

Abstract
Imaging mass spectrometry (IMS) enables the spatially targeted molecular assessment of biological tissues at cellular resolutions. New developments and technologies are essential for uncovering the molecular drivers of native physiological function and disease. Instrumentation must maximize spatial resolution, throughput, sensitivity, and specificity, because tissue imaging experiments consist of thousands to millions of pixels. Here, we report the development and application of a matrix-assisted laser desorption/ionization (MALDI) trapped ion-mobility spectrometry (TIMS) imaging platform. This prototype MALDI timsTOF instrument is capable of 10 μm spatial resolutions and 20 pixels/s throughout molecular imaging. The MALDI source utilizes a Bruker SmartBeam 3-D laser system that can generate a square burn pattern of <10 ×10 μm at the sample surface. General image performance was assessed using murine kidney and brain tissues and demonstrate that high-spatial-resolution imaging data can be generated rapidly with mass measurement errors <5 ppm and ~40 000 resolving power. Initial TIMS-based imaging experiments were performed on whole-body mouse pup tissue demonstrating the separation of closely isobaric [PC(32:0) + Na]+ and [PC(34:3) + H]+ (3 mDa mass difference) in the gas phase. We have shown that the MALDI timsTOF platform can maintain reasonable data acquisition rates (>2 pixels/s) while providing the specificity necessary to differentiate components in complex mixtures of lipid adducts. The combination of high-spatial-resolution and throughput imaging capabilities with high-performance TIMS separations provides a uniquely tunable platform to address many challenges associated with advanced molecular imaging applications.
Graphical Abstract
Matrix-assisted laser desorption/ionization (MALDI) imaging mass spectrometry (IMS) combines the sensitivity and molecular specificity of MS with the spatial fidelity of classical microscopy.1 IMS is routinely used to study a variety of analyte classes including pharmaceuticals,2,3 metabolites,4 lipids,5 nucleic acids, glycans,6,7 peptides,8,9 and proteins.10,11 In brief, samples are thinly sectioned, mounted onto conductive glass slides, and coated with a light absorbing matrix that assists in the desorption and ionization of endogenous molecules. Ions are generated by laser irradiation at each position (pixel) within a designated region of the tissue, resulting in the generation of a mass spectrum for each pixel. The spatial resolution of the image is defined by the size of the laser spot at the sample surface, the spacing between pixels in the array (i.e., pitch), and sample preparation processes. A specific molecular image is then generated for each m/z by plotting the intensity for the entire array of pixels. The thousands of ion images produced by a single IMS experiment can be compared to histologically stained sections providing molecular context to histological analysis.12
The defining performance characteristic of any image-based technology is the effective spatial resolution. Maximizing the resolution with minimal loss of sensitivity is paramount for understanding molecular processes that occur at cellular and subcellular levels13 and has motivated considerable efforts in developing new technologies for improving the spatial resolution of IMS. Although sample stage motor precision and matrix considerations are important, the primary means of improving MALDI spatial resolution is to minimize the laser spot size at the sample surface. For a Gaussian profile laser, the diffraction-limited spot size (Ds) is defined by
| (1) |
and is proportional to the beam quality factor (M), laser wavelength (λ), focal length of the final focusing lens (f), diameter of the input laser beam (Db), and incident angle of the laser at the sample surface (θ). Optimizing at least one of these parameters through the integration of pinholes,14 optical fibers,15 beam expanders,15 multiple focusing lenses, coaxial laser illumination,1 and transmission geometry optics17 results in higher-spatial-resolution IMS images. Typical commercial IMS platforms are limited to 20–30 μm spatial resolution,18 while advanced optical systems can achieve 5–10 μm spot sizes using front-side geometries and >1 μm using transmission geometry MALDI setups.15,19,20
IMS data acquisition throughput becomes critical when spatial resolution improves and a higher number of pixels is required. One approach to improve throughput is continuous raster sampling where the laser is fired continuously across the sample surface by moving the stage at a constant velocity. Although the exact location of each pixel can be difficult to determine, pixel size is approximated by accounting for the laser repetition rate, stage velocity, and number of laser shots at a pixel position. This approach can yield data acquisition rates of about 30–100 pixels/s.21–23 Alternatively, the Bruker SmartBeam 3-D laser system achieves high-throughput acquisition rates (50–60 pixels/s) while maintaining discrete pixels by incorporating galvo mirrors into the laser path. The additional mirrors allow laser redirection so that a defined number of laser shots can be collected from a single pixel location. Using this beam steering approach, the laser raster can be performed to effectively collect data from a square pixel with an area set by the operator.24
A significant challenge for any IMS experiment is adequately surveying the molecular complexity of tissue and identifying the many hundreds to thousands of chemical species within an individual pixel. Traditionally, chemical identification is performed by either combining high mass accuracy and resolving power to differentiate isobaric species, linking IMS data to offline liquid chromatography (LC)/MS of tissue extracts,25 or collecting fragmentation data directly from tissue. High-performance mass analyzers (e.g., FTICR and Orbitraps) enable high-spatial-resolution imaging with broad molecular coverage while providing putative identifications by mass accuracy.20,26 However, high mass accuracy alone cannot differentiate structural isomers observed in complex biological samples. By performing parallel IMS and LC–MS experiments,27 tandem MS data can be automatically interpreted by pre-existing databases and computational tools. However, LC–MS/MS has limited spatial information and throughput. Direct tissue MS/MS allows high spatial fidelity and tandem MS imaging experiments but is limited by the number of species that can be fragmented from a single pixel in addition to the limited quality of fragment information.28–30 Alternatively, ion mobility can be integrated into IMS platforms to enhance specificity, improve molecular coverage, and assist with molecular identification at an acquisition rate compatible with imaging.
Ion-mobility–MS provides 2-D separations dependent on the apparent size and shape and mass-to-charge ratio (m/z) of molecular species. While ion-mobility–MS is analogous to LC–MS, gas-phase ion-mobility separations are extremely rapid (μs-ms) and completely independent of MALDI ion generation. LC–MS offers enhanced peak capacity over ion-mobility–MS, although its lengthy separations (min–hr) make it incompatible with imaging experiments that require acquisition of thousands of pixels. Of particular importance for imaging mass spectrometry experiments, ion-mobility–MS enables qualitative identification of chemical class while suppressing noise for improved sensitivity.31,32 Multiple types of ion-mobility separations exist, including drift tube ion mobility (DTIM),33,34 traveling-wave ion-mobility spectrometry (TWIMS),35,36 field mobility spectrometry (FAIMS),37–39 and, more recently, trapped ion-mobility spectrometry (TIMS).40,41 The three former techniques have been previously utilized in the imaging domain, studying metabolites,42,43 lipids,44 peptides,45,46 and proteins,46,47 improving the separation, specificity, and identification of species. In recent years, however, TIMS has shown unparalleled resolving power, proving extremely popular in the -omics fields.48 TIMS separations are capable of resolving powers >200 and >300 for singly and multiply charged ions, respectively.41,49–52 Briefly, ion separation is achieved by using a carrier gas to send ions through a retardant electric field within the TIMS ion guide and is described by
| (2) |
where the mobility range of the analysis (ΔK) is proportional to the gas velocity (vg) and axial electric field gradient (ΔEx).49 The electric field gradient (EFG) axially traps and separates ions against the flow of carrier gas as a function of their mobility. Ions are then eluted from the TIMS analyzer by decreasing the retardant electric field.40 High-resolution gas-phase separations are critical for overcoming the challenges of molecular complexity associated with tissue imaging where LC-based fractionation is not practical.
Here, we report the development and application of a MALDI trapped ion-mobility spectrometry orthogonal time-of-flight (timsTOF) imaging platform capable of both standard qTOF MS and TIMS-activated qTOF MS. The development of this system enables high-performance gas-phase separations during image acquisition, enhancing the chemical information that can be acquired from a single tissue section. The MALDI integrated timsTOF combines high-spatial-resolution and throughput imaging capabilities with increased specificity afforded by TIMS separations.
METHODS
Materials
Acetic acid, trifluoroacetic acid (TFA), 1,5-diaminonaphthalene (DAN), ammonium formate, hematoxylin, and eiosin were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). HPLC-grade acetonitrile, methanol, ethanol, isopropanol, and chloroform were purchased from Fisher Scientific (Pittsburgh, PA, USA).
Sample Preparation
A 1 week old C57BL/6 control mouse was snap frozen at −80 °C, shaved over dry ice, cryo-sectioned at a 20 μm thickness using a CM3050 S cryostat (Leica Biosystems, Wetzlar, Germany), and thaw-mounted onto conductive indium tin oxide coated glass slides (Delta Technologies, Loveland, CO, USA). Rat kidney tissue (Pel-Freeze Biologicals, Rogers, AR, USA) was sectioned at 10 μm thickness and thaw-mounted onto a conductive ITO slide. Autofluorescence microscopy images were acquired using EGFP, DAPI, and DsRed filters on a Zeiss AxioScan Z1 slide scanner (Carl Zeiss Microscopy GmbH, Oberkochen, Germany) prior to matrix application. The DAN matrix was sublimated onto the tissue surface (~1.0 mg/cm2).53 Briefly, slides containing tissue were taped to the bottom of the condenser in a simple sublimation apparatus. Approximately 500 mg of DAN was sublimed at 130 °C and 24 mTorr for 3.5 min.
MALDI timsTOF IMS
The TIMS-based MALDI imaging platform was developed from an electrospray ionization (ESI) timsTOF Pro (Bruker Daltonik, Bremen, Germany) and is depicted in Figure 1. The instrument is equipped with a dual ESI/MALDI source with a SmartBeam 3D 10 kHz frequency tripled Nd:YAG laser (355 nm). The timsTOF Pro is equipped with a dual TIMS funnel, which can perform accumulation and analysis in parallel. The system can be operated in “qTOF” mode (TIMS deactivated) or utilizing TIMS for performing gas-phase separations during image acquisition. ESI of Agilent ESI-L Tune Mix was used to perform calibration of m/z and ion-mobility values. MALDI parameters in both qTOF and TIMS modes were optimized to maximize intensity by tuning ion optics, laser intensity, and laser focus. Additional experimental values include: an ion transfer time of 80 μs, a prepulse storage time of 10 μs, a collision RF of 2000 Vpp, a TIMS funnel 1 (accumulation) RF of 350 Vpp, a TIMS funnel 2 RF (analysis) of 400 Vpp, a multipole RF of 500 Vpp, and a collision cell entrance (in) voltage of 300 V.
Figure 1.

Prototype MALDI timsTOF mass spectrometer. (A) The developed imaging platform couples a MALDI source capable of high-spatial-resolution and throughput imaging with an ESI timsTOF Pro. (B) Ion-mobility separations are performed in a dual TIMS funnel in the first vacuum stage of the instrument. TIMS resolution is tunable by adjusting the EFG scan time, and throughput is maximized by performing accumulation and analysis in parallel on sequential pixels.
For ion-mobility experiments, TIMS is performed in the first vacuum stage of the mass spectrometer within an augmented ion funnel consisting of an entrance funnel, ion tunnel, and exit funnel. A tailored axial electric field gradient (EFG) is applied to the ion tunnel to trap ions with different mobilities (ΔK). Following the ion trapping event, the EFG is gradually reduced to allow sequential elution of trapped ions with ascending mobilities. Elongating the ion tunnel enables simultaneous ion accumulation and analysis, maximizing the duty cycle of the experiment (Figure 1). Further instrument details are provided in the Results and Discussion.
Images were acquired in both qTOF and TIMS modes of operation. qTOF mode images were collected at 10 and 15 μm spatial resolution with beam scan on using 400 laser shots per pixel and 30 and 35% laser power, respectively. Data were collected in both positive and negative ion modes from m/z 200–1500. Imaging data collected with TIMS activated were acquired at 20 μm spatial resolution with beam scan on using 400 laser shots per pixel and 42% laser power. The TIMS EFG scan time was set to 400 ms. To assess laser characteristics, pixel arrays were generated on sublimated DAN surfaces. With beam scan off, IMS was performed using 500 laser shots per pixel, 30% laser energy, and a 10 μm pitch. To better visualize the laser raster pattern, the pitch was increased to 30 μm with the beam scan activated with 500 laser shots per pixel, 30% laser energy, and beam scan set to 10 μm. Burn patterns were measured using the Nikon Eclipse 90i microscope (Nikon Instruments Inc., Melville, NY). All qTOF mode imaging data were visualized using SCiLS Lab Version 2019 (Bruker Daltonics, Bremen, Germany), and TIMS imaging data were analyzed using custom in-house developed software. All lipid identifications were determined based on mass accuracy using the LIPIDMAPS lipidomics gateway (lipidmaps.org)54,55 or by performing MS/MS in the collision cell of the protoype MALDI timsTOF platform (40 V CID).
MALDI FTICR IMS
To assist in molecular identification, high-mass-resolution imaging mass spectrometry of whole-body mouse pup tissue was performed using a 15T MALDI FTICR mass spectrometer (Bruker Daltonik, Billerica, MA, USA). The instrument is equipped with an Apollo II dual MALDI/ESI ion source and a Smartbeam II 2 kHz frequency tripled Nd:YAG laser (355 nm). Images were generated using the small laser setting (~50 μm) with a pixel spacing of 150 μm in both the x and y dimensions. Data was collected with 300 laser shots per pixel at 46% power from m/z 460–1500 with a data size of 4 MB per spectrum (transient length: 5.2 s) resulting in a resolving power of ~600 000 at m/z 760.
Data Processing
The mouse pup data was exported into a custom binary file format optimized for storage and analysis of ion-mobility–IMS data. Each individual spectrum contains between 10 000 and 100 000 centroid peaks that span the acquisition range of m/z 300–1200 that consists of 221 888 bins. The processing pipeline requires a common m/z axis and centroided peaks that were inserted at their appropriate bin positions while the missing values were set to zero. In addition, each bin has 4001 ion-mobility bins. Following preprocessing, a mean mass spectrum of the entire data set was generated and peak picked. The 1000 most intense species were then selected to avoid including chemical noise in the analysis. A mobility profile of each extracted ion was interrogated to identify species with single or multiple conformations.
Histology
Immediately following MALDI IMS experiments, matrix was removed from samples using 100% ethanol and rehydrated with graded ethanol and double distilled H2O. Tissue was then stained using a hematoxylin and eosin (H&E) stain. Brightfield microscopy of stained tissues was obtained at 20× magnification using a Leica SCN400 Brightfield Slide Scanner (Leica Microsystems, Buffalo Grove, IL, USA).
RESULTS AND DISCUSSION
Instrumentation Development
We have constructed a novel, TIMS-based molecular imaging platform by coupling a MALDI ion source with an ESI timsTOF Pro instrument (Figure 1). Broadly, this platform provides improved sensitivity, specificity, throughput, and spatial resolution over current commercial platforms. TIMS enables tunable, high-performance, gas-phase separations for differentiation of the many isobaric and isomeric species intrinsically present within biological systems. With respect to the original ESI-based instrument, the source housing has been modified to accommodate a MALDI stage, sample loading chamber, and laser optics. It is designed to load and unload a MALDI sample carrier through a vacuum isolated lock chamber similar to other MALDI platforms. The stage position is controlled by high-precision X,Y motors, drive screws, slide rails, and linear bearings to provide a stage position with a pitch of 5 μm, while encoders provide positional feedback with micrometer accuracy.
The MALDI source uses a Bruker SmartBeam 3-D laser system, consisting of a frequency tripled solid-state Nd:YAG laser (wavelength: 355 nm) with repetition rates ≤10 kHz and internal galvo mirrors that enable beam steering during acquisition. The beam exits the laser as parallel light and is focused by an aspherical lens stack before it enters the vacuum chamber through a window. Inside the vacuum chamber, there is a single flat mirror that directs the beam to the sample surface for generation of ions. The ions are transferred into the mass spectrometer using the TIMS funnel for mass analysis. ESI and MALDI experiments can be performed individually or simultaneously without any hardware adjustments.
The system can be operated in qTOF mode by deactivating the ion accumulation and EFG, allowing ions to simply pass through the TIMS funnel. Alternatively, ions can be stored in the accumulation region of the TIMS tunnel and then transferred to the analyzer region for ion-mobility separation. Throughput is maximized by accumulating and analyzing ions in parallel, as the TIMS analysis is performed during ion generation and accumulation of ions from the next pixel. Because of the high repetition rate of the laser (10 kHz), the EFG ramp time is typically the limiting factor for pixel acquisition speed. After ions leave the TIMS funnel, they pass through a mass selection quadrupole and collision cell prior to entering the ion pusher and reflection time-of-flight mass analyzer.
High-Spatial-Resolution Molecular Imaging
The effective spatial resolution of the MALDI timsTOF platform was assessed by imaging laser burn patterns. Although spot size measurements collected from tissue are expected to be smaller, the high-contrast burn patterns of sublimated DAN can be more accurately measured. Burn pattern arrays were collected using various laser conditions, such as laser fluence, shot number, and beam scan size, to mimic different imaging experimental setups commonly used for lipid imaging experiments. A burn pattern array was generated with a 10 μm pitch between pixels and the beam scan feature turned off to determine the laser spot size at the sample surface (Figure 2A).
Figure 2.

Optical measurements of laser burn patterns generated using the prototype MALDI timsTOF platform. (A) A burn pattern array produced using 500 laser shots, a 10 μm pitch between pixels, and the beam scan feature turned off showing a beam diameter of ~7.5 μm with a highly consistent pitch. (B) Burn patterns using 500 laser shots, a 30 μm pitch, and the beam scan set to 10 μm showed square-shaped laser ablation areas with an area of ~8.4 × 9.7 μm.
Optical measurements of the spot sizes show a beam diameter of ~7.5 μm in a regular pixel grid with highly consistent pitch. The unperturbed matrix between pixels suggests that 10 μm IMS experiments can be performed without oversampling. Instead of an inefficient circular footprint, approximated “square pixels” can be obtained using beam steering by rastering the laser within the defined area of the pixel, creating ~8.4 × 9.7 μm ablation marks (Figure 2B).
Ablating material and generating ions from square regions maximizes the sampled area during an imaging experiment. For example, circular ablation of a 10 × 10 μm target pixel dimension would sample a ~78.5 μm2 maximum area (assuming a 10 μm diameter). By steering the laser beam to sample a square area, the system can be tuned to ablate material closer to the entire 100 μm2 pixel area. Assuming a homogeneous distribution of molecules across the pixel, square pixels increase the number of sampled molecules by ~27%, resulting in improved sensitivity. Efficient sampling is of particular importance for high-spatial-resolution experiments, because as pixels become smaller, the number of molecules sampled decreases, and the measurement approaches the limit of detection of the instrument.
Normal, wild-type murine tissues were used to assess the performance of the MALDI TIMS system for molecular imaging in qTOF mode (Figure 3). Positive ion mode rat kidney lipid images were generated from m/z 500–1500 at 15 μm spatial resolution with the beam scan activated. Imaging data were collected from the entire kidney tissue section including the outer cortex, inner cortex, medulla, and renal pelvis (Figure 3A). Ions localizing to specific tissue subregions highlight the image quality and demonstrate the ability to chemically differentiate the kidney (Figure 3B). [SM(d42:3) + H]+ (m/z 811.6688, 0.12 ppm error), [PC(38:4) + H]+ (m/z 810.6009, 0.25 ppm error), and [PE(P-38:4) + K]+/[PE(O-38:5) + K]+ (m/z 790.5135, −1.5 ppm error) localized to the tubules of the inner cortex, outer cortex and renal pelvis, and medulla, respectively. The average mass spectrum for the entire imaging experiment is shown in Figure 3C. Over 2300 m/z peaks were detected with an S/N > 10, demonstrating the overall sensitivity and molecular coverage of the imaging system. The resolving power of the measurements was ~40 000 at m/z 800, enabling differentiation of nominally isobaric species.
Figure 3.

Positive ion mode lipid images collected from rat kidney tissue by MALDI timsTOF IMS operated in qTOF mode. (A) An optical microscopy image of the H&E stained tissue following IMS analysis shows the major anatomical regions of the tissue. (B) Overlayed and individual images for the selected ions m/z 811.6688 ([SM(d42:3) + H]+, 0.12 ppm error, pink), m/z 810.6009 ([PC(38:4) + H]+, 0.25 ppm error, yellow), and m/z 790.5135 ([PE(P-38:4) + K]+/[PE(O-38:5) + K]+, −1.5 ppm error, cyan) highlight the imaging performance of the platform and the unique distribution of each species. (C) The average spectrum for this experiment shows the spectral performance of the MALDI timsTOF MS, and selected m/z windows show the multiple isobars detected at each nominal mass. Ions selected for visualization in panel (B) are marked (*).
Negative ion mode images were also generated of the hind brain and hippocampus of transversally sectioned rat brain tissue to demonstrate the high-spatial-resolution and throughput capabilities of the system (Figure 4). Fine differentiation of the white matter, granular cell layer, and molecular layer of the cerebellum was achieved. Lipid images show specific localization to the various layers of the hippocampus including the pyramidal cell layer and the granule layer of the dentate gyrus. In this data set, 249 644 total pixels were acquired in ~3.5 h, producing an effective data acquisition rate of ~20 pixels/s. Similar to the rat kidney tissue analysis, data generated from rat brain tissue resulted in a mass measurement error <2 ppm and a resolving power of ~40 000 at m/z 800. Other imaging platforms can achieve similar spectral performance but require longer scan times or can generate data very rapidly but cannot achieve the same spatial or mass resolution.
Figure 4.

10 μm spatial-resolution negative ion mode lipid images collected at ~20 pixels/s. Data were generated from rat brain tissue by MALDI timsTOF IMS operated in qTOF mode. Ions were selected to show the variety of lipid distributions observed in the experiment: m/z 834.5437 ([PS(40:6) − H]−, −0.12 ppm error), m/z 883.5683 ([PI(38:5) − H]−, −0.45 ppm error), m/z 885.5498 ([PI(38:4) − H]−, −0.23 ppm 6.6431 ([SHexCer(t42:1) − H]−, −1.9 ppm error). The prototype imaging platform is able to resolve the various fine tissue structures of the cerebellum (e.g., white matter, granular cell layer, and molecular layer) and the hippocampus (e.g., stratum pyramidal layer and DG-granular layer).
MALDI TIMS Imaging Mass Spectrometry
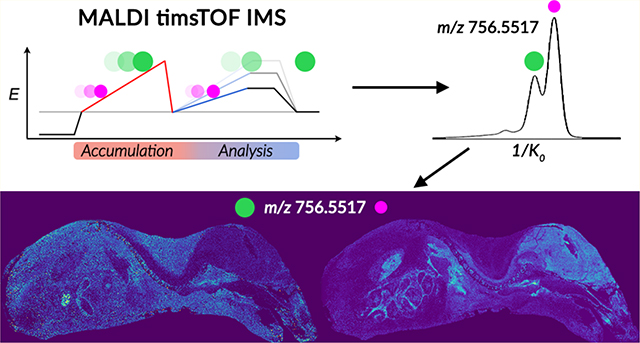
MALDI TIMS imaging of a whole-body mouse pup tissue was used to evaluate the effectiveness of the system. Positive ion mode TIMS lipid images were collected with a 50 μm spatial resolution and utilizing a 400 ms EFG scan time. Ion mobility provides drift time (1/K0) as an additional data dimension to typical imaging data (i.e., x/y locations and m/z value). Ion-mobility information for this system can be interpreted from an average mass spectrum or mobility heat map (m/z vs 1/K0, Supplemental Figure S1). The mobility heat map covers the entire collected m/z range for the mouse pup. Mobility decreases with larger m/z values, as relative collisional cross sections are proportionally related to the m/z value of the ion. The spectral data show the characteristic lipid distribution of biological samples with the most intense signals found between m/z 650–900. A preliminary survey of the data found that, of the 1000 most intense ions (overall mean spectrum), 289 species were detected that displayed multiple mobilities by TIMS. A prime example of this phenomena are the ions observed at m/z 756.5517 (Figure 5). The extracted ion mobilogram showed at least three different species resolved across the tissue sample, and interestingly, the spatial distributions vary dramatically between the different species. The ion detected at m/z 756.5517 with a mobility of 1.451.48 (1/K0) was detected throughout the animal but had a lower intensity within the brain and brain stem (Figure 5A). Conversely, the distribution of the isobar with the mobility of 1.48–1.53 (1/K0) was abundant within the brain, spine, and intestines (Figure 5B). However, the image of the average ion intensity covering both species (1/K0 of 1.45–1.53) is dominated by the more abundant species with a 1/K0 of 1.48–1.53 (Figure 5C). These ion images can only be differentiated because of the TIMS separation, and this observation would not be possible using mass analyzers with similar performance without ion mobility. The EFG scan time is tunable, allowing the ion-mobility resolution to vary based on experimental needs. Although longer scan times (>500 ms) are generally not compatible with LC-based experiments, imaging experiments provide more flexibility and can accommodate longer scan times. There is a significant loss of ion-mobility resolution and information at lower data acquisition rates, demonstrating a need for increasing scan time when possible. Imaging throughput can be adjusted without any reduction in spatial information, easily accommodating longer TIMS analysis times. This is analogous to imaging mass spectrometry performed on FTICR or Orbitrap systems where FTMS scan times are tuned to facilitate high-mass-resolution data acquisition. This example highlights the power of TIMS separations and ion mobility in general for imaging experiments to improve specificity and molecular coverage in direct tissue analysis.
Figure 5.

Ion-mobility IMS data collected from a whole-body mouse pup tissue collected at 50 μm spatial resolution and a 400 ms TIMS scan time. (A–C) Each panel highlights the positive ion mode images from selected mobility ranges for m/z 756.5517, and only one peak is detected in the selected mass window (orange), while multiple conformations are observed in the extracted ion mobilogram. (A) [PC(34:3) + H]+ is found throughout the tissue except the brain and spinal cord. (B) [PC(32:0) + Na]+ localizes in the brain, spine, and intestines. (C) Ion image taken from 1/K0 of 1.45–1.53 is dominated by the higher intensity [PC(32:0) + Na]+, mimicking results from a non-ion-mobility IMS experiment.
Off-line ultrahigh-mass-resolution MALDI FTICR MS and MS/MS were used to identify the species observed at m/z 756.5517 within the above TIMS IMS experiment. A serial whole-body mouse pup tissue section was imaged by MALDI FTICR MS at 150 μm spatial resolution and ~600 000 resolving power at m/z 760, uncovering two nominally isobaric species, [PC(32:0) + Na]+ (m/z 756.55087, −0.70 ppm error) and [PC(34:3) + H]+ (m/z 756.55325, −0.73 ppm error) at m/z 756.5 (Supplemental Figure 2). The spatial distributions of each lipid were used to link the FTICR MS data to the MALDI timsTOF images, and it was determined that the ion with a mobility of 1.48–1.53 was [PC(32:0) + Na]+, while [PC(34:3) + H]+ was the ion with a 1/K0 value of 1.45–1.48. This was confirmed by collecting MS/MS data from areas of adipose and brain tissue (Supplemental Figure 3) in a serial tissue section using the protoype MALDI timsTOF instrument. Although these ions are not isomers, they require resolving powers >300 000 at m/z 760 to resolve them solely with a mass analyzer. Resolutions of this magnitude are challenging to achieve and often require lengthy acquisition times for any mass analyzer. Even high-field FTMS platforms require long scan times (~1.5–3 s) for the necessary resolving power, making the experiment impractical for most imaging applications. However, TIMS imaging separated these species while maintaining a throughput >2 pixels/s, because these ions have different mobilities. The FTICR IMS experiment would require over 200 h to achieve baseline resolution of these two isobars at the same spatial resolution as acquired on the TIMS imaging experiment, leading to a 10-fold increase in analysis speed. Differentiating various ionizing adducts (e.g., H+, Na+, and K+) is important for imaging experiments, as they are regularly generated by MALDI analysis of tissue and cannot be removed without additional sample preparation. Unless MS/MS is performed, differences in the distributions of these species typically go unnoticed, potentially reducing the quality or accuracy of resulting IMS information.
CONCLUSIONS
The performance and application of a prototype MALDI timsTOF imaging mass spectrometer that is capable of 10 μm spatial resolution and 20 pixels/s throughput has been demonstrated. The orthogonal time-of-flight mass analyzer is capable of measuring ions with mass measurement errors <5 ppm and resolutions of ~40 000 while maintaining high-speed data acquisition. Imaging data collected with TIMS improves specificity and molecular coverage for direct tissue analysis compared to non-ion-mobility-based systems. As a proof-of-concept, it was shown that TIMS could separate nominally isobaric [PC(32:0) + Na]+ and [PC(34:3) + H]+ (Δ3 mDa) in a fraction of the time required to differentiate them using high-mass-resolution systems. The unique combination of spatial-resolution, throughput, spectral performance, and high-performance gas-phase separations is critical for advanced molecular imaging applications, particularly for metabolite and lipid analysis where mass redundancy and structural isomers are prevalent. By precisely defining the ion-mobility resolution and pixel size during MALDI timsTOF analysis, it is possible to vary molecular imaging experiments depending on experimental requirements and limitations, allowing for multidimensional specificity (e.g., spatial distribution, m/z, and high-resolution TIMS). The capabilities of the developed TIMS-based platform will provide a critical need to the imaging community by supplementing the lack of chromatographic separation in IMS studies. By broadly improving spatial resolution, speed, sensitivity and specificity, this imaging platform will significantly increase the ability to fully investigate the molecular drivers of biological processes.
Supplementary Material
ACKNOWLEDGMENTS
Instrument development was supported by the National Science Foundation Major Research Instrument Program through an instrument development grant awarded to J.M.S. and R.M.C. (CBET – 1828299). The 15T FTICR MS in the Mass Spectrometry Research Center at Vanderbilt University was acquired through the NIH Shared Instrumentation Grant Program (1S10OD012359). E.K.N. is supported by a National Institute of Environmental Health Sciences training grant (T32ES007028). Further support was provided by the NIH Common Fund and National Institute of Diabetes and Digestive and Kidney Diseases (U54DK120058 awarded to J.M.S. and R.M.C.), the NIH National Institute of Allergy and Infectious Diseases (R01AI138581 awarded to J.M.S. and Eric P. Skaar), and by the NIH National Institute of General Medical Sciences (2P41GM103391 awarded to R.M.C.). The authors also wish to thank Jens Hoehndorf and D. Shannon Cornett of Bruker Daltonik for their guidance and technical input as well as Terry Dermody from the University of Pittsburgh for providing the mouse pup and Daniel Ryan for sample preparation assistance.
Footnotes
The authors declare the following competing financial interest(s): A.F., J.S., N.G., and A.L. were employees of Bruker Daltonik GmbH for the duration of this study.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.9b03612.
Supplemental figures showing the MALDI timsTOF IMS average mass spectrum and ion-mobility heat map (Figure S1) and MALDI FTICR IMS data from whole-body mouse pup tissue (Figure S2) (PDF)
REFERENCES
- (1).Caprioli RM; Farmer TB; Gile J Anal. Chem 1997, 69 (23), 4751–60. [DOI] [PubMed] [Google Scholar]
- (2).Nilsson A; Goodwin RJ; Shariatgorji M; Vallianatou T; Webborn PJ; Andren PE Anal. Chem 2015, 87 (3), 1437–55. [DOI] [PubMed] [Google Scholar]
- (3).Huang JT; Hannah-Qiuhua L; Szyszka R; Veselov V; Reed G; Wang X; Price S; Alquier L; Vas G J. Mass Spectrom 2012, 47 (2), 155–62. [DOI] [PubMed] [Google Scholar]
- (4).Bhandari DR; Schott M; Rompp A; Vilcinskas A; Spengler B Anal. Bioanal. Chem 2015, 407, 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zemski Berry KA; Hankin JA; Barkley RM; Spraggins JM; Caprioli RM; Murphy RC Chem. Rev 2011, 111 (10), 6491–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Drake RR; Powers TW; Norris-Caneda K; Mehta AS; Angel PM Current Protocols in Protein Science 2018, 94 (1), No. e68. [DOI] [PubMed] [Google Scholar]
- (7).Angel PM; Mehta A; Norris-Caneda K; Drake RR Methods Mol. Biol 2017, 1788, 225–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Groseclose MR; Andersson M; Hardesty WM; Caprioli RM J. Mass Spectrom 2007, 42 (2), 254–62. [DOI] [PubMed] [Google Scholar]
- (9).Schober Y; Guenther S; Spengler B; Rompp A Rapid Commun. Mass Spectrom 2012, 26 (9), 1141–6. [DOI] [PubMed] [Google Scholar]
- (10).Deutskens F; Yang J; Caprioli RM J. Mass Spectrom 2011, 46 (6), 568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Seeley EH; Schwamborn K; Caprioli RM J. Biol. Chem 2011, 286 (29), 25459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).El-Aneed A; Cohen A; Banoub J Appl. Spectrosc. Rev 2009, 44 (3), 210–230. [Google Scholar]
- (13).Neumann EK; Do TD; Comi TJ; Sweedler JV Angew. Chem., Int. Ed 2019, 58 (28), 9348–9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zavalin A; Yang J; Caprioli R J. Am. Soc. Mass Spectrom 2013, 24 (7), 1153–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Feenstra AD; Dueñas ME; Lee YJ J. Am. Soc. Mass Spectrom 2017, 28 (3), 434–442. [DOI] [PubMed] [Google Scholar]
- (16).Guenther S; Koestler M; Schulz O; Spengler B Int. J. Mass Spectrom 2010, 294 (1), 7–15. [Google Scholar]
- (17).Zavalin A; Todd EM; Rawhouser PD; Yang J; Norris JL; Caprioli RM J. Mass Spectrom 2012, 47 (11), 1473–1481. [DOI] [PubMed] [Google Scholar]
- (18).Norris JL; Caprioli RM Proteomics: Clin. Appl 2013, 7 (11–12), 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Römpp A; Spengler B Histochem. Cell Biol 2013, 139 (6), 759–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kompauer M; Heiles S; Spengler B Nat. Methods 2017, 14, 90. [DOI] [PubMed] [Google Scholar]
- (21).Prentice BM; Chumbley CW; Caprioli RM J. Mass Spectrom 2015, 50 (4), 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Spraggins JM; Caprioli RM J. Am. Soc. Mass Spectrom 2011, 22 (6), 1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Barré F; Rocha B; Dewez F; Towers M; Murray P; Claude E; Cillero-Pastor B; Heeren R; Porta Siegel T Int. J. Mass Spectrom 2019, 437, 38–48. [Google Scholar]
- (24).Spraggins JM; Rizzo DG; Moore JL; Noto MJ; Skaar EP; Caprioli RM Proteomics 2016, 16 (11–12), 1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Théron L; Centeno D; Coudy-Gandilhon C; Pujos-Guillot E; Astruc T; Rémond D; Barthelemy J-C; Roche F; Feasson L; Hébraud M; Berhet D; Chambon C Proteomes 2016, 4 (4), 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smith DF; Kiss A; Leach FE; Robinson EW; Paša-Tolic L; Heeren RMA Anal. Bioanal. Chem 2013, 405 (18), 6069–6076. [DOI] [PubMed] [Google Scholar]
- (27).Dilillo M; Ait-Belkacem R; Esteve C; Pellegrini D; Nicolardi S; Costa M; Vannini E; Graaf E. L. d.; Caleo M; McDonnell LA Sci. Rep 2017, 7 (1), 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hansen RL; Lee YJ J. Am. Soc. Mass Spectrom 2017, 28 (9), 1910–1918. [DOI] [PubMed] [Google Scholar]
- (29).Dueñas ME; Larson EA; Lee YJ Front. Plant Sci 2019, 10, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kaletaş BK; van der Wiel IM; Stauber J; Dekker LJ; Güzel C; Kros JM; Luider TM; Heeren RMA Proteomics 2009, 9 (10), 2622–2633. [DOI] [PubMed] [Google Scholar]
- (31).Huang J-T; Hannah-Qiuhua L; Szyszka R; Veselov V; Reed G; Wang X; Price S; Alquier L; Vas G J. Mass Spectrom 2012, 47 (2), 155–162. [DOI] [PubMed] [Google Scholar]
- (32).Bhandari DR; Schott M; Römpp A; Vilcinskas A; Spengler B Anal. Bioanal. Chem 2015, 407 (8), 2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).May JC; Goodwin CR; Lareau NM; Leaptrot KL; Morris CB; Kurulugama RT; Mordehai A; Klein C; Barry W; Darland E; Overney G; Imatani K; Stafford GC; Fjeldsted JC; McLean JA Anal. Chem 2014, 86 (4), 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Valentine SJ; Koeniger SL; Clemmer DE Anal. Chem 2003, 75 (22), 6202–6208. [DOI] [PubMed] [Google Scholar]
- (35).Zhong Y; Hyung S-J; Ruotolo BT Analyst 2011, 136 (17), 3534–3541. [DOI] [PubMed] [Google Scholar]
- (36).Shvartsburg AA; Smith RD Anal. Chem 2008, 80 (24), 9689–9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Purves RW; Guevremont R Anal. Chem 1999, 71 (13), 2346–2357. [DOI] [PubMed] [Google Scholar]
- (38).Kolakowski BM; Mester Z Analyst 2007, 132 (9), 842–864. [DOI] [PubMed] [Google Scholar]
- (39).Guevremont R Journal of Chromatography A 2004, 1058 (1), 3–19. [PubMed] [Google Scholar]
- (40).Fernandez-Lima F; Kaplan DA; Suetering J; Park MA Int. J. Ion Mobility Spectrom 2011, 14 (2), 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Michelmann K; Silveira JA; Ridgeway ME; Park MA J. Am. Soc. Mass Spectrom 2015, 26 (1), 14–24. [DOI] [PubMed] [Google Scholar]
- (42).Chughtai K; Jiang L; Greenwood TR; Glunde K; Heeren RMA J. Lipid Res 2013, 54 (2), 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yan C; Parmeggiani F; Jones EA; Claude E; Hussain SA; Turner NJ; Flitsch SL; Barran PE J. Am. Chem. Soc 2017, 139 (4), 1408–1411. [DOI] [PubMed] [Google Scholar]
- (44).McLean JA; Ridenour WB; Caprioli RM J. Mass Spectrom 2007, 42 (8), 1099–1105. [DOI] [PubMed] [Google Scholar]
- (45).Hart PJ; Francese S; Woodroofe MN; Clench MR Int. J. Ion Mobility Spectrom 2013, 16 (2), 71–83. [Google Scholar]
- (46).Towers MW; Karancsi T; Jones EA; Pringle SD; Claude EJ Am. Soc. Mass Spectrom 2018, 29 (12), 2456–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kocurek KI; Griffiths RL; Cooper HJ J. Mass Spectrom 2018, 53 (7), 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ridgeway ME; Lubeck M; Jordens J; Mann M; Park MA Int. J. Mass Spectrom 2018, 425, 22–35. [Google Scholar]
- (49).Hernandez DR; DeBord JD; Ridgeway ME; Kaplan DA; Park MA; Fernandez-Lima F Analyst 2014, 139 (8), 19131921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Silveira JA; Ridgeway ME; Park MA Anal. Chem 2014, 86 (12), 5624–5627. [DOI] [PubMed] [Google Scholar]
- (51).Ridgeway ME; Silveira JA; Meier JE; Park MA Analyst 2015, 140 (20), 6964–6972. [DOI] [PubMed] [Google Scholar]
- (52).Jeanne Dit Fouque K; Ramirez CE; Lewis RL; Koelmel JP; Garrett TJ; Yost RA; Fernandez-Lima F Anal. Chem 2019, 91 (8), 5021–5027. [DOI] [PubMed] [Google Scholar]
- (53).Yang J; Caprioli RM Anal. Chem 2011, 83 (14), 57285734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Fahy E; Subramaniam S; Murphy RC; Nishijima M; Raetz CRH; Shimizu T; Spener F; van Meer G; Wakelam MJO; Dennis EA J. Lipid Res 2009, 50 (Supplement), S9–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Fahy E; Sud M; Cotter D; Subramaniam S Nucleic Acids Res. 2007, 35 (suppl_2), W606–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.