

Abstract
Primrose syndrome is characterized by variable intellectual deficiency, behavior disorders, facial features with macrocephaly, and a progressive phenotype with hearing loss and ectopic calcifications, distal muscle wasting, and contractures. In 2014, ZBTB20 variants were identified as responsible for this syndrome. Indeed, ZBTB20 plays an important role in cognition, memory, learning processes, and has a transcription repressive effect on numerous genes. A more severe phenotype was discussed in patients with missense single nucleotide variants than in those with large deletions. Here, we report on the clinical and molecular results of 14 patients: 6 carrying ZBTB20 missense SNVs, 1 carrying an early truncating indel, and 7 carrying 3q13.31 deletions, recruited through the AnDDI-Rares network. We compared their phenotypes and reviewed the data of the literature, in order to establish more powerful phenotype–genotype correlations. All 57 patients presented mild-to-severe ID and/or a psychomotor delay. Facial features were similar with macrocephaly, prominent forehead, downslanting palpebral fissures, ptosis, and large ears. Hearing loss was far more frequent in patients with missense SNVs (p = 0.002), ectopic calcification, progressive muscular wasting, and contractures were observed only in patients with missense SNVs (p nonsignificant). Corpus callosum dysgenesis (p = 0.00004), hypothyroidism (p = 0.047), and diabetes were also more frequent in this group. However, the median age was 9.4 years in patients with deletions and truncating variant compared with 15.1 years in those with missense SNVs. Longer follow-up will be necessary to determine whether the phenotype of patients with deletions is also progressive.
Subject terms: Development, Thyroid diseases, Type 2 diabetes, Neurological disorders, Cytogenetics
Introduction
Intellectual disability (ID) and multiple congenital anomalies (MCA) affect 1–3% of the population [1] and represent a large and heterogeneous group of disorders. The molecular bases still remain unresolved in a large proportion of cases due to this high heterogeneity, which makes their diagnosis challenging. Improvements in genomic investigation techniques in recent decades have allowed the identification of numerous genes in ID and MCA, first with array-comparative genomic hybridization (CGH), which gives a diagnostic yield of around 14% in ID/MCA [2], and more recently with exome sequencing (ES), which raised the diagnostic yield to around 28.8% [3]. For example, ES allowed the identification of about 555 genes between 2010 and 2015, including ZBTB20 [4].
The first description of Primrose syndrome was made by Primrose in 1982, in a 33-year-old male with ID, muscle weakness of the lower limbs, calcified ear flaps, bone abnormalities, and a torus palatinus. Today, it is characterized by a possibly recognizable but largely underdiagnosed entity that associates ID, autism spectrum disorders, facial features, ectopic calcifications, and distal muscle wasting. Other findings, such as corpus callosum anomalies, tall stature, bilateral cataract, hearing loss, hypothyroidism, and diabetes mellitus have been reported, in particular in older patients [5–8]. The implication of the ZBTB20 gene in this syndrome was established in [7], through the analysis of eight patients, all carrying heterozygous de novo missense variants in ZBTB20, detected by trio ES. Functional studies showed a dominant negative effect of missense variants affecting the DNA binding domain of this transcription factor. This gene plays a role in glucose metabolism, postnatal growth, and neurogenesis. It contains an N-terminal domain involved in protein–protein interactions and five zinc finger C2H2 domains binding the regulatory sites of α-fetoprotein promoters. It has been suggested that the ZBTB20 gene was strongly implicated in the phenotype of 3q13.31 deletion syndrome, which associates ID, corpus callosum agenesis, skeletal malformations, and facial features [9].
Here, we report on 14 new patients: 6 patients with intragenic ZBTB20 missense SNVs, 8 patients with either 3q13.31 microdeletions encompassing ZBTB20, and 1 with a ZBTB20 truncating indel, collected through a national collaboration based on the French AnDDI-Rares network (Figs. 1 and 2). The aim of this study was to collect and compare the clinical features observed in patients carrying ZBTB20 missense SNVs one the one hand, and ZBTB20 microdeletions or truncating variant on the other hand, in our series and a review of 37 patients in the literature, and to determine whether patients with haploinsufficiency of ZBTB20 are at risk of a progressive phenotype, as described in patients with missense variants.
Fig. 1. Pictures of the patients with ZBTB20 missense variants.

a Patient 1. b Patient 2. c Patient 3. d Patient 5.
Fig. 2. Pictures of the patients with ZBTB20 microdeletions.

a Patient 7. b Patient 8. c Patient 9. d Patient 11.
Patients and methods
Patients
Two groups were defined according to whether the proband carried a ZBTB20 missense SNV (dominant negative group) or a deletion involving ZBTB20/ZBTB20 truncating variant (haploinsufficiency group). The medical history and clinical features of each patient are detailed in Supplemental data.
Molecular and cytogenetic analyses
Written and informed consent were obtained from all patients or legal guardians. Peripheral blood samples were provided in a diagnosis context for the proband and both parents, when available.
Panels of different sizes were used for the diagnosis of patients 1, 3, 4, 5, 6, and 14. For patients 1, 3, 6, and 14, a panel comprising 456 genes involved in cognitive disorders was used (Strasbourg University Hospital). Sequence libraries preparation and coding region capture were performed [10] with in-solution enrichment methodology (Agilent XT2 or QXT SureSelect custom panel) and sequenced with an Illumina NextSeq 550 instrument (paired-end sequencing 2 × 150 bases). Reads were aligned on the hg19 human genome reference sequence, and SNVs and indels were called with the Genome Analysis Toolkit v.3.4.46 thanks to an in-house (Strasbourg Hospital University) pipeline (STARK) and following the Genome Analysis Toolkit (GATK) best practice. Annotation and analysis of the variants were performed using Varank [11]. For patients 4 and 5, the diagnostic panel test included 19 genes implicated in syndromes associating overgrowth and ID (Hôpital Necker, Paris). Sequencing was performed on a MiSeq instrument (Illumina), after multiplex PCR using the TruSeq Custom Amplicon (Illumina). Reads were aligned on the hg19 human genome reference sequence with the Burrows–Wheeler Aligner. Variant calling was done using the GATK. Annotation and analysis of the variants were performed via a local interface PolyWeb.
Exome capture and sequencing were performed for patient 2, in a research framework by the GAD team aiming to identify the molecular bases of marfanoid syndrome with ID. Sequence libraries preparation, coding region capture, and the in-house bioinformatics pipeline are detailed elsewhere [12].
In all cases, the presence of the ZBTB20 variant in the proband and in the parents was validated by Sanger sequencing.
Array-CGH was performed using Agilent microarray 44 K for patients 7 and 9, 60 K for patients 8, 12, and 13, 180 K for patient 10 and 11. In all cases, the deletion was confirmed by fluorescence in situ hybridization experiments in the proband using bacterial artificial chromosome (BAC) clones containing chromosome 3 specific sequences, in accordance with publicly available genome resources (NCBI Map Viewer, Santa Cruz Human Genome Browser). The BACs were obtained from the RP library (BACPAC Resources Center, CHORI, Oakland, CA, USA) and selected according to their positions on chromosome 3. The same method was used for segregation in the parental samples.
Identified variants have been submitted to freely accessible public databases: SNVs and indel to ClinVar (IDs SCV000930642 to SCV000930648) and CNVs to Decipher (IDs 388596, 388597, 388611, 388612 to 388615).
Statistical analysis
Clinical features were compared within the missense SNV group, with an expected dominant negative effect, and the group comprising CNVs and the truncating ZBTB20 variant, with an expected haploinsufficient effect, in patients gathered in this study and from the literature. χ2 tests and Fisher’s exact tests were used when appropriate. A p value below 0.05 was considered significant.
Results
Molecular and cytogenetic results
Molecular results of the SNV group are summarized in Table 1 and Fig. 3. All missense SNVs occurred de novo and impacted the zinc finger domains of the protein, at its C-terminal extremity. Of note, the variant of patient 2 (c.1862T>C; p.(Leu621Pro)), presented an amino-acid substitution in the same position as that in subject 11D5028 reported in 2014 [7] (c.1861C>T; p.(Leu621Phe)).
Table 1.
Molecular data of patients with ZBTB20 SNVs/indel reported in this series.
| Patient | Transcript | cDNA variant | Protein variation | Inheritance | ExAC-GnomAD |
|---|---|---|---|---|---|
| Missense | |||||
| 1 | NM_001164342.2 | c.1939A>C | p.(Ser647Arg) | de novo | Absent |
| 2 | NM_001164342.2 | c.1862T>C | p.(Leu621Pro) | de novo | Absent |
| 3 | NM_001164342.2 | c.1760T>G | p.(Phe587Cys) | de novo | Absent |
| 4 | NM_001164342.2 | c.1837C>T | p.(Arg613Cys) | de novo | Absent |
| 5 | NM_001164342.2 | c.1955A>G | p.(His652Arg) | de novo | Absent |
| 6 | NM_001348803.2 | c.1817A>C | p.(His606Pro) | de novo | Absent |
| Truncating | |||||
| 14 | NM_001164343.2 | c.172_178delinsAA | p.(Asp58Asnfs*24) | de novo | Absent |
Fig. 3.
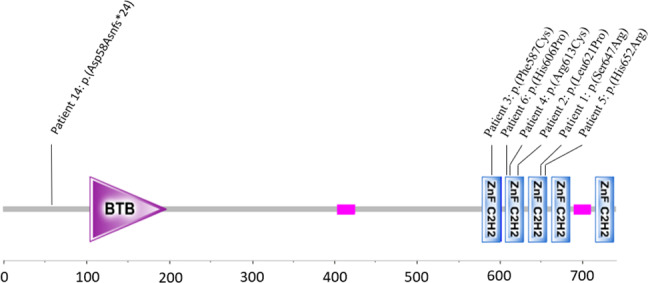
Position of the ZBTB20 intragenic SNVs and indel of the patients 1–6 and 14.
The extents of the CNVs are summarized in Table 2 and Fig. 4. The size of the 3q13.31 microdeletions ranged from 2.78 to 20.35 Mb, and contained 15 to 56 RefSeq genes, including 3–11 OMIM genes. The CNVs start and end positions listed in Table 2 only represent the minimal extent of the deletions detected by using CGH-array. All CNVs were found to be de novo.
Table 2.
Cytogenetic and molecular data of patients with ZBTB20 CNVs.
| Patient | Chromosome coordinates (GRCh37/hg19) | Size (Mb) | Array | Inheritance | Genes | DGV |
|---|---|---|---|---|---|---|
| 7 | chr3:g.(?_112152400)_(115507949_?)del | 3.35 | Agilent 44 K | de novo |
16 genes 4 OMIM: ATP6V1A, CFAP44, DRD3, ZBTB20 |
Absent |
| 8 | chr3:g.(?_ 112198129)_(114983673_?)del | 2.78 | Agilent 44 K | de novo |
15 genes 4 OMIM: ATP6V1A, CFAP44, DRD3, ZBTB20 |
Absent |
| 9 | chr3:g.(?_114522462)_(121094268_?)del | 6.6 | Agilent 60 K | de novo |
22 genes 4 OMIM ZBTB20, ARHGAPS1, POGLUT1, HGD |
Absent |
| 10 | chr3:g.(?_104884356)_(116229200_?)del | 11.34 | Agilent 180 K | de novo |
42 genes 5 OMIM: CD96, CFAP44, ATP6V1A, DRD3, ZBTB20 |
Absent |
| 11 | chr3:g.(?_109439522)_(117042142_?)del | 7.6 | Agilent 180 K | de novo |
29 genes 5 OMIM: CD96, CFAP44, ATP6V1A, DRD3, ZBTB20 |
Absent |
| 12 | chr3:g.(?_94473675)_(114825050_?)del | 20.35 | Agilent 60 K | de novo |
56 genes 11 OMIM: ARL6, CPOX, TBC1D23, TFG, IMPG2, TRMT10C, CD96, CFAP44, ATP6V1A, DRD3, ZBTB20 |
Absent |
| 13 | chr3:g.(?_94473675)_(114825050_?)del | 20.35 | Agilent 60 K | de novo |
56 genes 11 OMIM: ARL6, CPOX, TBC1D23, TFG, IMPG2, TRMT10C, CD96, CFAP44, ATP6V1A, DRD3, ZBTB20 |
Absent |
Fig. 4.

ZBTB20 deletions of patients 7–13 showed in UCSC Genome Browser.
Genotype–phenotype correlations
The clinical features of all patients of this study are presented in Table 3.
Table 3.
Clinical features of the 14 reported patients.
|
Patient ZBTB20 variant type |
1 Mis. SNV |
2 Mis. SNV |
3 Mis. SNV |
4 Mis. SNV |
5 Mis. SNV |
6 Mis. SNV |
7 Del |
8 Del |
9 Del |
10 Del |
11 Del |
12 Del |
13 Del |
14 Trunc. variant |
| Sex | F | F | M | F | M | F | M | M | F | F | M | M | M | M |
| Age (years) | 12 | 24 | 2 | 40 | 3 | 6 | 15 | 20 | 5 | 4 | 38 | 3 | 3 | 5 |
| Measurements | ||||||||||||||
| Birth weight (kg) | 2.93 | 3.5 | 3 | 3.6 | 3.74 | 3.33 | 3.45 | 3.85 | 3.34 | 2.7 | 3.9 | 2.19 | 2.39 | 3.84 |
| Birth length (cm) | 48 | 52 | 54 | 50 | 53 | 51 | 52.5 | 51 | 50 | 50 | 53 | 48 | 48 | 53 |
| Birth OFC (cm) | 36 | NA | 36 | 40 | 39 | 37 | 36.5 | 35 | 36 | 34 | 37 | 34 | 34 | 38.5 |
| Weight at last visit (SD) | NA | −3 | +0 | +0 | +1.5 | −2 | +0 | +3 | +3 | +1 | NA | +0 | −0.5 | +0 |
| Length at last visit (SD) | −3 | −3 | +0 | −0.5 | +2 | −1.5 | +0 | +2 | +3.5 | +1 | +2 | +1 | +0.5 | +1 |
| OFC at last visit (SD) | NA | +0.5 | +2.5 | +2 | +3 | +2.5 | −2 | +0 | +2 | +2 | +1.5 | +0.5 | +0.5 | +3 |
| Neurological | ||||||||||||||
| Developmental delay/ID | S | S | S | Mod | S | S | Mod | Mod | Mod | S | S | Mod | Mod | Mi |
| Ataxia | + | − | − | − | − | − | + | − | − | − | − | − | − | − |
| Fine motor disorders | − | − | − | + | − | − | − | − | − | + | − | − | − | − |
| Chronic pain | − | − | − | − | − | − | − | − | − | + | − | − | − | − |
| Corpus callosum anomaly | + | − | + | + | + | + | − | − | − | − | + | − | − | + |
| Cerebral calcifications | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Behavior disorders | − | − | − | − | − | + | − | − | + | − | + | − | − | + |
| Aggressivity | − | − | + | − | − | − | − | − | − | − | − | − | − | − |
| Anxiety | − | − | − | − | − | − | − | + | − | + | − | + | + | − |
| Motor stereotypies | + | − | − | − | + | + | − | − | − | − | + | + | + | + |
| Autism | − | − | − | − | − | − | − | − | − | + | − | − | − | − |
| Schizophrenia | − | − | − | + | − | − | − | − | − | − | − | − | − | − |
| Facial features | ||||||||||||||
| Plagiocephaly | − | − | − | − | − | − | − | − | + | + | − | + | − | + |
| Prominent forehead | − | − | + | + | − | + | − | − | + | + | − | − | − | − |
| Strabismus | + | − | + | − | − | + | − | − | − | − | − | − | − | + |
| Downslanted palpebral fissures | + | + | − | + | − | + | + | − | + | + | − | + | + | − |
| Ptosis | − | − | + | − | − | + | − | − | − | − | − | − | − | − |
| Epicanthus | + | − | + | + | + | + | − | − | + | + | − | − | − | − |
| Large ears | − | + | + | − | + | + | − | − | + | − | − | − | − | − |
| Depressed nasal bridge | − | − | − | − | − | − | − | − | − | + | − | − | − | − |
| Anteverted nares | − | − | − | + | − | + | + | − | − | + | − | − | − | − |
| Downturned corners of the mouth | + | − | − | − | − | + | − | − | − | + | − | − | − | − |
| Small mouth | − | + | + | − | − | + | − | − | − | − | − | − | − | − |
| High-arched palate | − | + | − | − | − | − | − | − | − | − | − | − | − | − |
| Retrognathia | − | − | − | − | − | − | − | − | + | − | + | − | − | − |
| Muscular | ||||||||||||||
| Hypotonia | + | + | + | − | + | + | + | − | − | + | + | + | + | + |
| Muscle wasting | − | + | − | − | − | − | − | − | − | − | − | − | − | − |
| Muscle contracture | − | + | − | − | − | − | − | − | − | − | − | − | − | − |
| Sensory | ||||||||||||||
| Bilateral hearing loss, perceptive or mixed | + | + | − | + | + | + | − | − | − | − | + | − | − | − |
| Orthopedic | ||||||||||||||
| Ossification delay | + | + | NA | NA | NA | + | NA | NA | NA | NA | NA | NA | NA | NA |
| Joint hyperlaxity | − | + | + | − | − | + | + | + | − | + | + | − | − | − |
| Knee malposition | − | valg | − | valg | − | − | − | recurv | valg | recurv | − | − | − | − |
| Hip dysplasia | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Spine anomalies | − | scol | − | − | − | − | − | kyphosis | − | − | scol | − | − | − |
| Long fingers | − | + | − | − | − | − | − | − | − | − | − | − | − | − |
| Feet malposition | valg | varus | valg | − | − | − | − | − | pes planus | valg, pes planus | − | − | − | − |
| Endocrinology | ||||||||||||||
| Hypothyroidism | − | − | + | − | − | + | − | − | − | − | − | − | − | − |
| Puberty delay | NC | + | NC | − | NC | NC | + | − | NC | NC | − | NC | NC | NC |
| Others | ||||||||||||||
| Constipation | + | + | + | − | − | + | − | − | − | + | − | + | − | − |
| Calcification of the external auditory canal | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Pinnae calcification | NA | NA | NA | + | NA | − | NA | NA | NA | NA | NA | NA | NA | NA |
| Bilateral cryptorchidism | NC | NC | − | NC | − | NC | − | + | NC | NC | − | − | − | − |
| Congenital cardiopathy | − | − | + | − | − | − | − | − | + | − | − | − | − | − |
Del deletion, Mi mild, Mis missense, Mod moderate, NA not available, NC not concerned, S severe, Trunc truncating, Recurv recurvatum, Valg valgus, Scol scoliosis.
The comparison between the missense SNV and CNVs/truncating variant groups was extended to patients reported in the literature (Table 4). The early truncating variant, located in the first exon above the BTB domain, prevents the synthesis of a functional protein. Therefore, this variant is expected to lead to haploinsufficiency. The median age was 15.1 years in patients with missense SNVs and 9.4 years in patients with CNVs/truncating variant. All the patients presented ID and/or mild-to-severe psychomotor delay. Behavioral features, stereotypies, and anxiety were found in both groups. One patient of each group developed schizophrenia, 2/23 with a missense SNV, and 5/34 patients in the CNVs/truncating variant group were diagnosed with autism. ADHD was found only in the CNVs/truncating variant group. Facial features of the two groups overlapped, with macrocephaly, prominent forehead, downslanting palpebral fissures, ptosis, and large ears. Ectopic calcifications, progressive muscle wasting, and contractures were observed only in patients with missense SNVs although clinical significance is reached for hearing loss (p = 0.002), observed in 18/23 in missense SNV group versus 1/8 in CNV/truncating variant group. In addition, corpus callosum anomalies, diabetes, and hypothyroidism were more frequent in this group. Again, clinical significance is reached only for corpus callosum anomalies (p = 0.00004) and hypothyroidism (p = 0.047). Skeletal malformation, mainly deformations of the knees, feet, and spine were present with no significant difference between the two groups (65.2% in SNVs and 56.3% in CNVs). However, the differential median age between the two groups requires caution in the interpretation of results.
Table 4.
Genotype–phenotype correlations in this series and patients of the literature.
| Missense SNV | CNV (and truncating varianta) | ||||||
|---|---|---|---|---|---|---|---|
| Patient/article | Patients study 1–6 | Mathijssen et al. [19] | Total missense SNV | Patients study 7–14 | Molin et al. [9] | Total CNV and truncating variant | p value SNV/CNV and truncating variant (Fisher’s test) |
| Dalal et al. [6] | Vuillaume et al. [20] | ||||||
| Carvalho et al. [21] | Lowther et al. [18] | ||||||
| Posmyk et al. [22] | Quintela et al. [23] | ||||||
| Mattioli et al. [8] | |||||||
| Casertano et al. [14] | |||||||
| Alby et al. [13] | |||||||
| Stellacci et al. [24] | |||||||
| Grímsdóttir et al. [25] | |||||||
| Cleaver et al. [26] | |||||||
| General | |||||||
| Number of patients | 6 | 17 | 23 | 8 | 26 | 34 | |
| M/F | 2/4 | 10/7 | 12/11 | 6/2 | 14/12 | 20/14 | p = NS |
| Mean age (years) | 14.5 | 15.4 | 15.1 | 11.6 | 8.7 | 9.4 | p = NS |
| Extreme ages (years) | (2–40) | (1.5–43) | (1.5–43) | (3–38) | (0–41) | (0–41) | |
| Measurement | |||||||
| Birth weight > +2 SD | 0/5 | 1/15 | 1/20 (5%) | 0/8 | 0/18 | 0/26 (0%) | p = NS |
| Birth length > +2 SD | 1/5 | 2/11 | 3/16 (18.8%) | 0/8 | 1/13 | 1/21 (4.8%) | p = NS |
| Birth OFC > +2 SD | 3/5 | 4/10 | 7/15 (46.7%) | 1/8 | 4/11 | 5/19 (26.3%) | p = NS |
| Last weight > +2 SD | 0/5 | 4/16 | 4/21 (19%) | 2/7 | 6/20 | 8/27 (29.6%) | p = NS |
| Last length > +2 SD | 0/6 | 2/17 | 2/23 (8.7%) | 3/8 | 6/21 | 9/29 (31.0%) | p = NS |
| Last OFC > +2 SD | 4/5 | 10/17 | 14/22 (63.6%) | 3/8 | 7/20 | 10/28 (39.7%) | p = NS |
| Neurological | |||||||
| Developmental delay/ID | 6/6 | 17/17 | 23/23 (100%) | 8/8 | 21/21 | 29/29 (100%) | p = NS |
| Language delay | 6/6 | 17/17 | 23/23 (100%) | 8/8 | 17/19 | 25/27 (92.6%) | p = NS |
| Behavioral disorder | 1/6 | 7/17 | 8/23 (34.8%) | 3/8 | 2/3 | 5/11 (45.5%) | p = NS |
| Ataxia | 2/6 | 0/17 | 2/23 (8.7%) | 1/8 | 1/3 | 2/11 (18.2%) | p = NS |
| Epilepsy | 0/6 | 0/17 | 0/23 (0%) | 0/8 | 2/26 | 2/34 (5.9%) | p = NS |
| Corpus callosum anomalies | 5/6 | 14/17 | 19/23 (82.6%) | 2/8 | 7/26 | 9/34 (26.5%) | p = 0.00004 |
| Ventriculomegaly | 3/6 | 1/17 | 4/23 (17.4%) | 0/8 | 3/26 | 3/34 (8.8%) | p = NS |
| Cerebral calcification | 0/NA | 3/NA | NA | 0/8 | 0/26 | 0/34 (0%) | NA |
| Anxiety | 2/6 | 1/17 | 3/23 (13%) | 4/8 | 1/3 | 5/11 (45.5%) | p = NS |
| Motor stereotypies | 4/6 | 1/17 | 5/23 (21.7%) | 4/8 | 1/3 | 5/11 (45.5%) | p = NS |
| Autism | 0/6 | 2/17 | 2/23 (8.7%) | 1/8 | 4/26 | 5/34 (14.7%) | p = NS |
| ADHD | 0/6 | 1/17 | 1/23 (4.3%) | 0/8 | 6/26 | 6/34 (17.6%) | p = NS |
| Schizophrenia | 1/6 | 0/17 | 1/23 (4.3%) | 0/8 | 1/26 | 1/34 (2.9%) | p = NS |
| Sleep disturbances | 0/6 | 1/17 | 1/23 (4.3%) | 1/8 | 1/3 | 2/11 (18.2%) | p = NS |
| Facial features | |||||||
| Skull malformation | 0/6 | 3/17 | 3/23 (13%) | 3/8 | 7/26 | 10/34 (29.4%) | p = NS |
| Hypertelorism | 0/6 | 2/17 | 2/23 (8.7%) | 0/8 | 8/17 | 8/25 (32%) | p = NS |
| Prominent forehead | 3/6 | 8/17 | 11/23 (47.8%) | 2/8 | 10/14 | 12/22 (54.5%) | p = NS |
| Strabismus | 3/6 | 4/17 | 7/23 (30,4%) | 0/8 | 6/15 | 6/23 (26.1%) | p = NS |
| Downslanting palpebral fissures | 4/6 | 9/17 | 13/23 (56.5%) | 5/8 | 7/13 | 12/21 (57.1%) | p = NS |
| Ptosis | 2/6 | 5/17 | 7/23 (30.4%) | 1/8 | 5/11 | 6/19 (31.6%) | p = NS |
| Epicanthus | 5/6 | 4/17 | 9/23 (39.1%) | 2/8 | 9/15 | 11/23 (47.8%) | p = NS |
| Larges ears | 4/6 | 0/17 | 4/23 (17.4%) | 1/8 | 5/15 | 6/23 (26.1%) | p = NS |
| Depressed nasal bridge | 0/6 | 5/17 | 5/23 (21.7%) | 1/8 | 0/4 | 1/12 (8.3%) | p = NS |
| Broad nasal bridge | 0/6 | 6/17 | 6/23 (26.1%) | 0/8 | 2/13 | 2/21 (9.5%) | p = NS |
| Anteverted nares | 2/6 | 2/17 | 4/23 (17.4%) | 2/8 | 3/10 | 5/18 (27.8%) | p = NS |
| Downturned corners of the mouth | 2/6 | 4/17 | 6/23 (26.1%) | 1/8 | ND | 1/8 (12.5%) | p = NS |
| Small mouth | 3/6 | 4/11 | 7/17 (41.2%) | 0/8 | ND | 0/8 (0%) | p = NS |
| High-arched palate | 1/6 | 3/17 | 4/23 (17.4%) | 0/8 | ND | 0/8 (0%) | p = NS |
| Micrognathia/retrognathia | 0/6 | 2/17 | 2/23 (8.7%) | 2/8 | ND | 2/8 (25%) | p = NS |
| Muscular | |||||||
| Hypotonia | 5/6 | 10/17 | 15/23 (65.2%) | 6/8 | 14/18 | 20/26 (76.9%) | p = NS |
| Muscle wasting | 1/6 | 5/17 | 6/23 (26.1%) | 0/8 | 0/2 | 0/10 (0%) | p = NS |
| Muscle contractures | 1/6 | 4/17 | 5/23 (21.7%) | 0/8 | ND | 0/8 (0%) | p = NS |
| Sensory | |||||||
| Bilateral hearing loss (perceptive or mixed) | 5/6 | 13/17 | 18/23 (78.3%) | 1/8 | ND | 1/8 (12.5%) | p = 0.002 |
| Cataract | 0/6 | 1/17 | 1/23 (4.3%) | 0/8 | 1/25 | 1/33 (3%) | p = NS |
| Calcification of the external auditory canal | 0/6 | 1/17 | 1/23 (4.3%) | 0/8 | ND | 0/8 (0%) | NA |
| Orthopedic | |||||||
| Ossification delay | 3/3 | 3/NA | NA | NA | NA | NA | NA |
| Joint hyperlaxity | 3/6 | 3/17 | 6/23 (26.1%) | 3/8 | 1/24 | 4/32 (12.5%) | p = NS |
| Knee deformation | 2/6 | 2/17 | 4/23 (17.4%) | 3/8 | 0/24 | 3/32 (9.4%) | p = NS |
| Hip dysplasia | 0/6 | 1/17 | 1/23 (4.3%) | 0/8 | 1/24 | 1/32 (3.1%) | p = NS |
| Spine deformation | 1/6 | 5/17 | 6/23 (26.1%) | 2/8 | 5/24 | 7/32 (21.9%) | p = NS |
| Long fingers | 1/6 | 0/17 | 1/23 (4.3%) | 0/8 | 2/24 | 2/32 (6.3%) | p = NS |
| Feet deformation | 3/6 | 2/17 | 5/23 (21.7%) | 2/8 | 7/24 | 9/32 (28.1%) | p = NS |
| Total skeletal malformation | 5/6 | 10/17 | 15/23 (65.2%) | 4/8 | 14/24 | 18/32 (56.3%) | p = NS |
| Endocrinology | |||||||
| Hypothyroidism | 2/6 | 3/17 | 5/23 (21.7%) | 0/8 | 1/23 | 1/31 (3.2%) | p = 0.047 |
| Diabetes mellitus | 0/6 | 2/14 | 2/20 (10%) | 0/8 | 1/23 | 1/31 (3.2%) | p = NS |
| Delayed puberty | 1/2 | 0/6 | 1/8 (12.5%) | 1/3 | ND | 1/3 (33.3%) | p = NS |
| Others | |||||||
| Pinnae calcification | 2/NA | 3/NA | NA | NA | NA | NA | NA |
| Inguinal hernia (males) | 0/2 | 3/10 | 3/12 (25%) | 1/6 | 10/17 | 11/23 (47.8%) | p = NS |
| Congenital cardiopathy | 1/6 | 3/17 | 4/23 (17.4%) | 1/8 | 2/26 | 3/34 (8.8%) | p = NS |
ND no data, NC not concerned, NS not significant.
aOnly one truncating SNV.
Discussion
Primrose syndrome is a rare syndrome, usually unrecognized in childhood. Indeed, patients display a specific association in late childhood or adulthood, with the emergence of progressive distinctive features, such as hearing loss, pinnae calcification, endocrine manifestations, and muscle wasting. Previous authors have mentioned that patients with missense SNVs may be more severely affected than patients with CNVs [7], but a large genotype–phenotype correlation was needed.
To date, the progression of the symptoms has not been described in patients with CNVs, but this finding has to be interpreted with caution since the mean age at the time of reports in patients with missense SNVs is significantly higher than the mean age of patients with CNVs (15.1 years for SNV patients and 9.4 years for CNVs/early truncating variant patients, including this series, and data from the literature). Indeed, patients with CNVs are now rapidly diagnosed since array-CGH is widely available and considered as a first-intention genetic test in developmental disorders. In the future, easier accessibility to large panels and ES should decrease the time to diagnosis for patients with SNVs.
In our study, ID and/or psychomotor delay were observed in all patients and ranged from mild to severe in both groups. Patients in the two groups shared the same facial features. Birth weight was generally normal in both groups. Overgrowth was a rare feature, but observed in both groups. The most distinctive difference was the presence of perceptive hearing loss, diagnosed in the majority of affected patients from the missense SNVs group, whereas in patients with CNVs only one patient developed hearing loss at the age of 14 months. The mean age at the diagnosis of the hearing loss cannot be determined from the literature data, but the youngest patient of the missense SNVs group, aged 2 years, had not yet been diagnosed with hearing loss. However, not all CNV patients had been screened, and this could make it difficult to draw definitive conclusions. Similarly, cerebral and pinnae calcifications have never been reported in ZBTB20-CNV patients and they therefore appear to be specific to patients carrying ZBTB20 missense SNVs. The age at onset of ear calcifications is unknown. In our study, it has been reported only in a 40-year-old female patient. An 11-year-old patient had been reported in 2018 with pinnae calcification [13]. However, this feature had not been explored by specific imaging in all cases. A higher rate of corpus callosum anomalies was found in the missense SNVs group, even though anomalies were observed in both groups [9]. Concerning endocrine complications, hypothyroidism was also more frequent in the missense SNVs group (21.7% vs 3.2%), with variable age of onset, from 2 years and 7 months in the present series to 43 years in the literature [6]. In addition, diabetes mellitus was mostly reported in patients with missense SNVs (10% vs 3.2% in the CNVs group). Diabetes mellitus was usually reported in adulthood (in a 43 and 23-year-old patients with missense SNV and in a 41-year-old patient with CNV), although an 8-year-old patient with glucose intolerance has been described [14]. Muscle wasting is often reported in association with missense SNVs (6/23), but never reported in patients with CNVs.
Among patients with ZBTB20 CNVs, the phenotype could also be explained by the deletion of other adjacent genes in the 3q13.31 region. Patients 9, 10, and 11 also had a deletion comprising the LSAMP gene. This gene encodes a limbic system-associated membrane protein. Significant associations between the LSAMP gene and schizophrenia or neuropsychiatric features have been found [15]. DRD3 is close to ZBTB20 and encodes a dopamine receptor. Polymorphisms in this gene are involved in susceptibility to schizophrenia and essential tremor [16, 17]. Altogether, 32 patients from the CNV group had a DRD3 deletion, and only one 41-year-old patient with a ZBTB20 and DRD3 complete deletion and partial deletion of LSAMP had schizophrenia [18]. Since one 40-year-old patient was also diagnosed with schizophrenia in the missense SNVs group, we cannot exclude that ZBTB20 could contribute to schizophrenia.
In data from the study and the literature, there were no findings present in patients with CNVs and absent in patients with missense SNVs, in favor of the major role of ZBTB20 in the 3q13.31 CNV phenotype. Functional studies performed by transfection experiments were in favor of a dominant negative impact of missense ZBTB20 SNVs [7], compatible with a more severe phenotype in patients with a missense ZBTB20 SNV than in patients with a ZBTB20 CNV. The difference in the mean age of patients remains an obstacle and makes it difficult to draw definitive conclusions since the majority of the differential features is progressive. The long-term follow-up of audiometric and endocrine parameters in patients with ZBTB20 CNVs is needed in order to determine risk and monitoring recommendations for these patients.
In conclusion, we compared the clinical features of six patients with missense ZBTB20 SNVs and eight patients with ZBTB20 CNVs or ZBTB20 early truncating variant, associated with a review of the literature. We highlighted the existence of a genotype–phenotype correlation between ZBTB20 missense SNVs and haploinsufficient variants, with a tendency toward a more severe and progressive clinical presentation in missense SNVs, although a longer follow-up of patients with CNVs is needed to draw further conclusions.
Supplementary information
Acknowledgements
We wish to thank the patients and their families involved in the study, and the physicians who referred their patients to our center. We also thank the University of Burgundy Centre de Calcul (CCuB) for technical support and management of the informatics platform. This work was supported by grants from Dijon University Hospital, the Regional Council of Burgundy Franche-Comté through the Plan d’Actions Régional pour l’Innovation (PARI 2016), and the European Union through the PO FEDER-FSE Bourgogne 2014/2020 programs and the FEDER 2016. This work was also partially funded by the French Ministry of Health (PHRC national 2008/2008-A00515-50) and the Regional Council of Burgundy/Dijon University hospital (PARI 2012). URLs DGV: http://dgv.tcag.ca/dgv/app/home. ClinVAR: https://www.ncbi.nlm.nih.gov/clinvar/. Decipher: https://decipher.sanger.ac.uk/. ExAC Browser: https://exac.broadinstitute.org, GnomAD: https://gnomad.broadinstitute.org. OMIM: https://www.omim.org. RefSeq: https://www.ncbi.nlm.nih.gov/refseq/. UCSC: https://genome.ucsc.edu.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-020-0582-3) contains supplementary material, which is available to authorized users.
References
- 1.Flore LA, Milunsky JM. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol. 2012;19:173–80. doi: 10.1016/j.spen.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2015;43:838–46. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18:696–704. doi: 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 4.Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The genetic basis of mendelian phenotypes: discoveries, challenges and opportunities. Am J Hum Genet. 2015;97:199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Battisti C, Dotti MT, Cerase A, Rufa A, Sicurelli F, Scarpini C, et al. The Primrose syndrome with progressive neurological involvement and cerebral calcification. J Neurol. 2002;249:1466–8. doi: 10.1007/s00415-002-0850-x. [DOI] [PubMed] [Google Scholar]
- 6.Dalal P, Leslie ND, Lindor NM, Gilbert DL, Espay AJ. Motor tics, stereotypies, and self-flagellation in primrose syndrome. Neurology. 2010;75:284–6. doi: 10.1212/WNL.0b013e3181e8e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cordeddu V, Redeker B, Stellacci E, Jongejan A, Fragale A, Bradley TE, et al. Mutations in ZBTB20 cause Primrose syndrome. Nat Genet. 2014;46:815–7. doi: 10.1038/ng.3035. [DOI] [PubMed] [Google Scholar]
- 8.Mattioli F, Piton A, Gérard B, Superti-Furga A, Mandel JL, Unger S. Novel de novo mutations in ZBTB20 in Primrose syndrome with congenital hypothyroidism. Am J Med Genet A. 2016;170:1626–9. doi: 10.1002/ajmg.a.37645. [DOI] [PubMed] [Google Scholar]
- 9.Molin AM, Andrieux J, Koolen DA, Malan V, Carella M, Colleaux L, et al. A novel microdeletion syndrome at 3q13.31 characterised by developmental delay, postnatal overgrowth, hypoplastic male genitals, and characteristic facial features. J Med Genet. 2012;49:104–9. doi: 10.1136/jmedgenet-2011-100534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Redin C, Gérard B, Lauer J, Herenger Y, Muller J, Quartier A, et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet. 2014;51:724–36. doi: 10.1136/jmedgenet-2014-102554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geoffroy V, Pizot C, Redin C, Piton A, Vasli N, Stoetzel C, et al. VaRank: a simple and powerful tool for ranking genetic variants. Peer J. 2015;3:e796. doi: 10.7717/peerj.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thevenon J, Duffourd Y, Masurel-Paulet A, Lefebvre M, Feillet F, El Chehadeh-Djebbar S, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet. 2016;89:700–7. doi: 10.1111/cge.12732. [DOI] [PubMed] [Google Scholar]
- 13.Alby C, Boutaud L, Bessières B, Serre V, Rio M, Cormier-Daire V, et al. Novel de novo ZBTB20 mutations in three cases with Primrose syndrome and constant corpus callosum anomalies. Am J Med Genet Part A. 2018;176A:1091–8. doi: 10.1002/ajmg.a.38684. [DOI] [PubMed] [Google Scholar]
- 14.Casertano A, Fontana P, Hennekam RC, Tartaglia M, Genesio R, Dieber TB, et al. Alterations in metabolic patterns have a key role in diagnosis and progression of Primrose syndrome. Am J Med Genet A. 2017;173:1896–902. doi: 10.1002/ajmg.a.38124. [DOI] [PubMed] [Google Scholar]
- 15.Koido K, Traks T, Balõtšev R, Eller T, Must A, Koks S, et al. Associations between LSAMP gene polymorphisms and major depressive disorder and panic disorder. Transl Psychiatry. 2012;2:e152. doi: 10.1038/tp.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crocq MA, Mant R, Asherson P, Williams J, Hode Y, Mayerova A, et al. Association between schizophrenia and homozygosity at the dopamine D3 receptor gene. J Med Genet. 1992;29:858–60. doi: 10.1136/jmg.29.12.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucotte G, Lagarde JP, Funalot B, Sokoloff P. Linkage with the Ser9Gly DRD3 polymorphism in essential tremor families. Clin Genet. 2006;69:437–40. doi: 10.1111/j.1399-0004.2006.00600.x. [DOI] [PubMed] [Google Scholar]
- 18.Lowther C, Costain G, Melvin R, Stavropoulos DJ, Lionel AC, Marshall CR, et al. Adult expression of a 3q13.31 microdeletion. Mol Cytogenet. 2014;7:23. doi: 10.1186/1755-8166-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathijssen IB, van Hasselt-van der Velde J, Hennekam RC. Testicular cancer in a patient with Primrose syndrome. Eur J Med Genet. 2006;49:127–33. doi: 10.1016/j.ejmg.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Vuillaume ML, Delrue MA, Naudion S, Toutain J, Fergelot P, Arveiler B, et al. Expanding the clinical phenotype at the 3q13.31 locus with a new case of microdeletion and first characterization of the reciprocal duplication. Mol Genet Metab. 2013;110:90–7. doi: 10.1016/j.ymgme.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 21.Carvalho DR, Speck-Martins CE. Additional features of unique Primrose syndrome phenotype. Am J Med Genet Part A. 2011;155:1379–83. doi: 10.1002/ajmg.a.33955. [DOI] [PubMed] [Google Scholar]
- 22.Posmyk R, Leśniewicz R, Chorąży M, Wołczyński S. New case of Primrose syndrome with mild intellectual disability. Am J Med Genet A. 2011;155A:2838–40. doi: 10.1002/ajmg.a.34257. [DOI] [PubMed] [Google Scholar]
- 23.Quintela I, Gomez-Guerrero L, Fernandez-Prieto M, Resches M, Barros F, Carracedo A, et al. Female patient with autistic disorder, intellectual disability, and co-morbid anxiety disorder: expanding the phenotype associated with the recurrent 3q13.2–q13.31 microdeletion. Am J Med Genet Part A. 2015;167A:3121–9. doi: 10.1002/ajmg.a.37292. [DOI] [PubMed] [Google Scholar]
- 24.Stellacci E, Steindl K, Joset P, Mercurio L, Anselmi M, Cecchetti S, et al. Clinical and functional characterization of two novel ZBTB20 mutations causing Primrose syndrome. Hum Mutat. 2018;39:959–64. doi: 10.1002/humu.23546. [DOI] [PubMed] [Google Scholar]
- 25.Grímsdóttir S, Hove HB, Kreiborg S, Ek J, Johansen A, Darvann TA, et al. Novel de novo mutation in ZBTB20 in Primrose syndrome in boy with short stature. Clin Dysmorphol. 2019;28:41–5. doi: 10.1097/MCD.0000000000000244. [DOI] [PubMed] [Google Scholar]
- 26.Cleaver R, Berg J, Craft E, Foster A, Gibbons RJ, Hobson E, et al. Refining the Primrose syndrome phenotype: a study of five patients with ZBTB20 de novo variants and a review of the literature. Am J Med Genet A. 2019;179(3):344–9. doi: 10.1002/ajmg.a.61024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.