WES is exome enrichment by PCR technology, some parts of the exome can be difficult to access and enrich because of the regional nucleotide composition (such as GC content) leading to limitations in exome coverage. In fact, ~400 disease-causing exonic variants reported in the Human Genome Mutation Database (HGMD) are not covered by the current methods (Caspar et al., 2017). Commercially available enrichment kits using high concentration of capture probes that cover difficult-to-enrich exome regions can improve yields (Lelieveld et al., 2015; Meienberg et al., 2015). Evolution of capture techniques has impacted DSD testing. Before 2014, less than half of the genes in our typical DSD-specific list used for exome variants filtering had their entire sequence captured by exome (i.e. had 100% coverage). A newer capture method allowed to reach full coverage for 83% of genes on the list, with another seven genes having upward of 97% sequence coverage (Délot et al., 2017).
On average exome sequencing identifies 21,000 coding variants in each case when compared to the reference genome (Lee et al., 2014), out of which ~500 are rare variants present in less than 1% of the population (Kernohan et al., 2018). Increasing availability of large ethnically and geographically diverse public databases of healthy controls (such as ExAC (Lek et al., 2016) and now gnomAD), is critical to accurately assess allele frequency. Under-representation of certain populations in such reference databases can lead to variant calling errors for patients belonging to such populations (eg. (Petrovski and Goldstein, 2016)). Manual curation of variants is highly labor intensive and automated in silico variant-prioritization platforms based on amino acid conservation and biochemical properties have been developed to interpret identified variants (Adzhubei et al., 2010; Kircher et al., 2014; Kumar et al., 2009; Schwarz et al., 2014; Smedley et al., 2015).
Sequencing of the two parents along with the proband helps prioritize variants (Wenger et al., 2017): Trio exome sequencing has been reported to increase the diagnostic yields from 22% to 33% (Lee et al., 2014) and 23.6% to 31% (Retterer et al., 2016) in comparison to proband-only sequencing. In the case of DSD, though, this brings additional interpretation challenges. For example, de novo variants in NR5A1 (the gene coding for SF1) are well known to cause developmental defects in gonads of both 46,XX and 46,XY individuals (reviewed in (Ferraz-de-Souza et al., 2011)). If the same variant was found in parents, automated algorithms would likely eliminate this variant as not pathogenic. However, many publications report as pathogenic novel heterozygous NR5A1 variants inherited from the (fertile) parent of the same karyotype as the patient (e.g. Philibert et al. 2010, Swartz et al. 2017), including the well-established, recurrent p.Arg92Trp variant (Baetens et al., 2016; Bashamboo et al., 2016). It has been suggested that the parent may present a milder phenotype, such as early menopause in the mother or progressive gonadal dysgenesis in the father, allowing them to reproduce and pass on the variant to their more severely affected child. Whether modifier genes and/or environmental influences are responsible for the variable expressivity of the phenotype remains to be determined. A recent publication suggests genotype/phenotype variability may be explained by an oligogenic mode of inheritance, a mechanism clinical exome sequencing is poorly equipped to detect at this time (Camats et al. 2018).
Identifed variants are classified for reporting using standardized nomenclature, following the American College of Medical Genetics and Genomics (ACMG) & the Association for Molecular Pathology recommendations (Richards et al., 2015). Variants are classified as pathogenic (previously reported in humans as the recognized cause of the specific DSD condition), likely pathogenic (e.g. previously unreported in a known DSD gene), benign (previously reported and recognized as a neutral variant), likely benign (e.g. previously unreported in DSD and is probably not predicted to affect protein funtions). However, the vast majority remain classified as variants of uncertain clinical significance (VUS).
The early adoption of exome sequencing by the academic institutions in the United States has helped to identify the genetic diagnosis in approximately 30% of cases for whom traditional molecular diagnostic methods were uninformative (Lee et al., 2014; Yang et al., 2014), a yield that has proven remarkably similar across countries, sequencing platforms, and conditions studied (Dong et al., 2016; Farwell et al., 2015; Sawyer et al., 2016; Soden et al., 2014; Yang et al., 2013). In many cases, the identified genetic diagnosis provided guidance for medical treatment and management. When performed early, exome sequencing is capable of not only increasing the diagnostic yields in DSD but also guiding clinical management of patients (e.g. Barseghyan et al., 2015; Baxter et al., 2015).
6. Gene-testing panels
Because interpretation of exome sequencing results beyond well-established variants in known DSD genes is challenging, clinical providers have developed gene panels. Gene panels are typically based on targeted next-generation sequencing (NGS) through either targeted capture or full exome capture followed by limited interpretation. They can provide high-throughput and accurate screening of multiple genes and different types of mutations in a highly efficient manner. Additional Sanger sequencing may be performed to fill in for genes with missing or insufficient read depth coverage. As gene panels test a limited set of genes, the utilization of NGS results in high read-depths enabling detection of non-reference alleles present at very low frequencies. This method facilitates detection of mosaics hypothesized to underlie variable phenotypic expression in DSD.
Comprehensive gene panels have been designed that report a high rate of diagnosis for 46,XY DSD: e.g. 43% using 64 genes (Eggers et al., 2016); 45% using 56 genes (Özen et al., 2017). Diagnosis rates are typically lower if undiagnosed 46, XX DSD cases are included (e.g. 29.5% of likely pathogenic or pathogenic variants found using a 67-gene panel (Kim et al., 2017). A study using a panel of 80 DSD genes demonstrated the superiority of the approach over single-gene testing performed on the same patient samples (Fan et al., 2017). Panels are also time- and cost-effective, with one study quoting 3 days vs. 6 years to diagnosis on average and reduction of costs by 2/3rds (Özen et al., 2017).
Many commercial providers offer clinical genetic testing of DSDs and many institutions have started developing their in-house tests. Prevention Genetics (www.preventiongenetics.com) provides a gene panel of 65 genes covering both syndromic and non-syndromic DSDs accounting for ~35% of cases of 46, XY DSDs. Using this panel they have reported deletions and duplications in known DSD genes such as SOX3, LHCGR, SRY, NR0B1, DMRT1, NR5A1, GATA4, WT1, WNT4, and FGFR2. Invitae (www.invitae.com) offers a panel of 8 genes (AR, DHH, MAP3K1, NR0B1, NR5A1, SRD5A2, SRY and WT1), expandable to 15, for genetic diagnosis of cases of 46, XY DSD, 46, XY complete gonadal dysgenesis and 46, XX testicular DSD. It combines full-gene sequencing and deletion/duplication analysis via NGS in clinically important genetic regions including exons and adjacent +/− 10 bp of intronic sequences. GeneDx (www.genedx.com) provides a 19-gene panel (AR, ARX, ATRX, CHD7, CYP11A1, CYP17A1, DHCR7, DHH, DYNC2H1, HSD17B3, HSD3B2, NEK1, NR5A1, POR, SOX9, SRD5A2, SRY, STAR, WT1) for diagnosis of prenatal and neonatal 46,XY DSDs through NGS and deletion/duplication analysis. Blueprint genetics (www.blueprintgenetics.com) offers a gene panel covering 49 genes that assesses coding as well as non-coding variants for patients suspected to harbour DSDs or congenital adrenal hyperplasia (CAH).
As is evident from the examples above, these panels are not standardized. With constantly evolving methods (capture kits, gene coverage percentage, sequencing platforms, and analysis pipelines) and variability of options, it is becoming increasingly difficult to ascertain what method was actually used to diagnose a patient and whether a different test might have found a different result.
7. Evaluation of genes on the DSD gene list
A challenge of both whole-exome and limited panels is deciding which genes make the gene list. A variety of criteria and resources are used, such as reported cases in PubMed (http://www.ncbi.nim.nih.gov/pubmed), the Online Mendelian Inheritance in Man (OMIM) database (http://www.ncbi.nlm.nih.gov/omim), GeneReviews, or clinical variant databases such as HGMD (http://www.hgmd.cf.ac.uk/ac/index.php) or ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). A typical clinical list includes 60–75 DSD genes. For research purposes, the list can be lengthened (to ~250) to include genes implicated in gonadal development, known molecular pathways (such as Hedgehog or WNT signaling, or AR-interacting proteins). However, the burden of proof available for assessing that a reported gene is indeed causative of DSD in humans varies greatly by gene. Some have had replication in multiple families and populations and are supported by extensive animal models and in vitro demonstration of the pathogenicity of specific variants, while others have been reported in single publications. As a proxy to assessing genetic evidence, we examined the variants reported in ClinVar, the widely used repository of clinical variants maintained and curated by the NIH. We extracted the variants classified as pathogenic and likely pathogenic in 69 genes, in cases reported with a DSD phenotype (Table 1). For each, we looked at the type of variant (missense, nonsense, frameshift, larger INDELs and rearrangements) and the publications quoted in support of the variant classification. We did not include the large CNVs (typically reported in batch from genome-wide scanning experiments) because whether deletion of the particular gene was indeed causative couldn’t be assessed.
Table 1: Pathogenic and likely pathogenic variants in 69 DSD genes listed in the ClinVar database:
The variants in 69 human DSD genes (column 1) deposited in the NIH-maintained ClinVar database and classified as pathogenic or likely pathogenic for a DSD condition were compiled. The total number (column 2) and type (column 3) of variants are shown (FS Del/Ins: deletion/insertion resulting in frameshift). Copy number variants that included multiple genes were not included, as pathogenicity could not be easily attributed to the single gene. If the genes were involved in several phenotypes, only the variants relevant to DSD were included. Variants for which phenotypic information was not available were not included. Selected references cited as evidence in the database are shown in column 4 (the complete list for each gene entry can be found at https://www.ncbi.nlm.nih.gov/clinvar/).
| Gene name | Number of variants | Type of variants | Example references |
|---|---|---|---|
| Sex Determination | |||
| BMP15 (POF4) | 3 | Missense, nonsense | (Di Pasquale et al., 2004; Dixit et al., 2006; Rossetti et al., 2009) |
| CBX2 | 2 | Missense | (Biason-Lauber et al., 2009) |
| DHH | 9 | FS Del, nonsense, missense | (Canto et al., 2008; Tajouri et al., 2018; Umehara et al., 2000; Werner et al., 2015) |
| DMRT1 | 1 | Missense | - |
| DMRT2 | 0 | - | - |
| FSHR | 17 | Missense | (Aittomäki et al., 1995; Beau et al., 1998; De Leener et al., 2008, 2006; Di Carlo et al., 1997; Doherty et al., 2002; Gromoll et al., 1996; Kuechler et al., 2010; Meduri et al., 2003; Montanelli et al., 2004; Smits et al., 2003; Vasseur et al., 2003) |
| GATA4 | 1 | Missense | (Lourenco et al., 2011) |
| HHAT | 0 | - | - |
| MAP3K1 | 9 | Missense, Splice site | (Baxter et al., 2015; Granados et al., 2017; Loke and Ostrer, 2012; Pearlman et al., 2010) |
| NR0B1 (DAX1) | 75 | FS del/ins, rearrangement, gene deletion, gene duplication, missense, nonsense, splice site | (Barbaro et al., 2007; Muscatelli et al., 1994; Jun Nakae et al., 1997; Schwartz et al., 1997; Yanase et al., 1996) |
| NR5A1 (SF1) | 26 | Missense, nonsense, in-frame deletion, FS del | (Achermann et al., 1999; Baetens et al., 2017; Bashamboo et al., 2010; Colson et al., 2017; Correa et al., 2004; Igarashi et al., 2017; Lin et al., 2007; Lourenço et al., 2009) |
| RSPO1 | 3 | Exon deletion, FS ins, Splice site | (Parma et al., 2006; Tomaselli et al., 2008) |
| SOX3 | 3 | Gene duplication, missense, in-frame duplication | (Karaca et al., 2015; Woods et al., 2005) |
| SOX9 | 14 | Promoter Del, Promoter Dup/Triplication, FS ins, FS del, missense, nonsense | (Benko et al., 2011; Cox et al., 2011; Foster et al., 1994; Kim et al., 2015; Kwok et al., 1995; Pop et al., 2005; Vetro et al., 2011) |
| SRY | 27 | Missense, FS del, in-frame del, nonsense | (Berta et al., 1990; Hawkins et al., 1992; Jäger et al., 1990; McElreavey et al., 1992; Nykamp et al., 2017a; Vilain et al., 1992) |
| STAG3 (POF8) | 4 | FS del, FS dup, Splice site, nonsense | (Caburet et al., 2014; He et al., 2018; Le Quesne Stabej et al., 2016) |
| WNT4 | 4 | Missense | (Biason-Lauber et al., 2007, 2004; Mandel et al., 2008; Philibert et al., 2008) |
| WT1 | 23 | Splice site, missense, nonsense, FS del, FS dup | (Barbaux et al., 1997; Barrera et al., 2016; Bruening et al., 1992; Little and Wells, 1997; Royer-Pokora et al., 2004; F. Wang et al., 2017) |
| WWOX | 0 | - | - |
| ZFPM2 | 3 | Missense | (Bashamboo et al., 2014; Royer-Pokora et al., 2004) |
| Sex differentiation | |||
| AKR1C2 | 4 | Missense | (Christa E. Flück et al., 2011) |
| AKR1C4 | 0 | - | - |
| AMH | 4 | FS Del, FS Ins, nonsense | (Carré-Eusèbe et al., 1992; Knebelmann et al., 1991; Lang-Muritano et al., 2001) |
| AMHR2 | 4 | FS Del, in-frame del, Splice site, missense | (Belville et al., 2009; Imbeaud et al., 1995, 1996; Messika-Zeitoun et al., 2001) |
| AR | 109 | Splice site, missense, FS Ins/del, nonsense, multi exon deletion | (Brown et al., 1988; Brüggenwirth et al., 1997; Choong et al., 1996; MacLean et al., 1993; Sammarco et al., 2000; Vilchis et al., 2003; Zhu et al., 1999; Zoppi et al., 1993) |
| ARX | 2 | Nonsense, FS Del | (Kato et al., 2004; Kitamura et al., 2002) |
| ATRX | 24 | Splice site, Nonsense, FS Del, missense | (Gibbons et al., 1995; Mitson et al., 2011a; Stevenson, 1993) |
| CYP11A1 | 10 | FS Del, FS Ins, in-frame insertion, missense | (Hiort et al., 2005; Katsumata et al., 2002; C. J. Kim et al., 2008; Tajima et al., 2001) |
| CYP17A1 | 35 | Splice site, in-frame deletion, FS del, FS Dup, rearrangement, missense, nonsense | (Ahlgren et al., 1992; Imai et al., 1992; Kagimoto et al., 1988; Oshiro et al., 1995; Schwab et al., 2005; van den Akker et al., 2002; Yamaguchi et al., 1997; Yanase et al., 1990; Yang et al., 2006) |
| CYP19A1 | 13 | fusion, insertion, deletion, missense SNV, intron SNV, missense SNV, nonsense SNV | (Deladoëy et al., 1999; Herrmann et al., 2002; Ito et al., 1993; Shozu et al., 2003; Tiulpakov et al., 2005) |
| CYP21A2 | 49 | Deletion, duplication, SNVs, insertion, intron SNV, missense SNV | (Billerbeck et al., 2002; Hayashi et al., 2013; Kaupert et al., 2016; Kirac et al., 2014; Livadas et al., 2015; C. Wang et al., 2017; Wang et al., 2016; Welzel et al., 2010; Yoo et al., 2013) |
| DHCR7 | 90 | Missense, FS Ins, in-frame deletion, nonsense, Splice site | (Lanthaler et al., 2015; Sparks et al., 2014; Wassif et al., 1998; Waterham and Hennekam, 2012; Waye et al., 2002; Yu et al., 2000) |
| FGFR2 | 3 | Intragenic deletion, missense | (Bagheri-Fam et al., 2015; Fonseca et al., 2008; Przylepa et al., 1996; Slavotinek et al., 2009) |
| FOXL2 (POF 3) | 5 | FS Del, FS Ins, in-frame deletion, missense | (Harris et al., 2002; Yang et al., 2018) |
| HSD17B3 | 23 | Missense, Splice site, FS Del, SNV deleting a stop codon | (Castro et al., 2012; Geissler et al., 1994; Lindqvist et al., 2001; Phelan et al., 2015) |
| HSD3B2 | 11 | FS ins, FS del, nonsense, missense | (Rhéaume et al., 1992; Simard et al., 1994; Welzel et al., 2008) |
| KDM5D | 0 | - | - |
| LHCGR | 32 | FS del, in-frame deletion, in-frame insertion, missense, nonsense, Splice site | (Boot et al., 2011; Gromoll et al., 2000; Laue et al., 1995; Misrahi et al., 1997; S. M. Wu et al., 1998) |
| MAMLD1 | 4 | Nonsense | (Fukami et al., 2006) |
| POR | 14 | Splice site, missense, FS Ins, FS Dup, deletion | (Arlt et al., 2004; Flück et al., 2004; Fukami et al., 2005; Huang et al., 2005; Oh et al., 2017) |
| SRD5A2 | 41 | FS Del, missense, nonsense, Splice site, exon loss deletion, in-frame deletion | (Bertelloni et al., 2016; Cai et al., 1996; Can et al., 1998; Eggers et al., 2016; Ko et al., 2010; Thigpen et al., 1992) |
| STAR | 33 | FS Del, FS Ins, Splice site, missense, nonsense | (Abdulhadi-Atwan et al., 2007; Bose et al., 1996; Christa E. Flück et al., 2011; Huang et al., 2016; J Nakae et al., 1997; Okuyama et al., 1997; Tee et al., 1995) |
| VAMP7 | 0 | - | - |
|
Central causes of hypogonadism | |||
| ARL6 (BBS3) | 10 | Missense, nonsense, gene deletion, FS Del, Splice site | (Chiang et al., 2004; Fan et al., 2004; Lindstrand et al., 2016) |
| CHD7 | 187 | Multi-exon deletion, FS Del, FS Ins, nonsense, Splice site, rearrangement, a few missense | (Jongmans et al., 2008; H.-G. Kim et al., 2008; Moccia et al., 2018; Udaka et al., 2007) |
| FGF8 | 7 | FS Del, Missense, 5’UTR SNV | (Costa-Barbosa et al., 2013; Falardeau et al., 2008) |
| FGFR1 | 27 | Missense, nonsense, FS Del, Splice site | (Dodé et al., 2003; Pitteloud et al., 2006; Trarbach et al., 2006; Xu et al., 2007) |
| FRAS1 | 17 | FS Ins, FS Del, nonsense splice site, in-frame deletion | (Cavalcanti et al., 2007; McGregor et al., 2003; Slavotinek et al., 2006) |
| FREM2 | 6 | Missense, FS Del, FS Ins, nonsense, Splice site | (Jadeja et al., 2005; Shafeghati et al., 2008; van Haelst et al., 2008) |
| GNRH1 | 1 | FS Ins | (Bouligand et al., 2009) |
| GNRHR | 20 | Missense, nonsense, Splice site | (Caron et al., 1999; Gianetti et al., 2012; Kottler et al., 2000; Layman et al., 1998; Meysing et al., 2004) |
| GRIP1 | 2 | Splice site, FS Del | (Vogel et al., 2012) |
| HESX1 | 14 | FS Del, FS Ins, missense, splice site, nonsense | (Brickman et al., 2001; Cohen et al., 2003; Dattani et al., 1998; Sobrier et al., 2006; Tajima et al., 2003; Thomas et al., 2001) |
| HFE | 11 | Missense, nonsense, FS Del | (Beutler et al., 2002; de Villiers et al., 1999; Gochee et al., 2002; Piperno et al., 2000) |
| KAL1/ANOS1 | 25 | Multi-exon deletion, FS Dup, FS Del, missense, nonsense, splice site | (Albuisson et al., 2005; Canto et al., 2008; Massin et al., 2003; Söderlund et al., 2002; Trarbach et al., 2006) |
| KISS1R | 7 | Intragenic deletion, missense, nonsense, FS SNV | (Brioude et al., 2013; Seminara et al., 2003; Tenenbaum-Rakover et al., 2007) |
| LEP | 4 | FS Del, missense | (Gibson et al., 2004; Strobel et al., 1998; Wabitsch et al., 2015) |
| LEPR | 2 | Splice site, FS Ins | (Clément et al., 1998) |
| LHX3 | 9 | Gene deletion, intragenic deletion, FS del, nonsense, missense | (Netchine et al., 2000; Pfaeffle et al., 2007; Rajab et al., 2008) |
| PCSK1 | 5 | Splice site, missense, nonsense, in-frame deletion | (Farooqi et al., 2007; Jackson et al., 2003, 1997) |
| PROK2 | 6 | FS Ins, FS Del, missense | (Cole et al., 2008; Dodé et al., 2006; Leroy et al., 2008; Pitteloud et al., 2007) |
| PROKR2 | 12 | Missense, FS Del | (Lindsay W. Cole et al., 2008a; Dodé et al., 2006b; Dodé and Rondard, 2013; McCabe et al., 2013; Monnier et al., 2009; Reynaud et al., 2012; Ruiz-Ferrer et al., 2011; Sarfati et al., 2013, 2010) |
| PROP1 | 31 | Nonsense, missense, FS Ins, FS Del, Splice site | (Lindsay W Cole et al., 2008; Johnny Deladoëy et al., 1999; Fofanova et al., 1998; Kelberman et al., 2009; Lemos et al., 2006; Reynaud et al., 2005; Sarfati et al., 2010; W. Wu et al., 1998) |
| PTPN11 | 80 | Missense, in-frame deletion | (Sobreira et al., 2010; Tartaglia et al., 2006; Thiel et al., 2009; Yoshida et al., 2004; Zenker et al., 2004) |
| SOS1 | 27 | Missense | (Lepri et al., 2011; Li et al., n.d.; Roberts et al., 2007) |
| TAC3 | 4 | Missense, FS Del | (Gianetti et al., 2010; Topaloglu et al., 2009) |
| TACR3 | 7 | Missense, nonsense | (Francou et al., 2011; Gianetti et al., 2010; Topaloglu et al., 2009) |
| TRIM32 | 6 | Missense, FS Del | (Chiang et al., 2006; Locke et al., 2009; Van Goor et al., 2014) |
| TTC8 | 4 | In-frame deletion, Splice site, 5’UTR deletion | (Ansley et al., 2003; Stoetzel et al., 2006) |
For several of the long-known DSD genes, the available proof of pathogenicity was obvious in the database, with the highest numbers of variants reported for AR (109), NR0B1/DAX1 (75), CYP21A2 (49), SRD5A2 (41), SRY (27), NR5A1/SF1 (26), WT1 (23). For example, many types of severe variants (splice site, multi-exon deletion, nonsense, frameshift-causing insertions or deletions) support the pathogenicity of potentially milder (missense) variants in AR causing Androgen Insensitivity Syndrome. The extensive number of publications in support of these variants is complemented by data uploads from reliable clinical testing labs such as GeneDx, Johns Hopkins or Invitae. However, even these can be underreported. The AR mutation database created at McGill University (last updated in 2014) had more than 550 variants (Gottlieb and Trifiro, 2017), while only 109 are found in ClinVar. Similarly, the number of variants reported in ClinVar for CYP21A2 is relatively low for such a frequent condition, and better resources exist elsewhere. A recent effort (Simonetti et al., 2018) curated existing variants from multiple sources (e.g. CYPAlleles and dbSNP databases) and compiled them in a new gene-specific database to support genetic counseling to families with Congenital Adrenal Hyperplasia.
Other “usual suspects” genes artificially show a low number of variants in ClinVar. In some cases, many more are actually in the database but without a phenotype reported they were excluded from the analysis, as “pathogenic” can only refer to a specific phenotype. In others cases, the explanation was less obvious. For example, AKR1C2 seems to have a similar amount of evidence as AMHR2 - 4 variants each - with possible slant in favor of AMHR2 due to more severe and diverse variants (e.g. frameshift, splice site, against only missense for AKR1C2). The AKR1C2 variants, however, are all from the same publication, and the finding hasn’t been replicated as widely. For AMHR2, one of the supporting publications quoted in the database (Belville et al., 2009) reports that 38 different mutations had been identified in patients with Persistent Müllerian Duct Syndrome by their team alone at that point, and describes the molecular effects of 10 of those variants. Those are yet to be included in ClinVar. Similarly, ARX, which causes an X-linked syndromic form of DSD in males, associating lissencephaly and abnormal genitalia, has only 2 ClinVar variants, one nonsense (Kato et al., 2004) and one deletion leading to a frameshift, but without a supporting reference. The quoted reference is actually the second paper from that team, which had reported at least another 15 variants in this gene. In these cases, the existing genetic proof is actually much stronger than reflected in the database.
This approach was also limited by the precision of phenotypic description in the database. For example, while pathogenicity of loss-of-function mutations in ATRX in the X-linked dominant alpha-thalassemia/MR syndrome is well documented with 24 variants in ClinVar, which of these mutations actually lead to DSD was more difficult to evaluate. The genital phenotype only affects males, while females have normal genitalia. However, variants are reported here without indication of whether they were found in XX or XY individuals. This is also an issue for syndromes that may include genital anomalies as a non-fully penetrant trait. Variants are reported in the databases under the umbrella of the syndrome, without specification of genital involvement. One striking example is DHCR7, the causative gene for CHARGE syndrome, which has one of the highest number of variants in the list (90). Clarifying which variants are associated with genital anomalies requires analyzing each original publication and is not possible in the case of variants reported by clinical testing labs, not linked to a publication. In these cases, genotype-phenotype correlations, if any exist, have to be manually curated.
Six genes in the table have no DSD-associated variants reported in ClinVar: AKR1C4, DMRT2, HHAT, KDM5D, VAMP7 and WWOX. We examined the existing evidence for including those in a primary gene list for exome filtering in human DSD diagnosis. Three of these genes have in common to have been identified as causative on the basis of CNVs. As in the case for other dosage-sensitive genes involved in DSD (such as SOX3, NR0B1/DAX1 or WNT4), variants are found in very few patients each and are difficult to both identify and validate.
Duplication of the distal region of the X chromosome Xq28, including VAMP7, was found using array CGH in 2/116 patients with genitourinary tract masculinization disorder (Tannour-Louet et al., 2010). That paper identified 11 other cases in the literature, 3 more by quantitative PCR in a different cohort, and demonstrated that transgenic mice overexpressing human VAMP7 mimicked the phenotype, possibly via potentiation of the estrogen signaling pathway through ESR1. The experimental evidence is solid, and awaits replication by others and identification of other types of variants in VAMP7.
A SNP array identified a 767-kb in-frame deletion of exons 6–8 of the WWOX gene in a patient with 46,XY DSD (White et al., 2012), and the corresponding truncated transcript was found in the patient’s cells. The paper reported that two different knock-out mouse strains showed Leydig cell function defects and gonadal abnormalities. Validation of WWOX as a human DSD gene awaits experimental demonstration of protein dysfunction and identification of variants in other patients.
Multiple reports have associated 46,XY gonadal dysgenesis with deletion of chromosome 9p24.3, without the other features of terminal 9p deletion syndrome (catalogued as 46,XY Sex Reversal Type 4 in OMIM). This led to the hypothesis that haploinsufficiency for DMRT1 and/or DMRT2 is causative of the genital phenotype. Embryonic expression of DMRT1 is gonad-specific and sexually dimorphic; DMRT2 shows only weak expression in the developing gonad. Both genes share homology with sex-determining genes in Drosophila (Doublesex) and C. Elegans (Mab3) but confirmatory evidence in humans is weak, with one missense (without supportive reference) in DMRT1 in ClinVar and none in DMRT2. However, at least two other types of variants have now been identified in DMRT1: a missense mutation (Chauhan et al., 2016) and a deletion of exons 3–4 (Ledig et al., 2012).
A short peptide derived from the KDM5D (lysine demethylase 5D) protein is the Y-chromosome-linked H-Y/HLA-B7 (aka HYA) minor histocompatibility antigen. GeneCards links KDM5D to “Spermatogenic Failure, Y-linked, Type 2”. UniProt/SwissProt data suggests that it may regulate transcriptional activity of AR. However, OMIM does not report a link between this gene and the phenotype and no clear evidence was found in a literature search. Since the azoospermia AZF locus has been shown to not include HYA, this gene should probably be removed from primary DSD gene lists.
A homozygous missense variant in HHAT, the Hedgehog acetyl transferase, was identified by exome sequencing as the cause of a familial autosomal recessive syndromic form of 46, XY DSD associated with chondrodysplasia and severe brain abnormalities (Callier et al., 2014). An XX sibling with normal ovaries shared the non-genital phenotype and the variant. Evidence included prediction of protein disruption by multiple algorithms and confirmation by in vitro experiments, expression of the protein in the developing gonad, and demonstration that palmitoylation of Hedgehog ligands is required for testes organogenesis in mouse. The experimental evidence is elegant, but awaits identification of further cases.
AKR1C4 is part of a complex encoded on chromosome 10p15.1 that play a role in metabolism of steroid hormones. Evidence for its involvement in human DSD rests on a single family with testicular and adrenal 17,20-desmolase deficiency resulting in a defect in androgen biosynthesis. In this family, a splice site variant in AKR1C4 segregated together with a missense in related gene AKR1C2 (Flück et al., 2011); another patient in the same report harbored a complex rearrangement of the region. Therefore AKR1C4 is likely best considered a modifier (as OMIM classifies it for 46,XY Sex Reversal Type 8) or in a digenic model (e.g. with AKR1C2) and searching for such a mode of inheritance in exome results might help identify further patients.
8. Whole genome sequencing
While exome sequencing is a clear improvement over single-gene testing in providing clinical diagnosis for DSD, it leads to a definitive molecular diagnosis in only 30% of all cases (Baxter et al., 2015; Lee et al., 2014; Yang et al., 2014). About 85% of disease-associated variants are found in coding regions (Abecasis et al., 2010), but this is likely to be an ascertainment bias. Metaanalysis of hundreds of genome-wide association studies showed that >90% of genetic variants associated with complex multigenic disorders lie within non-coding regions of the genome instead of the regions captured by exome sequencing (Maurano et al., 2012). Genome-wide screens of DSD patients have discovered variants in the regulatory non-coding regions of DSD associated genes, in particular SOX9 and SOX3 (Croft et al., 2018; Harrison et al., 2013; Sutton et al., 2011; White et al., 2011). Therefore a technique sequencing the whole genome rather than only the protein-coding regions has the potential to provide a much higher diagnostic yield than WES.
Whole-Genome Sequencing (WGS), which is also based on short-read sequencing but is not constrained by the exome capture step, overcomes limitations of WES such as lack of coverage of some GC rich parts of the exome because of the PCR enrichment step, or the preferential capture of reference sequence allele leading to an allele distribution bias (Heinrich et al., 2012; Meynert et al., 2014, 2013; Quail et al., 2008). WGS is more powerful in detecting disease-causing SNVs than WES, even in exomic regions. For example, a study comparing WES and WGS results for 6 unrelated individuals showed that both techniques identified a similar number of the total as well as high quality SNVs and INDELs, however, the distributions of coverage depth, genotype quality and minor read ratio were more uniform for WGS than for WES (Belkadi et al., 2015). Confirmation using Sanger sequencing showed a similar percentage of false positives for INDELs (44% vs. 46%). However, SNV false positives were dramatically higher (78%) in WES than in WGS (17%).
Studies comparing the diagnostic yield of WGS with that of clinically conventional genetic diagnostic techniques typically report higher yield. For example, a diagnostic yield of 34% in a cohort of children with various congenital anomalies, including developmental delay, represents a four-fold increase in diagnostic rate over CMA and two-fold increase over targeted gene sequencing (Stavropoulos et al., 2016). NGS-based targeted sequencing is being increasingly used in conjunction with CMA and WES to improve clinical diagnostics of DSD cases. For example, a combination of targeted NGS, WES and CMA was able to detect genetic defects in 85% of the families in a cohort of 63 Japanese patients with biochemically uncharacterized pediatric onset primary adrenal insufficiency, with some of them exhibiting 46,XY DSD phenotypes (Amano et al., 2017). In another report, array CGH and WGS were utilized to determine a structural variant caused by a ~774 kb insertional translocation from chromosome 1 into a palindromic sequence 84 kb distal of the SOX3 locus on chromosome X, leading to its upregulation in a 46,XX phenotypically male patient (Haines et al., 2015).
WGS is slowly being incorporated into the clinical diagnosis process throughout the world with the hope that providing physicians and patients with an interpreted genome sequence will reduce the uncertainty of clinical decision-making and improve clinical outcomes. However, the wide availability of WGS for clinical purposes is currently limited by several factors. With about 3 million variants compared to the reference genome in each individual, the complexity of variant filtering and prioritizing is exponentially amplified compared to exome sequencing. Diagnostic capacity and variant classification for non-coding variants is also limited by the availability of comprehensive, ethnically rich, reference databases of control and disease sequences. In addition, WGS has the potential to detect large SVs, CNVs, INDELs as well as SNPs across the genome, when de novo assembly becomes more routinely feasible. However, due to the utilization of short-reads, the discovery of SVs by WGS is compromized. Many bioinformatic tools have been developed for SV calling from NGS data using a combination of read-depth, read-pair, split-read methods, but the number and type of SVs identified by each of the tools vary significantly with low sensitivity, low concordance rates, and large false discovery (English et al., 2015;Tattini et al., 2015).
9. Long-read sequencing
The human genome is arguably the most complete mammalian reference genome assembly, but it still contained over 160 euchromatic gaps encompassing long runs of degenerate short tandem repeats, often several kilobases in length, embedded within GC-rich genomic regions (Chaisson et al., 2015; Sedlazeck et al., 2018a). Although NGS significantly reduced the cost of sequencing, the utilization of short-reads and amplification biases led to difficulties in identification of larger SVs, as well as problems in sequencing certain genomic regions (Chaisson et al., 2015). This limitation is being remedied with the advent of “third-generation”, long-read sequencing (LRS) methods such as Single Molecule Real Time (SMRT) sequencing (Pacific Biosciences, PacBio) and nanopore sequencing (Oxford Nanopore Technologies, ONT). These also promise to revolutionize transcriptome analysis by allowing the sequencing of full-length RNA/cDNA molecules in which exon skipping and isoform expression can be determined.
PacBio’s SMRT sequencing utilizes “sequencing by synthesis method” where incorporation of differentially fluorescently labelled nucleotides into immobilized individual DNA templates is recorded in real time, resulting in long reads that end only when the DNA polymerase dissociates from the template (Rank et al., 2009). The average read length from the current PacBio’s Sequel System is approximately 10,000 bp, but new improved chemistry promises to increase the read lengths to an average of 85,000 bp (Kosicki et al., 2018; Sedlazeck et al., 2018b).
Nanopore sequencing passes a single strand of DNA through a pore where the electric signal changes generated by the base-by-base movement of the molecule across the nanopore are converted into base-calls (Deamer et al., 2016; Jain et al., 2016, 2015). Some users have observed read-lengths in excess of 2 Mb (www.nanoporetech.com).
LRS has been employed for a number of purposes to overcome challenges faced by NGS technologies. For example, it was successfully used to sequence regions of microsatellite repeats in cases of neurodegenerative diseases such as ALS and frontotemporal dementia (FTD) as well as polyglutamine expansion in Huntington disease, dentatorubropallidoluysian atrophy, spinal and bulbar muscular atrophy (Ebbert et al., 2018; Liu et al., 2017). PacBio long-range RNA sequencing was used to sequence the chicken transcriptome, which helped resolve regions encompassing multiple-mapping loci, repeat-regions and ambiguous splice junctions across the coding as well as non-coding transcriptome (Kuo et al., 2017). PacBio and Nanopore sequencing were also utilized in conjunction with Illumina SRS to create de novo genome assembly of Saccharomyces cerevisiae (Jenjaroenpun et al., 2018). Application of LRS in clinical diagnosis in a case of two patients with congenital abnormalities detected novel chromothripsis rearrangements with details of breakpoints, parental origin and structure, as well as novel variants originating from retrotransposon insertions at higher efficiencies in comparison to Illumina SRS (Cretu Stancu et al., 2017).
In 2018, both Nanopore (Jain et al., 2018) and PacBio (pacbio.com press release, Oct. 2018) have reported complete de novo assembly of a human genome. However both techniques have severe limitations that will need to be overcome before widespread use in clinical practice is possible. Both require important compute time and bioinformatic resources. Nanopore sequencing is fast but shows a lack of base-calling accuracy; Pac-Bio is very accurate, but slow. Methods are being developed to combine the strengths of each to create accurate, isoform-level transcriptome and genome annotation (Volden et al., 2018), promising to fully exploit the power of LRS technologies to discover large SV and sequence variation in a single molecule, which can be the next paradigm for DSDs diagnosis.
10. Optical Genome Mapping
Many SVs are caused by non-allelic homologous recombination events between duplicated DNA sequences as large as 300 kbp in size. The repetitive nature of these regions makes them all but invisible to short-read sequencing platforms (Conrad et al., 2010; Sharp et al., 2005) or traditional cytogenetics methods such as CMAs, which can elucidate gain or loss of genetic material, but fail to detect the order/orientation of the SV and are virtually blind for detection of balanced events such as inversions and translocations. Recently, a novel method – optical genome mapping has been proposed as the “go to” method for identification of large/complex structural variants due to its high specificity and sensitivity (Hastie et al., 2017). SVs account for the largest portion of human to human genome variation (Sudmant et al., 2015; Weischenfeldt et al., 2013) and have been implicated in a variety of disease etiologies including DSD (Barseghyan et al., 2015), neuropsychiatric disorders (Brand et al., 2014) and heart disease (Brand et al., 2014).
Optical genome mapping (supported by the Bionano Genomics platform) uses specialized nanochannel arrays capable of housing immobilized, labeled megabase-size DNA molecules for optical imaging (Hastie et al., 2017). Prior to imaging, the long DNA molecules are fluorescently labeled at specific sequence motifs throughout the genome, using restriction enzymes (reviewed in (Barseghyan et al., 2018) or other proprietary enzymes (Figure 2). The resultant pattern of fluorescent tags within long DNA molecules is used for de novo assembly of each allele of the sample genome with scaffold length N50 values exceeding 60 Mbps (to compare with ~10.5 Mbps for Nanopore (Jain et al., 2018). Comparison of the sample-specific maps with the in silico-digested maps of the human genome reference allows for identification of large deletions, insertions, inversions, translocations and complex rearrangements. Optical mapping has been used for identification of potentially pathogenic SVs in cancer genomes (Dixon et al., 2018; Du et al., 2018; Jaratlerdsiri et al., 2017), population-specific genome assemblies (McCaffrey et al., 2017; Seo et al., 2016; Shi et al., 2016), as well as muscle-wasting disorders such as Duchenne and facioscapulohumeral muscular dystrophies (Barseghyan et al., 2017; Dai et al., 2018). In Duchenne Muscular Dystrophy, we showed the ability of whole-genome optical mapping to identify large deletions, insertions, and inversions in hemizygous and heterozygous states with 100% clinical diagnosis concordance rates (Barseghyan et al., 2017).
Figure 2: Preparation of long DNA molecules for optical mapping:
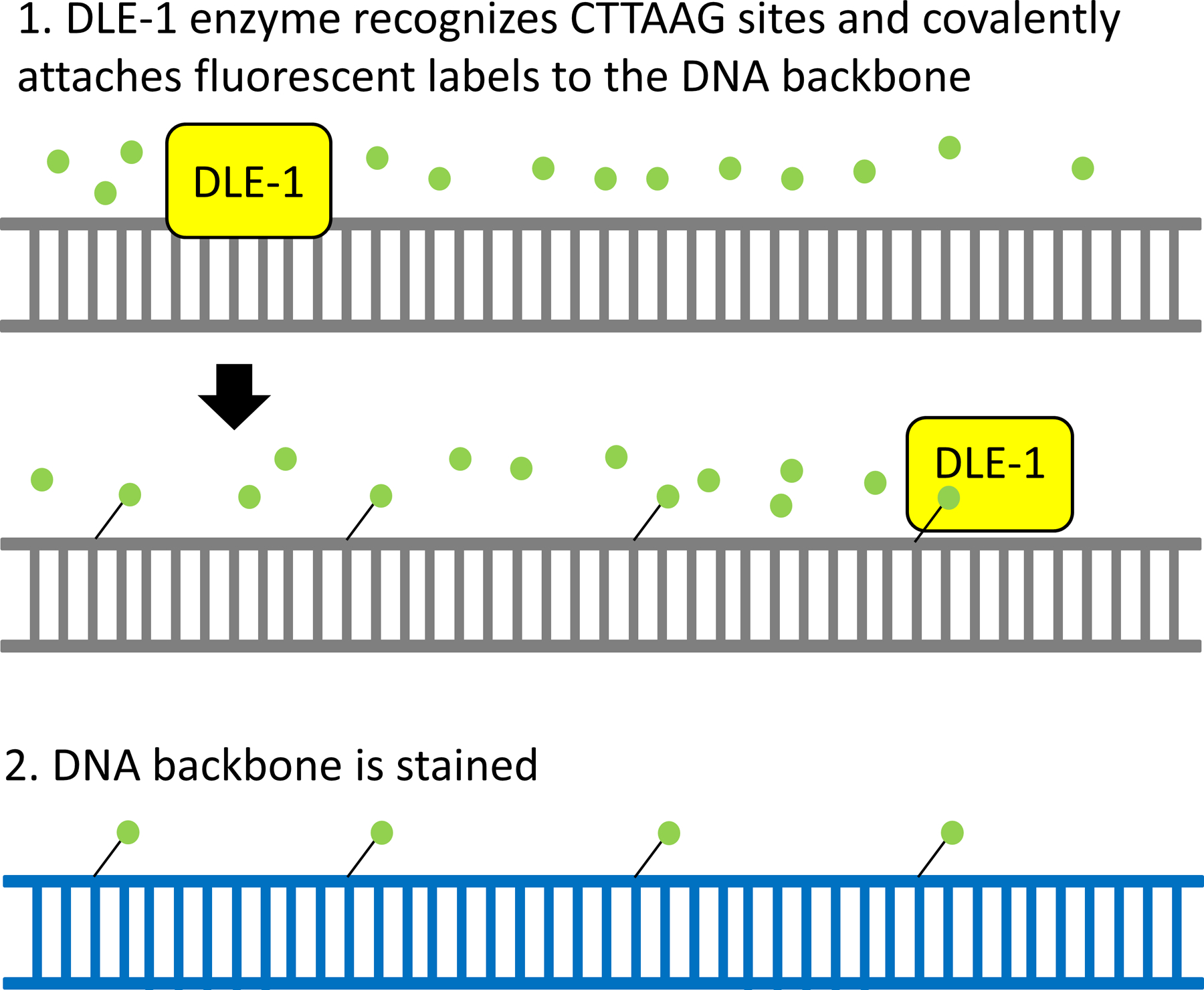
High molecular weight DNA is incubated with fluorescent labels and Direct Labeling Enzyme 1 (DLE1). The enzyme recognizes CTTAAG sequences throughout the genome and covalently attaches a fluorescent label (green). Subsequently, the DNA backbone is stained overnight and loaded onto the Bionano Genomics Chips containing millions of nanochannels for imaging. The green fluorescent labels are used to map DLE1 enzyme recognition patterns, whereas the blue staining of the DNA backbone is used to identify molecule sizes. Both are used for de novo genome assembly.
Although optical mapping brings key advantages over the clinically available cytogenetic technologies such as karyotype and microarray, mainly increased resolution and detection of balanced events, its adoption in clinic is still pending. Our group is working on bringing optical mapping to DSD diagnosis (Barseghyan et al., 2018). Integration of WGS and whole-genome optical mapping allows researchers to survey all of the different types of genomic variations: SNVs, INDELs, CNVs and large SVs. Recent, unpublished work suggests that integration of the two technologies tremendously improves WGS SV identification rates, while also decreasing optical mapping’s SV breakpoint uncertainty to less than 140 bp (originally ~3–5 kbp) (Porat et al., 2018). Combined integrated optical mapping and genome sequencing analysis is poised to provide higher diagnostic rates for DSD and other genetic disorders.
Identification of clinically relevant SVs will require integration of data sets obtained through all current technologies to ascertain both what is a true SV (variant calling) and which are pathogenic (variant classification). An early global map of 270 individuals using SNP arrays and array CGH identified ~1,500 copy number variable regions over 1 kb, covering 12% of the genome (Redon et al. 2006). Integration of SRS, LRS and optical mapping on a genome identified 10,000 putative SVs compared the hg19 reference assembly (English et al. 2015). The PacBio method identified >20,000 unique SVs (> 50 bp) and ~400,000 1–49 bp INDELs, 80% of which could not be resolved with current SRS techniques (Ameur et al., 2018a, 2018b; Sedlazeck et al., 2018b). A major difficulty remains assembling enough genomes to understand the complexity of pathogenic and non-pathogenic human genomic SV diversity.
11. Epigenetics
Finally, epigenetic variation may also represent a significant etiology in DSD. One of the most compelling pieces of evidence of the role of epigenetic changes governing sex determination is the promoter DNA methylation of the master regulator Sry (Nishino et al., 2004). Sry expression in male mice happens in a subset of gonadal somatic cells under tight spatiotemporal regulation around embryonic days 10.5–12.5 (DiNapoli and Capel, 2008; Hiramatsu et al., 2009). The promoter region of Sry was observed to be hypermethylated (associated with transcriptional repression) in gonadal somatic cells at 8.5 dpc and hypomethylated (associated with transcriptional activation) at 11.5 dpc while remaining hypermethylated in tissue not expressing Sry at the same time-point (Nishino et al., 2004). There have also been reports of differences in methylation patterns of key sex determination genes in cases of animal DSD that have a high congruence with human DSD genes. Persistent abnormal hypermethylation of the Sry gene (resulting in down-regulated mRNA and protein expression) was established as the cause of ovotestis and female phenotype in cloned Sry-positive XY dogs generated through somatic cell nuclear transfer (Jeong et al., 2016). In another example involving XX DSD dogs with ovotestis or testis phenotype, candidate gene promoter methylation bisulfite sequencing revealed hypomethylation in Sox9, hypermethylation in Wnt4 and hypermethylation of the Sox3 promoter at levels similar to that of control XY male dogs (Salamon et al., 2017). Both these examples show that epigenetic promoter methylation of important sex determination genes can be determinants of DSD in mammals.
Genomic DNA methylation at CpG sites can be detected by several technologies, including bisulphite conversion followed by SRS or SNP arrays (Kurdyukov and Bullock, 2016; Laird, 2010). Both PacBio and Oxford Nanopore LRS technologies potentially provide improved methylome sequencing capabilities (Fang et al., 2012; Murray et al., 2012) and Bionano is also developing a labeling method that will differentially detect unmethylated vs. methylated cytosines at the recognition site. All three have the advantage over current techniques to identify epigenetic marks and genomic sequence (PacBio, Nanopore) or mapping (Bionano) on the same, single molecule of DNA. These developing technologies bring the exciting prospect of enabling exploration of epigenetic modifications as a new and upcoming paradigm in diagnosis of DSDs.
12. References
- Abacl A, Çatll G, Berberoʇlu M, 2015. Gonadal malignancy risk and prophylactic gonadectomy in disorders of sexual development. J. Pediatr. Endocrinol. Metab 28, 1019–1027. 10.1515/jpem-2014-0522 [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Altshuler DL, Durbin RM, Bentley DR, Chakravarti A, Clark AG, Collins FS, De La Vega FM, Donnelly P, Egholm M, Flicek P, Gabriel SB, Gibbs RA, Knoppers BM, Lander ES, Lehrach H, Mardis ER, McVean GA, Nickerson DA, Peltonen L, Schafer AJ, Sherry ST, Wang J, Wilson RK, Deiros D, Metzker M, Muzny D, Reid J, Wheeler D, Wang SJ, Li J, Jian M, Li G, Li R, Liang H, Tian G, Wang B, Wang J, Wang W, Yang H, Zhang X, Zheng H, Ambrogio L, Bloom T, Cibulskis K, Fennell TJ, Jaffe DB, Shefler E, Sougnez CL, Bentley IDR, Gormley N, Humphray S, Kingsbury Z, Koko-Gonzales P, Stone J, Mc Kernan KJ, Costa GL, Ichikawa JK, Lee CC, Sudbrak R, Borodina TA, Dahl A, Davydov AN, Marquardt P, Mertes F, Nietfeld W, Rosenstiel P, Schreiber S, Soldatov AV, Timmermann B, Tolzmann M, Affourtit J, Ashworth D, Attiya S, Bachorski M, Buglione E, Burke A, Caprio A, Celone C, Clark S, Conners D, Desany B, Gu L, Guccione L, Kao K, Kebbel A, Knowlton J, Labrecque M, McDade L, Mealmaker C, Minderman M, Nawrocki A, Niazi F, Pareja K, Ramenani R, Riches D, Song W, Turcotte C, Wang S, Dooling D, Fulton L, Fulton R, Weinstock G, Burton J, Carter DM, Churcher C, Coffey A, Cox A, Palotie A, Quail M, Skelly T, Stalker J, Swerdlow HP, Turner D, De Witte A, Giles S, Bainbridge M, Challis D, Sabo A, Yu F, Yu J, Fang X, Guo X, Li Y, Luo R, Tai S, Wu H, Zheng H, Zheng X, Zhou Y, Marth GT, Garrison EP, Huang W, Indap A, Kural D, Lee WP, Leong WF, Quinlan AR, Stewart C, Stromberg MP, Ward AN, Wu J, Lee C, Mills RE, Shi X, Daly MJ, DePristo MA, Ball AD, Banks E, Browning BL, Garimella KV, Grossman SR, Handsaker RE, Hanna M, Hartl C, Kernytsky AM, Korn JM, Li H, Maguire JR, McKenna A, Nemesh JC, Philippakis AA, Poplin RE, Price A, Rivas MA, Sabeti PC, Schaffner SF, Shlyakhter IA, Cooper DN, Ball EV, Mort M, Phillips AD, Stenson PD, Sebat J, Makarov V, Ye K, Yoon SC, Bustamante CD, Boyko A, Degenhardt J, Gravel S, Gutenkunst RN, Kaganovich M, Keinan A, Lacroute P, Ma X, Reynolds A, Clarke L, Cunningham F, Herrero J, Keenen S, Kulesha E, Leinonen R, McLaren WM, Radhakrishnan R, Smith RE, Zalunin V, Korbel JO, Stütz AM, Humphray IS, Bauer M, Cheetham RK, Cox T, Eberle M, James T, Kahn S, Murray L, Ye K, Fu Y, Hyland FCL, Manning JM, Stephen FM, Peckham HE, Sakarya O, Sun YA, Tsung EF, Mark AB, Konkel MK, Walker JA, Albrecht MW, Amstislavskiy VS, Herwig R, Parkhomchuk DV, Agarwala R, Khouri HM, Morgulis AO, Paschall JE, Phan LD, Rotmistrovsky KE, Sanders RD, Shumway MF, Xiao C, Gil AM, Auton A, Iqbal Z, Lunter G, Marchini JL, Moutsianas L, Myers S, Tumian A, Knight J, Winer R, Craig DW, Beckstrom-Sternberg SM, Christoforides A, Kurdoglu AA, Pearson JV, Sinari SA, Tembe WD, Haussler D, Hinrichs AS, Katzman SJ, Kern A, Kuhn RM, Przeworski M, Hernandez RD, Howie B, Kelley JL, Melton SC, Li Y, Anderson P, Blackwell T, Chen W, Cookson WO, Ding J, Kang HM, Lathrop M, Liang L, Moffatt MF, Scheet P, Sidore C, Snyder M, Zhan X, Zöllner S, Awadalla P, Casals F, Idaghdour Y, Keebler J, Stone EA, Zilversmit M, Jorde L, Xing J, Eichler EE, Aksay G, Alkan C, Hajirasouliha I, Hormozdiari F, Kidd JM, CenkSahinalp S, Sudmant PH, Chen K, Chinwalla A, Ding L, Koboldt DC, McLellan MD, Wallis JW, Wendl MC, Zhang Q, Albers CA, Ayub Q, Balasubramaniam S, Barrett JC, Chen Y, Conrad DF, Danecek P, Dermitzakis ET, Hu M, Huang N, Matt EH, Jin H, Jostins L, Keane TM, Quang Le S, Lindsay S, Long Q, MacArthur DG, Montgomery SB, Parts L, Tyler-Smith C, Walter K, Zhang Y, Gerstein MB, Snyder M, Abyzov A, Balasubramanian S, Bjornson R, Grubert F, Habegger L, Haraksingh R, Khurana E, Lam HYK, Leng J, Mu XJ, Urban AE, Zhang Z, McCarroll SA, Zheng-Bradley X, Batzer MA, Hurles ME, Du J, Jee J, Coafra C, Dinh H, Kovar C, Lee S, Nazareth L, Wilkinson J, Coffey A, Scott C, Tyler-Smith C, Gharani N, Kaye JS, Kent A, Li T, McGuire AL, Ossorio PN, Rotimi CN, Su Y, Toji LH, Felsenfeld AL, McEwen JE, Abdallah A, Juenger CR, Clemm NC, Duncanson A, Green ED, Guyer MS, Peterson JL, 2010. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboura A, Dupas C, Tachdjian G, Portnoï M-F, Bourcigaux N, Dewailly D, Frydman R, Fauser B, Ronci-Chaix N, Donadille B, Bouchard P, Christin-Maitre S, 2009. Array Comparative Genomic Hybridization Profiling Analysis Reveals Deoxyribonucleic Acid Copy Number Variations Associated with Premature Ovarian Failure. J. Clin. Endocrinol. Metab 94, 4540–4546. 10.1210/jc.2009-0186 [DOI] [PubMed] [Google Scholar]
- ACMG Board of Directors, 2014. ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet. Med 17, 68. [DOI] [PubMed] [Google Scholar]
- Adam MP, Vilain E, 2017. Emerging issues in disorders/differences of sex development (DSD). Am. J. Med. Genet. Part C Semin. Med. Genet 175, 249–252. 10.1002/ajmg.c.31564 [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR, 2010. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SF, Hughes IA, 2002. The genetics of male undermasculinization. Clin Endocrinol 56, 1–18. [DOI] [PubMed] [Google Scholar]
- Aittomaki K, Lucena JL, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, Kaskikari R, Sankila EM, Lehvaslaiho H, Engel AR, Nieschlag E, Huhtaniemi I, de la Chapelle A, 1995. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 82, 959–968. [DOI] [PubMed] [Google Scholar]
- Allyse M, Michie M, 2013. Not-so-incidental findings: the ACMG recommendations on the reporting of incidental findings in clinical whole genome and whole exome sequencing. Trends Biotechnol 31, 439–441. 10.1016/j.tibtech.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano N, Narumi S, Hayashi M, Takagi M, Imai K, Nakamura T, Hachiya R, Sasaki G, Homma K, Ishii T, Hasegawa T, 2017. Genetic defects in pediatric-onset adrenal insufficiency in Japan. Eur. J. Endocrinol 177, 187–194. 10.1530/EJE-17-0027 [DOI] [PubMed] [Google Scholar]
- Amarillo IE, Nievera I, Hagan A, Huchthagowder V, Heeley J, Hollander A, Koenig J, Austin P, Wang T, 2016. Integrated small copy number variations and epigenome maps of disorders of sex development. Hum. genome Var 3, 16012 10.1038/hgv.2016.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameur A, Che H, Martin M, Bunikis I, Dahlberg J, Höijer I, Häggqvist S, Vezzi F, Nordlund J, Olason P, Feuk L, Gyllensten U, 2018a. De novo assembly of two swedish genomes reveals missing segments from the human GRCh38 reference and improves variant calling of population-scale sequencing data. Genes (Basel) 9, 1–16. 10.3390/genes9100486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameur A, Kloosterman WP, Hestand MS, 2018b. Single-Molecule Sequencing: Towards Clinical Applications. Trends Biotechnol xx, 1–14. 10.1016/j.tibtech.2018.07.013 [DOI] [PubMed] [Google Scholar]
- Aqeilan RI, Hagan JP, de Bruin A, Rawahneh M, Salah Z, Gaudio E, Siddiqui H, Volinia S, Alder H, Lian JB, Stein GS, Croce CM, 2009. Targeted ablation of the WW domain-containing oxidoreductase tumor suppressor leads to impaired steroidogenesis. Endocrinology 150, 1530–1535. 10.1210/en.2008-1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbodela VA, Vilain E, 2009. Disorders of Sex Development, in: Strauss J, Barbieri R (6th Ed.), Yen and Jaffe’s Reproductive Endocrinology. Saunders Elsevier, pp. 367–393. [Google Scholar]
- Arboleda VA, Fleming AA, Vilain E, 2010. Disorders of Sex Development, in: Weiss RE, Refetoff S (1st Ed.), Genetic Diagnosis of Endocrine Disorders. ScienceDirect, pp. 227–243. [Google Scholar]
- Arboleda VA, Fleming A, Barseghyan H, Delot E, Sinsheimer JS, Vilain E, 2014a. Regulation of Sex Determination in Mice by a Non-Coding Genomic Region. Genetics. 10.1534/genetics.113.160259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arboleda VA, Sandberg DE, Vilain E, 2014b. DSDs: genetics, underlying pathologies and psychosexual differentiation. Nat Rev Endocrinol 10, 603–615. 10.1038/nrendo.2014.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auchus RJ, Miller WL, 2012. Defects in androgen biosynthesis causing 46,XY disorders of sexual development. Semin Reprod Med 30, 417–426. 10.1055/s-0032-1324726 [DOI] [PubMed] [Google Scholar]
- Audí L, Ahmed SF, Krone N, Cools M, McElreavey K, Holterhus P-M, Greenfield A, Bashamboo A, Hiort O, Wudy SA, McGowan R, 2018. GENETICS IN ENDOCRINOLOGY: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 “DSDnet.” Eur. J. Endocrinol 179, R197–R206. 10.1530/EJE-18-0256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetens D, Stoop H, Peelman F, Todeschini A-L, Rosseel T, Coppieters F, Veitia RA, Looijenga LHJ, De Baere E, Cools M, 2016. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet. Med 19, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni B, Zanaria E, Guioli S, Floridia G, Worley KC, Tonini G, Ferrante E, Chiumello G, McCabe ER, Fraccaro M, et al. , 1994. A dosage sitive locus at chromosome Xp21 is involved in male to female sex reversal. Nat Genet 7, 497–501. 10.1038/ng0894-497 [DOI] [PubMed] [Google Scholar]
- Barseghyan H, Delot E, Vilain E, 2015. New Genomic Technologies : An Aid for Diagnosis of Disorders of Sex Development. Horm Metab Res 47, 312–320. [DOI] [PubMed] [Google Scholar]
- Barseghyan H, Délot EC, Vilain E, 2018. New technologies to uncover the molecular basis of disorders of sex development. Mol. Cell. Endocrinol 468, 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barseghyan H, Tang W, Wang RT, Almalvez M, Segura E, Bramble MS, Lipson A, Douine ED, Lee H, Delot EC, Nelson SF, Vilain E, 2017. Next-generation mapping: a novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med 9, 90 10.1186/s13073-017-0479-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashamboo A, Donohoue PA, Vilain E, Rojo S, Calvel P, Seneviratne SN, Buonocore F, Barseghyan H, Bingham N, Rosenfeld JA, Mulukutla SN, Jain M, Burrage L, Dhar S, Balasubramanyam A, Lee B, Members of, U.D.N., Dumargne MC, Eozenou C, Suntharalingham JP, de Silva K, Lin L, Bignon-Topalovic J, Poulat F, Lagos CF, McElreavey K, Achermann JC, 2016. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet 25, 3446–3453. 10.1093/hmg/ddw186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter RM, Arboleda VA, Lee H, Barseghyan H, Adam MP, Fechner PY, Bargman R, Keegan C, Travers S, Schelley S, Hudgins L, Mathew RP, Stalker HJ, Zori R, Gordon OK, Ramos-Platt L, Pawlikowska-Haddal A, Eskin A, Nelson SF, Delot E, Vilain E, 2015. Exome sequencing for the diagnosis of 46,XY disorders of sex development. J Clin Endocrinol Metab 100, E333–44. 10.1210/jc.2014-2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, Shang L, Boisson B, Casanova JL, Abel L, 2015. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A 112, 5473–5478. 10.1073/pnas.1418631112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belville C, Marechal J-D, Pennetier S, Carmillo P, Masgrau L, Messika-Zeitoun L, Galey J, Machado G, Treton D, Gonzales J, Picard J-Y, Josso N, Cate RL, di Clemente N, 2009. Natural mutations of the anti-Mullerian hormone type II receptor found in persistent Mullerian duct syndrome affect ligand binding, signal transduction and cellular transport. Hum. Mol. Genet 18, 3002–3013. 10.1093/hmg/ddp238 [DOI] [PubMed] [Google Scholar]
- Benko S, Gordon CT, Mallet D, Sreenivasan R, Thauvin-Robinet C, Brendehaug A, Thomas S, Bruland O, David M, Nicolino M, Labalme A, Sanlaville D, Callier P, Malan V, Huet F, Molven A, Dijoud F, Munnich A, Faivre L, Amiel J, Harley V, Houge G, Morel Y, Lyonnet S, 2011. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet 48, 825–830. 10.1136/jmedgenet-2011-100255 [DOI] [PubMed] [Google Scholar]
- Biason-Lauber A, 2012. WNT4, RSPO1, and FOXL2 in sex development. Semin Reprod Med 30, 387–395. 10.1055/s-0032-1324722 [DOI] [PubMed] [Google Scholar]
- Bowles J, Schepers G, Koopman P, 2000. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol 227, 239–55. 10.1006/dbio.2000.9883 [DOI] [PubMed] [Google Scholar]
- Brand H, Pillalamarri V, Collins RL, Eggert S, O’Dushlaine C, Braaten EB, Stone MR, Chambert K, Doty ND, Hanscom C, Rosenfeld JA, Ditmars H, Blais J, Mills R, Lee C, Gusella JF, McCarroll S, Smoller JW, Talkowski ME, Doyle AE, 2014. Cryptic and complex chromosomal aberrations in early-onset neuropsychiatric disorders. Am. J. Hum. Genet 95, 454–61. 10.1016/j.ajhg.2014.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonocore F, Achermann JC, 2016. Human sex development: targeted technologies to improve diagnosis. Genome Biol 17, 257 10.1186/s13059-016-1128-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caburet S, Arboleda VA, Llano E, Overbeek PA, Barbero JL, Oka K, Harrison W, Vaiman D, Ben-Neriah Z, Garcia-Tunon I, Fellous M, Pendas AM, Veitia RA, Vilain E, 2014. Mutant cohesin in premature ovarian failure. N Engl J Med 370, 943–949. 10.1056/NEJMoa1309635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callier P, Calvel P, Matevossian A, Makrythanasis P, Bernard P, Kurosaka H, Vannier A, Thauvin-Robinet C, Borel C, Mazaud-Guittot S, Rolland A, Desdoits-Lethimonier C, Guipponi M, Zimmermann C, Stévant I, Kuhne F, Conne B, Santoni F, Lambert S, Huet F, Mugneret F, Jaruzelska J, Faivre L, Wilhelm D, Jégou B, Trainor PA, Resh MD, Antonarakis SE, Nef S, 2014. Loss of Function Mutation in the Palmitoyl-Transferase HHAT Leads to Syndromic 46,XY Disorder of Sex Development by Impeding Hedgehog Protein Palmitoylation and Signaling. PLoS Genet 10, e1004340 10.1371/journal.pgen.1004340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camats N, Fernández-Cancio M, Audi L, Schaller A, Flück CE, 2018. Broad phenotypes in heterozygous NR5A1 46,XY patients with a disorder of sex development: an oligogenic origin? Eur J Hum Genet 26, 1329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G, 2017. Clinical sequencing: from raw data to diagnosis with lifetime value. Clin Genet 10.1111/cge.13190 [DOI] [PubMed] [Google Scholar]
- Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, Landolin JM, Stamatoyannopoulos JA, Hunkapiller MW, Korlach J, Eichler EE, 2015. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611. 10.1038/nature13907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman C, Cree L, Shelling AN, 2015. The genetics of premature ovarian failure: current perspectives. Int J Womens Heal 7, 799–810. 10.2147/IJWH.S64024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan V, Jyotsna V, Jain V, Khadgawat R, Dada R, 2016. Novel Heterozygous Genetic Variants in Patients with 46,XY Gonadal Dysgenesis. Horm. Metab. Res 49, 36–42. 10.1055/s-0042-114778 [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AWC, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Wellcome Trust Case Control Consortium, W.T.C.C., Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME, 2010. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–12. 10.1038/nature08516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JJ, Willatt L, Homfray T, Woods CG, 2011. A SOX9 duplication and familial 46,XX developmental testicular disorder. N Engl J Med 364, 91–93. 10.1056/NEJMc1010311 [DOI] [PubMed] [Google Scholar]
- Cox K, Bryce J, Jiang J, Rodie M, Sinnott R, Alkhawari M, Arlt W, Audi L, Balsamo A, Bertelloni S, Cools M, Darendeliler F, Drop S, Ellaithi M, Guran T, Hiort O, Holterhus P-M, Hughes I, Krone N, Lisa L, Morel Y, Soder O, Wieacker P, Ahmed SF, 2014. Novel associations in disorders of sex development: findings from the I-DSD Registry. J. Clin. Endocrinol. Metab 99, E348–55. 10.1210/jc.2013-2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu Stancu M, Van Roosmalen MJ, Renkens I, Nieboer MM, Middelkamp S, De Ligt J, Pregno G, Giachino D, Mandrile G, Espejo Valle-Inclan J, Korzelius J, De Bruijn E, Cuppen E, Talkowski ME, Marschall T, De Ridder J, Kloosterman WP, 2017. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun 8, 1–13. 10.1038/s41467-017-01343-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft B, Ayers K, Sinclair A, Ohnesorg T, 2016. Review disorders of sex development: The evolving role of genomics in diagnosis and gene discovery. Birth Defects Res. Part C - Embryo Today Rev 108, 337–350. 10.1002/bdrc.21148 [DOI] [PubMed] [Google Scholar]
- Croft B, Ohnesorg T, Sinclair AH, 2018. The Role of Copy Number Variants in Disorders of Sex Development. Sex. Dev 12, 19–29. 10.1159/000481896 [DOI] [PubMed] [Google Scholar]
- D’Aurizio R, Pippucci T, Tattini L, Giusti B, Pellegrini M, Magi A, 2016. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res 44 10.1093/nar/gkw695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Li P, Wang Z, Liang F, Yang F, Fang L, Huang Y, Huang S, 2018. Single-molecule optical mapping enables accurate molecular diagnosis of facioscapulohumeral muscular dystrophy (FSHD). bioRxiv 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paula GB, Barros BA, Carpini S, Tincani BJ, Mazzola TN, Sanches Guaragna M, Piveta C.S. da C., de Oliveira LC, Andrade JGR, Guaragna-Filho G, Barbieri PP, Ferreira NM, Miranda ML, Gonçalves EM, Morcillo AM, Viguetti-Campos NL, Lemos-Marini SHV, Silva R.B. de P., Marques-de-Faria AP, De Mello MP, Maciel-Guerra AT, Guerra-Junior G, 2016. 408 Cases of Genital Ambiguity Followed by Single Multidisciplinary Team during 23 Years: Etiologic Diagnosis and Sex of Rearing. Int. J. Endocrinol. 2016, 1–9. 10.1155/2016/4963574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deamer D, Akeson M, Branton D, 2016. Three decades of nanopore sequencing. Nat. Biotechnol 34, 518–524. 10.1038/nbt.3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Délot E, Vilain E, 2018. Disorders of Sex Development, in: Jerome Strauss Antonio Gargiulo RB (Ed.), Yen & Jaffe’s Reproductive Endocrinology. Elsevier, Philadelphia, PA. [Google Scholar]
- Délot EC, Papp JC, Délot EC, Fox M, Grody W, Lee H, Papp JC, Vilain E, Keegan C, Ramsdell L, Green J, Barseghyan H, Dorrani N, Mohnach L, Pearson MA, Diaz J, Leon E, Hopkin RJ, Johnson J, Saal H, Amarillo I, Adam M, Sandberg DE, Vilain E, 2017. Genetics of Disorders of Sex Development: The DSD-TRN Experience. Endocrinol. Metab. Clin. North Am 46, 519–537. 10.1016/j.ecl.2017.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Délot EC, Vilain EJ, 2015. Nonsyndromic 46,XX Testicular Disorders of Sex Development, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews Seattle (WA). [PubMed] [Google Scholar]
- Demirhan O, Tanrıverdi N, Süleymanova D, Cetinel N, Yasar Y, 2015. The Frequency and Types of Chromosomal Aberrations in the Patients with Hypogonadism. Hum. Genet. Embryol 05, 7–8. 10.4172/2161-0436.1000124 [DOI] [Google Scholar]
- DiNapoli L, Capel B, 2008. SRY and the Standoff in Sex Determination. Mol. Endocrinol 22, 1–9. 10.1210/me.2007-0250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardımcı GG, Chakraborty A, Bann DV, Wang Y, Clark R, Zhang L, Yang H, Liu T, Iyyanki S, An L, Pool C, Sasaki T, Rivera-Mulia JC, Ozadam H, Lajoie BR, Kaul R, Buckley M, Lee K, Diegel M, Pezic D, Ernst C, Hadjur S, Odom DT, Stamatoyannopoulos JA, Broach JR, Hardison RC, Ay F, Noble WS, Dekker J, Gilbert DM, Yue F, 2018. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet 50, 1388–1398. 10.1038/s41588-018-0195-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Yi Y, Yao H, Yang Z, Hu H, Liu J, Gao C, Zhang M, Zhou L, Asan Yi, X., Liang Z, 2016. Targeted next-generation sequencing identification of mutations in patients with disorders of sex development. BMC Med Genet 17, 23 10.1186/s12881-016-0286-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Mark D, Wappenschmidt B, Böckmann B, Pabst B, Chan S, Cao H, Morlot S, Scholz C, Auber B, Rhiem K, Schmutzler R, Illig T, Schlegelberger B, Steinemann D, 2018. A tandem duplication of BRCA1 exons 1–19 through DHX8 exon 2 in four families with hereditary breast and ovarian cancer syndrome. Breast Cancer Res. Treat 172, 561–569. 10.1007/s10549-018-4957-x [DOI] [PubMed] [Google Scholar]
- Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, McLaughlin IJ, Bowman B, Seetin M, Dejesus-Hernandez M, Jackson J, Brown PH, Dickson DW, Van Blitterswijk M, Rademakers R, Petrucelli L, Fryer JD, 2018. Long-read sequencing across the C9orf72 “GGGGCC” repeat expansion: Implications for clinical use and genetic discovery efforts in human disease. Mol. Neurodegener 13, 1–17. 10.1186/s13024-018-0274-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers S, Sadedin S, van den Bergen JA, Robevska G, Ohnesorg T, Hewitt J, Lambeth L, Bouty A, Knarston IM, Tan TY, Cameron F, Werther G, Hutson J, O’Connell M, Grover SR, Heloury Y, Zacharin M, Bergman P, Kimber C, Brown J, Webb N, Hunter MF, Srinivasan S, Titmuss A, Verge CF, Mowat D, Smith G, Smith J, Ewans L, Shalhoub C, Crock P, Cowell C, Leong GM, Ono M, Lafferty AR, Huynh T, Visser U, Choong CS, McKenzie F, Pachter N, Thompson EM, Couper J, Baxendale A, Gecz J, Wheeler BJ, Jefferies C, MacKenzie K, Hofman P, Carter P, King RI, Krausz C, van Ravenswaaij-Arts CM, Looijenga L, Drop S, Riedl S, Cools M, Dawson A, Juniarto AZ, Khadilkar V, Khadilkar A, Bhatia V, Dung VC, Atta I, Raza J, Thi Diem Chi N, Hao TK, Harley V, Koopman P, Warne G, Faradz S, Oshlack A, Ayers KL, Sinclair AH, 2016. Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol 17, 243 10.1186/s13059-016-1105-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers S, Sinclair A, 2012. Mammalian sex determination-insights from humans and mice. Chromosom. Res 20, 215–238. 10.1007/s10577-012-9274-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, Salerno WJ, Hampton OA, Gonzaga-Jauregui C, Ambreth S, Ritter DI, Beck CR, Davis CF, Dahdouli M, Ma S, Carroll A, Veeraraghavan N, Bruestle J, Drees B, Hastie A, Lam ET, White S, Mishra P, Wang M, Han Y, Zhang F, Stankiewicz P, Wheeler DA, Reid JG, Muzny DM, Rogers J, Sabo A, Worley KC, Lupski JR, Boerwinkle E, Gibbs RA, 2015. Assessing structural variation in a personal genome-towards a human reference diploid genome. BMC Genomics 16, 286 10.1186/s12864-015-1479-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Zhang X, Wang L, Wang R, Huang Z, Sun Y, Yao R, Huang X, Ye J, Han L, Qiu W, Zhang H, Liang L, Gu X, Yu Y, 2017. Diagnostic Application of Targeted Next-Generation Sequencing of 80 Genes Associated with Disorders of Sexual Development. Sci Rep 7, 44536 10.1038/srep44536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC, Jabado OJ, Deikus G, Clark TA, Luong K, Murray IA, Davis BM, Keren-Paz A, Chess A, Roberts RJ, Korlach J, Turner SW, Kumar V, Waldor MK, Schadt EE, 2012. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol 30, 1232–1239. 10.1038/nbt.2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, Baxter RM, Zeng W, Mroske C, Parra MC, Gandomi SK, Lu I, Li X, Lu H, Lu HM, Salvador D, Ruble D, Lao M, Fischbach S, Wen J, Lee S, Elliott A, Dunlop CL, Tang S, 2015. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 17, 578–586. 10.1038/gim.2014.154 [DOI] [PubMed] [Google Scholar]
- Ferraz-de-Souza B, Lin L, Achermann JC, 2011. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol. Cell. Endocrinol 336, 198–205. 10.1016/j.mce.2010.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flück CE, Meyer-Böni M, Pandey AV, Kempná P, Miller WL, Schoenle EJ, Biason-Lauber A, 2011. Why Boys Will Be Boys: Two Pathways of Fetal Testicular Androgen Biosynthesis Are Needed for Male Sexual Differentiation. Am. J. Hum. Genet 89, 201–218. 10.1016/j.ajhg.2011.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb B, Trifiro MA, 2017. Androgen Insensitivity Syndrome, GeneReviews University of Washington, Seattle. [PubMed] [Google Scholar]
- Granados A, Alaniz VI, Mohnach L, Barseghyan H, Vilain E, Ostrer H, Quint EH, Chen M, Keegan CE, 2017. MAP3K1-related gonadal dysgenesis: Six new cases and review of the literature. Am J Med Genet C Semin Med Genet 175, 253–259. 10.1002/ajmg.c.31559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG, American College of Medical, G., Genomics, 2013. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 15, 565–574. 10.1038/gim.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guercio G, Rey RA, 2014. Fertility issues in the management of patients with disorders of sex development. Endocr Dev 27, 87–98. 10.1159/000363633 [DOI] [PubMed] [Google Scholar]
- Haines B, Hughes J, Corbett M, Shaw M, Innes J, Patel L, Gecz J, Clayton-Smith J, Thomas P, 2015. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J. Clin. Endocrinol. Metab 100, E815–E820. 10.1210/jc.2014-4383 [DOI] [PubMed] [Google Scholar]
- Harrison SM, Campbell IM, Keays M, Granberg CF, Villanueva C, Tannin G, Zinn AR, Castrillon DH, Shaw CA, Stankiewicz P, Baker LA, 2013. Screening and familial characterization of copy-number variations in NR5A1 in 46,XY disorders of sex development and premature ovarian failure. Am. J. Med. Genet. Part A 161A, n/a-n/a. 10.1002/ajmg.a.36084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie AR, Lam ET, Pang AWC, Zhang X, Andrews W, Lee J, Liang TY, Wang J, Zhou X, Zhu Z, Anantharaman T, Zdzakula Z, Bocklandt S, Surti U, Saghbini M, Austin M, Borodkin M, Holmlin RE, Cao H, 2017. Rapid Automated Large Structural Variation Detection in a Diploid Genome by NanoChannel Based Next-Generation Mapping. bioRxiv 102764 10.1101/102764 [DOI] [Google Scholar]
- Heeley JM, Hollander AS, Austin PF, Merritt DF, Wesevich VG, Amarillo IE, Amarillo IE, 2018. Risk association of congenital anomalies in patients with ambiguous genitalia: A 22-year single-center experience. J. Pediatr. Urol 14, 153.e1–153.e7. 10.1016/j.jpurol.2017.09.027 [DOI] [PubMed] [Google Scholar]
- Heinrich V, Stange J, Dickhaus T, Imkeller P, Krüger U, Bauer S, Mundlos S, Robinson PN, Hecht J, Krawitz PM, 2012. The allele distribution in next-generation sequencing data sets is accurately described as the result of a stochastic branching process. Nucleic Acids Res 40, 2426–2431. 10.1093/nar/gkr1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu R, Matoba S, Kanai-Azuma M, Tsunekawa N, Katoh-Fukui Y, Kurohmaru M, Morohashi K. -i., Wilhelm D, Koopman P, Kanai Y, 2009. A critical time window of Sry action in gonadal sex determination in mice. Development 136, 129–138. 10.1242/dev.029587 [DOI] [PubMed] [Google Scholar]
- Hughes IA, Houk C, Ahmed SF, Lee PA, Group LC, Group EC, 2006. Consensus statement on management of intersex disorders. Arch Dis Child 91, 554–563. 10.1136/adc.2006.098319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson JM, Grover SR, O’Connell M, Pennell SD, 2014. Malformation syndromes associated with disorders of sex development. Nat. Rev. Endocrinol 10, 476–487. 10.1038/nrendo.2014.83 [DOI] [PubMed] [Google Scholar]
- Igarashi M, Dung VC, Suzuki E, Ida S, Nakacho M, Nakabayashi K, Mizuno K, Hayashi Y, Kohri K, Kojima Y, Ogata T, Fukami M, 2013. Cryptic genomic rearrangements in three patients with 46,XY disorders of sex development. PLoS One 8, e68194 10.1371/journal.pone.0068194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M, 2015. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 12, 351–356. 10.1038/nmeth.3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, Marriott H, Nieto T, O’Grady J, Olsen HE, Pedersen BS, Rhie A, Richardson H, Quinlan AR, Snutch TP, Tee L, Paten B, Phillippy AM, Simpson JT, Loman NJ, Loose M, 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol 36, 338–345. 10.1038/nbt.4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Olsen HE, Paten B, Akeson M, 2016. Erratum to: The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community [ Genome Biol. (2016), 17, 239] DOI:10.1186/s13059–016-1103–0. Genome Biol 17, 1–11. 10.1186/s13059-016-1122-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaratlerdsiri W, Chan EKF, Petersen DC, Yang C, Croucher PI, Bornman MSR, Sheth P, Hayes VM, 2017. Next generation mapping reveals novel large genomic rearrangements in prostate cancer. Oncotarget 8, 23588–23602. 10.18632/oncotarget.15802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenjaroenpun P, Wongsurawat T, Pereira R, Patumcharoenpol P, Ussery DW, Nielsen J, Nookaew I, 2018. Complete genomic and transcriptional landscape analysis using third-generation sequencing: A case study of Saccharomyces cerevisiae CEN.PK113–7D. Nucleic Acids Res 46, 1–15. 10.1093/nar/gky014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong YH, Lu H, Park CH, Li M, Luo H, Kim JJ, Liu S, Ko KH, Huang S, Hwang IS, Kang MN, Gong D, Park KB, Choi EJ, Park JH, Jeong YW, Moon C, Hyun SH, Kim NH, Jeung EB, Yang H, Hwang WS, Gao F, 2016. Stochastic anomaly of methylome but persistent SRY hypermethylation in disorder of sex development in canine somatic cell nuclear transfer. Sci. Rep 6, 1–11. 10.1038/srep31088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Das S, Petras K, Kitamura K, Morohashi K, Abuelo DN, Barr M, Bonneau D, Brady AF, Carpenter NJ, Cipero KL, Frisone F, Fukuda T, Guerrini R, Iida E, Itoh M, Lewanda AF, Nanba Y, Oka A, Proud VK, Saugier-Veber P, Schelley SL, Selicorni A, Shaner R, Silengo M, Stewart F, Sugiyama N, Toyama J, Toutain A, Vargas AL, Yanazawa M, Zackai EH, Dobyns WB, 2004. Mutations ofARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat 23, 147–159. 10.1002/humu.10310 [DOI] [PubMed] [Google Scholar]
- Kernohan KD, Hartley T, Alirezaie N, Robinson PN, Dyment DA, Boycott KM, 2018. Evaluation of exome filtering techniques for the analysis of clinically relevant genes. Hum. Mutat 39, 197–201. 10.1002/humu.23374 [DOI] [PubMed] [Google Scholar]
- Kim JH, Kang E, Heo SH, Kim GH, Jang JH, Cho EH, Lee BH, Yoo HW, Choi JH, 2017. Diagnostic yield of targeted gene panel sequencing to identify the genetic etiology of disorders of sex development. Mol Cell Endocrinol 444, 19–25. 10.1016/j.mce.2017.01.037 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J, 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–5. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon M, Fukami M, 2015. Submicroscopic copy-number variations associated with 46,XY disorders of sex development. Mol. Cell. Pediatr 2, 7 10.1186/s40348-015-0018-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, Bradley A, 2018. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol 36 10.1038/nbt.4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC, 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc 4, 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Kuo RI, Tseng E, Eory L, Paton IR, Archibald AL, Burt DW, 2017. Normalized long read RNA sequencing in chicken reveals transcriptome complexity similar to human. BMC Genomics 18, 1–19. 10.1186/s12864-017-3691-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdyukov S, Bullock M, 2016. DNA Methylation Analysis: Choosing the Right Method. Biology (Basel) 5, 3 10.3390/biology5010003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakou A, Lucas-Herald AL, McGowan R, Tobias AS, Ahmed SF, 2015. Disorders of sex development: advances in genetic diagnosis and challenges in management. Adv. Genomics Genet 2015, 165–177. 10.2147/AGG.S53226 [DOI] [Google Scholar]
- Laird PW, 2010. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet 11, 191–203. 10.1038/nrg2732 [DOI] [PubMed] [Google Scholar]
- Lecointre C, Pichon O, Hamel A, Heloury Y, Michel-Calemard L, Morel Y, David A, Le Caignec C, 2009. Familial acampomelic form of campomelic dysplasia caused by a 960 kb deletion upstream of SOX9. Am J Med Genet A 149A, 1183–1189. 10.1002/ajmg.a.32830 [DOI] [PubMed] [Google Scholar]
- Ledig S, Hiort O, Scherer G, Hoffmann M, Wolff G, Morlot S, Kuechler A, Wieacker P, 2010. Array-CGH analysis in patients with syndromic and non-syndromic XY gonadal dysgenesis: evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum Reprod 25, 2637–2646. 10.1093/humrep/deq167 [DOI] [PubMed] [Google Scholar]
- Ledig S, Hiort O, Wünsch L, Wieacker P, 2012. Partial deletion of DMRT1 causes 46,XY ovotesticular disorder of sexual development. Eur. J. Endocrinol 167, 119–24. 10.1530/EJE-12-0136 [DOI] [PubMed] [Google Scholar]
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, Fox M, Fogel BL, Martinez-Agosto JA, Wong DA, Chang VY, Shieh PB, Palmer CG, Dipple KM, Grody WW, Vilain E, Nelson SF, 2014. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 10.1001/jama.2014.14604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PA, Houk CP, Ahmed SF, Hughes IA, International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine, S., the European Society for Paediatric, E., 2006. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics 118, e488–500. 10.1542/peds.2006-0738 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C, 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelieveld SH, Spielmann M, Mundlos S, Veltman JA, Gilissen C, 2015. Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum. Mutat 36, 815–822. 10.1002/humu.22813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhang P, Wang D, Gu W, Wang K, 2017. Interrogating the “unsequenceable” genomic trinucleotide repeat disorders by long-read sequencing. Genome Med 9, 1–16. 10.1186/s13073-017-0456-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludes-Meyers JH, Kil H, Nunez MI, Conti CJ, Parker-Thornburg J, Bedford MT, Aldaz CM, 2007. WWOX hypomorphic mice display a higher incidence of B-cell lymphomas and develop testicular atrophy. Genes Chromosom. Cancer 46, 1129–1136. 10.1002/gcc.20497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul, Rajinder Stamatoyannopoulos JA, 2012. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science (80-.) 337, 1190–1195. 10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey J, Young E, Lassahn K, Sibert J, Pastor S, Riethman H, Xiao M, 2017. High-throughput single-molecule telomere characterization. Genome Res 27, 1904–1915. 10.1101/gr.222422.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meienberg J, Zerjavic K, Keller I, Okoniewski M, Patrignani A, Ludin K, Xu Z, Steinmann B, Carrel T, Röthlisberger B, Schlapbach R, Bruggmann R, Matyas G, 2015. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res 43, 1–14. 10.1093/nar/gkv216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meynert AM, Ansari M, FitzPatrick DR, Taylor MS, 2014. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics 15, 247 10.1186/1471-2105-15-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meynert AM, Bicknell LS, Hurles ME, Jackson AP, Taylor MS, 2013. Quantifying single nucleotide variant detection sensitivity in exome sequencing. BMC Bioinformatics 14, 195 10.1186/1471-2105-14-195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, Mu XJ, Nemesh J, Peckham HE, Rausch T, Scally A, Shi X, Stromberg MP, Stutz AM, Urban AE, Walker JA, Wu J, Zhang Y, Zhang ZD, Batzer MA, Ding L, Marth GT, McVean G, Sebat J, Snyder M, Wang J, Ye K, Eichler EE, Gerstein MB, Hurles ME, Lee C, McCarroll SA, Korbel JO, Genomes P, 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65. 10.1038/nature09708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Kojima Y, Kamisawa H, Moritoki Y, Nishio H, Nakane A, Kurokawa S, Kohri K, Hayashi Y, 2014. Elucidation of Distinctive Genomic DNA Structures in Patients with 46,XX Testicular Disorders of Sex Development Using Genome Wide Analyses. J. Urol 192, 535–541. 10.1016/j.juro.2014.02.044 [DOI] [PubMed] [Google Scholar]
- Moalem S, Babul-Hirji R, Stavropolous DJ, Wherrett D, Bagli DJ, Thomas P, Chitayat D, 2012. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am J Med Genet A 158A, 1759–1764. 10.1002/ajmg.a.35390 [DOI] [PubMed] [Google Scholar]
- Mohnach L, Fechner PY, Keegan CE, 2016. Nonsyndromic Disorders of Testicular Development, GeneReviews® University of Washington, Seattle. [PubMed] [Google Scholar]
- Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ, 2012. The methylomes of six bacteria. Nucleic Acids Res 40, 11450–11462. 10.1093/nar/gks891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscatelli F, Strom TM, Walker AP, Zanaria E, Recan D, Meindl A, Bardoni B, Guioli S, Zehetner G, Rabl W, et al. , 1994. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 372, 672–676. 10.1038/372672a0 [DOI] [PubMed] [Google Scholar]
- Nimkarn S, Gangishetti PK, Yau M, New MI, 2016. 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia, GeneReviews® University of Washington, Seattle. [PubMed] [Google Scholar]
- Nishino K, Hattori N, Tanaka S, Shiota K, 2004. DNA Methylation-mediated Control of Sry Gene Expression in Mouse Gonadal Development. J. Biol. Chem 279, 22306–22313. 10.1074/jbc.M309513200 [DOI] [PubMed] [Google Scholar]
- Norling A, Lindén Hirschberg A, Iwarsson E, Persson B, Wedell A, Barbaro M, 2013. Novel candidate genes for 46,XY gonadal dysgenesis identified by a customized 1 M array-CGH platform. Eur. J. Med. Genet 56, 661–668. 10.1016/j.ejmg.2013.09.003 [DOI] [PubMed] [Google Scholar]
- Ohnesorg T, Vilain E, Sinclair AH, 2014. The Genetics of Disorders of Sex Development in Humans. Sex Dev 10.1159/000357956 [DOI] [PubMed] [Google Scholar]
- Ono M, Harley VR, 2013. Disorders of sex development: New genes, new concepts. Nat. Rev. Endocrinol 9, 79–91. 10.1038/nrendo.2012.235 [DOI] [PubMed] [Google Scholar]
- Ono M, Harley VR, 2013. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol 9, 79–91. 10.1038/nrendo.2012.235 [DOI] [PubMed] [Google Scholar]
- Özen S, Onay H, Atik T, Solmaz AE, Özklnay F, Gökşen D, Darcan Ş, 2017. Rapid Molecular Genetic Diagnosis with Next-Generation Sequencing in 46,XY Disorders of Sex Development Cases: Efficiency and Cost Assessment. Horm. Res. Paediatr 87, 81–87. 10.1159/000452995 [DOI] [PubMed] [Google Scholar]
- Pearlman A, Loke J, Le Caignec C, White S, Chin L, Friedman A, Warr N, Willan J, Brauer D, Farmer C, Brooks E, Oddoux C, Riley B, Shajahan S, Camerino G, Homfray T, Crosby AH, Couper J, David A, Greenfield A, Sinclair A, Ostrer H, 2010. Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet 87, 898–904. 10.1016/j.ajhg.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Goldstein DB, 2016. Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol 17, 157 10.1186/s13059-016-1016-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert P, Polak M, Colmenares A, Lortat-Jacob S, Audran F, Poulat F, Sultan C, 2011. Predominant Sertoli cell deficiency in a 46,XY disorders of sex development patient with a new NR5A1/SF-1 mutation transmitted by his unaffected father. Fertil. Steril 95, 1788.e5–1788.e9. 10.1016/j.fertnstert.2010.11.035 [DOI] [PubMed] [Google Scholar]
- Porat Y, Lev O, Einhorn M, Shani O, Vilain E, Barseghyan H Bhattacharya S, Paz-Yaacov N, 2018. A novel approach for structural variant calling: Combining data from whole genome next-generation sequencing and optical mapping, in: 68th Annual Meeting of the American Society of Human Genetics, (October 16–20, 2018, San Diego, California). San Diego, California, p. Poster abstract/1643F. [Google Scholar]
- Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ, 2008. A large genome center’s improvements to the Illumina sequencing system. Nat. Methods 5, 1005–10. 10.1038/nmeth.1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinonez SC, Park JM, Rabah R, Owens KM, Yashar BM, Glover TW, Keegan CE, 2013. 9p partial monosomy and disorders of sex development: review and postulation of a pathogenetic mechanism. Am J Med Genet A 161A, 1882–1896. 10.1002/ajmg.a.36018 [DOI] [PubMed] [Google Scholar]
- Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S, 2009. Single Polymerase Molecules. Science (80-.) 323, 133–138. 10.1126/science.1162986 [DOI] [PubMed] [Google Scholar]
- Raymond CS, Parker ED, Kettlewell JR, Brown LG, Page DC, Kusz K, Jaruzelska J, Reinberg Y, Flejter WL, Bardwell VJ, Hirsch B, Zarkower D, 1999. A region of human chromosome 9p required for testis development contains two genes related to known sexual regulators. Hum. Mol. Genet 8, 989–96. [DOI] [PubMed] [Google Scholar]
- Refai O, Friedman A, Terry L, Jewett T, Pearlman A, Perle MA, Ostrer H, 2010. De novo 12;17 translocation upstream of SOX9 resulting in 46,XX testicular disorder of sex development. Am. J. Med. Genet. Part A 152A, 422–426. 10.1002/ajmg.a.33201 [DOI] [PubMed] [Google Scholar]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, McKnight D, Bai R, Suchy S, Friedman B, Tahiliani J, Pineda-Alvarez D, Richard G, Brandt T, Haverfield E, Chung WK, Bale S, 2016. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704. 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, 2015. Standards and guidelines for the interpretation of sequence variants : a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med 17, 1–20. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolston AM, Gardner M, van Leeuwen K, Mohnach L, Keegan C, Delot E, Vilain E, Sandberg DE, members of the, D.S.D.T.R.N.A., Advisory Network Accord, A., 2017. Disorders of sex development (DSD): Clinical service delivery in the United States. Am J Med Genet C Semin Med Genet 175, 268–278. 10.1002/ajmg.c.31558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti R, Ferrari I, Bonomi M, Persani L, 2017. Genetics of primary ovarian insufficiency. Clin. Genet 91, 183–198. 10.1111/cge.12921 [DOI] [PubMed] [Google Scholar]
- Salamon S, Flisikowski K, Switonski M, 2017. Methylation Patterns of SOX3, SOX9, and WNT4 Genes in Gonads of Dogs with XX (SRY -Negative) Disorder of Sexual Development. Sex. Dev 11, 86–93. 10.1159/000466712 [DOI] [PubMed] [Google Scholar]
- Sandberg DE, Callens N, Wisniewski AB, 2015. Disorders of Sex Development (DSD): Networking and Standardization Considerations. Horm Metab Res 47, 387–393. 10.1055/s-0035-1548936 [DOI] [PubMed] [Google Scholar]
- Sandberg DE, M.T., 2014. A Noncategorical Approach to the Psychosocial Care of Persons with DSD and Their Families. Kreukels B, Steensma T, Vries A Gend. Dysphoria Disord. Sex Dev. Focus Sex. Res
- Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, Bedford HM, Bernard G, Bernier FP, Brais B, Bulman DE, Warman Chardon J, Chitayat D, Deladoey J, Fernandez BA, Frosk P, Geraghty MT, Gerull B, Gibson W, Gow RM, Graham GE, Green JS, Heon E, Horvath G, Innes AM, Jabado N, Kim RH, Koenekoop RK, Khan A, Lehmann OJ, Mendoza-Londono R, Michaud JL, Nikkel SM, Penney LS, Polychronakos C, Richer J, Rouleau GA, Samuels ME, Siu VM, Suchowersky O, Tarnopolsky MA, Yoon G, Zahir FR, Consortium FC, Care4Rare Canada, C., Majewski J, Boycott KM, 2016. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet 89, 275–284. 10.1111/cge.12654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz RF, Trinh A, Sipos B, Brenton JD, Goldman N, Markowetz F, 2014. Phylogenetic Quantification of Intra-tumour Heterogeneity. PLoS Comput. Biol 10, e1003535 10.1371/journal.pcbi.1003535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Lee H, Darby CA, Schatz MC, 2018a. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet 19, 329–346. 10.1038/s41576-018-0003-4 [DOI] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, Von Haeseler A, Schatz MC, 2018b. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468. 10.1038/s41592-018-0001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J-S, Rhie A, Kim J, Lee S, Sohn M-H, Kim C-U, Hastie A, Cao H, Yun J-Y, Kim J, Kuk J, Park GH, Kim J, Ryu H, Kim J, Roh M, Baek J, Hunkapiller MW, Korlach J, Shin J-Y, Kim C, 2016. De novo assembly and phasing of a Korean human genome. Nature 538, 243–247. 10.1038/nature20098 [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, Oseroff VV, Albertson DG, Pinkel D, Eichler EE, 2005. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet 77, 78–88. 10.1086/431652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Guo Y, Dong C, Huddleston J, Yang H, Han X, Fu A, Li Q, Li N, Gong S, Lintner KE, Ding Q, Wang Z, Hu J, Wang D, Wang F, Wang L, Lyon GJ, Guan Y, Shen Y, Evgrafov OV, Knowles JA, Thibaud-Nissen F, Schneider V, Yu C-Y, Zhou L, Eichler EE, So K-F, Wang K, 2016. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun 7, 12065 10.1038/ncomms12065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemizu D, Momozawa Y, Abe T, Morizono T, Boroevich KA, Takata S, Ashikawa K, Kubo M, Tsunoda T, 2015. Performance comparison of four commercial human whole-exome capture platforms. Sci. Rep 5, 1–8. 10.1038/srep12742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, Espeche LD, Buzzalino ND, Nadra AD, Dain L, 2018. CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum. Mutat 39, 5–22. 10.1002/humu.23351 [DOI] [PubMed] [Google Scholar]
- Słowikowska-Hilczer J, Hirschberg AL, Claahsen-van der Grinten H, Reisch N, Bouvattier C, Thyen U, Cohen Kettenis P, Roehle R, Köhler B, Nordenström A, Kohler B, Cohen-Kettenis P, de Vries A, Arlt W, Wiesemann C, Slowikowska-Hilczer J, de la Perriere AB, Sultan C, Paris F, Bouvattier C, Thyen U, Reisch N, Richter-Unruh A, Claahsen-van der Grinten H, Nordenstrom A, Pienkowski C, Szarras-Czapnik M, 2017. Fertility outcome and information on fertility issues in individuals with different forms of disorders of sex development: findings from the dsd-LIFE study. Fertil. Steril 108, 822–831. 10.1016/j.fertnstert.2017.08.013 [DOI] [PubMed] [Google Scholar]
- Smedley D, Jacobsen JOB, Jäger M, Köhler S, Holtgrewe M, Schubach M, Siragusa E, Zemojtel T, Buske OJ, Washington NL, Bone WP, Haendel MA, Robinson PN, 2015. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc 10, 2004–2015. 10.1038/nprot.2015.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyk M, Berg JS, Pursley A, Curtis FK, Fernandez BA, Bien-Willner GA, Lupski JR, Cheung SW, Stankiewicz P, 2007. Male-to-female sex reversal associated with an ∼250 kb deletion upstream of NR0B1 (DAX1). Hum. Genet 122, 63–70. 10.1007/s00439-007-0373-8 [DOI] [PubMed] [Google Scholar]
- Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, LePichon JB, Miller NA, Thiffault I, Dinwiddie DL, Twist G, Noll A, Heese BA, Zellmer L, Atherton AM, Abdelmoity AT, Safina N, Nyp SS, Zuccarelli B, Larson IA, Modrcin A, Herd S, Creed M, Ye Z, Yuan X, Brodsky RA, Kingsmore SF, 2014. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 6, 265ra168 10.1126/scitranslmed.3010076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, Meyer-Bahlburg HFL, Miller WL, Montori VM, Oberfield SE, Ritzen M, White PC, Endocrine Society, 2010. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab 95, 4133–4160. 10.1210/jc.2009-2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulos DJ, Merico D, Jobling R, Bowdin S, Monfared N, Thiruvahindrapuram B, Nalpathamkalam T, Pellecchia G, Yuen RKC, Szego MJ, Hayeems RZ, Shaul RZ, Brudno M, Girdea M, Frey B, Alipanahi B, Ahmed S, Babul-Hirji R, Porras RB, Carter MT, Chad L, Chaudhry A, Chitayat D, Doust SJ, Cytrynbaum C, Dupuis L, Ejaz R, Fishman L, Guerin A, Hashemi B, Helal M, Hewson S, Inbar-Feigenberg M, Kannu P, Karp N, Kim RH, Kronick J, Liston E, MacDonald H, Mercimek-Mahmutoglu S, Mendoza-Londono R, Nasr E, Nimmo G, Parkinson N, Quercia N, Raiman J, Roifman M, Schulze A, Shugar A, Shuman C, Sinajon P, Siriwardena K, Weksberg R, Yoon G, Carew C, Erickson R, Leach RA, Klein R, Ray PN, Meyn MS, Scherer SW, Cohn RD, Marshall CR, 2016. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. npj Genomic Med 1 10.1038/npjgenmed.2015.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH-Y, Konkel MK, Malhotra A, Stütz AM, Shi X, Casale FP, Chen J, Hormozdiari F, Dayama G, Chen K, Malig M, Chaisson MJP, Walter K, Meiers S, Kashin S, Garrison E, Auton A, Lam HYK, Mu XJ, Alkan C, Antaki D, Bae T, Cerveira E, Chines P, Chong Z, Clarke L, Dal E, Ding L, Emery S, Fan X, Gujral M, Kahveci F, Kidd JM, Kong Y, Lameijer E-W, McCarthy S, Flicek P, Gibbs RA, Marth G, Mason CE, Menelaou A, Muzny DM, Nelson BJ, Noor A, Parrish NF, Pendleton M, Quitadamo A, Raeder B, Schadt EE, Romanovitch M, Schlattl A, Sebra R, Shabalin AA, Untergasser A, Walker JA, Wang M, Yu F, Zhang C, Zhang J, Zheng-Bradley X, Zhou W, Zichner T, Sebat J, Batzer MA, McCarroll SA, 1000 Genomes Project Consortium, R.E., Mills RE, Gerstein MB, Bashir A, Stegle O, Devine SE, Lee C, Eichler EE, Korbel JO, 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sund KL, Rehder CW, 2014. Detection and reporting of homozygosity associated with consanguinity in the clinical laboratory. Hum Hered 77, 217–224. 10.1159/000362448 [DOI] [PubMed] [Google Scholar]
- Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, Rogers N, Knower K, Rowley L, Eyre H, Rizzoti K, McAninch D, Goncalves J, Slee J, Turbitt E, Bruno D, Bengtsson H, Harley V, Vilain E, Sinclair A, Lovell-Badge R, Thomas P, 2011. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest 121, 328–341. 10.1172/JCI42580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz JM, Ciarlo R, Guo MH, Abrha A, Diamond DA, Chan Y-M, Hirschhorn JN, 2017. Two Unrelated Undervirilized 46,XY Males with Inherited NR5A1 Variants Identified by Whole-Exome Sequencing. Horm. Res. Paediatr 87, 264–270. 10.1159/000448754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannour-Louet M, Han S, Corbett ST, Louet JF, Yatsenko S, Meyers L, Shaw CA, Kang SH, Cheung SW, Lamb DJ, 2010. Identification of de novo copy number variants associated with human disorders of sexual development. PLoS One 5, e15392 10.1371/journal.pone.0015392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannour-Louet M, Han S, Louet JF, Zhang B, Romero K, Addai J, Sahin A, Cheung SW, Lamb DJ, 2014. Increased gene copy number of VAMP7 disrupts human male urogenital development through altered estrogen action. Nat Med 20, 715–724. 10.1038/nm.3580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattini L, D’Aurizio R, Magi A, 2015. Detection of Genomic Structural Variants from Next-Generation Sequencing Data. Front. Bioeng. Biotechnol 3, 1–8. 10.3389/fbioe.2015.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias ES, McElreavey K, 2014. Next generation sequencing for disorders of sex development. Endocr. Dev 27, 53–62. 10.1159/000363615 [DOI] [PubMed] [Google Scholar]
- Vetro A, Ciccone R, Giorda R, Patricelli MG, Della Mina E, Forlino A, Zuffardi O, 2011. XX males SRY negative: a confirmed cause of infertility. J Med Genet 48, 710–712. 10.1136/jmedgenet-2011-100036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetro A, Dehghani MR, Kraoua L, Giorda R, Beri S, Cardarelli L, Merico M, Manolakos E, Parada-Bustamante A, Castro A, Radi O, Camerino G, Brusco A, Sabaghian M, Sofocleous C, Forzano F, Palumbo P, Palumbo O, Calvano S, Zelante L, Grammatico P, Giglio S, Basly M, Chaabouni M, Carella M, Russo G, Bonaglia MC, Zuffardi O, 2015. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet 23, 1025–1032. 10.1038/ejhg.2014.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volden R, Palmer T, Byrne A, Cole C, Schmitz RJ, Green RE, Vollmers C, 2018. Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA. Proc. Natl. Acad. Sci 115, 9726–9731. 10.1073/pnas.1806447115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr A, Robert C, Hume D, Archibald A, Deeb N, Watson M, 2015. Exome Sequencing: Current and Future Perspectives. G3: Genes|Genomes|Genetics 5, 1543–1550. 10.1534/g3.115.018564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Symmons O, Spitz F, Korbel JO, 2013. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet 14, 125–38. 10.1038/nrg3373 [DOI] [PubMed] [Google Scholar]
- Weiss J, Meeks JJ, Hurley L, Raverot G, Frassetto A, Jameson JL, 2003. Sox3 is required for gonadal function, but not sex determination, in males and females. Mol Cell Biol 23, 8084–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger AM, Guturu H, Bernstein JA, Bejerano G, 2017. Systematic reanalysis of clinical exome data yields additional diagnoses: Implications for providers. Genet. Med 19, 209–214. 10.1038/gim.2016.88 [DOI] [PubMed] [Google Scholar]
- White S, Hewitt J, Turbitt E, Van Der Zwan Y, Hersmus R, Drop S, Koopman P, Harley V, Cools M, Looijenga L, Sinclair A, 2012. A multi-exon deletion within WWOX is associated with a 46,XY disorder of sex development. Eur. J. Hum. Genet 20, 348–351. 10.1038/ejhg.2011.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S, Ohnesorg T, Notini A, Roeszler K, Hewitt J, Daggag H, Smith C, Turbitt E, Gustin S, van den Bergen J, Miles D, Western P, Arboleda V, Schumacher V, Gordon L, Bell K, Bengtsson H, Speed T, Hutson J, Warne G, Harley V, Koopman P, Vilain E, Sinclair A, 2011. Copy number variation in patients with disorders of sex development due to 46,XY gonadal dysgenesis. PLoS One 6, e17793 10.1371/journal.pone.0017793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM, 2013. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369, 1502–1511. 10.1056/NEJMoa1306555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, Veeraraghavan N, Hawes A, Chiang T, Leduc M, Beuten J, Zhang J, He W, Scull J, Willis A, Landsverk M, Craigen WJ, Bekheirnia MR, Stray-Pedersen A, Liu P, Wen S, Alcaraz W, Cui H, Walkiewicz M, Reid J, Bainbridge M, Patel A, Boerwinkle E, Beaudet AL, Lupski JR, Plon SE, Gibbs RA, Eng CM, 2014. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879. 10.1001/jama.2014.14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Franco HL, Rodriguez KF, Brown PR, Tsai MJ, Tsai SY, Yao HH, 2017. Elimination of the male reproductive tract in the female embryo is promoted by COUP-TFII in mice. Science (80-.) 357, 717–720. 10.1126/science.aai9136 [DOI] [PMC free article] [PubMed] [Google Scholar]