

SUMMARY
Protein ensembles control genome function by establishing, maintaining, and deconstructing cell-type-specific chromosomal landscapes. A plethora of small molecules orchestrate cellular functions and therefore may link physiological processes with genome biology. The metabolic enzyme and hemoglobin cofactor heme induces proteolysis of a transcriptional repressor, Bach1, and regulates gene expression post-transcriptionally. However, whether heme controls genome function broadly or through prescriptive actions is unclear. Using assay for transposase-accessible chromatin sequencing (ATAC-seq), we establish a heme-dependent chromatin atlas in wild-type and mutant erythroblasts lacking enhancers that confer normal heme synthesis. Amalgamating chromatin landscapes and transcriptomes in cells with sub-physiological heme and post-heme rescue reveals parallel Bach1-dependent and Bach1-independent mechanisms that target heme-sensing chromosomal hotspots. The hotspots harbor a DNA motif demarcating heme-regulated chromatin and genes encoding proteins not known to be heme regulated, including metabolic enzymes. The heme-omics analysis establishes how an essential biochemical cofactor controls genome function and cellular physiology.
Graphical Abstract
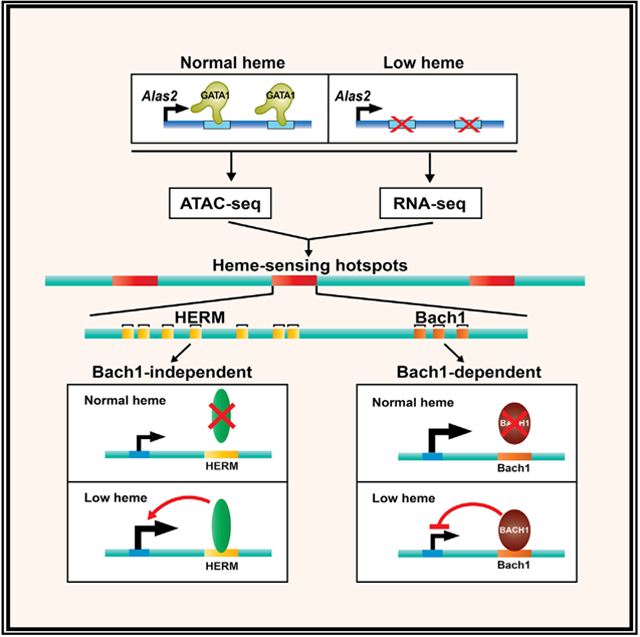
In Brief
Liao et al. generate a heme-regulated chromatin atlas by amalgamating ATAC-seq and RNA-seq datasets from cells with normal and sub-physiological heme, and they identify parallel Bach1-dependent and Bach1-independent mechanisms that target heme-sensing chromatin hotspots. The hotspots harbor a DNA motif demarcating heme-regulated chromatin and genes not known to be heme regulated.
INTRODUCTION
Maps depicting sites of protein residency on chromosomes constitute an invaluable resource for discovering physiological mechanisms and how genetic variation and epigenetic aberrations predispose to, or trigger, pathogenesis. Elucidating mechanisms that dynamically establish, maintain, and deconstruct chromatin landscapes has enormous potential to unveil cell biological paradigms. In this regard, much remains to be discovered on how endogenous small molecules (e.g., protein cofactors, metabolites, and metals) link cellular physiology with genome function.
The iron-containing protoporphyrin heme is a critical prosthetic group for metabolic enzymes and hemoglobin. Heme homeostasis is established and maintained by modulating heme synthesis and transport (Dailey and Meissner, 2013; Yuan et al., 2013). In erythroid cells, the transcription factor GATA1 increases heme synthesis by activating two ALAS2 intronic enhancers, which increases expression of the heme biosynthetic enzyme aminolevulinic acid synthase 2 (Tanimura et al., 2016). ALAS2 intron 1 enhancer mutations disrupt heme synthesis and cause X-linked sideroblastic anemia (Campagna et al., 2014; Kaneko et al., 2014). Mutation of the murine enhancers abrogates GATA1 occupancy at these sites, decreasing Alas2 expression and heme synthesis, thus causing severe anemia and embryonic lethality (Zhang et al., 2017). Heme synthesis is also controlled by erythropoietin-activated protein kinase A, which phosphorylates and activates the heme synthesis enzyme ferrochelatase (FECH) (Chung et al., 2017).
By regulating intracellular heme levels, plasma membrane transporters prevent excessive or inadequate heme accumulation, both deleterious to cells (Yuan et al., 2013). Depletion of the heme exporter Flvcr1 increases intracellular heme and causes anemia (Doty et al., 2015; Keel et al., 2008). Increased heme during erythroid maturation downregulates GATA1 as a negative feedback loop (Doty et al., 2019). The C. elegans importers HRG-1 and HRG-4 mediate heme uptake, and the zebra-fish HRG-1 ortholog Hrg1 promotes erythropoiesis (Rajagopal et al., 2008). The murine HRG-1 ortholog SLC48A1 localizes to lysosomal membranes and promotes heme-iron recycling in macrophages during erythrophagocytosis and erythroid maturation (White et al., 2013).
Beyond its canonical functions as an enzyme cofactor and hemoglobin component, heme regulates genes transcriptionally and post-transcriptionally. Though rigorous studies have established heme mechanisms that control transcription and translation, many questions remain unanswered regarding how heme controls fundamental processes such as genome function. Heme deficiency promotes autophosphorylation and activation of the eIF2α kinase heme-regulated inhibitor (HRI) (Chen and London, 1995; Han et al., 2001). HRI-mediated phosphorylation of eIF2α inhibits globin and ribosomal protein translation (Zhang et al., 2019). In heme deficiency, transcriptional and translational mechanisms coordinate globin protein and heme synthesis, preventing cytotoxicity elicited by excess globin polypeptides. Heme binding to the repressor Bach1 induces its degradation via the proteasome (Hira et al., 2007; Ogawa et al., 2001; Zenke-Kawasaki et al., 2007). In low heme, Bach1 accumulates and represses target genes, including Hmox1 encoding heme oxygenase 1 that restricts heme cytotoxicity and Hbb-b1 encoding b-globin (Sun et al., 2002; Tanimura et al., 2016). Heme also regulates a gene cohort by amplifying GATA1-regulated transcription in a Bach1-independent manner (Tanimura et al., 2016).
Heme-dependent transcriptional control, exemplified by Bach1 downregulation (Ogawa et al., 2001), activation of the cytoprotective transcription factor Nrf2 (Motohashi and Yamamoto, 2004), amplification of GATA1 activity (Tanimura et al., 2018; Tanimura et al., 2016), and HRI repressing γ-globin transcription (Grevet et al., 2018), is critical in physiology and pathology. In principle, these activities may impact genome function extensively, yet how heme acts at the genomic level is largely unexplored. It is instructive therefore to ask if heme controls genome function broadly or via prescriptive actions. We used assay for transposase-accessible chromatin sequencing (ATAC-seq) (Buenrostro et al., 2013) to establish a genome-wide atlas of heme-regulated chromatin in a genetic complementation system with GATA1 null erythroblast cells (G1E-ER-GATA1) and a subline with Alas2 enhancer GATA motif deletions that reduce heme ~30-fold (Tanimura et al., 2018; Tanimura et al., 2016). Amalgamating heme-regulated chromatin and transcriptomic landscapes, coupled with bioinformatics and functional genomics (“heme-omics”), revealed parallel Bach1-dependent and Bach1-independent genome-regulatory mechanisms, a DNA motif that predicts heme-regulated chromatin targeting and heme-sensing chromosomal hotspots harboring genes that unveil how an essential biochemical cofactor controls genome and cellular functions.
RESULTS
Genome-wide Atlas of GATA1- and Heme-Regulated Chromatin Accessibility
To elucidate a mechanism for how a genome senses alterations in heme levels, ATAC-seq was deployed in the mouse G1E-ER-GATA1 cell genetic complementation system. GATA1-null G1E proerythroblasts stably express an estradiol-inducible ER-GATA1 allele, and estradiol treatment induces erythroid maturation, recapitulating early stages of mouse and human erythrocyte development (Gregory et al., 1999; Welch et al., 2004). Previously, we generated G1E-ER-GATA1-derived clonal lines harboring GATA motif mutations in Alas2 intron 1 and eight enhancers that abrogated ER-GATA1 occupancy, decreasing Alas2 expression and reducing heme by ~30-fold (Tanimura et al., 2016, 2018). The sub-physiological heme dysregulates a gene cohort, and 5-aminolevulinate (ALA) rescues heme, restoring expression at a subset of these genes. ATAC-seq was conducted with untreated and β-estradiol-treated wild-type or mutant cells (Figure 1A), sequences were mapped to mm9 mouse genome assembly using Bowtie (Langmead et al., 2009), and peaks were called with model-based analysis of ChIP-seq (MACS) (Zhang et al., 2008). Comparison of untreated and β-estradiol-treated wild-type cells revealed GATA1-induced and GATA1-repressed ATAC-seq peaks. For example, GATA1 activated Slc4a1 with GATA1-induced peaks −10.4 and +1.2 kb relative to the start site (Figures 1B and 1C). GATA1-repressed Il1rl1 with a GATA1-repressed peak at −11.1 kb.
Figure 1. GATA1- and Heme-Regulated Chromatin Landscape.
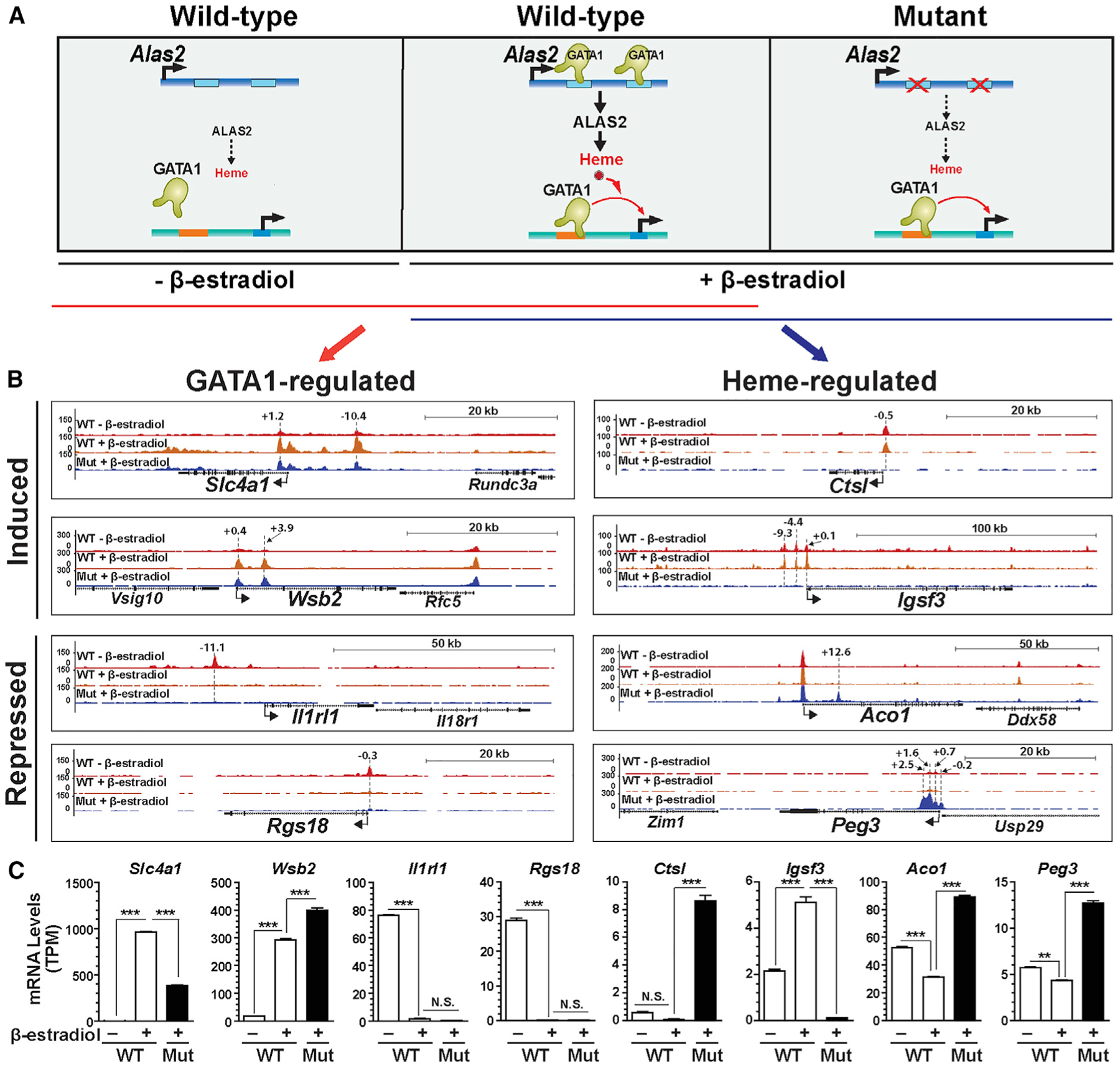
(A) Genetic complementation assay. β-Estradiol activation of ER-GATA1 in G1E-ER-GATA1 cells increases Alas2 expression, which elevates heme in wild-type (WT) cells. Deletion of Alas2 enhancers in mutant cells severely reduces Alas2 expression and heme.
(B) Representative ATAC-seq profiles of GATA1- or heme-regulated chromatin loci. The numbers indicate the location of ATAC-seq peak centers relative to the transcription start sites (in kb).
(C) RNA expression of genes associated with GATA1- or heme-regulated loci in B (n = 3, mean ± SE). p values were calculated by two-tailed unpaired Student’s t test (**p < 0.01 and ***p < 0.001; N.S., not significant).
We compared ATAC-seq profiles of β-estradiol-treated wild-type and mutant G1E-ER-GATA1 cells to identify heme-induced or heme-repressed accessibility sites. A comparison of untreated wild-type and mutant cells was also conducted and will be discussed later. ER-GATA1 induced accessibility −0.2 to +2.5 kb relative to the Peg3 transcription start site only in mutant cells. Peg3 mRNA levels were higher (2.9 fold) in β-estradiol-treated mutant versus wild-type cells, and the nearest-neighbor genes were not expressed. Ctsl and Igsf3 expression was higher and lower, respectively, in mutant cells, and accessibility at both loci was abrogated in mutant cells. GATA1- or heme-dependent accessibility changes were not invariably concordant with RNA expression. Discordance was exemplified by heme-induced accessibility at the Ctsl promoter upon repression (Figures 1B and 1C), consistent with prior reports of accessibility preceding transcriptional activation and not invariably predicting a state of ongoing transcription (Daugherty et al., 2017; Jiménez et al., 1992; Voigt et al., 2013) .
To determine the relationship between GATA1- and heme-regulated accessibility, we amalgamated overlapping ATAC-seq peaks from three biological replicates of all samples analyzed to yield 59,483 total (master) peaks. The read counts under each master peak were used to quantify the peak for each sample. Differential analyses of the master peaks were conducted with DEseq2 (Love et al., 2014). Peaks with | log2 fold change| > = 1 and adjusted p value ≤ 0.05 were deemed differentially accessible. This strategy identified 11,340 GATA1- (Figure 2A) and 1,222 heme-regulated peaks (Figure 2B). GATA1- and heme-regulated peaks were parsed based on whether the peak signal increased or decreased (Figure 2C). The majority of peaks were intronic or intergenic, while ~6% of GATA1- and ~20% of heme-regulated peaks resided within 1 kb of promoters (Figure 2D). These results conform to GATA1 (Cheng et al., 2009; Fujiwara et al., 2009; Kang et al., 2012; Yu et al., 2009) and Bach1 (Warnatz et al., 2011) chromatin immunoprecipitation sequencing (ChIP-seq) data, in which the majority of occupied sites are intronic or intergenic. To establish how many GATA1-regulated ATAC-seq peaks overlapped with GATA1 occupancy, GATA1-regulated ATAC-seq peaks were compared with GATA1 ChIP-seq peaks from G1E-ER-GATA1 cells and Ter119+ primary erythroblasts. Of 11,340 GATA1-regulated ATAC-seq peaks, 2,721 and 2,911 overlapped with GATA1 occupancy in G1E-ER-GATA1 and primary erythroblasts, respectively (Figure 2E), suggesting that GATA1 directly controls accessibility at this large cohort of genomic loci. Other GATA1-induced accessibility changes did not overlap with GATA1 occupancy, suggesting long-range GATA1 actions or indirect influences of GATA1 action at other loci.
Figure 2. Principles Governing GATA1 and Heme Function through Chromatin.
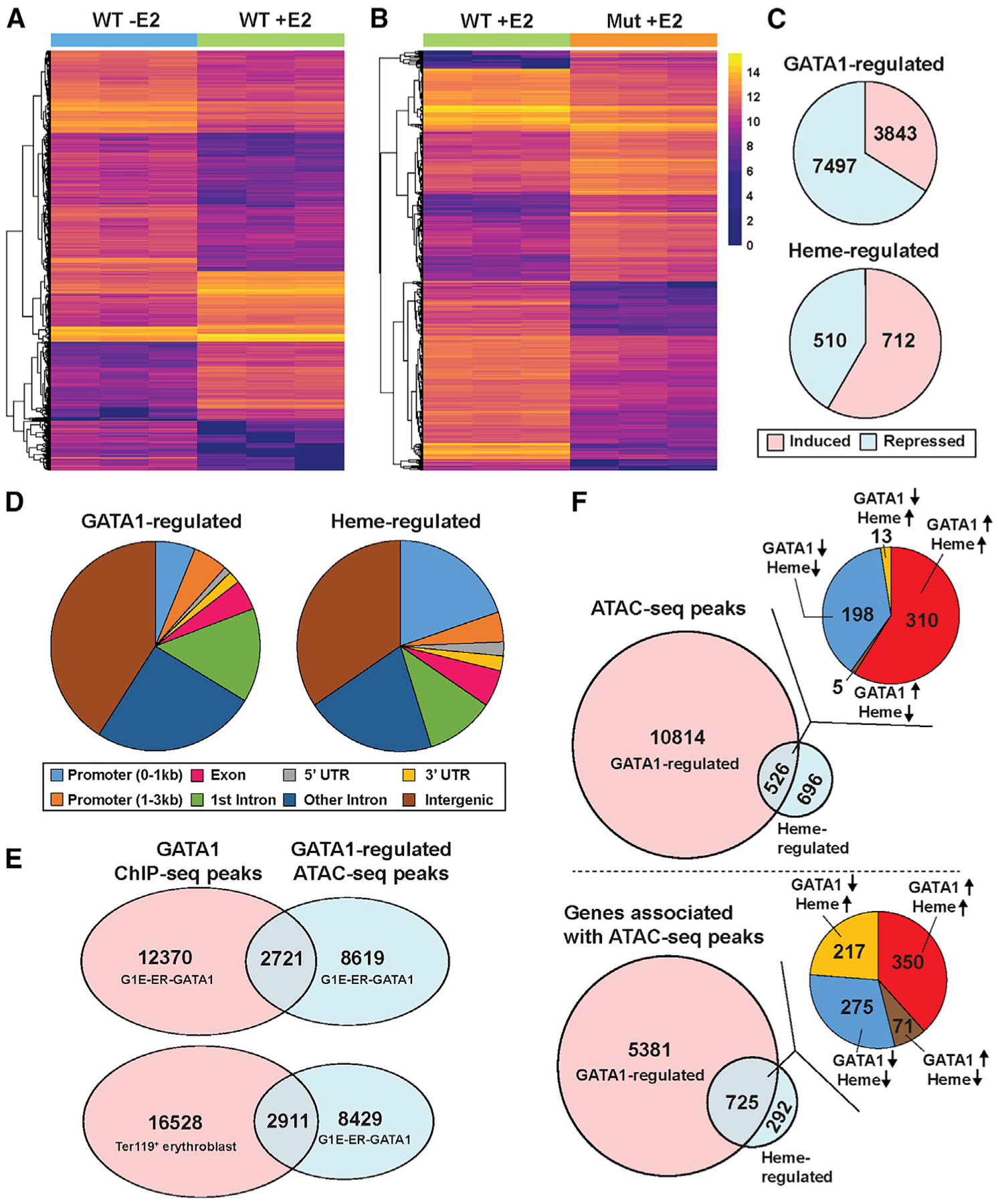
(A) Heatmap depicting all GATA1-regulated ATAC-seq peaks. The color indicates normalized read count for each peak. E2, β-estradiol.
(B) Heatmap depicting all heme-regulated ATAC-seq peaks. The color indicates normalized read count for each peak.
(C) Pie charts depicting the number of induced or repressed peaks in GATA1 or heme-regulated ATAC-seq peaks.
(D) Pie charts illustrating the GATA1- or heme-regulated ATAC-seq peak distribution over the genome.
(E) Venn diagram depicting overlap between GATA1-regulated ATAC-seq peaks and GATA1 ChIP-seq peaks in different cell types.
(F) Top: Venn diagram depicting overlap between GATA1- and heme-regulated ATAC-seq peaks, with a pie chart showing the number of peaks with different regulatory modes. The arrows indicate the direction of changes in chromatin accessibility by GATA1 or heme. Bottom: Venn diagram depicting overlap between genes associated with GATA1- or heme-regulated ATAC-seq peaks, with a pie chart illustrating the number of genes with different modes of regulation. The arrows indicate the direction of changes in gene expression by GATA1 or heme.
Heme facilitates or antagonizes GATA1 activity to regulate transcription. For a gene regulated by both GATA1 and heme, GATA1 and heme might function through the same or distinct sites at the locus. We identified 526 GATA1/heme-co-regulated ATAC-seq peaks (Figure 2F, upper panel), which parsed into four groups: (1) GATA1 induced, heme induced; (2) GATA1 induced, heme repressed; (3) GATA1 repressed, heme repressed; or (4) GATA1 repressed, heme induced. As the majority of the co-regulated peaks conformed to groups 1 and 3, heme amplification of GATA1 activity to control accessibility was the most common mechanism. Since a gene can contain multiple ATAC-seq peaks, we enumerated genes harboring GATA1- and heme-regulated peaks. 526 of the GATA1/heme-co-regulated peaks resided in 489 of the 725 genes associated with GATA1- and heme-regulated peaks. The remaining 236 genes have distinct GATA1- and heme-regulated peaks (Figure 2F, lower panel). Thus, GATA1/heme co-regulation of 526 endogenous loci involves multiple regulatory modes, and GATA1 and heme function at the same or distinct sites of a given gene.
A Hallmark Sequence Motif Within Heme-regulated Chromatin
Using 1,222 heme-regulated ATAC-seq peaks (Table S2) and de novo motif finding with MEME (multiple EM for motif elicitation) (Bailey et al., 2009), we analyzed the relationship between heme-regulated chromatin targeting and the plethora of genomic DNA motifs. This analysis identified AGATAA within the top 1,000 GATA1-regulated ATAC-seq peaks (Figure 3A, left). De novo motif finding revealed anenriched 15-bp motif in heme-regulated peaks. Comparison to known motifs with TOMTOM (Gupta et al., 2007) identified it as a Bach1 motif (Figure 3A, middle). Analysis of 526 GATA1/heme-co-regulated peaks revealed a similar motif (Figure 3A, right). As < 30% of the 1,222 heme-regulated peaks contained a Bach1 motif, either heme also targets chromatin via a Bach1-independent mechanism, heme regulates the genes indirectly or heme/Bach1 function over a long distance not predictable by linear gene configurations. Our prior study involving a very limited cohort of heme-regulated genes demonstrated that not all were sensitive to Bach1 downregulation (Tanimura et al., 2016).
Figure 3. A Novel Motif, HERM, Is Enriched at Heme-Regulated Chromatin Sites.
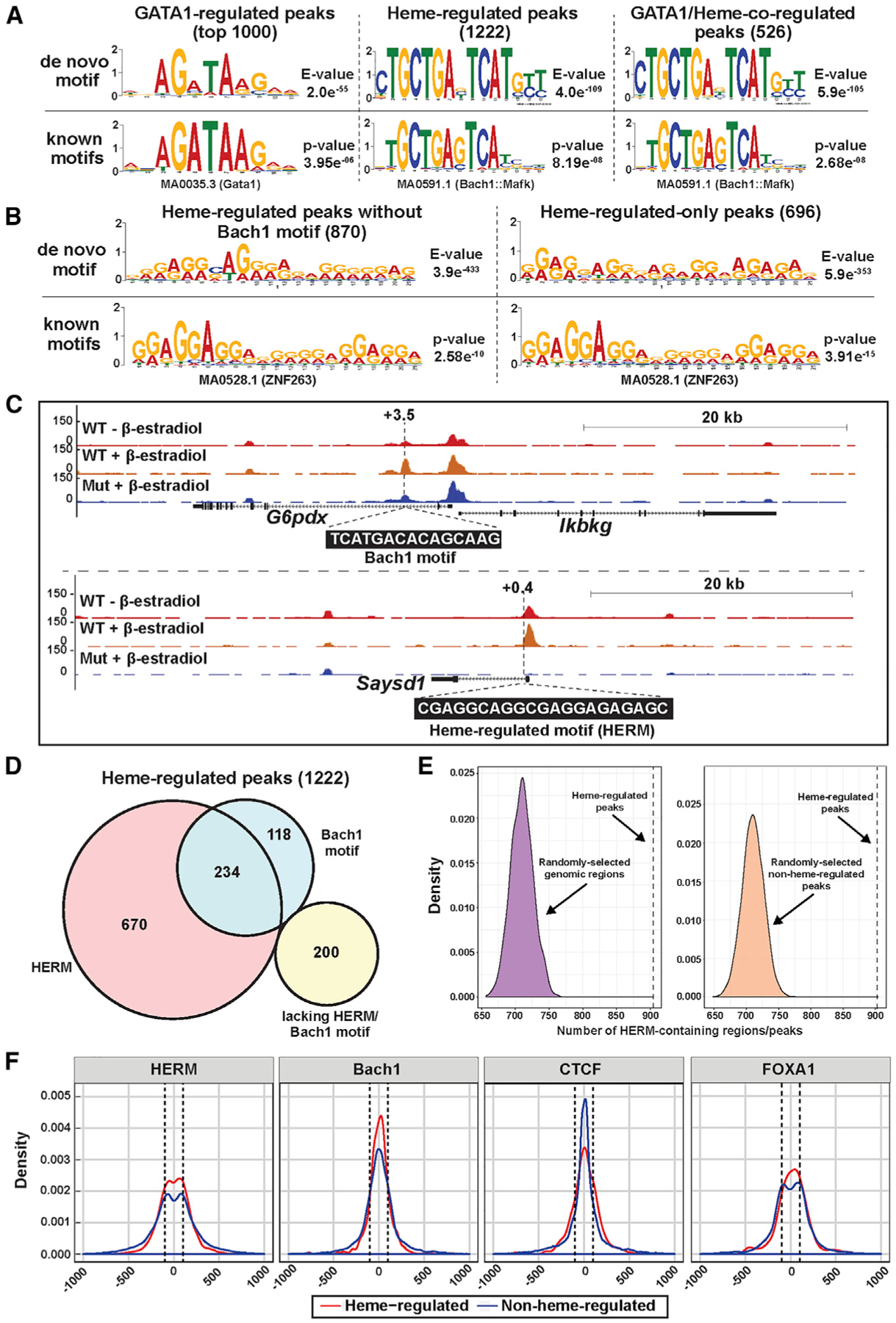
(A) De novo motifs identified by MEME analysis of the top 1,000 GATA1-regulated peaks, 1,222 heme-regulated peaks, or 526 GATA1/heme-co-regulated peaks. A known motif matching each de novo motif is shown below.
(B) De novo motifs were identified by MEME using 870 heme-regulated peaks without Bach1 motif or 696 heme-regulated-only peaks. One known motif matching each de novo motif is shown below. “Heme-regulated-only” peaks were not GATA1 regulated. (C) ATAC-seq tracks for representative heme-regulated peaks with Bach1 motif or HERM. The numbers indicate the location of Bach1 motif or heme-regulated motif (HERM) relative to the transcription start sites (in kb).
(D) Venn diagram depicting the number of heme-regulated peaks that contain Bach1 motifs and/or HERMs.
(E) Density distribution plots depicting the number of HERM-containing genomic regions from 10,000 repeats of random sampling of the mouse genome (left) or non-heme-regulated ATAC-seq peaks (right) with number and length matching that of heme-regulated ATAC-seq peaks. The dotted lines indicate the number of heme-regulated ATAC-seq peaks that contain HERMs.
(F) Density distribution plot depicting the distance of HERM, Bach1 motif, CTCF, and FoxA1 motifs from the ATAC-seq peak center. Dotted lines, 100 bp from the peak center.
See also Figure S1.
To unveil how the apparent Bach1-independent heme mechanism targets chromatin globally, we conducted de novo motif finding using 870 heme-regulated ATAC-seq peaks (including heme-regulated and GATA1/heme-co-regulated) lacking Bach1 motifs, or 696 heme-regulated, but not GATA1-regulated, peaks. The analysis from the 870 peaks revealed a 21-bp A-G-rich motif (Figure 3B) that we deemed the heme-regulated motif (HERM). De novo motif finding using an alternative tool (Hyper-geometric Optimization of Motif EnRichment [HOMER]), or using heme-induced or heme-repressed peaks, respectively, revealed similar motifs (Figures S1A and S1B). Figure 3C depicts representative genes with heme-regulated peaks harboring a Bach1 motif or HERM. 904 of 1,222 (74.0%) heme-regulated peaks contain HERMs, 352 (28.8%) contain Bach1 motifs, 234 (19.2%) contain both, and 200 (16.4%) contain neither (Figure 3D). Of the 904 heme-regulated peaks containing HERMs, 520 are heme induced and 384 are heme repressed. Whereas 6.8% of heme-regulated peaks containing Bach1 motifs, but not HERMs, resided within 1 kb of promoters, 26% of HERM-containing, but not Bach1-motif-containing, heme-regulated peaks resided near promoters (Figure S1C) (p < 1e−5). By contrast to 74% of heme-regulated peaks containing HERM, only 11,511 of 58,261 (19.8%) non-heme-regulated peaks contained HERM (Figure S1D, left). Thus, HERMs were more prevalent in heme- versus non-heme-regulated peaks (p < 2.2e−16, chi-square test). The Bach1 motif was also more frequent in heme- versus non-heme-regulated peaks (28.8 versus 19.6%, p = 2.531e−15) (Figure S1D, right), although the chi-square statistics were considerably less than that of HERM (62.6 versus 2127).
To determine the prevalence of HERMs in mammalian genome, we conducted motif searching with mouse genome sequences, which revealed 405,266 nonoverlapping HERMs (Figure S1E). Weasked if HERM distinguishes heme-regulatedpeaks from other genomic regions. We randomly sampled the mouse genome with the same number and length of DNA fragments as heme-regulated peaks and conducted motif searching in these fragments. This analysis resulted in a median of 710 HERM-containing regions, contrasting with 904 from heme-regulated peaks (p value = 0; Figure 3E, left). We applied a similar strategy to the 58,261 non-heme-regulated peaks, which identified a median of 710 HERM-containing peaks (p value = 0, Figure 3E, right). These analyses provide further evidence for HERM enrichment in heme-regulated peaks.
To analyze how heme targets chromatin via Bach1 motifs or HERMs, we analyzed the motif distance to the center of each heme- or non-heme-regulated ATAC-seq peak. Unlike Bach1 or CCCTC-binding factor (CTCF; analyzed as a control) motifs enriched centrally within peaks, HERM was distributed biphasically with two peaks ~100 bp (dotted line) away from the peak center (Figure 3F). The distance between the two HERM peaks (~200 bp) matches the length of nucleosomal plus linker DNA. The FoxA1 transcription factor DNA-binding domain resembles the linker histone globular domain and binds inactive chromatin, replacing the linker histone and facilitating chromatin opening (Cirillo et al., 1998). Though HERM and FoxA1 motifs were distributed biphasically within ATAC-seq peaks, they lack sequence similarity, and unlike HERM, FoxA1 motif frequency was not enriched in heme- versus non-heme-regulated peaks (17.4 and 15.4%, respectively). In aggregate, this analysis led to the discovery of a new DNA motif that demarcates heme-regulated chromatin with greater fidelity than the Bach1 motif.
Parallel Bach1-Dependent and Bach1-Independent Heme Mechanisms of Genome Targeting
To evaluate the relationship between GATA1 and heme-regulated chromatin accessibility and gene expression, we amalgamated ATAC-seq data with our G1E-ER-GATA1 RNA-sequencing (RNA-seq) data (Tanimura et al., 2016) to identify GATA1 and/or heme-regulated genes harboring GATA1 and/or heme-regulated ATAC-seq peaks. Of 7,212 GATA1-regulated genes, 2,624 genes contained GATA1-regulated ATAC-seq peaks. The 2,624 genes parsed into the following groups: (1) chromatin accessible, expression higher; (2) chromatin accessible, expression lower; (3) chromatin inaccessible, expression higher; and (4) chromatin inaccessible, expression lower. The similar gene segregation in these groups implies that qualitative accessibility changes do not predict the GATA1-regulatory mode (Figure 4A). 169 of 1,340 heme-regulated genes contained heme-regulated ATAC-seq peaks (Figure 4B). Of 169 genes with heme-regulated chromatin/mRNA, 88 (52%) exhibited acces sibility and elevated expression. Since heme-dependent chromatin transitions occurred at a minority of heme-regulated genes, regulatory sites at most heme-dependent genes might preexist as open chromatin. Alternatively, many transitions may be <2-fold or involve long-range mechanisms. 904 of 1,171 heme-regulated genes, without heme-dependent accessibility changes, harbor at least one ATAC-seq peak, 116 of which are associated with a significant, but <2-fold, accessibility change.
Figure 4. Integrating ATAC-Seq and RNASeq Datasets Reveals Mechanisms Underlying GATA1/Heme-Mediated Establishment of Genetic Networks.
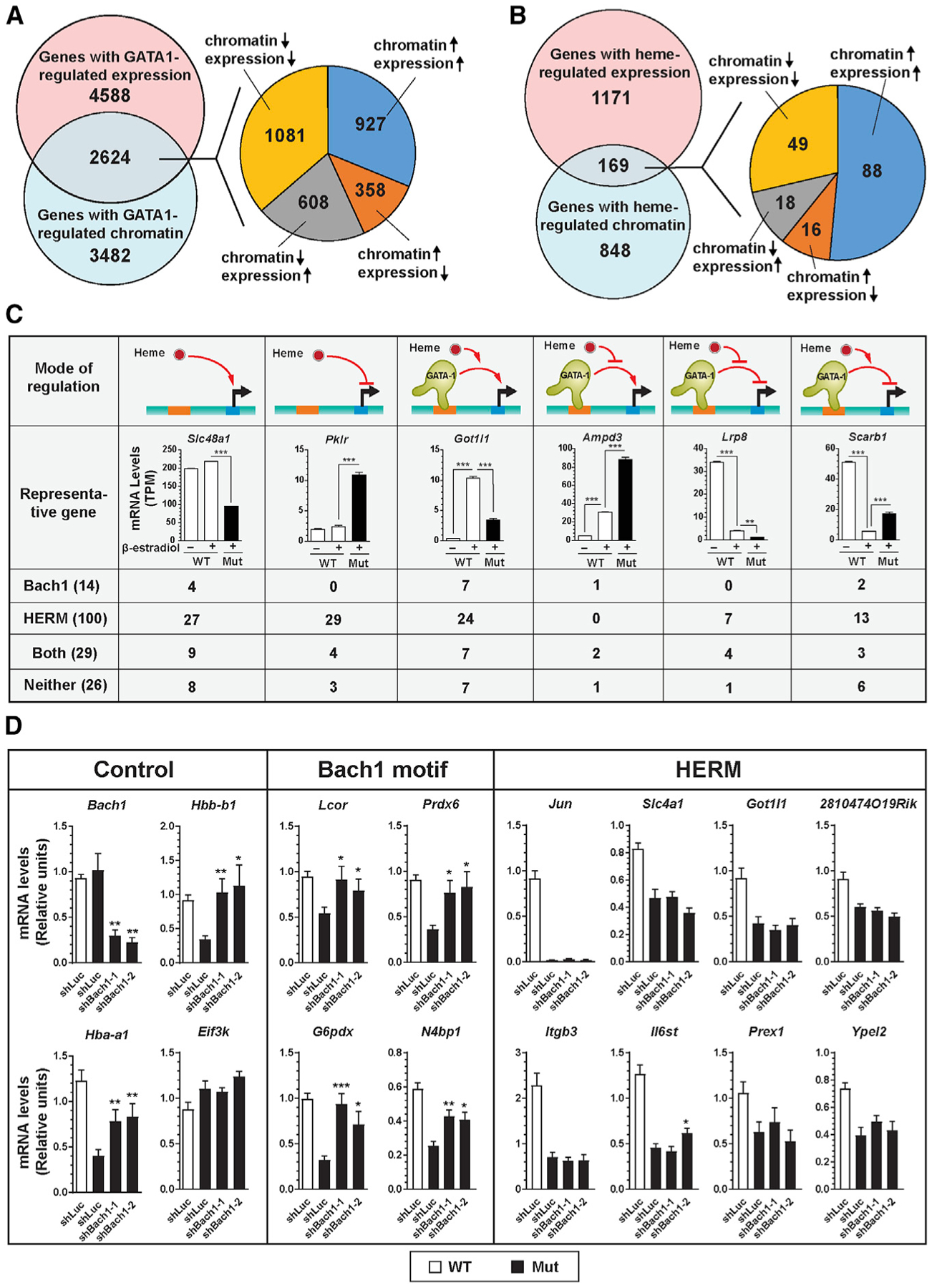
(A) Venn diagram illustrating overlap between genes with GATA1-regulated RNA expression and GATA1-regulated chromatin. The pie chart depicts the number of genes with distinct modes of regulation.
(B) Venn diagram depicting overlap between genes with heme-regulated RNA expression and heme-regulated chromatin. The pie chart illustrates the number of genes with distinct regulatory modes.
(C) GATA1/heme gene regulatory modes. Expression of representative genes (n = 3, mean ± SE; p values were calculated by two-tailed unpaired Student’s t test; ***p < 0.001), and the number of genes containing HERM and/or Bach1 motifs in each cohort.
(D) shRNA knockdown of Bach1 in b-estradiol-induced mutant G1E-ER-GATA1 cells. mRNA was quantified by qRT-PCR (n = 6, mean ± SE). p values were calculated relative to shLuc-infected mutant cells by a two-tailed paired Student’s t test (*p < 0.05, **p < 0.01, and ***p < 0.001).
See also Figures S2, S3, and S5.
Since heme binding induces Bach1 proteolysis (Zenke-Kawasaki et al., 2007), in mutant cells with low heme, Bach1 levels are high and Bach1 target genes are repressed (Tanimura et al., 2016). We tested whether Bach1 motifs or HERMs predict the GATA1/heme-regulatory mode. The 169 genes (Figure 4B) conformed to six regulatory modes (Figure 4C, top row). Based on the presence or absence of Bach1 motifs and HERMs within heme-regulated ATAC-seq peaks, each group was parsed into four subgroups (Figure 4C). As genes containing both motifs resided in each group, these motifs do not predict the regulatory mode.
GATA1/heme co-regulated genes can contain Bach1 and/or HERM motifs. Heme may therefore target chromatin via multiple mechanisms, including a sequential process involving heme downregulation of Bach1, upregulation of Bach1 motif-containing genes, and subsequently, upregulated proteins regulate HERM-containing genes. Alternatively, a Bach1-independent mechanism of regulating HERM-containing genes may operate in parallel with Bach1 function at distinct loci or distinct sites at a heme-regulated locus. To test sequential and parallel models, we downregulated Bach1 with two small hairpin RNAs (shRNAs) in mutant G1E-ER-GATA1 cells and quantified heme-regulated transcription (Figure 4D). The shRNAs decreased Bach1 expression by 70%–80%, which induced Bach1-repressed genes Hbb-b1 and Hba-a1; the negative control Eif3k was unaltered (Figure 4D, left). We compared sensitivities of four Bach1-motif-containing (Figure 4D, middle) and eight HERM-containing genes (Figure 4D, right) from the GATA1/heme-co-activated cohort. Expression of all genes was lower in mutant versus wild-type cells. While Bach1 downregulation in mutant cells by both shRNAs increased expression of Bach1-motif-containing genes, none of the HERM-containing genes were rescued by both shRNAs. Bach1-independent expression of HERM-containing genes indicates that Bach1 and HERM can function in parallel at distinct loci to establish a heme-dependent genetic network. One cannot exclude, however, context-dependent Bach1 and HERM co-function at the same locus.
To further compare Bach1-dependent and independent mechanisms of heme-regulated transcription, we analyzed heme-dependent chromatin targeting at Bach1-dependent and Bach1-independent genes in uninduced G1E-ER-GATA1 cells. Since GATA1 induces Bach1, untreated G1E-ER-GATA1 cells have little to no active ER-GATA1 and Bach1 (Tanimura et al., 2016). Thus, a Bach1-dependent gene might have identical chromatin in untreated wild-type and mutant cells. Consistent with this prediction, ATAC-seq peaks at Bach1-dependent genes (Prdx6 and G6pdx) were indistinguishable in untreated wild-type and mutant cells, despite their heme regulation in β-estradiol-treated cells (Figure S2A, left). By contrast, heme-regulated peaks at HERM-containing genes Jun and Slc4a1 differed between wild-type and mutant cells in vehicle- and β-estradiol-treated conditions (Figure S2A, right). Heme-regulated accessibility at select HERM-containing genes in cells with little to no GATA1 and Bach1 activities further supports parallel heme-sensing mechanisms.
To determine the scope of heme-regulated, Bach1-independent chromatin transitions, we identified ATAC-seq peaks differentially regulated in untreated wild-type versus mutant cells with little to no active ER-GATA1 and Bach1 and determined how many overlap with differentially regulated peaks in β-estradiol-treated cells. Comparing untreated wild-type and mutant cells revealed 533 heme-regulated peaks (Figure S2B). While 321 were heme-regulated in untreated and β-estradiol-treated wild-type versus mutant cells, 898 were differentially regulated only in β-estradiol-treated cells. Each cohort contains peaks with HERMs or Bach1 motifs, further indicating that the motifs do not predict the heme-regulatory mode (Figure S3). Bach1 motifs are more enriched in heme-regulated peaks from β-estradiol-treated versus untreated cells (30.2 versus 17.4%, chi-square statistic = 13.85, p value = 0.0002), suggesting that heme-regulated genes in GATA1-expressing cells are more likely to be Bach1 dependent (Figures S3C and S3D).
The 321 heme-regulated peaks in vehicle- and β-estradiol-treated cells (Figure S2B) reside at 312 genes; heme regulated expression of 65 (group 1). The 898 peaks regulated only in β-estradiol-treated cells reside at 725 genes; heme regulated expression of 104 (group 2) (Figure S2C). Gene Ontology analysis revealed that group 1 and 2 genes are linked to signal transduction. Group 2 genes were also linked to endocytosis, gas transport, and myeloid development (Figure S2D).
To determine if the magnitude of heme-regulated mRNA changes predicts whether a locus exhibits heme-regulated chromatin in the absence of GATA1 and Bach1 activities, we quantified group 1 and 2 gene expression in β-estradiol-treated wild-type versus mutant cells and β-estradiol-treated mutant versus b-estradiol- and 5-ALA-treated mutant cells (Figure S2E). While the average fold change of heme-regulated expression (Figure S2E, upper) was indistinguishable between groups 1 and 2, 5-ALA-regulated expression (Figure S2E, lower) was 1.7-fold higher in group 2 than in group 1 (p = 0.0106). Group 2 genes were more likely to be 5-ALA rescued, and group 1 genes in mutant cells may adopt a repressive chromatin state in the absence of Bach1 that renders them refractory to rescue.
To elucidate mechanistic relationships between Bach1-regulated genes and genes permissive for 5-ALA rescue, we analyzed 25 heme-regulated genes at mRNA and chromatin levels. We quantified expression upon 5-ALA treatment and Bach1 knockdown in β-estradiol-treated mutant cells. 5-ALA-responsiveness correlated with sensitivity of gene expression to Bach1 knockdown (rs = 0.5808, p = 0.0023, Spearman’s rank correlation) (Figure S2F), suggesting that restoring heme with 5-ALA is more likely to rescue Bach1-dependent genes. Thus, heme controls chromatin and transcription through multiple mechanisms. The Bach1-regulated mechanism repressed transcription in a GATA1-dependent manner in low heme, and elevating heme with 5-ALA rescued expression. The Bach1-independent mechanism induced or repressed transcription in GATA1-deficient cells with low heme, and the chromatin state persisted despite heme restoration.
Heme-omics Unveils New Dimensions of Genome Biology and Cellular Regulation
To further establish how heme controls genome function, we analyzed the chromosomal distribution of heme-regulated ATAC-seq peaks. Peaks were enriched in a restricted cohort of “hotspots” (Figure S4A). The heme-regulated peaks were associated with six of eight human red blood cell traits (Ulirsch et al., 2019) (p < 0.01) (Figure S4B). To quantify enrichment of heme-regulated peaks and genes at specific chromosomal neighborhoods, we divided each chromosome into 10-Mb bins and enumerated heme-regulated ATAC-seq peaks and genes in each bin. Seven hotspots harboring ≥8 heme-regulated peaks and ≥15 heme-regulated genes within a bin were identified (Figure 5A). This clustering of heme-regulated peaks and genes is not likely to result from coincidence, as random selection of ATAC-seq peaks and genes 10,000 times led to a median of 1 hotspot (p value = 0) (Figure S4C). Of the seven hotspots, chromosome 4 (chr4; 40–50 Mb) and chr16 (0–10 Mb) contained predominantly heme-repressed peaks, whereas >80% of the heme-regulated peaks on chr3 (80–90 Mb) and chr8 (70–80 Mb) were heme induced. Strikingly, 21 out of 22 heme-regulated genes on chr16 (0–10 Mb) were heme-repressed and downregulated upon erythroid maturation (Figure 5C). Heme-regulated genes at other hotspots exhibited variable expression patterns. ATAC-seq profiles for the 21 genes were clustered in a 2-Mb region (chr16, 3.5–5.5 Mb) with 19 heme-repressed ATAC-seq peaks (Figure 5B). A Bach1-motif- and HERM-containing peak located ~1 Mb downstream was abrogated in mutants. As this heme-regulated gene cohort had not been linked to heme, it is attractive to propose that the encoded machinery transduces altered heme levels into changes in cellular physiology.
Figure 5. Heme-Regulated Genomic Hotspots.
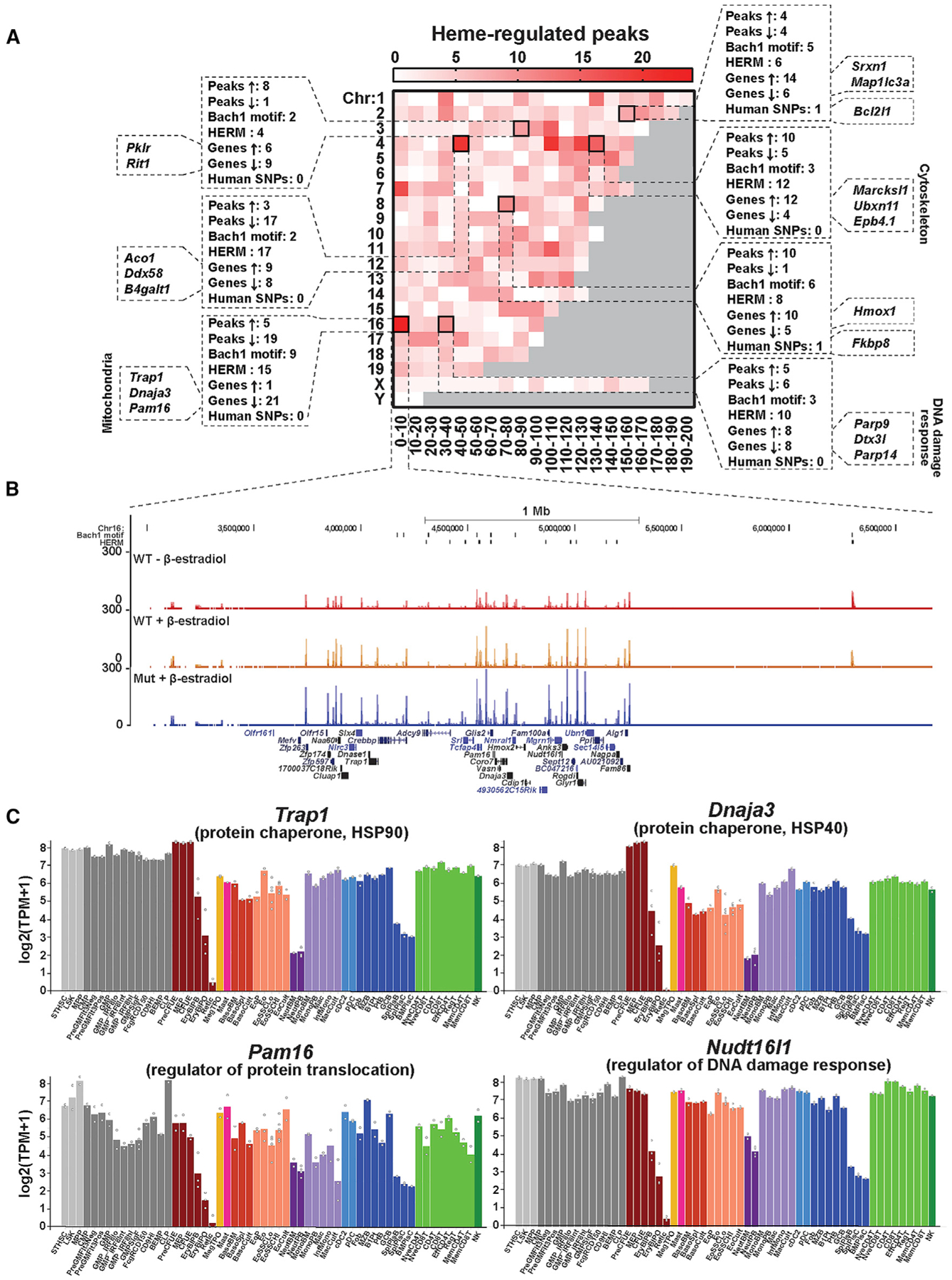
(A) Heatmap depicting the number of heme-regulated peaks that fall into each 10-Mb bin over the mouse genome. Hotspots that contain ≥8 heme-regulated peaks and ≥15 heme-regulated genes are highlighted.
(B) ATAC-seq profiles for the chromosome 16 (chr16) 0–10 Mb hotspot. Heme-regulated genes are labeled in black fonts.
(C) Expression profiles in mouse hematopoietic lineages for representative heme-regulated genes in the chr16 0–10 Mb hotspot. The data were obtained from Haemosphere (https://www.haemosphere.org/).
See also Figure S4.
To rigorously identify heme-regulated genes, we applied stringent criteria based on differential expression in wild-type and mutant cells and 5-ALA rescue in mutant cells. Of 169 heme-regulated genes (chromatin and mRNA) (Figure 4B), 5-ALA rescued 25 (Figure S5A, left). 12 of the 25 genes (Figure S5A, left, black fonts) were validated by qRT-PCR for GATA1/heme/5-ALA regulation. Proteins encoded by 10 of the genes were detected in our prior proteomic analysis (Tanimura et al., 2018) and were heme regulated (Figure S5A, right). Heme-regulated genes validated by qRT-PCR and/or proteomics included known GATA1/heme targets (Tanimura et al., 2018) encoding hemoglobins (Hbb-b1 and Hbb-y), redox proteins (Txnrd1 and Prdx6), and a ubiquitination/proteasome component that regulates erythroid differentiation (Ube2o) (Nguyen et al., 2017). Previously undescribed heme-regulated genes included transporters (nucleoside transporter Slc28a3 and intracellular heme transporter Slc48a1), signaling factors (calcium-dependent kinase Camk1d, and Ras family small GTPase Rit1), and cell-cycle regulators (CDK4/6 inhibitor Cdkn2b and microtubule depolymerization regulator during mitosis Kif2a). This gene cohort was downregulated in β-estradiol-treated mutant G1E-ER-GATA-1 cells and rescued with 5-ALA (Figure S5B). GATA1 activated four of these six genes, consistent with heme-facilitated GATA1 function. Bach1 downregulation in β-estradiol-treated mutant cells with two shRNAs restored expression of five of the genes (Figure S5C), implicating Bach1 in the heme mechanism.
To evaluate mechanistic conservation, we utilized a human erythroid differentiation system (Tanimura et al., 2018). Mononu-clear cells from granulocyte colony-stimulating factor (G-CSF)-mobilized peripheral blood were expanded for 4 days and differentiated for 12 days. Cells were treated with the heme synthesis inhibitor succinylacetone (SA) at day 13 and analyzed on days 14–16. SA repressed the same five genes as in the mouse (Figure S5D).
Our analyses revealed new heme-repressed genes including Pklr encoding an erythroid- and liver-specific pyruvate kinase isoform crucial for glycolysis (Demina et al., 1998). Pklr resided in the chr3 (80–90 Mb) hotspot (Figure 5A) and contained heme-repressed ATAC-seq peaks in β-estradiol-treated cells at −3.4 and +0.6 kb. β-Estradiol induced the −3.4 kb peak that overlapped with GATA1 ChIP-seq peaks in G1E-ER-GATA1 cells and primary erythroblasts (Figure 6A). The +0.6-kb peak lacking GATA1 occupancy contains two HERMs 29 bp apart (Figure 6A). Although GATA1 activated Pklr in wild-type and mutant cells, the induction was 86- to 119-fold higher in mutant cells. Restoring heme in mutant cells repressed Pklr 3.7- to 4.6-fold (Figure 6B). Lowering Bach1 in β-estradiol-treated mutants had no impact, indicating Bach1-independent repression (Figure 6C).
Figure 6. GATA1/Heme Control of Cellular Metabolism during Erythroid Differentiation.
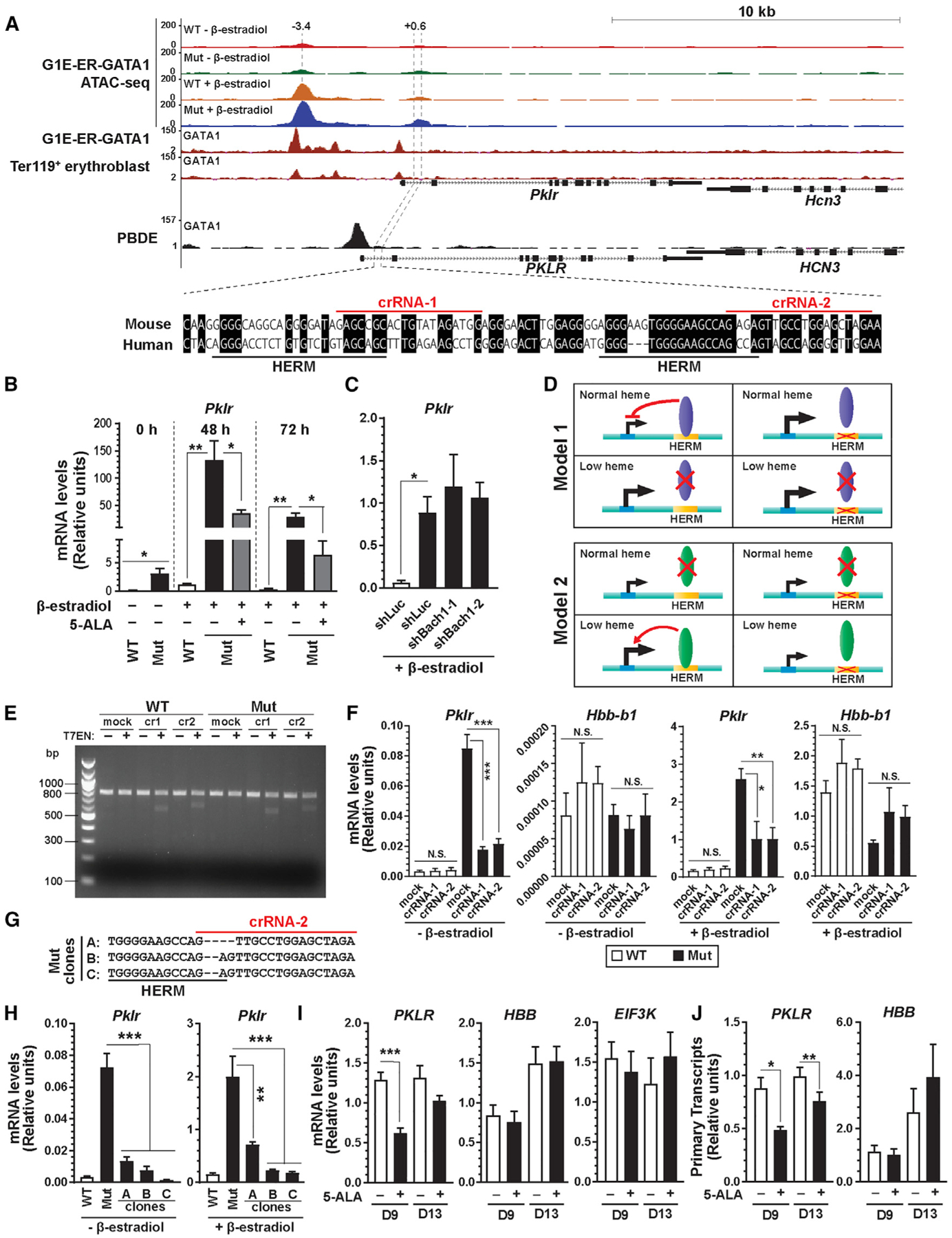
(A) ATAC-seq and GATA1 ChIP-seq profiles for Pklr locus in mouse and human cells showing GATA1 and heme-regulated chromatin loci. The numbers indicate the location of ATAC-seq peak centers relative to the transcription start sites (in kb). Sequence conservation under the intronic ATAC-seq peak between mouse and human and the locations of HERMs and crRNAs used in (E)–(H) are shown below.
(B) Pklr mRNA in G1E-ER-GATA1 cells treated with b-estradiol and 5-ALA for 48 or 72 h was quantified by qRT-PCR (n = 5, mean ± SE). p values were calculated by two-tailed paired Student’s t test (*p < 0.05 and **p < 0.01).
(C) shRNA knockdown of Bach1 was conducted in b-estradiol-induced mutant G1E-ER-GATA1 cells, and Pklr mRNA was quantified by qRT-PCR (n = 4, mean ± SE). p values were calculated relative to shLuc-infected mutant cells by a two-tailed paired Student’s t test (*p < 0.05).
(D) Models for HERM as a mediator of heme-dependent transcriptional repression.
(E) CRISPR-Cas9-mediated editing of HERMs within intron 1 of Pklr gene using two distinct crRNAs shown in (A). WT or Alas2-enhancer mutant G1E-ER-GATA1 cells were mock treated or treated with RNP complexes containing Cas9 and two distinct crRNAs, respectively. The editing efficiency of the targeted locus was assessed by T7 endonuclease assay.
(F) WT or Alas2-enhancer mutant G1E-ER-GATA1 cells were treated as in (E). mRNA levels for Pklr and Hbb-b1 in uninduced and β-estradiol-treated cells were quantified by qRT-PCR (n = 4, mean ± SE). p values were calculated by a two-tailed paired Student’s t test (*p < 0.05, **p < 0.01, and ***p < 0.001; N.S, not significant).
(G) DNA sequences of HERM mutant clones generated from Alas2-enhancer mutant cells.
(H) mRNA levels for Pklr in HERM mutant clones shown in (G) were quantified by qRT-PCR (n = 4, mean ± SE). p values were calculated by a two-tailed unpaired Student’s t test (**p < 0.01 and ***p < 0.001).
(I) Relative expression of representative genes with 5-ALA treatment in comparison with vehicle-treated human mononuclear cells from G-CSF-mobilized peripheral blood at days 9 and 13 (n = 6, mean ± SE). p values were calculated by two-tailed paired Student’s t test (***p < 0.001).
(J) Cells were treated as in (I). PKLR and HBB primary transcripts were quantified by qRT-PCR (n = 6, mean ± SE). p values were calculated by two-tailed paired Student’s t test (*p < 0.05 and **p < 0.01).
See also Figure S6.
We tested models to explain how HERM functions in heme mechanisms. A heme-dependent repressor may become limiting in low heme (model 1). Disrupting HERM in normal heme would de-repress target gene expression. Alternatively, a heme-dependent activator may be limiting in normal heme, and disrupting HERM would downregulate target gene expression only in low heme (model 2) (Figure 6D). We used CRISPR-Cas9 with CRISPR RNAs (crRNAs) to target the HERMs (Figure 6A). T7 endonuclease assay identified indels, and TIDE analysis (Brinkman et al., 2014) estimated a 13%–22% editing efficiency (Figure 6E). The editing decreased Pklr expression in untreated and β-estradiol-treated mutant cells 2.5-fold and 3.9- to 4.7-fold, respectively; Pklr expression in wild-type cells was unaffected (Figure 6F). Expression of the control Hbb-b1 gene, which is induced upon differentiation, was unaffected by editing (Figure 6F). Clonal lines harboring a 2- or 4-bp deletion of HERM were derived from mutant cell pools (Figure 6G). Pklr expression declined in these lines by 5.4- to 52-fold and 2.8-to 11-fold in comparison with vehicle- and β-estradiol-treated mutant cells, respectively (Figure 6H). Thus, Pklr upregulation in low heme required HERM.
Since Pklr controls glycolysis, we investigated whether its upregulation in low heme correlates with altered glycolysis. As a metric of glycolytic rate, we quantified lactate secretion using liquid chromatography-mass spectrometry (LC-MS) (Fan et al., 2014) (Figure S6A). Consistent with GATA1 downregulation of 11 of 19 glycolytic enzymes (Figures S6D and S6E), lactate secretion in undifferentiated and differentiated wild-type cells decreased upon differentiation, consistent with reduced glycolysis. GATA1 induced differentiation of both wild-type and mutant cells, and lactate secretion declined in both cell types. The rate of decline was slower in mutant cells with upregulated expression of Pklr and other glycolytic genes than in wild-type cells, suggesting that low heme attenuated a glycolytic decline (Figures S6B and S6C). Although six genes besides Pklr were heme repressed, only PKLR protein was upregulated in mutants (Figures S6E and S6F). To test if heme regulation of Pklr is conserved, we treated human primary erythroblasts with 1 mM 5-ALA for 48 h at day 9. 5-ALA decreased PKLR expression 2.0-fold without affecting EIF3K or HBB (Figure 6I). PKLR primary transcripts decreased similarly (Figure 6J). Consistent with heme-dependent repression, PKLR expression decreased gradually as erythroblasts matured (An et al., 2014) (Figure S6G). These results demonstrated a HERM-dependent mechanism that regulates erythroid cell metabolism under low-heme conditions.
DISCUSSION
Amalgamating our genome-wide atlas of GATA1- and heme-regulated chromatin sites with transcriptomic data generated a heme-omics resource that revealed a multicomponent mechanism of heme-regulated genome activity (Figure 7). This mechanism illustrates how an essential biochemical cofactor controls genome and cell function. Beyond the paradigm of heme-depen dent Bach1 proteolytic destruction that de-represses genes containing Bach1 motifs, we provide evidence for a prevalent mode of heme function involving chromatin targeting independent of Bach1 motifs and insensitive to Bach1 downregulation. In low heme, genes regulated by the Bach1-independent mechanism were commonly rendered refractory to rescue. Thus, pathological heme deficiency may generate a deleterious epigenetic memory, an indelible mark that disrupts local genome function and impedes reversal even when heme levels are normalized.
Figure 7. Model of Parallel Bach1-Dependent and Bach1-Independent Mechanisms of Heme-Dependent Genome Regulation.
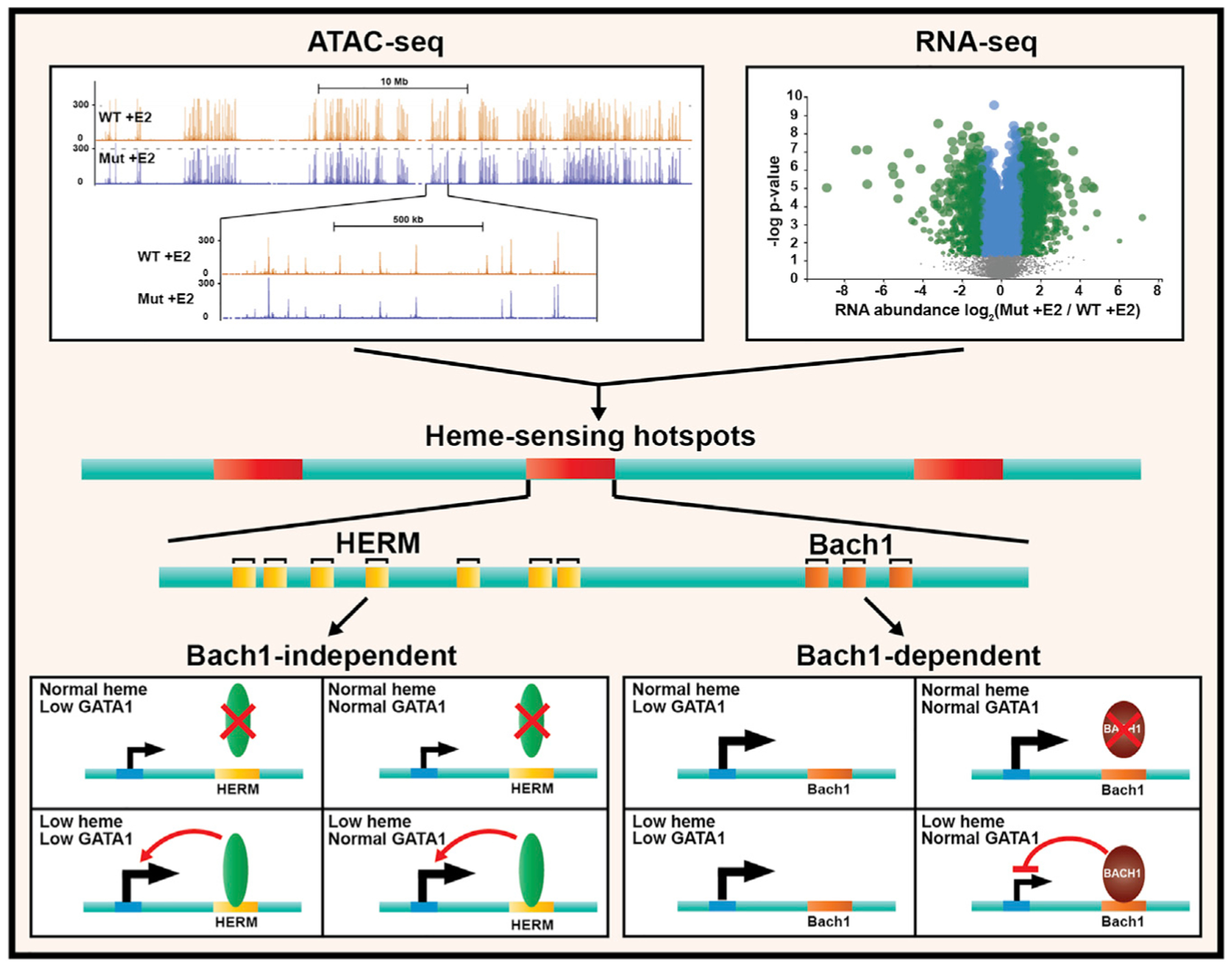
Heme-dependent chromatin transitions occurred in cells containing little to no Bach1 and GATA1. The Bach1-independent mechanism involved HERM that lacks sequence similarity with the Bach1 motif and was enriched at heme-regulated chromatin loci. Comparison of HERM to known motifs revealed similarity to a ZNF263 motif inferred from a ChIP-seq study (Frietze et al., 2010). ZNF263 (and murine Zfp263) mRNA was not detected in the cells studied, and the proteins were undetectable by proteomics, inconsistent with ZNF263 mediating HERM function. In principle, HERM function may be independent of, or dependent on, transcription factor binding, and future studies will establish the requisite protein components.
The vital importance of the Bach1-dependent mechanism of heme-regulated transcription has been rigorously established (Brand et al., 2004; Haldar et al., 2014; Ogawa et al., 2001; Zenke-Kawasaki et al., 2007). Integrating genome-wide chromatin and transcriptomic data provided evidence that the heme-regulated, Bach1-independent and Bach1-dependent mechanisms operate in parallel to establish a critical genetic network. Since heme deficiency and excess cause or contribute to human disease etiology, including myelodysplastic syndrome (Yang et al., 2016), our resource may inform diverse biological and pathological problems. In this regard, heme-omics identified genes not linked previously to heme mechanisms. These genes included Rit1, Cdkn2b, and Pklr. Rit1 encodes a Ras-like small GTPase that regulates neuronal cell morphogenesis and survival (Mir et al., 2017). RIT1 mutations occur in 5%–9% of Noonan syndrome patients and other Ras-driven malignancies, including lung adenocarcinoma and myeloid leukemia (Aoki et al., 2013; Cavé et al., 2016). RIT1 switch II region mutations disrupt its interaction with an E3 ubiquitin ligase via LZTR1, upregulating RIT1 and dysregulating growth factor signaling (Castel et al., 2019). GATA1/heme induced Rit1 in erythroid cells, suggesting involvement of Rit1-linked signaling in erythroid maturation. Cdkn2b (p15Ink4b) belongs to a CDK4/6 inhibitor family (p15, p16, p18, and p19) (Hannon and Beach, 1994). Silenced CDKN2B expression occurs in 60%–80% of myelodysplastic syndromes (MDSs) and leukemia, which correlates with leukemic transformation and poor prognosis (Quesnel et al., 1998; Uchida et al., 1997). Heme activated Cdkn2b, and in low heme, Bach1 repressed Cdkn2b. CDKN2B is highly expressed by human erythroblasts. Based on CDKN2B cell-cycle-inhibitory activity, heme-dependent CDKN2B induction may contribute to cessation of cell-cycle progression as a step in differentiation.
Pklr encodes two pyruvate kinase isoforms expressed primarily in liver and red blood cells. PKLR coding region and splice site mutations cause pyruvate kinase deficiency and nonspherocytic hemolytic anemia (Baronciani and Beutler, 1993; Tanaka et al., 1962). Our results, and a PKLR GATA motif mutant in K562 erythroleukemia cells (Wakabayashi et al., 2016), demonstrated that GATA1 activates Pklr expression. In low heme, Pklr and Bpgm expression were elevated, which would be predicted to elevate 2,3-bisphosphoglycerate (2,3-BPG). 2,3-BPG binds deoxygenated hemoglobin with high affinity and promotes oxygen release from hemoglobin (Agarwal et al., 2007). The attenuated decline in glycolytic rate in low heme may alleviate symptoms of iron-deficiency anemia by promoting oxygen release in tissues as a cytoprotective mechanism. Furthermore, as heme accumulates in differentiating erythroblasts, it is attractive to propose that heme-facilitated repression of glycolytic enzyme expression may lower glycolysis. In low heme, elevated glycolytic enzyme expression may favor precursor expansion versus maturation, averting severe consequences of a globin chain-heme imbalance. Such a mechanism may operate in parallel with Bach1-dependent repression of globin transcription (Brand et al., 2004; Sun et al., 2004; Tanimura et al., 2016).
In summary, heme controls chromatin and genome function via parallel Bach1-dependent and Bach1-independent pathways. Heme-regulated chromatin sites are enriched in a motif unlike others linked to heme (or any other small molecule) function. Our analysis unveiled heme target genes linked to erythroid development and erythrocyte function and those not associated with these processes. Our heme-omics resource, which identified how an essential biochemical cofactor controls genome function, will enable a wide swath of studies to elucidate physiological and pathological mechanisms involving disrupted GATA1 and/or heme mechanisms involving, but not limited to, the hematopoietic system. Furthermore, our heme-omics strategy can be extrapolated to fill in critical knowledge gaps on how a broad spectrum of small molecules impact genome function.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Emery H. Bresnick (ehbresni@wisc.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
The ATAC-seq datasets generated during this study are available at GEO: GSE114996.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
G1E-ER-GATA-1 cells (Grass et al., 2003) derived from GATA1 mutant male murine ES cells (Weiss et al., 1997) were cultured in Is-cove’s modified Dulbecco’s medium (IMDM; GIBCO) containing 15% FBS (Gemini), 1% penicillin-streptomycin (Gemini), 2 U/ml erythropoietin (Amgen), 120 nM monothioglycerol (Sigma), and 0.6% conditioned medium from a Kit ligand-producing CHO cell line, and 1 μg/ml puromycin (Gemini). ER-GATA-1 activity was induced by adding 1 μM β-estradiol (Steraloids) to media. To rescue heme synthesis, 1 mM 5-aminolevulinic acid hydrochloride (5-ALA; Sigma) was added to media.
Primary human mononuclear cells were isolated from G-CSF-mobilized peripheral blood using Histopaque (Sigma). The sex of the primary cells used for experiments is not available. Cells were maintained in StemSpan SFEM medium (Stem Cell Technologies) supplemented with 1x CC100 cytokine mix (Stem Cell Technologies) for 4 days. To induce differentiation, cells were cultured in basal differentiation media [IMDM containing 15% FBS, 2 mM L-glutamine, 1% BSA (Sigma), 500 μg/ml holo human transferrin (Sigma), and 10 μg/ml insulin (Sigma)] supplemented with 1 μM dexamethasone (Sigma), 1 μM β-estradiol (Steraloids), 5 ng/ml IL-3, 100 ng/ml SCF, and 6 U/ml erythropoietin (Amgen) for 5 days, followed by four days in basal differentiation media supplemented with 50 ng/ml SCF and 6 U/ml erythropoietin and then three days in basal differentiation media containing 2 U/ml erythropoietin. Cells were cultured in a humidified 5% CO2 incubator at 37°C.
METHOD DETAILS
ATAC-seq
ATAC-seq was performed as previously described with modifications (Liu et al., 2017). Specifically, 5 × 104 cells were washed twice in PBS and resuspended in 500 μL cell lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, pH 7.4). Nuclei were harvested by centrifuge at 500 x g for 10 min at 4°C, resuspended in 50 μL of tagmentation mix (10 mM TAPS (Sigma), 5 mM MgCl2, pH 8.0 and 2.5 μl Tn5), and incubated at 37°C for 30 min. Tagmentation was terminated by incubating nuclei at room temperature for 2 min followed by adding 10 mL of 0.2% SDS and incubating at 55°C for 7 min. Tn5 transposase-tagged DNA was purified using QIA-quick MinElute PCR Purification kit (QIAGEN), amplified using KAPA HiFi Hotstart PCR Kit (KAPA).
ATAC-seq reads were sequenced to 75 base pairs in the single-end mode on the NextSeq500 system and aligned to the mouse genome (mm9) using Bowtie version 1.0.0 (Langmead et al., 2009) with parameters,–best–strata -k 1 -m 1. PCR duplicates were removed by Picard (https://github.com/broadinstitute/picard) using the default parameter setting. Significantly enriched ATAC-seq peaks were detected by Model-based Analysis of ChIP-seq (MACS) (Zhang et al., 2008). Genomic track figures of the ATAC-seq peaks (Figures 1B, 3C, 5B, 6A, 7, and S2A) were modified from the visualizations on the UCSC Genome Browser (http://genome.ucsc.edu/).
qRT-PCR
Total RNA was purified from 0.5–2 × 106 cells with TRIzol (Life Technologies). 1 μg RNA was treated with DNase I (Life Technologies) for 15 min at room temperature. DNase I was inactivated by addition of EDTA and heating at 65°C for 10 min. To synthesize cDNA, DNase I-treated RNA was incubated with 125 ng of a 5:1 mixture of oligo-dT primers and random hexamer at 68°C for 10 min. RNA/primers were incubated with Moloney MLV reverse transcriptase (Life Technologies), 10 mM DTT, RNAsin (Promega), and 0.5 mM deoxynucleoside triphosphates (dNTP) at 42°C for 1 h, and then heat inactivated at 98°C for 5 min. Real-time PCR (qRT-PCR) was conducted with Power SYBR Green Master Mix (Applied Biosystems) using ViiA 7 Real-Time PCR system (Applied Biosystems).
shRNA-mediated knockdown
Two distinct shRNAs targeting Bach1 transcripts were designed and cloned into MSCV-PIG (IRES-GFP) vector (Tanimura et al., 2016). Transfecting 293T cells with 15 μg of MSCV-PIG vector and pCL-Eco packaging vector produced retrovirus expressing shRNA targeting luciferase or Bach1. G1E-ER-GATA-1 cells (2 × 105) were added to 100 μl viral supernatant, polybrene (8 μg/ml), and HEPES buffer and then spinoculated at 2,800 rpm for 90 min at 30°C. Three days post-infection, cells were treated with β-estradiol, and after 48 h, cells were harvested for qRT–PCR analyses.
Gene editing with CRISPR-Cas9
Guide sequences targeting the intronic HERMs of murine Pklr gene were designed using online tools (http://www.idtdna.com/). Chemically modified crRNAs and tracrRNA were purchased from IDT and Cas9 protein was purchased from Aldevron. 100 pmole of crRNA was mixed with 100 pmole of tracrRNA and incubated at 37°C for 30 min. 8 μg of Cas9 protein was added to the mixture and incubated at 37°C for 15 min to form RNP complex. 2 × 105 G1E-ER-GATA1 cells were resuspended in 20 μL P3 buffer with 22% supplement (Lonza) and added to the RNP complex. Electroporation was done using EO-100 program on Nucleofector 4D (Lonza). 72 h after nucleofection, genomic DNA was extracted from the cell population and subjected to T7 endonuclease assay to detect mutation. Genomic DNA flanking the target site was amplified by PCR and amplicons were denatured, reannealed, and incubated with T7 endonuclease I (New England BioLabs) for 15 min at 37°C. The amplicons were sequenced and analyzed by TIDE to estimate editing efficiency.
Glycolysis measurement
Glycolytic rate of G1E-ER-GATA1 cells was quantified by measuring lactate secretion in the culture media. Cells were seeded in fresh media and cultured for 24 or 48 h. Media was collected by spinning cells down at 1,200 rpm for 5 min, diluted 1:5 in ice cold LC-MS grade methanol, and centrifuged at 20000 x g for 10 min at 4°C to remove proteins. 20 ml supernatant was then mixed with 180 ml 90% acetonitrile and lactate concentration was quantified by LC-MS.
QUANTIFICATION AND STATISTICAL ANALYSIS
ATAC-seq peak quantification
For detecting differentially accessible peaks under different experimental conditions, a master peak list was created by merging all ATAC-seq peaks from three biological replicates of various conditions. If any of the individual peaks overlapped with each other, the master peak will take the union regions of both peaks. Read counts within each master peak region were retrieved from the aligned bam files by R package chromVAR (Schep et al., 2017). A count matrix was built to summarize the ATAC-seq read count for each biological sample in each master peak region across all experimental conditions. Heatmap figures of the count matrix (Figures 2A and 2B) were generated by R package pheatmap (Kolde and Vilo, 2015), and the master peaks were clustered by hierarchical clustering using the inherited R function hcluster in the pheatmap package with the default parameter setting. Differential peak signals between any pair of conditions were identified by R package DESeq2 (Love et al., 2014), where the count matrix was normalized first to eliminate the sequencing depth bias and fit into a negative binomial model. The resulting p values were adjusted by Benjamini-Hochberg procedures to control the overall False Discovery Rate. The final differential ATAC-seq peaks were deemed significant if the |log2FoldChange| > = 1 and the adjusted p value ≤ 0.05. Moreover, R package ChIPseeker (Yu et al., 2015) was utilized to annotate the genomic features of the significant differentially accessible peaks where the maximum range of promoter to transcription start site was set at 3 kb. The significant differentially accessible peaks were assigned to the nearest genes based on the distance of peak region to the start site, to build associations between ATAC-seq peaks and genes.
Motif analyses
We utilized MEME (Multiple EM for Motif Elicitation) Suite (Bailey et al., 2009) version 4.12.0 (http://meme-suite.org/) for motif-based sequence analyses. Motif discoveries (Figures 3A and 3B, upper panel; Figure S1B) were implemented by MEME using searching mode “anr” assuming that each peak sequence may contain any number of nonoverlapping occurrences of each motif. MEME detects the most significant motif first, and the significance is evaluated by the E-value, an estimate of the expected number of motifs with the given log-likelihood ratio. We compared recognized motifs with motif databases such as JASPAR by TOMTOM (Gupta et al., 2007) and got the top significantly similar motif ranked by the p value (Figures 3A and 3B; lower panel). Alternatively, we used HOMER (Heinz et al., 2010) findMotifs.pl function to find motifs in heme-regulated ATAC-seq peaks and searched for up to 3 motifs with a length of ~21 bp (Figure S1A). The remaining parameters were set as default.
We used FIMO (Grant et al., 2011) to locate motifs in ATAC-seq peak sequences using default settings (Figure 3C–F). To search for a match to a HERM, the following position-specific weight matrix can be used:
> GGAGGAGGAGGGGGAGGAGGA HERM
0.316092 0.095402 0.417241 0.171264
0.417241 0.058621 0.442529 0.081609
0.341379 0.072414 0.564368 0.021839
0.587356 0.004598 0.358621 0.049425
0.341379 0.027586 0.616092 0.014943
0.413793 0.014943 0.548276 0.022989
0.171264 0.455172 0.368966 0.004598
0.717241 0.020690 0.010345 0.251724
0.188506 0.006897 0.802299 0.002299
0.458621 0.039080 0.482759 0.019540
0.455172 0.048276 0.463218 0.033333
0.480460 0.041379 0.467816 0.010345
0.286207 0.198851 0.471264 0.043678
0.506897 0.111494 0.249425 0.132184
0.370115 0.044828 0.539080 0.045977
0.404598 0.041379 0.497701 0.056322
0.390805 0.125287 0.457471 0.026437
0.436782 0.048276 0.441379 0.073563
0.281609 0.185057 0.527586 0.005747
0.572414 0.031034 0.285057 0.111494
0.219540 0.067816 0.598851 0.113793
FIMO was also employed for genome-wide motif searching in mouse (Figure S1E). To control the overall false discovery rate, a stringent filtering threshold was applied. Only regions that have q-values ≤ 0.0001 were kept, instead of using the default threshold setting of p value ≤ 0.0001. q-values are the p values adjusted by Benjamini-Hochberg correction.
Due to the repetitive pattern of the HERM motif, multiple motifs that are 2 or 4 bp away may be detected separately. To avoid over-representation, we aligned the motif ranges and merged the overlapping motifs to produce a simplified region set. The frequency of the motif is also summarized based on the non-overlapping motif regions.
Random sampling
By matching the number of peaks and peak lengths on each chromosome with the heme-regulated peaks, we randomly selected 1222 peak regions on the mouse genome and scanned for HERM using FIMO with default parameters. Using similar approach, we randomly sampled 1,222 peaks among 58,261 non-heme-regulated ATAC-seq peaks matching the number of peaks on each chromosome with the heme-regulated peaks and searched for HERM using FIMO. Such random sampling was conducted 10,000 times, and for each run, the number of peaks overlapping with HERM was recorded. Eventually, the 10,000 overlapping results were compared to the observed number of heme-regulated peaks containing HERM (904). There were zero sampled results with more than 904 peaks overlapping HERM; hence, the p value was 0 (Figure 3E).
To quantify the enrichment of the heme-regulated peaks or genes, the mouse genome was binned by 10 Mb windows, and bins were considered hotspots if they have ≥ 8 heme-regulated ATAC-seq peaks or ≥ 15 heme-regulated genes or both. Then we conducted random sampling of the ATAC-seq peaks in the mouse genome while matching the number of heme-regulated peaks and the peak length on each chromosome. Similarly, genes were randomly selected, while matching the number of heme-regulated genes on each chromosome. The above permutation was carried out 10,000 times. For each permutation, the number of hotspots that met the filtering criteria was collected. Such permutation results were compared to the observed number of hotspots from heme-regulated peaks and genes. Since zero sampled result led to more hotspots than the observed result, the p value was 0 (Figure S4C).
Lifting over human genetic variants to mouse genome
To link human genetic variation to heme-regulated ATAC-seq peaks, we lifted over human GWAS SNPs related to blood cell traits (Ulirsch et al., 2019) to the mouse genome (Figure S4B). The chain file, hg19ToMm9.over.chain, essential for this analysis, was downloaded from http://hgdownload.cse.ucsc.edu/goldenpath/hg19/liftOver/. An iterative lifting over strategy was constructed, allowing more human SNPs lifted over to the mouse genome and in a more accurate way. SNPs were first lifted over on its own (1 bp), and those that failed were extended 5 bp upstream and downstream to form a 10 bp sequence around the original location. These extended sequences were lifted over again. Among those that still do not yield a mapping location on the mouse genome, the window size was increased to 100 bp and then 1000 bp. SNPs that were lifted over using 1, 10, 100 and 1000 bp were merged for further analysis. The lifting over is conducted by the liftover() function in R package rtracklayer (Lawrence et al., 2009). After lifting over, syntenic regions can be intermittent; hence need to be merged if such regions are in proximity (≤10 bp). For SNPs that are lifted to multiple regions on the mouse genome, even after merging the syntenic regions, the widest subregion was selected as the final lifted-over region. We overlapped the lifted-over region on the mouse genome with heme-regulated ATAC-seq peaks and quantified whether the lifted-over regions reside in the peaks more than expected by chance. The significance of such overlapping was established by bootstrapping syntenic regions while preserving the chromosome constitution and the genetic conservation score. A statistical significance was assigned to the observed ATAC-seq peak enrichment to quantify the association between heme-regulated ATAC-seq peaks and blood cell traits.
Other statistical analyses
The exact numbers of biological replicates conducted and statistical analysis used for each experiment are stated in the figure legends. The data were presented as mean ± SE, and no data were excluded from the analysis. Statistical analyses were performed with GraphPad Prism 7. To compare the difference between two groups, unpaired or paired two-tailed Student’s t tests were conducted. For multiple comparisons, one-way ANOVA was used, followed by Tukey’s test.
Supplementary Material
Table 3.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| JM109 competent cells | Promega | L2001 |
| Biological Samples | ||
| G-CSF-mobilized human peripheral blood | University of Wisconsin-Madison | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-Aminolevulinic acid hydrochloride (5-ALA) | Sigma | A7793 |
| 4,6-Dioxoheptanoic acid (Succinylacetone) | Sigma | D1415 |
| Recombinant Human IL-3 | PeproTech | 200–03 |
| Recombinant Human SCF | PeproTech | 300–07 |
| sNLS-spCas9-sNLS | Aldevron | 9212–0.25MG |
| Critical Commercial Assays | ||
| KAPA HiFi Hotstart PCR Kit | KAPA | KK2502 |
| P3 Primary Cell 4D-Nucleofector– X Kit S | Lonza | V4XP-3032 |
| Deposited Data | ||
| GATA1 ChIP-seq (Ter119+ erythroblast) | Ross Hardison (Penn State University), ENCODE | GEO: GSM923575 |
| GATA1 ChIP-seq (G1E-ER-GATA1 cells) | Ross Hardison (Penn State University), ENCODE | GEO: GSM923572 |
| GATA1 ChIP-seq (PBDE) | Kang et al. (2012); ENCODE | GEO: GSM935465 |
| RNA-seq (G1E-ER-GATA1 cells) | Tanimura et al. (2016) | GEO: GSE74371 |
| ATAC-seq (G1E-ER-GATA1 cells) | This paper | GEO: GSE114996 |
| Experimental Models: Cell Lines | ||
| Mouse: G1E-ER-GATA-1 cells | Tanimura et al. (2016) | N/A |
| Oligonucleotides | ||
| Primers for qRT-PCR and PCR, see Table S1 | This paper | N/A |
| crRNA1: GAGCCGCACTGTATAGATGG | This paper | N/A |
| crRNA2: TCTAGCTCCAGGCAACTCTC | This paper | N/A |
| Recombinant DNA | ||
| pMSCV-PIG-shBach1 | Tanimura et al. (2016) | N/A |
| Software and Algorithms | ||
| Bowtie (version 1.0.0) | Langmead et al. (2009) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS | Zhang et al. (2008) | https://github.com/macs3-project/MACS |
| chromVAR | Schep et al. (2017) | https://github.com/GreenleafLab/chromVAR |
| pheatmap | Kolde and Vilo (2015) | https://cran.r-project.org/web/packages/pheatmap/index.html |
| DESeq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MEME suite (version 4.12.0) | Bailey et al. (2009) | http://meme-suite.org/index.html |
| HOMER (version 4.11) | Heinz et al. (2010) | http://homer.ucsd.edu/homer/ |
| rtracklayer | Lawrence et al. (2009) | https://bioconductor.org/packages/release/bioc/html/rtracklayer.html |
| Prism 7 | GraphPad | https://www.graphpad.com/ |
Highlights.
Heme-omics resource is generated by amalgamating ATAC-seq and RNA-seq datasets
Parallel Bach1-dependent and independent heme mechanisms regulate genome function
A unique DNA motif demarcates heme-regulated chromatin sites
Heme-sensing hotspots reveal new dimensions in genome biology and cellular regulation
ACKNOWLEDGMENTS
This study was supported by National Institute of Health grant DK50107 to E.H.B., HG003747 to S.K., and University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j. celrep.2020.107832.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agarwal N, Gordeuk RV, and Prchal JT (2007). Genetic mechanisms underlying regulation of hemoglobin mass. Adv. Exp. Med. Biol 618, 195–210. [DOI] [PubMed] [Google Scholar]
- An X, Schulz VP, Li J, Wu K, Liu J, Xue F, Hu J, Mohandas N, and Gallagher PG (2014). Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 123, 3466–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, Ogata T, Takada F, Yano M, Ando T, et al. (2013). Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am. J. Hum. Genet 93, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baronciani L, and Beutler E (1993). Analysis of pyruvate kinase-deficiency mutations that produce nonspherocytic hemolytic anemia. Proc. Natl. Acad. Sci. USA 90, 4324–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M, Ranish JA, Kummer NT, Hamilton J, Igarashi K, Francastel C, Chi TH, Crabtree GR, Aebersold R, and Groudine M (2004). Dynamic changes in transcription factor complexes during erythroid differentiation revealed by quantitative proteomics. Nat. Struct. Mol. Biol 11, 73–80. [DOI] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagna DR, de Bie CI, Schmitz-Abe K, Sweeney M, Sendamarai AK, Schmidt PJ, Heeney MM, Yntema HG, Kannengiesser C, Grand-champ B, et al. (2014). X-linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA-binding site mutations. Am. J. Hematol 89, 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel P, Cheng A, Cuevas-Navarro A, Everman DB, Papageorge AG, Simanshu DK, Tankka A, Galeas J, Urisman A, and McCormick F (2019). RIT1 oncoproteins escape LZTR1-mediated proteolysis. Science 363, 1226–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavé H, Caye A, Ghedira N, Capri Y, Pouvreau N, Fillot N, Trimouille A, Vignal C, Fenneteau O, Alembik Y, et al. (2016). Mutations in RIT1 cause Noonan syndrome with possible juvenile myelomonocytic leukemia but are not involved in acute lymphoblastic leukemia. Eur. J. Hum. Genet 24, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, and London IM (1995). Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Biochem. Sci 20, 105–108. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Wu W, Kumar SA, Yu D, Deng W, Tripic T, King DC, Chen K-B, Zhang Y, Drautz D, et al. (2009). Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 19, 2172–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Wittig JG, Ghamari A, Maeda M, Dailey TA, Bergonia H, Kafina MD, Coughlin EE, Minogue CE, Hebert AS, et al. (2017). Erythropoietin signaling regulates heme biosynthesis. eLife 6, e24767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY, Clark KL, Burley SK, and Zaret KS (1998). Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 17, 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey HA, and Meissner PN (2013). Erythroid heme biosynthesis and its disorders. Cold Spring Harb. Perspect. Med 3, a011676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty AC, Yeo RW, Buenrostro JD, Greenleaf WJ, Kundaje A, and Brunet A (2017). Chromatin accessibility dynamics reveal novel functional enhancers in C. elegans. Genome Res. 27, 2096–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demina A, Varughese KI, Barbot J, Forman L, and Beutler E (1998). Six previously undescribed pyruvate kinase mutations causing enzyme deficiency. Blood 92, 647–652. [PubMed] [Google Scholar]
- Doty RT, Phelps SR, Shadle C, Sanchez-Bonilla M, Keel SB, and Abkowitz JL (2015). Coordinate expression of heme and globin is essential for effective erythropoiesis. J. Clin. Invest 125, 4681–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doty RT, Yan X, Lausted C, Munday AD, Yang Z, Yi D, Jabbari N, Liu L, Keel SB, Tian Q, and Abkowitz JL (2019). Single-cell analyses demonstrate that a heme-GATA1 feedback loop regulates red cell differentiation. Blood 133, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, and Rabinowitz JD (2014). Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frietze S, Lan X, Jin VX, and Farnham PJ (2010). Genomic targets of the KRAB and SCAN domain-containing zinc finger protein 263. J. Biol. Chem 285, 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, O’Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, Choi K, Farnham PJ, and Bresnick EH (2009). Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol. Cell 36, 667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass JA, Boyer ME, Pal S, Wu J, Weiss MJ, and Bresnick EH (2003). GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc. Natl. Acad. Sci. USA 100, 8811–8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory T, Yu C, Ma A, Orkin SH, Blobel GA, and Weiss MJ (1999). GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 94, 87–96. [PubMed] [Google Scholar]
- Grevet JD, Lan X, Hamagami N, Edwards CR, Sankaranarayanan L, Ji X, Bhardwaj SK, Face CJ, Posocco DF, Abdulmalik O, et al. (2018). Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, and Noble WS (2007). Quantifying similarity between motifs. Genome Biol. 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar M, Kohyama M, So AY, Kc W, Wu X, Briseño CG, Satpathy AT, Kretzer NM, Arase H, Rajasekaran NS, et al. (2014). Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 156, 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, Fleming M, Leboulch P, Orkin SH, and Chen JJ (2001). Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 20, 6909–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon GJ, and Beach D (1994). p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 371, 257–261. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hira S, Tomita T, Matsui T, Igarashi K, and Ikeda-Saito M (2007). Bach1, a heme-dependent transcription factor, reveals presence of multiple heme binding sites with distinct coordination structure. IUBMB Life 59, 542–551. [DOI] [PubMed] [Google Scholar]
- Jiménez G, Griffiths SD, Ford AM, Greaves MF, and Enver T (1992). Activation of the beta-globin locus control region precedes commitment to the erythroid lineage. Proc. Natl. Acad. Sci. USA 89, 10618–10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K, Furuyama K, Fujiwara T, Kobayashi R, Ishida H, Harigae H, and Shibahara S (2014). Identification of a novel erythroid-specific enhancer for the ALAS2 gene and its loss-of-function mutation which is associated with congenital sideroblastic anemia. Haematologica 99, 252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YA, Sanalkumar R, O’Geen H, Linnemann AK, Chang CJ, Bouhassira EE, Farnham PJ, Keles S, and Bresnick EH (2012). Autophagy driven by a master regulator of hematopoiesis. Mol. Cell. Biol 32, 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, et al. (2008). A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 319, 825–828. [DOI] [PubMed] [Google Scholar]
- Kolde R, and Vilo J (2015). GOsummaries: an R package for visual functional annotation of experimental data. F1000Res. 4, 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, and Carey V (2009). rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Y, Chen Y, Li M, Zhou F, Li K, Cao H, Ni M, Liu Y, Gu Z, et al. (2017). In situ capture of chromatin interactions by biotinylated dCas9. Cell 170, 1028–1043.e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir S, Cai W, Carlson SW, Saatman KE, and Andres DA (2017). IGF-1 mediated neurogenesis involves a novel RIT1/Akt/Sox2 cascade. Sci. Rep 7, 3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, and Yamamoto M (2004). Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med 10, 549–557. [DOI] [PubMed] [Google Scholar]
- Nguyen AT, Prado MA, Schmidt PJ, Sendamarai AK, Wilson-Grady JT, Min M, Campagna DR, Tian G, Shi Y, Dederer V, et al. (2017). UBE2O remodels the proteome during terminal erythroid differentiation. Science 357, eaan0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, Hayashi N, Yamamoto M, Shibahara S, Fujita H, and Igarashi K (2001). Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 20, 2835–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel B, Guillerm G, Vereecque R, Wattel E, Preudhomme C, Bauters F, Vanrumbeke M, and Fenaux P (1998). Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood 91, 2985–2990. [PubMed] [Google Scholar]
- Rajagopal A, Rao AU, Amigo J, Tian M, Upadhyay SK, Hall C, Uhm S, Mathew MK, Fleming MD, Paw BH, et al. (2008). Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 453, 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, et al. (2002). Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 21, 5216–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, and Igarashi K (2004). Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA 101, 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka KR, Valentine WN, and Miwa S (1962). Pyruvate kinase (PK) deficiency hereditary nonspherocytic hemolytic anemia. Blood 19, 267–295. [PubMed] [Google Scholar]
- Tanimura N, Miller E, Igarashi K, Yang D, Burstyn JN, Dewey CN, and Bresnick EH (2016). Mechanism governing heme synthesis reveals a GATA factor/heme circuit that controls differentiation. EMBO Rep. 17, 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura N, Liao R, Wilson GM, Dent MR, Cao M, Burstyn JN, Hematti P, Liu X, Zhang Y, Zheng Y, et al. (2018). ). GATA/heme multi-omics reveals a trace metal-dependent cellular differentiation mechanism. Dev. Cell 46, 581–594.e584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida T, Kinoshita T, Nagai H, Nakahara Y, Saito H, Hotta T, and Mu-rate T (1997). Hypermethylation of the p15INK4B gene in myelodysplastic syndromes. Blood 90, 1403–1409. [PubMed] [Google Scholar]
- Ulirsch JC, Lareau CA, Bao EL, Ludwig LS, Guo MH, Benner C, Satpathy AT, Kartha VK, Salem RM, Hirschhorn JN, et al. (2019). Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet 51, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P, Tee WW, and Reinberg D (2013). A double take on bivalent promoters. Genes Dev. 27, 1318–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi A, Ulirsch JC, Ludwig LS, Fiorini C, Yasuda M, Choudhuri A, McDonel P, Zon LI, and Sankaran VG (2016). Insight into GATA1 transcriptional activity through interrogation of cis elements disrupted in human erythroid disorders. Proc. Natl. Acad. Sci. USA 113, 4434–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnatz HJ, Schmidt D, Manke T, Piccini I, Sultan M, Borodina T, Balzereit D, Wruck W, Soldatov A, Vingron M, et al. (2011). The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J. Biol. Chem 286, 23521–23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MJ, Yu C, and Orkin SH (1997). Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol. Cell. Biol 17, 1642–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, Blobel GA, Chodosh LA, and Weiss MJ (2004). Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 104, 3136–3147. [DOI] [PubMed] [Google Scholar]
- White C, Yuan X, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, Hall C, Bishop K, Calicchio ML, Lapierre A, et al. (2013). HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 17, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Keel SB, Shimamura A, Liu L, Gerds AT, Li HY, Wood BL, Scott BL, and Abkowitz JL (2016). Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med 8, 338ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, Yu D, Hardison R, Weiss MJ, Orkin SH, et al. (2009). Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol. Cell 36, 682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Yuan X, Fleming MD, and Hamza I (2013). Heme transport and erythropoiesis. Curr. Opin. Chem. Biol 17, 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, Tokunaga F, Iwai K, and Igarashi K (2007). Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol 27, 6962–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, An W, Wan Y, Ma S, Yin J, Li X, Gao J, Yuan W, Guo Y, et al. (2017). Intron 1 GATA site enhances ALAS2 expression indispensably during erythroid differentiation. Nucleic Acids Res. 45, 657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Macias-Garcia A, Ulirsch JC, Velazquez J, Butty VL, Levine SS, Sankaran VG, and Chen JJ (2019). HRI coordinates translation necessary for protein homeostasis and mitochondrial function in erythropoiesis. eLife 8, e46976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ATAC-seq datasets generated during this study are available at GEO: GSE114996.