

Supplemental Digital Content is available in the text.
Keywords: genome-wide association study, inheritance patterns, long QT syndrome
Background:
Long QT syndrome (LQTS) is a rare genetic disorder and a major preventable cause of sudden cardiac death in the young. A causal rare genetic variant with large effect size is identified in up to 80% of probands (genotype positive) and cascade family screening shows incomplete penetrance of genetic variants. Furthermore, a proportion of cases meeting diagnostic criteria for LQTS remain genetically elusive despite genetic testing of established genes (genotype negative). These observations raise the possibility that common genetic variants with small effect size contribute to the clinical picture of LQTS. This study aimed to characterize and quantify the contribution of common genetic variation to LQTS disease susceptibility.
Methods:
We conducted genome-wide association studies followed by transethnic meta-analysis in 1656 unrelated patients with LQTS of European or Japanese ancestry and 9890 controls to identify susceptibility single nucleotide polymorphisms. We estimated the common variant heritability of LQTS and tested the genetic correlation between LQTS susceptibility and other cardiac traits. Furthermore, we tested the aggregate effect of the 68 single nucleotide polymorphisms previously associated with the QT-interval in the general population using a polygenic risk score.
Results:
Genome-wide association analysis identified 3 loci associated with LQTS at genome-wide statistical significance (P<5×10−8) near NOS1AP, KCNQ1, and KLF12, and 1 missense variant in KCNE1(p.Asp85Asn) at the suggestive threshold (P<10−6). Heritability analyses showed that ≈15% of variance in overall LQTS susceptibility was attributable to common genetic variation (h2SNP 0.148; standard error 0.019). LQTS susceptibility showed a strong genome-wide genetic correlation with the QT-interval in the general population (rg=0.40; P=3.2×10−3). The polygenic risk score comprising common variants previously associated with the QT-interval in the general population was greater in LQTS cases compared with controls (P<10−13), and it is notable that, among patients with LQTS, this polygenic risk score was greater in patients who were genotype negative compared with those who were genotype positive (P<0.005).
Conclusions:
This work establishes an important role for common genetic variation in susceptibility to LQTS. We demonstrate overlap between genetic control of the QT-interval in the general population and genetic factors contributing to LQTS susceptibility. Using polygenic risk score analyses aggregating common genetic variants that modulate the QT-interval in the general population, we provide evidence for a polygenic architecture in genotype negative LQTS.
Clinical Perspective.
What Is New?
A genome-wide association study in long QT syndrome (LQTS) patients establishes and quantifies the role of common genetic variation in susceptibility to LQTS.
Genetic overlap exists between control of QT-interval in the general population and susceptibility to LQTS.
Polygenic risk score analyses based on common genetic variants that modulate the QT-interval in the general population provide evidence for a polygenic architecture in LQTS patients that remains genetically elusive despite genetic testing of established genes (ie, genotype negative).
What Are the Clinical Applications?
These findings enhance the understanding of the genetic basis of LQTS and underscore the genetic relationship between QT-interval in the general population and susceptibility to LQTS.
Increasing burden of QT-prolonging common variants is associated with higher susceptibility for LQTS.
Polygenicity in genotype negative LQTS patients implies that risk is not primarily attributable to 1 genetic factor inherited from 1 of the biological parents as is the case for autosomal dominant LQTS.
Future clinical utility of genetic testing based on polygenic inheritance necessitates the availability of polygenic risk scores with high discriminative capacity.
Editorial, see p 399
Long QT syndrome (LQTS) is a rare inherited disorder of ventricular repolarization characterized by prolongation of the QT interval on the ECG.1,2 LQTS has a prevalence of approximately 1 in 2500, and is a major and often preventable cause of sudden cardiac death in the young.3,4 Multiple genes have been implicated in LQTS and clinical genetic testing is now performed to identify causative rare genetic variants.5 Disease-causing variants (ie, mutations) in the 3 major LQTS genes (ie, KCNQ1 [LQT1], KCNH2 [LQT2], and SCN5A [LQT3]), account for up to 80% of LQTS cases overall and >95% of genotype positive LQTS.2
Studies in families with multiple mutation carriers have shown that disease penetrance (proportion of carriers that manifest with a prolonged QT-interval) can be low,6–8 and that among those with disease manifestations, there can be broad variability in the types of symptoms and severity thereof (variable expression).2,6–8 These observations suggest that, like other Mendelian disorders, allocating the disease in the individual patient exclusively to a rare variant at a single locus (ie, monogenetic) might be an oversimplification of biological phenomena. It is likely that a combination of genetic and nongenetic modifying factors underlies this clinical variability. A comprehensive knowledge of such risk factors that affect penetrance and expressivity of disease-causing variants in LQTS will improve the predictive accuracy of genetic testing in the individual patient and enable personalized clinical interventions. While many clinical risk factors such as sex, hypokalemia, or bradyarrhythmia, have been implicated as modulators of the clinical manifestations of LQTS,9 modulatory genetic factors remain largely unexplored with the exception of a few proof-of-concept studies using a candidate gene approach.10–14
Besides variability in disease manifestations among carriers of pathogenic variants, an outstanding issue in LQTS is the fact that in ≈20% of patients, an underlying causal rare genetic variant remains unidentified after extensive panel-based genetic testing.15 This complicates cascade screening in families and the presymptomatic identification of affected relatives. Although a small proportion of such patients with genotype negative LQTS could have a yet unknown Mendelian defect, another possibility is that a more complex inheritance pattern underlies the disorder in a subset of these patients.
Previous work has shown that a genome-wide association study (GWAS) comparing cases of a rare arrhythmia syndrome with unaffected controls can define modulators of disease susceptibility and suggest a polygenic etiology.16 We report here a GWAS in ≈1700 unrelated patients with LQTS, of European or Japanese ancestry, identifying common genetic variants implicated in LQTS disease susceptibility, and providing a quantification of the contribution of common genetic variants to LQTS predisposition. Using polygenic risk score analyses aggregating common genetic variants that modulate the QT-interval in the general population, we provide evidence for a polygenic architecture in genotype negative LQTS.
Methods
The summary statistics generated in this study are available from the corresponding author on request or on the Cardiovascular Disease Knowledge Portal (http://www.broadcvdi.org/).
Study Population
We established an international consortium allowing recruitment of 1781 unrelated patients with LQTS: 1344 cases of European ancestry from 23 referral centers in Europe, New Zealand, and North America, as well as 437 patients of East Asian ancestry from 4 referral centers in Japan (Table I in the Data Supplement). Included unrelated individuals were probands (97%) except when DNA was not available, in which case 1 other affected family member was included instead. Included patients had a clinical diagnosis of LQTS5 and were classified as “genotype positive” if they carried a single rare variant in 1 of the 3 established major LQTS genes (KCNQ1 [LQT1], KCNH2 [LQT2] and SCN5A [LQT3]), or “genotype negative” if no rare variant was identified in genes unequivocally associated with nonsyndromic LQTS (KCNQ1, KCNH2, SCN5A, CALM1-3, and TRDN).17–19 A rare variant was defined as a protein sequence altering (ie, missense, nonsense, frameshift deletion, in-frame deletion, large deletion, and duplication) or splice-site variant with an allele frequency <1×10−4 in the Genome Aggregation Database.20–22 Genetic testing and variant curation as per the American College of Medical Genetics and Genomics and Association of Molecular Pathology guidelines23 was conducted as described in the Methods section in the Data Supplement. All subjects or their guardians provided informed consent, and the study was approved by the appropriate ethical review boards.
Phenotypic Characterization and Measurement of the QT-Interval
Clinical data were collected at each of the participating centers. We collected a baseline ECG for each patient, preferably not during β-blocker use. The QT-interval duration was measured as previously described (Figure I in the Data Supplement, and Methods in the Data Supplement).24 In genotype negative patients, a LQTS diagnosis was additionally curated by 2 clinicians (NL, RT) and in case of uncertainty, 2 senior LQTS experts (AAW, PJS) were consulted. As per international guidelines,5 we only included genotype negative patients with a LQTS risk score≥3.5 or with a resting QTc≥500ms in repeated 12-lead ECGs, in the absence of a secondary cause for QT prolongation.
Genome-Wide Array Genotyping, Quality Control, and Imputation
We performed genome-wide genotyping for all European cases on the Illumina HumanOmniExpress array and for all Japanese cases on the Illumina Global Screening Array. Genotypic data of 8219 control individuals of European ancestry and 1671 individuals of Japanese ancestry were obtained from different cohorts (Table II in the Data Supplement). Quality control,25 imputation and association analysis were performed separately in the European and Japanese datasets. All genetic variants were mapped to and reported using Genome Reference Consortium Human genome build 37.
After quality control (see Methods in the Data Supplement for details), we performed genome-wide imputation using Eagle2 phasing, Minimac3 and the Haplotype reference consortium (HRCr1.1) panel implemented on the Michigan Imputation Server for both the European and Japanese datasets.26 After imputation, only single nucleotide polymorphisms (SNPs) with minor allele frequency >0.01 and a Minimac3 imputation score of R2>0.3 were included in further analyses.
Genome-Wide Association Analysis
We performed genome-wide association analyses to assess the role of common variants in LQTS susceptibility (case–control) and severity (QTc within the cases). Case–control association of alternate allele dosage with LQTS was performed using logistic regression correcting for genotypic principal components 1 to 10. Quantitative trait analyses for QTc were conducted using a linear regression model correcting for age, β-blocker use at ECG, LQTS type (KCNQ1 [LQT1], KCNH2 [LQT2], SCN5A [LQT3], or genotype negative), sex, and principal components 1 to 10. Genome-wide association analyses were carried out separately for the European and Japanese LQTS cohorts, followed by meta-analysis using an inverse variance weighted fixed effect model, implemented in METAL (version 2011-03-25).27 Genome-wide statistical significance and suggestive thresholds were set to P<5×10−8 and P<1×10−6, respectively. Summary statistics were uploaded to FUMA (Functional Mapping and Annotation of GWAS) for generation of Manhattan, quantile-quantile, and regional association plots for risk loci.28
Survival Analyses
Time to life-threatening arrhythmic events (LAE) survival analyses were performed in the LQTS cases. Follow-up started at birth and stopped at the date of a documented LAE, the last visit, or the 41st birthday, whichever came first. LAE were defined as out of hospital cardiac arrest, hemodynamically unstable ventricular tachycardia/ventricular fibrillation, or appropriate implantable cardioverter-defibrillator therapy. The effect of genotype positive versus genotype negative status was estimated using Cox proportional hazards regression with/without adjustment for classic risk factors (ie, sex and QTc≥500 ms). To examine possible differences in effect of these well-recognized risk factors in genotype positive and genotype negative LQTS cases, interactions between these risk factors and genotype status were included in the model. In addition, puberty and a sex × puberty interaction were included to model the modifying effect of puberty on the effect of sex. Puberty was included as time-varying covariate and the age of puberty was set at 16 years in both sexes (ie, during the follow-up period before the age of 16, puberty was coded as 0, whereas puberty was coded as 1 during the remainder of the follow-up period). Kaplan Meier curves were created to illustrate the cumulative event free survival and log rank tests were used to compare the survival curves.
Polygenic Risk Scores
For all cases and controls, we calculated a weighted QT polygenic risk score (PRSQT) comprising 68 SNPs that had been associated with the QT-interval in the general population at genome-wide statistical significance, in a study primarily including Europeans.29 All 68 SNPs were included in the European dataset analyses whereas only 60/68 SNPs were well-imputed and included in the Japanese dataset analyses (Table III in the Data Supplement). PRSQT was calculated by multiplying the alternate allele dosage by the associated effect size (β) in the published QT GWAS for each of the 68 SNPs. Then, the PRSQT was normalized to a mean of 0 and standard deviation of 1. We used logistic regression to test for association of PRSQT with case–control status, correcting for principal components 1 to 10. We also used P value thresholding and R2 pruning with P values of 5×10−8, 1×10−5, 1×10−4, 1×10−3, and 1×10−2 and R2 of 0.2 and 0.1 on summary statistics from a European29 and Japanese30 descent general population QT-interval GWAS. The resulting 10 models were used to calculate a European and Japanese PRSQT. The association of PRSQT with LQTS was assessed using a logistic regression for the European and Japanese cases separately. The best model was selected based on the maximal C-statistic, as recently performed.31 No other covariate was used to avoid model overfitting.
The odds ratios (ORs) associated with quartile 2, 3, and 4 were calculated using the first PRSQT quartile as the reference. The association of PRSQT and known QT predictors with QTc was performed using a univariable linear regression followed by multivariable analysis, including in the final model only those variables with a P<0.05 in the univariable analyses. The association of PRSQT quartiles with time to LAEs was assessed using Cox proportional hazards regression with/without adjustment for classic risk factors. Association analyses of PRSQT with case–control status, QTc, and time to LAE were performed separately in the European and Japanese datasets, followed by a fixed-effects model meta-analysis.
Common Variant Heritability
We used the generalized restricted maximum likelihood (GREML) approach of GCTA (GCTA-GREML)32 to estimate how much of the variance in LQTS susceptibility could be attributed to common genetic variants (SNP-based heritability, h2SNP). Before heritability estimation, we conducted additional stringent genetic quality control, as previously suggested (Methods in the Data Supplement).33 We estimated the SNP-heritability on the liability scale assuming a 0.04% prevalence with principal components 1-10 as covariates.1 We assessed the robustness of heritability estimates from GCTA-GREML using the GREML and phenotype-correlation genotype-correlation regression34 analyses implemented in LDAK.35 We estimated h2SNP in the overall LQTS and genotype positive LQTS dataset in the both European and Japanese ancestries. Because of small sample size we were not able to estimate h2SNP in genotype negative patients with LQTS using the approaches implemented in GCTA or LDAK.
Genetic Correlation With Other Traits
We used bivariate linkage disequilibrium score regression36 to evaluate the genetic correlation between LQTS susceptibility (as obtained in the European descent case–control GWAS) and other cardiac electric traits,2 namely PR, QRS, QT, heart rate (HR) at rest, HR in response to exercise and recovery, and atrial fibrillation (see Methods in the Data Supplement for origin of summary statistics). We used Bonferroni correction to account for multiple testing (P=0.05/7=0.0071). We did not constrain the bivariate regression intercepts in any of these analyses given the potential for (modest) sample overlap and population stratification.
Results
Clinical Characteristics of the Case Cohort
Demographic and clinical characteristics of the unrelated LQTS cases are presented in Table IV in the Data Supplement separately for the European and Japanese datasets and in Table 1 for the combined cohort. We included a total of 1781 unrelated patients with LQTS of European (n=1344, mean QTc±SD: 484±48ms) and Japanese descent (n=437, QTc: 485±49ms). A total of 1584 cases (89%) were genotype positive, carrying a rare variant in KCNQ1 (LQT1, n=800), KCNH2 (LQT2, n=661), or SCN5A (LQT3, n=123), while in 197 (11%) no disease causing variant was identified (ie, genotype negative) despite extensive genetic testing.
Table 1.
Clinical Characteristics of All Unrelated LQTS Cases
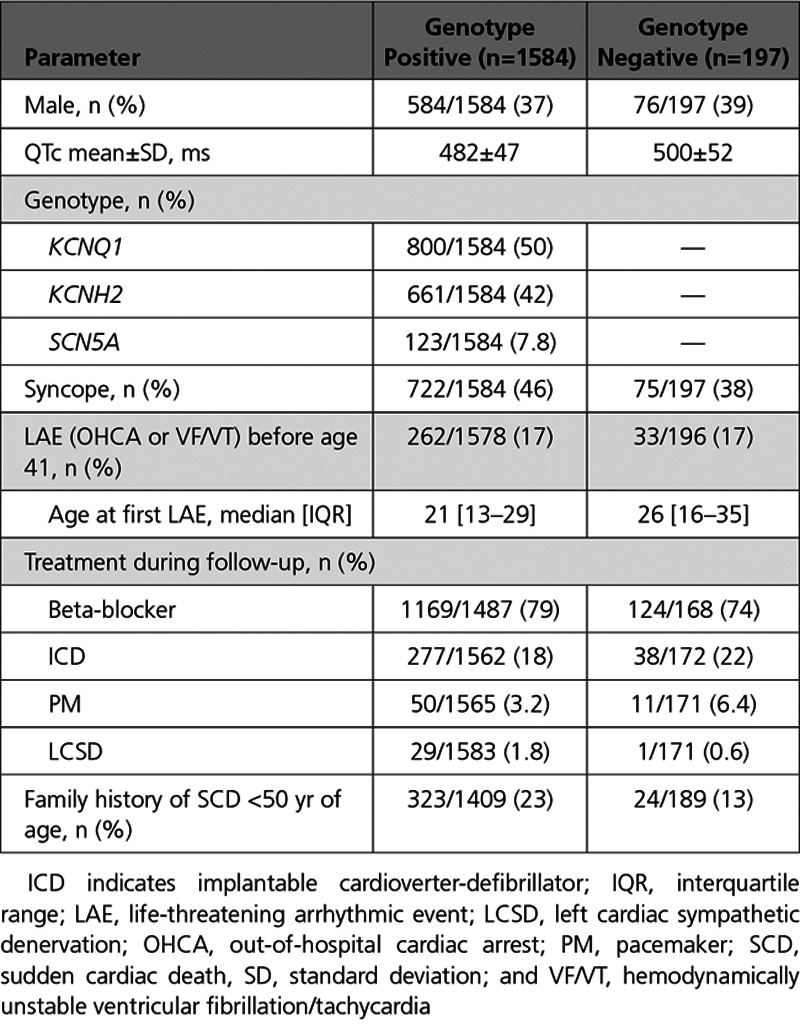
The mean QTc interval in genotype negative cases was higher in comparison with genotype positive ones (500±52ms vs 482±47ms, P=2×10−5) and in genotype negative cases a family history of sudden cardiac death at <50 years of age in 1st and 2nd degree relatives was less frequent compared with genotype positive ones (12.7% vs 22.9%, P=0.001). Of the 1584 genotype positive cases, 1333 (84%) carried a pathogenic or likely-pathogenic variant according to American College of Medical Genetics and Genomics and Association of Molecular Pathology guidelines, and the remainder had a variant of unknown significance. The QTc did not significantly differ between carriers of variants of unknown significance and those with a pathogenic or likely-pathogenic variant (P=0.9).
In total, 429 cases (24%) had an LAE at a median age of 28 years (interquartile range, 17 to 46 years), with 295 cases (17%) having such an event by age 40. LAE-free survival did not significantly differ between genotype negative and positive cases (P=0.8) or between European and Japanese cases (P=0.053; Figure 1). In a multivariable Cox proportional hazard model, male sex (OR 1.9; P=0.004), QTc>500ms (OR 1.8; P=4×10−6) and Japanese ancestry (OR 1.4; P=0.03) were independent risk factors for LAE (Table V in the Data Supplement). We found a significant sex-puberty interaction (P=1×10−6), where males were at higher risk of LAE in the prepubertal years but lower risk thereafter (Figure II in the Data Supplement). The effect of the conventional risk factors sex (Pinteraction=0.3) and QTc≥500ms (Pinteraction=0.7) did not differ between genotype positive and genotype negative cases. Genotype (KCNQ1, KCNH2, SCN5A, or negative) significantly affected time to LAE (log-rank test P<0.001; Figure III in the Data Supplement). Cases with a rare variant in KCNQ1 had a lower risk of LAE compared with KCNH2, SCN5A, and genotype negative ones (P<0.01 for all comparisons). None of the other post hoc pairwise comparisons reached statistical significance. Time to LAE did not differ between cases with a variant of unknown significance and those with pathogenic or likely-pathogenic variant (Figure IV in the Data Supplement).
Figure 1.

Kaplan-Meier life-threatening arrhythmic event–free survival curves stratified by ancestry. EU indicates European LQTS cases; Geno-, genotype negative LQTS cases; Geno+, genotype positive LQTS cases; JP, Japanese LQTS cases; LAE, life-threatening arrhythmic event (defined as the composite of out of hospital cardiac arrest or hemodynamically unstable ventricular tachycardia/arrhythmia; and LQTS, long QT syndrome. Log-rank test P=0.3.
Case–Control GWAS
We conducted a case–control GWAS separately in European (1238 cases vs 8219 controls, genomic test inflation (λ)=1.024) and Japanese (418 cases vs 1617 controls, λ=1.034) cases using ancestry-matched controls (Figures V and VI in the Data Supplement), followed by transethnic meta-analysis (λ=1.028). This uncovered 3 loci reaching the threshold for genome-wide statistical significance (Table 2, Figure 2, and Figures VII and VIII in the Data Supplement). The most significant association was obtained for rs12143842 (OR=1.32 [95% CI, 1.21–1.42]; P=1.09×10−11) located upstream of NOS1AP (Figure VIIIA in the Data Supplement). The lead SNP at the second locus was located in an intron of KCNQ1 (rs179405, OR=1.38 [95% CI, 1.23–1.54]; P=1.92×10−8; Figure VIIIB in the Data Supplement). At the third locus, the lead SNP, rs17061696 (OR=1.25 [95% CI, 1.15–1.35]; P=4.33×10−8), was located in an intron of KLF12 (Figure VIIIC in the Data Supplement). All 3 loci had been previously associated with the QT-interval duration, a measure of myocardial repolarization on the ECG, in the general population (Table 1).29 The low-frequency missense variant in KCNE1, p.Asp85Asn (rs1805128, OR=2.78 [95% CI, 1.67–3.90]; P=5.31×10−7; Figure VIIID in the Data Supplement) reached the suggestive statistical significance threshold in the European case–control analysis. This variant, which is rare and not well imputed in the Japanese dataset (minor allele frequency=0.001; R2<0.3), has the largest reported effect size among the 68 independent SNPs (hereafter referred to as QT-SNPs) previously associated with QT-interval in the general population (7.4ms increase per minor allele).29 Of note, The KCNE1-p.Asp85Asn variant had a more pronounced effect in genotype negative (OR=7.64 [95% CI, 3.66–15.95]; P=5.99×10−8) than in genotype positive LQTS (OR=2.28 [95% CI, 1.46–3.54]; P=2.59×10−4).
Table 2.
Significant Loci in LQTS Case–Control GWAS

Figure 2.
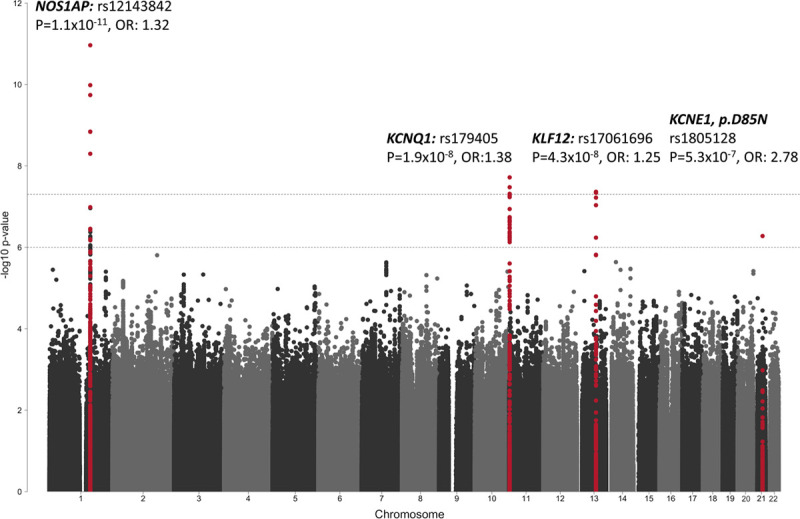
Manhattan plot of long QT syndrome case–control meta-analysis. Manhattan plot displaying the base-pair position of each of the tested single nucleotide polymorphisms (SNPs; each dot represents an individual SNP) along the chromosomes on the x axis and the corresponding −log10 transformed association P value on the y axis. The association P values from the meta-analysis of the 2 genome-wide association studies conducted separately in European and Japanese cases and controls, respectively, are displayed. The upper and lower dashed lines indicate the genome-wide significance (P<5×10–8) and suggestive significance (P<1×10–6) thresholds, respectively. SNPs at genomic regions that reached the genome-wide or suggestive significance thresholds, are marked in red, whereas SNPs from other regions are marked in black or grey. The association for variant rs1805128 (KCNE1:p.Asp85Asn) is solely driven by the European analysis because it is not well imputed and rare (R2<0.3. minor allele frequency=0.001) in the Japanese dataset.
Genetic Overlap Between LQTS and QT-Interval in the General Population
The identification of SNPs previously associated with QT-interval in the general population is in line with the fact that QT-interval prolongation on the ECG (representing prolonged cardiac repolarization) is the central intermediate phenotype underlying LQTS. In fact, 23 of the 68 QT-SNPs previously associated with QT-interval in the general population, were associated with LQTS at nominal significance (ie, P<0.05), while only 4 would be expected under the null hypothesis (Table VI in the Data Supplement). We observed a strong positive correlation between the effect that each of the 68 QT-SNPs had on the QT-interval in the general population29 and the risk they conferred for LQTS in the current study. This effect was consistent across both the European (Figure 3A; R2=0.67; P=2.04×10−17) and the Japanese (Figure 3B; R2=0.52; P=1.43×10−10) datasets. Overlap between genetic risk for LQTS and genetic determinants of the QT-interval in the general population29 was further demonstrated by genome-wide bivariate linkage disequilibrium score regression,36 which detected a significant positive genetic correlation (rg=0.40, SE=0.14; P=3.2×10−3) between these phenotypes. No significant correlation was found for other cardiac electric traits (Figure IX in the Data Supplement).
Figure 3.
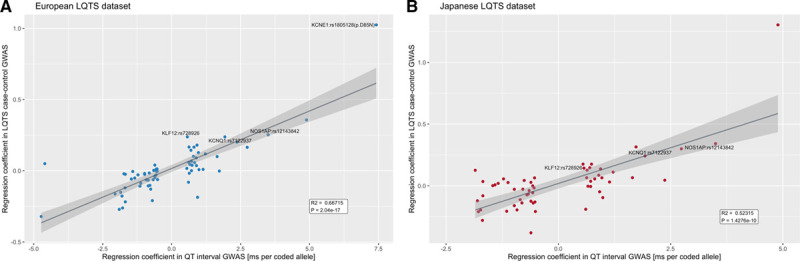
Correlation of effect size of QT-associated single nucleotide polymorphisms with their effect size in long QT syndrome genome-wide association study. The x axis represents the effect estimates from the QT-interval genome-wide association study (GWAS) conducted in the general population (milliseconds per alternative allele) and the y axis the effect of each of these QT-interval associated alleles on disease risk of long QT syndrome (LQTS; [Ln(OR)]) in the European (A) and Japanese (B) datasets. All 68 SNPs associated with QT in the general population were assessed in Europeans, whereas 60 SNPs were properly imputed in the Japanese dataset. In the LQTS-GWAS meta-analysis, 23/68 SNPs previously associated with the QT in the general population reached nominal significance (see Table VI in the Data Supplement). Loci that reached genome-wide significance in the LQTS case–control transethnic meta-analysis NOS1AP-rs12143842, KCNQ1-rs179405, KLF12-rs728926, and KCNE1-rs1805128 are identified with text.
Analysis of PRSQT in LQTS Disease Susceptibility
We then tested the aggregate effect of the 68 QT-SNPs (PRSQT) on susceptibility to LQTS by means of PRS analysis (Table III in the Data Supplement). PRSQT was significantly associated with a diagnosis of LQTS in the European set, the Japanese set, and in the meta-analysis of both datasets (Figure 4A and 4C; Table 3; meta-analysis β=0.34, SE=0.03; P=1.1×10−38, heterogeneity P=0.15). Similar results were obtained when we excluded common variants located at the known Mendelian LQTS loci from the PRS (Table VII in the Data Supplement). Ten different PRS derived by the pruning and thresholding method on summary statistics from the European descent general population QT-interval GWAS did not significantly outperform the PRSQT in discriminating case–control status (Table VIIIA in the Data Supplement). Similarly, Japanese ancestry-specific PRS derived from summary statistics of a small Japanese QT-interval GWAS30 had less discriminative accuracy in the Japanese case–control dataset compared with the European-derived PRSQT, likely because of the small size of the Japanese QT-interval GWAS (Table VIIIB in the Data Supplement).
Figure 4.
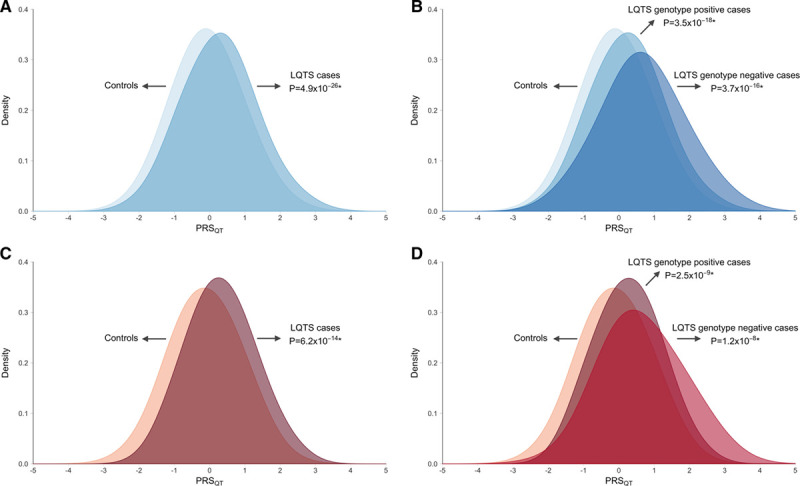
Distribution of QT polygenic score in controls, long QT syndrome, and genotype positive and negative subgroups. The x axis represents the QT polygenic score (PRSQT) in the European (A and B; blue) and Japanese (C and D; red) long QT syndrome (LQTS) case–control datasets. In A and C, all LQTS cases are grouped regardless of whether they are genotype positive or negative, whereas in B and C, cases have been stratified in genotype positive and negative LQTS subgroups. PRSQT was normalized to a mean of 0 and standard deviation of 1. Reported P values refer to the effect of PRSQT in a logistic regression correcting for the first 10 principal components. *Refers to case–control association. Comparison of PRSQT between genotype negative versus genotype positive LQTS uncovered a significantly higher PRSQT in genotype negative patients. This effect was consistently observed in both the European (P=5.1×10−6) and the Japanese (P=2.0×10−3) patients (Table 3).
Table 3.
Association of QT Polygenic Score With Long QT Syndrome
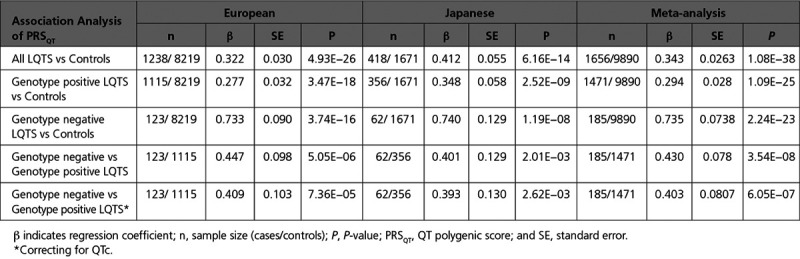
We next explored whether the genetic architecture of genotype negative patients (ie, those lacking a rare variant after extensive genetic testing of the established LQTS disease genes) differed from that of genotype positive patients. This was done by comparing PRSQT between both groups, uncovering a significantly higher PRSQT in genotype negative patients, pointing to a more prominent role for common variants in disease susceptibility in these patients. This effect was consistently observed in both the European (P=5.1×10−6, Figure 4B) and the Japanese (P=2.0×10−3, Figure 4D) datasets (Table 3). Similar results were obtained in a sensitivity analysis correcting for QT-interval, ensuring that enrichment of QT prolonging alleles in the genotype negative patients was not driven by differences in QT-interval (P=7.4×10−5 in Europeans; P=2.6×10−3 in Japanese, Table 3). These associations remained statistically significant when we restricted the analysis to patients with a pathogenic or likely-pathogenic variant according to American College of Medical Genetics and Genomics and Association of Molecular Pathology guidelines (ie, excluding cases with a rare variant of unknown significance; Methods in the Data Supplement and Tables IX and X in the Data Supplement). Increasing PRSQT quartiles were associated with a significantly higher disease susceptibility for genotype negative LQTS compared with the lowest quartile (Figure 5;Table XI in the Data Supplement). It is notable that, using a PRSQT percentile threshold of 80, 90, and 95, individuals above the threshold compared with those below have an OR (95%CI) of 2.9 (2.2–4.0), 4.1 (2.9–5.8), and 5.7 (3.9–8.4), respectively, for genotype negative LQTS. Of interest, the higher PRSQT in genotype negative patients compared with genotype positive patients was reflected by the larger difference in PRSQT between genotype negative patients versus controls (Table 3; meta-analysis β=0.735, SE=0.074; P=2.24×10−23) compared with genotype positive versus controls (Table 3; meta-analysis β=0.294, SE=0.028; P=1.09×10−25).
Figure 5.
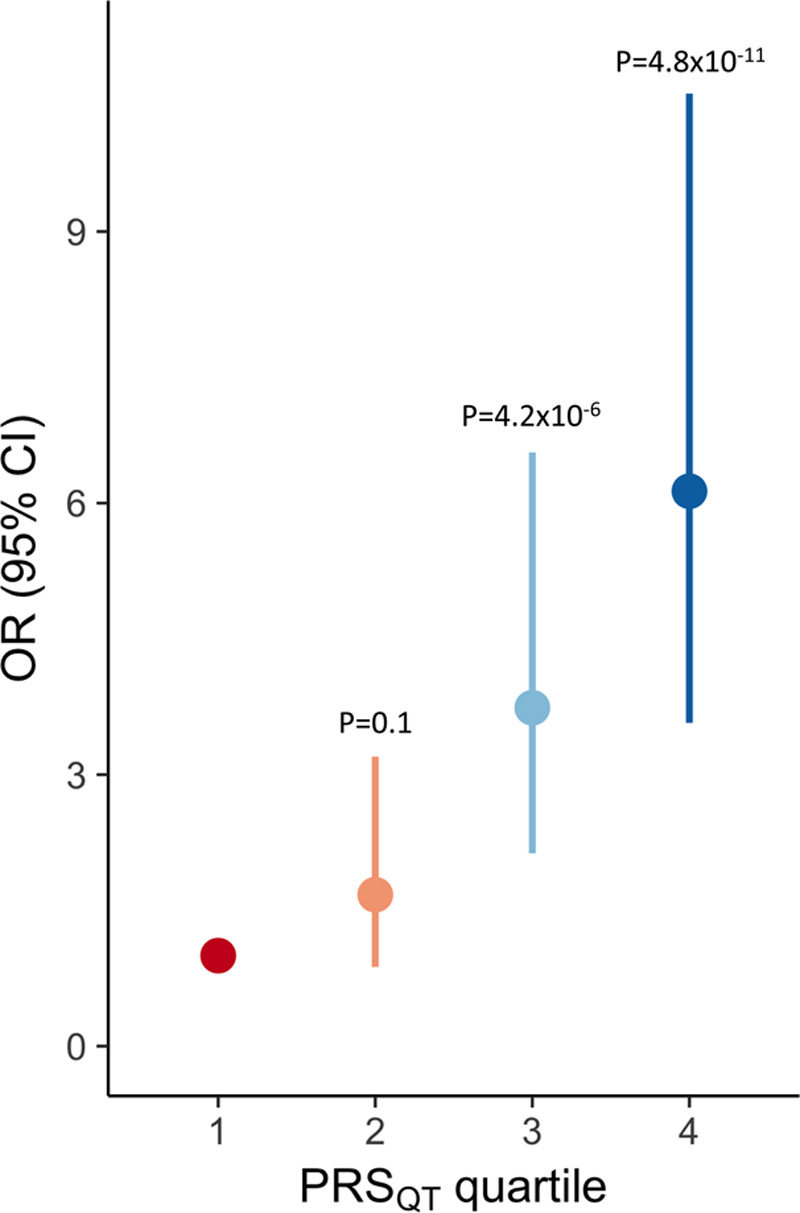
Increasing long QT syndrome risk with increasing QT polygenic score quartiles. Odds ratio (OR) for genotype negative long QT syndrome (LQTS; filled circles) and 95% CI (vertical bars) associated with each QT polygenic score (PRSQT) quartile taking the first PRSQT quartile as the reference. Data shown correspond to a meta-analysis of effects computed separately in the European and Japanese datasets. P values refer to comparison of each quartile against the first quartile.
Common Variant Heritability of LQTS
To evaluate the proportion of variance in LQTS susceptibility explained by common genetic variants (h2SNP) we used GCTA-GREML.32,37 Assuming a disease prevalence of 0.04%,1 the SNP heritability estimate on the liability scale was h2SNP=0.148 (SE=0.019 [95% CI, 0.111–0.185]; P=5.0×10−18) in the overall European LQTS dataset. h2SNP was similar when the analysis was restricted to genotype positive patients with LQTS. Similar results were also observed in the Japanese dataset and when using the phenotype-correlation genotype-correlation regression34 and the GREML estimation implemented in LDAK,35 as well as when we restricted h2SNP analyses to only patients with a pathogenic or likely-pathogenic variant (Table XII in the Data Supplement).
Association Analyses of Single SNPs and PRSQT With LQTS Severity
To identify genetic modifiers of disease severity we conducted a GWAS for QT-interval within the LQTS cases which did not uncover any genome-wide significant loci (Figure X in the Data Supplement). None of the 68 SNPs previously associated with QTc in the general population29 showed association with QTc after Bonferroni correction. PRSQT showed a weak positive correlation with QTc in the European cases (correlation coefficient [r]=0.06; P=0.042; Figure XI in the Data Supplement). In a multivariable linear regression model including clinical covariates associated with QTc (age at ECG recording, LQTS type, and sex), PRSQT was not significantly associated with QTc (Table XIII in the Data Supplement). Similarly, in a subanalysis restricted to probands (comprising 97% of the total of unrelated LQTS cases) using the multivariable linear regression model, PRSQT was not significantly associated with QTc (data not shown). In exploratory subgroup analyses, PRSQT was independently associated with QTc in KCNH2 rare variant carriers but not in KCNQ1 rare variant carriers (Table XIII in the Data Supplement). This result was not replicated in the Japanese LQTS dataset. PRSQT was not significantly associated with time to LAE in neither Europeans nor Japanese cases (Figure XII in the Data Supplement).
Discussion
Our findings establish an important role for common genetic variation in LQTS susceptibility and support a complex (polygenic) architecture in genotype negative LQTS. Case–control GWAS identified 3 genome-wide significant risk loci near NOS1AP, KCNQ1, and KLF12. Heritability analysis demonstrated that ≈15% of LQTS disease liability is attributable to common genetic variation. PRS analysis testing the aggregate effect of SNPs previously associated with QT-interval in the general population (PRSQT) identified a higher PRSQT in LQTS cases compared with controls and higher PRSQT in genotype negative versus genotype positive LQTS.
Shared Genetics of LQTS and QT-Interval in the General Population
The case–control GWAS uncovered 3 genetic LQTS susceptibility loci at genome-wide statistical significance near NOS1AP, KCNQ1, and KLF12, and 1 missense variant in KCNE1 at the suggestive threshold (Figure 2). The association of SNPs at KCNQ1 points to the involvement of common variants acting alongside rare variants in these genes in mediating disease susceptibility, akin to what was previously reported for common and rare variation in and around the SCN5A gene in Brugada syndrome.16 All 4 risk loci had been previously implicated in genetic control of the QT-interval by GWAS in the general population.29 For the 68 SNPs associated with QT-interval in the general population, we noted a strong positive correlation between their effect on QT-interval (obtained in the general population) and their OR for LQTS susceptibility, indicating, as expected, that the larger the effect a SNP has on the QT-interval, the more it increases LQTS susceptibility (Figure 3). The strong genetic correlation between LQTS susceptibility and QT-interval in the general population provides quantitative support for genetic overlap (Figure IX in the Data Supplement).
The association with the highest effect in the case–control GWAS was found for the p.Asp85Asn missense variant in KCNE1 (rs1805128). This variant increased susceptibility for LQTS in the overall cohort but had a more prominent effect in genotype negative LQTS with an OR of ≈7 versus an OR of 2 in genotype positive patients. This variant has an allele frequency of ≈1.2% in non-Finnish Europeans and ≈0.5% in East Asians, and has the largest effect size on the QT in the general (European descent) population (7.42 ms per minor [Asn] allele).29 It has been shown to be enriched in patients with drug-induced torsades de pointes.38
Genetic Architecture of Genotype Positive LQTS
LQTS has traditionally been viewed as a monogenic disorder mostly attributed to a rare variant with a drastic effect on ion channel function. We now demonstrate that a considerable extent (≈15%) of disease liability is attributable to common genetic variation. In genotype positive LQTS families, where the penetrance of pathogenic variants may be low for certain variants,8 the contribution of common variants to disease susceptibility may also contribute to variable disease penetrance. It has been well established that LQTS probands have a longer QT-interval and greater arrhythmic risk compared with family members carrying the same variant.7,13,39 This observed increased penetrance in probands may result from a greater burden of common QT-prolonging variants compared with other, less-severely affected, or unaffected mutation-carriers. However, because this study comprised only unrelated patients, this remains to be determined. Whether the PRSQT could discriminate between affected versus unaffected mutation carrier family members is intuitively appealing but remains to be formally demonstrated.
Genotype Negative LQTS, A Polygenic Subtype of LQTS?
PRS analysis, testing the aggregate effect of SNPs previously associated with QT-interval in the general population (PRSQT), identified a higher PRSQT in genotype negative versus genotype positive patients. This observation points to genotype negative LQTS, comprising ≈10% of patients with LQTS, as a polygenic subtype of the disorder where the underlying etiology involves, at least in part, a high burden of common QT prolonging alleles. As such, genetic susceptibility in genotype negative patients may not be determined to a large extent by 1 strong genetic factor as occurs in genotype positive patients but results from the accumulation of multiple variants (polygenic inheritance). The lower rate of family history of sudden cardiac death in genotype negative patients with LQTS is in line with polygenic inheritance. Our observations corroborate findings in other heritable phenotypes, such as familial hypercholesterolemia, where patients without a disease-causing variant in the LDLR, APOB, and PCSK9 genes have a higher PRS based on low-density lipoprotein modulating variants in comparison with those with rare familial hypercholesterolemia causing genetic variants.40 As such, the accumulation of multiple discrete common variants may confer risk similar to a monogenic mutation. This was recently demonstrated for common disorders such as coronary artery disease and atrial fibrillation, where individuals at the upper extreme of the PRS distribution had a risk of developing the disease reportedly comparable with carriers of a monogenic mutation.31 The overlap in the PRSQT distributions among genotype negative LQTS cases and controls (Figure 4) suggests that other factors are involved, possibly including low-frequency genetic variants with intermediate effect sizes as well as other common variants with smaller effect sizes.
In addition to providing insight into the genetic architecture of genotype negative LQTS, we here also describe for the first time the natural history in these patients. All ≈200 genotype negative patients with LQTS met diagnostic criteria for definite LQTS (ie, QTc>500ms or LQTS score≥3.5) and underwent sequencing of the unequivocal nonsyndromic LQTS genes. Genotype negative patients with LQTS had a higher QTc in comparison with patients with LQT1–3 but similar event-free survival as their genotype positive counterparts (Figure 1). The effect of established clinical risk factors, for example sex and QTc-duration, did not significantly differ between genotype positive and negative (no interaction effect) suggesting they may also be used to stratify risk of events in genotype negative LQTS.
Common Variants Do Not Contribute to LQTS Severity Within Probands
We sought to identify genetic modifiers of LQTS. In contrast to the case–control GWAS, GWAS for QTc and arrhythmic events within the unrelated LQTS cases did not uncover any genome-wide significant locus. PRSQT was also not significantly associated with QTc nor with the occurrence of events. At first glance, this may seem contradictory to previous studies in LQTS that demonstrated a modulatory effect of SNPs at NOS1AP on the QTc and arrhythmic events,10,11,13 as well as a study in the general population that showed a modulatory effect of PRS derived from previous GWAS on QT-interval.41 For example, a study we previously conducted in patients with LQT2 uncovered strong associations with large effect sizes (>12 ms/allele) for SNPs at NOS1AP.13 An important difference however, is that the current study did not include family members but only 1 patient per family (97% probands), whereas the previous studies considered both probands and genotype positive relatives. Conceptually, inclusion of both probands and relatives results in greater variation in QTc and is thus expected to increase statistical power for detection of modulatory effects. Moreover, the different rare variants in the patients we studied here are associated with biophysical defects of varying severity. As such, they are also expected to contribute to interindividual variability which is difficult to account for. For instance, patients with LQT2 with pore-region variants are known to be more severely affected than other patients with LQT2.42 Indeed, in a subanalysis, restricted to European patients with LQT2, where we accounted for the mutation location (pore versus nonpore), we detected an association of PRSQT with QTc. In sum, our data show that common variants do not affect disease severity across all probands studied. Further studies are needed to explore their predictive role in family members.
Potential Clinical Implications
In genotype negative LQTS, disease susceptibility estimation for relatives does not follow a Mendelian pattern. In our cohort, a positive family history of sudden cardiac death was less often observed in genotype negative individuals compared with genotype positive ones, suggesting that risk for family members in genotype negative patients may be lower. Polygenicity in genotype negative individuals implies that risk is not primarily attributable to 1 genetic factor inherited from 1 of the biological parents as is the case for autosomal dominant LQTS. In such cases, cascade screening may necessitate clinical evaluation of both maternal and paternal family members. Future clinical utility of genetic testing based on polygenic inheritance necessitates the availability of polygenic risk scores with high discriminative capacity. The discriminative capacity of a PRS based on QT modulating SNPs is expected to improve as knowledge concerning variants that modulate the QT-interval become known, for example through larger GWAS studies, or by combining it with nongenetic modifiers. In a recent study, a PRS based on 61 QT-SNPs (a subset of the 68 QT modulating SNPs included in the PRSQT used herein) explained a substantial proportion of QT-interval response to QT-prolonging drugs in a trial of 3 QT-prolonging drugs conducted in healthy individuals, as well as risk of torsade de pointes in a case–control study.43 This provides further support to a liability threshold model whereby multiple factors, genetic and nongenetic, impact on cardiac repolarization and determine arrhythmic risk. In this respect, calculation of PRSQT for the purpose of preventive avoidance of QT-prolonging drugs may be desirable for relatives of genotype negative LQTS. It is clear that further studies are needed to address how testing for polygenic susceptibility may become clinically useful.
Study Limitations
Although in genotype negative patients with LQTS we performed sequencing of the coding region of nonsyndromic LQTS genes, this may have missed copy number variation or disease-causing variants in the noncoding region44 of established genes as well as mutations in yet unknown disease genes. This may have blunted the differences between genotype negative and genotype positive patients and thus would not affect the study conclusions. Despite being the largest international dataset of unrelated patients with LQTS published to date, the study had limited statistical power to detect lower effect associations at GWAS significance threshold. The prespecified design of meta-analyzing European and Japanese GWAS may also miss disease loci with differences in haplotype structure among European and East Asian chromosomes. Nonetheless, GWAS in separate ancestries did not detect any association at GWAS threshold. Last, studies in larger patient sets are required to further refine our understanding of the genetic architecture underlying LQTS in genotype negative patients.
Conclusions
This work establishes an important role for common genetic variation in susceptibility to LQTS. Common genetic variation affecting the QT-interval in the general population contributes to disease susceptibility in both genotype positive and genotype negative LQTS. The role of common variants is predominant in genotype negative LQTS, suggesting that the latter may constitute a polygenic form of LQTS. Increasing burden of QT-prolonging common variants (eg, PRSQT) is associated with higher susceptibility for LQTS but is not associated with disease severity within LQTS probands. Further studies are needed to assess the role of polygenic risk within LQTS families.
Acknowledgments
The authors thank Estelle Baron, Simon Lecointe, Annabelle Rajalu, Aurélie Thollet, and Florence Kyndt for their help in DNA sample preparation and regulatory processes at the Nantes center, the Nantes biological resource center for biobanking (CHU Nantes, Hôtel Dieu, Center de Resources Biologiques, Nantes, F-44093, France [BRIF: BB-0033-00040]), and the National Referral Center for Inherited Cardiac Arrhythmias of Nantes and its associated competence centers. We thank Professor Pier D. Lambiase and Professor Patricia B. Munroe for sharing summary statistics. We thank Dr Nicky Whiffin for help with using CardioClassifier. The KORA research platform (KORA, Cooperative Research in the Region of Augsburg) was initiated and financed by the Helmholtz Zentrum München–German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data are available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113, 085475, and 090355.
Sources of Funding
Najim Lahrouchi was supported by the Kenneth M. Rosen Fellowship in Cardiac Pacing and Electrophysiology Scholarship of the Heart Rhythm Society, the CVON-PREDICT Young Talent Program of the Dutch Heart Foundation, and the SADS Foundation Courts K. Cleveland Jr. Young Investigator Award in Cardiac Channelopathy Research. Drs Bezzina, Lahrouchi, Wilde, van Tintelen, and Tan acknowledge the support from the Dutch Heart Foundation (CVON 2018-30 PREDICT2 project to Drs Bezzina, Tan, and Wilde) and the Netherlands Organization for Scientific Research (VICI fellowship, 016.150.610, to Dr Bezzina). Dr Tan has received funding from the European Union’s Horizon 2020 research and innovation program under acronym ESCAPE-NET (The European Sudden Cardiac Arrest network toward Prevention, Education, New Effective Treatment), registered under grant agreement No 733381. Dr Tadros received support from the Canadian Heart Rhythm Society (George Mines Award), the European Society of Cardiology (research award), and the Philippa and Marvin Carsley Cardiology Chair and is currently a clinical research scholar of the Fonds de Recherche du Québec—Santé. Drs Shaw and Crotti acknowledge the support of the Leducq Foundation for cardiovascular research grant 18CVD05: Toward Precision Medicine with Human iPSCs for Cardiac Channelopathies. Drs Probst and Gourraud were supported by a grant from Hopitaux Universitaires du Grand Ouest and Fondation Maladies Rares (RC17_0357). Dr Breckpot was supported by the H2020-MSCA-IF-2014 Program of the European Commission (RISTRAD-661617) and by the Regional Council of Pays-de-la-Loire (Etoile montante: REGIOCARD). Dr Schott was supported by the Fondation pour la Recherche Médicale (DEQ20140329545) and by the National Agency for Research (ANR-GENSUD-14-CE10-0001). Drs Rydberg and Diamant are supported by The Swedish Heart-Lung Foundation. Dr Behr is supported by the Higher Education Funding Council for England and the British Heart Foundation and acknowledges support from Cardiac Risk in the Young. Dr Wijeyeratne had received support through an Academic Clinical Fellowship from the National Institute of Health Research. Drs Wijeyeratne and Behr gratefully acknowledge funding and ongoing support from the James Lancaster Memorial Fund sponsored by McColl’s RG Ltd. This work was partly supported by a Health and Labor Sciences Research Grant from the Ministry of Health, Labor, and Welfare of Japan (H22-032, H24-033, H26-040, and H29-055) to Dr Yoshinaga. Dr Makita is supported by AMED (19kk0305011h0001). Drs Shimizu and Aung acknowledge support from a Health Science Research Grant from the Ministry of Health, Labor and Welfare of Japan for Clinical Research on Measures for Intractable Diseases (H24-033, H26-040, and H27-032). Dr Arbelo is supported by Instituto de Salud Carlos III (FIS PI16/01203 and PI17/01690) cofunded by ERDF/ESF, “Investing in Your Future.” This work was supported by the John and Birthe Meyer Foundation, The Hallas-Møller Emerging Investigator Novo Nordisk (NNF17OC0031204). The iPSYCH study was funded by the Lundbeck Foundation Initiative for Integrative Psychiatric Research. This research has been conducted using the Danish National Biobank resource. The Cardiac Inherited Disease Registry has been supported by Cure Kids. AW is supported by the Hugh Green Foundation. Dr Ellinor is supported by the Fondation Leducq (14CVD01), by grants from the National Institutes of Health (1RO1HL092577, R01HL128914, K24HL105780) and the American Heart Association (18SFRN34110082). SAL is supported by National Institutes for Health grant 1R01HL139731 and American Heart Association 18SFRN34250007. Dr Loeys is supported by a European Research Council consolidator grant. Dr Roden is supported by P50 GM115305 and MBS by K23 HL127704. Dr Antzelevitch is supported by grants from the National Institutes for Health, HL47678 and HL138103. Dr Postema is supported by The Swedish Heart-Lung Foundation, grant #20180444. Drs Turkowski, Tester, Bos, and Ackerman were supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program.
Disclosures
Dr Behr received previous research funds from Biotronik and consulting for Medtronic. Dr Arbelo received speaking fees from Biosense Webster. Dr Lubitz receives sponsored research support from Bristol Myers Squibb/Pfizer, Bayer AG, and Boehringer Ingelheim, and has consulted for Bristol Myers Squibb/Pfizer and Bayer AG. Dr Weeke is supported by a grant from Bayer AG to the Broad Institute focused on the genetics and therapeutics of cardiovascular diseases. Dr Weeke has also served on advisory boards or consulted for Bayer AG, Quest Diagnostics, and Novartis. Dr Cusi is a scientific consultant for Bio4Dreams. Dr Ackerman is a consultant for Audentes Therapeutics, Boston Scientific, Gilead Sciences, Invitae, Medtronic, Myokardia, and St. Jude Medical. Dr Ackerman and Mayo Clinic have an equity/royalty-based licensing agreement with AliveCor. The other authors report no conflicts.
Supplemental Materials
Methods
Data Supplement Figures I–XII
Data Supplement Tables I–XIII
References 45–57
Supplementary Material
Footnotes
Drs Lahrouchi and Tadros contributed equally to the study and share first authorship.
Sources of Funding, see page 336
The Data Supplement, podcast, and transcript are available with this article at https://www.ahajournals.org/doi/suppl/10.1161/circulationaha.120.045956.
References
- 1.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, et al. Prevalence of the congenital long-QT syndrome. Circulation 20091201761–1767doi: 10.1161/CIRCULATIONAHA.109.863209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res 20151161919–1936doi: 10.1161/CIRCRESAHA.116.304030 [DOI] [PubMed] [Google Scholar]
- 3.Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol 200749240–246doi: 10.1016/j.jacc.2006.10.010 [DOI] [PubMed] [Google Scholar]
- 4.Lahrouchi N, Raju H, Lodder EM, Papatheodorou E, Ware JS, Papadakis M, Tadros R, Cole D, Skinner JR, Crawford J, et al. Utility of post-mortem genetic testing in cases of sudden arrhythmic death syndrome. J Am Coll Cardiol 2017692134–2145doi: 10.1016/j.jacc.2017.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013101932–1963doi: 10.1016/j.hrthm.2013.05.014 [DOI] [PubMed] [Google Scholar]
- 6.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation 199999529–533doi: 10.1161/01.cir.99.4.529 [DOI] [PubMed] [Google Scholar]
- 7.Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol 20115751–59doi: 10.1016/j.jacc.2010.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazzanti A, Maragna R, Vacanti G, Monteforte N, Bloise R, Marino M, Braghieri L, Gambelli P, Memmi M, Pagan E, et al. Interplay between genetic substrate, QTc duration, and arrhythmia risk in patients with long QT syndrome. J Am Coll Cardiol 2018711663–1671doi: 10.1016/j.jacc.2018.01.078 [DOI] [PubMed] [Google Scholar]
- 9.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, et al. Risk stratification in the long-QT syndrome. N Engl J Med 20033481866–1874doi: 10.1056/NEJMoa022147 [DOI] [PubMed] [Google Scholar]
- 10.Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ, George AL., Jr. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation 20091201657–1663doi: 10.1161/CIRCULATIONAHA.109.879643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomás M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, Bellazzi R, Arking DE, Marban E, Chakravarti A, et al. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol 2010552745–2752doi: 10.1016/j.jacc.2009.12.065 [DOI] [PubMed] [Google Scholar]
- 12.Duchatelet S, Crotti L, Peat RA, Denjoy I, Itoh H, Berthet M, Ohno S, Fressart V, Monti MC, Crocamo C, et al. Identification of a KCNQ1 polymorphism acting as a protective modifier against arrhythmic risk in long-QT syndrome. Circ Cardiovasc Genet 20136354–361doi: 10.1161/CIRCGENETICS.113.000023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolder ICRM, Tanck MWT, Postema PG, Barc J, Sinner MF, Zumhagen S, Husemann A, Stallmeyer B, Koopmann TT, Hofman N, et al. Analysis for genetic modifiers of disease severity in patients with long-QT syndrome type 2. Circ Cardiovasc Genet 20158447–456doi: 10.1161/CIRCGENETICS.114.000785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz PJ, Crotti L, George AL. Modifier genes for sudden cardiac death. Eur Heart J 2018393925–3931doi:10.1093/eurheartj/ehy502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, et al. ; Heart Rhythm Society (HRS); European Heart Rhythm Association (EHRA) HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011131077–1109doi: 10.1093/europace/eur245 [DOI] [PubMed] [Google Scholar]
- 16.Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 2013451044–1049doi: 10.1038/ng.2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adler A, Novelli V, Amin AS, Abiusi E, Care M, Nannenberg EA, Feilotter H, Amenta S, Mazza D, Bikker H, et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 2020141418–428doi: 10.1161/CIRCULATIONAHA.119.043132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giudicessi JR, Rohatgi RK, Tester DJ, Ackerman MJ. Variant frequency and clinical phenotype call into question the nature of minor, nonsyndromic long-QT syndrome-susceptibility gene-disease associations. Circulation 2020141495–497doi: 10.1161/CIRCULATIONAHA.119.043131 [DOI] [PubMed] [Google Scholar]
- 19.Roberts JD, Asaki SY, Mazzanti A, Bos JM, Tuleta I, Muir AR, Crotti L, Krahn AD, Kutyifa V, Shoemaker MB, et al. An international multicenter evaluation of type 5 long QT syndrome: a low penetrant primary arrhythmic condition. Circulation 2020141429–439doi: 10.1161/CIRCULATIONAHA.119.043114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016536285–291doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi Y, Yang S, Nykamp K, Garcia J, Lincoln SE, Topper SE. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017;9:13. doi: 10.1186/s13073-017-0403-7. doi: 10.1186/s13073-017-0403-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotta F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2016 doi: 10.1038/gim.2016.90. doi:10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. ; ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 201517405–424doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vink AS, Neumann B, Lieve KVV, Sinner MF, Hofman N, El Kadi S, Schoenmaker MHA, Slaghekke HMJ, de Jong JSSG, Clur SB, et al. Determination and interpretation of the QT interval. Circulation 20181382345–2358doi: 10.1161/CIRCULATIONAHA.118.033943 [DOI] [PubMed] [Google Scholar]
- 25.Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT. Data quality control in genetic case-control association studies. Nat Protoc 201051564–1573doi: 10.1038/nprot.2010.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. Next-generation genotype imputation service and methods. Nat Genet 2016481284–1287doi: 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010262190–2191doi: 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826. doi: 10.1038/s41467-017-01261-5. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, Koopmann TT, Sotoodehnia N, Rossin EJ, Morley M, Wang X, et al. ; CARe Consortium; COGENT Consortium; DCCT/EDIC; eMERGE Consortium; HRGEN Consortium Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet 201446826–836doi: 10.1038/ng.3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sano M, Kamitsuji S, Kamatani N, Hong KW, Han BG, Kim Y, Kim JW, Aizawa Y, Fukuda K; Japan Pharmacogenomics Data Science Consortium (JPDSC) Genome-wide association study of electrocardiographic parameters identifies a new association for PR interval and confirms previously reported associations. Hum Mol Genet 2014236668–6676doi: 10.1093/hmg/ddu375 [DOI] [PubMed] [Google Scholar]
- 31.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018501219–1224doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 20118876–82doi: 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Zeng J, Goddard ME, Wray NR, Visscher PM. Concepts, estimation and interpretation of SNP-based heritability. Nat Genet 2017491304–1310doi: 10.1038/ng.3941 [DOI] [PubMed] [Google Scholar]
- 34.Golan D, Lander ES, Rosset S. Measuring missing heritability: inferring the contribution of common variants. Proc Natl Acad Sci U S A 2014111E5272–E5281doi: 10.1073/pnas.1419064111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weissbrod O, Flint J, Rosset S. Estimating SNP-based heritability and genetic correlation in case-control studies directly and with summary statistics. Am J Hum Genet 201810389–99doi: 10.1016/j.ajhg.2018.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, Duncan L, Perry JR, Patterson N, Robinson EB, et al. ; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3 An atlas of genetic correlations across human diseases and traits. Nat Genet 2015471236–1241doi: 10.1038/ng.3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 201042565–569doi: 10.1038/ng.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kääb S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze-Bahr E, Guicheney P, Bishopric NH, et al. A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet 2012591–99doi: 10.1161/CIRCGENETICS.111.960930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A., Jr. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 1991841136–1144doi: 10.1161/01.cir.84.3.1136 [DOI] [PubMed] [Google Scholar]
- 40.Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, Harrison SC, Li K, Drenos F, Karpe F, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet 20133811293–1301doi: 10.1016/S0140-6736(12)62127-8 [DOI] [PubMed] [Google Scholar]
- 41.Rosenberg MA, Lubitz SA, Lin H, Kosova G, Castro VM, Huang P, Ellinor PT, Perlis RH, Newton-Cheh C. Validation of polygenic scores for QT interval in clinical populations. Circ Cardiovasc Genet. 2017;10 doi: 10.1161/CIRCGENETICS.117.001724. doi:10.1161/CIRCGENETICS.117.001724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, Towbin JA, Keating MT, Priori SG, Schwartz PJ, et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 2002105794–799doi: 10.1161/hc0702.105124 [DOI] [PubMed] [Google Scholar]
- 43.Strauss DG, Vicente J, Johannesen L, Blinova K, Mason JW, Weeke P, Behr ER, Roden DM, Woosley R, Kosova G, et al. Common genetic variant risk score is associated with drug-induced QT prolongation and torsade de pointes risk: a pilot study. Circulation 20171351300–1310doi: 10.1161/CIRCULATIONAHA.116.023980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bagnall RD, Ingles J, Dinger ME, Cowley MJ, Ross SB, Minoche AE, Lal S, Turner C, Colley A, Rajagopalan S, et al. Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 201872419–429doi: 10.1016/j.jacc.2018.04.078 [DOI] [PubMed] [Google Scholar]
- 45.Whiffin N, Walsh R, Govind R, Edwards M, Ahmad M, Zhang X, Tayal U, Buchan R, Midwinter W, Wilk AE, et al. CardioClassifier: disease- and gene-specific computational decision support for clinical genome interpretation. Genet Med 2018201246–1254doi: 10.1038/gim.2017.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, Li N, Felkin L, Ingold N, Govind R, et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. 2019;11:5. doi: 10.1186/s13073-019-0616-z. doi: 10.1186/s13073-019-0616-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, Herron TJ, McCarthy S, Schmidt EM, Sveinbjornsson G, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet 2018501234–1239doi: 10.1038/s41588-018-0171-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 20129671–675doi: 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Postema PG, Wilde AA. The measurement of the QT interval. Curr Cardiol Rev 201410287–294doi: 10.2174/1573403x10666140514103612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, Barton PJR, Funke B, Cook SA, MacArthur D, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med 2017191151–1158doi: 10.1038/gim.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA, Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 200961297–1303doi: 10.1016/j.hrthm.2009.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ware JS, Walsh R, Cunningham F, Birney E, Cook SA. Paralogous annotation of disease-causing variants in long QT syndrome genes. Hum Mutat 2012331188–1191doi: 10.1002/humu.22114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapplinger JD, Tseng AS, Salisbury BA, Tester DJ, Callis TE, Alders M, Wilde AA, Ackerman MJ. Enhancing the predictive power of mutations in the C-terminus of the KCNQ1-encoded Kv7.1 voltage-gated potassium channel. J Cardiovasc Transl Res 20158187–197doi: 10.1007/s12265-015-9622-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Setten J, Brody JA, Jamshidi Y, Swenson BR, Butler AM, Campbell H, Del Greco FM, Evans DS, Gibson Q, Gudbjartsson DF, et al. PR interval genome-wide association meta-analysis identifies 50 loci associated with atrial and atrioventricular electrical activity. Nat Commun. 2018;9:2904. doi: 10.1038/s41467-018-04766-9. doi: 10.1038/s41467-018-04766-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sotoodehnia N, Isaacs A, de Bakker PI, Dörr M, Newton-Cheh C, Nolte IM, van der Harst P, Müller M, Eijgelsheim M, Alonso A, et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet 2010421068–1076doi: 10.1038/ng.716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.den Hoed M, Eijgelsheim M, Esko T, Brundel BJ, Peal DS, Evans DM, Nolte IM, Segrè AV, Holm H, Handsaker RE, et al. ; Global BPgen Consortium; CARDIoGRAM Consortium; PR GWAS Consortium; QRS GWAS Consortium; QT-IGC Consortium; CHARGE-AF Consortium Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet 201345621–631doi: 10.1038/ng.2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramírez J, Duijvenboden SV, Ntalla I, Mifsud B, Warren HR, Tzanis E, Orini M, Tinker A, Lambiase PD, Munroe PB. Thirty loci identified for heart rate response to exercise and recovery implicate autonomic nervous system. Nat Commun. 2018;9:1947. doi: 10.1038/s41467-018-04148-1. doi: 10.1038/s41467-018-04148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.