

Abstract
Background
The resistance of Streptococcus pneumoniae to macrolides is becoming an increasingly important issue and thus it is important to understand the genetics related to adaptation of this species to the widespread use of antibiotics in Europe. The 58 isolates of S. pneumoniae belonging to sequence type (ST) 416 and serotype 19A and to several different phenotypes originated from Italy, Portugal and Czech Republic were thus sequenced on Illumina MiSeq. The aim of the study was to describe genetical origine of isolates, investigate their macrolide resistance and suggest reasons for spread of ST416 in the Czech Republic.
Results
Investigation of genes associated with serotype determined serotype switch between 15B and 19A serotypes and core genome multilocus sequence typing (cgMLST) confirmed the origine of concerned isolates in Netherlands15B-37 clone. Inspected genomes proved variability of genes associated with the macrolide resistance even within closely genetically relative isolates.
Conclusions
Participation of 19A/ST416 on the spread of Netherlands15B-37 is accompanied by serotype switch between 19A and 15B serotypes and with acquisition of genes involved in macrolide resistance to the clone that was originally macrolide susceptible. There is evident tendency to interchanging and modifications of these and surrounding genes, that could lead to accelerate spreading of this sequence type in regions with high macrolide consumption.
Background
Streptococcus pneumoniae is a leading cause of community-acquired pneumonia, meningitis, acute otitis media, acute sinusitis, and bacteremia. Pneumococcal infections are mainly prevalent in infants, elderly, and immunocompromised patients. It is estimated that 0.8 million children die every year from pneumonia in which pneumococcus is considered the major pathogen [1]. The main reservoir of S. pneumoniae in humans is the nasopharynx. Carriage rates are highest in children of preschool age (up to 50%), in adults the carriage rates ranges between less than 5% to more than 20% depending on factors such as age, immune status and antibiotic use [2]. Although the colonization of the nasopharynx is usually asymptomatic, carriage can progress to invasive respiratory or systemic disease [3]. Pneumococci are encapsulated, the polysaccharide capsule which protects pneumococci from phagocytosis, is considered as the major virulence factor. To date, at least 97 diverse capsular types have been described [4], although only a limited number of serotypes cause the vast majority of pneumococcal infections, some of them are also associated with antibiotic resistance.
The current prevention strategies of S. pneumoniae infections include the use of pneumococcal conjugate vaccines (PCV) which cover a subset of serotypes selected based on their contribution to pneumococcal infections. The first conjugated vaccine PCV7 targeting seven serotypes (4, 6B, 9 V, 18C, 19F, and 23F) was implemented in 2000 in USA [4]. The use of vaccines lead to decrease of invasive pneumococcal diseases (IPD). However, due to the limited number of serotypes included in the vaccines, an increase of occurrence of non-vaccine serotypes was reported ([4]). The serotype, which was globally associated with replacement disease after PCV7 use is 19A [5–9]. Currently, PCV10 (PCV7 plus serotypes 1, 5, and 7F) and PCV13 (PCV10 plus serotypes 3, 6A, and 19A) are used. Use of PCV10 and PCV13 led to an additional reduction in IPD caused by the additional serotypes included in the vaccines, but the cross-protection of PCV10 against vaccine-related serotypes 6A and 19A seems variable in different studies [10–12].
A substantial part of serotype 19A isolates are also resistant to antibiotics. Antibiotic resistance is in general caused by changes in core genes, or by acquisition of accessory genes, it is presumable that the later vary within clones more than the previous. Resistance to penicillin is in pneumococci caused predominantly by changes in penicillin-binding proteins situated near the capsular polysaccharide locus (cps locus). On the other hand, resistance to macrolides is caused by acquisition of particular genes (ermB, tetM). Distribution of clonal complexes (CC) vary in different countries, CC320 (Taiwan19F-14 clone) is spread worldwide, and it is the most frequent 19A clone in Asia and South America, CC199 (Netherlands15B-37 clone) is the predominant clone in North America and in Europe. There is a number of other less populated clones, from which the most frequent are CC695 in North America, and CC230 (Denmark14–32 clone) or CC156 (Spain9V-3 clone) in Europe [13–19].
In the Czech Republic, vaccination using PCV10 or PCV13 has been included to the routine childhood immunization programme since January 2010, both vaccines sharing the market equally. The vaccine uptake is continuously declining, from 81% vaccinated children in 2011 to 68% in 2017. In the post-PCV era, 19A isolates were found in carriage and respiratory samples and occurred also in IPD cases in all age cohorts [20]. The overall prevalence of the 19A serotype increased from 2.7% in 2010 to 9.9% in 2015, with 19A being the second most frequent invasive serotype reported in the Czech Republic [21]. Sequencing identified ST416, a double-locus variant (DLV) of the Netherlands15B-37 clone (CC199), as the dominant emerging genotype within serotype 19A [22]. Multilocus sequence typing (MLST) analysis of penicillin and/or macrolide resistant isolates of serogroup 19 revealed CC199 represented by ST416 in 64% of the serotype 19A isolates, other minor CCs were CC230 (Denmark14–32 clone), and CC193 (Greece21–30 clone). Participation of 19A/ST416 in the serogroup 19 was 44% in 2014 and 68% in 2017 (the second were 19F/ST1464 representing 25% of serogroup 19 in 2014 and 19A/ST320 12% in 2017 respectively) Isolates belonging to ST416 were penicillin susceptible, but erythromycin, clindamycin, and tetracycline resistant [22, 23].
Contribution of ST416 on increase of 19A serotype has been described in other European countries. In Italy, proportion of 19A isolates from IPD increased from 8% in in 2001–2003 to 17% in 2006–2009, and ST416 from 6 to 10% respectively [7, 8]. Increase of ST416 was accompanied with growth (in Italy) or introduction (in Germany) of macrolide resistance within CC199, which was originally susceptible to macrolides. According to public database of sequence types (PubMLST [24]), the isolates showing ST416 are present in Great Britain, Germany, Italy, and Spain from 2000, single isolates appeared in Australia, USA, Canada and China after year 2007.
Results
SNP phylogeny
Mapping of Illumina reads to the Netherlands15B-37 representative contigs gave consensus sequences with percentages of unambiguously calling bases in the range 91–98%. The input alignment for maximum likelihood phylogeny after the recombination sequences were removed consisted of 1,631,892 sites from which were 7367 polymorphic.
The dendrogram (Fig. 1) differentiated two clades, one was represented by a homogenous group of 46 isolates (Clade I), while the other included a more divergent group of 12 isolates (Clade II). There were isolates from Portugal, Italy and the Czech Republic isolated in 2002–2015 in Clade I. Four strains from Clade I were susceptible to erythromycin, clindamycin, and tetracycline, two were susceptible only to clindamycin, one only to tetracycline and the remaining isolates were resistant to erythromycin, clindamycin, and tetracycline. The topology demonstrates separations of the branches according to countries of origin. Clade II consisted of strains from Portugal isolated in 2002–2011, and it was differentiated into a few distinct sub-branches, with antibiotic profiles characteristic for each sub-branch.
Fig. 1.

SNP phylogeny of 58 Streptococcus pneumoniae ST416 isolates with Netherlands15B-37 as reference sequence. Branches are labelled with strain numbers and year of isolation. They are coloured according to the country of origin: in red those from Italy, in green from Portugal and in blue from the Czech Republic
Core genome MLST
There were found 1177 groups of orthologs that were presented once in each genome. From these were 450 represented by an identical allele in each genome and 7 by different alleles in more than ¾ of genomes. cgMLST scheme was developed on basis of the rest 720 genes. Minimum-spanning tree (Fig. 2) demonstrates that Netherlads15B-37 differs from the nearest genome of Clade II in 145 loci and that differs from the nearest genome of Clade I in 130 loci. The longest distance between two genomes within a clade was 23 loci in Clade I and 116 loci in Clade II.
Fig. 2.
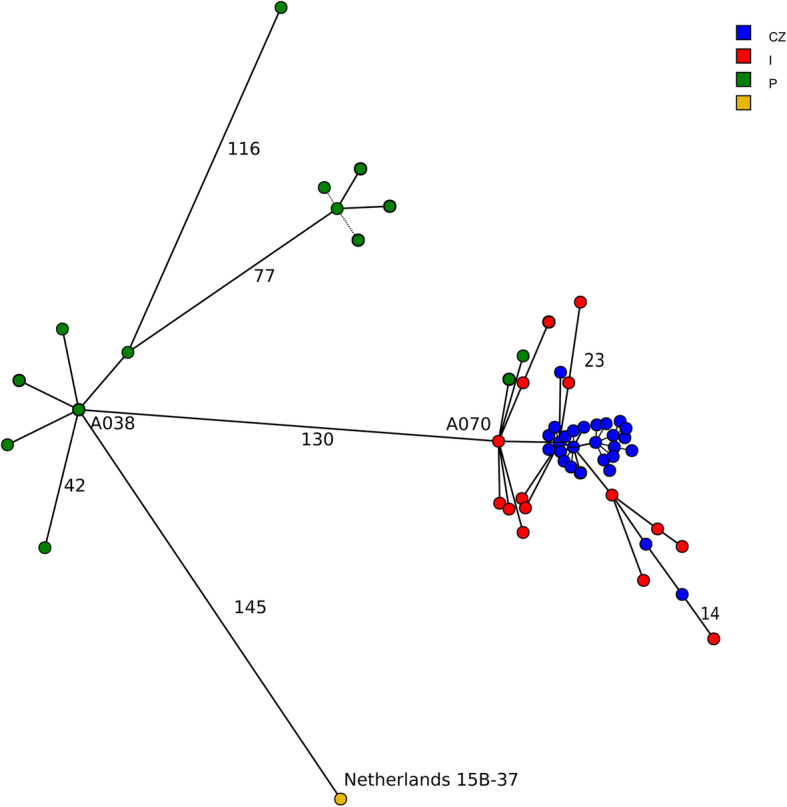
Core genome MLST of collection consisting of 58 Streptococcus pneumoniae ST416 isolates and Netherlands15B-37. Lengths of branches correspond to number of different loci. Nodes are coloured according to the country of origin: in red those from Italy, in green from Portugal and in blue from the Czech Republic
Relatedness between ST416 and reference strain Netherlands15B-37
Netherlands15B-37 (ATCC BAA-1663) represents ST199 which is a DLV of ST416. They differ in the aroE allele by one single nucleotide polymorphism (SNP), and in the xpt allele by six SNPs. According criteria previously defined, the change in aroE most probably arose by a point mutation and while that in xpt arose by recombination [25].
De novo assemblies could not be used neither for reconstruction of cps locus and flanking sequences nor for transposable elements, as involved adjacent genes were rarely included in the same contig, that was probably caused by presence of repetitive sequences.
Detailed investigation of cps locus and flanking genes (Fig. 3) showed a close relationship between the 19A cps loci within the collection, as well as regions of high similarity to the sequences surrounding Netherlands15B-37 cps locus. Sequences of dexB, and MraY situated upstream the cps locus were identical within the collection, dexB was 98% identical to the Netherlands15B-37 representative, the other 100% identical. Sequences of remaining genes situated between pbp2x and the beginning of the cps locus had greater variability when compared to other genes from the region. The wzg, wzh, wzd, wze, wchA, wchO, wchP, and wchQ genes from the beginning of cps locus were identical within the collection, the remaining genes of the cps locus, as well as pbp2x, pbp1a, and flanking genes differed in a few isolates. Sequences of pbp2x, and pbp1a of most isolates were 100% identical with the corresponding sequences in Netherlands15B-37 strain, sequence of gene engBF coding endo-alpha-N-acetylgalactosaminidase situated upstream pbp1a differed from reference in most of isolates by the same recombination positioned at the beginning of the sequence (from 1 to 1,309 bp). Sequence of oligopeptide-binding protein aliA were 100% identical with the corresponding sequences in Netherlands15B-37 strain in 42 isolates, in remaining 16 isolates were present 5 different alleles of aliA. High variability in particular genes indicates that these areas are changing very quickly within the clone, and those events could be associated with serotype switches.
Fig. 3.

Relatedness between sequences of particular genes involved in serotype switch and the corresponding genes in references. Genome diagram compiled from assemblies with three reference sequences: region between start of pbp1a and end of dexB in Netherlands15B-37 (coloured in dark brown), 19A cps locus (coloured in light brown) and region between start of aliA and end of pbp1a (coloured in light brown). Genes with sequences identical between particular strain and corresponding gene in the genome of Netherlands15B-37 are coloured in dark brown, those identical to the sequences from cps locus in brown. (SP0310, SPAR147_310; SP0340, SPAR147_0340) Genes with sequences different to reference and to all samples in the collection are shown in orange. Genes with sequences differing to reference in one SNP same in > 1 sample are coloured in green, those differing in the same SNPs > 1 sample are represented in shades of blue, genes which were not found in particular genome are grey. Isolates are ordered and labelled likewise in Fig. 1
Genes of resistance to erythromycin, clindamycin and tetracycline
The genes coding macrolide resistance, mef(A)/mel, mef(E), erm(A) and erm(TR) were not found in any of the isolates belonging to ST416. Both erm(B) and tet(M) were present in genomes of 47 isolates, from these were 37 phenotypically resistant to erythromycin, tetracycline, and clindamycin, 7 were phenotypically resistant to erythromycin and tetracycline, showing inducible resistance to clindamycin. Three isolates were phenotypically susceptible to tetracycline and resistant to other two examined antibiotics. Alignments of tetM show deletion of a single base leading to a frameshift mutation in the beginning of gene (position 650 resp. 653) in two genomes, phenotypical susceptibility of the third is not explainable by presented methods. Sequences of erm(B) and tet(M) were not identical within the collection, there were four different alleles of tet(M), and three different alleles of erm(B).
All genomes containing erm(B) and tet(M) carried all genes constituting Tn6002 as well. These were identical to reference in 29 of 47 isolates (Fig. 4.). They differed from reference either in 1–2 separate genes (11 isolates) or in most of genes (7 isolates). The later were apparently carried by three different mobile genetics elements.
Fig. 4.

SNPs within genes presented in Tn6002 transposon. Genes with sequences identical between a particular strain and the corresponding gene in the Tn6002 are coloured in dark brown. Genes with sequences differing to reference in one SNP same for more samples are coloured in shades of green, these differing in more SNPs same for more samples in shades of blue. Genes which were not found in particular genomes are coloured in grey. Country of origin and resistance (R, resistant; S, susceptible) are indicated alongside the strain identifiers. Inducible clindamycin resistance is marked with “!” in the last column. Labelled are genes associated with resistance and gene transfer. (TR, transcriptional regulator; CJG, conjugal transfer protein; TTD, tetracycline resistance determinant leader peptide; tps, transposase; tls, translocase)
Discussion
After introduction of PCV7, serotype 19A ranked globally among the top non-PCV7 serotypes associated with serotype replacement [26–29]. Molecular analysis of 19A post-PCV7 isolates revealed, that majority of isolates belong to 3 clonal complexes: CC199, CC320/271, and CC695 [26]. While CC320/271 arose by a serotype switch from ancestral ST’s 271 and 236 of serotype 19F, representing a “vaccine-escape” event, those representing CC695 arose from recombination involving both non-PCV7 and PCV7 serotypes since the ST695 serotype 19A originated from a penicillin susceptible ST695 serotype 4 and a penicillin intermediate ST199 serotype 19A [30]. It was suggested that the observed capsular switches were mainly driven by selective pressure of antibiotic consumption, as altered pbp genes associated with penicillin non-susceptibility have been identified in isolates belonging to those clones. Clonal complex CC199, represented by ST199, is widely geographically distributed as penicillin non-susceptible serotype 19A or penicillin susceptible serotype 15B/C [31, 32].
Likely, penicillin susceptible isolates of serotype 15B/C served as progenitor for penicillin non-susceptible ST199 serotype 19A. Study of serotype switches in CC199 in isolates from pre-PCV13 era in Germany identified at least two serotype switches from 19A to 15B of ST199, suggesting that capsular switches in ST199 is not unusual [33].
All assemblies of sequences surrounding cps locus presented in the study confirm serotype switch, although used methods do not allow to identify position, and orientation of genes. Nevertheless, the high variability of particular sequences indicates that these areas are changing very quickly within the clone and could be associated with multiple serotype switch events.
The results indicate that there is a recent common ancestor between the Netherlands15B-37 representative and the isolates included in the present study. This conjecture is supported by the SNP phylogeny and by the match in the sequences associated with the cps locus. The specific evidence is that the entire 15B cps locus present in the Netherlands15B-37 between the pbp2x and pbp1a genes was replaced in the studied isolates by the 19A cps locus. It is worthy of note that the engBF sequence situated downstream the locus is 100% identical in most of the isolates within the collection (with individual SNPs in three isolates). The engBF sequence consists of two parts: the first part (the base pairs 1.1309) is the same in all studied isolates whilst showing 97.6% identity to the corresponding sequence in Netherlands15B-37 representative and the second part (the remaining base pairs after 1309) are 100% identical to Netherlands15B-37. It is very likely that the present isolates share the beginning of the sequence with the conjectured donor of the 19A cps locus. If this is the case, the aliA gene, situated between the engBF and cps locus, should also originate from this donor. It is remarkable that whilst the sequence of aliA is quite variable within the entire studied collection there is a substantial group (a majority) of isolates that have the aliA sequence 100% identical to the Netherlands15B-37 representative. This observation suggests that, when the conjectured serotype switch occurred, the donor had the aliA alleles identical to the Netherlands15B-37 representative. A similar situation is apparent upstream of the cps locus, where the dexB gene originates probably from the same donor. It is very likely that the serotype switch took place once and included the sequences surrounding the cps locus, but not the penicillin binding proteins. In addition to the common donor it seems that the genomes of a few of the isolates included in the present study were changed by recombinations in different genes afterwards. It is possible that some isolates, especially those which phylogenically differ from rest of the collection, originate from different serotype switches. Nevertheless the sequences where the exchange actually happened (engF and dexB) are identical within the present collection and it is unlikely that they would originate from distinct serotype switches.
An increase of serotype 19A after introduction of PCV’s (PCV10 and PCV13) in the Czech Republic was due to an expansion of ST416 which is a DLV of ST199. It was proposed that an emergence of this subclone was facilitated by the presence of pilus 1 (PI-1), as group founder ST199 was found to be PI-1 negative [22]. The ST416 was identified in other European countries (Italy, Portugal, Germany), but it’s contribution to the increase of serotype 19A varies [5, 7, 8]. Antibiotic consumption may enhance the spread of certain drug-resistant clones. It is a plausible explanation that increased use of macrolides favoured the spread of this macrolide-resistant clone. Despite a decrease of macrolide consumption in Italy and Portugal during last decade, consumption of macrolides is high in both countries, and it shows a significant increasing trend in Czech Republic [34].
The isolates in the study represent two different forms of genetic diversification within ST416. Topology of the tree (Fig. 1) suggests that the strains present in Clade II possibly reflect and old lineage that has had time to diversify. The Clade I on the other hand, consists of strains with very similar core genomes although originating in different countries within a relatively long timescale, suggesting the rapid spread of a highly successful lineage that seems to be adapting mostly through recombination.
Recombination events led to the alteration of sequence type and it was followed by acquisition of a transposable elements carrying erm(B) and tet(M) genes in particular subclones and to variability within accessory genes. Sequences associated with transposable elements were not successfully assembled, because de-novo assembly frequently fails in repetitive sequences. Aligning reads to representative sequences of genes of interest gives a good overview of variability within isolates but does not inform about positioning and orientation of genes, not even about gene compositions of a given mobile element and accurate identification of transposable elements. It is also the reason, why inducible resistance to clindamycin in 7 isolates carrying erm(B) gene cannot be explained in this study, as it is associated with inverted repeat sequences located in the region upstream the gene [35]. Gene erm(B) is usually associated with constitutive resistance to clindamycin, but inducible resistance was also reported [36]. Isolates resistant to erythromycin belonging to ST199 were reported to carry mef genes [37], however mef genes were not present in the genome of any isolate in the study, neither in the genome of Netherlands15B-37.
Conclusions
The elimination of vaccine serotypes through the use of PCVs promotes the spread of pre-existing minor clones as well as of novel clones arising from genetic exchange. Using whole genome sequencing we described the properties of ST416 serotype 19A, and the relationship between ST416 and ST199. Our study shows that ST416 developed from the Netherlands15B-37 clone and that it can now be found in several European countries. Comparing to the ancestral ST199 genotype, isolates of ST416 have additional accessory genes coding for antibiotic resistance. Whereas the capsular switch apparently featured only once, high variability of sequences in accessory genes confirms repeated horizontal gene transfer of mobile genetic elements carrying drug resistant genes.
Methods
We used a collection of 58 isolates of serotype 19A of ST416 recovered between 2002 to 2016 in the Czech Republic, Italy, and Portugal to analyse their sequence diversity, and to investigate whether the spread of ST416 could be related to transfer of genes associated with antibiotic resistance.
Bacterial isolates
Altogether, 58 isolates of Streptococcus pneumoniae of serotype 19A of ST416 were analysed (Additional file 1). Pneumococcal isolates (n = 29) were collected by the National Reference Laboratory for Antibiotics (National Institute of Public Health, Czech Republic) from 2010 to 2015, 15 isolates were obtained from the Department of Infectious Diseases (National Institute of Health, Rome, Italy) between 2002 to 2016 and 14 isolates were collected by the Faculdade de Medicina (Universidade de Lisboa, Lisbon, Portugal) between 2002 and 2011. The majority (n = 50; 86.2%) of isolates was collected from blood (n = 47) or cerebrospinal fluid (n = 3), Suppl. Table 1. The isolates were characterised by MLST, serotyping was performed by Quellung reaction as previously described [5, 7, 8]. MICs of penicillin, cefotaxime, erythromycin, clindamycin, tetracycline, chloramphenicol, trimethoprim/sulfamethoxazole were determined by broth microdilution method according to the EUCAST recommendations [38].
WGS analysis
All isolates presented in the study were sequenced in the Laboratory of Antibiotic Resistance and applications of Mass Spectrometry in Microbiology (Charles University, Faculty of Medicine in Pilsen, Biomedical Centre Pilsen) using the Illumina MiSeq platform for paired end reads. Reads were trimmed by Trimmomatic v. 0.36 [39]. Trimmed reads were assembled de novo by SPAdes v 3.9.1 [40] and the resulting contigs were annotated with PROKKA v 1.12 [41]. De novo assemblies were uploaded to PubMLST [24] for confirmation of sequence types. PubMLST was also used for estimation of relatedness between sequence types 416 and 199.
Core genes of collection consisting of genomes under study and that of Netherlands15B-37 were set by Orthofinder v. 2.0.0 [42]. Coding sequences of orthologs were used to set core genome multilocus sequence typing (cgMLST) scheme for the collection with BLAST v. 2.2.31. Minimum spanning tree was constructed with Bionumeric v. 7.6.
Trimmed reads were aligned to particular reference sequences (Netherlands15B-37, 19A cps locus, Tn6002, erm(A), erm(TR), and mefA/mel gene) using SMALT (SMALT is Copyright (C) 2010–2015 Genome Research Ltd). Consensus sequences were reconstructed using SAMtools [43] and VCFtools [44]. Only unambiguously calling bases with minimum values for DP (depth) > 4, MQ (mapping quality) > 50, and QUAL (position quality) > 50 were accepted.
Consensus sequences obtained by mapping to reference strain Netherlands15B-37 (ATCC BAA-1663) were used for construction of a phylogeny tree based on single nucleotide polymorphisms (SNP). The reference sequence was obtained by concatenation of contig sequences downloaded from GenBank (BioSample: SAMN00792697). SNP phylogeny based on the Netherlands15B-37 reference enabled elimination of the sites involved in recombination and that resulted in more accurately branching tree. Recombination sequences were predicted on the basis of the SNPs densities in the sliding window of 101 sites in length. The cut-off was set for each sequence separately as ratio of number of SNPs and length of sequence. Sites where recombination emerged at least in one genome were cut out from all the genomes. RaxML in default setting, which calculates ICA and consequently TCA scores, was used to generate unrooted tree with model GAMMA. Lengths (L) of branches correspond to probability that mutation occurs on a site, e.g. two nodes differ in every 1/Lth site.
Sequences positioned between genes coding for penicillin binding proteins (pbp), pbp2x and pbp1a were obtained by mapping reads to corresponding sequences in the genome of Netherlads15B-37 (pbp2x – dexB, aliA – pbp1a), and to the sequence of S. pneumoniae serotype 19A capsular locus (accession number JF9111520). Consensus sequences were inspected manually. Differences between sequences coding particular genes situated between pbp2x and pbp1a in the genome of reference sequence, and in the genome of every isolate, were described according to number of variants within collection and the origin of change (point mutation or recombination, Fig. 2b) [25].
Presence of particular genes coding for resistance to antibiotics (macrolides, tetracycline) was determined by mapping reads to sequences of mef(A)/mel (accession number EF042094.1), mef(E) (accession number U83667.1), erm(A) (accession number KT803896848263.1), erm(TR) (accession number AF002716) and to the transposon Tn6002 (accession number AY898750.1) which included genes erm(B), and tet(M).
Supplementary information
Additional file 1. List of Streptococcus pneumoniae clinical isolates included in the study. Table contains data of origin and properties of the isolates. (CZ, the Czech Republic; ITA, Italy; POR, Portugal; F, female; M, male; PEN, penicillin; CTX, cefotaxime; ERY, erythromycin; CLI, clindamycin; TET, tetracycline; CMP, chloramphenicol; SXT, trimethoprim/sulfamethoxazole; Induction, inducible clindamycin resistance).
Acknowledgements
We are grateful to Mario Ramirez from Instituto de Microbiologia, Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Lisboa, Portugal for providing isolates and kind advice.
Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures.
Abbreviations
- CC
Clonal complex
- cgMLST
core genome multilocus sequence typing
- cps
caplular polysaccharide
- DLV
Double-locus variant
- IPD
Invasive pneumococcal disease
- MIC
Minimal inhibitory concentration
- MLST
Multilocus sequence typing
- PCV
Pneumococcal conjugate vaccines
- SNP
Single nucleotide polymorphism
- ST
Sequence type
Authors’ contributions
HZ and JH designed the study, AP and MdG provided data, CCP and IB performed whole genome sequencing. LM, MM and JK performed all other laboratory tests. VJ was involved in interpretation of results. PS analysed data and drafted the text. All authors read through and approved the manuscript.
Funding
This work was supported by the Czech Health Research Council, Ministry of Health of the Czech Republic, grant no. 16-27109A.
Availability of data and materials
Whole genome sequencing data are available in the NCBI Short Read Archive under BioProject accession PRJNA600380.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have not competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12866-020-01909-1.
References
- 1.O’Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet. 2009;374(9693):893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 2.Regev-Yochay G, Raz M, Dagan R, Porat N, Shainberg B, Pinco E, et al. Nasopharyngeal carriage of Streptococcus pneumoniae by adults and children in community and family settings. Clin Infect Dis. 2004;38(5):632–639. doi: 10.1086/381547. [DOI] [PubMed] [Google Scholar]
- 3.Bogaert D, de Groot R, Hermans PWM. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis. 2004;4(3):144–154. doi: 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 4.Geno KA, Gilbert GL, Song JY, Skovsted IC, Klugman KP, Jones C, et al. Pneumococcal capsules and their types: past, present, and future. Clin Microbiol Rev. 2015;28(3):871–899. doi: 10.1128/Cmr.00024-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguiar SI, Pinto FR, Nunes S, Serrano I, Melo-Cristino J, Sa-Leao R, et al. Denmark (14)-230 clone as an increasing cause of pneumococcal infection in Portugal within a background of diverse serotype 19A lineages. J Clin Microbiol. 2010;48(1):101–108. doi: 10.1128/Jcm.00665-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore MR, Gertz RE, Woodbury RL, Barkocy-Gallagher GA, Schaffner W, Lexau C, et al. Population snapshot of emergent Streptococcus pneumoniae serotype 19A in the United States, 2005. J Infect Dis. 2008;197(7):1016–1027. doi: 10.1086/528996. [DOI] [PubMed] [Google Scholar]
- 7.Del Grosso M, Camilli R, D'Ambrosio F, Petrucci G, Melchiorre S, Moschioni M, et al. Increase of pneumococcal serotype 19A in Italy is due to expansion of the piliated clone ST416/CC199. J Med Microbiol. 2013;62:1220–1225. doi: 10.1099/jmm.0.061242-0. [DOI] [PubMed] [Google Scholar]
- 8.van der Linden M, Reinert RR, Kern WV, Imohl M. Epidemiology of serotype 19A isolates from invasive pneumococcal disease in German children. BMC Infect Dis. 2013;13:70. doi: 10.1186/1471-2334-13-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaur R, Casey JR, Pichichero ME. Emerging Streptococcus pneumoniae strains colonizing the nasopharynx in children after 13-valent pneumococcal conjugate vaccination in comparison to the 7-valent era, 2006-2015. Pediatr Infect Dis J. 2016;35(8):901–906. doi: 10.1097/Inf.0000000000001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domingues CMAS, Verani JR, Renoiner EIM, Brandileone MCD, Flannery B, de Oliveira LH, et al. Effectiveness of ten-valent pneumococcal conjugate vaccine against invasive pneumococcal disease in Brazil: a matched case-control study. Lancet Respir Med. 2014;2(6):464–471. doi: 10.1016/S2213-2600(14)70060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naucler P, Galanis I, Morfeldt E, Darenberg J, Ortqvist A, Henriques-Normark B. Comparison of the impact of pneumococcal conjugate vaccine 10 or pneumococcal conjugate vaccine 13 on invasive pneumococcal disease in equivalent populations. Clin Infect Dis. 2017;65(11):1780–1789. doi: 10.1093/cid/cix685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quirk SJ, Haraldsson G, Hjalmarsdottir MA, van Tonder AJ, Hrafnkelsson B, Bentley SD, et al. Vaccination of Icelandic children with the 10-valent pneumococcal vaccine leads to a significant herd effect among adults in Iceland. J Clin Microbiol. 2019;57(4):1. doi: 10.1128/JCM.01766-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muñoz-Almagro C, Jordan I, Gene A, Latorre C, Garcia-Garcia JJ, Pallares R. Emergence of invasive pneumococcal disease caused by nonvaccine serotypes in the era of 7-valent conjugate vaccine. Clin Infect Dis. 2008;46(2):174–182. doi: 10.1086/524660. [DOI] [PubMed] [Google Scholar]
- 14.Tyrrell GJ. The changing epidemiology of Streptococcus pneumoniae serotype 19A clonal complexes. J Infect Dis. 2011;203(10):1345–1347. doi: 10.1093/infdis/jir056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng X, Arya G, Memari N, Mackenzie R, MacMullin G, Low DE, et al. Genetic analysis of invasive pneumococcal isolates from children in Ontario, Canada, 2007-2012. Pediatr Infect Dis J. 2015;34(6):594–598. doi: 10.1097/INF.0000000000000697. [DOI] [PubMed] [Google Scholar]
- 16.Sadowy E, Kuch A, Gniadkowski M, Hryniewicz W. Expansion and evolution of the Streptococcus pneumoniae Spain9V-ST156 clonal complex in Poland. Antimicrob Agents Chemother. 2010;54(5):1720–1727. doi: 10.1128/AAC.01340-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramos V, Parra EL, Duarte C, Moreno J. Characterization of Streptococcus pneumoniae invasive serotype 19A isolates recovered in Colombia. Vaccine. 2014;32(7):755–758. doi: 10.1016/j.vaccine.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 18.Mott MP, Caierão J, Cunha GR, Del Maschi MM, Pizzutti K, d’Azevedo P, et al. Emergence of serotype 19A Streptococcus pneumoniae after PCV10 associated with a ST320 in adult population, in Porto Alegre, Brazil. Epidemiol Infect. 2019;147:e93. doi: 10.1017/S0950268819000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao C, Li Z, Zhang F, Zhang X, Ji P, Zeng J, et al. Serotype distribution and antibiotic resistance of Streptococcus pneumoniae isolates from 17 Chinese cities from 2011 to 2016. BMC Infect Dis. 2017;17(1):804. doi: 10.1186/s12879-017-2880-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vancikova Z, Trojanek M, Zemlickova H, Blechova Z, Motlova J, Matejkova J, et al. Pneumococcal urinary antigen positivity in healthy colonized children: is it age dependent? Wien Klin Wochenschr. 2013;125(17–18):495–500. doi: 10.1007/s00508-013-0405-4. [DOI] [PubMed] [Google Scholar]
- 21.Kozakova J, Sebestova H, Krizova P. Invasive pneumococcal disease in the Czech Republic in 2015. Bull Centre Epidemiol Microbiol. 2016;25:100–107. [Google Scholar]
- 22.Zemlickova H, Malisova L, Spanelova P, Jakubu V, Kozakova J, Musilek M, et al. Molecular characterization of serogroup 19 Streptococcus pneumoniae in the Czech Republic in the post-vaccine era. J Med Microbiol. 2018;67:1003–1011. doi: 10.1099/jmm.0.000765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malisova L, Urbaskova P, Jakubu V, Spanelova P, Kozakova J, Musilek M, et al. Surveillance of antibiotic resistance of Streptococcus pneumoniae in the Czech Republic, respiratory study results, 2010–2017. Epidemiol Microbiol Immunol. 2019;2:75–81. [PubMed] [Google Scholar]
- 24.Public databases for molecular typing and microbial genome diversity. https://pubmlst.org/ Accessed January 2020.
- 25.Feil EJ, Smith JM, Enright MC, Spratt BG. Estimating recombinational parameters in Streptococcus pneumoniae from multilocus sequence typing data. Genetics. 2000;154(4):1439–1450. doi: 10.1093/genetics/154.4.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beall BW, Gertz RE, Hulkower RL, Whitney CG, Moore MR, Brueggemann AB. Shifting genetic structure of invasive serotype 19A pneumococci in the United States. J Infect Dis. 2011;203(10):1360–1368. doi: 10.1093/infdis/jir052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Croucher NJ, Hanage WP, Harris SR, McGee L, van der Linden M, de Lencastre H, et al. Variable recombination dynamics during the emergence, transmission and ‘disarming’ of a multidrug-resistant pneumococcal clone. BMC Biol. 2014;12:49. doi: 10.1186/1741-7007-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hulten KG, Kaplan SL, Lamberth LB, Barson WJ, Romero JR, Lin PL, et al. Changes in Streptococcus pneumoniae serotype 19A invasive infections in children from 1993 to 2011. J Clin Microbiol. 2013;51(4):1294–1297. doi: 10.1128/JCM.00058-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenoll A, Granizo JJ, Giménez MJ, Yuste J, Aguilar L. Secular trends (1990-2013) in serotypes and associated non-susceptibility of S. pneumoniae isolates causing invasive disease in the pre−/post-era of pneumococcal conjugate vaccines in Spanish regions without universal paediatric pneumococcal vaccination. Vaccine. 2015;33(42):5691–5699. doi: 10.1016/j.vaccine.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 30.Brueggemann AB, Pai R, Crook DW, Beall B. Vaccine escape recombinants emerge after pneumococcal vaccination in the United States. PLoS Pathog. 2007;3(11):1628–1636. doi: 10.1371/journal.ppat.0030168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gertz RE, McEllistrem MC, Boxrud DJ, Li ZY, Sakota V, Thompson TA, et al. Clonal distribution of invasive pneumococcal isolates from children and selected adults in the United States prior to 7-valent conjugate vaccine introduction. J Clin Microbiol. 2003;41(9):4194–4216. doi: 10.1128/Jcm.41.9.4194-4216.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gladstone RA, Devine V, Jones J, Cleary D, Jefferies JM, Bentley SD, et al. Pre-vaccine serotype composition within a lineage signposts its serotype replacement - a carriage study over 7 years following pneumococcal conjugate vaccine use in the UK. Microb Genomics. 2017;3(6):000119. doi: 10.1099/mgen.0.000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makarewicz O, Lucas M, Brandt C, Herrmann L, Albersmeier A, Ruckert C, et al. Whole genome sequencing of 39 invasive Streptococcus pneumoniae sequence type 199 isolates revealed switches from serotype 19A to 15B. PLoS One. 2017;12(1):e0169370. doi: 10.1371/journal.pone.0169370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.ECDC. Antimicrobial consumption database (ESAC-Net). https://www.ecdc.europa.eu/en/antimicrobial-consumption/surveillance-and-disease-data/database Accessed Nov. 2019 2019.
- 35.de Vries LE, Christensen H, Agerso Y. The diversity of inducible and constitutively expressed erm (C) genes and association to different replicon types in staphylococci plasmids. Mob Genet Elem. 2012;2(2):72–80. doi: 10.4161/mge.20109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kataja J, Huovinen P, Seppala H, Grp MRS. Erythromycin resistance genes in group a streptococci of different geographical origins. J Antimicrob Chemother. 2000;46(5):789–792. doi: 10.1093/jac/46.5.789. [DOI] [PubMed] [Google Scholar]
- 37.Rudolph K, Bruce MG, Bulkow L, Zulz T, Reasonover A, Harker-Jones M, et al. Molecular epidemiology of serotype 19A Streptococcus pneumoniae among invasive isolates from Alaska, 1986–2010. Int J Circumpol Heal. 2013;72:611–617. doi: 10.3402/ijch.v72i0.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasselmann C, European Soc Clinical Microbiology Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin Microbiol Infect. 2003;9(8):1–7. [Google Scholar]
- 39.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nurk S, Bankevich A, Antipov D, Gurevich A, Korobeynikov A, Lapidus A, et al. Assembling genomes and mini-metagenomes from highly chimeric reads. In: Deng M, Jiang R, Sun F, Zhang X, et al., editors. Research in computational molecular biology. Berlin, Heidelberg: Springer Berlin Heidelberg; 2013. pp. 158–170. [Google Scholar]
- 41.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 42.Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20(1):238. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. List of Streptococcus pneumoniae clinical isolates included in the study. Table contains data of origin and properties of the isolates. (CZ, the Czech Republic; ITA, Italy; POR, Portugal; F, female; M, male; PEN, penicillin; CTX, cefotaxime; ERY, erythromycin; CLI, clindamycin; TET, tetracycline; CMP, chloramphenicol; SXT, trimethoprim/sulfamethoxazole; Induction, inducible clindamycin resistance).
Data Availability Statement
Whole genome sequencing data are available in the NCBI Short Read Archive under BioProject accession PRJNA600380.