

Abstract
More than 50 years have passed since Haszeldine reported the first addition of a trifluoromethyl radical to an allene; in the intervening years, both the chemistry of allenes and the reactivity of single-electron species have become topics of intense interest. In this Review, we provide an overview of the fundamentals of radical additions to allenes and highlight the emergence of theoretical and experimental evidence that reveals unique reactivity patterns for radical additions to allenes as compared with other unsaturated compounds. Factors capable of exerting control over the chemo-, regio-, and stereoselectivities of the attack of carbon- and heteroatom-based radicals at each of the three potential reactive sites in an allene substrate are described. These include reaction conditions, the nature of the attacking radical, the substitution pattern of the allene, and the length of the linker between the radical center and the proximal allene carbon in the substrate. Cycloaddition reactions between allenes and partners containing π-bonds, which are likely to proceed through radical pathways, are presented to highlight their ability to rapidly access complex polycyclic scaffolds. Finally, the synthetic utility of the products arising from these chemistries is described, including their applications to the construction of complex molecules.
Graphical Abstract
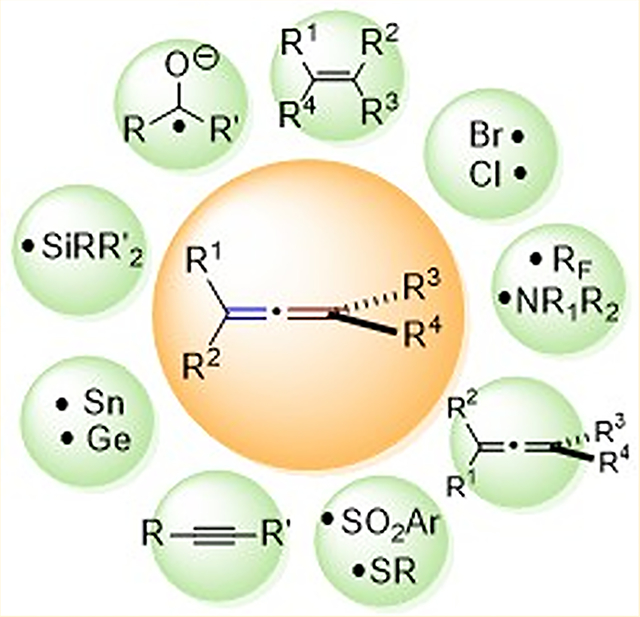
1. INTRODUCTION
Since the first synthesis of an allene by Burton and Pechmann in 1887,1 significant efforts have been devoted to studying the reactivities of this unique type of unsaturated compound.2–16 More than 50 years have passed since Haszeldine reported the first addition of a trifluoromethyl radical to an allene,17 yet there continues to be great interest in both the mechanistic aspects and the synthetic applications of radical reactions involving allenes.18 This is due in part to the expanding diversity and ease of new methods to synthesize increasingly complex allenes19–24 as well as ongoing reports of applications that harness the unique reactivity of allenes as compared to alkenes and alkynes.9,10,13,14 Because of their structural differences and unique bonding properties, the reactivities of allenes toward different types of radicals is less predictable than the trends observed in reactions with alkenes and alkynes.25 In the intervening decades since Haszeldine’s report, a handful of reviews dedicated specifically to radical reactions of allenes have appeared,6,7 most notably a pair of reviews by Ma et al.7 and Hartung and Kofp,25 both published in 2004. More recently, and during the preparation of this manuscript, Wu and coworkers published a short review on the functionalization of allenes via radical processes.18 The goal of this Review is to present a comprehensive picture of the development of radical additions to allenes since 1950 to the present day, even if aspects have been previously covered. This is key to enabling the reader to directly compare and contrast reactivity patterns in the additions of diverse radicals to allenes, as well as gaining a more complete picture of the mechanistic aspects and broad synthetic applications of radical reactions of allenes. A section highlighting the power of radical additions to allenes for the preparation of complex molecular scaffolds present in bioactive natural products is also presented in the hope that it will inspire continuing investigation into this fascinating research topic.
Allenes contain two cumulated π-bonds, with the double bonds located in mutually perpendicular planes (Figure 1). The C1 and C3 carbons of an allene are sp2-hybridized, whereas the central C2 carbon is sp-hybridized. To simplify discussions throughout this Review, the positions and π-bonds have been labeled as illustrated in Figure 1.
Figure 1.
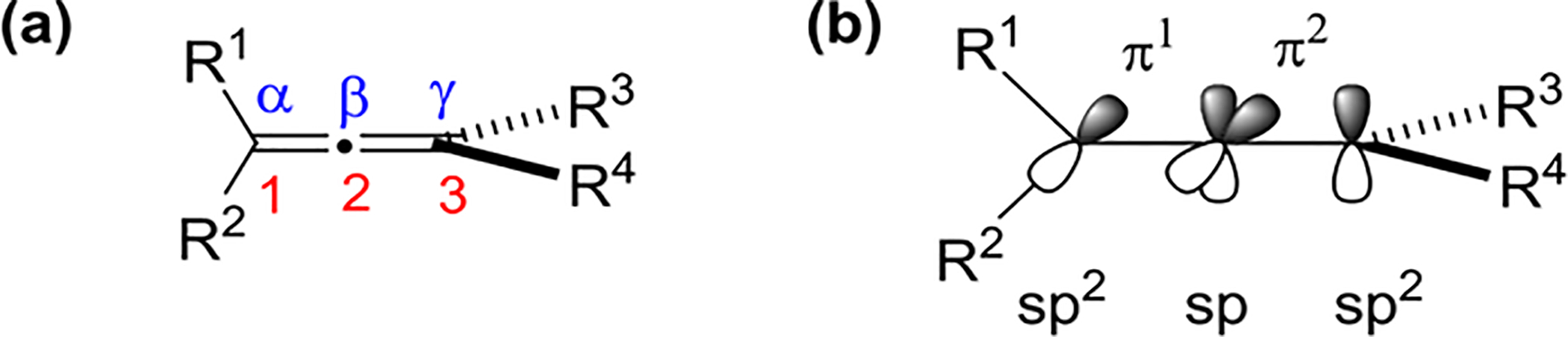
Cumulated π-bond system of allenes.
The ability to achieve radical attack on an allene in a selective manner can be both challenging and synthetically powerful. This is illustrated in Schemes 1 and 2, where the intermolecular addition of a radical to an unsymmetrically substituted allene can occur at three potential sites: terminal attack at either C1 or C3 or central attack at C2.
Scheme 1.

Regio- and Stereoselectivity in Intermolecular Radical Addition to C1 or C3
Scheme 2.
Regio- and Stereoselectivity in Intermolecular Radical Addition to C2
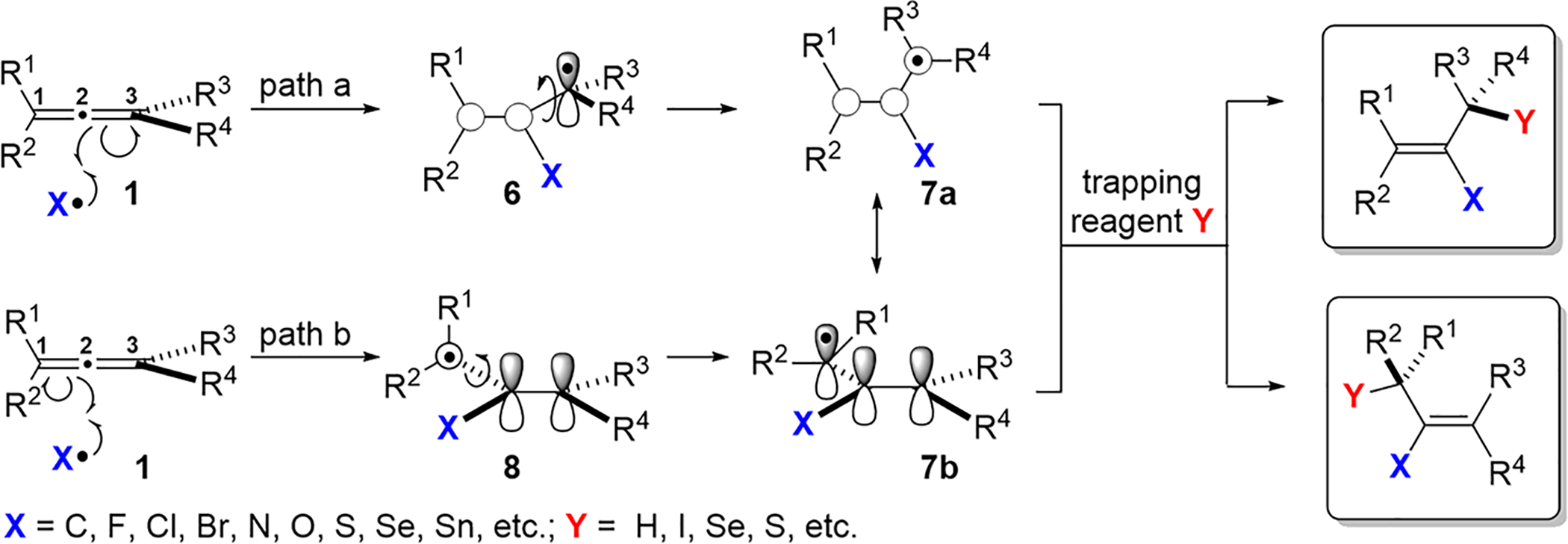
Attack at the C1 or C3 of allene 1 results in a pair of rapidly interconverting, nonlinear radical intermediates 3a,b and 5a,b,25 where the attack is reversible under certain circumstances26–30 (Scheme 1). The initial vinyl radical intermediate 2 or 4 first adopts a π-type configuration, which then isomerizes to the more energetically favored σ-type radicals 3a,b and 5a,b. The isomerization of the E/Z vinyl radicals has a low activation barrier,31–34 with the ratio of E/Z products largely affected by steric effects; these are covered in more detail in section 2.
Attack on the C2 of allene 1 initially results in a π-type alkyl radical 6 or 8, which rotates around the C2–C3 bond of 6 or the C1–C2 bond of 8 to assume the appropriate geometry for resonance-stabilized allylic radical 7a or 7b (Scheme 2). Multiple research groups have demonstrated that the activation barrier for this rotation is negligible.28,29,35,36
In general, reactions of radicals with an unsubstituted allene (propadiene) typically occur at the α carbon, irrespective of the type and polarity of the radical intermediate (Scheme 1). In contrast, substituted allenes are generally attacked at the central (β) carbon to give allylic radical intermediates (Scheme 2); indeed, selectivity can often be determined by assessing the relative stabilities of these intermediates. Thus, thermodynamics largely controls the selectivity in radical additions to allenes, as opposed to the use of frontier molecular orbital (FMO) theory to explain the selectivities of radical additions to alkenes and aromatic π-systems. However, further studies are needed to better understand the factors that contribute to the differences between alkenes and allenes in terms of their behavior toward radicals. Such investigations will undoubtedly expand the synthetic utility of these reactions and enable tunable control over site-selectivity.
2. INTERMOLECULAR ADDITIONS OF RADICALS TO ALLENES
2.1. Halogen Radicals
2.1.1. Chlorine Radicals.
Reactions of chlorine radicals with allenes have only recently been studied by a handful of research groups. A greater understanding of the processes involved in the chlorination of low-molecular-weight hydrocarbons, including allenes, may facilitate better methods for the synthesis of chlorinated compounds, efficient biomass combustion, and the incineration of hazardous waste. Two possible pathways were proposed for the reaction between a chlorine radical and propadiene37 (Scheme 3): (1) H-atom abstraction of the allenic proton to form an allenic radical 9 and (2) chlorine addition to C2 of propadiene to form an allylic radical 11, which could undergo the elimination of HCl to form the propargyl radical 10.
Scheme 3.

Two Potential Reaction Pathways between Chlorine Radical and Propadiene
Mixtures of propadiene and Cl2 were photolyzed at 355 nm, and the absolute rate coefficients of these reactions were measured over a temperature range of 292 to 850 K and a pressure range of 4–10 Torr. These results led Farrell et al. to the discovery that the addition of the chlorine radical to propadiene constitutes an important pathway up to T ≈ 800 K.37 On the basis of ab initio calculations, the 2-chloroallyl radical 11 was suggested to be the major product. These calculations were carried out at the G2 level of theory, indicating that the chlorine radical attack on the C2 of propadiene is 20 kcal/mol lower in enthalpy than the attack on C1 or C3. Nonetheless, the possibility of an initial bond formation between the chlorine and C1 or C3, followed by isomerization to the more stable allylic radical via a 1,2-Cl shift could not be ruled out.
As the temperature of the reaction of propadiene and Cl2 is increased to >800 K, HCl formation is the major pathway. The larger pre-exponential factor extracted from the Arrhenius plot, compared with those of other exothermic hydrogen abstraction reactions between chlorine radicals and hydrocarbons, indicates that an addition–elimination mechanism might be the major pathway at higher temperatures.
By irradiating mixtures, under an inert atmosphere, of Cl2, and propadiene at 351 nm, Atkinson et al. provided further spectral evidence that a C3H4Cl radical was produced upon irradiation.38 This study was also briefly discussed in a previous review by Hartung and Kopf.25 Ab initio calculations supported the proposal that the addition of a chlorine radical to propadiene should readily occur at room temperature. The addition of a chlorine radical to both the C2 and C1/C3 of propadiene would form incipient ensembles of 2-chloroallyl and 3-chloro-1-propene-2-yl radicals 11 and 12 (Scheme 4), with the internal energy of each radical enabling isomerization to establish an equilibrium mixture of radicals. However, the allylic radical 11 was proposed as the dominant species in the reaction due to its stability.
Scheme 4.

Cl Radical Addition to the C2 and C1/C3 Carbons of Propadiene in the Gas Phase
Studies by Hudgens et al. showed results consistent with previous experimental and computational studies. Ab initio calculations supported a higher barrier for H-atom abstraction compared with the exothermicity of chlorine radical addition to propadiene. An equilibrium between 11 and 12 is established by the rapid transfer of a chlorine atom between the C1/C3 and C2 atoms.36 The possibility of the isomerization of 11 and 12 via hydrogen and chlorine atom transfers was deemed irrelevant at ambient temperature based on ab initio calculations. For a discussion on this study in a previous review, see the review by Hartung and Kopf.25
Zhao and Murphy carried out the chlorination of allenes using the hypervalent iodine reagent 1-chloro-1,2-benziodoxol-3-one. The regioselective and chemoselective dichlorination of various aryl-substituted allenes 1339 furnished the corresponding 2,3-dichlorides 14 in moderate to good yield, with the Z isomer as the major isomer (Scheme 5a). The reaction was proposed to proceed via a radical mechanism, which was supported by the inhibition of the reaction in the presence of 1.5 equiv of TEMPO (Scheme 5b). The homolytic cleavage of the I–Cl bond in 1-chloro-1,2-benziodoxol-3-one provides a chlorine radical, which attacks the C2 of allene 13. When R = alkyl in 13, the 1,3-diene 15 is proposed to form from the same allylic radical intermediate 16 by a competing hydrogen atom abstraction step (Scheme 5c).
Scheme 5.

Dichlorination of Aryl-Substituted Allenes 13 with 1-Chloro-1,2-benziodoxol-3-one
2.1.2. Bromine Radicals.
An early report of the homolytic addition of HBr to an allene was published by Kovachic et al. in 196140 and was previously discussed in the review by Griesbaum.2 Mixtures of HBr or DBr were irradiated with deuterated or nondeuterated propadienes 17 to synthesize deuterated propenes (Scheme 6). The major products 18 and 19 were brominated on the central carbon and hydrogenated/deuterated on the terminal carbon. Only a small amount of 3-bromopropene was observed.
Scheme 6.

HBr/DBr Addition to Deuterated/Nondeuterated Propadienes
In 1964, Griesbaum et al. published a study on the free-radical addition of HBr to propadiene, where several reaction parameters were varied, including the HBr/propadiene ratio, the reaction temperature, the reaction phase, the reaction medium, and other conditions.26 When the reaction was carried out in the gas phase under UV irradiation at room temperature, an equimolar mixture of HBr and propadiene generated 2-bromopropene 20a as the major product in 85% yield, with trace amounts of 1,2-dibromopropane 21 and 2,2-dibromopropane 22 (Scheme 7).
Scheme 7.
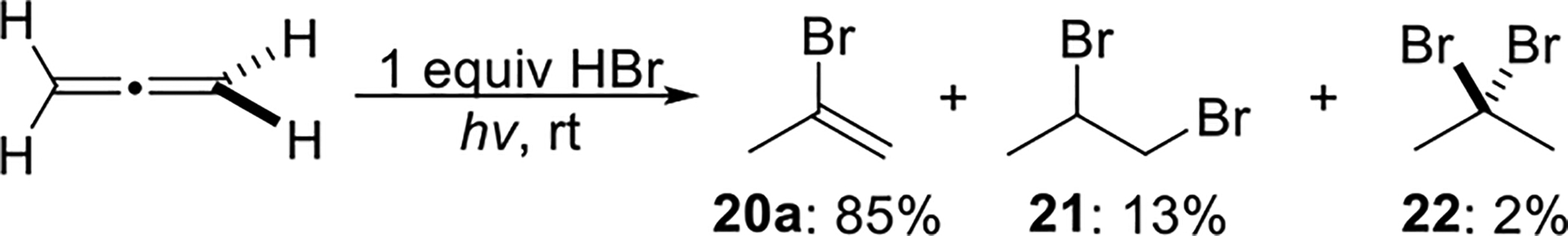
Equimolar HBr Addition to Propadiene in the Gas Phase
By lowering the reaction temperature to −40 °C and changing to the liquid phase, Griesbaum found that the same adducts were formed, but in varying ratios (Scheme 8). As the temperature was decreased, the amount of terminal addition product increased, along with a concomitant decrease in the extent of central addition product. Increased amounts of the dibromo adducts 21 and 23 were also noted.
Scheme 8.

Equimolar HBr Addition to Propadiene in the Liquid Phase
The use of AIBN as the initiator decreased the reaction time by a factor of three; however, the amount of 20b and 23 also increased (Scheme 8). When the reaction was carried out in a 1 M solution of propane, the amount of terminal addition product resulting from the attack of the radical on the terminal allene carbon was increased by 10%. In the presence of 10 equiv of HBr, the major products were various dibrominated adducts, including 23 (43%), 21 (34%), and 20b (23%). When excess propadiene was utilized, the amount of the terminal addition product 20b decreased, leading to 20a as the major product (Scheme 9).
Scheme 9.

HBr Addition to Excess Propadiene in the Liquid Phase
On the basis of the above experimental data, Griesbaum proposed the reaction pathway illustrated in Scheme 10. The regioselectivity of bromine radical addition to propadiene was strongly dependent on the reaction temperature and reactant ratio; lowered temperatures and excess HBr favored the formation of a terminal attack product. Griesbaum stated that the experimental results described above do not reflect the initial regioselectivity of bromine attack on the central versus the terminal allene carbon. This was proposed to be due to the ability of allenes to form bridged radical intermediates, followed by fast rearrangement to yield the corresponding allylic or vinylic radical intermediates (Scheme 10).41 For a discussion on this study in previous reviews, see refs 2, 3, and 25.
Scheme 10.

Rearrangements among Allylic, Vinylic, and Bridged Bromine Radical Intermediates
In 1964, Abell et al. published studies on the gas-phase addition of HBr to propadiene, in which several reaction parameters were varied, including the reaction temperature (from 50 to 150 °C), the reactant ratio (propadiene/HBr from 10:1 to 1:10), and the pressure (from 10 to 80 mm).3,4,25,30 The major product was 2-bromopropene 20a, in accordance with the findings of Kovachic and Griesbaum. The reaction was first-order in HBr concentration but independent of propadiene concentration, with a positive activation energy (~4.5 kcal/mol). By utilizing the available experimental data and making approximations using a group contribution method, it was deduced that the addition of bromine radical to propadiene was exothermic. The authors proposed that the initial point of attack of the bromine atom was on the C1/C3 of the allene, followed by rearrangement to the more stable 2-bromoallyl radical due to the small reaction barrier. Reviews by Griesbaum2 and Taylor3 also discuss this study.
In 1966, Heiba et al. published studies on the kinetics of the free-radical addition of HBr to propadiene, which provided an explanation for the observed ratios of terminal versus central attack on the allene, as well as the effects of the reaction temperature and HBr/propadiene ratio on the product distribution.35 A mechanism was proposed that was consistent with these results (Scheme 11) based on the observation that the ratio of terminal:central addition products 20b:20a was constant when the HBr concentration was high but dropped when the HBr concentration was low. The dependence of the product ratio on the HBr concentration ruled out the mechanism initially proposed by Abell.27
Scheme 11.

Proposed Mechanism for the Free-Radical Addition of HBr to Propadiene
Kinetic studies also carried out by Heiba showed that the ratio of rate constants k1/k2 in Scheme 11 is equal to 2. Bromine addition to both the terminal and central carbons is exothermic; however, the addition to the terminal carbon is reversible, even at −78 °C, whereas the addition to the central allene carbon is irreversible. The observation that more 20a is produced as the temperature increases was explained by the higher activation energy of the reverse reaction of the Br radical addition (k−1) as compared with the hydrogen-transfer reaction rate k3. At constant temperature, the ratio 20b:20a depends only on the HBr concentration.
In 1969, Tien and Abell studied the relative energetics and the stereochemical outcome of free-radical additions of HBr to various methyl-substituted allenes in the gas phase.4,25,27 By varying the concentration of either the allene or HBr, a first-order rate dependence on HBr concentration was established. Activation energies were obtained for the free-radical HBr addition to the allenes in Scheme 12 by studying the reaction kinetics at temperatures of 40–120 °C. The tetramethyl-substituted allene 25 gave the lowest values, and propadiene 17c gave the highest values, albeit with <1 kcal/mol difference in energy. Competition experiments of allenes with limited amounts of HBr were conducted to compare the relative reactivities of allenes in Scheme 12. The relative reactivities were found to be 1:1.36:1.31:1.56:1.65 (17c/26/24/27/25) at 60 °C (the same trend holds at higher temperatures), with 25 displaying the fastest rate. The increased reactivity with increased methyl substitution was proposed to result from the additional stabilization of the 2-bromoallyl radical intermediates due to increased hyperconjugation. The major product for each allene has Br attached to the central allene carbon.
Scheme 12.

Free-Radical HBr Addition to Methyl-Substituted Allenes
There has been a recent surge in studies directed toward the synthetic applications of the addition of bromine radicals to allenes. Forty years after Moorthy and Devaprabhakara’s initial studies, the Ryu group reported their findings on the regioselective radical bromoallylation of allenes 28 for the synthesis of 2-bromosubstituted 1,5-dienes 30.42 By employing AIBN as the initiator and an electron-deficient allyl bromide 29 as the radical acceptor, regioselective radical bromination and new carbon–carbon bond formation were achieved in one pot (Scheme 13). This method tolerated monosubstituted, 1,1-disubstituted, and 1,3-disubstituted allenes, as well as functional groups that include OTIPS, NPhth, and CO2Et. Interestingly, allenes containing C1 CO2Et substituents exclusively gave the E alkene. Both 2-(bromomethyl)acrylate and 2-(bromomethyl)-acrylonitrile gave excellent yields.
Scheme 13.

Scope of Regioselective Radical Bromoallylation of Allenes 28
The Ryu group proposed these reactions proceed via a radical chain mechanism involving generation of 2-bromoallyl radical 31a,b, followed by an SH2′ reaction (SH = homolytic substitution) to yield the addition product and regenerate the bromine radical (Scheme 14). The E selectivity was explained by the steric repulsion between the R group and Br in the allyl radical precursor 31b required for formation of the Z product.
Scheme 14.

Proposed Mechanism of Regioselective Radical Bromoallylation of Allenes
In 2014, the Ryu group published a three-component coupling strategy to synthesize 2-bromo-1,7-dienes by adding an electron-deficient alkene coupling partner to a mixture of allene and allyl bromide43 (Scheme 15a). This one-pot method involves the initial attack of a bromine radical on the allene central carbon to form a 2-bromoallyl radical intermediate; this nucleophilic species selectively attacks the electron-deficient alkene to form an electrophilic alkyl radical. The alkyl radical then attacks the allyl bromide to furnish the product and a Br radical. Interestingly, when the alkylidenecyclopropane 32 was subjected to the reaction conditions, two products were isolated, the coupling product 34 and a methylenecyclopentane 35. The two products have a common precursor, 33, that can either attack the allyl bromide to form the major product 34 or undergo 5-exo cyclization, followed by β-fission, to form the minor product 35 (Scheme 15b).
Scheme 15.

Br-Radical-Mediated Three-Component Reaction for the Synthesis of 2-Bromo-1,7-dienes
In summary, the regioselectivity in the addition of halogen radicals (Cl, Br) to allenes strongly depends on the reaction conditions and the allene substitution pattern. In general, reactions of Cl and Br radicals with allenes yield the product resulting from the addition to the central carbon of the allene. In the case of both Cl and Br radicals, the temperature has been shown to affect both the reaction pathway and the regioselectivity. After the initial formation of a 2-chlorinated allylic radical, temperatures >800 K lead to the elimination of HCl (Scheme 3, vide infra). Decreasing the reaction temperature of the free-radical HBr addition to propadiene increases the amount of addition to the terminal carbon and decreases the addition to the central allene carbon. The identity of the radical initiator and a decreased concentration can also increase the amount of terminal addition product. In contrast, a higher allene/HBr ratio favors the central addition product. Multiple research groups also noted that the final product distribution does not reflect the initial regioselectivity in the addition of halogen radicals to the allene. Computational analysis on the addition of a Cl radical to propadiene at room temperature indicate that it can occur at both the C2 and the C1/C3 of the allene to give a mixture of radicals that interconvert via 1,2-halogen transfer. The radical addition of a Br radical to any carbon of an allene is exothermic; however, addition to the terminal allene carbon is reversible, whereas the addition to the central carbon is irreversible.
Recent studies have shifted focus from understanding the physical organic chemistry of the addition of halogen-based radicals to allenes to broadening the synthetic applications of these types of reactions. More complex allenes have been converted to motifs that include 1,2-dichlorides, 1,5-dienes, and 1,7-dienes under mild conditions, showcasing the great potential of the regioselective addition of halogen radicals to allenes to furnish stereodefined vinyl halides containing additional synthetic handles for further transformations.
2.2. Carbon-Centered Radicals
2.2.1. Perfluoroalkyl Radicals.
Haszeldine et al. reported the first addition of a carbon-centered radical to an allene in 1954.17 By irradiating a mixture of propadiene and CF3I with UV light, a high yield of a compound with the formula CF3(C3H4)I 36 was obtained. The spectral (UV and IR) and chemical evidence supported the CF3 radical addition to the C1/C3 carbons of propadiene (Scheme 16). The abstraction of the allenic hydrogen was not deemed to be a major pathway because only trace amounts of trifluoromethane were detected. Reviews by Griesbaum2 and Taylor3 also discuss this study.
Scheme 16.

Free-Radical Addition of CF3I to Propadiene
Szwarc et al. utilized the photolysis of hexafluoroazomethane to study the kinetics of the CF3 radical addition to olefins or allenes, as previously discussed in other reviews.3,25,44 In Scheme 17, k2 denotes the rate of CF3 radical addition to either olefins or allenes, whereas k1 is the rate of hydrogen abstraction of the iso-octane solvent by the CF3 radical. Higher ratios of k2/k1 indicate the higher reactivity of the CF3 radical toward the substrate. Because the k2/k1 of penta-2,3-diene (1,3-dimethylallene) was higher than that of, it was surmised that penta-2,3-diene is more reactive than propadiene toward the electrophilic CF3 radical due to the increased electron density on the terminal allene carbons induced by the additional methyl groups.
Scheme 17.

Ratio of Rate Constants k2/k1 at 65 °C for Allenes and CF3 Radical
Abell and Meunier studied the kinetics of the photoaddition of CF3I to propadiene by irradiating propadiene and CF3I at different ratios, reaction temperatures, total pressures, and light intensities.3,25,45 The sole product was 36, resulting from the addition of the CF3 radical to the terminal allene carbon, consistent with previous reports.46,47 On the basis of kinetic data and studies showing that the reaction rate is inversely dependent on temperature, the mechanism in Scheme 18 was proposed.
Scheme 18.

Proposed Mechanism of Free Radical Addition of CF3I to Propadiene
To the best of our knowledge, after Abell’s 1967 report, no published studies on the intermolecular addition of carbon-centered radicals to allenes appeared in the literature until after 2000. In 2001, Ogawa and Hirao46,47 described a photoinduced, regioselective radical addition of perfluoroalkyl iodides to various substituted allenes 38 to form monoadducts of the form 39. The perfluoroalkyl radical added to the terminal carbon of the allene to form a vinylic radical, which abstracts an iodine atom from the perfluoroalkyl iodide (Scheme 19). Trace amounts of products formed from the attack of the perfluoroalkyl radical on the central allene carbon were also observed for 1,3- and 1,1-disubstituted allenes. Nonpolar solvents, including toluene or halogen-containing solvents (C6F6, PhCF3, and CHCl3), are the most effective for this iodoperfluoroalkylation protocol. The E/Z ratios are likely controlled by the A1,3-strain present in the intermediate vinylic radical.
Scheme 19.

Photoinitiated Radical Additions of Perfluoroalkyliodides to Allenes
The Ma group described a Na2S2O4-initiated free-radical addition of perfluoroalkyl iodides to monosubstituted allenes 40 to form adducts 41 (Scheme 20).48,49 The regioselectivities mirror those in Scheme 19, with the perfluoroalkyl radical adding to the least-substituted terminal carbon and the iodine atom adding to the central carbon of the allene. Compared with the photoinduced formation of perfluoroalkyl radicals, the major isomer was Z, albeit with low selectivity.
Scheme 20.
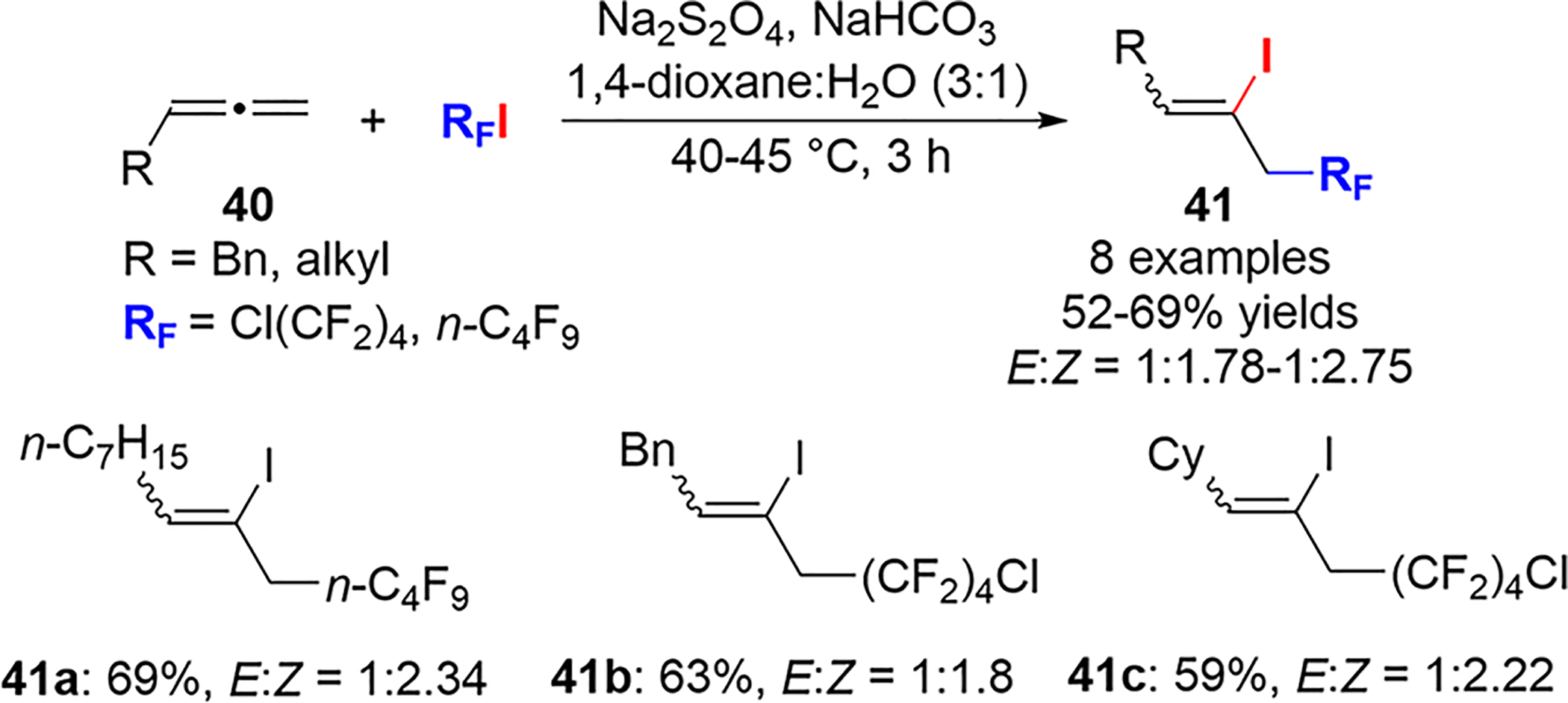
Na2S2O4-Initiated Free-Radical Addition of Perfluoroalkyliodides to Allenes
Liu et al. also described the use of Na2S2O4 to initiate a stereo- and regioselective radical coupling between perfluoroalkyl iodides and electron-deficient allenes 42 substituted with phosphine oxide, phosphonate, or ester groups50 (Scheme 21). Vinyl iodides of the E configuration 43 were exclusively obtained; however, when the allene was substituted with an alkyl group, mixtures of E/Z isomers were observed. The excellent stereoselectivity in some products was proposed to arise from the disfavored interaction between the vinyl radical electron and the lone pairs on the double-bonded oxygen in the intermediate alkene substituents P(O)Ph2, P(O)(OEt)2, and CO2Et (Scheme 21).
Scheme 21.

Na2S2O4-Initiated Radical Addition of Perfluoroalkyliodides to Allenes with Electron-Withdrawing Groups
In 2011, the Ma group described a similar protocol51 using Na2S2O4/Na2CO3 as the initiator for the radical addition of perfluoroalkyl iodides to allenes. The scope was expanded from their previous work48 (Scheme 22) to encompass 2,3-allenols of the form 44. In general, this protocol gives 3-iodo-4-perfluoroalkyl-substituted allylic alcohols 45 in moderate to good yields with moderate to good stereoselectivity. The 1,1-disubstituted allenes gave lower yields as compared with the monosubstituted allenes (Scheme 22), whereas tri- and tetrasubstituted allenols gave complicated mixtures and no evidence of the desired products.
Scheme 22.

Na2S2O4-Initiated Radical Addition of Perfluoroalkyliodides to 2,3-Allenols 44
In 2013, the Ma group described a Cu(I)-catalyzed synthesis of β-trifluoromethyl butenolides 47 via the oxytrifluoromethylation of 2,3-allenoic acids 46 in the presence of the Togni Reagent II (Scheme 23).52 In contrast with the previous examples described in this section, the CF3 radical is ultimately installed on the central carbon of the original allene carbon. The reaction required the presence of at least one aromatic group at C1 of 46; however, further alkyl and aryl substitution was tolerated at C1 and C3.
Scheme 23.

Cu(II)-Catalyzed formation of β-Trifluoromethyl Butenolides from 2,3-Allenoic Acids 46
To better understand the mechanism leading to this reversal in regioselectivity, the Ma group carried out the experiments described in Scheme 24. The addition of TEMPO to the reaction mixture (Scheme 24a) resulted in no butenolide product formation; only the TEMPO–CF3 adduct 49 was observed. In a second experiment, employing an enantioenriched allene 50 as the starting material yielded only the racemic product (Scheme 24b). Finally, when the carboxylic acid functionality was replaced with an ester, none of the desired product was produced, and 82% of the precursor 52 was recovered (Scheme 24c). The same observation was made when the phenyl substituent on the allene 52 was replaced with an n-hexyl group; the lack of reaction suggests that benzylic stabilization is key for the successful addition of the CF3 radical to the allene.
Scheme 24.

Evidence Supporting the Presence of a CF3 Radical Intermediate
On the basis of these experimental results, Ma proposed two possible mechanisms for Cu(II)-catalyzed radical cyclizations of 2,3-allenoic acids (Scheme 25). In Pathway I, the activated catalyst 53 and the Togni Reagent II react to form the radical intermediate 54, which releases a CF3 radical and 55. The CF3 radical attacks the central carbon of the allene 46 to form an allylic radical intermediate, which is then oxidized by 55 to generate 56. The subsequent cyclization of 56 forms the product 47 and 2-iodobenzoic acid. The alternative Pathway II involves the coordination of 46 and 55 to form a complex 57, which is then attacked by the CF3 radical to form 58. The reductive elimination of 58 furnishes the final product.
Scheme 25.

Cu(II)-Catalyzed Formation of β-Trifluoromethyl Butenolides from 2,3-Allenoic Acids
Liu et al. developed a Cu(I)-catalyzed, intermolecular, regioselective oxytrifluoromethylation of allenes 59 that also employs the Togni Reagent II53 as the source of a trifluoromethyl radical (Scheme 26). The majority of the substrates are monosubstituted allenes, which give moderate to high yield of 60; similar to Ma’s work, the trifluoromethyl group is installed at the central C2 carbon of the allene precursor. In contrast, a more sterically hindered 1,3-disubstituted allene gave only 14% yield.
Scheme 26.

Cu(I)-Catalyzed Synthesis of Vinyl Trifluorides from Allenes with Togni Reagent II
To probe the mechanism of this transformation, TEMPO was added to the reaction mixture, resulting in the formation of 46% yield of a TEMPO–CF3 adduct. The addition of BHT (butylated hydroxytoluene) as the radical scavenger inhibited the reaction and decreased the overall yield by 20%. On the basis of these experimental results, which suggest the intermediacy of radical species, two reaction pathways were proposed (Scheme 27). In Pathway I, an interaction between Togni II and CuCl generates a CF3 radical, which attacks the central carbon of allene 59 to form the allylic radical intermediate 62. Intermediate 63, formed from 62, undergoes reductive elimination to give the desired product 60. Alternatively, Pathway II involves the cationic species 64, generated from Togni II and CuCl. This is proposed to react with allene 59 to give an iodine(III) intermediate 65. The attack of the carbonyl oxygen on the terminal allene carbon in the more stable allyl cation 66 eventually affords the desired product 60. In both mechanisms, the heteroatom linkage (O, N, S) on the allene 59 is important for the effective stabilization of proposed intermediates in pathway I (62) and pathway II (66).
Scheme 27.

Cu(I)-Catalyzed Synthesis of Vinyl Trifluorides from Allenes with Togni Reagent II
The Wang and Liu groups reported Cu(I)-catalyzed syntheses of CF3-containing allyl azides and thiocyanates from 1,1-disubstituted aryl allenes 67 in the presence of Togni’s Reagent.54 This method afforded the trifluoromethylazidation products 68 with excellent regioselectivity. In these examples, the CF3 also exclusively adds to the central allene carbon and furnishes good stereoselectivity for the E isomer. However, substrates containing bulky R groups or ortho-substituted aryl rings gave lower E/Z selectivities, indicating that the chemistry is sensitive to steric hindrance. The scope was also limited because monosubstituted, 1,3-disubstituted, and 1,1-dialkyl substituted allenes result in low yield and with poor regioselectivity (Scheme 28).
Scheme 28.

Cu(I)-Catalyzed Trifluoromethylazidation of Allenes With Togni’s Reagent
The trifluoromethylazidation protocol was extended to trifluoromethylthiocyanation (Scheme 29) to provide the desired products 70 in good to excellent yield. The E/Z selectivity was lower as compared to trifluoromethylazidation, although the major isomer was also of the E configuration.
Scheme 29.
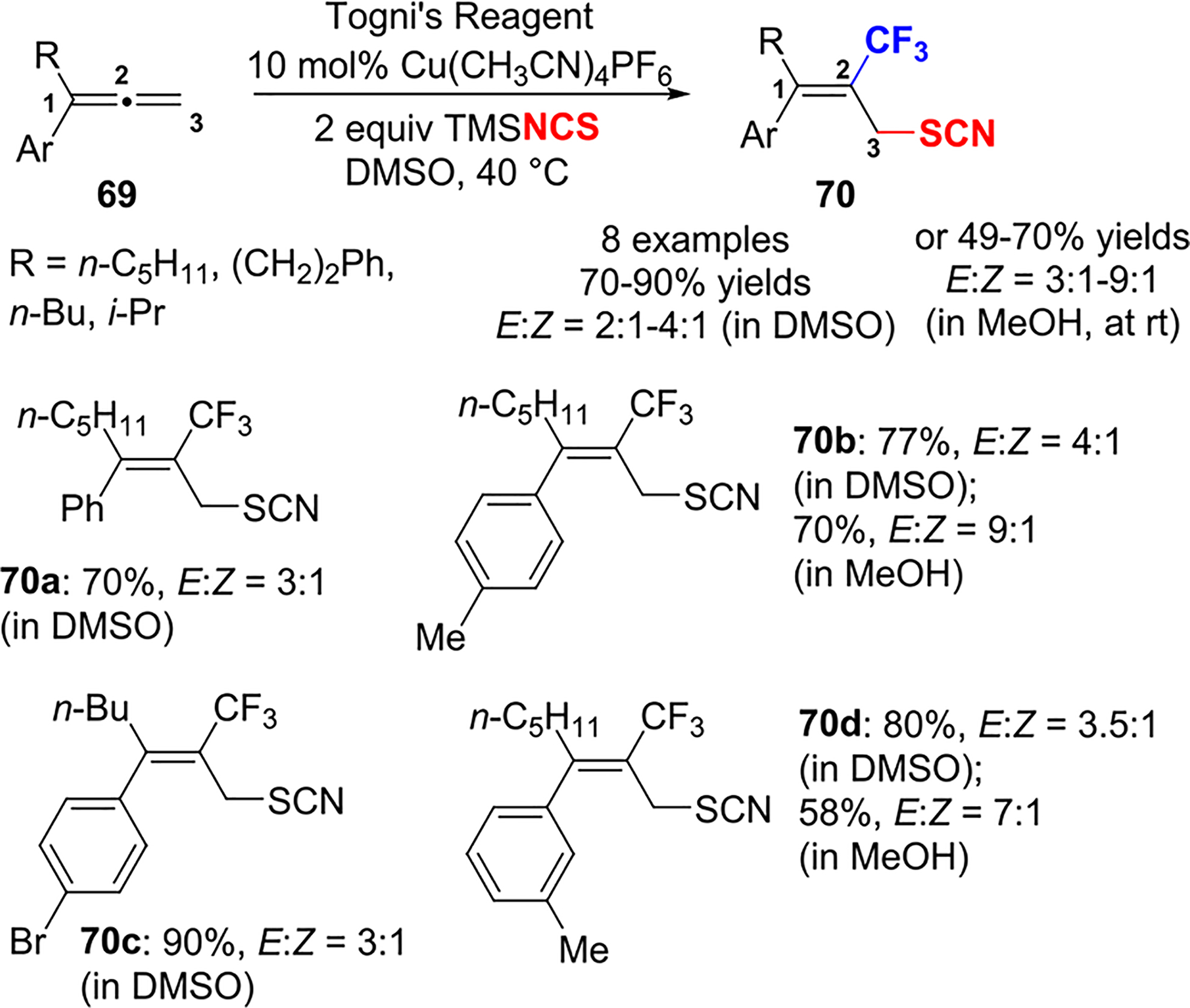
Cu(I)-Catalyzed Trifluoromethylthiocyanation of Allenes with Togni’s Reagent
On the basis of the experimental results, a mechanism was proposed for the trifluoromethylthiocyanation (Scheme 30). The CF3 radical is generated from 71, which itself forms from the attack of Togni’s reagent by TMSNu. The addition of the CF3 radical to allene 67/69 affords allylic radical 72, which is proposed to be more stable than its Z isomer due to the steric hindrance between the aryl group and the adjacent CF3 group. A nucleophilic radical, produced from the reduction of the corresponding anionic species by Cu(II), is proposed to couple to the allyl radical to form the desired product 68/70. Pathway II involves generation of the Cu(III) intermediate 73 from a combination of 72, Cu(II), and the nucleophilic anion. The reductive elimination of 73 affords the final product.
Scheme 30.

Proposed Pathway for Cu(I)-Catalyzed Trifluoromethylazidation and Trifluoromethylthiocyanation of 1,1-Disubstituted Aryl Allenes with Togni’s reagent
In 2017, the Ma group reported a Cu(II)-catalyzed formation of CF3-substituted vinylic cyclopropanes 75 from 2,3-butadienyl malonates 74 in the presence of Togni’s reagent II (Scheme 31).55 The CF3 radical, generated in situ, selectively attacks the central carbon of the allene. High regioselectivity was achieved by employing 1,1,3-trisubstituted allenes to prevent CF3 radical attack on the terminal carbons of the allene.
Scheme 31.
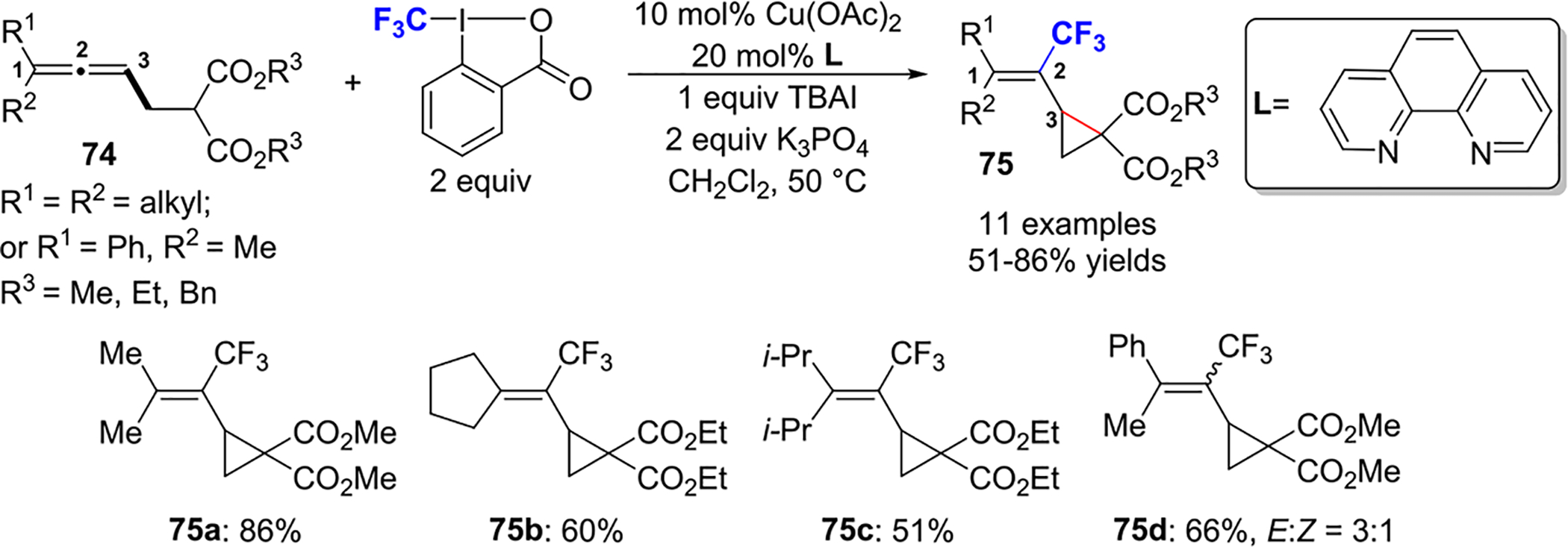
Cu(II)-Catalyzed Formation of CF3-Substituted Vinylic Cyclopropanes 75 from 2,3-Butadienyl Malonates 74 with Togni Reagent II
Radical scavengers, including 1,4-dinitrobenzene and benzoquinone, either suppressed or completely shut down the reaction. TEMPO–CF3 and BHT–CF3 adducts were formed when TEMPO or BHT was added to the reaction mixture, supporting the presence of CF3 radicals. Additionally, 31% of byproduct 76 was isolated. Control experiments showed that 76 does not form in the absence of the Cu(II) catalyst. On the basis of the experimental results, the Ma group proposed the mechanism in Scheme 32. The key step includes the production of a CF3 radical by release from the radical intermediate 77 (generated from the reaction between active catalyst 78 and Togni Reagent II).
Scheme 32.

Proposed Mechanism of Cu(II)-Catalyzed Formation of CF3-Substituted Vinylic Cyclopropanes 75 from 2,3-Butadienyl Malonates 74 with Togni Reagent II
In 2017, the Akita group56 described a Ru(II)-catalyzed oxytrifluoromethylation of allenes 79 to form 2-CF3-substituted allyl acetates 80 in the presence of the Umemoto reagent, which functions as an electrophilic source of CF3. Under irradiation with blue LED, CF3-containing allyl acetates are synthesized in a regioselective and stereoselective manner from 1,1-di-, 1,3-di-, and 1,1,3-trisubstituted allenes (Scheme 33). Monosubstituted allenes give low regio- and stereoselectivities, whereas tetrasubstituted allenes are not compatible under the reaction conditions. Alkyl-substituted allenes (such as cyclohexylallene) give unidentifiable mixtures of products.
Scheme 33.
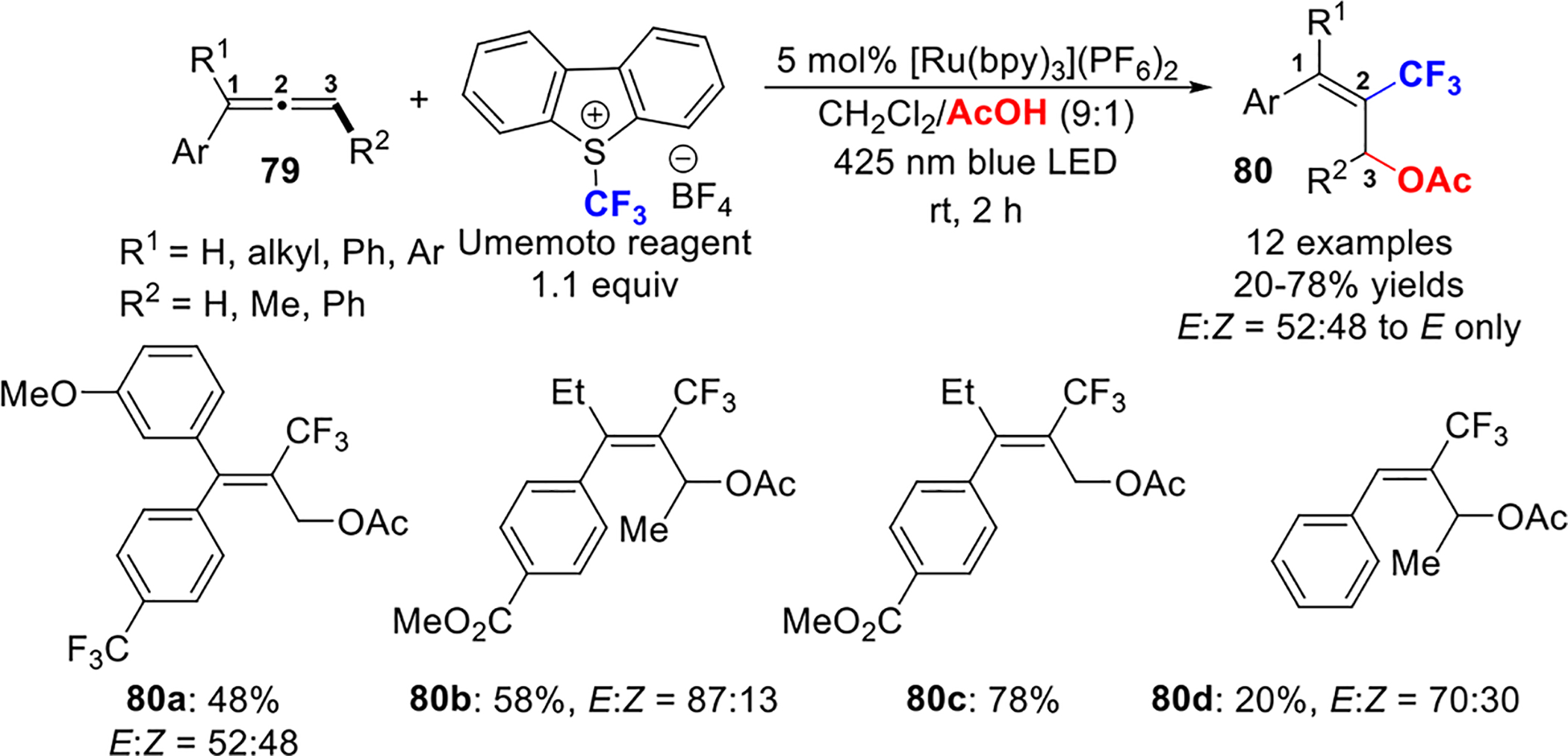
Ru(II)-Catalyzed Formation of CF3-Substituted Allyl Acetates from Allenes
On the basis of the experimental results, a mechanism was proposed (Scheme 34). Upon irradiation, the ground-state Ru(II) catalyst is promoted to the excited state, which reduces the Umemoto reagent and generates a CF3 radical. The addition of the radical to the allene furnishes allylic radical 81, which is oxidized by Ru(III) to regenerate Ru(II) and form allyl cation 82. The thermodynamically more stable conformation of 82 avoids the steric clashing between CF3 and aryl groups. Nucleophilic attack of AcOH on 82 yields the product 80.
Scheme 34.

Ru(II)-Catalyzed Formation of CF3-Substituted Allyl Acetates from Allenes/Umemoto Reagent
The synthesis of α-CF3 acroleins 84 from the Ag(I)-catalyzed regioselective trifluoromethylation of allenes 83 in the presence of the Langlois reagent was described by Zanoni and Maiti in 2018 (Scheme 35).57
Scheme 35.
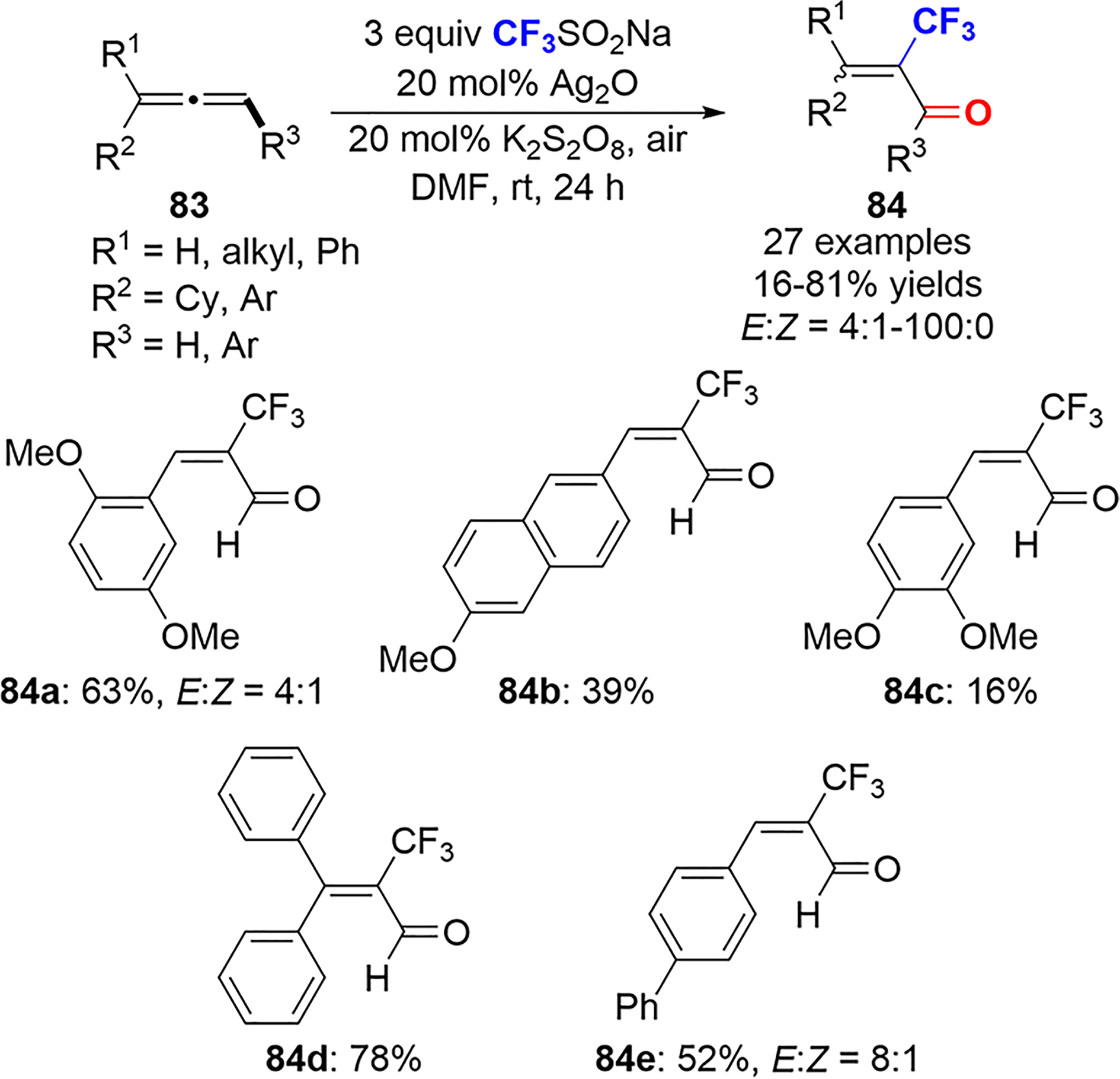
Ag(I)-Catalyzed Synthesis of α-CF3 Acroleins from Allenes with Langlois Reagent
Three key experiments helped to show the importance of air for a successful reaction, which requires the presence of a CF3 radical intermediate. First, the addition of TEMPO to the reaction mixture completely inhibits the reaction, with a TEMPO–CF3 adduct detected by GC–MS. When the reaction is conducted under N2, dimer 85 is isolated in 90% yield; however, when the reaction is carried out under O2, the oxidation of the aryl allene to the corresponding aryl aldehyde 86 (R = H) is observed (Scheme 36a), leading to the proposed mechanism shown in Scheme 36b. Ag(I) is reduced by persulfate to Ag(0), which reduces the Langlois reagent to generate the CF3 radical. The addition of the CF3 radical to the allene forms an allylic radical intermediate 87; oxidation at C3 furnishes the carbonyl product. The authors propose that the spin density of the allene carbons in the allylic radical intermediate determines the stability and reactivity of the corresponding radical, with a lower spin density equating to a more reactive radical species. Benzylic conjugation on C1 increases the spin density, whereas computational analysis determined that C3 always displays a lower spin density, regardless of the identity of the aryl group, leading to the observed oxidation at this site.
Scheme 36.

Experimental Evidence and Proposed Mechanism for Ag(I)-Catalyzed Formation of α-CF3 Acroleins from Allenes with Langlois Reagent
In conclusion, both the source of the perfluoroalkyl radical and the substitution pattern of the allenes greatly affect the regioselectivity and chemoselectivity of the RF radical addition to the allene precursor. Using perfluoroalkyl iodides (RFI) as the radical source favors the addition of the RF radical to the terminal allene carbon. (See Schemes 16 and 19–22.) In addition, the addition of RFI is generally site-selective, with RFI added to the less substituted allene double bond, regardless of the allene substitution pattern. (See Schemes 19–22.) Stereoselectivity in these reactions, irrespective of the RF radical source, has been shown to be affected by the presence of carbonyl-containing substituents on the allene, giving the E isomer as the only product. (See Schemes 21 and 35.)
Recently, the transition-metal-catalyzed incorporation of CF3 groups into allene substrates has attracted significant attention from multiple research groups. In these trifluoromethylation reactions, transition metals that include Cu(I), Cu(II), Ag(I), and Ru(II) have been used in conjunction with common trifluoromethylation reagents such as Togni’s Reagent, the Togni Reagent II, the Umemoto reagent, and the Langlois reagent. In contrast with the use of perfluoroalkyl iodides as the radical source, these reactions showed a drastic change in regioselectivity, with the CF3 radical exclusively adding to the central allene carbon. However, this regioselectivity could be achieved only via substrate control. For example, the reaction of Cu(I)/Togni II and allenes proceeded regioselectively only with aryl or heteroatom-substituted allenes. Trifluoromethylation carried out with Umemoto/Ru(II) or Langlois/Ag(I) proceeded regioselectively only with aryl allene substrates. This unique regioselectivity was largely attributed to the stabilization of the allylic radical or cationic intermediate enabled by the presence of either an aryl group or a heteroatom substituent; alternatively, the CF3 radical attack on the terminal allene carbon could be blocked by increasing the steric hindrance through the installation of alkyl substituents. Future work in this area should focus on methods to achieve catalyst control over the regioselectivity of CF3 radical addition to allenes without having to rely solely on substrate control.
2.2.2. Other Carbon-Centered Radicals.
Szwarc et al. studied the relative reactivities of methyl radicals in addition to isolated, conjugated, and cumulated dienes at various temperatures.58 In the presence of decomposing acetyl peroxide, the ratio of k2/k1 was measured, where k2 denotes the rate of a CH3 radical addition to the diene and k1 denotes the rate of hydrogen abstraction of the solvent molecule (iso-octane) (Scheme 37). A larger ratio of k2/k1 indicates a higher rate of radical addition under the same reaction conditions.
Scheme 37.
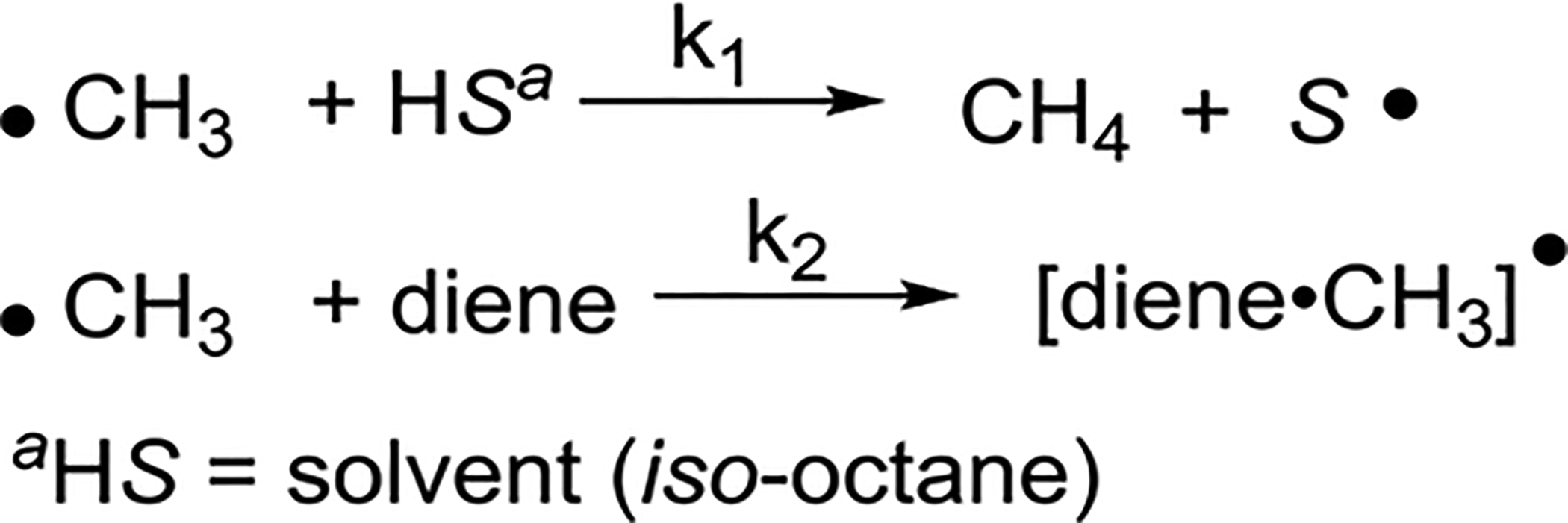
Measuring the Ratio of Rate Constants k2/k1
Selected values of k2/k1 for the addition of a CH3 radical to five allenes are presented in Scheme 38. Compared with k2/k1 values for the corresponding isomers of isolated dienes or conjugated dienes (e.g., 1,4-pentadiene = 60; trans-1,3-pentadiene ≈ 840 at 65 °C), the reactivity of allenes toward methyl radicals is decreased. Additionally, alkyl or phenyl substitution on the C1 and C3 of the allene precursors did not affect the reactivity, in contrast with the “steric blocking effect” present in noncumulated dienes. These observations led Szwarc to propose that the initial site of attack for the CH3 radical is on C2. Interestingly, the presence of phenyl groups in 89 does not obstruct the attack of the CH3 radical and increases the reactivity three-fold, as compared with propadiene 17c. In considering the reported attack of CF3 radicals on the terminal carbons of allenes,58 Szwarc reasoned that the electrophilic CF3 radical should attack the more electron-rich C1 and C3 allene carbons, whereas the nucleophilic methyl radical should favor attack at C2 due to the better matching of the electronics of the allene and the radical. By comparing the frequency factor A and the activation energies of reactions between CH3 radicals and allenes as well as noncumulated dienes, the decreased reactivity of allenes was proposed to arise from a low A factor, as opposed to a higher activation energy.
Scheme 38.

Selected Values of k2/k1 of Allenes at 65 °C
Experimental evidence45 that a CH3 radical adds to the terminal carbon of propadiene was presented by the Abell group in 1967. After irradiating a mixture of CH3I and propadiene in the gas phase at 35 and 75 °C, 90 was isolated as the only product (Scheme 39). This result refuted previous predictions that a methyl radical would only attack the central allene carbon, indicating that the polar effects of the radicals do not play a role in determining the regioselectivity of the reaction.
Scheme 39.

Free-Radical Addition of CH3I to Propadiene
In 1972, Caserio and coworkers investigated the factors governing the regioselectivity of the free-radical addition of methane derivatives, as briefly described in a previous review by Hartung and Kopf,25,29 to penta-2,3-diene 27, 2-methyl-penta-2,3-diene 91, and cyclonona-1,2-diene 101. The methane derivatives studied included CF3I, CH3I, and CCl3Br. The yields were not provided, but the ratios of the isolated adducts and identifiable byproducts are given in Scheme 40.
Scheme 40.

Free-Radical Additions of CF3I, CH3I, and CCl3Br to Allenes
Scheme 40a,b shows that the ground-state electron distribution of the allenes does not dictate the regioselectivity of the attacking radicals, as a more electrophilic CF3 radical does not show a stronger preference for terminal attack when compared with the nucleophilic CH3 radical (Scheme 40a). Steric factors, on the contrary, are thought to play a role in regioselectivity, as the larger CCl3 radical favors attack at the central carbon in allenes that are more heavily substituted (such as 96 as compared with 27) (Schemes 40b,c). The irreversible addition of a CCl3 radical to an allene is shown by the recovery of the enantiomerically enriched (S)-(+)-27. A small net rotation is observed in 94 and 95, indicating that the addition of the CCl3 radical is not stereospecific. The reaction of cyclic allene 101 with CCl3Br proceeds in a regiospecific fashion, with only 102 being observed.
In 1981, the Boldt group published their findings25,59 on the free-radical addition of bromomalonitrile to allenes with alkyl substitution. They envisioned two pathways to the addition product, where the attack of the malonitrile radical occurs at C2 or at C1/C3 of the allene precursor (Scheme 41). Selected experimental results in Scheme 41 show that allenes with more alkyl substituents (such as 24 and 96) favor the attack of the malonitrile radical at the central allene carbon.
Scheme 41.
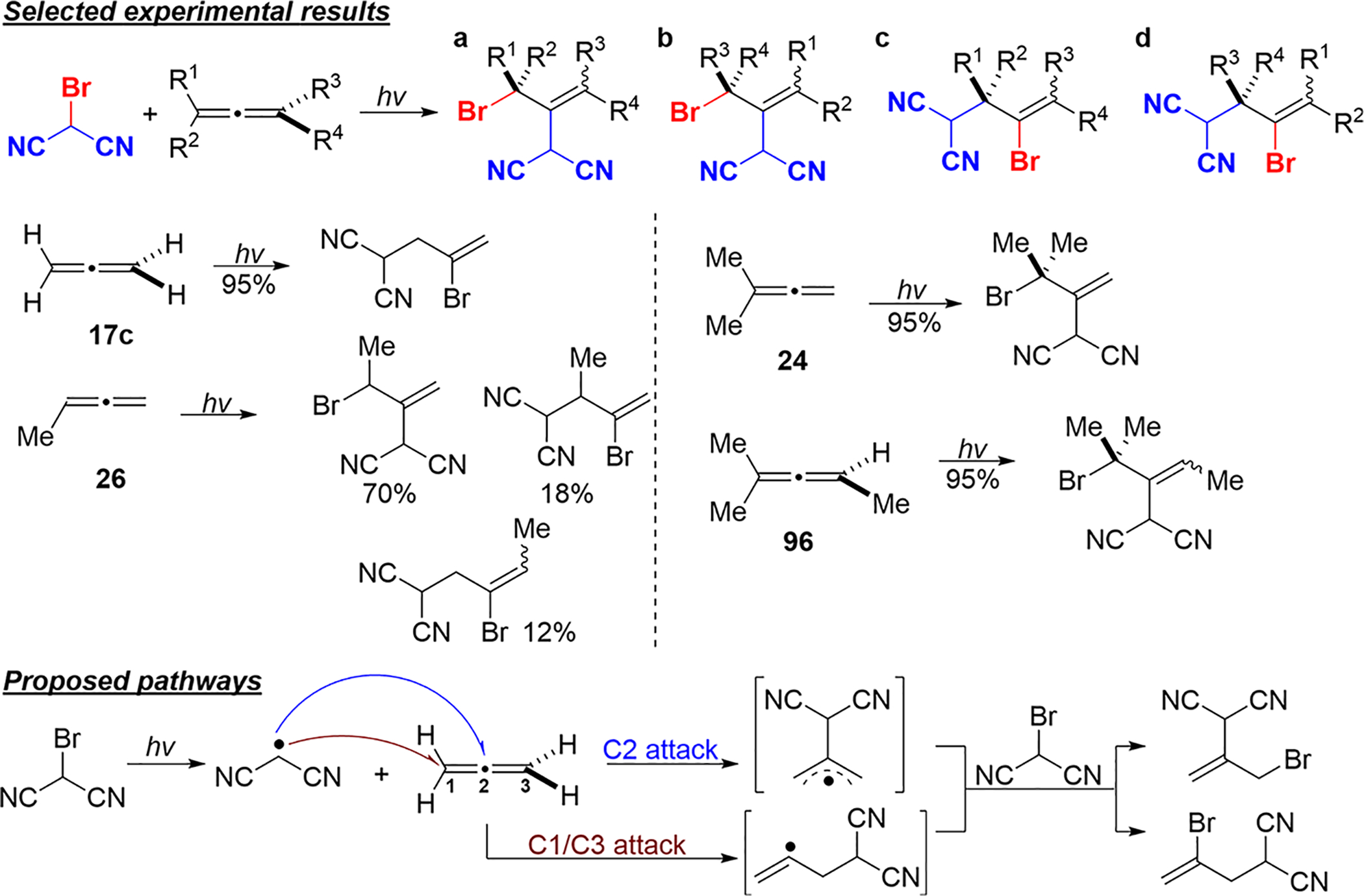
Proposed Pathways of Free-Radical Addition of Bromomalonitrile to Propadiene
In 2003, Reissig and coworkers reported a SmI2-induced ketyl–allene coupling reaction to yield 4-hydroxy-1-enol ethers 104 (Scheme 42).60 Various carbonyl compounds 103, including cyclic and acyclic ketones, as well as aldehydes, were coupled to methoxyallene to furnish 104 in moderate to good yield. The key intermediate in this reaction is proposed to be the ketyl radical anion 105, which is generated by electron transfer between SmI2 and the carbonyl group. The regioselective addition of the ketyl species to the terminal allene carbon furnishes the vinyl radical intermediate 106. This regioselectivity is attributed to the match between the more electrophilic character of the distal allene double bond and the nucleophilic samarium ketyl.61,62
Scheme 42.
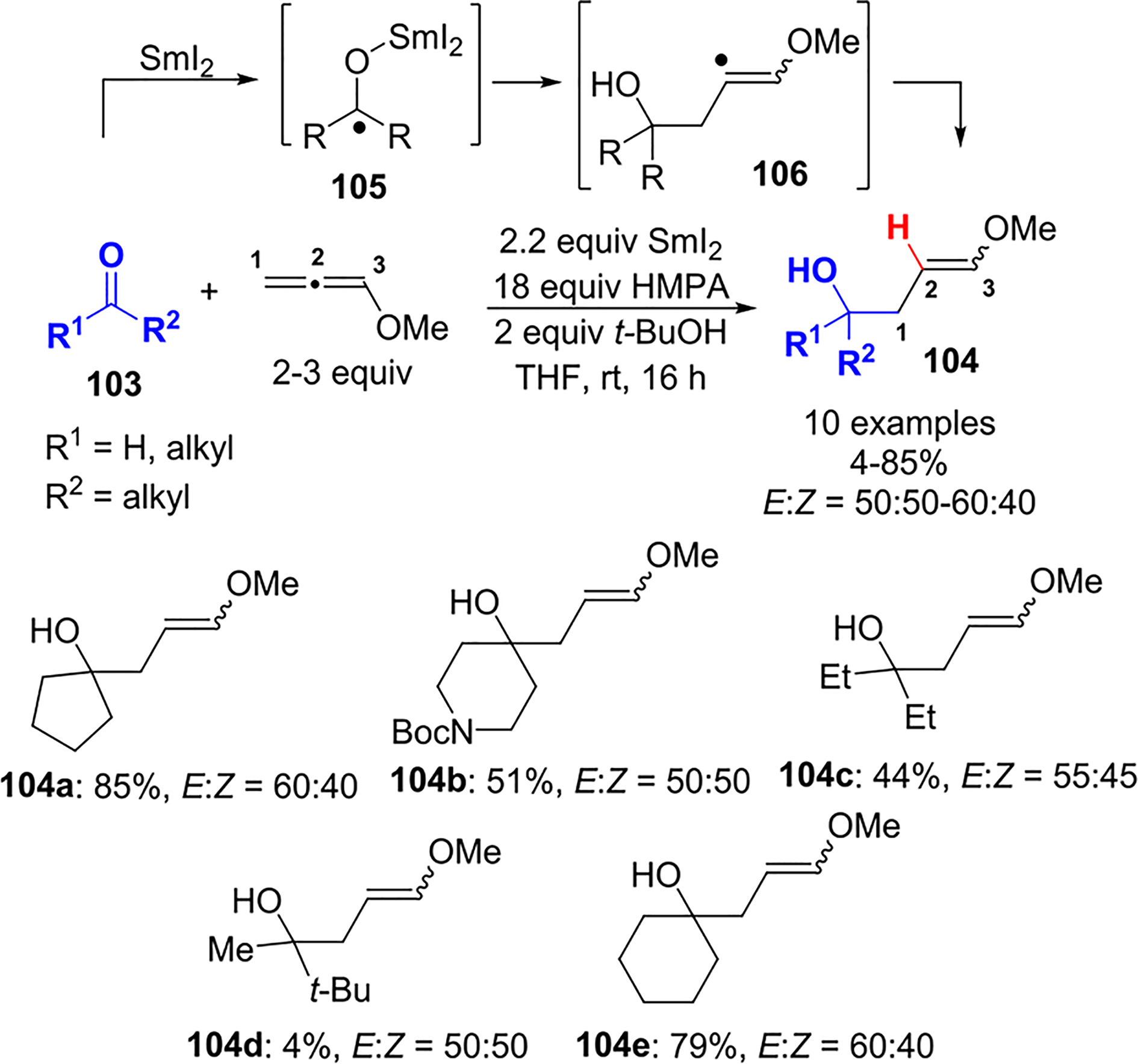
Free-Radical Addition of Samarium Ketyl to Methoxyallene
In 2007, the Huang group developed a Mn(OAc)3-mediated oxidative free-radical addition of dimethyl malonate or ethyl cyanoacetate to allenes 107 to furnish furan-2(5H)-ones or dimethyl 2-(2-oxoethylidene)malonates 108 (Scheme 43) as the major products.63
Scheme 43.

Mn(OAc)3-Mediated Oxidative Free-Radical Reaction of Allenes with Malonate and Cyanoacetate
In the presence of 4.5 equiv of Mn(OAc)3·2H2O, as opposed to 2.5 equiv, the allene 109 was converted to the noncyclized product 110 (Scheme 44).
Scheme 44.

Mn(OAc)3-Mediated Oxidative Free-Radical Reaction of Aryl-Substituted Allenes with Malonates
On the basis of experimental results and previous literature reports, a mechanism for the Mn-mediated cyclization was proposed, as shown in Scheme 45. Mn(OAc)3 oxidizes dimethyl malonate or cyanoacetate to the corresponding alkyl radical 111. The intermediate 111 adds the allene at C2 to form resonance-stabilized 112, which upon oxidation by Mn(III) forms a resonance-stabilized carbocation 113. When R = alkyl, the cyclization is followed by the reaction with H2O and a 1,3-H shift to furnish the product 108. However, when R = H, H2O attacks the benzylic carbocation to form 114, which is oxidized by Mn(III) to the corresponding carbonyl group. A final 1,3-H shift of the carbonyl yields the product 110.
Scheme 45.

Proposed Mechanism of Mn(OAc)3-Mediated Oxidative Free-Radical Reaction of Allenes with Dimethyl Malonate and Cyanoacetate
In 2013, Yoshida et al. developed a regio- and stereoselective synthesis of functionalized hydroxyhexa-hydrocyclopenta[b]-furancarboxylates from alkenes.64 The method was extended to a single example of an aryl-substituted allene 115 (Scheme 46). The diastereoselectivity was proposed to result from the steric hindrance between the aryl and ester group.
Scheme 46.

Oxidative Radical Cyclization of Cyclic β-Keto Esters with Allene 115
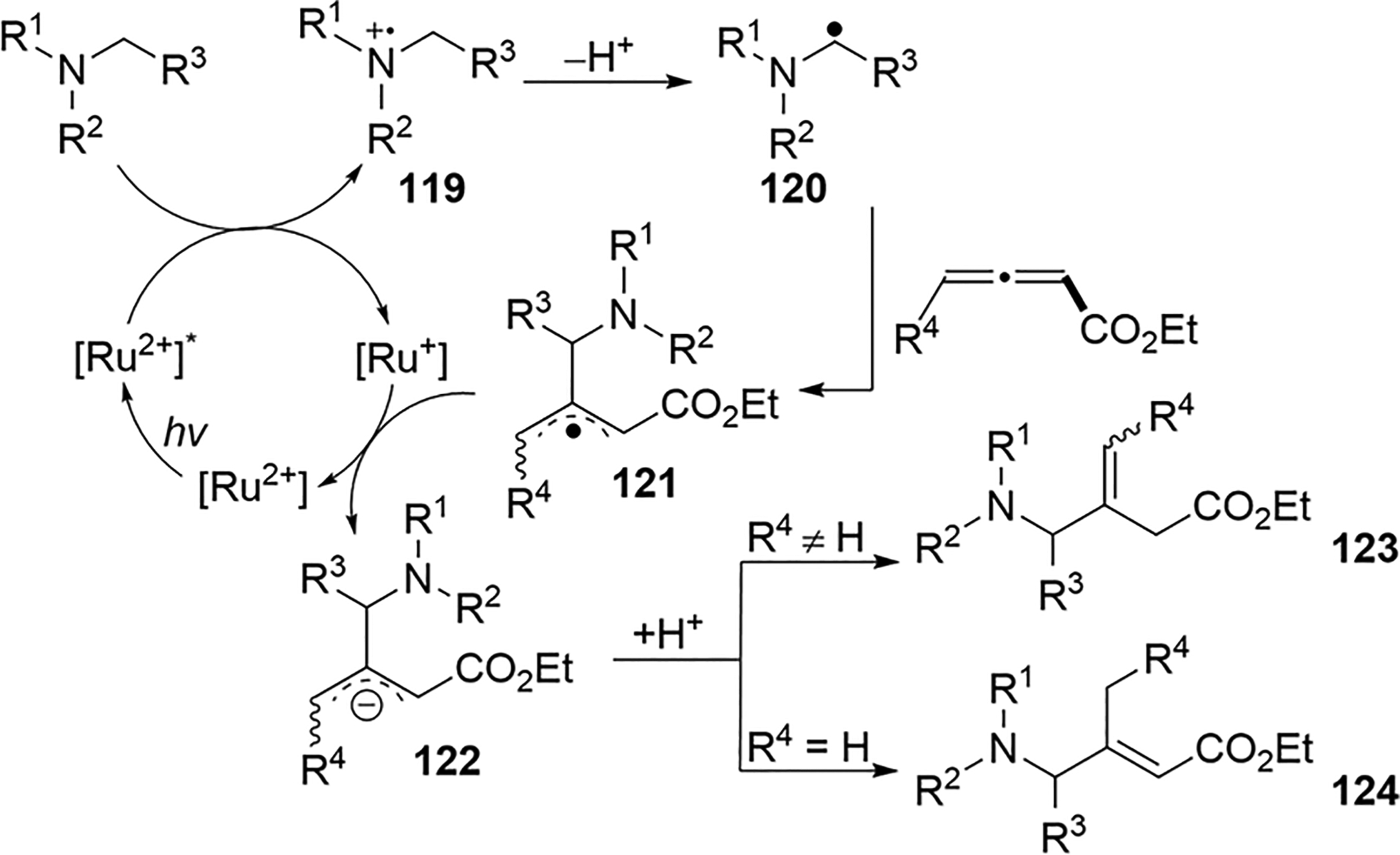
In 2015, the Xu and Li groups reported a photo-redoxcatalyzed addition of an α-aminoalkyl radical to 2,3-allenoates 116.65 In the presence of a catalytic amount of Ru(bpy)3(BF4)2, the α-aminoalkyl radical is regioselectively added to the central carbon of the 2,3-allenoate to produce unsaturated γ-aminobutyric ester derivatives in moderate yield (Scheme 47). The electron-deficient nature of 2,3-allenoates enables them to function as efficient coupling partners in reactions with nucleophilic α-aminoalkyl radicals. It is worth noting that only 117 is obtained when at least one substituent is present on the C1 of the allene. The reported yields are low (<35%) for 1,1-disubstituted-2,3-allenoates, likely due to steric effects. On the basis of experimental results and literature precedent, a mechanism was proposed for the radical coupling (Scheme 48). The excited Ru(II) catalyst oxidizes the amine to the corresponding radical cation 119, which is deprotonated by K2HPO4·3H2O to form an α-aminoalkyl radical 120. The addition of 120 to the central allene carbon generates an allylic radical 121, which is reduced by Ru(I) to the corresponding allylic anion 122. The protonation of the anion affords the final products 123 and 124.
Scheme 47.

Ru(II)-Catalyzed α-Aminoalkyl Radical Addition to 2,3-Allenoates 116
Scheme 48.
Proposed Mechanism of Ru(II)-Catalyzed α-Aminoalkyl Radical Addition to 2,3-Allenoates
Overall, general rules to predict the regio-, chemo-, and stereoselectivities of additions of carbon-centered radicals to allenes are difficult to ascertain due to the greater diversity of carbon versus halogen radicals and the relatively limited scope of allenes that have been studied. However, some conclusions can be drawn as to the regioselectivity in certain cases. First, the sterics of the attacking radical play an important role in directing the regioselectivity. With the same methylated allene substrate, the larger CCl3 radical favors attack on C2, whereas a smaller CF3 radical prefers to attack at C1/C3. In addition, allene sterics also affect the regioselectivity in the attack on a malonitrile radical. Allenes containing more methyl substituents divert the regioselectivity of malonitrile radical to the less hindered C2 of the highly substituted allene. Future efforts should focus on examining these reactivity patterns with a larger range of carbon-centered radicals and allenes to provide a more comprehensive picture of the selectivity trends that will improve the synthetic utility of this chemistry.
2.3. Nitrogen-, Phosphorus-, Oxygen-, and Sulfur-Centered Radicals
2.3.1. Nitrogen Radicals.
Research on the addition of nitrogen-centered radicals to allenes has been explored with aminium, nitro, and N-sulfonimide radicals. Among these, the nitro radical has been the most heavily studied due to its facile conversion to other functional groups and its potential to access useful building blocks such as nitroolefins, which are widely used in a biological or pharmaceutical context.66–68 These reactions proceed in a regioselective fashion to attack at C2 of allenes that contain alkyl, aryl, or silyl substitution.
In 1967, Neale published a study on the free-radical addition of dialkyl N-chloramines to unsaturated hydrocarbons, including 1,3-dienes, terminal olefins, acetylenes, and allenes.69 The irradiation or addition of a Fe(II) catalyst to a mixture of dialkyl N-chloramine and allene 125 in sulfuric and acetic acids generated the corresponding 1:1 chloramine adducts 126 via a free-radical chain mechanism (Scheme 49). The major product was formed from the initial attack of an aminium radical on the terminal allene carbon.
Scheme 49.
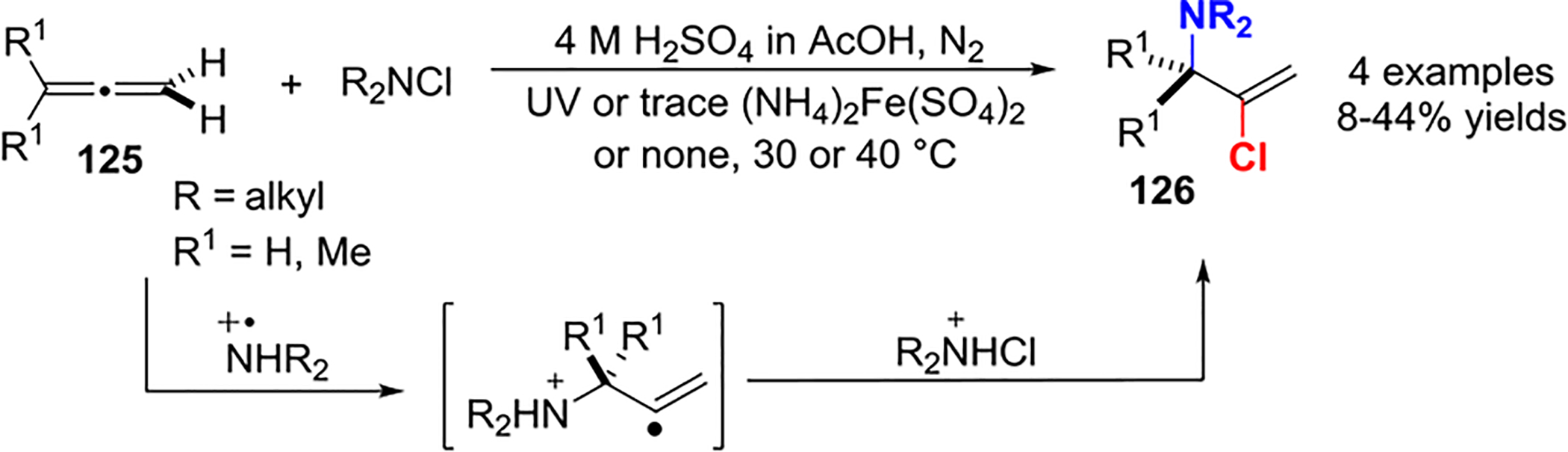
Free-Radical Addition of Dialkyl N-Chloramines to Allenes 125
Almost 50 years after Neale’s publication on the intermolecular aminium radical attack on allenes, the Lee group developed a regioselective synthesis of α-nitro-α,β-unsaturated silyl oximes 128 by employing NaNO2 and AcOH as a nitrogen dioxide radical source and silyl allenes 127 as radical acceptors70 (Scheme 50). After testing various 1,1,3-trisubstituted silyl allenes, it was determined that the size of the silyl group did not affect the yields or E/Z ratios of 128. However, replacing a silyl with a tert-butyl group gave decomposition, indicating the importance of the silyl group to the success of the reaction. Unsaturated functionalities, including alkenes or aromatic rings, were compatible with the method, suggesting that the silyl allenyl moiety is more reactive toward the NO2 radical. No reaction was noted with 1,3-disubstituted silyl allenes, possibly due to the formation of an unstable secondary carbon-centered radical 130 (R1 = H). The treatment of nitroalkene derivatives 128 with TBAF initiates 5-endo-dig cyclization onto the alkene carbon; the subsequent Nef reaction71,72 gives isooxazolidinones 129 in moderate yield.
Scheme 50.

Nitro Radical Addition to Silyl Allenes and Conversion to Isooxazolidinone Derivatives
In addition to the protocol in Scheme 50, the Lee group also described the chloronitration of allenes 131 employing Fe(NO3)3 as a nitrite source (Scheme 51). It is noteworthy that allene 131b, which lacks a silyl substituent, is also a viable substrate for Fe(NO3)3 nitration; however, silyl allenes do give better regioselectivity, with 132 noted as the major regioisomer.
Scheme 51.
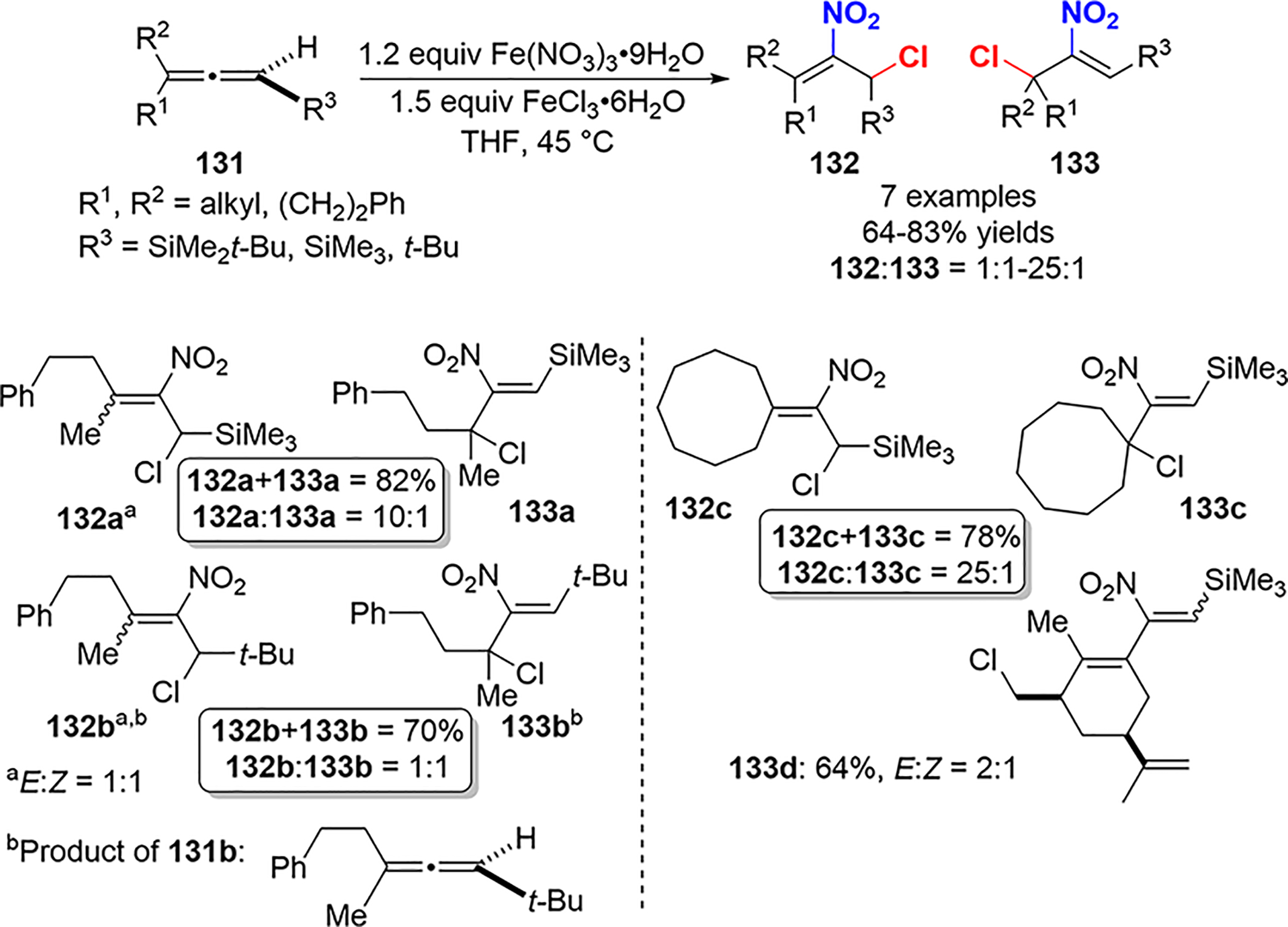
Chloronitration of Allenes 131 Using Fe(NO3)3 and FeCl3
On the basis of experimental results, the Lee group proposed the mechanism shown in Scheme 52. The nitrogen dioxyl radical is generated from heating a mixture of NaNO2 and AcOH or by combining Fe(NO3)3 and FeCl3. The addition of the NO2 radical to allene 127 gives the allylic radical intermediate 134, which is either trapped by the nitroso radical to form 135 or reacts with FeCl3 via electron transfer to form the regioisomers 132 and 133. The subsequent tautomerization of 135 gives 128.
Scheme 52.

Proposed Mechanism for Nitroso Nitration and Chloronitration of Silyl Allenes
In 2014, Ma reported a regio- and stereoselective nitrooxoamination of allenes 137 with AgNO2 and TEMPO.73 Various monosubstituted allenes gave nitroalkenes of the form 138, with the E configuration as the major product (Scheme 53). In general, allenes substituted with aryl and heterocyclic groups gave higher yields compared with alkyl substitution. This chemistry shows mechanistic similarities to Lee’s method (Scheme 52), where a nitrogen dioxyl radical adds to the central allene carbon to form an allylic radical intermediate.
Scheme 53.

Regio- and Stereoselective Nitro-Oxoamination of Allenes 137
In 2015, Zhang published a regioselective synthesis of allenamides and fluorinated tetrasubstituted alkenes from allenes and NFSI or its derivatives.74 A Cu(I) catalyst supported by a neocuproine (NC) ligand furnished allenamides from 1,1- and 1,3-disubstituted aryl allenes (Scheme 54a). However, 1-aryl-substituted and 1,1-dialkyl-substituted allenes were not viable substrates. Various aryl-substituted N–F reagents were also well-tolerated in this protocol.
Scheme 54.
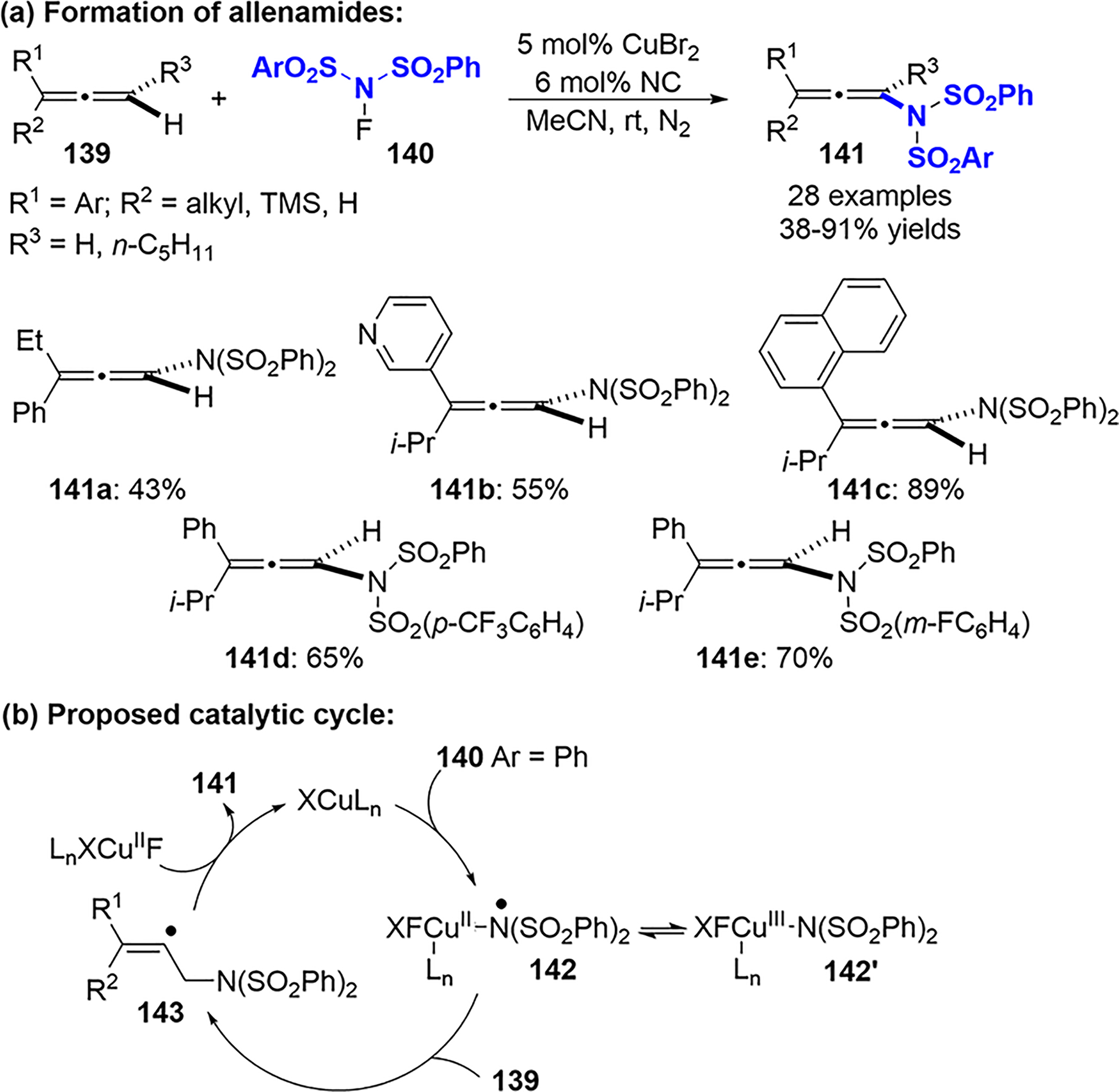
Synthesis of Allenamides via Oxidative Coupling of Allenes and N-Fluoroaryl-sulfonimides
A mechanism was proposed (Scheme 54b) based on the experimental results and previous literature.75–77 The initial oxidation of the Cu(I) catalyst by 140 generates a Cu(III) species 142′, which is in equilibrium with a Cu(II)-stabilized nitrogen radical species 142 through redox isomerization. The addition of 142 to allene 139 affords a vinyl radical intermediate that yields the allenamide product 141 and regenerates the Cu(I) catalyst following β-hydride elimination.
In the presence of an Ag(I) catalyst, a mixture of allene 144 and the N–F reagent 140 yields fluorinated tetrasubstituted alkenes 145 via a radical aminofluorination process (Scheme 55). In general, 1,1-disubstituted and monosubstituted allenes give the corresponding fluorinated alkenes in moderate to excellent yield. Electron-deficient allenes with an ester group at C3 do not react, even at 100 °C. The stereoselectivity of the alkene products is strongly affected by the sterics of the alkyl substituent on the allene, with bulky groups giving higher stereoselectivity in favor of the E isomer. The fluorinated alkene product was proposed to form from fluorine atom transfer to the vinyl radical intermediate 143′.
Scheme 55.

Ag(I)-Catalyzed Aminofluorination of Allenes 144
2.3.2. Phosphorus Radicals.
In 1965, Goldwhite reported the first example of the free-radical addition of phosphine to an allene by irradiating an equimolar mixture of phosphine and propadiene under UV light.2,3,78 The product mixture included a trace amount of a 1:1 adduct, plus large amounts of polymeric materials. The adduct was characterized as isopropenylphosphine (Scheme 56).
Scheme 56.

Phosphine Addition to Propadiene by UV Irradiation
In 1991, Mitchell published a synthesis of vinyl phosphines by the free-radical addition of diphenylphosphine to allenes and alkynes.79 The major products were generally vinyl phosphines 147 (Scheme 57), generated from diphenylphosphine radical attack on the central allene carbon, regardless of the substituents on the allene precursor. In most cases, allyl phosphines 149, which are formed from phosphine radical attack on the terminal allene carbon, are observed in small amounts.
Scheme 57.

Product Distribution of Diphenylphosphine Addition to Allenes 146
2.3.3. Oxygen Radicals.
Studies of oxygen-centered radical attack on allenes are rare, with the majority of interest centering on atmospheric chemistry. For example, reactions of allenes with hydroxyl radicals has been extensively studied80,81 because the hydroxyl radical is an important reactive intermediate in combustion processes. It is also present in the atmosphere of a few planets and the interstellar medium.82 However, because these studies were not directed toward synthetic applications of radical additions to allenes, they are not discussed here.
2.3.4. Sulfur Radicals.
In 2014, the Renaud group published a review article describing the regio- and stereoselectivities of thiyl radical attack on allenes.83 Therefore, this Review will cover only sulfonyl radical and other sulfur-centered radical additions to allenes; the reader is referred to the Renaud review for further details.
2.3.4.1. Sulfonyl Radicals.
Free-radical reactions of aryl/alkyl sulfonyl halides and allenes have been studied since the late 1960s. Recently, a surge in difunctionalization of allene double bonds (with one of the functionalities being the sulfonyl group) has attracted the attention of multiple research groups due to the synthetic versatility of sulfonyl groups as well as the abundance of biological molecules containing a vinyl sulfone moiety.84–86 In general, a sulfonyl radical adds regioselectively to allenes, with terminal attack giving rise to the major product in reactions with propadiene; however, for more hindered allenes, the sulfonyl radical tends to attack the central carbon. Other sulfur-centered radicals, such as the SCF3 radical, also favor attack on central allene carbon.
Truce and Wolf published studies on the free-radical addition of sulfonyl iodides to allenes in the late 1960s and early 1970s.87,88 The irradiation of mixtures of alkyl or aryl sulfonyl iodides and various allenes yielded 1:1 adducts in moderate to good yield (Scheme 58a). The regioselectivity and stereoselectivity depended on the substituents present on the allenes as well as the identity of the sulfonyl iodides. The use of propadiene gave mixtures of products, irrespective of the sulfonyl iodide employed in the reaction. The four products 151–154 were proposed to arise from the mechanistic pathways depicted in Scheme 58b. Irradiation initiates the homolytic cleavage of the S–I bond to generate the corresponding sulfonyl radical and an iodine atom. The addition of the sulfonyl radical to either the central or terminal allene carbon gives the corresponding allyl or vinyl radical intermediates; these undergo iodine abstraction with sulfonyl iodide to generate products 152 and 151, respectively.
Scheme 58.

Free-Radical Addition of Sulfonyl Iodides to Propadiene
As shown in Scheme 59, 156–158 were the only products observed when allenes 24, 27, and 155 were subjected to the same reaction conditions described in Scheme 58. Any substitution on the allene strongly favored the attack of the sulfonyl radical on the central allene carbon due to the lower activation energy to yield a stable allylic radical intermediate. In the case of phenyl allene 155, sulfonyl radical attack favored the formation of the product with the iodine attached to C3. The preference of iodine abstraction by the C3 radical was attributed to less steric hindrance at C3 compared with C1 as well as the increased stability of the intermediate resulting from conjugation with the styrene (Scheme 59). The trans stereoselectivity in the addition of sulfonyl iodide to phenyl allene 155 was rationalized by the relief of the steric strain between the relatively large sulfonyl and phenyl groups in the allylic radical intermediate.25
Scheme 59.

Free-Radical Addition of Sulfonyl Iodides to Allenes
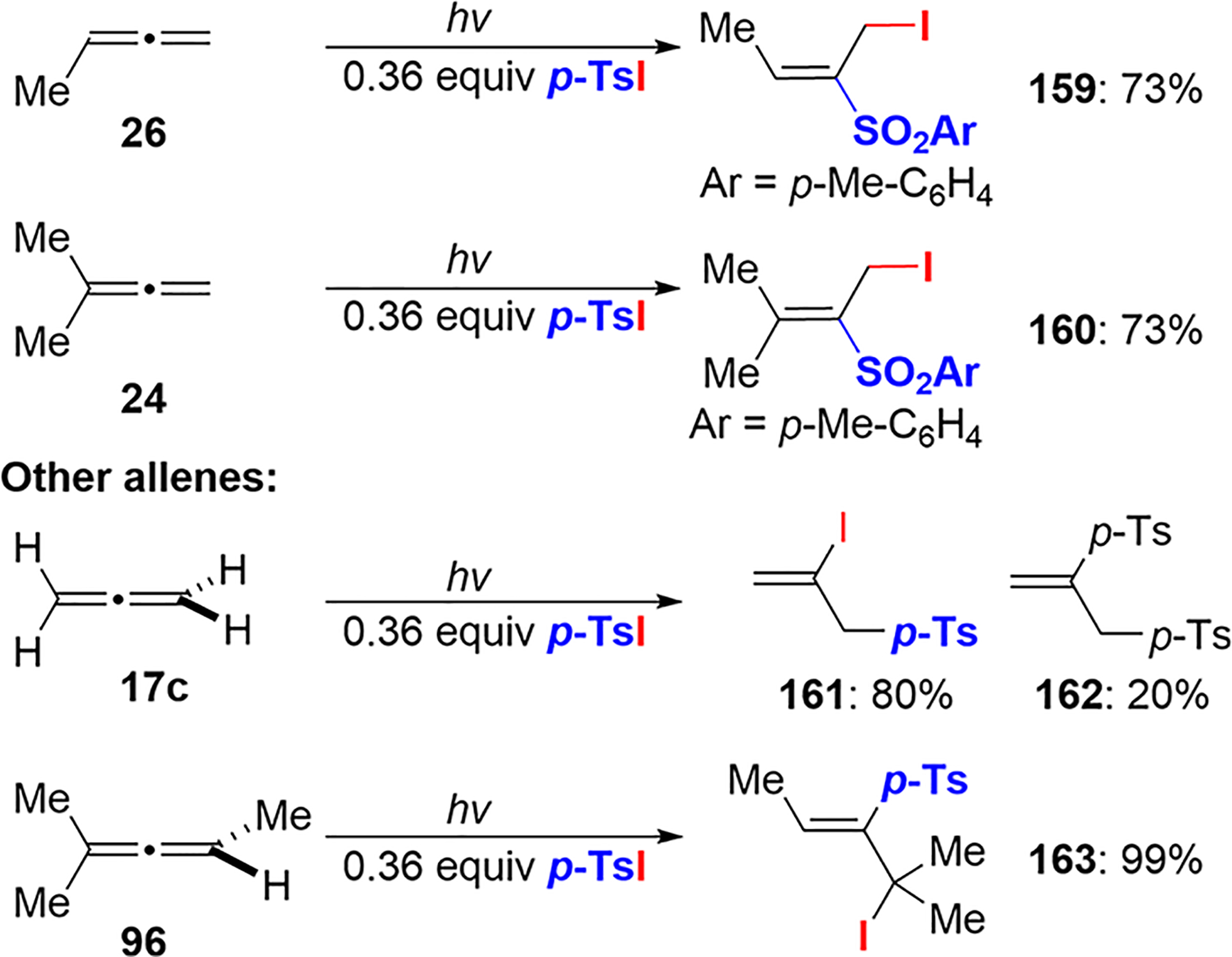
The Caserio group studied the regio- and stereoselectivities of tosyl iodide addition to various allenes, including optically active examples.29 Similar to the experimental results obtained by Truce and Wolf, the tosyl radical preferentially attacked the terminal carbon of propadiene but preferred to attack the central carbon of methyl-substituted allenes 24 and 26 (Scheme 60) and 1,2-cyclononadiene (Scheme 61). Iodine abstraction furnished the most substituted alkene, with the exception of 2-methyl-2,3-pentadiene 96, which delivered 163.
Scheme 60.
Free-Radical Addition of Tosyl Iodide to Allenes
Scheme 61.

Tosyl Iodide Addition to Optically Active Allenes 27 and 101
When optically active allenes (S)-(+)-27 and (S)-(−)-1,2-101 were irradiated in the presence of tosyl iodide, the allylic iodide product 164 was found to be racemic (Scheme 61). The loss of optical activity indicated that the reaction proceeds through a planar, delocalized allylic radical intermediate, as opposed to a nonplanar, localized radical. Racemization also suggested that 90° rotation about the C2–C3 bond is faster than iodine abstraction. The recovered 2,3-pentadiene (S)-(+)-27 retained its optical activity, implying that the tosyl radical adds to the central carbon of 2,3-pentadiene in an irreversible fashion. However, in the case of 1,2-cyclononadiene (S)-(−)-1,2-101, the addition of the tosyl radical to the central carbon proved reversible. The intermediate radical was proposed to be nonplanar, favoring a conformation that relieves torsional strain and nonbonded interactions in the nine-membered carbocycle. The lack of allylic resonance stabilization promoted reversibility in this thermoneutral process, as described in a previous review.25
In 2001, Kang et al. achieved regio- and stereospecific radical additions of p-TsBr or p-TsI to allenic alcohols in the presence of catalytic amounts of AIBN89 (Scheme 62). The p-Ts radical was found to attack the central carbon of the allene, with the (E)-alkene observed as the sole product for allene 166a. Depending on the length of the tether bearing the hydroxy group, products can arise from radical addition only (168, 172) or radical addition, followed by cyclization (168–170).
Scheme 62.

Radical Addition of p-TsI/p-TsBr to Allenic Alcohols
When allenic sulfonamides 173 were subjected to the same conditions using p-TsBr as the radical source, the Br was installed at the less substituted terminal double bond of the allene. The corresponding cyclization products 175 and 176 were obtained by subjecting the initial addition products 174 to K2CO3 in DMF (Scheme 63).
Scheme 63.

Radical Addition of p-TsBr to Allenic Sulfonamides
Kang also published a similar study on the regio- and stereospecific syntheses of (E)-p-Ts-substituted allylic bromides using the same protocol90 (Scheme 64). By subjecting the allylic bromide product 177 to different nucleophiles, the regioselective synthesis of α,β-unsaturated sulfone derivatives 178 was achieved in good to excellent yield.
Scheme 64.

Radical Addition of p-TsBr to Monosubstituted Allenes
The same group employed α-allenic alcohols to accomplish regio- and stereospecific syntheses of β-sulfonyl-α,β-unsaturated ketones 179 via p-TsBr radical addition to allenes, followed by base-promoted 1,4-elimination91 (Scheme 65).
Scheme 65.

Radical Addition of p-TsBr to α-Allenic Alcohols and 1,4-Elimination of the Radical Adducts
In 2018, Lu et al. published a regio- and stereoselective synthesis of (E)-α-halomethyl vinylsulfones 181 and 183 via a Cu(I)-catalyzed difunctionalization of allenes 180 and 182.92 A 1.5:1 mixture of allene and sulfonyl iodide was subjected to a catalytic amount of CuI and 1,10-phenanthroline in dichloro-methane at room temperature to produce various 1:1 adducts in moderate to excellent yield (Scheme 66).
Scheme 66.
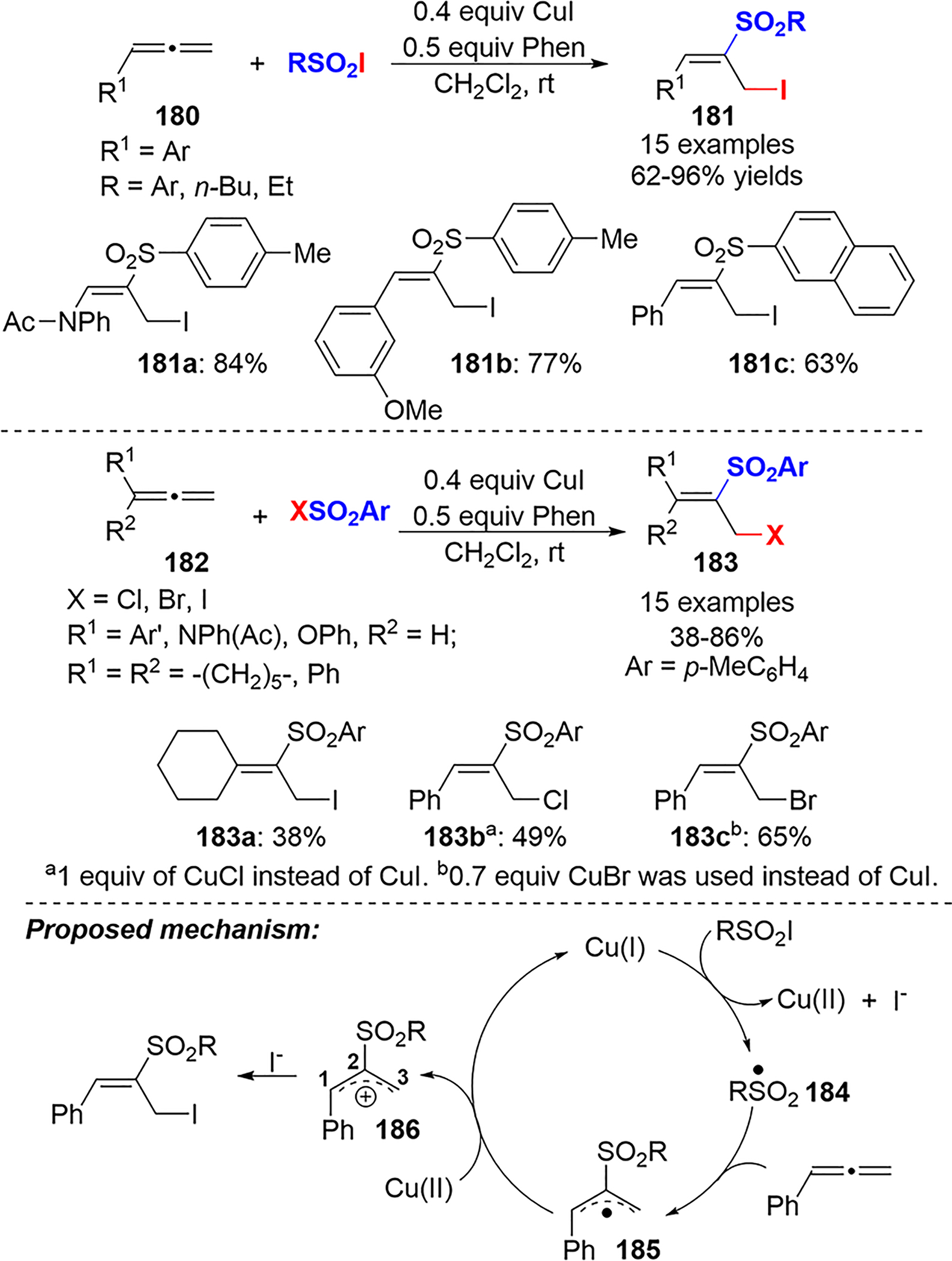
Cu(I)-Catalyzed Sulfur Iodide Addition to Allenes and a Proposed Mechanism
TEMPO and galvinoxyl radical trapping experiments gave reduced yields, suggesting the presence of radical intermediates. On the basis of previous literature and their experimental results, Lu et al. proposed the mechanism in Scheme 66. The oxidation of Cu(I) to Cu(II) by the sulfonyl iodide reagent gives a sulfonyl radical 184 and iodide. The attack of the sulfonyl radical on the central carbon of the allene affords the corresponding allylic radical 185, which was oxidized to the allylic cation 186 by Cu(II) to regenerate Cu(I). The regioselective attack of the iodide on C3 furnished the final product. Density functional theory (DFT) computational analysis was used to rationalize the regioselectivity of the iodide attack; the analysis showed a higher NPA charge on C3 than C1, indicating a higher electrophilicity at C3.93
Recently, Wu described a Cu(II)-catalyzed cyclic arylsulfonylation of 2,3-allenoic acids 187 to afford 4-sulfonylated furan-2(5H)-ones 188 in moderate to good yield (Scheme 67).94 The aryldiazonium tetrafluoroborate likely reacts with DABCO-(SO2)2 to form an aryl sulfonyl radical, which regioselectively attacks C2 of the 2,3-allenoic acid. Na2S2O5 could also be used as a SO2 source but gives lower yields of 188 than DABCO·(SO2)2. Electron-rich and electron-deficient aryl groups are tolerated by both methods.
Scheme 67.
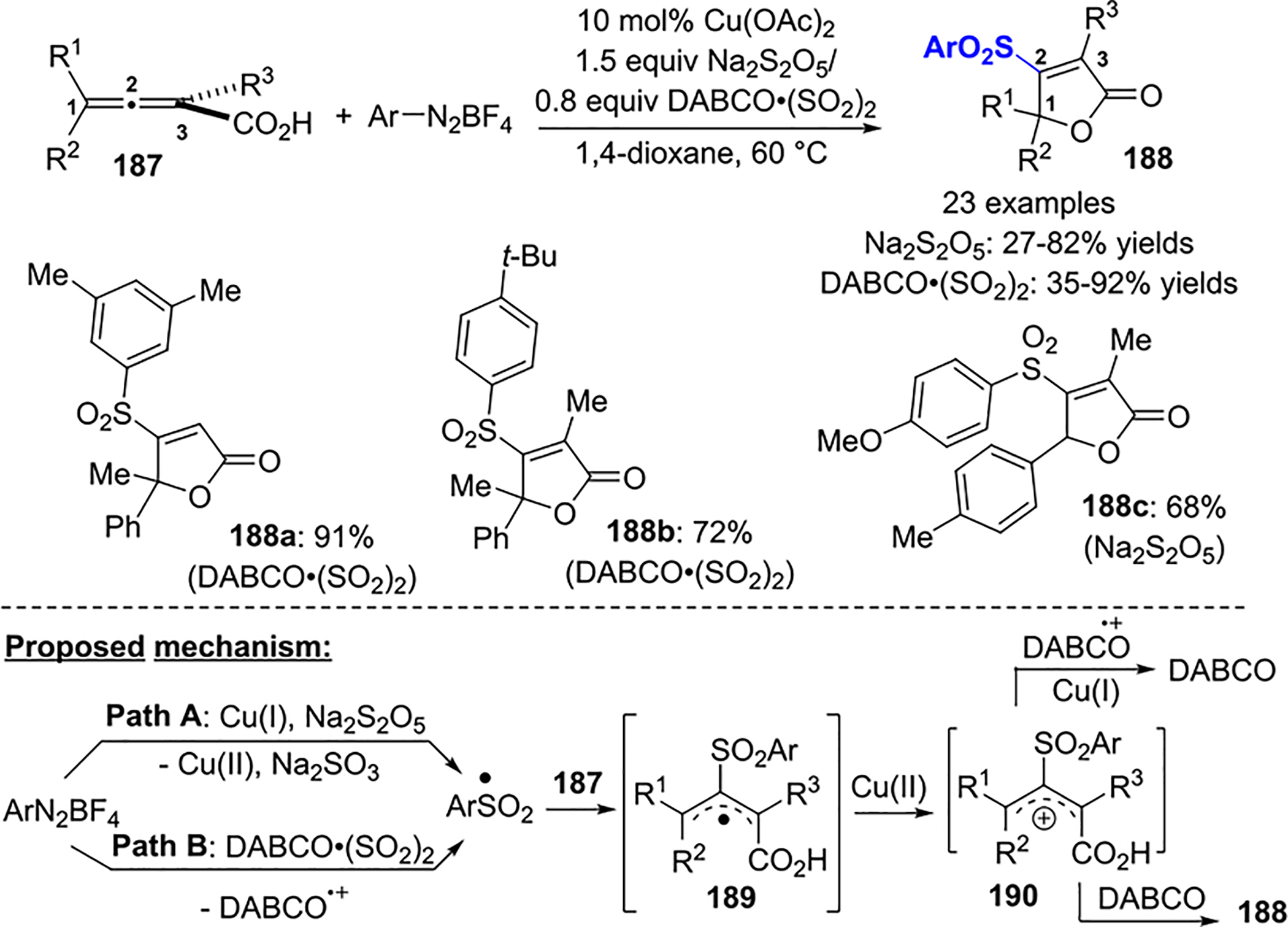
Arylsulfonylation of 2,3-Allenoic Acids and a Proposed Mechanism
The addition of 2 equiv of TEMPO completely inhibited the reaction, indicating the presence of radical intermediates. Wu proposed the mechanism in Scheme 67. In Path A, the aryl sulfonyl radical resulting from the addition of SO2 to the aryl radical is generated from single electron transfer between Cu(I) and ArN2BF4. In Path B, the aryl sulfonyl radical is formed from DABCO·(SO2)2 and ArN2BF4. The addition of the aryl sulfonyl radical to the central allene carbon gives the allylic radical intermediate 189, which is oxidized by Cu(II) to the corresponding carbocation 190. The cyclization of the carboxylate yields the final product 188.
2.3.4.2. Other Sulfur-Centered Radicals.
In 2016, Lei and coworkers described a highly regio- and stereoselective oxysulfonylation of monosubstituted allenes 191 to form 2-sulfonyl allylic alcohols 192 in moderate to good yield.95 Various arylsulfinic acids were examined, with electron-rich arylsulfinic acids giving higher yields than electron-deficient ones. A 1,1-diphenyl allene gave an inferior yield compared with monosubstituted allenes (Scheme 68). Allenes substituted with ester groups 193 furnish olefinic sulfonyl compounds 194 as the major products.
Scheme 68.

Regio- and Stereoselective Oxysulfonylation of Allenes 191 and 193
The addition of TEMPO and BHT inhibited the reaction, suggesting a mechanism involving radical intermediates. The use of 18O2 in place of air results in 86% of isotope incorporation into 192, indicating the participation of O2 in the reaction (Scheme 69a). On the basis of these experimental data, Lei proposed the mechanism in Scheme 69b. The initial deprotonation of phenyl sulfinic acid gives 195, which undergoes oxidation to form the conjugated sulfonyl radicals 196 and 197.96 The regioselective addition of the sulfonyl radical to the allene produces allylic radical 198, which is trapped by O2 to yield 199. The subsequent reduction by 195, protonation by the pyridinium ion, and a final reduction give the product 200.
Scheme 69.

Proposed Mechanism for Oxysulfonylation of Allenes
In 2017, the Qing group published a Cu(I)-catalyzed cyclic oxytrifluoromethylthiolation and Cu(II)-catalyzed bis-trifluorothiolation of 2,3-allenoic acids.97 Various 1-, 1,1-di-, and 1,3-substituted allenoic acids were heated with CuCN, (NH4)2S2O8, and AgSCF3 to afford trifluoromethylthiolated butenolides 202 (Scheme 70). Most 2-alkyl-4-arylbuta-2,3-allenoic acids 201 gave moderate to good yield, with the exception of 202e′, which gave only trace amounts of bis-trifluoromethylthiolated byproduct. Product 202 could be transformed to trifluoromethylthiolated furan derivatives by employing either LDA/Ac2O or NaHDMS/ClP(O)(OPh)2.
Scheme 70.

Cu(I)-Catalyzed Cyclic Oxytrifluoromethylthiolation of 2,3-Allenoic Acids 201
In the same paper, Qing described a catalytic Cu(OAc)2-mediated bis-trifluoromethylthiolation of 2,3-allenoic acids 205. The corresponding ditrifluoromethylthiolated carboxylic acids 206 were obtained in moderate yield in most cases (Scheme 71).
Scheme 71.
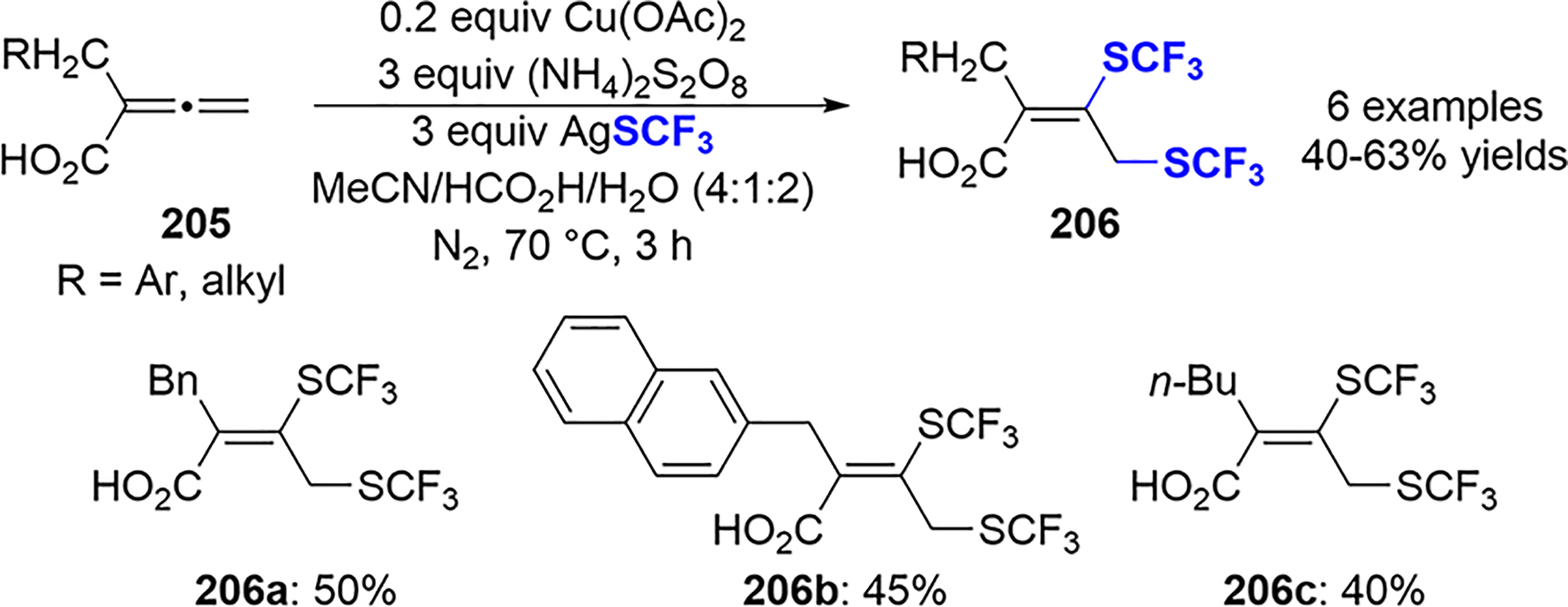
Cu(II)-Catalyzed Bis-trifluoromethylthiolation of 2,3-Allenoic Acids 205
To probe the mechanism of the reaction, Qing and coworkers added radical scavengers to the reaction, including BHT or 1,4-benzoquinone, and found only trace amounts of 202. In addition, replacing carboxylic acid in 205 with an ethyl ester gave only trace amounts of desired bis-trifluoromethylthiolation products. Qing proposed the mechanism shown in Scheme 70, where the oxidation of AgSCF3 by peroxydisulfate anion forms the Ag(II)SCF3 species that serves as the source of either the SCF3 radical or (SCF3)2. The regioselective addition of the SCF3 radical to the central allene carbon gives the allylic radical intermediate 203. The oxidation of 203 by Cu(II) or S2O8− yields the corresponding allylic cation 204, which eventually cyclizes to the desired product 202. For similar work that employs Cu(OAc)2/S2O8− to accomplish the sulfenylation, sulfonylation, and selenylation of 2,3-allenoic acids with disulfides or diselenides, see ref 98.
2.4. Other Metal-Centered Radicals (Sn, Ge, Se, Te, In)
2.4.1. Sn and Ge Radicals.
In 1965, Fish et al. published the first study99,100 on AIBN-catalyzed Me3SnH addition to allenes 17c, 14, 26, 27, and 96. In all cases, an excess of allene was used. The reaction proceeds via a free-radical chain mechanism to yield 1:1 adducts from trimethyltin radical attack on either the central or terminal allene carbon (Scheme 72). Theoretically, two types of products can be formed from attack of the trimethyltin radical on either the central or the terminal carbon of the allene. When the allene is unsymmetrical, four potential products (207–210) are possible.
Scheme 72.

Free-Radical Addition of Me3SnH to Allenes
Fish et al. compared the amounts of products resulting from attack of the central allene carbon on an ethanethiyl radical versus the attack of a nucleophilic trimethyltin radical on an allene substrate. Products resulting from the reaction at the central carbon of the allene increased with increasing methyl substitution on the substrate (Scheme 73). Thus it was concluded that the regioselectivities of ethanethiyl and trimethyltin radical additions to allenes are not determined by polarity but rather by the favored formation of an allylic radical intermediate as the methyl substitution increases on the allene. This was ascribed to the increased steric hindrance in the approach of the trimethyltin radical and the increased stability of the resulting radical intermediate due to hyperconjugation.
Scheme 73.
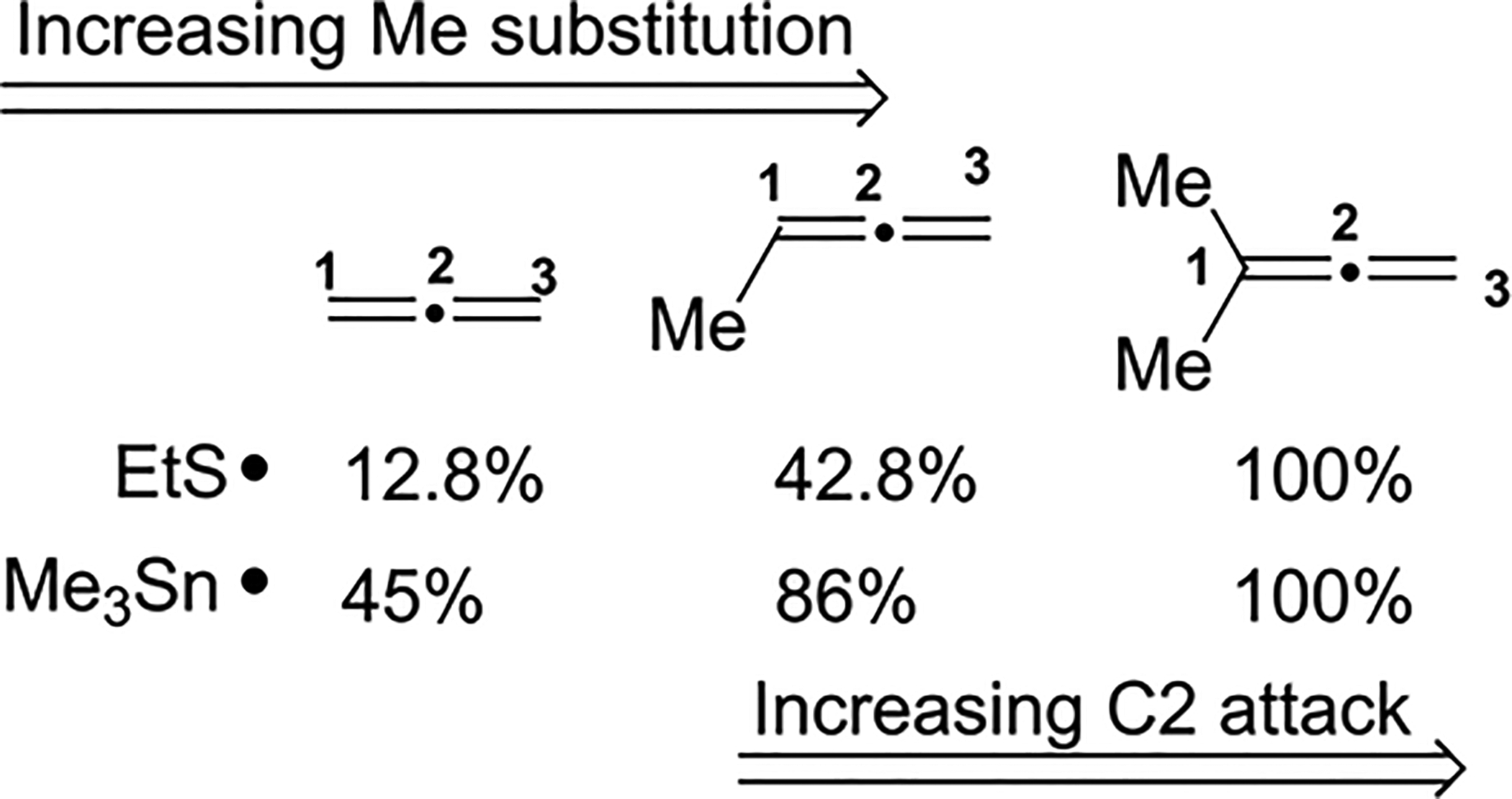
Increased C2 Attack with Increasing Methyl Substitution on the Allene
The product distribution for allene 96 by considering the stability of allylic radicals 211 and 211′ leads to the formation of 207–96 and 208–96 (Scheme 74). Both allylic radical intermediates 211 and 211′ display strain resulting from steric interactions between the SnMe3 and vicinal methyl group(s) or 1,3-dimethyl interactions. Rotation around the C1–C2 or C2–C3 bond minimizes strain while also maintaining maximum π-orbital overlap. Rotation around the C1–C2 bond in both 211 and 211′ is more favorable than rotation around the C2–C3 bond due to the preference for a vinyl-substituted tertiary radical over a secondary radical. Therefore, hydrogen abstraction occurs preferentially at C1 as opposed to C3 to furnish 207–96 as the major product.
Scheme 74.

Intermediates 211 and 211′ Leading to the Formation of 207–96 and 208–96
There is a preference for the formation of 208-26-(E) over 208-26-(Z) for allene 26 by considering the stability of radical intermediates 212 and 212′, as shown in Scheme 75. Both 212 and 212′ were formed from the attack of the SnMe3 radical on the C2 of allene 26. It was reasoned that 212 was produced as the major isomer due to the absence of the strain between C1-Me and C2-SnMe3 that is present in 212′.
Scheme 75.
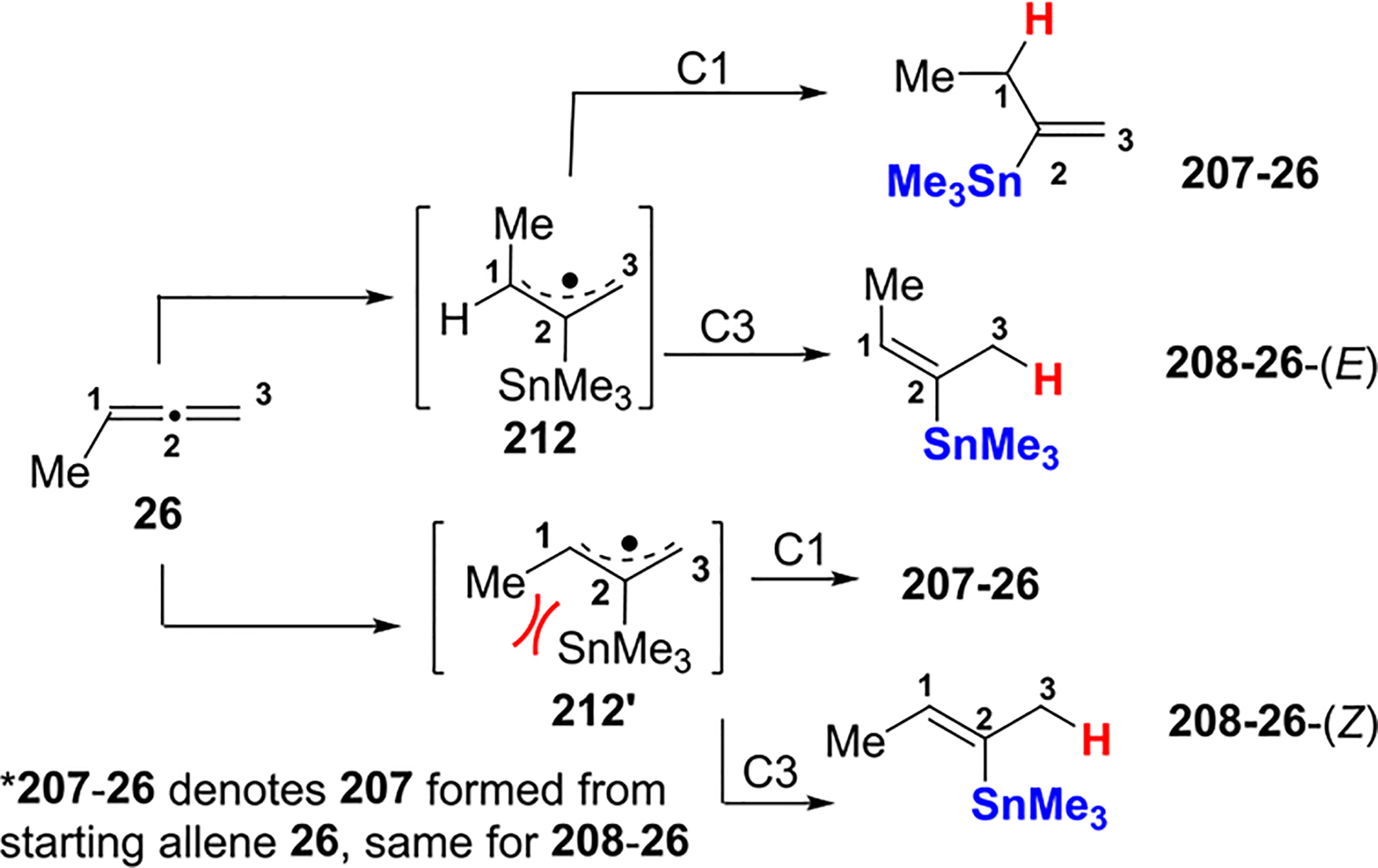
Intermediates 212 and 212′ Leading to the Formation of 208–26-(E) and 208–26-(Z)
Fish proposed that the predominant formation of 207-27-(Z) over 207-27-(E) can be explained as described in Scheme 76. The allylic radical intermediate has two isomeric forms, represented by 213 and 213′. Isomer 213′ is expected to be the major form of the radical because the interaction between C2-SnMe3 and C3-Me in 213′ is estimated to be lower in energy than the corresponding interaction between C1-Me and C3-Me in 213. The latter interaction in 213 is estimated to be 5.4 kcal, similar to values for 1,3-diaxial interactions between methyl groups in cyclohexane. However, the interaction energy between C2-SnMe3 and C3-Me in 213′ is estimated to be <4.3 kcal (the computed value for the cis interaction between Me and tBu attached to ethylene).101 The longer C–Sn bond in 213′, as compared with the C–C bond of a tBu group (2.17 vs 1.54 Å for the C–C bond), is largely responsible for a lower interaction energy between the SnMe3 and its proximal groups. Similar arguments may be applied to explain the regio- and stereoselectivities for the addition of Me3SnH to 26 under radical conditions.
Scheme 76.

Intermediates 213 and 213′ leading to the formation of 207–27-(E) and 207–27-(Z)
In 1988, Oshima et al. described a BEt3-induced radical addition of Ph3SnH and Ph3GeH to allenes to access vinylic triphenylstannanes (Scheme 77) and vinylic triphenylger-manes102 (Scheme 78). For the unsubstituted allene 17c, the Ph3Sn radical attacks both the terminal and central allene carbons, favoring the formation of allyltin 215 but also producing significant amounts of the vinyltin 216. For monoor 1,3-disubsituted allenes 214a–c and 101, the Ph3Sn radical attacks the central carbon in a regioselective manner to give predominantly the vinyltin products 216 and 217.
Scheme 77.

BEt3-Induced Radical Addition of Ph3SnH to Allenes
Scheme 78.

BEt3-Induced Radical Addition of Ph3GeH to Allenes
In general, allylic germanes were formed as the major product in Scheme 78. Because no E/Z ratio was provided by the authors, the stereoselectivity of this hydrogermylation method has not been established. The Ph3Ge radical is less reactive than the Ph3Sn radical, as exemplified by its reluctance to react with 6,7-tridecadiene and 1,2-cyclononadiene.
In 1991, Mitchell et al. studied the radical addition of Me3SnH to allenes, demonstrating an expanded substrate scope compared with the work of Fish et al. (Scheme 79).103 Two conditions were employed, where the reaction was initiated by either AIBN or light. Overall, the results showed that these two different modes of initiation furnish different ratios of products yet still display the same preference for attack at either the central or the terminal carbon if the same allene precursor is employed.
Scheme 79.

Free-Radical Addition of Me3SnH to Allenes
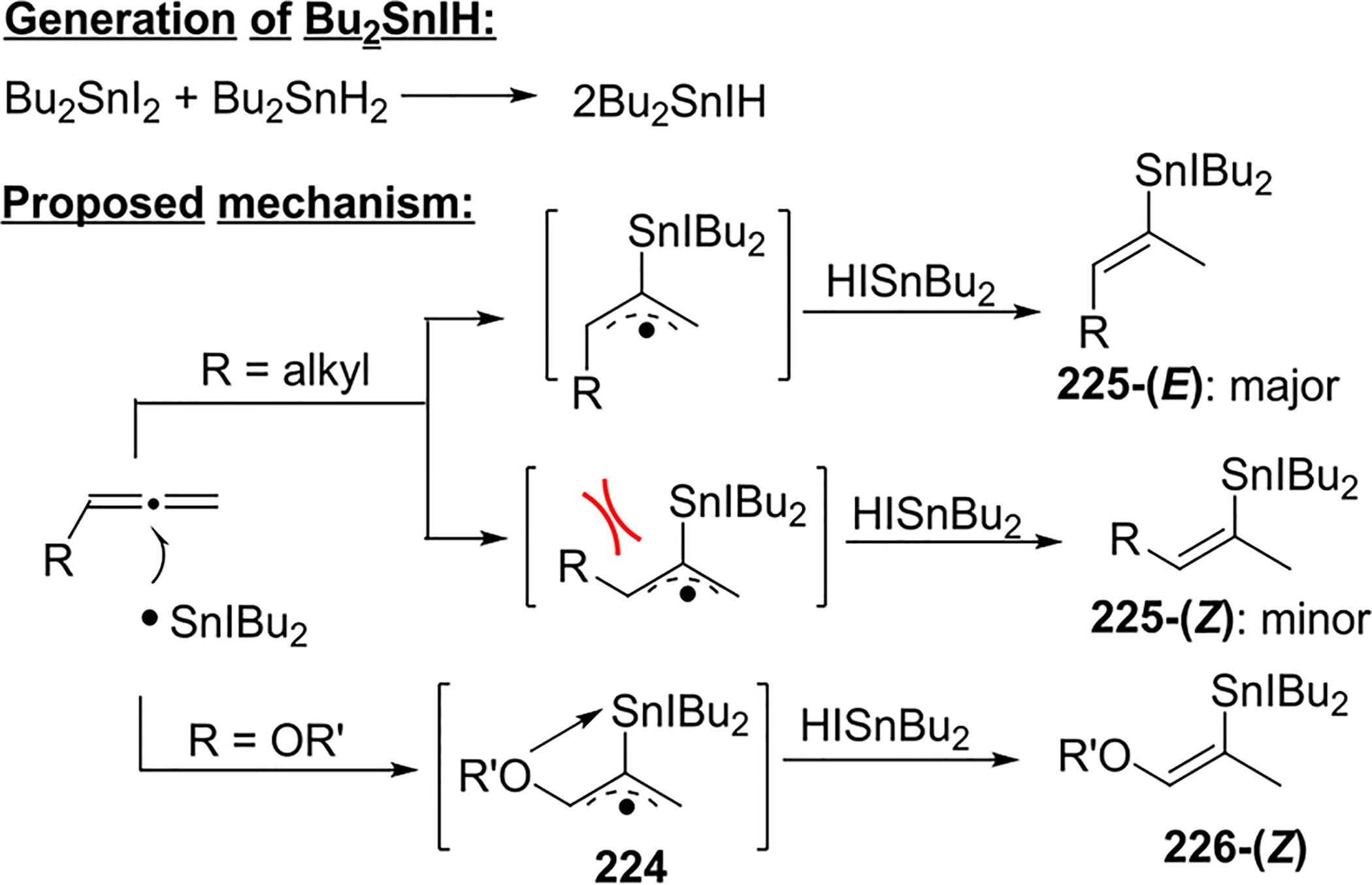
In 2007, Shibata and Baba accomplished a regio- and stereoselective hydrostannation of allenes by using dibutyliodotin hydride generated in situ.104 A mixture of Bu2SnH2 and Bu2SnI2, in combination with monosubstituted or disubstituted allenes, predominantly delivered the (E)-vinyl tin products at room temperature in moderate to good yield (Scheme 80). Substituting Me3SnH (Scheme 79) with Bu2SnIH increased the regioselectivity by predominantly favoring attack at the central carbon, with little product observed from attack at the terminal carbon. It is noteworthy that the stereoselectivity was improved with a bulky R substituent on the allene (e.g., compare 222a with 222b). Allenes with oxygen substituents (222c,d) yielded Z alkenes preferentially, in contrast with allenes bearing alkyl substituents. The reaction was completely suppressed in the presence of the radical scavenger galvinoxyl.
Scheme 80.

Free-Radical addition of Bu2SnIH to Allenes 221
The reaction was proposed to proceed through a free-radical chain mechanism (Scheme 81). The redistribution of Bu2SnI2 and Bu2SnH2 generates the Bu2ISn radical, which attacks the central allene carbon to form the allylic radical intermediate 224. Because of the steric repulsion between the alkyl and the tin substituents, the E alkene 225-(E) is the major product following hydrogen abstraction by Bu2SnIH on the less substituted allene carbon. When an alkoxy substituent was present on the allene, the coordination between the oxygen and the iodotin center favored the formation of the Z alkene product 226-(Z).
Scheme 81.
Proposed Mechanism for Bu2SnIH Addition to Allenes
In summary, the addition of Sn-based radicals to allenes is sensitive to the sterics of the substituents present on the precursor. For both SnPh3 and SnMe3 radicals, the addition to the terminal carbon is favored if the reactant allene is propadiene; however, for methylated allenes, a preference to add to the central carbon was observed. In general, regardless of the source of the Sn radical, the hydrostannation is chemoselective, with the less substituted double bond of the allene undergoing reaction. Ge radicals, on the contrary, favor addition to the terminal allene carbon. However, hydrogermylation displays the same chemoselectivity as radical hydrostannation, with the less substituted allene double bond undergoing the preferential reaction.
2.4.2. Se and Te Radicals.
Interest in introducing Se into an allene carbon primarily originated from the known synthetic applications of vinylselenides.105 Reports on the intermolecular addition of Se, Te, and In radicals to allenes have demonstrated that the addition proceeds in a regioselective fashion, with a new bond formed between the heteroatoms and C2 of the allene. In addition, the radical hydroselenation of allenes is chemoselective, with the less substituted allene double bond undergoing the preferential reaction.
Sonoda et al. developed a regioselective hydroselenation of mono- and disubstituted allenes 227 to afford vinyl selenides 228 in good to excellent yield using benzene selenol as the selenium source.106 The reaction was proposed to occur via a radical chain mechanism with O2 as the initiator. The selenyl radical selectively attacks the central allene carbon to yield 1:1 to 2:1 E/Z mixtures of vinyl selenides (Scheme 82). Selenyl radical addition to allenes is more regioselective than its sulfur counterpart, as a phenyl thiyl radical was reported to afford a 3:1 mixture of products from the attack at the central and terminal allene carbons, respectively.25,107
Scheme 82.

O2-Initiated Radical Addition of Benzene Selenol to Allenes
In 1990, Ogawa published a study on photoinduced radical additions of Ph2Se2 to allenes.108 Various mono-, 1,1-di-, and 1,3-disubstituted allenes 229 were converted to 1-(phenylselenomethyl)vinyl selenides 230 in moderate to excellent yield and with moderate to excellent E/Z ratio (Scheme 83). The phenylselenyl radical, generated from the homolytic dissociation of (PhSe)2, regioselectively adds to the central allene carbon to form an allylic radical 231, which is trapped by a phenylseleno group by the approach of (PhSe)2 from the less substituted terminal allene carbon.
Scheme 83.

Free-Radical Addition of Diphenyl Diselenide to Allenes
Interestingly, replacing Ph2Se2 with Ph2S2 gave a complex mixture of products, presumably due to the inferior ability of Ph2S2 to capture carbon radicals as compared with diselenide. For comparison, the rate constant for homolytic substitution between diphenyl disulfide and a primary carbon radical is 7.6 × 104 L·mol−1·s−1 in benzene at 80 °C compared with 1.2 × 107 L·mol−1·s−1 for diphenyl diselenide under the same conditions.109 In 1993, Ito published a kinetic study on the addition of arylthiyl and phenylselenyl radicals to allenes using flash photolysis (Scheme 84).25,110 Both ArS and PhSe radicals add to olefins, acetylenes, and dienes in a reversible fashion. By studying the decay profile of ArS and PhSe radicals in aerated and degassed cyclohexane solutions of 3-methyl-1,2-butadiene and 2,4-dimethyl-2,3-hexadiene, Ito concluded that these radicals add to the central allene carbon in an irreversible manner. The rate of ArS and PhSe radical addition to the central allene carbon was not affected by increasing the methyl substitution on the allene. In general, the PhSe radical is less reactive than the ArS radical toward 1,3-diene, styrene, and phenylacetylene. However, this trend is reversed in reactivities toward allenes, as the PhSe radical was 1.3 to 1.8 times more reactive than the PhS radical. This suggests an increase in the rate of addition to the central carbon as the orbital size of the unpaired electron on the attacking radical increases.
Scheme 84.
Photoinduced Thioselenation of Allenes 232 by Using the (PhS)2–(PhSe)2 Binary System
In 1998, Ogawa published a photoinduced regioselective thioselenation of allenes 232 using a (PhS)2–(PhSe)2 binary system.111 Upon irradiation at wavelengths >300 nm, β-selenoallylic sulfides 233 were afforded in moderate to excellent yield (Scheme 84). 1H NMR monitoring of the reaction showed the initial formation of diselenide 234, followed by the slow conversion of 234 to 233. In addition, subjecting 234 to (PhS)2 under the same photoinduced conditions generated the thioselenation product 233 over time, suggesting the mechanism illustrated in Scheme 84. At λ > 300 nm, the homolysis of (PhSe)2 was favored over (PhS)2 because the absorption maximum of (PhS)2 is 250 and 330 nm for (PhSe)2. The PhSe radical regioselectively adds to the central carbon of allene to form an allylic radical intermediate, which is in equilibrium with the diselenide adduct 234. Meanwhile, the allylic radical is also converted to the more stable thioselenation product 233 upon reaction with PhSSePh.
The dithiolation of allenes was also accomplished by Ogawa by employing a (PhS)2–(PhTe)2 binary system. Interestingly, when 4,4-dimethylpenta-1,2-diene 146g (Scheme 85) was subjected to an equimolar amount of (PhS)2 in an attempt to obtain the dithiolation product, a complex mixture of products was formed instead, likely due to allene oligomerization. This result was attributed to the inferior ability of (PhS)2, as compared with (PhSe)2 and (PhTe)2, to capture the carbon-centered radical intermediate. However, the addition of (PhS)2 and (PhTe)2 to 4,4-dimethylpenta-1,2-diene did not afford the expected thiotelluration product and instead gave the dithiolation product 235 in good yield and with good stereoselectivity. Ogawa proposed that the presence of (PhTe)2 prevented competing oligomerization.
Scheme 85.

Photoinduced Dithiolation of 4,4-Dimethylpenta-1,2-diene 146g with a (PhS)2–(PhTe)2 Binary System
Ogawa also developed regioselective selenophosphinations of allenes and alkynes to introduce diphenylphosphino and phenylseleno groups simultaneously to the double or triple bond of allenes or alkynes, respectively.112 The irradiation of an equimolar mixture of allene 236, (Ph2P)2, and (PhSe)2 in degassed CDCl3 in a sealed NMR tube afforded the selenophosphination product 237 in good to excellent yield with a moderate E/Z ratio (Scheme 86). Upon oxidation by air during workup, the corresponding phosphine oxide products 238 were generated in moderate yield. Seleno and phosphino groups were found to add to the less substituted double bond of the allene, with the seleno group ultimately attached to the central allene carbon. Mono- and 1,1-disubstituted allenes were tolerated in this protocol; however, cyclononadiene gave no selenophosphination product.
Scheme 86.

Selenophosphination of Allenes 236
A control experiment run in the dark gave trace amounts of the addition product, suggesting a radical mechanism (Scheme 87). At wavelengths >350 nm, only the (PhSe)2 was homolytically cleaved to generate the phenylselenyl radical. In situ formed Ph2PSePh could also be employed as another source of the phenylselenyl radical, which regioselectively attacks the central allene carbon to form an allylic radical intermediate 239. Subsequent trapping by Ph2PSePh (SH2 homolytic substitution) gives the final product.
Scheme 87.

Proposed Mechanism for Selenophosphination of Allenes
In 2016, Kumaraswamy et al. described visible-light-induced regio- and stereoselective chalcogenyl oxyfunctionalizations of mono-, 1,1-di-, and 1,3-disubstituted allenes 240, 242, and 244.113 In the presence of a diphenyl dichalcogen reagent and LiCl, allenes were converted to Z-substituted α,β-unsaturated aldehydes or ketones as the major product in good to excellent yield (Scheme 88b). When (PhTe)2 was used in place of (PhSe)2, the corresponding vinyl tellurium product 241b was obtained in good yield and with good stereoselectivity. However, no reaction occurred when (PhS)2 was used (Scheme 88a). For allenes 244, the oxidation stopped in the alcohol stage to furnish the allylic alcohol products (Scheme 88c).
Scheme 88.

Visible-Light-Initiated Phenylchalcogenyl Oxygenation of Allenes
It is noteworthy that the phenoxy allene 244a and the allenyl N-tosylbenzenamide 244b gave phenol and aniline sulfonamide as the only products (Scheme 89). This was attributed to captodative stabilization114–116 or the stabilization of the radical cation 246 by the allene functioning as an adjacent π-acceptor. The resulting highly stabilized neutral radical 247 gives the corresponding phenol and aniline sulfonamide 248 or 249 upon hydrogen abstraction.
Scheme 89.

Photoinduced Formation of Phenol and Aniline Sulfonamide
Kumaraswamy et al. proposed the mechanism as shown in Scheme 90 based on the experimental results and previous literature.117,118 Upon irradiation, (PhSe)2 is promoted to the singlet excited state, which is then reduced by the ground-state allene via a single electron transfer to form exciplex 250. LiCl and exciplex 250 yield the solvent-separated ions 251 and 252. The attack of the radical anion 251 on the radical cation 252 gives the allylic radical intermediate 253, which, upon trapping of O2, forms the peroxy radical 254. The disproportionation of 254 gave the aldehyde, ketone, or allylic alcohol product depending on the starting allene.
Scheme 90.

Proposed Mechanism of Photoinduced Chalcogenyl Oxyfunctionalization of Allene
2.4.3. In Radicals.
Baba and Shibata published the first regioselective hydroindation of allenes 255 in 2008 by using HInCl2 generated in situ as an indium radical source.119 Sequential protonation converted the vinylindium product 256 and 257 to the corresponding olefins 258 and 259 in moderate to good yield (Scheme 91). The regioselectivity of the indium radical attack was confirmed by adding iodine to the hydroindation products 256 and 257. The isolation of the vinyl iodide product confirmed the preference for the indium radical to attack the central allene carbon. The addition of the radical inhibitor galvinoxyl completely suppressed the reaction, suggesting a radical mechanism. The intermediacy of an indium radical in similar reactions has been described.120–124
Scheme 91.

Hydroindation and Sequential Protonation of Allenes
3. INTRAMOLECULAR RADICAL ADDITIONS TO ALLENES
3.1. Carbon-Centered Radicals
3.1.1. Ketyl Radical Anions.
In 1983, the Pattenden group developed a reductive cyclization of allenic ketones via electrolysis to furnish functionalized fused bicyclic ring systems (Scheme 92).125 The cyclization introduces both unsaturation and a bridgehead hydroxyl group into the product, a motif common in marine terpenoids.126 Allenes examined in the annulation reaction all favored exo-cyclization to form five-membered rings. Allene 262 produced 263, with the vinyl group located trans to the hydroxyl group and hydrogen atom.
Scheme 92.

Reductive Cyclization of Allenic Ketones by Electrolysis
A detailed discussion of the reaction results was provided in 1985.126 Two results are particularly noteworthy: First, when a tethered cyclohexanone (260b in Scheme 92) was employed, changing the reaction condition from electrolysis to sodium naphthalenide changed the cyclization mode from 5-exo-dig to 5-endo-dig (compare 261b with 266); however, in the case of the tethered cyclopentanone 260a, altering the reaction conditions did not affect the cyclization mode. The Pattenden group reasoned that efficient orbital overlap between the singly occupied molecular orbital (SOMO) of the ketyl radical and the lowest unoccupied molecular orbital (LUMO) of the central allene carbon, which is influenced by the size of the ketone ring, was an important consideration (Scheme 93). The ketyl radical 267 overlaps efficiently with only one LUMO on the central sp-hybridized carbon, forming the 5-exo-dig product 261a with both sets of reaction conditions. However, in 268, the closer proximity between the radical center and the central allene carbon enables a sufficient overlap between the SOMO and both LUMOs of the central allene carbon. In addition, the difference in potentials between the sodium naphthalenide anion and the electrode (−3.09 vs −2.43 V (vs Ag/Ag(I)) proved important. The electrochemically generated ketyl radical is of lower energy and cannot overcome the activation energy barrier to form the thermodynamic endo product. Pattenden reasoned that this is due to the greater potential of the sodium naphthalenide radical anion (−3.09 V vs Ag/Ag(I)) as compared with the electrode potential of −2.43 V under the experimental conditions. The trans relationship between the hydroxyl and vinyl groups results from the minimization of the repulsion between the electron density on the oxyanion and the vinyl radical in the transition state.
Scheme 93.

Annulation via a 5-exo-dig versus 5-endo-dig Pathway
In 1986, the Crandall group described an alternative to Pattenden’s strategy for the reductive radical cyclization of allenic ketones, where the ketyl radical is generated from a dissolving metal reduction (Scheme 94).127 For allene 269, cyclization onto the central carbon was the major pathway, resulting in the same regioselectivity as the cyclopentanone analogue 260 in Scheme 92 and its carbon-centered radical analogue 313a in Scheme 103. When n = 2, cyclization gave the five-membered ring as the major product, resulting from the regioselective radical attack on the proximal allene carbon. Compared with its analogue 313b in Scheme 103, the regioselectivity of the ketyl radical anion is improved.
Scheme 94.

Dissolving Metal-Assisted Reductive Cyclizations of Allenic Ketones
Scheme 103.

Cyclizations of Carbon-Centered Allenyl Radicals Generated from n-Bu3SnH
Cyclization is the major pathway for allenyl ketones of the form 269, regardless of the reaction conditions. However, ketone 270 seems to be less competitive in cyclization, as it does not cyclize under Birch reduction conditions. In contrast, the reduction with sodium naphthalenide gave stereoselective cyclization to five-membered rings. Compared with 270, the analogous aldehyde 271 favored the reduction to the acyclic alcohols 276–278 over cyclization under sodium naphthalenide reduction conditions.
Cossy et al. published their findings on the intramolecular additions of ketyl radical anions to unsaturated systems, including alkenes, alkynes, allenes, and nitriles via photochemically induced electron transfer.128 Electron donors utilized in the reaction include HMPA and Et3N (Scheme 95). Compared with electroreductive cyclizations of allenic ketones (Scheme 92), the overall yield is higher; however, products resulted from both 5-endo- and 5-exo-dig cyclization modes.
Scheme 95.

Photochemically Induced Reductive Cyclization of Allenic Ketones in the Presence of Et3N
In 1993, Gillmann described a regioselective reductive cyclization of ester-substituted allenic aldehydes mediated by SmI, 129 2 where the regioselectivity parallels previous results under similar conditions127 (Scheme 96). The high 284/285 diastereoselectivity for allene 280 could be explained by the chelation of the samarium to both the ester and ketyl radical oxygen atoms. Replacing SmI2 with n-Bu3SnH and AIBN lowered the yield to 37% and the dr to 5:1. For allene 281, the competing reduction of either the aldehyde or the allene double bond could not be suppressed.
Scheme 96.

SmI2-Mediated Regioselective Reductive Cyclization of Ester-Substituted Allenic Aldehydes
In 2005, the Molander group described a SmI2-mediated regioselective reductive cyclization of allenic ketones and aldehydes to afford carbocycles and heterocycles (Scheme 97).130 For allenes bearing a cyclic ketone substituent, [3.3.0], [3.4.0], and [4.4.0] ring systems were obtained in low to good yield. Uncyclized alcohol resulting from ketone reduction was an unavoidable byproduct in some cases. The lower cyclization yields and increased reduction products using β-keto esters, as compared with their ketone counterparts, were attributed to the lower energy of the SOMO in the ketyl radical anion of the former, leading to a larger SOMO–LUMO gap and retarding the rate-determining cyclization step. Heteroatoms, including nitrogen, were tolerated in the tether, resulting in moderate to excellent yield of the cyclized products.
Scheme 97.

SmI2-Mediated Reductive Cyclization of Allenic Aldehydes and Ketones
In 2012, the Procter group described reductive radical cyclizations and cyclization cascades of unsaturated allenyl lactones in the presence of SmI2–H2O to afford fused [5.3.0] bicycles (Scheme 98).131 When allenyl lactones were treated with 8 equiv of SmI2–H2O at room temperature, cycloheptan-1,4-diols containing three new stereocenters were produced with excellent dr and in moderate to excellent yield. The use of 4 equiv of SmI2–H2O furnished the corresponding hemiketal products in modest to good yield and with high dr. Allenyllactones gave greater diastereoselectivity than their alkenyl and alkynyl counterparts; the higher dr and preference for the formation of the cycloheptanol and hemiketal products are explained further in Scheme 99.
Scheme 98.
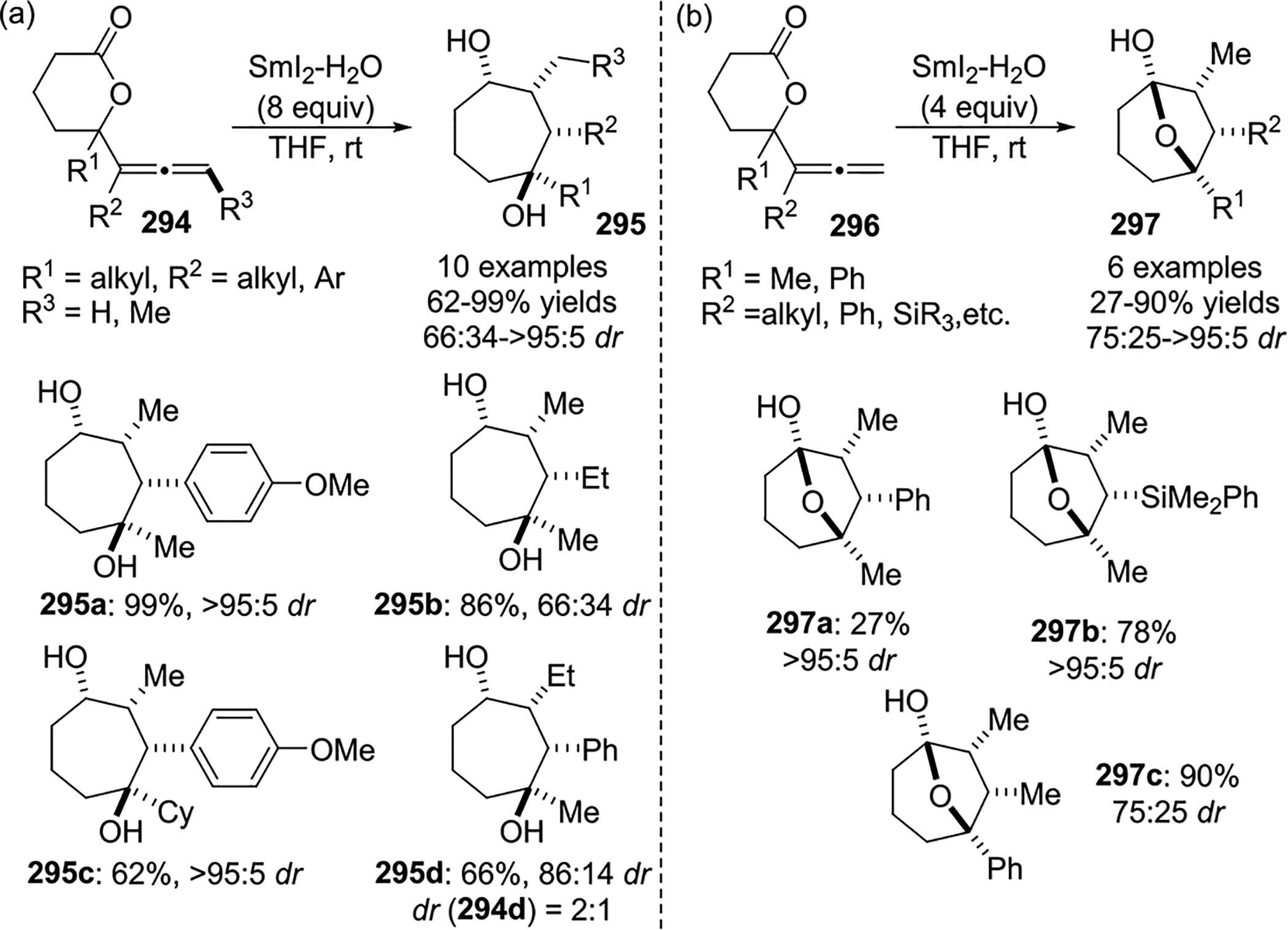
Reductive Radical Cyclizations of Allenyl Lactones in the Presence of SmI2–H2O
Scheme 99.

Proposed Mechanism for Formation of Cycloheptan-1,4-diols and Hemiketals
The reduction of the ester group in lactone 294 or 296 by SmI2–H2O affords a ketyl radical anion 298, where the radical resides in the axial position, due to the stabilization by the anomeric effect. A 5-exo-dig cyclization, reduction, and protonation affords hemiketal 299, which ring-opens to enone 300. Another single-electron reduction of 300 yields the ketyl radical anion 301. The diastereoselective protonation and reduction of 301′ furnish enone 302, which undergoes diastereoselective protonation to deliver 303. In the presence of excess SmI2–H2O, 303 is converted to a cycloheptan-1,4-diol; otherwise, the process stops at the hemiketal 299. Alkenyllactones result in a lower dr, as the diastereoselectivity is established during the formation of the seven-membered ring, as opposed to “post-cyclization”.
The Procter group further demonstrated reductive cyclization cascades of lactones that bear appropriately positioned allenes and alkenes to afford [5.3.0]- and [4.2.1]-bicyclic ring systems (Scheme 100). The [5.3.0] bicycles, containing four new stereocenters, were obtained in good to excellent yield and with moderate to excellent dr, whereas the [4.2.1] bicycles were obtained in moderate yield and with moderate dr.
Scheme 100.

Reductive Cyclization Cascades of cis- and trans-Allenyllactones
3.1.2. Other Carbon-Centered Radicals.
The Crandall group extensively studied the intramolecular addition of carbon-centered radicals to allenes during the 1980s and early 1990s. In 1982, they reported their first study of the reactivity of homoallenyl radicals generated from homoallenyl iodides in the presence of n-Bu3SnH.132 Theoretically, four products 305–308 could be generated, as illustrated in Scheme 101.
Scheme 101.

Reaction of Homoallenyl Iodides with n-Bu3SnH
In most cases, reactions of homoallenyl iodides with n-Bu3SnH predominantly gave the corresponding acyclic allene from hydrogen abstraction. Only vinyl cyclopropanes result from cyclization; no 307 or 308 was observed. For allenes studied by Crandall, the preference for 3-exo-trig cyclization is higher than that for the analogous olefinic counterpart (cyclopropylcarbinyl radical), which tends to rearrange to the corresponding cyclopropyl radical. (See Scheme 102.) For allene 304c, the cyclization proved more facile when compared with allenes that lack substituent branching. The ring-opening process was readily reversible, as evidenced by small amounts of unbranched acyclic allenes formed from the isomerization of cyclopropyl vinyl radical intermediates.
Scheme 102.

Reactions Studying the Equilibrium between Homoallenyl and Vinyl Cyclopropyl Radicals
The reversibility of the 3-exo-trig cyclization of a homoallenyl radical was tested by subjecting deuterated allene 309 to n-Bu3SnH in mineral oil at room temperature. More than 95% of the deuterium was retained at the homoallenyl position, suggesting that the cyclization is irreversible.
The cyclic vinyl iodide 310 was subjected to different reaction conditions to study the interconversion between the homoallenyl and the vinyl cyclopropane radical. On the basis of the product ratios, it was proposed that ring opening is slower than H-atom abstraction at room temperature. When the temperature was increased to 80 °C, the amount of ring-opened product 311 increased, indicating a large activation barrier for isomerization between the two radicals. In contrast, the interconversion between the cyclocarbinyl radical and the homoallylic radical is fast; no n-Bu3SnH-trapped cyclopropane product was observed.133,134
In 1984, the Crandall group used allenic bromides and iodides 313a–d as precursors to generate carbon-centered radicals.135 The tether length between the halide atom and the proximal allene carbon was altered to determine the impact on the cyclizations of these reactive intermediates (Scheme 103). Four products were isolated, where 314 and 315 result from the cyclization of an allylic radical generated from the initial attack of the carbon radical on the central allene carbon. The cyclization product 317 is formed from the vinyl radical intermediate resulting from the attack on the proximal allene carbon. Finally, 316 forms from the competing reduction of the homoallenic radical by n-Bu3SnH.
As shown in Scheme 103, the regioselectivity and cyclization efficiencies were strongly dependent on the tether length and the size of the resulting ring. As expected, as the chain length increases, the cyclization efficiencies decrease. The regioselectivity was not dependent on the stability of the radical intermediate, similar to previous reports on the intermolecular radical attack of allenes.136
When n = 1 (313a), cyclization was faster than H-atom abstraction, giving a low yield of 316 under neat conditions. Crandall suggested an early transition state for the cyclization, where conformational features dictate whether the radical attacks the central or the proximal allene carbon. The absence of bond angle distortions in the transition state when attack occurs at the central allene carbene favors the formation of 314 and 315 over 316.
When n = 2 in 313b, the carbon radical cyclizes slowly as compared with 313a; this was confirmed by the generation of 316, even in dilute solution. The formation of 317 was 1.6 times faster than the formation of 314 and 315 based on the product ratio. The preference for the formation of the smaller ring was rationalized using the same arguments proposed by Beckwith for the cyclization of 5-hexenyl radicals.137
Increasing the tether to n = 3 (allene 313c) resulted in faster rates of hydrogen abstraction. Cyclization favored the formation of 317 over 314 and 315; the 1,5-hydrogen transfer in the acyclic allenic radical accounted for the minor product 2-methyl-2,4-nonadiene. When n = 4 (allene substrate 313d), hydrogen abstraction was the dominant pathway, with minor products observed from acid-catalyzed isomerization of the allene.
In 1991, the Crandall group published another study on the reduction of allenyl iodides with n-Bu3SnH.138 In contrast with the precursors utilized in their previous study,135 these substrates contained radical stabilizing groups attached to the proximal allene carbon, including P(O)Ph2, S(O)Ph, and SO2Ph. The introduction of radical stabilizing groups had a significant impact on the regioselectivity and the cyclization rate. As shown in Scheme 104, four products could potentially be formed from the allenyl radical: attack at the central allene carbon to afford 321 and 322, on the proximal carbon to yield 324, and hydrogen abstraction to give 323.
Scheme 104.
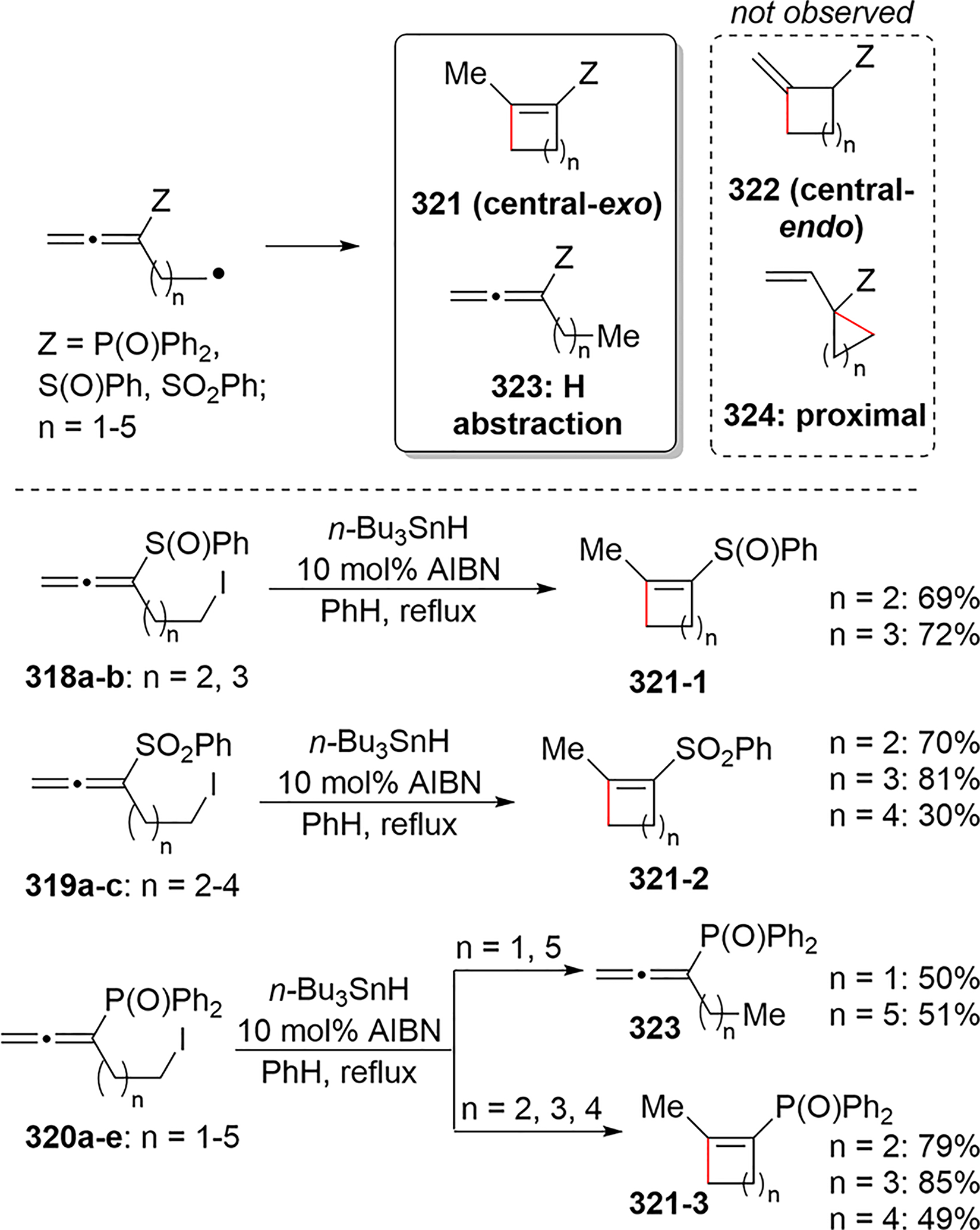
Potential Reaction Pathways and Product Distributions for n-Bu3SnH Reduction of Allenes
When n = 1 (allene 320a), the product distribution parallels that of analogues 304a and 304b (Scheme 101); the major product was 323. Any observed cyclization products resulted from a 3-exo-trig cyclization mode. When substrates with n = 2 (allene 318a, 319a, and 320b) were compared with analogue 313a (Scheme 103), a preference to attack the central allene carbon was maintained, but allenes substituted with electron-withdrawing Z groups (Scheme 104) preferred 5-exo-dig over the 5-endo-dig cyclization mode. The cyclization rate was increased, and no H-atom abstraction product 323 was observed. When n = 3, the radical formed in the analogue 313b attacked both the central and proximal allene carbons, whereas Z-substituted allenes 318b, 319b, and 320c gave products resulting only from attack at the central allene carbon. The ratio of cyclization was increased, and no H-atom abstraction product 323 was observed.
When n = 4, the analogue 313c mainly gave the hydrogen abstraction product 316. Attack on the proximal allene carbon was favored to furnish cyclization product 317. Again, the addition of an electron-withdrawing Z substituent (allenes 319c, 320d) completely altered the regioselectivity to favor attack on the central allene carbon to form a seven-membered ring in 321. When n = 5 (allene 320e), cyclization did not occur, and the major product was the acyclic allene 323 from hydrogen atom abstraction.
In 1994, Balasubramanian et al. reported a regioselective synthesis of 3-ethenyl-2,3-dihydrobenzofurans and 3-ethenyl-2,3-dihydroindoles from o-haloaryl allenyl methyl ethers and amines.139 In the presence of n-Bu3SnH, aryl halides readily undergo 5-exo-trig cyclization at the proximal carbon of the allene 325 to afford the corresponding products 326 in good yield (Scheme 105). For allenyl ether substrates, <10% of products formed from 6-exo-dig cyclization were detected. In all cases, no reduction products were observed.
Scheme 105.

n-Bu3SnH-Mediated Radical Cyclization of OHaloaryl Allenylmethyl Ethers and Amines
In 1998, Renaud and coworkers developed a diastereoselective Ueno–Stork-type radical cyclization of bromoacetals, where diastereoselectivity is controlled by the acetal center.25,140 The protocol is applicable to both alkene- and allene-tethered bromoacetals, with an allene example illustrated in Scheme 106. The chemistry is initiated by tributyltin hydride and triethylborane-oxygen to generate a carbon-centered radical, which subsequently undergoes 5-exo-trig cyclization. The cis diastereoselectivity is best explained by intermediate 328, in which the alkoxy group preferentially occupies the axial position. Employing the (1R,2S)-2-phenyl-cyclohexyl group as a chiral auxiliary enabled the enantiomerically and diastereomerically pure allene 327 to be converted to the enantiomerically pure lactone 328 in 50% overall yield in a cyclization/hydrolysis/oxidation sequence.
Scheme 106.

Ueno–Stork-Type Radical Cyclization of Bromoacetal-Containing Allene 327
In 2004, Sha published a photoinduced regioselective radical cyclization of α-iodocycloalkanones 330 bearing an allenic substituent, to give spirocyclic rings 331 in moderate to good yield141 (Scheme 107a). The addition of (Bu3Sn)2 to the reaction affords the same products, albeit in lower yield for allenes 332b–d. Extending the scope to the formation of [3.3.0]- and [4.3.0]-bicyclic ring systems of the form 333 and 334 gave products in moderate yield and with moderate dr (Scheme 107b). A 1,5-H transfer in vinyl radical 336, formed from the 5-exo-trig cyclization of 335, was proposed as a key step in the formation of the allylic iodide isomer (Scheme 107c).
Scheme 107.
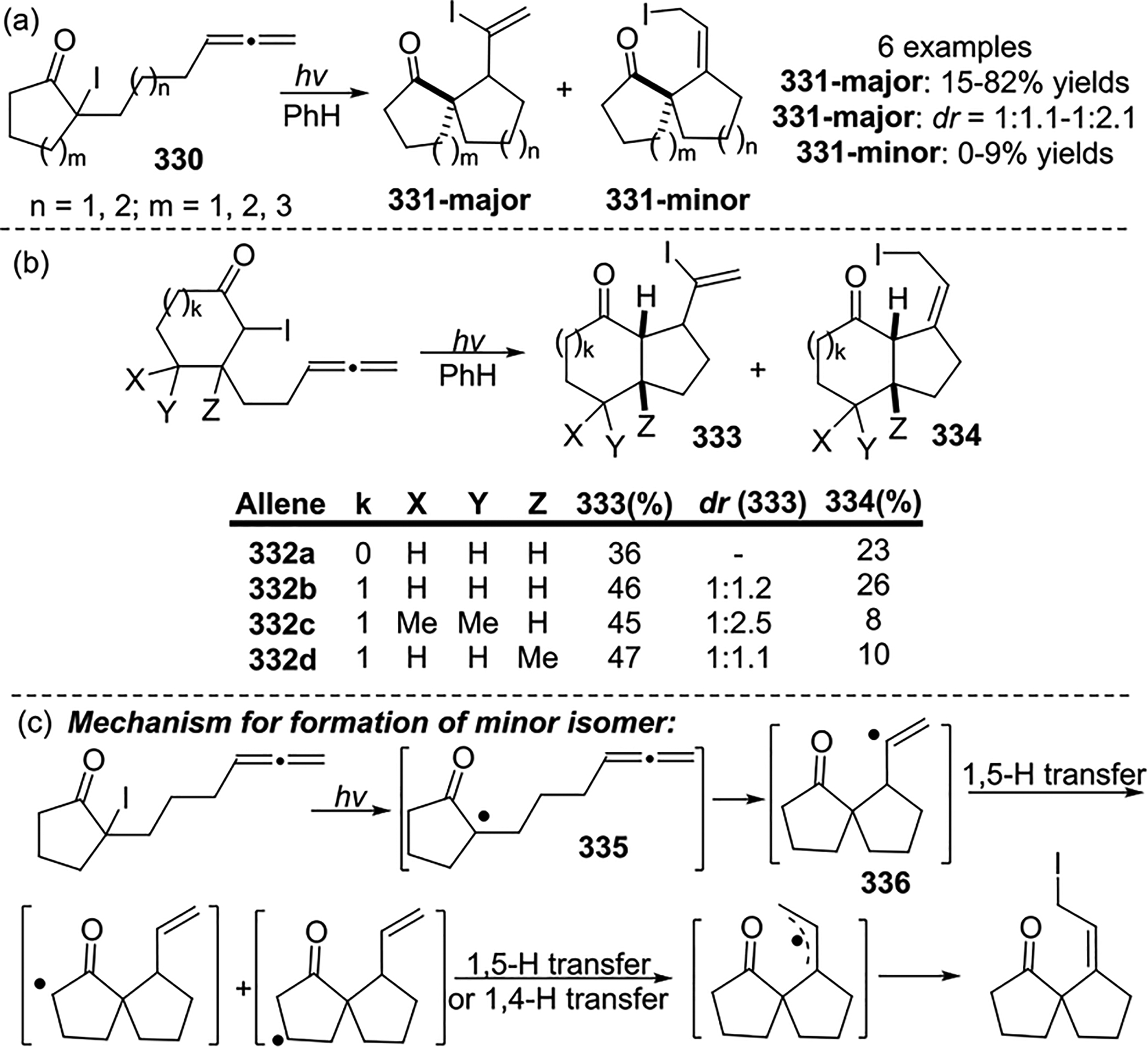
Photoinduced Radical Cyclizations of α-Iodoallenic Ketones
In 2005, the Hsung group developed a highly regioselective radical cyclization of allenamides mediated by n-Bu3SnH.142 Aryl iodides were employed as radical precursors to synthesize derivatives of isoquinoline, indane, naphthalene, and isoindole scaffolds (Scheme 108). The intermediate aryl radical selectively attacks the central carbon of the allene in all cases. However, when a carbonyl group was inserted adjacent to the nitrogen, as in 337 and 340, attack at the proximal allene carbon was favored, albeit with moderate selectivity and yield (Scheme 108). Hsung proposed that this change results from the ability of the carbonyl group to alter the trajectory of attack via structural constraints. It is worth noting that the introduction of a chiral auxiliary into allene 340 furnished only one diastereomer as the product.
Scheme 108.
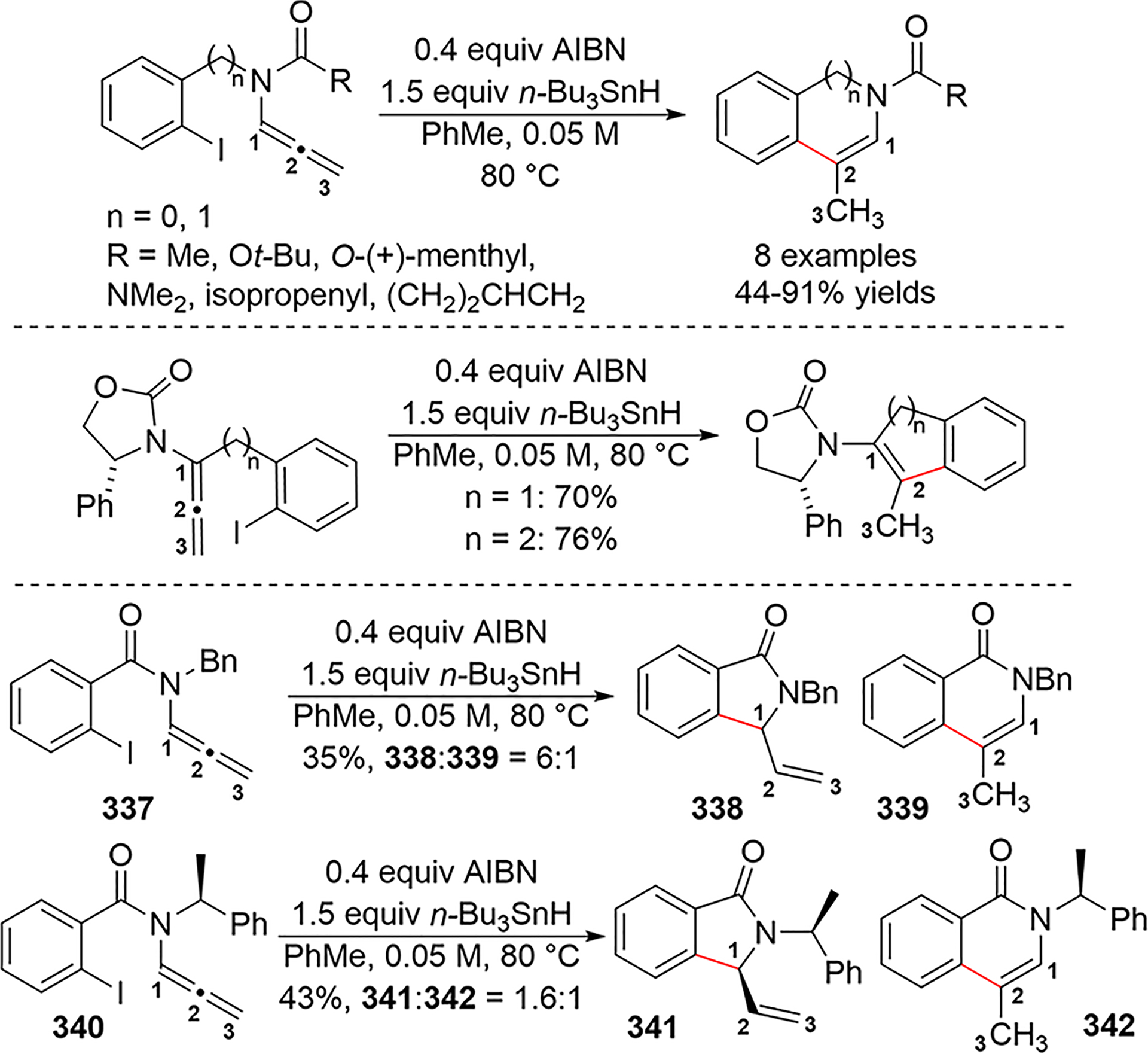
n-Bu3SnH-Mediated Radical Cyclization of Allenamides
In 2007, Guo et al. published the first computational study describing the factors that control the regiochemistry in radical cyclizations of allenes.143 The ONIOM(QCISD(T)/6–311+G-(2df,2p)UB3LYP/6–311+G-(2df,2p)) method was utilized and gave computational results consistent with experimental results (Schemes 101 and 103). The reliability of using this computational method to handle radical cyclization reactions was demonstrated in their previous work.144 In all cases, distal attack on C1 was neither kinetically nor thermodynamically favored. On the basis of the data presented in Scheme 109, when X = CH2 and n = 2 (allene 343b), the product 347 that results from the attack on the central carbon C2 was predicted to be both thermodynamically and kinetically preferred. When n = 3 (allene 343c), the small difference (11.6 – 10.7 = 0.7 kcal/mol) in ΔG‡ between 346 and 347 indicated the potential to form a mixture of 346 and 347 despite the fact that ΔG is lower for the formation of 347. For allene 343a, the case was further complicated by the fact that 346 is kinetically favored, whereas 347 is thermodynamically favored. However, the calculated free energies of activation for the hydrogen abstraction of Me3SnH with allene 343a and cyclopropyl radical 348 (13.8 and 11.6 kcal/mol) were both lower than the ΔG‡ value of 347 (17.9 kcal/mol), suggesting a preference for proximal attack, which is consistent with the experimental results. (See Scheme 101.)
Scheme 109.

Calculated ΔG‡ and ΔG for the Addition of C-, N-, and O-Centered Radicals to Allenes
When n = 2 in the allenes 344a and 345a, attack at the central C2 carbon to form 347 is kinetically and thermodynamically favored for both nitrogen- and oxygen-centered radicals. For allenes 344b and 345b with n = 3, both proximal and central attack products were expected for nitrogen- and oxygen-centered radicals, as computations predicted ΔG‡ of <1.6 kcal/mol in both cases.
Intrinsic and thermodynamic energy barriers were calculated from activation energy barriers using the Marcus theory.145–147 The regioselectivity was predicted by balancing intrinsic and thermodynamic barriers (Scheme 110). Guo et al. discovered that ΔGintrin values were always lower for the formation of 346 as compared with 347, whereas a lower ΔGtherm term predicts a preference to proceed through allylic radical intermediate 347.
Scheme 110.

Intrinsic and Thermodynamic Barriers for Carbon-Centered Radical Cyclizations of Allenes
Lastly, Guo et al. calculated ΔG‡ values for the radical cyclizations of allenes 343b and 343c to model the effects on the regioselectivity in the presence of various substituents on C1 or C3, including Ph, OMe, Cl, CN, and C(O)Me. When n = 2 (allene 343b), substitution on C1 or C3 did not affect the regioselectivity of the preferred radical attack on the central allene carbon. However, when n = 3 (allene 343c), certain substituents at either C1 or C3 were able to reverse the regioselectivity. The substitution of CN or C(O)Me at C1 reversed the preference for vinyl radical formation to favor the formation of the allylic radical. For C3 substitution, all of the groups examined reversed the regioselectivity to favor attack of the central carbon, providing computational support for the results obtained from Crandall’s 1991 paper. (See Scheme 104.)
In 2007, Alcaide and Almendros described a regioselective radical cyclization of enantiopure and racemic 2-azetidinones tethered to bromovinyl and haloaryl allenes to afford fused bi- and tricyclic β-lactams in moderate to good yield148 (Scheme 111). The vinyl and aryl radicals selectively attacked the central allene carbon to form nonconventional β-lactams.
Scheme 111.

Radical Cyclization of Enantiopure and Racemic 2-Azetidinones Tethered to Bromovinyl and Haloaryl Allenes
In 2008, Huang et al. reported Ti(III)-induced intramolecular radical cyclizations of epoxyallene ethers and amines for the synthesis of tetrahydrofuran and pyrrolidine derivatives.149 Various monosubstituted allenes 349 and 351 were converted to the desired products 350 and 352 in good yield. The epoxide C–O bond was homolytically cleaved to form an alkyl radical 353, which regioselectively adds to the proximal allene carbon to form the five-membered ring (Scheme 112).
Scheme 112.

Ti(III)-Induced Intramolecular Radical Cyclizations of Epoxyallene Ethers and Amines
In 2014 and 2015, Yamashita et al. described a regioselective radical cyclization of allenylanilines 354150 and allenyl aryl iodides 356 to afford indole and dihydroindole derivatives in moderate to good yield.151 A mixture of the single-electron reductant SmI2 and an aryl iodide generated the corresponding aryl radical, which cyclized in a regioselective fashion at either the central or the proximal carbon, depending on the length of the tether between the proximal allene carbon and the aryl iodide (Scheme 113). The regioselectivity was consistent with previously reported literature examples (see Scheme 108), albeit displaying a different radical initiation pathway. Yamashita et al. also developed an oxidation protocol employing DDQ, which converts the dihydroindole derivatives 357 to the corresponding hydroindoles 358 in modest to good yield.
Scheme 113.

SmI2-Mediated Radical Cyclization of Allenylanilines 354 and Allenyl Aryl Iodides 356
3.2. Nitrogen- and Oxygen-Centered Radicals
Compared with intramolecular radical cyclizations of allenes induced by carbon-centered radicals, instances of reactions initiated by nitrogen-centered radicals are scarce. Nonetheless, two examples have been reported by Hatem, one of which152 is discussed in more detail in section 4.3. In this report, the chemoselectivity of an in-situ-generated stannyl radical was complicated by attack at both the central allene carbon and the oxime benzoyl group. In 2001, Hatem described a method to improve the chemoselectivity of the stannyl attack by employing a more stannophilic dithiosemicarbazide precursor, resulting in successful syntheses of 3H-pyrroles and alkylidenepyrrolines153 (Scheme 114a). Refluxing a mixture of allenyl dithiosemicarbazide 359, n-Bu3SnH, and AIBN in cyclohexane afforded the unsaturated five-membered rings 360 and 361 in good yield. Because of the instability of certain products on silica gel and poor separations, a one-pot cyclization/reduction/protection protocol was developed to convert allenes 362 directly to pyrrolidine derivatives 363 (Scheme 114b). Hatem proposed that the unpaired electron on the iminyl nitrogen occupies a 2p orbital orthogonal to the C–N double bond; this predicts that 5-exo cyclization should be favored over the 5-endo mode due to the better overlap of the unpaired electron orbital with the π* orbital of the distal allene double bond.
Scheme 114.

n-Bu3SnH-Mediated Radical Cyclizations of Allenes 359
In 2010, Cramer and coworkers developed a heteroatom-induced Grob-type fragmentation of vinyl triflates to form allenes containing a terminal carbonyl functionality;154 one example is illustrated in Scheme 115a. Cramer applied this fragmentation method to 364 to form the hydroxamic acid 366 in good yield. Subjecting 366 to intramolecular oxidative radical cyclization conditions induced a 6-exo-trig cyclization to form the vinyl radical intermediate 368, which was trapped by molecular oxygen to form the hydroperoxide 369. The rearrangement of 369 gave the α-hydroxy ketone 367 in moderate yield (Scheme 115b).
Scheme 115.
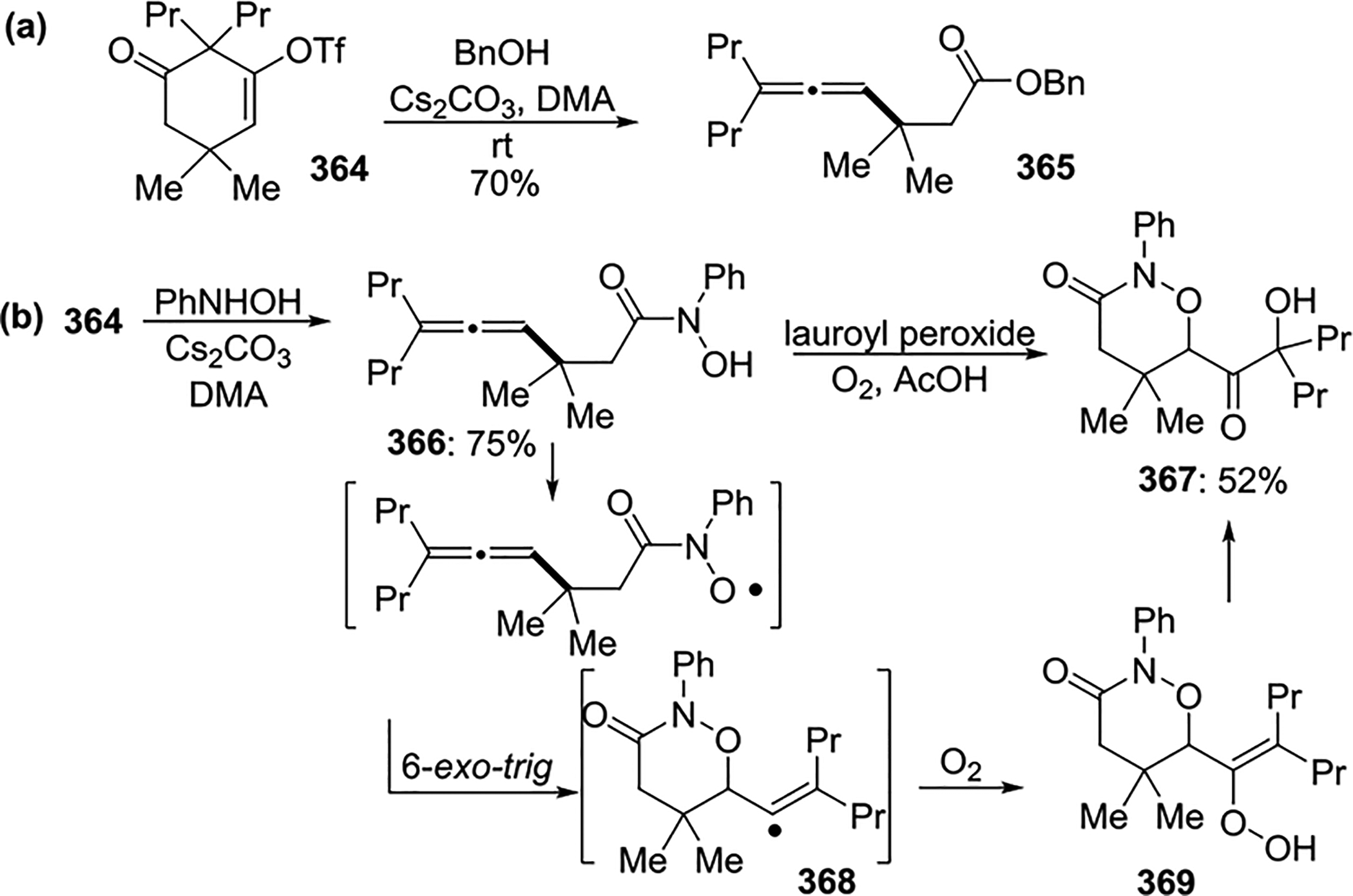
Heteroatom-Nucleophile-Induced Grob-Type Fragmentation and Intramolecular Oxidative Radical Cyclization of the Fragmentation Product 366
In 2015, Liu et al. developed a one-pot synthesis of α-O-, S-, and N-substituted 4-methylquinoline derivatives via a Cu(I)-catalyzed aerobic oxidative radical cyclization of N-hydroxyaminoallenes 370 in the presence of various nucleophiles (Scheme 116).155 The first step of the synthesis proceeds through a radical cyclization pathway to afford tautomer 371 or 372 (or, in some cases, tautomeric mixtures of 371 and 372), which then cyclizes to the corresponding 4-methylquinolines 373 in moderate to good yield upon the treatment with polyphosphoric acid (PPA) in hot toluene.
Scheme 116.

One-Pot Synthesis of α-O-, S-, and N-Substituted 4-Methylquinoline Derivatives 373
Selected mechanistic studies, as shown in Scheme 117, indicate a radical-type pathway for this chemistry. In the presence of 10 mol % TEMPO under a N2 atmosphere, N-hydroxyaminoallene 374 gave a mixture of tautomers 375 and 376 in 80% yield, together with 4% of the TEMPO-trapped product. Liu proposed that the amidoxy radical 378 was produced from a Cu(I)/O2 oxidation of 374. The oxygen-centered radical of 378 attacks the central allene carbon to form an allylic radical intermediate 379, which abstracts a hydrogen atom from 374 to yield 380. The Cu(I)/O2 oxidation of 380 gives 381, which upon nucleophilic attack generates 382; a final 1,2-H shift of 382 gives the ring-opened product.
Scheme 117.

Mechanism for Cu(I)-Catalyzed Aerobic Oxidative Cyclization of N-Hydroxyaminoallenes 374
Overall, the regio-, site-, and stereoselectivities of the intramolecular addition of carbon-, nitrogen-, and oxygen-centered radicals to allenes depend on the specific substrates and radical precursors. Nonetheless, the regioselectivity typically responds to the tether length between the attacking radical and the allene carbon. A general rule of thumb for predicting the regioselectivity is summarized in Scheme 118. When n = 1, the C-, N-, and O-centered radicals add to the C2 of allene to favor the formation of a five-membered ring. When n ≠ 1, addition to C3 become the major pathway, with the only exception being shown in Scheme 108; in this case, n = 2, but products formed from C2 addition are reported as the major product. To reverse the addition of C-centered radicals from C3 to C2 (n = 2, 3, 4), radical stabilizing groups are required at C3, including P(O)Ph2, S(O)Ph, and SO2Ph (see Scheme 104).
Scheme 118.
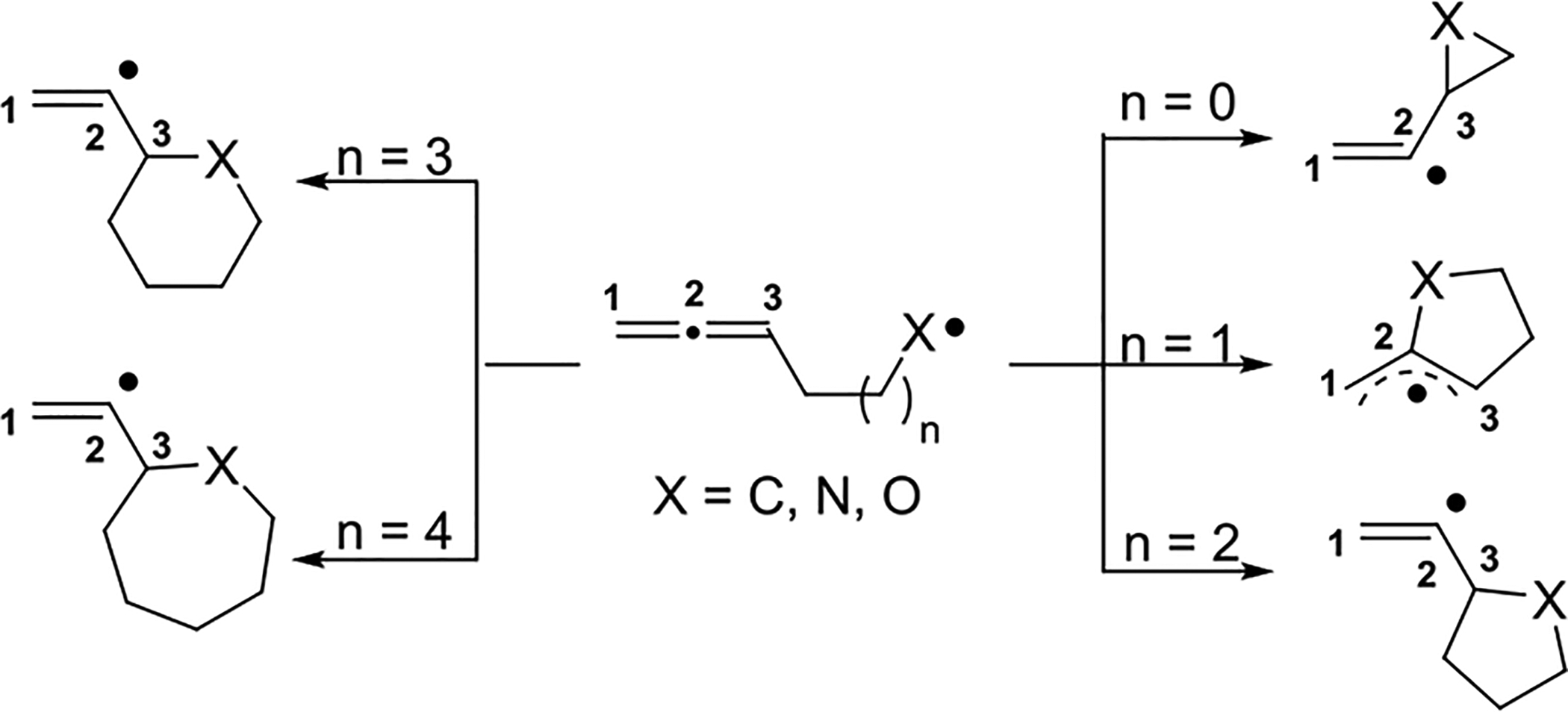
Prediction in the Regioselectivity of C-, N-, and O-Centered Radical Additions to Allenes
4. TANDEM RADICAL CYCLIZATION REACTIONS
This section reviews tandem radical cyclization reactions of allenes, which typically display more complicated chemo-, regio-, and stereoselectivities due to the presence of extra sites of unsaturation tethered to the allene functionality. Typical substrates employed for such tandem reactions include allene-enes, allene-ynes, bis(allenes), aryl allenes, and their derivatives. Tandem reactions of allenes can be broadly divided into two types. In the first type (Type 1, Scheme 119), the radical adds either to the allene double bond (path a) or to the unsaturated functionality tethered to the allene (path b) to form a carbon-centered radical intermediate; the radical then attacks the unsaturated functionality of the allene tether or the allene itself, respectively. In the second type (Type 2), initiation first generates a radical in the allene tether; this intermediate radical species then either adds to unsaturated functionality in the allene tether (path a) or to the allene itself (path b); the resultant carbon radical then adds to any remaining unsaturated functionality.
Scheme 119.

Simplified Schemes of Tandem Radical Cyclization Reactions Involving Allenes
4.1. Tandem Radical Cyclizations Initiated by Carbon-Centered Radicals
In 2005, the Hsung group explored tandem radical cyclizations with allenamides 383 and 389 (see Scheme 108;142 Scheme 120). Products of the tandem radical cyclization of 383 include 386 and 388, whereas allenamide 389 did not undergo tandem cyclization, furnishing 390 instead. The aryl radical generated from 389 regioselectively attacks C2; 6-exo-dig cyclization establishes the 1,2-dihydroisoquinoline core in 390.
Scheme 120.
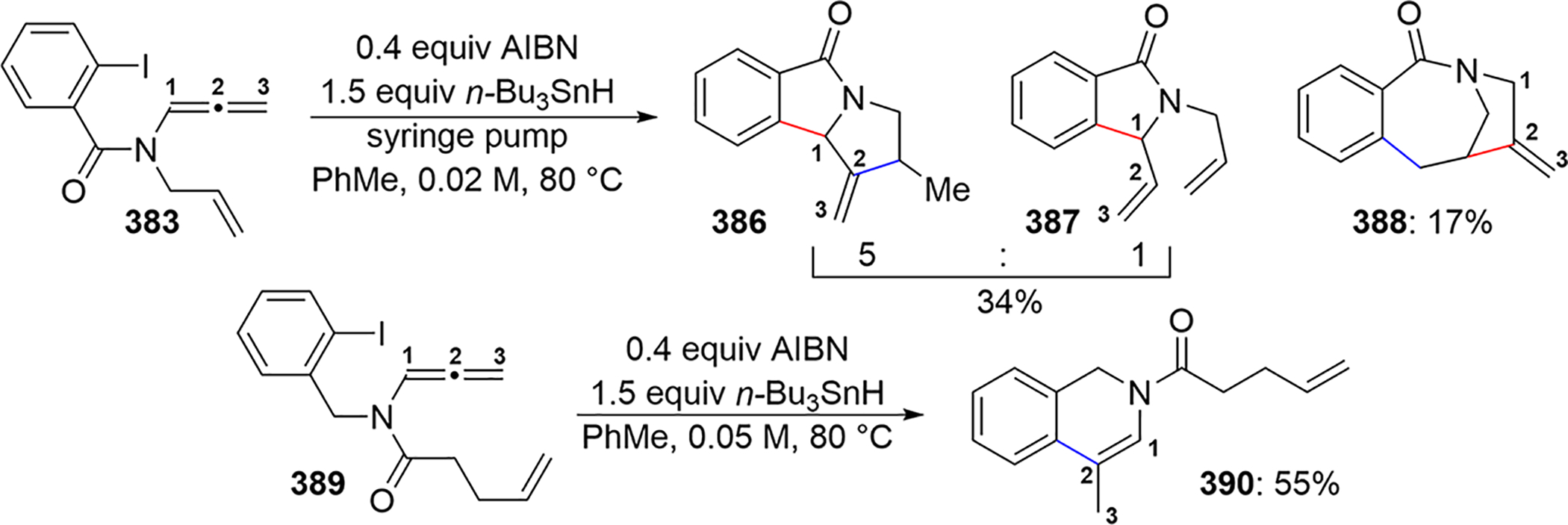
Tandem Radical Cyclizations of Allenamides 383 and 389
Treatment of the aryl iodide 383 with n-Bu3SnH and AIBN produced the corresponding aryl radical 384, which attacks the proximal C1 of the allene to afford 385. The vinyl radical 385 either undergoes 5-exo-trig cyclization or hydrogen abstraction to yield 386 or 387, respectively. Alternatively, rotamer 384′ may undergo 7-exo-trig cyclization onto the pendant alkene to form 391. This intermediate is responsible for the formation of the bridged heterocycle 388 via attack at C2 of the allene (Scheme 121). Hsung reasoned that the allenamide 389 does not participate in tandem cyclization due to faster hydrogen abstraction by the stable allylic radical 392.
Scheme 121.

Proposed Mechanisms for the Formation of 386–388 and 390
In 2012, the Ma group developed a tandem radical addition/cyclization reaction of allene-enes.156 Traditionally, radicals add in a chemoselective fashion to an allene over a competing alkene group, proceeding in an “allene-to-alkene” fashion.119,157 In contrast, Ma reported that a perfluoroalkyl radical, generated from perfluoroalkyl iodide, chemoselectively attacks the alkene moiety of 393; the subsequent 5-exo-trig cyclization of the resulting alkyl radical occurs at C1 of the proximal allene (Scheme 122) to give vinyl iodides of the form 394, which undergo dehydroiodination with TBAF to form the corresponding allenes 395.
Scheme 122.

Tandem Radical Addition/Cyclization of Allene-enes 393 and Dehydroiodination of Vinyl Iodides 394
Wang et al. reported the synthesis of functionalized phenanthrenes 398 via the oxidative cyclization of 2-allenyl-1,1′-biphenyls 396 with α-carbonyl alkyl bromides 397 in the presence of catalytic amounts of CuCl and excess Ag2CO3.158 Various alkyl bromides bearing α-ester, amide, and ketone groups were tested, resulting in moderate yield of the corresponding phenanthrene products. Allenes and 1′-phenyl rings substituted with a variety of groups were also examined to showcase the functional group compatibility (Scheme 123).
Scheme 123.

Phenanthrenes via Oxidative Cyclization of 2-Allenyl-1,1′-biphenyl 396 and α-Carbonyl Alkyl Bromide 397
In the presence of stoichiometric amounts of TEMPO or BHT, only trace amounts of the desired phenanthrenes were obtained. Products derived from the trapping of 397 with TEMPO or BHT were observed by GC–MS. On the basis of previous literature precedent and experimental evidence, Wang proposed the mechanism shown in Scheme 124. The reduction of 399 by Cu(I) and Ag(I) yields radical species 400, which attacks the less substituted terminal allene carbon in a regioselective fashion to afford vinyl radical 401. The cyclization of 401 assembles the phenanthrene scaffold of 402, which is subsequently oxidized to 403 by Cu(II) and Ag(II). The deprotonation of 403 gives the final product 404.
Scheme 124.

Mechanism for Oxidative Cyclization of 2-Allenyl-1,1′-biphenyl and α-Carbonyl Alkyl Bromide
4.2. Tandem Reactions Initiated by Sulfonyl Radicals
In 1995, Hatem described the chemoselective radical addition of tosyl bromide to allene-enes of the form 405 to afford vinyl sulfones 406.157 The tosyl radical selectively adds to C2 to yield an allylic radical, which adds to the alkene moiety in a 5-exo-trig fashion in the majority of the allenes that were examined (Scheme 125).
Scheme 125.

Vinyl Sulfone Formation via Radical Addition/Cyclization of Tosyl Bromide to Allene-enes 405
In the case of allene 407, an alternative pathway involves the dehydrobromination of the addition/cyclization product to yield the alkene 408. For allene 409, 6-endo-trig cyclization competes with the 5-exo-trig pathway to deliver products 411 and 412 (Scheme 126).
Scheme 126.

Radical Addition/Cyclization of Tosyl Bromide to Allene-enes 407 and 409
In 1999, Hatem described radical addition/cyclization of tosyl bromide to β-allenyl ketoximebenzoates of the form 413159 (Scheme 127). The allenes examined in the reaction furnished products 414–417 through the mechanisms proposed in Scheme 128. The addition of the in-situ-generated tosyl radical to the allene occurred in the same manner as described in previous reports (see Scheme 125), involving the chemoselective addition to the central allene carbon to form the allylic radical 418. Depending on the substitution pattern of the β-allenyl ketoximebenzoates, differing reaction pathways give rise to the cyclization products 415–417. It is noteworthy to point out that 417 is produced via the 6-endo-trig cyclization of 418, where the regioselectivity is attributed to steric factors that disfavor attack by the C1 radical on the oxime carbon.
Scheme 127.

Radical Addition/Cyclization of p-TsBr to β-Allenyl Ketoximebenzoates 413
Scheme 128.

Proposed Mechanistic Pathways for Radical Intermediate 418
Kang et al. reported tandem radical addition/cyclizations of bis(allenes) 419 initiated by p-tosyl radicals to afford the more stable trans-fused cyclopentanes 420 bearing vinyl sulfone, bromide, or selenide substitution160 (Scheme 129). The reaction presumably proceeds in a manner similar to that of previous reports (see Scheme 61), with regioselective attack of the p-tosyl radical occurring on one of the central allene carbons to yield an allylic radical intermediate, which then attacks the proximal carbon of the remaining allene.
Scheme 129.

p-Ts-Radical-Initiated Tandem Radical Addition/Cyclization Reactions of Bis(allenes) 419
In 2017, Li et al. reported a diastereoselective synthesis of polycyclopentanoids, including diquinanes and triquinanes, that proceeds via tandem radical addition/cyclization of allenes bearing electron-deficient olefins; these reactions are initiated by thiyl radicals161 (Scheme 130). A catalytic amount of ABVN (2,2′-azobis(2,4 dimethylvaleronitrile)) generates a phenyl thiyl radical, which attacks the central allene carbon in a chemo- and regioselective manner to form a stabilized carbon-centered radical 421. Intermediate 421 undergoes a tandem 5-exo-trig/5-endo-trig cyclization to establish the diquinane scaffold. Two quaternary bridgehead stereogenic centers are installed in a single step.
Scheme 130.

Mechanism for Phenyl-Thiyl-Radical-Initiated Tandem Radical Addition/Cyclization of Allene-enes
Various 1,1-disubstituted allenes, bearing aldehyde or ester groups on the olefin, were tested to afford diquinanes 423 in moderate to good yield and with good to excellent dr (Scheme 131a). Aryl thiols bearing various electron-donating and electron-withdrawing groups (EDGs and EWGs) were also examined to test the effect of electronics on the reaction (Scheme 131a). Triquinanes 425 and 427 were prepared in a similar fashion by employing cyclo-alkyl-substituted allenes 424 and the enantiomerically enriched allene 426 (Scheme 131b).
Scheme 131.

Substrate Scope for Aryl-Thiyl-Radical-Initiated Tandem Addition/Cyclizations of Allene-enes
4.3. Tandem Reactions Initiated by Sn Radicals
In 1992, the Hatem group described a tandem addition/cyclization of β-allenyl O-methyloximes 428 initiated by a tributyltin radical. The resultant products were cyclopentenes of the form 429, bearing an amine substituent and a vinyl stannyl functionality.162,163 A HCl-facilitated destannylation transforms 429 to 430 (Scheme 132). A limited scope of alkyl-substituted allenes was examined; however, in these cases, the tributyltin radical regioselectively attacks the central allene carbon to form an allylic radical, which then cyclizes onto the oxime carbon to establish the cyclopentene backbone.
Scheme 132.
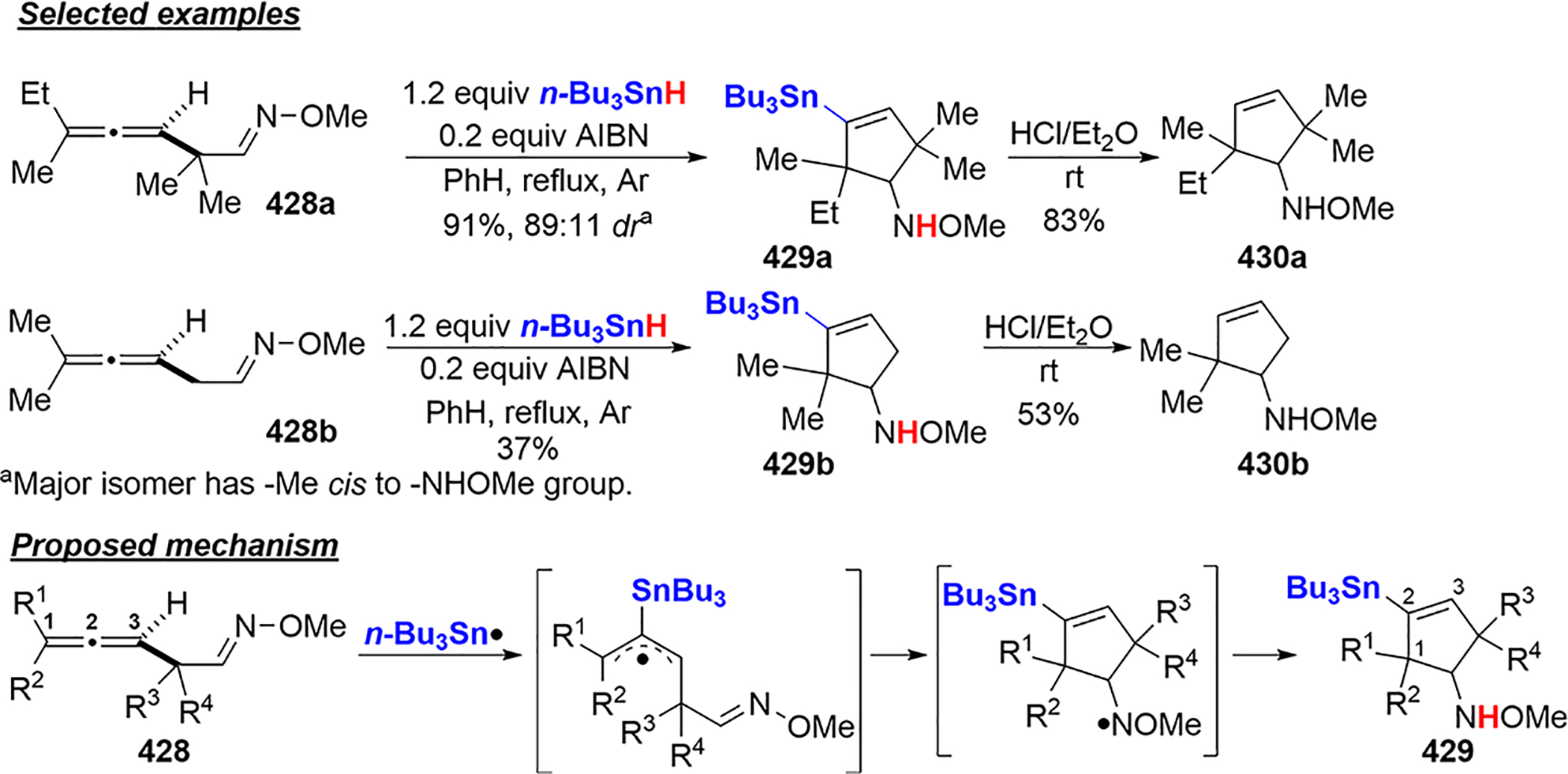
n-Bu3Sn-Radical-Initiated Tandem Addition/Cyclization of β-Allenyl O-Methyloximes 428
In 1994, the Hatem group described a similar tandem addition/cyclization initiated by a tributyltin radical using N,N-dimethyl β-allenyl hydrazones 431 as radical acceptors152,163 (Scheme 133a). Hydrostannylation was observed in most cases to give 432 as the major product. The triene 433, arising from tributyltin radical addition to C3 of 431, followed by 4-exo-trig cyclization and fragmentation, was noted in some instances. Hatem also explored the potential for intramolecular stereo-control by installing a SAMP chiral auxiliary on the hydrazone. In this case, subjecting 434 to the same conditions gave 435 in a de of 50% (Scheme 133b).
Scheme 133.

n-Bu3Sn-Radical-Initiated Tandem Addition/Cyclization of N,N-Dimethyl β-Allenyl Hydrazones
In 1997, the Hatem group presented two additional examples of the tandem addition/cyclization of allene-tethered O-benzyl oximes 436a,b initiated by tributyl tin radicals.163 Increasing the length of the tether between C3 of the allene and oxime by one carbon (Scheme 134) in 436 showed that the regioselectivity of the initial n-Bu3Sn radical attack still favors the less substituted allene C1. However, minor amounts of product arising from the attack of the n-Bu3Sn radical at C2 of the allene were noted. The authors reasoned that the exclusive C1 attack in allenes with n = 1 was due to the disfavored 4-exo or 6-exo modes of the cyclization of intermediate 442 as compared with the favored 5-exo cyclization of 441. In addition, the attack of the n-Bu3Sn radical on the sp-hybridized central allene carbon leads to more electron repulsion as compared with the attack on the sp2-hybridized carbon.
Scheme 134.

n-Bu3Sn-Radical-Initiated Cyclization of Allene-Tethered O-Benzyl Oximes 436
In 2000, Hatem subjected β-allenylbenzoyloximes 443 to n-Bu3SnH under radical conditions and found that the product distribution depended strongly on the allene substitution pattern. Both steric and polar effects influenced the reaction pathway.152 For example, when R3 = H in 443, cyclopentene 444 was isolated exclusively, suggesting that the major pathway involved the chemo- and regioselective addition of the tin radical to the central allene carbon, followed by 5-exo-trig cyclization (Scheme 135a). When n-Bu3SnH was employed, the yield of 444 decreased when C1 of 443 was sterically hindered; when R3 = Me, the cyclopentene was produced in good yield in some cases (Scheme 135b). When R3 = Ph or P(O)(OEt)2, the 6-endo-trig cyclization product 446 dominates with allenes, where C1 is sterically hindered. Chemoselectivity is an issue, as indicated by the competitive formation of the iminyl radical intermediate 447, which furnishes 448 and 449 (Scheme 135c,d). Notably, for allenes bearing an EWG (phenyl and phosphonate groups) on the oxime carbon, the 6-endo process outcompetes the 5-exo ring closure. Hatem attributed this change in regioselectivity to a β-substituent effect; that is, a nucleophilic radical preferentially attacks the carbon that is not substituted with the EWG.164
Scheme 135.

n-Bu3Sn-Radical-Initiated Cyclization of β-Allenylbenzoyloximes 443
In 2001, Hatem reported a nBu3Sn-radical-initiated tandem addition/cyclization of allene-tethered hydrazones and oximes using identical reaction conditions to previous reports.165 As discussed in Scheme 136, the five-membered ring of 451 arises from attack of the tin radical at C2, followed by 5-exo cyclization of the attack of the resulting nucleophilic carbon radical on the oxime carbon. In contrast, the formation of 452 occurs via the initial attack of the Sn radical at C3, followed by 4-exo cyclization onto the oxime carbon. The attack of the Sn radical on C2 is followed by the nucleophilic attack of a carbon radical on the oxime nitrogen to deliver 453. Regioselectivity in the carbon-based radical cyclization was rationalized by the steric and electronic effects of the R groups as well as the electron-withdrawing power of the Y group. (When σ > 0, Y is electron-withdrawing.)
Scheme 136.

n-Bu3Sn-Radical-Initiated Tandem Addition/Cyclization of Allene-Tethered Hydrazones and Oximes
A comparison of the σ values of the three Y groups (Scheme 136) indicates that the C=N of the oxime is most activated for attack by a nucleophilic radical when Y = OBz and most deactivated when Y = N(Me)2. In precursors of the form 450 that bear the same Y group, increasing the sterics of R disfavors the 5-exo mode of cyclization. When the sterics of the R groups are similar (e.g., i-Pr vs CF3), polar effects play a bigger role in determining the regioselectivity; for example, a C=N bond that is more polarized by virtue of being attached to a polar group favors 5-exo cyclization. When Y = OBz, the favored 5-exo cyclization to the activated C=N is less sensitive to polar effects exerted by less hindered R groups, such as H, Me, and CF3.
In 2003, Alcaide and coworkers described a tandem reaction of 2-azetidinone-tethered allenynes 454 and 456. The reaction features an initial attack of a triphenyltin radical on an alkyne, followed by the attack of the resulting vinyl radical on the central allene carbon (Scheme 137).166 Both racemic and enantiopure allenynes were subjected to Ph3SnH, furnishing bicyclic β-lactams in moderate yield and with excellent stereoselectivity. Compounds 455 and 457 were proposed to form via the initial addition of a triphenyltin radical to the terminal alkyne carbon to give a vinyl radical intermediate, which subsequently cyclizes at C2 of the allene.
Scheme 137.

Ph3Sn-Radical-Induced Cyclization of 2-Azetidinone-Tethered Allenynes
In 2007, Alcaide and coworkers extended their previous reports of triphenyltin-radical-promoted cyclizations of 2-azetidinone-tethered allenynes to bromovinyl and halo-aryl allenes 458–464, providing unusual bi- and tricyclic β-lactams 465–471 in a regio- and stereoselective fashion.148 Subjecting 458–464 to Ph3SnH and catalytic AIBN converts them to the corresponding vinyl or aryl radicals, which regioselectively attack the central allene carbon to form fused seven-membered rings (Scheme 138).
Scheme 138.
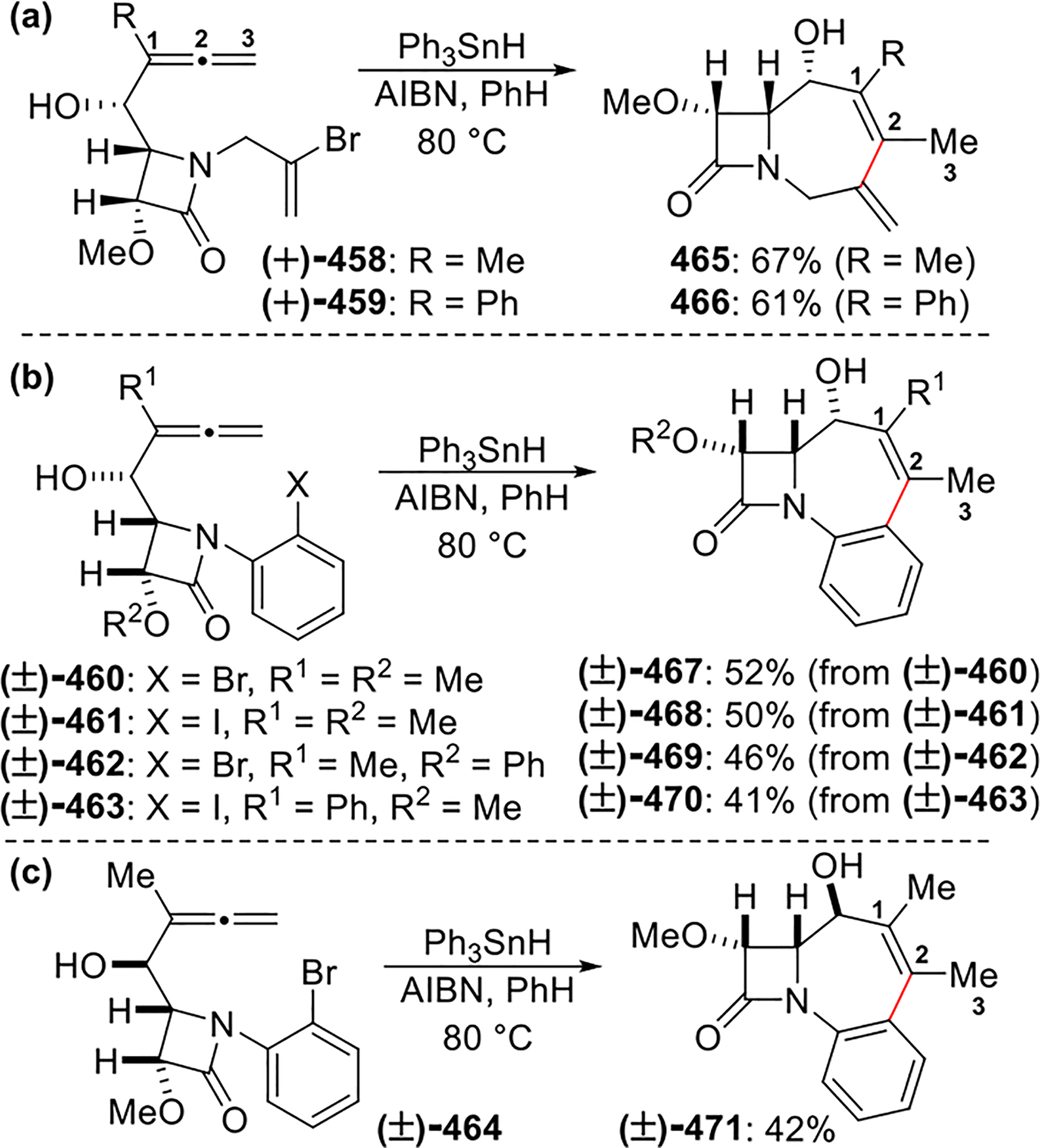
Tin-Radical-Promoted Cyclization of Bromovinyl and Halo-aryl Allenes 459–464
In contrast with previous substrates that contained no olefin tethered to the allene, Hayashi subjected the allene-ene 472 to a tin radical;119 473 was obtained in moderate yield, resulting from the regio- and chemoselective addition of a dibutyliodotin radical to the central allene carbon (Scheme 139).
Scheme 139.

Dibutyliodotin-Radical-Induced Tandem Addition/Cyclization of Allene-ene 472
4.4. Tandem Reactions Initiated by Other Metal and Nonmetal Radicals
4.4.1. Silyl Radicals.
The Pattenden group developed a tris(trimethylsilyl)silyl-radical-promoted cyclization of alkenyl allenecyclopropanes 475 and 476 to produce [5.6.0]- and [6.6.0]-1,3-dienes 480 and 481, which are desilylated by TBAF to form 482 and 483, respectively.167 The reaction pathway was proposed as shown in Scheme 140. The regioselective addition of the silyl radical to the terminal alkene carbon gave the alkyl radical 477, which cyclized at C1 of the allene to form vinyl radical 479. Fragmentation and radical ring opening of the cyclopropane ultimately furnished allene 479 in good yield. Interestingly, when n = 1, a prolonged reaction time converted 479 to its isomer 480/481. Alternatively, heating pure 479 in the presence of AIBN gives 480/481 in similar yield. An allenecyclopropane fused with a cyclohexane (n = 2) was also compatible with the reaction conditions, giving 480/481 and 482/483 in moderate yields.
Scheme 140.
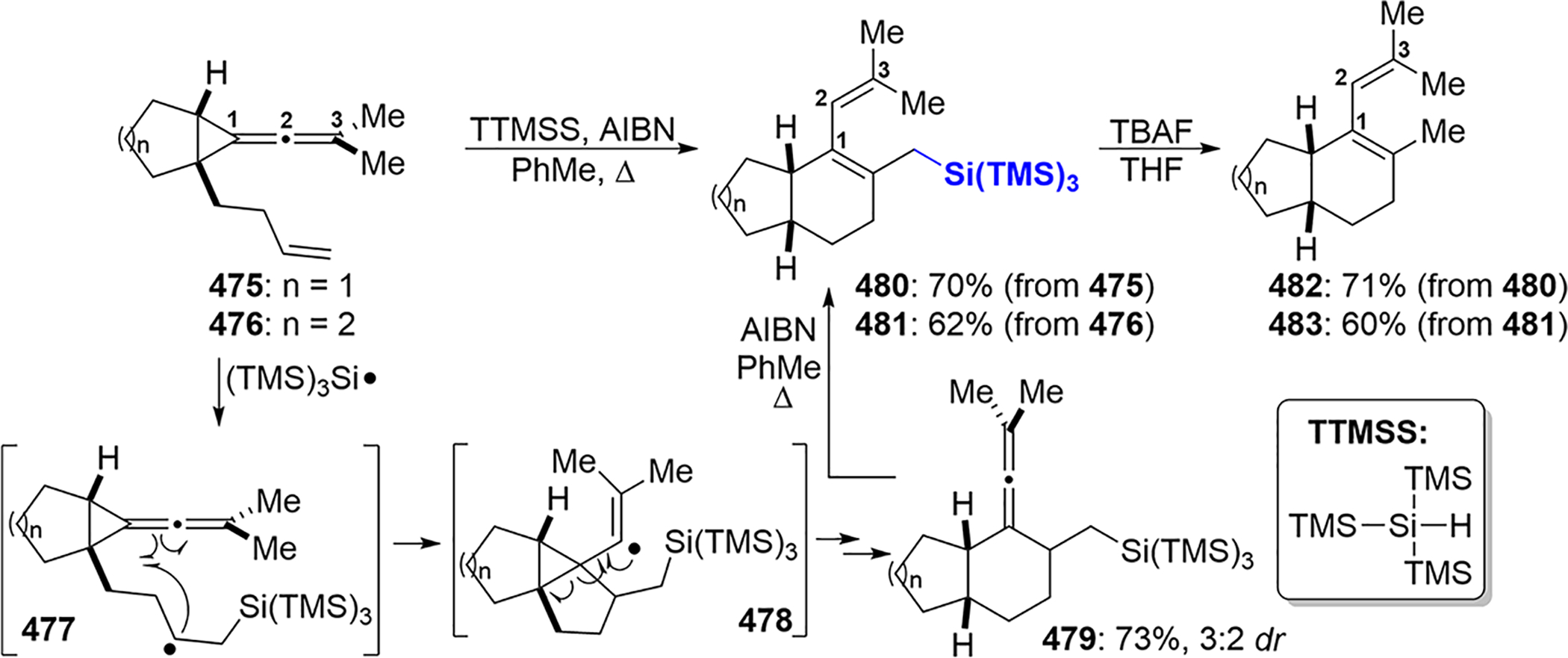
Tris(trimethyl)silyl Radical Promoted Cyclization of Alkenyl Allenecyclopropanes 475 and 476
The Pattenden group studied the effect of the tether length between C1 of the allene and the internal alkene carbon (Scheme 141). When n = 3, no formation of seven-membered ring was observed; rather, 485 was isolated in 80% yield. When n = 1, only 486 was recovered; the lack of product formation was attributed to the steric demand of the bulky tris(trimethylsilyl) group. To study the chemistry of the system when n = 1, a similar primary alkyl radical was generated from iodide 487. Instead of undergoing unfavorable 4-exo cyclization, the radical intermediate engages in 1,4-H transfer and cyclopropane ring opening to give 490 in 43% yield.
Scheme 141.

Effect of Tether Length on Silyl-Radical-Promoted Cyclization of 484 and 486
4.4.2. Indium Radicals.
In 2008, Hayashi et al. described an indium-radical-initiated hydroindation of allene-enes 491 to produce cyclic 492 bearing a vinyl indium functionality.119 Subsequent protonation of 492 yields the corresponding alkene 493–497 in good-to-excellent yield and with moderate-to-good dr (Scheme 142a). The reaction was proposed to proceed through the initial addition of the indium radical to C2 of allene 491 to form the allylic radical 500, which then undergoes a 5-exo-trig cyclization. Hydrogen abstraction of 501 by HInCl2 regenerates the indium radical and furnishes the product 492.
Scheme 142.
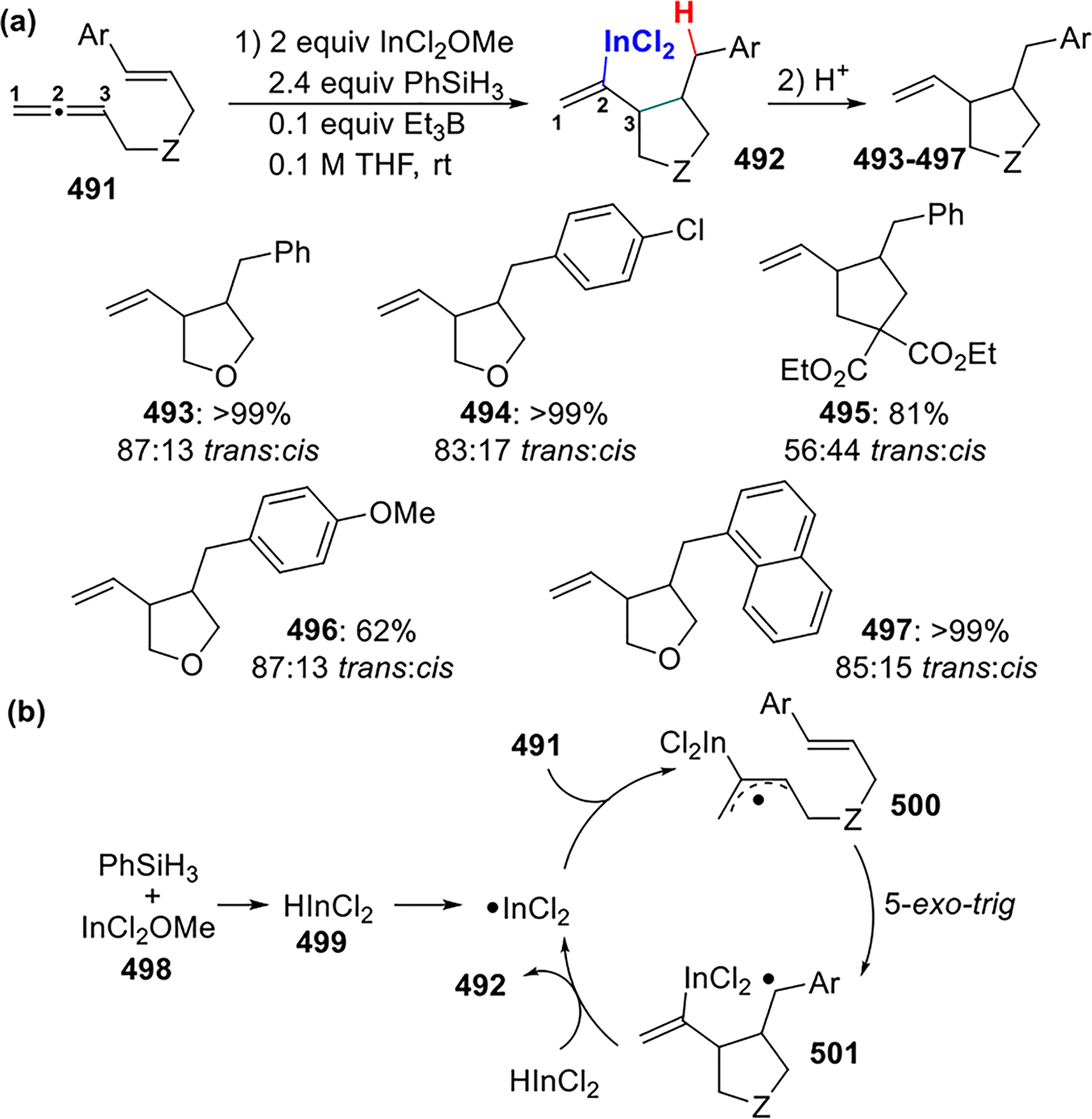
Indium Radical-Initiated Tandem Addition/Cyclization of Allene-enes 491
5. ALLENE CYCLIZATION INVOLVING POTENTIAL DIRADICAL INTERMEDIATES
Whereas sections 1–4 discuss the addition of carbon- and heteroatom-based radicals to allenes, there are other reactions of allenes that are likely to involve radical intermediates. As methods for the preparation of allenes continue to be reported on a regular basis, there is a high likelihood that these compounds will be of significant interest as key building blocks for the construction of complex polycylic scaffolds, explorations of new chemical space, and the development of new reaction methodologies. Thus section 5 is focused on methods involving the generation of presumed diradical intermediates with allenes in reactions that include [2 + 2] cycloadditions, enyne–allene reactions, and several others. Reactions and mechanistic experiments supporting a stepwise diradical mechanism will be discussed, along with challenges and opportunities in improving the regio- and stereochemical control in these potentially powerful transformations.
5.1. [2 + 2] Cycloadditions
Methods that employ two alkenes in formal or concerted [2 + 2] cycloadditions are well-established, with several excellent reviews available that focus on reactions involving diradical mechanisms.168–170 In contrast, methods that couple an allene, alkene, or alkyne to an allene in a [2 + 2] fashion are less common but have been recently gaining attention because the methylenecyclobutane derivatives can be employed as useful precursors to natural products and other complex molecules.169 Known [2 + 2] reactions involving an allene and another π-system are readily categorized by subdividing these transformations into three categories: thermal cyclizations, photo-cyclizations, and metal-catalyzed cyclizations. The next section focuses on aspects of thermal [2 + 2] cycloadditions; whereas the bulk of the reactions are proposed to proceed through a diradical mechanism, an improved understanding of this chemistry through additional mechanistic studies is desirable. Nonetheless, useful insight into reactivity patterns does emerge from a discussion of how the substrate structure influences the regio-, chemo-, and stereoselectivities of the [2 + 2] reaction as well as isomerization of the precursor allenes, competing dimerization, and other side reactions.
5.1.1. With Allenes.
Allene [2 + 2] cycloadditions have been demonstrated under thermal conditions. However, recent reports are scarce, likely due to the harsh temperatures and further reactions of the initial products, as discussed in more detail later. Despite these limitations, the mechanisms of allene–allene [2 + 2] reactions remain a topic of debate that may be resolved by further experimental investigations. Two mechanistic possibilities include a concerted [π2s + π2a] cycloaddition or a stepwise diradical [2 + 2] cycloaddition. Evidence disfavoring the concerted pathway, as discussed in section 5.2, arises from experimental results showing that the regioselectivity of the observed products aligns better with a diradical mechanism. The high level of stereospecificity can be rationalized by either the fast ring closure of the diradical intermediate or the generation of such species via individual stereospecific steps.171 This section focuses on the rationale for diradical mechanisms involving allene dimerization reactions.
Lebedev was the first to report the products of allene dimerization in the early 20th century (Scheme 143).172,173 Depending on the reaction conditions, dimers 502 and 503, trimers 504–507, or oligomers from reactions of 504–507 with 17c, were observed. Yields of 503–507 are reduced when the thermally induced dimerization is carried out in dilute solutions of benzene, giving 502 in excellent yield.174
Scheme 143.

Various Oligomeric Byproducts from Thermal Dimerization of Allenes
Analogous to thermal [2 + 2] reactions of allenes and alkenes with alkynes, allene–allene cycloadditions are proposed to proceed via stepwise diradical pathways, as opposed to concerted [π2s + π2a] mechanisms, through plausible intermediates highlighted in Scheme 144. Center–center attack (C2–C2) involving 17c results in a perpendicular, freely rotating diradical 508 that contains two stabilized allylic radicals. Minimization of steric interactions in the transition state suggests that the process occurs though a “cross-like configuration”, eventually resulting in a perpendicular arrangement of the allyl radicals in 508. Radical recombination results in ring closure to 502. An alternative pathway to 502 involves the terminal–terminal attack (C3–C3) of 17c to give a bisvinyl diradical intermediate 509. A minor byproduct, 1,3-dimethyle-necyclobutane 503, is often observed under thermal conditions and is proposed to arise from a C2–C3 attack of 17c to give 510, followed by ring closure.
Scheme 144.
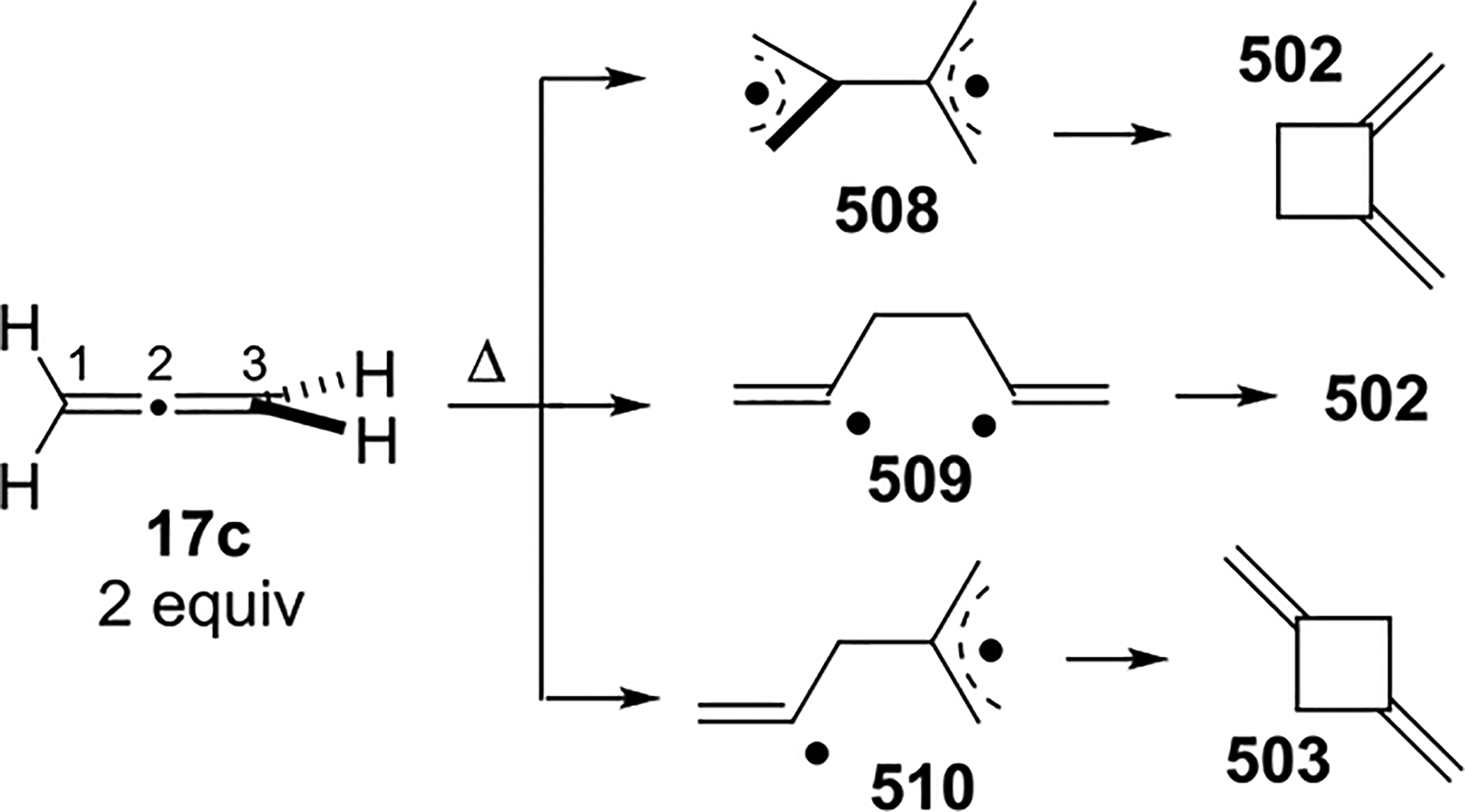
Plausible Diradical Intermediates and Resulting Dimers Formed via Allene Dimerization
Support for a diradical pathway and investigation into the plausibility of intermediates 508–510 has been demonstrated both computationally and experimentally. Johnson has carried out computational analysis at the CCSD(T)/6–311+G(d,p)//B3LYP/6–311+G(d,p) level and identified two possible transition states for 508 (Scheme 144) that differ in the geometry of the approach of the allenes.175 Furthermore, a concerted transition state was not located. The energetically lower transition state to form 508 results from an orthogonal arrangement of the reacting allene (calculated energy barrier of 34.5 kcal/mol); the alternative transition state arises from the “side-on” reaction of the two allenes, resulting in a steric clash of the hydrogens in 17c and a higher energy barrier (40.3 kcal/mol). The barrier for the formation of diradical 510 was calculated to be 41.5 kcal/mol, much greater than C2–C2 attack. The higher energy barrier is attributed to the instability of the vinyl radical in 510. Presumably, the presence of two vinyl radicals imparts an even higher energy barrier to 509 as compared with 510; therefore, 502 is assumed to arise solely from 508.
Experimental evidence of stepwise diradical pathways has been provided by several groups.176,177 Kiefer compared the ratio of products obtained from the thermal dimerization of 2-methyl-2,3-butadiene 24 and the thermolysis of 4,5-dimethylene-3,3,6,6-tetramethyl-3,4,5,6-tetrahydropyridazine 511 (Scheme 145). The dimerization of 24 yields regioisomers 512–514, with 512 as the major component. To investigate if the thermal dimerization of allenes proceeds through a bisallylic diradical intermediate, 511 was subjected to either thermal or photochemical conditions. Under thermal conditions, the product ratios obtained with 511 were identical to those obtained using 24, indicating that the bisallylic diradical 508 (Scheme 144) is a likely intermediate.
Scheme 145.

Dimethylenecyclobutane Derivatives Obtained via a Presumed Diradical Intermediate
Other molecules have been used to probe the involvement of diradicals in a [2 + 2] reaction of allenes (Scheme 146). For example, Gajewski demonstrated that the pyrolysis of 1,2-bis(dideuteriomethylene)cyclobutane 515 results in a skeletal rearrangement to 518 and 519, potentially via 516.178,179 Four fates for 516 were proposed; at low conversions, the ratio of 518/519 was close to 2:1, indicating the internal rotation about the central bond of 516. The pyrolysis of a 1:1 mixture of 515 and 1,2-bis(methylene)cyclobutane 502 gave little to no dideuterated material, indicating that the ring closure of 516 is faster than cleavage, as no 517 was observed.
Scheme 146.

Pyrolysis of Bis(dideuteriomethylene)cyclobutane 515
To investigate the potential for diradical pathways in intramolecular allene [2 + 2] cycloadditions, Becher et al. utilized meso and racemic bisallenes 520–521 (Scheme 147).180 Central–central attack in both 520 and 521 gives 522 and 523, respectively. These intermediates undergo ring cleavage to furnish 526 and 529 from 522 and 523, respectively. In addition, 522 and 523 can interconvert to 524 and 525 to establish planarity and increase alkene conjugation. Whereas 524 and 525 undergo ring closure to 527 and 528, ring strain and high reaction temperatures can trigger ring opening back to the diradicals. The observation of 524–529 in the thermolysis of 520 and 521 indicates that bond rotation is competitive with ring closure as well as cleavage of the 1,2-dimethylenecyclobutane. This is supported by noting that estimates of the energetics of reaction/activation energies for ring closure and bond rotation give similar values (12–14 kcal/mol rotation barrier for allylic radicals and 10 kcal/mol for ring closure of 1,2-dimethylenecyclobutane).
Scheme 147.

Thermal Intramolecular Dimerization of Racemic and Meso Bisallenes 520 and 521 at 1 mmHg
5.1.2. With Alkenes.
In contrast with allene–allene [2 + 2] cycloadditions, thermal alkene–allene reactions have been extensively studied. Competing allene–allene dimerization is prevented by performing these reactions under dilute conditions or by using an excess of the alkene. As discussed later, regiocontrol in the addition has been demonstrated intramolecularly to form polycyclic scaffolds. However, intermolecular reactions suffer from poor regioselectivity and are ideal for investigating the mechanistic aspects of this chemistry.
Analogous to allene dimerization, [2 + 2] allene–alkene cycloaddition may proceed through either a concerted or a stepwise pathway (Scheme 148).181,182 Concerted reactions likely involve transition states 537 → 540, including a [π2s + π2a] arrangement to yield a stereospecific cyclization. However, it is postulated that the transition states 537 and 538 of a [π2s + π2a] process will significantly differ in their respective heats of formation, leading to different energetic barriers for the formation of 532 and 533, respectively. Variable temperature experiments show similar 532/533 ratios across a range of temperatures, indicating that a concerted pathway is not likely involved. Furthermore, the expected chemoselectivity for a [π2s + π2a] process does not match the experimental outcome, where an electron-deficient alkene reacts with the more electron-rich olefin of the allene and vice versa.
Scheme 148.

Mechanistic Aspect of Thermal Allene–Alkene [2 + 2] Cycloadditions
A concerted [π2s + (π2s + π2s)] process (539 and 540, Scheme 148) was also considered,183 where the alkene interacts with the allene in a six-electron transition state. The expected and observed chemoselectivities are congruent, as the cyclobutane formation occurs at the less hindered site of an alkyl-containing allene. However, other factors, including reaction rates, kinetic isotope effect (KIE) experiments, and product ratios, weaken the support for a concerted pathway in favor of a stepwise diradical process. Furthermore, concerted [π2s + π2s] processes are forbidden according to the Woodward–Hoffmann rules.184
Two stepwise pathways are typically considered for [2 + 2] alkene–allene cycloadditions, diradical and ionic, with an ionic mechanism favored in metal-catalyzed reactions.180 Potential intermediates are highlighted in Scheme 148, where a new σ-bond forms at the central carbon of 530 to give 534 (via C2) or at one of the terminal allene carbons to furnish 535 (via C1) or 536 (via C3). Diradical 534 is proposed as the sole intermediate because this mimics the reactivity of carbon-based free-radical additions to allenes, which predominantly react at the central carbon (C2).185
Taylor et al. published reports181,182 using fluorinated alkenes to provide evidence of a stepwise diradical mechanism in addition to 1,3-dienes (Scheme 149). The reactions of fluorinated alkene 542 and 1,3-diene 541 are proposed to yield diradical intermediates 543 and 544 to ultimately deliver 1,2- and 1,4-addition products 545 and 546, respectively.186 Compound 547 would be expected if the reaction was concerted; however, no 547 was noted. Taylor further employed symmetric and unsymmetric fluorinated alkenes to evaluate the plausibility of diradicals in thermal allene–alkene [2 + 2] cycloadditions (Scheme 150). The reaction of 24 with 548a–b and 542 results in four cyclobutanes (550a–c, 551a–c, 553a–c, and 554a–c) via 549 and 552. Presumably, 549 arises from the formation of a σ-bond between the alkene and the central allene carbon. The preferred geometry for 549 orients the allene and alkene perpendicular to one another, where the rotation of the alkene toward the substituted side of the allene results in the radical recombination to 551a–c and rotation toward the least-substituted carbon gives 550a–c. Vinyl cyclobutane derivatives 553a–c and 554a–c result from 552 via a 1,3-rearrangement of the starting allene 24.
Scheme 149.

Previous Evidence of Diradical Intermediate
Scheme 150.

Proposed Reaction Pathways for Allene/Alkene Cycloaddition under Thermal Conditions
Variable-temperature experiments181 reveal key distinctions between concerted and stepwise pathways in terms of their respective potential energy surfaces. The concerted reaction of 24 and 548a (Scheme 150) would entail two significantly different reaction barriers to furnish 550a and 551a; thus changing the temperature would likely influence the 550a/551a ratio. In contrast, a stepwise pathway involves rate-limiting diradical formation, followed by relatively small barriers for ring closure of 549. Thus changing the temperature would not be expected to alter the ratio of 550a/551a if the reaction is stepwise. Experimentally, only small variations in the 550/551 ratios are observed, which is more consistent with a diradical, stepwise path.
Pasto and coworkers showed that reactions of monoalkylallenes 26 and 555a,b with 542 (Scheme 151)187 delivered E/Z stereoisomers 559 and 560. In theory, either a concerted or diradical mechanism could be inferred from ratios of 559/560. A concerted [π2s + (π2s + π2s)] process predicts 559 as the major product (even as R increases in size) due to steric effects and the preferred direction of the bond rotation about the terminal-substituted allene carbon. Experimentally, it was found that increasing the steric bulk in 26 and 555a,b gives 560 as the major product. The reaction pathway (Scheme 151), supported by KIE experiments, suggests that 542 reacts with the more substituted olefin of 26 and 555a,b to give transition state 556. Pathway A rotates the large substituent in 556 toward the incoming alkene, resulting in a steric clash. However, pathway B rotates the large substituent away from the incoming alkene, minimizing steric interactions to give diradical 558. This explains the observed product ratios, where increased steric interactions in 556 increase the formation of 560 at the expense of 559. Ring closure is favored at the less hindered C3 of the allene, yielding higher ratios of 559 and 560, as compared with competing closure at C1 to give 561.
Scheme 151.

Diradical Pathway for the Allene/Alkene [2 + 2] Cycloaddition To Furnish Methylenecyclobutanes
The above results and reasoning in Scheme 151 are used to infer if radicals are present in other [2 + 2] allene–alkene cycloadditions.187 For example, N-phenylmaleimide 563 (Scheme 152), an electron-poor (LUMO controlled) reagent, could be envisaged to undergo a concerted reaction with the electron-rich olefin of allene 562. The reaction of 563 with allenes (24, 562a,b, R = CH3, CH2CH3, C(CH3)3) under thermal conditions183 showed chemoselectivity consistent with either a concerted [π2s + (π2s + π2s)] or a stepwise diradical process. A [π2s + π2a] pathway was ruled out because an electron-deficient alkene should react with the more electron-rich olefin. Compound 568, a 1:2 adduct, presumably arises from the 1,5-hydrogen atom transfer (1,5-HAT) of the alkyl radical in the diradical intermediate, followed by radical recombination to form the alkene. The resulting diene would then presumably undergo a [4 + 2] cycloaddition to result in the Diels–Alder product 568.
Scheme 152.

Product Distribution from Reaction of Allene with N-Phenylmaleimide
Pasto performed follow-up experiments on previous work carried out by the Kiefer group.188 Diethyl fumarate and diethyl maleate were heated with dimethylallene to give methylenecyclobutanes. High stereospecificity (up to 99%) was observed and used to support a concerted mechanism. However, in this instance and other similar cases, the argument could be made that a high stereospecificity in radical cyclizations can be obtained if the bond formation/ring closure is faster than the bond rotation or competing processes that would give the expected lower stereospecificity. In the reaction outlined in Scheme 153, a proposed diradical intermediate would be at an energy minimum, as represented by 576–579, when the new C–C is marginally shifted from a perpendicular arrangement. This “tilting” toward the allyl radical allows for a better orbital overlap in the ring closure. Of the four possible diradical intermediates, 576 and 578 are expected to be lower in energy because they minimize pseudo-1,3-diaxial interactions in the transition state. This proposal agrees with the experiment in terms of the relative ratios of products 571–575. The alkyne 575 results from the reaction of the alkene at the less substituted carbon of 26, 569, and 146g.
Scheme 153.

Formation of Products from Reaction of Diethyl Fumarate and Alkyl-Containing Allenes via Diradical Pathways
To better understand the nature of diradical intermediates involved in this chemistry, Pasto and coworkers studied reactions of 570 with dimethyl allene 24 and diethyl maleate 583 with 24 (Scheme 154).189 The reaction of 24 with 570 led to trans diesters 580 and 581, arising from a [2 + 2] cycloaddition, plus a cyclohexene 582 that results from the [1,3]-sigmatropic rearrangement of 24 and the subsequent [4 + 2] cyclization. In contrast, the reaction of 24 with 583 gives the expected cis diesters 584–586; however, 580–582 are also observed. The presence of 580–582 is attributed to an internal rotation in diradical intermediates 587 and 589 to minimize steric interactions caused by eclipsing ester groups. In addition to the presence of trans ester products, a significant isomerization of 583 to 570 is observed, with ratios up to 5:1 (583/570) with 24. Control experiments without 24 show no isomerization, suggesting that 24 is required for this process. Thus it was postulated that 570 arises from the cleavage of diradical intermediates 588 and 590. The internal rotation and cleavage events are rationalized by an increase in energy of intermediates 587 and 589 due to increased steric interactions as compared with 588 and 590. This gives a smaller energy barrier for rotation and cleavage in the reaction of 24 and 583. The ring closure event itself is unaffected; therefore, competition between internal rotation, cleavage (from 588 to give 24 and 570), and ring closure is dictated by the relative energies of diradical intermediates. Increased strain in either the allylic or aliphatic radical (for example, when tert-butylallene is used instead of 24) results in increased internal rotation and cleavage, as compared with desired ring closure. The stabilization of diradical intermediates (for example, using methoxyallene instead of 1,1-dimethylallene 24) leads to increased rates of ring closure relative to internal rotation and cleavage.
Scheme 154.

Evaluating Internal Rotation and Cleavage Properties of Diradical Intermediates in Allene–Alkene [2 + 2] Cycloadditions
As shown in the examples discussed above, 1:1 adducts are often the major products if alkyl allenes are employed; however, functional groups other than alkyls can result in additional adducts from cyclodimerization processes or increased reactivity.181 Using a methoxy-substituted allene 146e (Scheme 155a), Pasto noted that 1:1 adducts 591, 592, and 593 comprise the majority of the product mixture.190 The remainder is composed of 1:2 adduct 594 and 2:1 adducts resulting from the allene dimerization of 595 or 596. Chloro-substituted allene 597 (Scheme 155b) allows for the formation of additional adducts as compared with 146e. This includes the formation of the 2:2 adduct 601, 2:1 adducts 600a,b, presumably arising from 599a,b, and the 2:0 adduct 599c. Analogous to reactions of 146e, the reaction of 597 also results in 1:1 adducts 598a–c and the 1:2 adduct 594.
Scheme 155.

Reaction of Methoxy- and Chloro-Substituted Allenes with 542
As already mentioned, intermolecular [2 + 2] allene–alkene cycloadditions suffer from poor regioselectivity (proximal vs distal π-bond of the allene) and side reactions. On the contrary, intramolecular reactions display higher regioselectivity and are tunable to some extent. Padwa reported that the terminal C1–C2 olefin of (phenylsulfonyl)allene 602 (Scheme 156) undergoes a [2 + 2] cycloaddition with a tethered alkene despite the presence of a more activated internal C2–C3 olefin of the allene.191 This stereospecificity was unexpected, as C2–C3 was thought to be more reactive than C1–C2; regioselectivity was attributed to the stability of radical intermediate 603 or 605. When R3 = H (602), the reaction proceeds through diradical 603 to furnish 604. The preference for 6-exo-trig, as opposed to 7-endo-trig, cyclization is consistent with previous work involving the intramolecular addition to an alkene via a carbon radical.192 A switch in regioselectivity was achieved by replacing the H at R3 with Me or Br (602). The nonallylic radical in 605 is tertiary (when R3 = Me) and gains extra stability from hyperconjugation and kinetic stability from steric hindrance. In this case, the two radicals in 605 form a fused cyclopentane–cyclobutane, as exemplified by 606. A further investigation of substitution on the alkene (602, R3 = H) showed that faster reaction rates are achieved with alkyl substitution at R1 and R2, presumably due to the higher stability of the nonallylic radical.
Scheme 156.

Intramolecular [2 + 2] Addition of an Alkene to a (Phenylsulfonyl)allene
The nature of the tether in 602 (Scheme 156) could be altered at C5 to give bicyclic rings of different functionalities. The introduction of an oxygen at position five in 607 (Scheme 157) led to the formation of bicyclic pyran derivative 609 in 74% yield. A 60% yield of 612 is obtained when the oxygen of 607 is replaced by a methylene bridge in 610. The placement of a bis(phenylsulfonyl)methane group at position five results in a good yield (69%) and a faster reaction rate, consistent with the Thorpe–Ingold effect. Increasing the allene tether by one methylene group (616) led to an unexpected product, where a nonallylic radical 617 undergoes a 1,5-HAT (hydrogen atom transfer) to result in a radical situated β to the SO2Ph group, which combines with the allylic radical to deliver trans-diene 618.
Scheme 157.

Effect of Tether Variations on the Thermal Reaction of (Phenylsulfonyl)allenes
Alcaide and coworkers demonstrated an intramolecular allene–alkene [2 + 2] cycloaddition with excellent positional selectivity (Scheme 158).193 Utilizing 2-azetidinone-tethered enallenes 619a–c under thermal conditions results in 621a–c as the sole product. Intermediate 620 arises from formation of a new σ-bond between the allene central carbon and the internal alkene carbon. Radical recombination of the diradical 620 furnishes the tricyclic β-lactam 621, which has utility as a potential antibacterial agent. However, 623 was the authors’ expected outcome based on the likelihood of forming a more stable secondary alkyl radical in 622; however, this leads to a larger ring in 622 as compared with intermediate 620, which is apparently unfavorable.
Scheme 158.

Synthesis of Tricyclic β-Lactams from [2 + 2] Cycloaddition
5.1.3. With Alkynes.
Alkynes, similar to alkenes and allenes, undergo thermal [2 + 2] cycloadditions with allenes. The products are methylenecyclobutenes, which are utilized in materials chemistry as dyes194 and organic semiconductors.195 Whereas the mechanisms of allene–alkene [2 + 2] reactions have been well studied, investigations involving alkynes as coupling partners have not received much attention. Reactions suffer from poor control of regio- and stereoselectivities, high reaction temperatures, and the formation of undesired byproducts. Scattered reports that address these issues and provide some mechanistic insights are discussed in this section.
The product of the [2 + 2] cycloaddition between 624 (R′ = H) and allene 625 (R = H) gives methylene cyclobutene 626 (Scheme 159). Regio- and stereoisomers 627 and 628 are obtained with 625 (R = Me). Additional methylene cyclobutene products 629–632 arise when the substitution is increased on the allene 625 (R = Me) and alkyne 624 (R′ = Me). Analogous to alkenes, the product ratios and reactivity of alkyne–allene [2 + 2] cycloadditions best reflect a diradical mechanism, as opposed to a concerted mechanism involving 633–636. Diradical intermediates 637–640 result from formation of a new σ-bond at the central carbon or at the terminal carbon to give bisvinyl radicals 641–644. Presumably the [2 + 2] cycloaddition proceeds through the allylic biradical intermediate due to energetic considerations; both inter- and intramolecular variants have been explored experimentally and computationally, as described in more detail later.
Scheme 159.

Potential Products of Intermolecular [2 + 2] Cycloaddition of Alkynes and Allenes
Pasto investigated relative reactivities of alkene 646 versus alkyne 645 with allene 24 through an intermolecular competition experiment (Scheme 160).196 Under thermal conditions, the alkyne reacts faster than the alkene, and no observable reaction with 646 was detected. Four products are observed: two 1:1 adducts (647–648) resulting from [2 + 2] cycloaddition and trace amounts of 2:1 (alkyne/allene) adducts 649–650.197 This observed reactivity is opposite that expected for free-radical additions, as alkenes typically react faster than alkynes. It is postulated that the reaction proceeds through a late transition state that depends on the relative stabilization of the radical by attached functional groups. The diradical resulting from the reaction of 24 and 645 is stabilized by functional groups on the allene and alkyne and is considered an endothermic process. A closer look at the energetics to generate two new radical centers and a new C–C bond, while cleaving a C–C π-bond, agrees with this classification. The greater reactivity was proposed to result from a decrease in the heat of formation of the diradical from the alkyne. The heats of formation of acetylene and ethene are 54.2 and 12.5 kcal/mol, respectively, and thus a lower barrier exists for the formation of a diradical intermediate from the alkyne.
Scheme 160.

Competition Experiment Involving an Alkyne, Alkene, and Allene under Thermal Conditions
In 2010, Brummond and Tantillo reported a mechanistic study of the thermal intramolecular [2 + 2] reaction of alleneynes (Scheme 161).198 A concerted process would entail a [π2s + π2a] arrangement in the transition state; this was ruled out based on geometric restrictions. Two diradical pathways (a) and (b) were considered, differing in the size of the initial ring formation. The pathway forming the small ring (a) has a lower barrier for cyclization than the competing pathway (b) due to the formation of a stabilized allyl and vinyl radical 652 when compared with bisvinyl radical 654. In agreement with experimental results, the barrier for the formation of the diradical via the distal π-bond of the allene is lower than for the proximal π-bond. Thus regioselectivity is governed by the stability of diradical intermediates (allyl vs vinyl); the initial σ-bond formation occurs at the central allene carbon, analogous to [2 + 2] cycloadditions involving a π-bond and an allene.
Scheme 161.

Intramolecular [2 + 2] Reaction Pathways Involving the Distal π-Bond of the Allene
Different alkyne substituents affect the outcome of intramolecular reactions from both experimental and computational standpoints.198 Aromatic substituents are often attached to the alkyne, as the resulting benzylic radical is stabilized by delocalization into the aromatic ring. Computational analysis shows that the intermediate radical is almost linear in nature, contributing to a more planar π-system and a lower energy intermediate. In contrast, carbonyl groups α to the alkyne have only a small stabilizing effect. Experiments involving the use of cyclopropyl groups on the alkyne have been carried out by Brummond and Tantillo (Scheme 162).199 Three alkynyl cyclopropyl derivatives 655a–c were used to probe the presence of diradical intermediates. Compound 655a underwent a [2 + 2] cycloaddition with no observed ring opening; however, 655b furnished ring-opened 657–658, supporting the hypothesis that radical intermediates are involved.199 Performing the reaction with 655c also supported the likelihood of radical over cation intermediates. Electrostatic potential maps suggested that diradical pathways are more likely than zwitterionic pathways200 due to the small difference in charge build-up in the intermediates for the conversion of 655a to 656 and 655c to 659–660.
Scheme 162.

Investigating the Possibility of Diradical Intermediates Using Cyclopropyl Probes
In addition to their work in [2 + 2] intramolecular alkene/allene cycloadditions, Aragoncillo and coworkers reported an alkyne variant that furnishes 665a–e (Scheme 163).201 Subjecting 661 to thermal conditions gave a new σ-bond between the proximal alkynyl carbon and the central allene carbon to yield allylic/vinyl radical 664. Whereas the distal alkynyl carbon could potentially form a new σ-bond with the central allene carbon, a higher energy barrier for the formation of 662 relative to 664 is likely; this, coupled to a lack of stabilization in the resulting vinyl radical, disfavors this process. Thus 663a–e are not observed in the reaction.
Scheme 163.
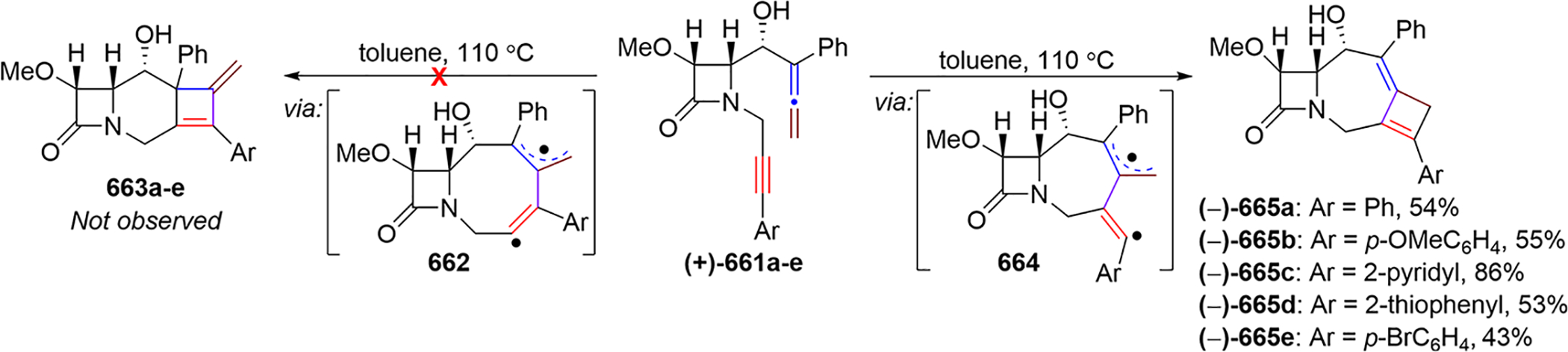
Intramolecular [2 + 2] Cycloaddition of Allenynes
The use of the diyne–diallene 666 allows for a double [2 + 2] cycloaddition, presumably via a tetraradical intermediate 667 (Scheme 164). Stopping the reaction prematurely results in the recovery of starting material and the bis(β-lactam-allenyne) 668, indicating that both cyclizations may occur concurrently.
Scheme 164.

Intramolecular [2 + 2] of Diallene–Diynes To Result in Tricyclic β-Lactam Dimers
In summary, allenes have been utilized in thermal [2 + 2] reactions with molecules containing π-bonds, including other allenes, alkenes, and alkynes; however, the intermolecular control of regioselectivity in these reactions is usually poor. In contrast, intramolecular variants often display excellent selectivity, depending on the substituents installed on the allene, and are synthetically useful. Whereas the field of allene [2 + 2] cycloaddition has been largely dominated by thermal processes, focus has more recently shifted to metal-catalyzed [2 + 2] cycloaddition reactions, which are not thought to proceed through diradical mechanisms. Despite their limitations, non-metal-catalyzed [2 + 2] cycloadditions are still of great interest due to their functional group tolerance, the retention of unsaturation in the product for further functionalizations, the “green” aspects of this approach, and the general interest in the mechanistic details. In addition, diradical intermediates have been implicated in the cleavage of double-stranded deoxyribonucleic acid (DNA) to influence disease pathways and thus continue to be pertinent to the medicinal chemistry community.147 Future opportunities include the development of useful intermolecular cycloaddition reactions of allenes with improved selectivities and yields, advancing the photochemical variants of this chemistry and the applications of electrochemical variants. To the best of our knowledge, the addition of electrochemically generated radicals to allenes has not been developed, but such variants could further the general understanding of the mechanism and provide insight into other similar systems.
5.2. Enyne–Allene Radical Cyclizations
Since the initial report of the Bergman cyclization, diradical intermediates have been of great interest due to their antitumor activities and their ability to engage in cascade reactions.202 In the Bergman cyclization, a diphenyl radical intermediate is proposed to result from a corresponding enediyne under thermal conditions. A benzene ring results upon trapping with hydrogen. Antibiotics such as esperamicin and calicheamicin can be synthesized via this method, with the enediyne core furnishing the diradical intermediate in situ under thermal conditions. These diradical intermediates are indicated in the cleavage of double-stranded DNA and remain of great interest to medicinal chemists. In most cases, the Bergman cyclization of acyclic enediynes requires high reaction temperatures (>130 °C); however, the analogous reaction of cyclic enediynes can occur at much lower temperatures (37 °C) due to increased ring strain and are more applicable to drug design.
Following Bergman’s seminal report, variations on this chemistry have been explored, including reactions of enyne–allenes, such as the Myers–Saito and Schmittel cyclizations (Scheme 165). The Schmittel cyclization involves the formation of a new bond between C2 and C6, resulting in a fulvenyl diradical intermediate. On the contrary, the Myers–Saito cyclization involves the reaction of the central allene carbon C2 and the terminal C7 carbon of the alkyne to form an intermediate α,3-tolyl diradical. There are several excellent sources for interested readers to seek further information on the Myers–Saito and Schmittel cyclizations.203–206
Scheme 165.
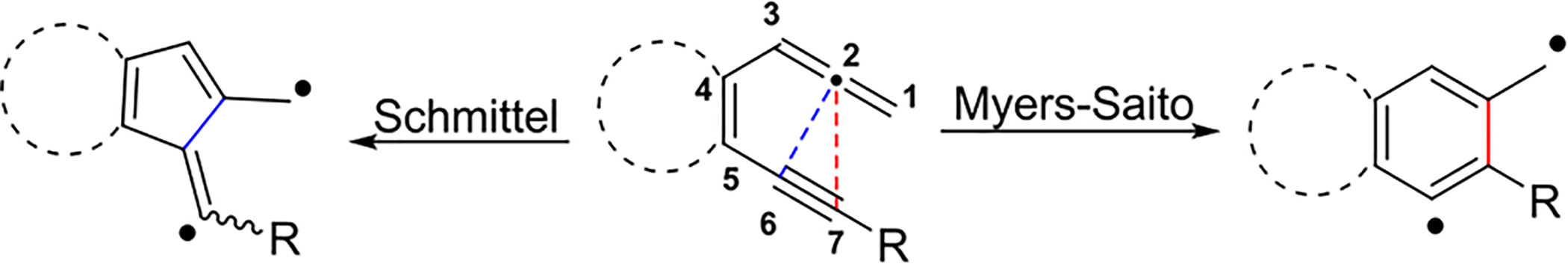
General Depiction of the Schmittel and Myers–Saito Cyclizations
5.2.1. Myers–Saito Cyclization.
The Myers–Saito cyclization was independently reported by Myers and Saito in the late 20th century (Scheme 165).204,205 Saito postulated that the Bergman cyclization requires high temperatures due to the bonding distance between the two carbons that form the initial C–C σ-bond. It was predicted that utilizing enyne–allenes would lower the activation barrier for the reaction due to a decrease in the bonding distance between C2 and C7. Accordingly, mild reaction temperatures can be utilized with acyclic enyne–allenes (R = H, alkyl) to furnish α,3-didehydrotoluene derivatives. Studies describing the applications of this useful transformation are discussed in the following sections.
5.2.1.1. Mechanism of the Myers–Saito Cyclization.
The mechanism of the Myers–Saito cyclization has been studied both experimentally and computationally. Saito showed that performing the reaction in cyclohexadiene (1,4-CHD) led to 672, along with 673 and dimer 674 (Scheme 166).204 DNA cleavage studies indicate that enyne–allenes react similarly to enediynes and proceed through diradical intermediates. However, the nature of the intermediate of the Myers–Saito reaction is solvent-dependent.205 In protic solvents, diradical 671 displays ionic character and is best represented by 670. In nonpolar solvents, such as 1,4-CHD, 671 possesses more diradical character and participates in a stepwise pathway. Overall, the intermediate of the Myers–Saito cyclization is best described by the resonance structures 670 and 671 and is dependent on solvation effects, with polar protic solvents stabilizing charged intermediates. Finn published a comprehensive report on various functional groups that favor 670 or 671.207
Scheme 166.

Radical Trapping Experiments Using 1,4-CHD
Utilizing 675 in methanol or 1,4-CHD results in different ring-opened products, 677 (via 676) and 679 (via 678), respectively (Scheme 167). Shibuya reported ionic character could be minimized by introducing electron-withdrawing groups on the ring, resulting in more cleavage of double-stranded DNA via diradical intermediates.208 Computational analysis was used to further investigate why the Myers–Saito proceeds at much lower temperature than the Bergman cyclization. Morokuma reported the lower activation energy of the Myers–Saito (21–23 kcal/mol) versus the Bergman (32 kcal/mol) cyclization is due to smaller four-electron repulsion and smaller overlap between the two in-plane π-bonds.209 In addition, as the C–H bonds are out-of-plane in the transition state, the methylene radical is perpendicular to aromatic π-system, suggesting conjugation does not contribute to the observed lower activation energy.
Scheme 167.
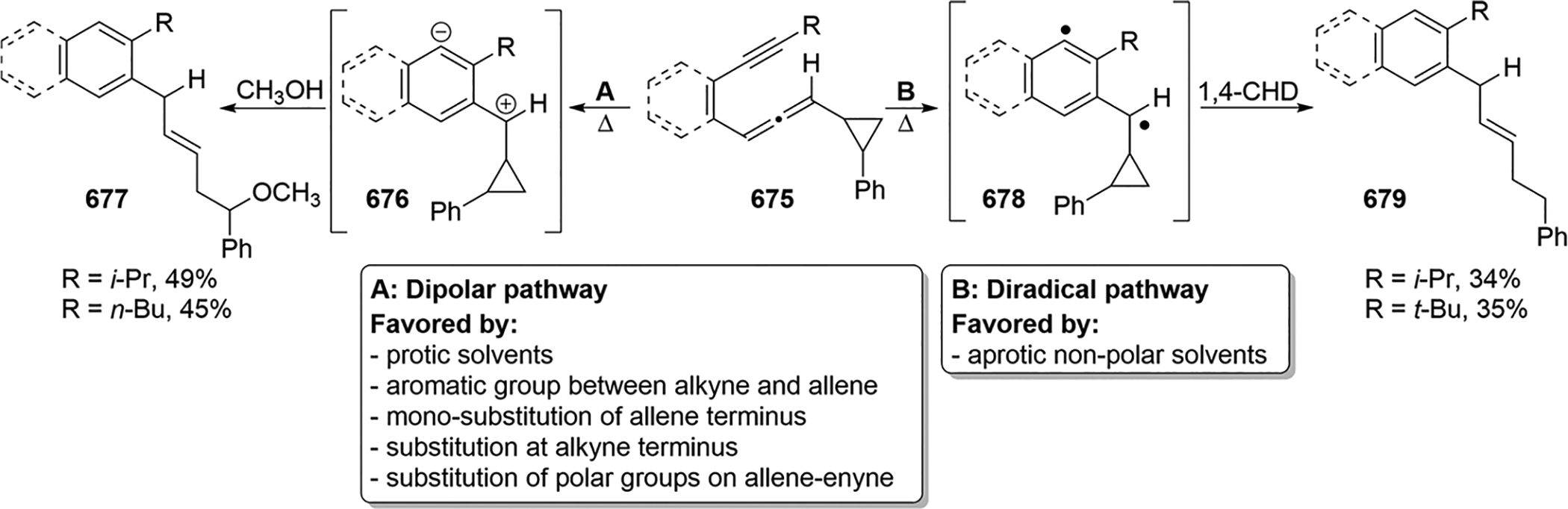
Cyclopropyl Trapping Experiments from the Dipolar and Diradical Pathways
5.2.1.2. Generation of Enyne–Allenes and Substituent Effects on Myers–Saito Cyclization.
The enyne–allene core 682 is accessed via [2,3]- and [3,3]-sigmatropic rearrangements of 681.210 Saito showed that a [2,3]-sigmatropic rearrangement of 681a generates allenylphosphine 682a (Scheme 168); 682a can be trapped with 1,4-CHD to give 684a. Saito later expanded the scope of the [2,3]-sigmatropic rearrangement to include the sulfoxide 681b and vinyl ether 681d.211
Scheme 168.

Generation of a Variety of Allene Precursors from [2,3]-Sigmatropic Rearrangement
Other allene substitution patterns have been investigated (Scheme 169). Grissom utilized aromatic enyne–allenes with various substituents at C3 in 685 (R = P(O)Ph2) and observed no cyclization under thermal conditions, presumably due to the A1,3-strain between the C3 substituent and the ortho substituent of the aromatic ring. In addition, Grissom also investigated substitution at C1 in 686, including a phosphine and a phenylsulfoxide group.212
Scheme 169.

Various Enyne–Allene Substitution Patterns
As mentioned previously, Finn summarized the effects of different substituents on the outcome of the Myers–Saito cyclization.207 Early work by Saito indicated that decreasing the distance between the reactive centers is critical to achieving increased rates relative to Bergman cyclization.204 Substituent effects support this hypothesis: Large bulky groups on the alkyne, such as t-butyl (Scheme 169), dramatically decrease the rate due to the increase in distance between the reactive centers (3.25–3.3 Å). However, if the reactive centers in the enyne–allene are too close (3.18–3.19 Å), then the compounds are unstable. Substituents at the alkyne terminus show little electronic effects on the cyclization rates, based on observations using substrates with electronically differentiated para-substituted aromatic rings on the alkyne. Electronically differentiated groups on the allene terminus of 685 also have little effect on the rate of cyclization. These results provide support for an early transition state, where the nascent benzylic radical or cation is not in conjugation with the aromatic system but resides orthogonal to it.
5.2.1.3. Cascade Reactions.
Diradical intermediates in the Myers–Saito reaction can be intercepted with a variety of radical trapping groups or acceptors (Scheme 170). Wang and coworkers developed several cascade reactions using this α,3-didehydro-toluene diradical intermediate.213,214 For example, the intermediate 690 that results from reaction of 689 can undergo a 5-exo-trig cyclization involving the phenyl radical. The resultant alkyl radical undergoes 1,5-HAT to give 692, which engages in a 6-endo-trig cyclization and radical recombination to deliver 693. The Myers–Saito cyclization of enyne–allene 697 results in 698; a 6-exo-dig cyclization, followed by trapping of the benzylic radical in 699 results in 700. Grissom demonstrated the 5-exo-trig cyclization of 695 (from enyne–allene 694) to gain access to substituted bicyclic scaffolds 696,215–217 whereas Moore engaged α,3-didehydrotoluene diradical intermediates in polymerization reactions.218
Scheme 170.

Radical Cascade Reactions Utilizing the Myers–Saito Cyclization
The Myers–Saito cyclization provides a mild strategy for the formation of α,3-didehydrotoluene diradicals that have found uses in synthetic, materials, and medicinal chemistry. The diradical may undergo either inter- and intramolecular reactions to furnish polycyclic scaffolds that can be subjected to additional functionalizations. The generation of this type of diradical intermediate through either photochemical or electrochemical means could unlock other useful application of this chemistry, whereas the reduction or oxidation of the benzyl radical can be envisaged as an alternative pathway for further reactivity.
5.2.2. Schmittel Cyclization.
The Schmittel cyclization involves the formation of a new bond between C2 and C6 in 701 and is the predominant pathway when the alkyne is substituted with a large bulky group, such as a tert-butyl or trialkylsilyl group (Scheme 171).219 In addition, computational analysis suggests that the Schmittel cyclization occurs via orbital interactions between vertical and side π-orbitals, whereas the Myers–Saito reaction occurs via vertical–vertical and side–side π-orbital interactions.220 In a classical physical organic sense, either a concerted or stepwise pathway is possible from 703 (Scheme 172). A concerted pathway involves either inter- or intramolecular H-atom transfer in 704 via a six-membered transition state, whereas a stepwise pathway involves the formation of diradical 705, followed by H-atom abstraction to furnish 706. Whereas many of these types of cyclizations are defined by classical views, the Schmittel cyclization is more complicated. Theoretical studies, radical trapping experiments,221 and DNA cleavage studies have all provided compelling evidence of a diradical intermediate. However, as described in the following section, recent mechanistic studies indicate that dynamic effects need to be considered and incorporated into mechanistic explanations for these cyclizations.
Scheme 171.
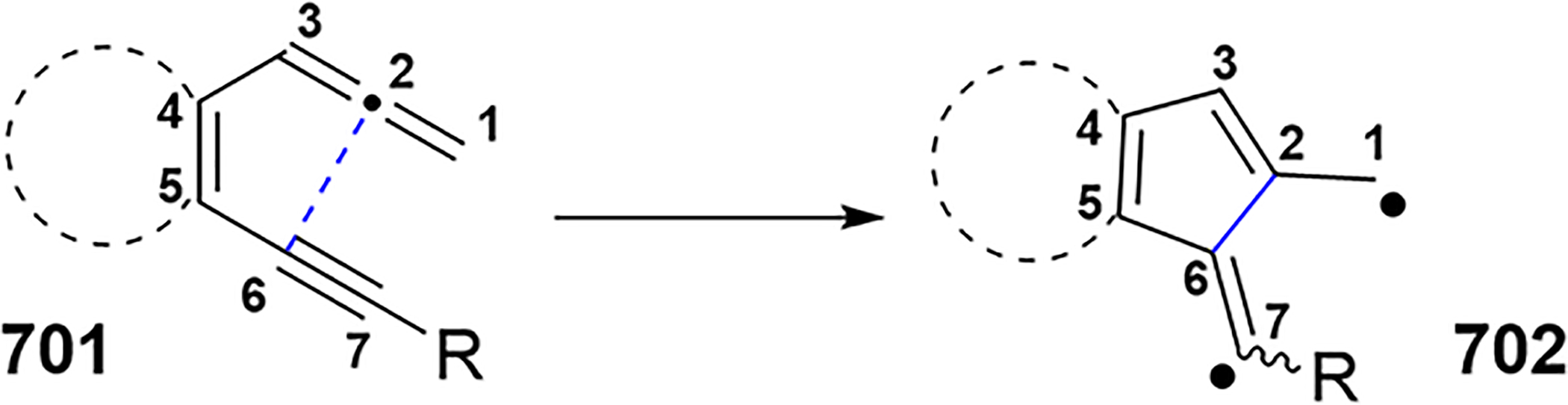
General Depiction of the Schmittel Pathway
Scheme 172.

Proposed Pathways for the Schmittel Cyclization
5.2.2.1. Dynamic Trajectories and KIE Experiments.
Singleton and coworkers have explored the mechanism of the Schmittel cyclization through computations and KIE experiments.222 They postulated that both concerted and stepwise pathways result from the same initial transition state due to their inability to identify a transition state for the diradical intermediate and computed geometric similarities in the initial stages of the reaction. To explain the concerted nature of the reaction, Carpenter’s views on dynamic effects were incorporated.223 Carpenter has discussed multiple examples where a mechanism appears to be concerted but is actually stepwise in nature. In these cases, the barrier of the second step is overcome by an excess of kinetic energy generated by the formation of the intermediate, leading directly to the product. In addition, Engels has found via computational analysis that stepwise and concerted mechanisms have approximately the same free energy of activation.224
Schmittel reported a comprehensive mechanistic study involving a detailed examination of the reactivity in cyclizations where various substituents have been installed at different positions in the enyne–allenes 707a–c (Scheme 173).225 When radical stabilizing groups are present at the allene or alkyne terminus (or both), the stepwise/dynamic mechanism is favored, as evidenced by an observed KIE near unity with 707a,b. This can also be explained by Carpenter’s dynamic model, where the intermediate overcomes the reaction barrier of the second step by an excess buildup of kinetic energy in the first step; this explains the lack of discrimination between hydrogen versus deuterium abstraction. In the absence of a radical stabilizing group, the mechanism displays a greater concerted character, as indicated by a KIE value much greater than unity (707c).
Scheme 173.

Intramolecular KIE Experiments for Investigating the Mechanism of the Schmittel Cyclization
5.2.2.2. Thermal and Photochemical Radical Clock Experiments.
Further experimental investigations of the mechanism of the Schmittel reaction have been carried out using radical clock experiments (Scheme 174).226,227 Schmittel demonstrated thermal and photochemical ring openings of diphenyl cyclopropane enyne–allene 712. Because the ring opening of pendant cyclopropanes is generally accepted as evidence of a diradical intermediate, experimental observations of ring-opened products 710 and 713 support the presence of a stepwise mechanism.
Scheme 174.

Thermal and Photochemical Investigation of the Schmittel Cyclization with Cyclopropanes
5.2.2.3. Schmittel Cascades.
The Schmittel cyclization can be used as a powerful tool to create polycyclic aromatic compounds in a single step. Wang and coworkers engaged an Ireland–Claisen rearrangement to generate the corresponding enyne–allene from propargylic acetates (Scheme 175).228 The Schmittel cyclization of 714 results in the corresponding benzofulvene biradicals 717 and 719, which undergo further cyclization to result in six-membered or four-membered rings 716 and 718, respectively. In an additional report, Wang described access to polycyclic spirocyclic or highly twisted polycyclic molecules via an SNi′ reaction by treating the corresponding propargylic alcohol with thionyl chloride.229
Scheme 175.

Synthesis of Polycyclic Molecules via Schmittel Cyclization
The Schmittel cyclization enables the formation of fulvenyl diradical intermediates that can lead to diverse polycyclic compounds. Similar to suggestions for future explorations of the Myers–Saito reaction, the use of electrochemical methods to reduce or oxidize the diradical intermediate would unlock different reactivities and potentially provide shorter routes to target molecules useful for exploring bioactive chemical space. In addition, identifying new and mild procedures to prepare the enyne–allenes in situ is highly desirable and may find useful applications in biorthogonal chemistry.
5.3. Diradical Intermediates in Other Allene Reactions
Whereas there has been extensive work on thermal and photochemical [2 + 2] cycloadditions of allenes and enyne reactions involving diradical intermediates, there are other reactions of allenes proposed to proceed through similar species that are worth of mentioning. For example, Kamigata studied sulfur- and selenium-containing allenes under thermal230 and photochemical conditions.231 Acyclic 1,3-bis(alkylthio)allenes 721 (R = H or Ph) gave several products under thermal conditions (Scheme 176). Path 1 illustrates the formation of methylenecyclobutanes 723–724 through an allene–allene [2 + 2] thermal dimerization that proceeds via bisallylic radical 722. Path 2 provides an explanation for the formation of 727 and 729–730 and is proposed to proceed through diradical 725. The addition of an α-thioradical on the ortho position of the aromatic ring of 725 gives 726. A 1,5-hydrogen migration furnishes 727 and restores aromaticity. Thiophenes 729–730 are proposed to result from 725, where radical recombination gives 728; this then eliminates RCH2SH (R = H, Ph) to deliver the major components of the mixture (729–730). Path 3 involves homolytic cleavage of the C–S bond of 721, presumably due to the stability of the resulting benzylic and sulfur (731) radicals. Rearrangement of 731 to 732, followed by H-atom abstraction and hydrolysis furnishes 736, whereas the conversion of 731 to 733, followed by H-atom abstraction, gives 734 in 5% yield.
Scheme 176.
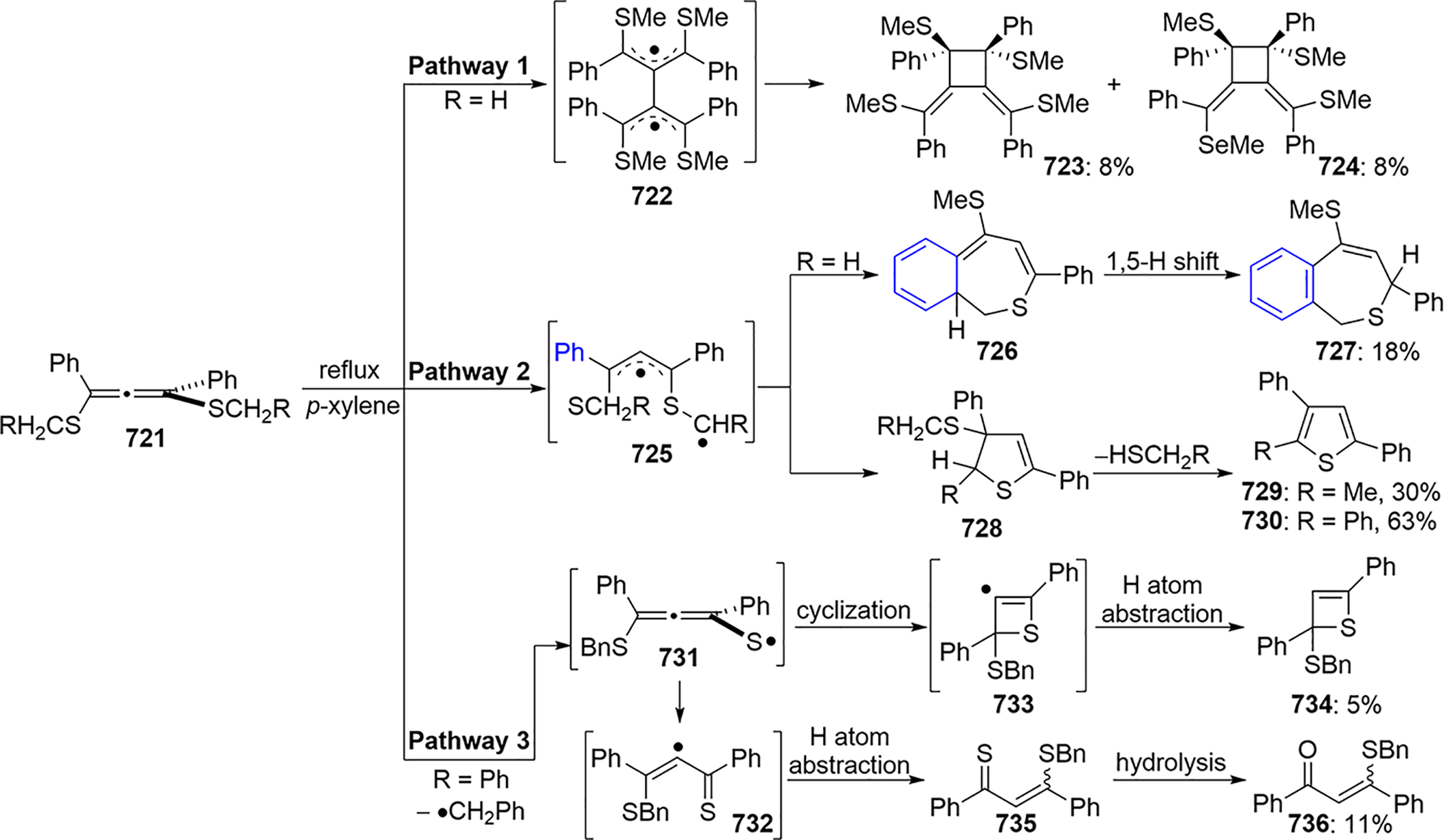
Potential Intermediates and Products from Thermal Reactions of Acyclic 1,3-Bis(alkylthio)allenes
The irradiation of 1,3-bis(alkylthio)allenes by Kamigata and coworkers furnished rearranged products 740/745 and 741/746 (Scheme 177). The mechanism is proposed to proceed through a diradical/carbene pathway (from 738/743 to 739/744), as opposed to a photoinduced [1,3]-sigmatropic rearrangement. A 1,2-migration of 737/742, followed by carbene formation to 739/744 and subsequent rearrangement furnishes alkynes 740/745. Monothiosubstituted compounds 741 and 746 result from the irradiation of 740/745 under the reaction conditions.
Scheme 177.

Irradiation of 1,3-Bis(alkylthio)allenes
Under thermal conditions, meso-tetrathiocyclic bisallene 747 furnishes the tricyclic compound 749, presumably via diradical intermediate 748 (Scheme 178). Allene dimerization becomes significant when the temperature exceeds 130 °C in refluxing xylene; under these conditions, 747 results in intermediate 748, which closes to furnish dimethylenecyclobutane 749. Under photochemical conditions, the rearranged products 750–751 are obtained, along with the stereoisomer of the starting material 752. Presumably, 750–751 arise from the same pathway as was observed in the photochemical reaction of acyclic bisalkylthioallenes (Scheme 178).
Scheme 178.

Reactions of Cyclic Bisalkylbissulfur Allene under Photochemical and Thermal Conditions
Under thermal conditions, bisseleniumalkylallenes display a different reactivities from their sulfur–chalogen counterparts.232 Kamigata postulated that this is due to the smaller bond dissociation energy (BDE) of the C–Se bond compared with that of the C–S bond. Running the reaction in the presence of galvinoxyl gave lower yields, providing support for a radical mechanism (Scheme 179). Route 1 is proposed to lead to 756, which results from the dimerization of acyclic allene 753 to give bisallylic biradical intermediate 754. The radical recombination of 754, followed by the loss of diphenyl selenide from 755, delivers 756. Route 2 involves the sequential loss of two phenyl selenium radicals from 753 to give carbene 759; dimerization of 759 furnishes 760–761.
Scheme 179.

Reaction of Bisseleniumallenes under Thermal Chemical Conditions
Under photochemical conditions, acyclic bisselenium allenes primarily result in (E)- and (Z)-1,3,4,6-tetraphenyl-3-hexen-1,5-diynes 760–761 (Scheme 180; also see Scheme 179).233 These products evolve from the 1,2-photorearrangement of 762 to yield biradical intermediate 763, which is in resonance with carbene 764 and ultimately undergoes 1,2-migration to 765. Homolytic C–Se bond cleavage to 766, followed by dimerization yields 767, which loses RSe–SeR to deliver 760/761. Carrying out the reaction under O2 bubbling (Scheme 181) produced no 760/761, giving 770 as the major product, supporting the reaction pathway outlined in Scheme 180.
Scheme 180.

Reaction of Bisseleniumalkylallenes under Photochemical Conditions
Scheme 181.

Support for a Radical Mechanism
Photochemical irradiation of cyclic bisallene 771 yields 760/761 (Scheme 182). In addition, 772 is obtained as a minor product, presumably via the homolytic cleavage of the C–Se bond in 771, followed by the radical recombination of the selenium radicals. As in the case of cyclic sulfur bisallenes, the presence of the other stereoisomer of the starting material as meso-771 suggests a photoinduced isomerization. The thermal pathway (Scheme 182, bottom) yields products 773–774 and 760–761 through the indicated mechanistic pathway.
Scheme 182.

Thermal and Photochemical Reactions of Cyclic Bissulfur Allenes
Houk carried out computational analyses of the addition of butadiene and benzene (aromatic-containing diene) to propadiene (Scheme 183).234 Potential products include methylenecyclobutanes 783 from a formal [2 + 2] cycloaddition or cyclohexene 776 from a [4 + 2] cycloaddition. The formation of 776 could be explained by a concerted mechanism proceeding through 775 or by a stepwise cis-diradical pathway proceeding through 777/778. Calculations indicate that both mechanisms (concerted and stepwise) involve the same transition state, computed to have a barrier of 27.7 kcal/mol. The transition state involved in the formation of 781 was calculated to have an energy barrier of 28.1 kcal/mol, which can compete with the transition state leading to 775 or 777.
Scheme 183.

Mechanistic Pathways for the Reaction of 1,3-Butadiene with Propadiene
Computational analysis indicates that the reaction of propadiene with benzene under thermal conditions likely occurs via a concerted pathway (Scheme 184). A stepwise pathway would entail the loss of aromaticity, which would counteract any favorable allylic radical stabilization in intermediate 785. Accordingly, the transition state 784 for a stepwise pathway is calculated to be greater than that of the concerted transition state 789 (42.1 vs 37.1 kcal/mol, respectively). The concerted reaction goes through an aromatic transition state, which mitigates the disruption of the aromaticity of benzene. Despite the lower barrier for the concerted reaction (37.1 kcal/mol), the barrier is still higher than that of allene dimerization (32.9 kcal/mol) and thus allene dimerization would be expected to outcompete other reactions.235
Scheme 184.

Mechanistic Pathways for the Reaction of Propadiene with Benzene
Goré showed that reactions of α-allenic hydrazines 791a–c give 3-pyrroline 798 or azetidine 795 via radicals 796 and 793, respectively (Scheme 185).236 The ratio of 798:795 varies with temperature; at higher temperatures and long reaction times, 798 is the major product (45 vs 20 h). This led the authors to speculate that the formation of 795 is reversible at higher temperatures. Utilizing hydrazines 791a–c resulted in different reactivities and different product ratios as the temperature was varied. Substituent effects, including the installation of a more basic terminal nitrogen of the hydrazine, result in a higher 3-pyrroline/azetidine ratio. Dimethylamine- and piperidine-containing hydrazines (791a) display enhanced reactivity when compared with a morpholine-containing hydrazine 791b. An exception to this trend is the SAMP hydrazine 791c, due to the coordination of the methoxy group to the lithium, which increases the basicity of the terminal nitrogen in 791c. Additional tunability could be achieved by the selective positioning of radical stabilizing groups in the precursors to favor 792 or 796.
Scheme 185.

Mechanistic Pathway and Ratio of Products from Cyclization of Allene Hydrazines
In terms of the mechanism, the hydrazide 791 could undergo two possible pathways. At lower temperatures, the corresponding azetidine 795 is obtained, first through the loss of an electron, followed by 4-exo-dig cyclization at the central carbon to give stabilized allylic radical 793. This radical is reduced to the corresponding anion 794, whereas azetidine 795 is obtained via the protonation of 794. When the reaction is carried out at higher temperatures, the formation of 795 is proposed to be reversible, giving 3-pyrroline as the major product. A separate reaction pathway is hypothesized to occur from 791, where electron transfer to the allene results in intermediate 796. The radical recombination of 796 yields the five-membered ring 797. Protonation of the anion results in the corresponding 3-pyrroline 798.
6. SYNTHESIS OF NATURAL PRODUCTS
The Hart group described a synthesis of pyrrolizidinones and indolizidinones utilizing α-acylamino radical additions to allenes 799–800.237 The phenylselenyl lactams, arising from 799 and 800, were converted to the indolizidinones and pyrrolizidinones 802–803 and 804–805, respectively, in moderate yields (Scheme 186).
Scheme 186.

n-Bu3Sn-Radical-Initiated Cyclization of Allenes
The pyrrolizidine alkaloid (±)-supinidine,238–241 which has reported antitumor properties, could be prepared from either 804 or 805, as shown in Scheme 187. The conversion of 804 to the allylic acetate 806 was achieved in one step by SeO2-mediated allylic oxidation. On the contrary, 805 was transformed to 806 via a sequence of four steps in a higher 41% yield, as compared with the 14% yield obtained utilizing 804 as the precursor.
Scheme 187.

Synthesis of (±)-Supinidine from 804 and 805
In 1988, the Hart group published a synthesis of (+)-heliotridine 812 from (S)-3-acetoxysuccinimide 808242 (Scheme 188) by employing a protocol similar to that used for the preparation of (±)-supinidine.237 The key step in building the [3.3.0]-fused bicyclic ring system involved a n-Bu3SnH-initiated free-radical cyclization of allene 809, obtained from enantiomerically enriched 808 in good yield. The exo-cyclization product 810 was afforded in 40% yield following the radical cyclization step and was converted to the diacetate 811 in two additional steps. Lithium aluminum hydride reduction of 811 gave the pyrrolizidine alkaloid (+)-heliotridine in an overall 4.3% yield from 808.
Scheme 188.

Synthesis of (+)-Heliotridine via n-Bu3SnH-Initiated Free-Radical Cyclization
In 1993, the Meyers group published the first synthesis of (±)-7,8-epoxy-4-basmen-6-one 813, a diterpenoid tobacco isolate that possesses a tricyclic ring system composed of three fused rings A, B, and C243 (Scheme 189). The key annulation step to form rings A–C involved an AIBN-mediated radical cyclization of the allene precursor 816. The synthesis of 813 commenced with commercially available neryl acetone, which was converted to the alcohol 814 in quantitative yield. A sequence of nine steps furnished the allenyl macrocycle 815 in 20% overall yield from 814. An additional four steps gave precursor 816 for the key radical cyclization.
Scheme 189.

Synthesis of (±)-7,8-Epoxy-4-basmen-6-one 813
Irradiating a mixture of the cyclization precursor 816, N-methylcarbazole, and 1,4-cyclohexadiene in THF/H2O for 5 h at 55 °C using Pyrex-filtered light from a medium-pressure mercury lamp yielded the isomeric tricyclic fused rings 817–818 in 51% yield. From a mechanistic perspective, the 5–8–5 tricyclic ring backbone is formed via two consecutive 5-exo cyclizations from intermediate I (Scheme 194). A 5-exo-trig cyclization of I was proposed to lead to II. Theoretical studies described by Houk and Spellmeyer244 support the observed regio- and chemoselectivity of this cyclization, indicating a ≥4.2 kcal/mol preference for this pathway over alternative cyclization pathways. The formation of radical intermediate IV was proposed to result from 5-exo-dig cyclization of intermediate III instead of II, as calculations predicted that it was energetically more favorable by ~4 kcal/mol. The isomeric mixture of 817–818 was subsequently heated in a 1:3 mixture (v/v) of thiophenol and heptane in the presence of a catalytic amount of AIBN to promote equilibration to deliver 91% of 817 as a single isomer. Intermediate 817 was then converted to (±)-7,8-epoxy-4-basmen-6-one 813 in an additional 12 steps in 18% overall yield.
Scheme 194.

Radical Cyclization of Allene 840 for the Total Synthesis of Azadirachtin
In 1996, Chen and coworkers demonstrated a radical cyclization leading to the synthesis of (±)-chamigrene 824, a spirocyclic sesquiterpene containing a spiro[5,5]undecane motif245 (Scheme 190). The α-allenic nitroketone radical precursor 820 was obtained in three steps from 1-methyl-4-nitrocyclohexene 819, which itself was prepared via a known procedure. The treatment of 820 with n-Bu3SnH and a catalytic amount of AIBN in refluxing benzene gave 37% of the 6-exo-dig cyclization product 821, along with 26% of an enone byproduct 822. The reduction of the carbonyl group in 821 was achieved by dithiolation and a subsequent dethiolation employing sodium in liquid ammonia.
Scheme 190.

Synthesis of (±)-α-Chamigrene
In 1999, Renaud and coworkers published a synthesis of (±)-botryodiplodin 829, which is a mycotoxin that exhibits antibiotic and antileukemic activities246 by employing a Ueno–Stork reaction of allene 825. This reaction involves a stereoselective radical cyclization of the α-bromoacetal moiety of an allene alcohol247 (Scheme 191). The stereoselectivity at C3 was controlled by the acetal center; however, the stereochemistry at C2 proved difficult to control. To overcome this stereoselectivity issue, the α-dibromoacetal 825 was employed to deliver 826 after the addition of the first equivalent of n-Bu3SnH. The immediate addition of the bulky reductant TTMSS to 826 in situ yielded the all-syn product 827 in good yield. The cis stereoselectivity at C2 was attributed to the bulky reductant approaching from the face opposite to the C2 and C3 substituents. Hydrolysis of 827, followed by Wacker oxidation, gave 829 as a mixture of anomers.
Scheme 191.

Renaud’s Synthesis of (±)-Botryodiplodin
In 2003, Renaud and coworkers published the synthesis of enantiomerically pure (−)-botryodiplodin by utilizing (1R,2S)-2-phenylcyclohexanol as a chiral auxiliary.248 The enantio- and diastereomerically pure allene 832 was prepared in four steps from the allylated chiral auxiliary 831 (Scheme 192). The radical cyclization of 832, followed by acetal hydrolysis and Wacker oxidation gave 830 in 15% yield.
Scheme 192.
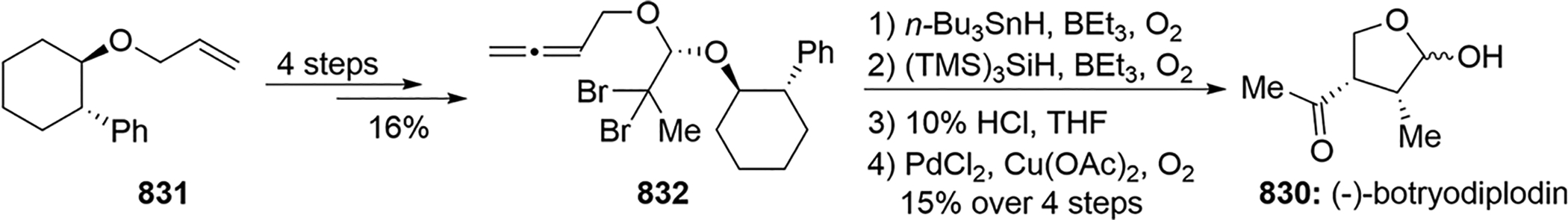
Renaud’s Synthesis of (−)-Botryodiplodin
In 2002, the Ley group published a synthesis of the framework of azadirachtin (Figure 2), a compound belonging to the C-seco-limonoid group of triterpenoids. Azadirachtin is a potent insect antifeedant, although its biochemical mode of action is not yet fully understood.249 Ley’s strategy involved a highly diastereoselective Claisen rearrangement of a propargylic enol ether to build the C8–C14 (azadirachtin numbering) bond, whereas a radical cyclization of an allene was used to establish the bicyclo[3.2.1] ring system of the natural product.
Figure 2.

Natural product azadirachtin.
In the synthesis, propargylic enol ether 835 was prepared from the pyran derivative 833 in eight steps in 31% overall yield (Scheme 193). After the extensive optimization of the conditions for the Claisen rearrangement, Ley discovered that the silyl deprotection of 834 to 835 afforded a better substrate for a microwave-assisted Claisen rearrangement. The irradiation of 835 with 15 consecutive 60 s pulses in 1,2-dichlorobenzene gave allene 836 in high yield. The radical cyclization of 836 first necessitated the protection of the hydroxyl groups at C1 and C3. However, TES protection at C3 failed due to the steric congestion at this site. The bis-xanthate 837 was designed as an alternative radical precursor and was obtained in good yield from 836. Refluxing 837 in toluene in the presence of n-Bu3SnH and AIBN exclusively gave an 81% yield of the exo cyclization product 838. The absence of any endo cyclization product 839 was attributed to the steric inaccessibility of the tin hydride to form the tertiary radical center.
Scheme 193.

Claisen Rearrangement and Allene Radical Cyclization as an Approach to Azadirachtin
In 2008, Ley et al. completed the first total synthesis of azadirachtin.250 A different precursor (cf. 838 in Scheme 193) was used to construct the bicyclo[3.2.1] ring system, but the same strategy involving a radical cyclization of an allene was employed to convert 840 to 841 in high yield (Scheme 194).
In 2009, Hart et al. employed allene 851 as a radical acceptor in an attempt to develop a route to C19 quassinoid polyandranes, such as 842–844251 (Scheme 195a). Previous attempts using the radical precursors 845 or 848 resulted in undesired byproducts 847, 849, and 850. Products 847 and 850 were produced from the 1,6-hydrogen atom transfer of the vinyl radical intermediates I and III, whereas 849 was generated from the early quenching of the vinyl radical intermediate II by tin hydride (Scheme 195b). Allene 851 was considered to be an alternate substrate, as geometric constraints would forbid any 1,6-H transfer to form the undesired intermediate 853. Indeed, the authors achieved a 95% yield of 852, as demonstrated in Scheme 195c.
Scheme 195.

Free-Radical Cyclizations of Alkynes and Allenes as Routes to the Synthesis of Polyandranes
In 2015, Jahn et al. developed a radical cyclization approach to furnish diverse bridged diketopiperazines with variable ring sizes in good to excellent yield and with moderate to excellent dr from alkoxyamines;252 one example is shown in Scheme 196a. Jahn applied this protocol to allene precursor 855 (Scheme 196b), prepared from a known dichloroacetamide 854 in three steps. The key bicyclo[4.2.2]piperazinedione ring system in 856 was furnished via an 8-exo-trig cyclization, where the carbon-centered radical regioselectively attacks the central allene carbon. Compound 856 was converted to 857 in 79% yield over two steps; 857 has been previously transformed to bicyclomycin in four additional steps by Williams.253,254
Scheme 196.

Synthesis of Bridged Diketopiperazines and Applications to the Synthesis of Bicyclomycin
7. CONCLUSIONS AND FUTURE PROSPECTS
The presence of two orthogonal double bonds in allenes imparts unique reactivity profiles to these unsaturated compounds, as compared with their alkene and alkyne counterparts. The rapid increase in the number of new synthetic methods available to prepare allenes bearing a diverse array of substitution patterns and structural complexities, often in an enantioenriched form, offers exciting opportunities to investigate the underexplored chemistry of these fascinating molecules. Within the context of allene reactivity, the addition of radicals has certainly been studied using a range of heteroatom- and carbon-based radical species for well over 50 years. Nonetheless, significant challenges remain in understanding the “rules” that govern the addition of radicals to allenes, despite the fact that our understanding of radical additions to alkenes and alkynes is fairly well-established. Several features of allenes contribute to this challenge, including the differing electron density distributions in allenes, especially between the two cumulated double bonds that are significantly affected by substituents installed directly (and even remote to) on the allene carbon(s). The orthogonality of the two π systems also presents difficulties in achieving predictable control over the regio-, site-, and stereoselectivities of allene functionalizations, in particular, in intermolecular reactions. This currently limits the synthetic utility of these building blocks in radical chemistry, and thus increasing our understanding of how to achieve both substrate and catalyst control over the selectivity is an enabling opportunity that will inspire the development of new methodologies with useful applications to complex and bioactive molecule synthesis.
In terms of what is known about intermolecular additions of halogen radicals to substituted allenes, addition typically occurs at the central carbon. However, little is known as to whether the nature of the solvent can alter the behavior of these reactions independent of substrate control; the advent of ionic liquids and supercritical solvents offers new opportunities in this area. Further insight into reactivity patterns in the additions of halogens to allenes would also benefit from more careful kinetic and mechanistic studies employing a broader range of electronically and sterically distinct allenes now that the methodology is in place to secure such precursors in a convenient manner. Recent improvements in spectroscopic techniques to detect and interpret the behavior of radical intermediates as well as improved computational methods could also be brought to bear on updated versions of reactions that were carried out before these resources were available.
The ability to selectively control the addition of a carbon-based radical to an allene represents a powerful way to forge new C–C bonds while leaving significant functionality intact for further transformations. Thus far, reports surrounding the reactivity and selectivity of carbon-centered radicals show a heavy dependence on the substitution pattern of both the allene and the radical species. This is far from ideal, yet little has been done to date to explore whether the generation of carbon-based radicals using tunable, visible-light photocatalysts or electrochemical processes might offer more predictable control over the key C–C bond-forming process. These methods for generating radicals are certainly much milder than those traditionally used in these kinds of transformations. In addition, the ability to alter the lifetime of the intermediate carbon radical resulting from the addition to the allene could offer the chance to transfer axial chirality in the allene precursor to the product. In cases where stereochemical information from the allene substrate is lost due to the formation of achiral intermediates, a better understanding of the behavior of such reactive species could lead to asymmetric methods for the enantioselective construction of adjacent stereogenic carbons.
The introduction of heteroatoms into allenes by the addition of nitrogen-, phosphorus-, sulfur-, tin-, indium-, and selenium-based radicals is another powerful strategy to rapidly increase the structural complexity in products resulting from such reactions. Work thus far shows that heteroatom-based radicals prefer to attack at the central allene carbon, although changes in the substitution pattern of either reactant can change the product distribution. Nonetheless, the field requires that we explore a broader scope of substituted allenes to achieve a predictable and synthetically useful reactivity, particularly in intermolecular reactions. Easy access to silylated, borylated, and halogenated allenes offers potential ways to target the delivery of a heteroatom radicals to the distal allene double bond in intermolecular reactions. Intramolecular reactions largely depend on the tether length between the radical and the allene in the reported results, but the ability to flexibly control the ring size in these types of reactions through either substrate or catalyst control would be valuable. The identity of the radical could also be better understood, in particular, in the context of nitrogen radicals, which form extremely useful C–N bonds in reactions with allenes. This area might benefit greatly from the recent availability of photochemical and electrochemical methods to generate nitrogen radicals under mild and functional-group-tolerant conditions. Enantioselective methods to add nitrogen, phosphorus, and sulfur radicals to allenes could prove to be especially popular for rapid access to functionalized building blocks for bioactive chemical space.
Whereas there are some very nice examples showcasing the ability of tandem radical allene cyclizations to furnish complex molecular architectures, including application in the total syntheses of natural products that include the pyrrolizidine alkaloids supinidine and heliotridine and (−)-botryodiplodin and, notably, Ley’s first total synthesis of azadirachtin, we feel that the potential of such chemistry is greatly underexplored and will certainly benefit from advances in methodology.
In another class of reactions that potentially involve radical intermediates, cycloadditions of allenes with alkenes, alkynes, and other allenes have emerged as strategies for the rapid construction of polycyclic scaffolds. However, these reactions have not yet achieved broad synthetic utility and instead serve as valuable probes for mechanistic studies. Nonetheless, enyne–allene radical cyclizations are capable of generating significant molecular complexity through diradical intermediates that are of synthetic and biological interest. The key is to continue to explore allenes of diverse substitution patterns to identify transformations that are capable of participating in regio- and stereoselective reactions. Whereas this has been addressed to some extent in intramolecular reactions, many opportunities remain to address these challenges in intermolecular reactions. Whereas there are scattered reports of visible-light photo-catalysis involving allenes, further investigations in this area may prove fruitful, especially in the execution of tandem cyclizations to efficiently generate polycyclic scaffolds, ideally with excellent control over the stereochemical outcome of the reaction. All told, the field seems ripe for chemists to reinvestigate many old reactions involving the additions of radicals to allenes using modern catalysts, mild methods to generate radicals, the expanded range of readily available allenes, improved computational methods, and access to more advanced spectroscopic techniques for studying the behavior of radical intermediates.
ACKNOWLEDGMENTS
We acknowledge ACS PRF 58268-ND1 for funding to support the research in our group related to this topic.
Biographies
Biographies
Lu Liu received her Bachelor’s degrees in chemistry and in English language and literature from Northern State University in 2014, where she carried out research with Prof. George Nora on synthesis of quinabactin. She then moved to UW-Madison, where she received her Ph.D. degree in chemistry with Prof. Jennifer M. Schomaker. Her research focuses on the synthesis of fluorinated amine stereotriads via allene amination and nitrogen-centered radical cyclizations of allenes.
Robert M. Ward received his B.S. degree in chemistry and a minor in biology from the University of Alberta in 2016, where he carried out research with Prof. Dennis G. Hall on boronic acid catalysis and the design of kinase/phosphatase inhibitors. He received an M.Sc. in chemistry from the University of Toronto under the supervision of Prof. Mark S. Taylor in 2017. He then moved to UW-Madison, where he is pursuing his Ph.D. in chemistry with Prof. Jennifer M. Schomaker. His research focuses on nitrogen radical cyclizations of allenes and tandem reactions involving copper-catalyzed 1,3-halomigration.
Jennifer M. Schomaker received her Ph.D. in 2006 from Michigan State University, working under the supervision of Prof. Babak Borhan. After completing an NIH postdoctoral fellowship at UC-Berkeley under Prof. Robert Bergman and F. Dean Toste, she began her independent career at the University of Wisconsin-Madison in 2009. Her research interests include catalyst-controlled C–H oxidations, new methods for the syntheses of new N-heterocycle chemical space, and the total synthesis of complex bioactive molecules.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Burton BS; von Pechmann H Ueber die Einwirkung von Chlorphosphor auf Acetondicarbonsäureäther. Ber. Dtsch. Chem. Ges 1887, 20, 145–149. [Google Scholar]
- (2).Griesbaum K Progress in the Chemistry of Allene. Angew. Chem., Int. Ed. Engl 1966, 5, 933–946. [Google Scholar]
- (3).Taylor DR The Chemistry of Allenes. Chem. Rev 1967, 67, 317–359. [Google Scholar]
- (4).Pasto DJ Recent Developments in Allene Chemistry. Tetrahedron 1984, 40, 2805–2827. [Google Scholar]
- (5).Wang KK Cascade Radical Cyclizations via Biradicals Generated from Enediynes, Enyne-Allene, and Enyne-Ketenes. Chem. Rev 1996, 96, 207–222. [DOI] [PubMed] [Google Scholar]
- (6).Modern Allene Chemistry; Krause N, Hashmi ASK, Eds.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- (7).Feng P; Fu C; Ma S Radical Reactions of Allenes. Youji Huaxue 2004, 24, 1168–1190. [Google Scholar]
- (8).Ma S Some Typical Advances in the Synthetic Applications of Allenes. Chem. Rev 2005, 105, 2829–2872. [DOI] [PubMed] [Google Scholar]
- (9).Yu S; Ma S Allenes in Catalytic Asymmetric Synthesis and Natural Product Syntheses. Angew. Chem., Int. Ed 2012, 51, 3074–3112. [DOI] [PubMed] [Google Scholar]
- (10).Alcaide B; Almendros P Progress in Allene Chemistry. Chem. Soc. Rev 2014, 43, 2886–2887. [DOI] [PubMed] [Google Scholar]
- (11).Zimmer R; Dinesh CU; Nandanan E; Khan FA Palladium-Catalyzed Reactions of Allenes. Chem. Rev 2000, 100, 3067–3126. [DOI] [PubMed] [Google Scholar]
- (12).Sydnes LK Allenes from Cyclopropanes and Their Use in Organic Synthesis–Recent Developments. Chem. Rev 2003, 103, 1133–1150. [DOI] [PubMed] [Google Scholar]
- (13).Alcaide B; Almendros P; Aragoncillo C Exploiting [2 + 2] Cycloaddition Chemistry: Achievements with Allenes. Chem. Soc. Rev 2010, 39, 783–816. [DOI] [PubMed] [Google Scholar]
- (14).Krause N; Winter C Gold-Catalyzed Nucleophilic Cyclization of Functionalized Allenes: A Powerful Access to Carbo- and Heterocycles. Chem. Rev 2011, 111, 1994–2009. [DOI] [PubMed] [Google Scholar]
- (15).Pasto DJ; L’Hermine G Experimental and Theoretical Study of the Radical-Chain Addition of Benzenethiol to Heterosubstituted Allenes. J. Org. Chem 1990, 55, 685–694. [Google Scholar]
- (16).Furet P; Matcha RL; Fuchs R Ab Initio Studies of Substituent Effects on Allene Stability. J. Phys. Chem 1986, 90, 5571–5573. [Google Scholar]
- (17).Haszeldine RN; Leedham K; Steele BR Addition of Free Radicals to Unsaturated Systems. Part IX. The Direction of Free-Radical Addition to Allene and Ally1 Chloride. J. Chem. Soc 1954, 2040–2042. [Google Scholar]
- (18).During the preparation of this Review, a review on a similar topic was published:; Qiu G; Zhang J; Zhou K; Wu J Recent Advances in the Functionalization of Allenes via Radical Process. Tetrahedron 2018, 74, 7290–7301 [Google Scholar]; For other articles on radical reactions of allenes that were published after 2004, see the references within this Review..
- (19).Chu W-D; Zhang Y; Wang J Recent Advances in Catalytic Asymmetric Synthesis of Allenes. Catal. Sci. Technol 2017, 7, 4570–4579. [Google Scholar]
- (20).Neff R; Frantz DE Recent Advances in the Catalytic Syntheses of Allenes: A Critical Assessment. ACS Catal 2014, 4, 519–528. [Google Scholar]
- (21).Qian D; Wu L; Lin Z; Sun J Organocatalytic Synthesis of Chiral Tetrasubstituted Allenes from Racemic Propargylic Alcohols. Nat. Commun 2017, 8, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Song S; Zhou J; Fu C; Ma S Catalytic Enantioselective Construction of Axial Chirality in 1,3-Disubstituted Allenes. Nat. Commun 2019, 10, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hashimoto T; Sakata K; Tamakuni F; Dutton MJ; Maruoka K Phase-Transfer-Catalysed Asymmetric Synthesis of Tetrasubstituted Allenes. Nat. Chem 2013, 5, 240–244. [DOI] [PubMed] [Google Scholar]
- (24).Tejedor D; Mendez-Abt G; Cotos L; Garcia-Tellado F Propargyl Claisen Rearrangement: Allene Synthesis and Beyond. Chem. Soc. Rev 2013, 42, 458–471. [DOI] [PubMed] [Google Scholar]
- (25).Hartung J; Kopf T Fundamentals and Application of Free Radical Addition to Allenes In Modern Allene Chemistry; Krause NA, Hashmi SK, Eds.; Wiley-VHC: Weinheim, Germany, 2004; Vol. 2, pp 701–726. [Google Scholar]
- (26).Griesbaum KG; Oswald AA; Hall DN Allene Chemistry. II. Free-Radical Addition of Hydrogen Bromide to Allene. J. Org. Chem 1964, 29, 2404–2408. [Google Scholar]
- (27).Tien RY; Abell PI Kinetics and Stereochemistry of the Gas-Phase Addition of HBr to Methyl-Substituted Allenes. J. Org. Chem 1970, 35, 956–960. [Google Scholar]
- (28).Heiba E-AI The Chemistry of Allene. I. Factors Governing the Orientation of Free-Radical Addition. J. Org. Chem 1966, 31, 776–780. [Google Scholar]
- (29).Byrd LR; Caserio MC Stereochemistry of Addition Reactions of Allenes. VI. Orientation and Stereochemistry of Radical Addition. J. Org. Chem 1972, 37, 3881–3891. [Google Scholar]
- (30).Abell PI; Anderson RS The Gas Phase Addition of Hydrogen Bromide to Allenes. Tetrahedron Lett 1964, 5, 3727–3731. [Google Scholar]
- (31).Curran DP; Porter NA; Giese B Stereochemistry of Radical Reactions: Concepts, Guidelines and Synthetic Applications; Wiley-VCH Verlag: Weinheim, Germany, 1996. [Google Scholar]
- (32).Goumans TPM; van Alem K; Lodder G Photochemical Generation and Structure of Vinyl Radicals. Eur. J. Org. Chem 2008, 2008, 435–443. [Google Scholar]
- (33).Nagai S; Ohnishi S; Nitta I ESR Spectra of Vinyl Radicals Stabilized on Silica Gel Surfaces. Chem. Phys. Lett 1972, 13, 379–381. [Google Scholar]
- (34).Jenkins PR; Symons MCR; Booth SE; Swain CJ Why Is Vinyl Anion Configurationally Stable but a Vinyl Radical Configurationally Unstable? Tetrahedron Lett 1992, 33, 3543–3546. [Google Scholar]
- (35).Heiba E-AI; Haag WO The Chemistry of Allene. II. The Kinetics of Free-Radical Addition of Hydrogen Bromide. J. Org. Chem 1966, 31, 3814–3817. [Google Scholar]
- (36).Hudgens JW; Gonzalez C Chlorination Chemistry. 3. Ab Initio Study of the Reaction of Chlorine Atom with Allene. J. Phys. Chem. A 2002, 106, 1739–1745. [Google Scholar]
- (37).Farrell JT; Taatjes CA Infrared Frequency-Modulation Probing of Cl + C3H4 (Allene, Propyne) Reactions: Kinetics of HCl Production from 292 to 850 K. J. Phys. Chem. A 1998, 102, 4846–4856. [Google Scholar]
- (38).Atkinson DB; Hudgens JW Chlorination Chemistry. 2. Rate Coefficients, Reaction Mechanism, and Spectrum of the Chlorine Adduct of Allene. J. Phys. Chem. A 2000, 104, 811–818. [Google Scholar]
- (39).Zhao Z; Murphy GK Chlorination of Phenylallene Derivatives with 1-Chloro-1,2-benziodoxol-3-one: Synthesis of Vicinal-dichlorides and Chlorodienes. Beilstein J. Org. Chem 2018, 14, 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kovachic D; Leitch LC Organic Deuterium Compounds. XXIII. Synthesis of Deuterated Olefins. Can. J. Chem 1961, 39, 363–374. [Google Scholar]
- (41).Skell PS; Tuleen DL; Readio PD Stereochemical Radicals of Bridged Radicals. J. Am. Chem. Soc 1963, 85, 2849–2850. [Google Scholar]
- (42).Kippo T; Fukuyama T; Ryu I Regioselective Radical Bromoallylation of Allenes Leading to 2-Bromo-Substituted 1,5-Dienes. Org. Lett 2011, 13, 3864–3867. [DOI] [PubMed] [Google Scholar]
- (43).Kippo T; Ryu I A Bromine-Radical Mediated Three-Component Reaction Comprising Allenes, Electron-Deficient Alkenes and Allyl Bromides: Facile Synthesis of 2-Bromo-1,7-dienes. Chem. Commun 2014, 50, 5993–5996. [DOI] [PubMed] [Google Scholar]
- (44).Stefani AP; Herk L; Szwarc M Kinetics of Addition of CF3 Radicals to Olefins and Their Derivatives. J. Am. Chem. Soc 1961, 83, 4732–4730. [Google Scholar]
- (45).Meunier HG; Abell PI Kinetics of the Photoaddition of Trifluoromethyl Iodide to Allene. J. Phys. Chem 1967, 71, 1430–1435. [Google Scholar]
- (46).Ogawa A; Imura M; Kamada N; Hirao T Highly Regioselective Iodoperfluoroalkylation of Allenes with Perfluoroalkyl Iodides upon Irradiation with Near-UV Light. Tetrahedron Lett 2001, 42, 2489–2492. [Google Scholar]
- (47).Tsuchii K; Imura M; Kamada N; Hirao T; Ogawa A An Efficient Photoinduced Iodoperfluoroalkylation of Caron-Carbon Unsaturated Compounds with Perfluoroalkyl Iodides. J. Org. Chem 2004, 69, 6658–6665. [DOI] [PubMed] [Google Scholar]
- (48).Ma S; Ma Z Na2S2O4-Promoted Radical Addition Reaction of Perfluoroalkyl Iodides with Allenes and the Pd(0)-Catalyzed Stereoselective Sonogashira Coupling Reaction of Addition Products with Propargyl Alcohol. Synlett 2006, 2006, 1263–1265. [Google Scholar]
- (49).Ma Z; Ma S Stereoselective Synthesis of 2-Iodo-1-perfluoroalkyl-2(Z)-alkenes and E or Z-4-Perfluoroalkylmethyl-4-en-2-ynol via Na2S2O4-Promoted Radical Addition Reaction of Perfluoroalkyl Iodides with Allenes and the Palladium-Catalyzed Kinetic Resolution with Sonogashira Coulping Reaction. Tetrahedron 2008, 64, 6500–6509. [Google Scholar]
- (50).Mei Y-Q; Liu J-T; Liu Z-J Regio- and Stereoselective Addition of Perfluoroalkyl Iodide to Allenes Conjugated with Carbon-Oxygen or Phosphorus-Oxygen Double Bonds. Synthesis 2007, 2007, 739–743. [Google Scholar]
- (51).Ma Z; Zeng R; Fu C; Ma S Studies on the Radical Addition Reaction of Perfluoroalkyl Iodides with 2,3-Allenols and the Pd-Catalyzed Kinetic Resolution via Sonogashira Coupling Reaction. Tetrahedron 2011, 67, 8808–8818. [Google Scholar]
- (52).Yu Q; Ma S Copper-Catalyzed Cyclic Oxytrifluoromethylation of 2,3-Allenoic Acids to Trifluoromethylated Butenolides. Chem. - Eur. J 2013, 19, 13304–13308. [DOI] [PubMed] [Google Scholar]
- (53).Wang Y; Jiang M; Liu J-T Copper-Catalyzed Regioselective Oxytrifluoromethylation of Allenes Using a CF3-Transfer Reagent. Adv. Synth. Catal 2014, 356, 2907–2912. [Google Scholar]
- (54).Zhu N; Wang F; Chen P; Ye J; Liu G Copper-Catalyzed Intermolecular Trifluoromethylazidation and Trifluoromethylthiocyanation of Allenes: Efficient Access to CF3-Containing Allyl Azides and Thiocyanates. Org. Lett 2015, 17, 3580–3583. [DOI] [PubMed] [Google Scholar]
- (55).Tang Y; Yu Q; Ma S Efficient Trifluoromethylation via the Cyclopropanation of Allenes and Subsequent C-C Bond Cleavage. Org. Chem. Front 2017, 4, 1762–1767. [Google Scholar]
- (56).Tomita R; Koike T; Akita M Photoredox-Catalyzed Oxytrifluoromethylation of Allenes: Stereoselective Synthesis of 2-Trifluoromethylated Allyl Acetates. Chem. Commun 2017, 53, 4681–4684. [DOI] [PubMed] [Google Scholar]
- (57).Brochetta M; Borsari T; Gandini A; Porey S; Deb A; Casali E; Chakraborty A; Zanoni G; Maiti D Trifluoromethylation of Allenes: An Expedient Access to α-Trifluoromethylated Enones at Room Temperature. Chem. - Eur. J 2019, 25, 750–753. [DOI] [PubMed] [Google Scholar]
- (58).Rajbenbach A; Szwarc M Addition of Methyl Radicals to Isolated, Conjugated and Cumulated Dienes. Proc. R. Soc. London A 1959, 251, 394–406. [Google Scholar]
- (59).Bartels HM; Boldt P Radikalische Addition von Brommalononitril an Allene. Liebigs Ann. Chem 1981, 1981, 40–46. [Google Scholar]
- (60).Holemann A; Reissig H-U Samarium Diiodide-Induced Couplings of Carbonyl Compounds with Methoxyallene Leading to 4-Hydroxy-1-enol Ethers. Org. Lett 2003, 5, 1463–1466. [DOI] [PubMed] [Google Scholar]
- (61).Holemann A; Reissig H-U Regioselective Samarium Diiodide Induced Couplings of Carbonyl Compounds with 1,3-Diphenylallene and Alkoxyallenes: A New Route to 4-Hydroxy-1-enol Ethers. Chem. - Eur. J 2004, 10, 5493–5506. [DOI] [PubMed] [Google Scholar]
- (62).Berndt M; Gross S; Holemann A; Reissig H-U New Samarium Diiodide-Induced Ketyl Couplings–From Analogous Reactions to Serendipitously Discovered Processes. Synlett 2004, 422–438. [Google Scholar]
- (63).Wu Z; Huang X Intermolecular Mn(OAc)3-Mediated Oxidative Free-Radical Reaction of Dimethyl Malonate or Ethyl Cyanoacetate with Allenes: An Efficient Synthesis of Furan-2(5H)-ones or Dimethyl 2-(2-Oxoethylidene)malonates. Synthesis 2007, 2007, 45–50. [Google Scholar]
- (64).Yoshida M; Takai H; Yodokawa S; Shishido K Regio- and Diastereoselective Synthesis of Functionalized Hydroxyhexahydrocyclopenta[b]furancarboxylates by Oxidative Radical Cyclization of Cyclic β-Keto Esters with Alkenes. Tetrahedron 2013, 69, 5273–5280. [Google Scholar]
- (65).Dai X; Mao R; Guan B; Xu X; Li X Visible Light Photoredox Catalysis: Regioselective Radical Addition of Aminoalkyl Radicals to 2,3-Allenoates. RSC Adv 2015, 5, 55290–55294. [Google Scholar]
- (66).Kaap S; Quentin I; Tamiru D; Shaheen M; Eger K; Steinfelder HJ Structure-activity Analysis of the Pro-apoptotic, Antitumor Effect of Nitrostyrene Adducts and Related Compounds. Biochem. Pharmacol 2003, 65, 603–610. [DOI] [PubMed] [Google Scholar]
- (67).Kim JH; Kim JH; Lee GE; Lee JE; Chung IK Potent Inhibition of Human Telomerase by Nitrostyrene Derivatives. Mol. Pharmacol 2003, 63, 1117–1124. [DOI] [PubMed] [Google Scholar]
- (68).Reddy MA; Jain N; Yada D; Kishore C; Vangala JR; Surendra RP; Addlagatta A; Kalivendi SV; Sreedhar B Design and Synthesis of Resveratrol-based Nitrovinylstilbenes as Antimitotic Agents. J. Med. Chem 2011, 54, 6751–6760. [DOI] [PubMed] [Google Scholar]
- (69).Neale RS The Chemistry of Nitrogen Radicals. V. The Free-Radical Addition of Dialkyl-N-chloramines to Olefinic and Acetylenic Hydrocarbons. J. Org. Chem 1967, 32, 3263–3273. [Google Scholar]
- (70).Sabbasani VR; Lee D Nitration of Silyl Allenes to Form Functionalized Nitroalkenes. Org. Lett 2013, 15, 3954–3597. [DOI] [PubMed] [Google Scholar]
- (71).Ballini R; Petrini M Recent Synthetic Developments in the Nitro to Carbonyl Conversion (Nef Reaction). Tetrahedron 2004, 60, 1017–1047. [Google Scholar]
- (72).Ballini R; Petrini M The Nitro to Carbonyl Conversion (Nef Reaction) New Perspectives for a Classical Transformation. Adv. Synth. Catal 2015, 357, 2371–2402. [Google Scholar]
- (73).Xue C; Fu C; Ma S Highly Regio- and Stereoselective Nitrooxoamination of Mono-Substituted Allenes. Chem. Commun 2014, 50, 15333–15336. [DOI] [PubMed] [Google Scholar]
- (74).Zhang G; Xiong T; Wang Z; Xu G; Wang X; Zhang Q Highly Regioselective Radical Amination of Allenes: Direct Synthesis of Allenamides and Tetrasubstituted Alkenes. Angew. Chem., Int. Ed 2015, 54, 12649–12653. [DOI] [PubMed] [Google Scholar]
- (75).Zheng G; Li Y; Han J; Xiong T; Zhang Q Radical Cascade Reaction of Alkynes with N-fluoroarylsulfonimides and Alcohols. Nat. Commun 2015, 6, 7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Zhang H; Pu W; Xiong T; Li Y; Zhou X; Sun K; Liu Q; Zhang Q Copper-Catalyzed Intermolecular Aminocyanation and Diamination of Alkenes. Angew. Chem., Int. Ed 2013, 52, 2529–2533. [DOI] [PubMed] [Google Scholar]
- (77).Zhang H; Song Y; Zhao J; Zhang J; Zhang Q Regioselective Radical Aminofluorination of Styrenes. Angew. Chem., Int. Ed 2014, 53, 11079–11083. [DOI] [PubMed] [Google Scholar]
- (78).Goldwhite H Free-Radical Addition of Phosphine to Allene. J. Chem. Soc 1965, 3901–3904. [Google Scholar]
- (79).Mitchell TN; Heesche K Preparation of Vinylphosphines by Means of Free Radical Addition of Diphenylphosphine to Alkynes and Allenes. J. Organomet. Chem 1991, 409, 163–170. [Google Scholar]
- (80).Atkinson R; Perry RA; Pitts JN Jr. Absolute Rate Constants for the Reaction of OH Radicals with Allene, 1,3-Butadiene, and 3-Methyl-1-butene over the Temperature Range 299–424 K. J. Chem. Phys 1977, 67, 3170–3174. [Google Scholar]
- (81).Liu A; Mulac WA; Jonah CD Rate Constants for the Gas-Phase Reactions of OH Radicals with 1,3-Butadiene and Allene at 1 Atm in Ar and over the Temperature Range 305–1173 K. J. Phys. Chem 1988, 92, 131–134. [Google Scholar]
- (82).Daranlot J; Hickson KM; Loison J-C; Mereau R; Caralp F; Forst W; Bergeat A Gas-Phase Kinetics of the Hydroxyl Radical Reaction with Allene: Absolute Rate Measurements at Low Temperature, Product Determinations, and Calculations. J. Phys. Chem. A 2012, 116, 10871–10881. [DOI] [PubMed] [Google Scholar]
- (83).Denes F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- (84).Meadows DC; Sanchez T; Neamati N; North TW; GervayHague J Ring Substituent Effects on Biological Activity of Vinyl Sulfones as Inhibitors of HIV-1. Bioorg. Med. Chem 2007, 15, 1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Frankel BA; Bentley M; Kruger RG; McCafferty DG Vinyl sulfones: Inhibitors of SrtA, a Transpeptidase Required for Cell Wall Protein Anchoring and Virulence in Staphylococcus Aureus. J. Am. Chem. Soc 2004, 126, 3404–3405. [DOI] [PubMed] [Google Scholar]
- (86).Santos MM; Moreira R Michael Acceptors as Cysteine Protease Inhibitors. Mini-Rev. Med. Chem 2007, 7, 1040–1050. [DOI] [PubMed] [Google Scholar]
- (87).Truce WE; Wolf GC Additions of Sulphonyl Iodides to Acetylenes and Allenes. J. Chem. Soc. D 1969, 0, 150–150. [Google Scholar]
- (88).Truce WE; Heuring DL; Wolf GC Additions of Sulfonyl Iodides to Allenes. J. Org. Chem 1974, 39, 238–244. [Google Scholar]
- (89).Kang S-K; Ko B-S; Ha Y-H Radical Addition of p-Toluenesulfonyl Bromide and p-Toluenesulfonyl Iodide to Allenic Alcohols and Sulfonamides in the Presence of AIBN: Synthesis of Heterocyclic Compounds. J. Org. Chem 2001, 66, 3630–33. [DOI] [PubMed] [Google Scholar]
- (90).Kang S-K; Seo H-W; Ha Y-H Highly Regioselective and Stereoselective Radical Addition of p-TsBr to Substituted Terminal Allenes and Their Nucleophilic Substitutions: Synthesis of α,β-Unsaturated Sulfones. Synthesis 2001, 2001, 1321–1326. [Google Scholar]
- (91).Kang S-K; Ko B-S; Lee D-M Highly Regioselective and Stereoselective Radical Addition of p-TsBr to α-Allenic Alcohols and Their Elimination Reaction: Synthesis of β-p-Tosyl-Substituted α,β-Unsaturated Ketones. Synth. Commun 2002, 32, 3263–3271. [Google Scholar]
- (92).Lu N; Zhang Z; Ma N; Wu C; Zhang G; Liu Q; Liu T Copper-Catalyzed Difunctionalization of Allenes with Sulfonyl Iodides Leading to (E)-α-Iodomethyl Vinylsulfones. Org. Lett 2018, 20, 4318–4322. [DOI] [PubMed] [Google Scholar]
- (93).Mandal D; Mondal B; Das AK Nucleophilic Degradation of Fenitrothion Insecticide and Performance of Nucleophiles: A Computational Study. J. Phys. Chem. A 2012, 116, 2536–2546. [DOI] [PubMed] [Google Scholar]
- (94).Zhou K; Zhang J; Qiu G; Wu J Copper(II)-Catalyzed Reaction of 2,3-Allenoic Acids, Sulfur Dioxide, and Aryldiazonium Tetrafluoroborates: Route to 4-Sulfonylated Furan-2(5H)-ones. Org. Lett 2019, 21, 275–278. [DOI] [PubMed] [Google Scholar]
- (95).Huang Z; Lu Q; Liu Y; Liu D; Zhang J; Lei A Regio- and Stereoselective Oxysulfonylation of Allenes. Org. Lett 2016, 18, 3940–3943. [DOI] [PubMed] [Google Scholar]
- (96).Rohokale RS; Tambe SD; Kshirsagar UA Eosin Y Photoredox-catalyzed Net Redox Neutral Reaction for Regioselective Annulation to 3-Sulfonlylindoles vis Anion Oxidation of Sodium Sulfinate Salts. Org. Biomol. Chem 2018, 16, 536–540. [DOI] [PubMed] [Google Scholar]
- (97).Pan S; Huang Y; Xu X-H; Qing F-L Copper-Assisted Oxidative Trifluoromethylthiolation of 2,3-Allenoic Acids with AgSCF3. Org. Lett 2017, 19, 4624–4627. [DOI] [PubMed] [Google Scholar]
- (98).Xin Y-X; Pan S; Huang Y; Xu X-H; Qing F-L Copper-Catalyzed Sulfenylation, Sulfonylation, and Selenylation of 2,3-Allenoic Acids with Disulfides or Diselenides. J. Org. Chem 2018, 83, 6101–6109. [DOI] [PubMed] [Google Scholar]
- (99).Kuivila HG; Rahman W; Fish RH Free-Radical Addition of Trimethyltin Hydride to Allenes. J. Am. Chem. Soc 1965, 87, 2835–2840. [Google Scholar]
- (100).Jacobs TL; Illingworth GE Jr. The Addition of Thiyl Radicals to Allenic Hydrocarbons. J. Org. Chem 1963, 28, 2692–2698. [Google Scholar]
- (101).Turner RB; Nettleton DE; Perelman M Heats of Hydrogenation. VI. Heats of Hydrogenation of Some Substituted Ethylenes. J. Am. Chem. Soc 1958, 80, 1430–1433. [Google Scholar]
- (102).Ichinose Y; Oshima K; Utimoto K Hydrostannation and Hydrogermylation of Allenes. Bull. Chem. Soc. Jpn 1988, 61, 2693–2695. [Google Scholar]
- (103).Mitchell TN; Schneider U Free Radical and Palladium-Catalyzed Hydrostannation of Allenes: a Comparison. J. Organomet. Chem 1991, 405, 195–199. [Google Scholar]
- (104).Hayashi N; Kusano K; Sekizawa S; Shibata I; Yasuda M; Baba A Regio- and Stereoselective Hydrostannation of Allenes Using Dibutyliodotin Hydride (Bu2SnIH) and Successive Coupling with Aromatic Halides. Chem. Commun 2007, 4913–4915. [DOI] [PubMed] [Google Scholar]
- (105).Perin G; Lenardao EJ; Jacob RG; Panatieri RB Synthesis of Vinyl Selenides. Chem. Rev 2009, 109, 1277–1301. [DOI] [PubMed] [Google Scholar]
- (106).Masawaki T; Ogawa A; Kambe N; Ryu I; Sonoda N Oxygen Induced Free-Radical Addition of Benzeneselenol to Allenes. Chem. Lett 1987, 16, 2407–2408. [Google Scholar]
- (107).Pasto DJ; Warren SE; Morrison MA Radical-Chain Addition of Benzenethiol to Allenes. Analysis of Steric Effects and Reversibility. J. Org. Chem 1981, 46, 2837–2841. [Google Scholar]
- (108).Ogawa A; Yokoyama K; Yokoyama H; Sekiguchi M; Kambe N; Sonoda N Photo-Initiated Addition of Diphenyl Diselenide to Allenes. Tetrahedron Lett 1990, 31, 5931–5934. [Google Scholar]
- (109).Russell GA; Tashtoush H Free-Radical Chain-Substitution Reactions of Alkylmercury Halides. J. Am. Chem. Soc 1983, 105, 1398–1399. [Google Scholar]
- (110).Ito O Kinetics for Reaction of ArS· and PhSe· with Methyl-Substituted Allenes in Solution. J. Org. Chem 1993, 58, 1466–1471. [Google Scholar]
- (111).Ogawa A; Obayashi R; Doi M; Sonoda N; Hirao T A Novel Photoinduced Thioselenation of Allenes by Use of a Disulfide-Diselenide Binary System. J. Org. Chem 1998, 63, 4277–4281. [Google Scholar]
- (112).Kawaguchi S-I; Shirai T; Ohe T; Nomoto A; Sonoda M; Ogawa A Highly Regioselective Simultaneous Introduction of Phosphino and Seleno Groups into Unsaturated Bonds by the Novel Combination of (Ph2P)2 and (PhSe)2 upon Photoirradiation. J. Org. Chem 2009, 74, 1751–1754. [DOI] [PubMed] [Google Scholar]
- (113).Kumaraswamy G; Vijaykumar S; Ankamma K; Narayanarao V Visible-Light-Induced Phenylchalcogenyloxygenation of Allenes Having Aryl or Electron Withdrawing Substituents with Ambient Air as a Sole Oxidant. Org. Biomol. Chem 2016, 14, 11415–11425. [DOI] [PubMed] [Google Scholar]
- (114).Smith JRL; Mead LAV Amine Oxidation. Part VII. The Effect of Structure on the Reactivity of Alkyl Tertiary Amines Towards Alkaline Potassium Hexacyanoferrate(III). J. Chem. Soc., Perkin Trans. 2 1973, 2, 206–210. [Google Scholar]
- (115).Smith JRL; Masheder DJ Amine Oxidation. Part IX. The Electrochemical Oxidation of Some Tertiary Amines: The Effect of Structure on Reactivity. J. Chem. Soc., Perkin Trans. 2 1976, 1, 47–51. [Google Scholar]
- (116).Viehe HG; Merényi R; Stella Z; Janousek Z Capto-dative Substituent Effects in Syntheses with Radicals and Radicophiles. Angew. Chem., Int. Ed. Engl 1979, 18, 917–932. [Google Scholar]
- (117).Mizuno K; Ichinose N; Tamai T; Otsuji Y Electron-Transfer Mediated Photooxygenation of Biphenyl and Its Derivatives in the Presence of Mg(ClO4)2. Tetrahedron Lett 1985, 26, 5823–5826. [Google Scholar]
- (118).Mizuno K; Ichinose N; Otsuji Y Cis-Trans Photo-isomerization and Photooxygenation of 1,2-Diarylcyclopropanes, Salt Effects on the Photoinduced Electron Transfer Reactions. Chem. Lett 1985, 14, 455–458. [Google Scholar]
- (119).Hayashi N; Hirokawa Y; Shibata I; Yasuda M; Baba A Hydroindation of Allenes and Its Application to Radical Cyclization. Org. Biomol. Chem 2008, 6, 1949–1954. [DOI] [PubMed] [Google Scholar]
- (120).Inoue K; Sawada A; Shibata I; Baba A Indium Hydride: A Novel Radical Inhibitor in the Reduction of Organic Halides with Tributyltin Hydride. Tetrahedron Lett 2001, 42, 4661–4663. [Google Scholar]
- (121).Inoue K; Sawada A; Shibata I; Baba A Indium(III) Chloride-Sodium Borohydride System: A Convenient Radical Reagent for an Alternative to Tributyltin Hydride System. J. Am. Chem. Soc 2002, 124, 906–907. [DOI] [PubMed] [Google Scholar]
- (122).Hayashi N; Shibata I; Baba A Triethylsilane-Indium(III) Chloride System as a Radical Reagent. Org. Lett 2004, 6, 4981–4983. [DOI] [PubMed] [Google Scholar]
- (123).Hayashi N; Shibata I; Baba A Inter- and Intramolecular Radical Couplings of Ene-ynes or Halo-alkenes Promoted by an InCl3/MeONa/Ph2SiH2 System. Org. Lett 2005, 7, 3093–3096. [DOI] [PubMed] [Google Scholar]
- (124).Hayashi N; Honda H; Yasuda M; Shibata I; Baba A Generation of Allylic Indium by Hydroindation of 1,3-Dienes and Onepot Reaction with Carbonyl Compounds. Org. Lett 2006, 8, 4553–4556. [DOI] [PubMed] [Google Scholar]
- (125).Pattenden G; Robertson GM Reductive Cyclization of Allenic Ketones. A New Approach to Functionalized Five-Membered Rings. Tetrahedron Lett 1983, 24, 4617–4620. [Google Scholar]
- (126).Pattenden G; Robertson GM Synthesis of Functionalized Cyclopentanes by Intramolecular Radical-Mediated Cyclizations of Terminal Allenic Ketones. Tetrahedron 1985, 41, 4001–4011. [Google Scholar]
- (127).Crandall JK; Mualla M Reductive Cyclizations of Allenic Ketones by Dissolving Metals. Tetrahedron Lett 1986, 27, 2243–2246. [Google Scholar]
- (128).Belotti D; Cossy J; Pete JP; Portella C Synthesis of Bicyclic Cyclopentanols by Photoreductive Cyclization of δ, ε-Unsaturated Ketones. J. Org. Chem 1986, 51, 4196–4200. [Google Scholar]
- (129).Gillmann T Samarium (II) Iodide-Mediated Intramolecular Reductive Coupling of Aldehydes with Allenic Esters. Tetrahedron Lett 1993, 34, 607–610. [Google Scholar]
- (130).Molander GA; Cormier EP Ketyl-Allene Cyclizations Promoted by Samarium(II) Iodide. J. Org. Chem 2005, 70, 2622–2626. [DOI] [PubMed] [Google Scholar]
- (131).Parmar D; Matsubara H; Price K; Spain M; Procter DJ Lactone Radical Cyclizations and Cyclization Cascades Mediated by SmI2-H2O. J. Am. Chem. Soc 2012, 134, 12751–12757. [DOI] [PubMed] [Google Scholar]
- (132).Crandall JK; Tindell GL; Manmade A Homoallenyl Radicals. Tetrahedron Lett 1982, 23, 3769–3772. [Google Scholar]
- (133).Castaing M; Pereyre M; Ratier M; Blum PM; Davies AG Regioselectivity in the Ring-Opening of 2-Methylcyclopropylcarbinyl and 2-Methylcyclobutylcarbinyl Radicals. J. Chem. Soc., Perkin Trans. 2 1979, 0, 287–292. [Google Scholar]
- (134).Beckwith ALJ; Moad G Ring-Opening of Some Radicals Containing the Cyclopropylmethyl System. J. Chem. Soc., Perkin Trans. 2 1980, 0, 1473–1482. [Google Scholar]
- (135).Apparu M; Crandall JK Cyclizations of ω-Allenyl Radicals. J. Org. Chem 1984, 49, 2125–2130. [Google Scholar]
- (136).Caserio MC Selective Organic Transformations; Thyagarajan BS, Ed.; Wiley-Interscience: New York, 1970; Vol. 1, p 239. [Google Scholar]
- (137).Beckwith ALJ Regioselectivity and Stereoselectivity in Radical Reactions. Tetrahedron 1981, 37, 3073–3100. [Google Scholar]
- (138).Crandall JK; Ayers TA Radical Cyclizations of Functionalized Allenes. Tetrahedron Lett 1991, 32, 3659–3662. [Google Scholar]
- (139).Balasubramanian T; Balasubramanian KK Radical Cyclization of o-Haloaryl Allenylmethyl Ethers and Amines: Synthesis of 3-Ethenyl-2,3-dihydrobenzofurans and 3-Ethenyl-2,3-dihydroin-doles. Synlett 1994, 1994, 946–948. [Google Scholar]
- (140).Villar F; Renaud P Diastereoselective Radical Cyclization of Bromoacetals (Ueno-Stork Reaction) Controlled by the Acetal Center. Tetrahedron Lett 1998, 39, 8655–8658. [Google Scholar]
- (141).Lin H-H; Chang W-S; Luo S-Y; Sha C-K Photoinduced Atom-Transfer Cyclization of α-Iodocycloalkanones Bearing an Allenyl Side Chain. Org. Lett 2004, 6, 3289–3292. [DOI] [PubMed] [Google Scholar]
- (142).Shen L; Hsung RP Highly Regioselective Radical Cyclizations of Allenamides. Org. Lett 2005, 7, 775–778. [DOI] [PubMed] [Google Scholar]
- (143).Shi J; Zhang M; Fu Y; Liu L; Guo Q-X Regiochemistry in Radical Cyclization of Allenes. Tetrahedron 2007, 63, 12681–12688. [Google Scholar]
- (144).Wang Y-M; Fu Y; Liu L; Guo Q-X Substituent Effects on the Ring Opening of 2-Aziridinylmethyl Radicals. J. Org. Chem 2005, 70, 3633–3640. [DOI] [PubMed] [Google Scholar]
- (145).Marcus RA On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys 1956, 24, 966–978. [Google Scholar]
- (146).Marcus RA Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem 1964, 15, 155–196. [Google Scholar]
- (147).Marcus RA Theoretical Relations among Rate Constants, Barriers, and Brønsted Slopes of Chemical Reactions. J. Phys. Chem 1968, 72, 891–899. [Google Scholar]
- (148).Alcaide B; Almendros P; Aragoncillo C; Redondo MC Carbonyl Allenylation/Free Radical Cyclization Sequence as a New Regio- and Stereocontrolled Access to Bi- and Tricyclic β-Lactams. J. Org. Chem 2007, 72, 1604–1608. [DOI] [PubMed] [Google Scholar]
- (149).Xu L; Huang X Titanium(III)-Induced Intramolecular Radical Cyclization of Epoxyallene Ethers: an Efficient Method for Synthesis of Multifunctional Tetrahydrofurans. Tetrahedron Lett 2008, 49, 500–503. [Google Scholar]
- (150).Iwasaki H; Suzuki K; Yamane M; Yoshida S; Kojima N; Ozeki M; Yamashita M Indole Synthesis from N-allenyl-2-iodoanilines under Mild Conditions Mediated by Samarium(II) Diiodide. Org. Biomol. Chem 2014, 12, 6812–6815. [DOI] [PubMed] [Google Scholar]
- (151).Suzuki K; Iwasaki H; Ichiyoshi F; Tominaga M; Kojima N; Ozeki M; Yamashita M Synthesis of 3-Ethenylindoles via Intramolecular Cyclization of Aryl Radical with Allene Generated by Samarium(II) Diiodide. Heterocycles 2015, 91, 1244–1255. [Google Scholar]
- (152).Depature M; Diewok J; Grimaldi J; Hatem J Tin-Mediated Free-Radical Cyclization of α-Allenylbenzoyloximes. Eur. J. Org. Chem 2000, 2000, 275–280. [Google Scholar]
- (153).Depature M; Grimaldi J; Hatem J 3H-Pyrroles, Alkylidene-Pyrrolines and Functionalized Pyrrolidines by Radical Cyclization of β-Allenyliminyl Radicals. Eur. J. Org. Chem 2001, 2001, 941–946. [Google Scholar]
- (154).Saget T; Cramer N Heteroatom-Nucleophile-Induced C-C Fragmentations: Synthesis of Allenes and Entry to Domino Reactions. Angew. Chem., Int. Ed 2010, 49, 8962–8965. [DOI] [PubMed] [Google Scholar]
- (155).Sharma P; Liu R-S Cu-Catalyzed Aerobic Oxidative Cyclizations of 3-N-Hydroxyamino-1,2-propadienes with Alcohols, Thiols, and Amines to Form α-O-, S-, and N-Substituted 4-Methylquinoline Derivatives. Chem. - Eur. J 2015, 21, 4590–4594. [DOI] [PubMed] [Google Scholar]
- (156).Zeng R; Fu C; Ma S Formal Alkylation of Allenes through Highly Selective Radical Cyclizations of Allene-Enes. Angew. Chem., Int. Ed 2012, 51, 3888–3891. [DOI] [PubMed] [Google Scholar]
- (157).El Gueddari F; Grimaldi JR; Hatem JM Tosyl Mediated Radical Cyclization of Allylallenes. Tetrahedron Lett 1995, 36, 6685–6688. [Google Scholar]
- (158).Wang C-Y; Pan G-H; Chen F; Li J-H Oxidative Cyclization of 2-Allenyl-1,1’-biphenyls with α-Carbonyl Alkyl Bromides: Facile Access to Functionalized Phenanthrenes. Chem. Commun 2017, 53, 4730–4733. [DOI] [PubMed] [Google Scholar]
- (159).Depature M; Siri D; Grimaldi J; Hatem J; Faure R 3,6-Dihydropyridines from 6-endo Radical Cyclization onto Nitrogen in β-Allenyl Ketoximebenzoates. Tetrahedron Lett 1999, 40, 4547–4550. [Google Scholar]
- (160).Kang S-K; Ha Y-H; Kim D-H; Lim Y; Jung J Toluene-p-sulfonyl-mediated Radical Cyclization of Bis(allenes) Utilizing p-TsBr and p-TsSePh. Chem. Commun 2001, 1306–1307. [Google Scholar]
- (161).Li S; Zhang P; Li Y; Lu S; Gong J; Yang Z Diastereoselective Synthesis of Diquinanes and Triquinanes Bearing Vicinal Quaternary Carbon Stereocenters from Acyclic Allene-Based Precursors via a Cascade Reaction. Org. Lett 2017, 19, 4416–4419. [DOI] [PubMed] [Google Scholar]
- (162).Hatem J; Henriet-Bernard C; Grimaldi J; Maurin R Radical Cyclization of β-Allenic Oxime Ethers. Tetrahedron Lett 1992, 33, 1057–1058. [Google Scholar]
- (163).Marco-Contelles J; Balme G; Bouyssi D; Destabel C; Henriet-Bernard CD; Grimaldi J; Hatem JM Tributyltin Hydride-Mediated Free-Radical Cyclization of Allene-Tethered Oxime Ethers and Hydrazones. J. Org. Chem 1997, 62, 1202–1209. [Google Scholar]
- (164).Bernard-Henriet CD; Grimaldi JR; Hatem JM Radical Cyclization of β-Allenic Hydrazones. An Asymmetric Approach. Tetrahedron Lett 1994, 35, 3699–3702. [Google Scholar]
- (165).Depature M; Hatem J Tributyltin Hydride-mediated Free Radical Cyclization of Allene-tethered Oxime Ethers, Oxime Esters and Hydrazones. Influence of the Substituents Borne by the C=N Double Bond on the Course of the Reaction. C. R. Acad. Sci., Ser. IIc: Chim 2001, 4, 523–529. [Google Scholar]
- (166).Alcaide B; Almendros P; Aragoncillo C Structurally Novel Bi- and Tricyclic β-Lactams via [2 + 2] Cycloaddition or Radical Reactions in 2-Azetidinone-Tethered Enallenes and Allenynes. Org. Lett 2003, 5, 3795–3798. [DOI] [PubMed] [Google Scholar]
- (167).Jones NR; Pattenden G Alkene-Allenecyclopropane Radical Cyclizations Promoted by Tris-(trimethylsilyl)silane. Tetrahedron Lett 2009, 50, 3527–3529. [Google Scholar]
- (168).Poplata S; Tröster A; Zou Y-Q; Bach T Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev 2016, 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Namyslo JC; Kaufmann DE The Application of Cyclobutane Derivatives in Organic Synthesis. Chem. Rev 2003, 103, 1485–1537. [DOI] [PubMed] [Google Scholar]
- (170).Alcaide B; Almendros P; Aragoncillo C Exploiting [2 + 2] Cycloaddition Chemistry: Achievements with Allenes. Chem. Soc. Rev 2010, 39, 783–816. [DOI] [PubMed] [Google Scholar]
- (171).Moore WR; Bach AD; Ozretich TM Dimerization of Racemic and Optically Active 1,2-Cyclononadiene. J. Am. Chem. Soc 1969, 91, 5918–5919. [Google Scholar]
- (172).Weinstein B; Fenselau AH Oligomers of Allene. Part II. Dimers and Trimers Formed in the Thermal Polymerisation of Liquid Allene. J. Chem. Soc. C 1967, 0, 368–372. [Google Scholar]
- (173).Lebedev SV Velocity of Polymerization of Hydrocarbons of the Divinyl and Allene Series. J. Russ. Phys. Chem. Soc 1913, 45, 1357–1372. [Google Scholar]
- (174).Dai S-H; Dolbier WR Jr. Secondary Deuterium Isotope Effects in Allene Cycloadditions. J. Am. Chem. Soc 1972, 94, 3946–3952. [Google Scholar]
- (175).Skraba SL; Johnson RP A Computational Model for the Dimerization of Allene. J. Org. Chem 2012, 77, 11096–11100. [DOI] [PubMed] [Google Scholar]
- (176).Levek TL; Kiefer EF The Mechanism of Allene Cycloaddition. 111. Thermal and Photochemical Generation of 2,2’-Bis(1,1-dimethylallyl) Biradical from an Azocyclane Precursor. J. Am. Chem. Soc 1976, 98, 1875–1879. [Google Scholar]
- (177).Roth WR; Heiber M; Erker G Zum Mechanismus der Allendimerisierung. Angew. Chem 1973, 85, 511–512. [Google Scholar]
- (178).Gajewski JJ; Shih CN Hydrocarbon Thermal Degenerate Rearrangements. V. Stereochemistry of the 1,2-Dimethylenecyclobutane Self-Interconversion and Its Relation to the Allene Dimerization and the Rearrangements of Other C6H8 Isomers. J. Am. Chem. Soc 1972, 94, 1675–1682. [Google Scholar]
- (179).Gajewski JJ; Shih CN Characterization of the Dimethyl-1,2-dimethylenecylclobutanes from the Methylallene Thermal Dimerization. J. Org. Chem 1972, 37, 64–68. [Google Scholar]
- (180).Becher G; Skattebøl L Thermal Reactions of Meso- and Racemic Deca-2,3,7,8-tetraene. Tetrahedron Lett 1979, 20, 1261–1264. [Google Scholar]
- (181).Taylor DR; Warburton MR; Wright DB Allene Cycloadditisns. Part 1. Reaction of Tetraf luoroethylene with Allenic Hydrocarbons. J. Chem. Soc. C 1971, 385–391. [Google Scholar]
- (182).Taylor DR; Wright DB Allene Cycloadditions. Part II. Reaction of a Dialkylallene with Unsymmetrical Fluoro-Olefins: Evidence for a Two-Step [2 + 2] Cycloaddition. J. Chem. Soc. C 1971, 391–397. [Google Scholar]
- (183).Pasto DJ; Heid PF; Warren SE Cycloaddition Reactions of Allenes with N-Phenylmaleimide. A Two-Step, Diradical-Intermediate Process. J. Am. Chem. Soc 1982, 104, 3676–3685. [Google Scholar]
- (184).Woodward RB; Hoffmann R The Conservation of Orbital Symmetry. Angew. Chem., Int. Ed. Engl 1969, 8, 781–853. [Google Scholar]
- (185).Pasto DJ; Warren SE Isotope Effects and Relative Reactivities in the Radical-Chain Addition of Benzenethiol to Substituted Allenes. J. Org. Chem 1981, 46, 2842–2846. [Google Scholar]
- (186).Swenton JS; Bartlett PD Cycloaddition. VI. Competitive 1,2 and 1,4 Cycloaddition of 1,1-Dichloro-2,2-difluoroethylene to Butadiene. J. Am. Chem. Soc 1968, 90, 2056–2058. [Google Scholar]
- (187).Pasto DJ; Warren SE Cycloaddition of Substituted Allenes with 1,1-Dichloro-2,2-difluoroethene. A Model for the Two-Step, Diradical-Intermediate Cycloaddition of Allenes. J. Am. Chem. Soc 1982, 104, 3670–3676. [Google Scholar]
- (188).Kiefer EF; Okamura MY Evidence for a Concerted Mechanism for Allene Cycloaddition. J. Am. Chem. Soc 1968, 90, 4187–4189. [Google Scholar]
- (189).Pasto DJ; Yang SH Detection and the Analysis of the Factors Affecting the Competition between Ring Closure, Internal Rotation, and Cleavage in Diradical Intermediates Formed in Allene Cycloaddition Reactions. J. Am. Chem. Soc 1984, 106, 152–157. [Google Scholar]
- (190).Pasto DJ; Yang SH Cycloaddition of Chloro-, Cyano-, Methoxy-, and (Pheny1thio)allene with l,l-Dichloro-2,2-difluoroethene (1122). Competitive Cyclodimerization and Trapping of the Cyclodimers with 1122. J. Org. Chem 1986, 51, 3611–3619. [Google Scholar]
- (191).Padwa A; Meske M; Murphree SS; Watterson SH; Ni Z (Phenylsulfony1)allenes as Substrates for Cycloaddition Reactions: Intramolecular Cyclizations onto Unactivated Alkenes. J. Am. Chem. Soc 1995, 117, 7071–7080. [Google Scholar]
- (192).Spellmeyer DC; Houk KN A Force-Field Model for Intramolecular Radical Additions. J. Org. Chem 1987, 52, 959–974. [Google Scholar]
- (193).Alcaide B; Almendros P; Aragoncillo C Structurally Novel Bi- and Tricyclic β-Lactams via [2 + 2] Cycloaddition or Radical Reactions in 2-Azetidinone-Tethered Enallenes and Allenyne. Org. Lett 2003, 5, 3795–3798. [DOI] [PubMed] [Google Scholar]
- (194).Paterno GM; Barbero N; Galliano S; Barolo C; Lanzani G; Scotognella F; Borrelli R Excited State Photophysics of Squaraine Dyes for Photovoltaic Applications: an Alternative Deactivation Scenario. J. Mater. Chem. C 2018, 6, 2778–2785. [Google Scholar]
- (195).Parkhurst RR; Swager TM Synthesis and Optical Properties of Phenylene-Containing Oligoacenes. J. Am. Chem. Soc 2012, 134, 15351–15356. [DOI] [PubMed] [Google Scholar]
- (196).Pasto DJ; Kong W Comparison of the Relative Rates of Radical Addition versus Diradical Intermediate Formation in [2 + 2] Cycloaddition Reactions of Similarly Substituted Alkenes and Alkynes. J. Org. Chem 1989, 54, 3215–3216. [Google Scholar]
- (197).Pasto DJ; Kong W Cycloaddition Reactions of 1,1-Dimethylallene with Substituted Alkynes. J. Org. Chem 1988, 53, 4807–4810. [Google Scholar]
- (198).Siebert MR; Osbourn JM; Brummond KM; Tantillo DJ Differentiating Mechanistic Possibilities for the Thermal, Intramolecular [2 + 2] Cycloaddition of Allene-Ynes. J. Am. Chem. Soc 2010, 132, 11952–11966. [DOI] [PubMed] [Google Scholar]
- (199).Newcomb M; Le Tadic-Biadatti M-H; Chestney DL; Roberts ES; Hollenberg PF A Nonsynchronous Concerted Mechanism for Cytochrome P-450 Catalyzed Hydroxylation. J. Am. Chem. Soc 1995, 117, 12085–12091. [Google Scholar]
- (200).Newcomb M; Chestney DL A Hypersensitive Mechanistic Probe for Distinguishing between Radical and Carbocation Intermediates. J. Am. Chem. Soc 1994, 116, 9753–9754. [Google Scholar]
- (201).Alcaide B; Almendros P; Aragoncillo C; Gómez-Campillos G Expeditious Entry to Enantiopure Mono- and Bis(tricyclic) β-lactams by Single or Double [2 + 2] Cycloaddition of Allenynes. Eur. J. Org. Chem 2011, 2011, 364–370. [Google Scholar]
- (202).Jones RR; Bergman RG p-Benzyne. Generation as an Intermediate in a Thermal Isomerization Reaction and Trapping Evidence for the 1,4-Benzenediyl Structure. J. Am. Chem. Soc 1972, 94, 660–661. [Google Scholar]
- (203).Wang KK Enyne-Allenes In Modern Allene Chemistry; Krause NA, Hashmi SK, Eds.; Wiley-VHC: Weinheim, Germany, 2004; Vol. 2, pp 1091–1126. [Google Scholar]
- (204).Nagata R; Yamanaka H; Okazaki E; Saito I Biradical Formation from Acyclic Conjugated Eneyne-Allene System Related to Neocarzinostatin and Esperamicin-Calichemicin. Tetrahedron Lett 1989, 30, 4995–4998. [Google Scholar]
- (205).Myers AG; Dragovich PS; Kuo EY Studies on the Thermal Generation and Reactivity of a Class of (σ,π)-1,4-Biradicals. J. Am. Chem. Soc 1992, 114, 9369–9386. [Google Scholar]
- (206).Schmittel M; Strittmatter M; Kiau S Switching from the Myers Reaction to a New Thermal Cyclization Mode in Enyne-Allenes. Tetrahedron Lett 1995, 36, 4975–4978. [Google Scholar]
- (207).Finn MG; Dopico PG Synthesis and Cycloaromatization Kinetics of Aromatic Allene Enynes. Tetrahedron 1999, 55, 29–62. [Google Scholar]
- (208).Suzuki I; Wakayama M; Shigenaga A; Nemoto H; Shibuya M Synthesis of Enediyne Model Compounds Producing Toluene Diradicals Possessing a Highly Radical Character via Enyne–Allene Intermediates. Tetrahedron Lett 2000, 41, 10019–10023. [Google Scholar]
- (209).Koga N; Morokuma K Comparison of Biradical Formation between Enediyne and Enyne-Allene. Ab Initio CASSCF and MRSDCI Study. J. Am. Chem. Soc 1991, 113, 1907–1911. [Google Scholar]
- (210).Grissom JW; Klingberg D; Huang D; Slattery BJ Tandem Enyne Allene–Radical Cyclization: Low-Temperature Approaches to Benz[e]indene and Indene Compounds. J. Org. Chem 1997, 62, 603–626. [DOI] [PubMed] [Google Scholar]
- (211).Nagata R; Yamanaka H; Murahashi E; Saito I DNA Cleavage by Acyclic Eneyne-Allene Systems Related to Neocarzinostatin and Esperamicin-Calicheamicin. Tetrahedron Lett 1990, 31, 2907–2910. [Google Scholar]
- (212).Grissom JW; Klingberg D Tandem Enyne Allene-Radical Cyclization via Base-Catalyzed Isomerization of Enediyne Sulfones. Tetrahedron Lett 1995, 36, 6607–6610. [Google Scholar]
- (213).Andemichael YW; Huang Y; Wang KK Thermally-Induced One-Step Construction of the Tetracyclic Steroidal Skeleton from Acyclic Enyne-Allenes. J. Org. Chem 1993, 58, 1651–1652. [Google Scholar]
- (214).Wang KK; Zhang H-R; Petersen JL Thermolysis of Benzoenyne–Allenes To Form Biradicals and Subsequent Intramolecular Trapping with a Tetraarylallene To Generate Two Triarylmethyl Radical Centers. J. Org. Chem 1999, 64, 1650–1656. [DOI] [PubMed] [Google Scholar]
- (215).Grissom JW; Huang D Tandem Eneyne Allene-Radical Cyclization via [3,3] Sigmatropic Rearrangements. J. Org. Chem 1994, 59, 5114–5116. [Google Scholar]
- (216).Grissom JW; Huang D Low-Temperature Tandem Enyne Allene Radical Cyclizations: Efficient Synthesis of 2, 3-Dihydroindenes from Simple Enediynes. Angew. Chem., Int. Ed. Engl 1995, 34, 2037–2039. [Google Scholar]
- (217).Grissom JW; Slattery BJ Tandem Eneyne Allene-Radical Cyclization via [2,3] Sigmatropic Shifts. Tetrahedron Lett 1994, 35, 5137–5140. [Google Scholar]
- (218).Rule JD; Moore JS Polymerizations initiated by diradicals from cycloaromatization reactions. Macromolecules 2005, 38, 7266–7273. [Google Scholar]
- (219).Gaudel-Siri A; Campolo D; Mondal S; Nechab M; Siri D; Bertrand MP Theoretical Study to Explain How Chirality Is Stored and Evolves throughout the Radical Cascade Rearrangement of Enyneallenes. J. Org. Chem 2014, 79, 9086–9093. [DOI] [PubMed] [Google Scholar]
- (220).Sakai S; Nishitani M Theoretical Studies on Myers–Saito and Schmittel Cyclization Mechanisms of Hepta-1,2,4-triene-6-yne. J. Phys. Chem. A 2010, 114, 11807–11813. [DOI] [PubMed] [Google Scholar]
- (221).Engels B; Lennartz C; Hanrath M; Schmittel M; Strittmatter M Regioselectivity of Biradical Cyclizations of Enyne-Allenes: Influence of Substituents on the Switch from the Myers–Saito to the Novel C2–C6 Cyclization. Angew. Chem., Int. Ed 1998, 37, 1960–1963. [Google Scholar]
- (222).Bekele T; Christian CF; Lipton MA; Singleton DA Concerted” Transition State, Stepwise Mechanism. Dynamics Effects in C2-C6 Enyne Allene Cyclizations. J. Am. Chem. Soc 2005, 127, 9216–9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Carpenter BK Dynamic Behavior of Organic Reactive Intermediates. Angew. Chem., Int. Ed 1998, 37, 3340–3350. [DOI] [PubMed] [Google Scholar]
- (224).Musch PW; Engels B The Importance of the Ene Reaction for the C2–C6 Cyclization of Enyne–Allenes. J. Am. Chem. Soc 2001, 123, 5557–5562. [DOI] [PubMed] [Google Scholar]
- (225).Vavilala C; Bats JW; Schmittel M Classical Versus Nonstatistical Behavior in the C2-C6/Ene Cyclization of Enyne-Allenes: Intramolecular Kinetic Isotope Effects and Radical Clock Reactions. Synthesis 2010, 2010, 2213–2222. [Google Scholar]
- (226).Schmittel M; Mahajan AA; Bucher G; Bats JW Thermal C2–C6 Cyclization of Enyne–Allenes. Experimental Evidence for a Stepwise Mechanism and for an Unusual Thermal Silyl Shift. J. Org. Chem 2007, 72, 2166–2173. [DOI] [PubMed] [Google Scholar]
- (227).Bucher G; Mahajan AA; Schmittel M The Photochemical C2–C6 Cyclization of Enyne–Allenes: Interception of the Fulvene Diradical with a Radical Clock Ring Opening. J. Org. Chem 2009, 74, 5850–5860. [DOI] [PubMed] [Google Scholar]
- (228).Yang Y; Petersen JL; Wang KK Polycyclic Aromatic Compounds via Radical Cyclizations of Benzannulated Enyne-Allenes Derived from Ireland-Claisen Rearrangement. J. Org. Chem 2003, 68, 8545–8549. [DOI] [PubMed] [Google Scholar]
- (229).Yang Y; Petersen JL; Wang KK Cascade Radical Cyclizations of Benzannulated Enyne–Allenes. Unusual Cleavage of a Benzene Ring Leading to Twisted 1,1’-Dialkyl-9,9’-bifluorenylidenes and Spiro[1H-cyclobut[a]indene-1,9’-[9H]fluorenes]. J. Org. Chem 2003, 68, 5832–5837. [DOI] [PubMed] [Google Scholar]
- (230).Shimizu T; Sakamaki K; Kamigata N Preparation and Thermal Reaction of 1,3-Bis(alkylthio)allenes. Tetrahedron Lett 1997, 38, 8529–8532. [Google Scholar]
- (231).Shimizu T; Sakamaki K; Miyasaka D; Kamigata N Preparation and Reactivity of 1,3-Bis(alkylthio)allenes and Tetrathia-cyclic Bisallenes. J. Org. Chem 2000, 65, 1721–1728. [DOI] [PubMed] [Google Scholar]
- (232).Shimizu T; Miyasaka D; Kamigata N Synthesis and Thermal Reaction of 1,3-Bis(alkylseleno)allenes. Org. Lett 2000, 2, 1923–1925. [DOI] [PubMed] [Google Scholar]
- (233).Shimizu T; Miyasaka D; Kamigata N Synthesis and Reactivity of Allenes Substituted by Selenenyl Groups at 1- and 3-Positions. J. Org. Chem 2001, 66, 1787–1794. [DOI] [PubMed] [Google Scholar]
- (234).Pham HV; Houk KN Diels–Alder Reactions of Allene with Benzene and Butadiene: Concerted, Stepwise, and Ambimodal Transition States. J. Org. Chem 2014, 79, 8968–8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Skraba SL; Johnson RP A Computational Model for the Dimerization of Allene. J. Org. Chem 2012, 77, 11096–11100. [DOI] [PubMed] [Google Scholar]
- (236).Breuil-Desvergnes V; Goré J Reaction of the Lithio-Derivative of Methoxyallene with Hydrazones. Part 2: Formation of 3-Pyrrolines and Azetidines; Synthetic and Mechanistic Aspects. Tetrahedron 2001, 57, 1951–1960. [Google Scholar]
- (237).Burnett DA; Choi J-K; Hart DJ; Tsai Y-M Pyrrolizidinone and Indolizidinone Synthesis: Generation and Intramolecular Addition of α-Acylamino Radicals to Olefins and Allenes. J. Am. Chem. Soc 1984, 106, 8201–8209. [Google Scholar]
- (238).Tufariello JJ; Tette JP Synthesis in the Pyrrolizidine Class of Alkaloids. dl-Supinidine. J. Org. Chem 1975, 40, 3866–3869. [DOI] [PubMed] [Google Scholar]
- (239).Hudlicky T; Frazier JO; Kwart LD Intramolecular [4 + 1] Pyrroline Annulation Approach to Pyrrolizidine Alkaloids. Formal Total Synthesis of (±)-Supinidine. Tetrahedron Lett 1985, 26, 3523–3526. [Google Scholar]
- (240).Hassner A; Singh S; Sharma R; Maurya R A Synthesis of (−)-Supinidine and Its Regioisomer by Intramolecular Oxime Olefin Cycloaddition. Tetrahedron 1993, 49, 2317–2324. [Google Scholar]
- (241).Takahata H; Banba Y; Momose T An Asymmetric Total Synthesis of (−)-Supinidine. Tetrahedron 1991, 47, 7635–7644. [Google Scholar]
- (242).Dener JM; Hart DJ α-Acylamino Radical Cyclizations Using Allenes and Vinylsilanes as Addendends: Applications to the Synthesis of (+)-Heliotridine and (−)-Dihydroxyheliotridine. Tetrahedron 1988, 44, 7037–7046. [Google Scholar]
- (243).Myers AG; Condroski KR Synthesis of (±)-7,8-Epoxy-4-basmen-6-one by a Transannular Cyclization Strategy. J. Am. Chem. Soc 1993, 115, 7926–7927. [Google Scholar]
- (244).Spellmeyer DC; Houk KN A Force-Field Model for Intramolecular Radical Additions. J. Org. Chem 1987, 52, 959–977. [Google Scholar]
- (245).Chen Y-J; Wang C-Y; Lin W-Y Radical Cyclization of α-Allenic Ketone: A New Approach to the Synthesis of (+)-α-Chamigrene. Tetrahedron 1996, 52, 13181–13188. [Google Scholar]
- (246).Moreau S; Lablache-Combier A; Biguet J; Foulon C; Delfosse M Botryodiplodin, a Mycotoxin Synthesized by a Strain of P. roqueforti. J. Org. Chem 1982, 47, 2358–2359. [Google Scholar]
- (247).Villar F; Andrey O; Renaud P Diastereoselective Radical Cyclization of Bromoacetals: Efficient Synthesis of (±)-Botryodiplodin. Tetrahedron Lett 1999, 40, 3375–3378. [Google Scholar]
- (248).Nouguier R; Gastaldi S; Stien D; Bertrand M; Villar F; Andrey O; Renaud P Synthesis of (±)- and (−)-Botryodiplodin Using Stereoselective Radical Cyclizations of Acyclic Esters and Acetals. Tetrahedron: Asymmetry 2003, 14, 3005–3018. [Google Scholar]
- (249).Ley SV; Denholm AA; Wood A The Chemistry of Azadirachtin. Nat. Prod. Rep 1993, 10, 109–157. [Google Scholar]
- (250).Ley SV; Abad-Somovilla A; Anderson JC; Ayats C; Bänteli R; Beckmann E; Boyer A; Brasca MG; Brice A; et al. Chem. -Eur. J 2008, 14, 10683–10704. [DOI] [PubMed] [Google Scholar]
- (251).Yang D; Cwynar V; Donahue MG; Hart DJ; Mbogo G Two Approaches to Diverting the Course of a Free-Radical Cyclization: Application of Cyclopropylcarbinyl Radical Fragmentations and Allenes as Radical Acceptors. J. Org. Chem 2009, 74, 8726–8732. [DOI] [PubMed] [Google Scholar]
- (252).Amatov T; Pohl R; Cisarova I; Jahn U Synthesis of Bridged Diketopiperazines by Using the Persistent Radical Effect and a Formal Synthesis of Bicyclomycin. Angew. Chem., Int. Ed 2015, 54, 12153–12157. [DOI] [PubMed] [Google Scholar]
- (253).Williams RM; Armstrong RW; Dung J-S Stereocontrolled Total Synthesis of (±)- and (+)-Bicyclomycin: New Carbon-Carbon Bond-Forming Reactions on Electrophilic Glycine Anhydride Derivatives. J. Am. Chem. Soc 1984, 106, 5748–5750. [Google Scholar]
- (254).Williams RM; Armstrong RW; Dung J-S Stereocontrolled Total Synthesis of (±)- and (+)-Bicyclomycin. J. Am. Chem. Soc 1985, 107, 3253–3266. [Google Scholar]