

Abstract
Artificial intelligence (AI) and machine learning, in particular, have gained significant interest in many fields, including pharmaceutical sciences. The enormous growth of data from several sources, the recent advances in various analytical tools, and the continuous developments in machine learning algorithms have resulted in a rapid increase in new machine learning applications in different areas of pharmaceutical sciences. This review summarizes the past, present, and potential future impacts of machine learning technologies on different areas of pharmaceutical sciences, including drug design and discovery, preformulation, and formulation. The machine learning methods commonly used in pharmaceutical sciences are discussed, with a specific emphasis on artificial neural networks due to their capability to model the nonlinear relationships that are commonly encountered in pharmaceutical research. AI and machine learning technologies in common day-to-day pharma needs as well as industrial and regulatory insights are reviewed. Beyond traditional potentials of implementing digital technologies using machine learning in the development of more efficient, fast, and economical solutions in pharmaceutical sciences are also discussed.
KEY WORDS: artificial intelligence, machine learning, artificial neural networks, pharmaceutical sciences, pharmaceutical industry
INTRODUCTION: BIG DATA IN PHARMACEUTICAL SCIENCES
There has been a remarkable increase in the amount of data—including pharmaceutical data—that are generated each day. The term “big data” has gained increasing interest in various research areas. In addition, data-driven companies currently show how various industries are able to profit from the massive generation of data. Several definitions have been proposed for the term “big data.” One of the widely recognized definitions used is the “4 Vs” definition. The definition was first proposed by Douglas Laney and encompasses “3 Vs” which consist of volume, velocity, and variety (1,2). This definition was later extended by IBM to include the fourth “V” for veracity (3). However, the reported definitions of “big data” usually lack consistency and quantification.
Because of its potential value, data has been considered as the new oil (4,5). Textbooks and publications, social media, user-generated content, electronic health records, genomics, sensor networks, and many other types of data all form “big data” and contribute to its diversity and complexity. The remarkable increase in the amount of data can be attributed to advancements in data storage and innovative technologies (6). Almost 2.5 million new scientific papers are published annually (7). In addition, there were more than 15,000 PubMed-reported publications on “pharmaceutical sciences” in 2019 only (8).
Thus, “big data” in pharmaceutical sciences can be viewed as both a challenge and an opportunity. The evolution of artificial intelligence (AI), particularly machine learning technologies in which computers can “learn” and perform tasks, has improved the potential of using big data in pharmaceutical sciences. The scope of this review is specific to machine learning because, among all AI branches, machine learning is the most currently used AI technology in the field of pharmaceutical sciences. Other AI fields, such as natural language processing (NLP), expert systems, and robotics, are becoming very popular in many healthcare settings, such as in the diagnosis of diseases, patient monitoring, and robotic surgeries (9,10). These methods, however, have not yet received as much attention as machine learning in pharmaceutical sciences settings. The aim of this review is to summarize the past, present, and potential future impacts of machine learning on different areas of pharmaceutical sciences, including drug design and discovery, preformulation, and formulation. This review covers different machine learning algorithms that are commonly implemented in different areas of pharmaceutical sciences, with a special emphasis on the use of artificial neural networks (ANNs). Notably, compared with other machine learning methods, ANNs have displayed superior performance in various pharmaceutical settings, as will be discussed in the following sections.
PRINCIPLES OF AI AND MACHINE LEARNING
Despite its long history, as will be discussed below, there is still no standard definition of AI. However, mimicking human intelligence using computer systems is the basic concept of AI. The physiology and function of neurons in the brain inspired Warren McCulloch and Walter Pitts (1943) to propose a computational model of artificial neurons. Similar to human neurons, artificial neurons are characterized by being “on” or “off” in response to sufficient stimulation from neighboring neurons (11). The term “artificial intelligence” was officially introduced by John McCarthy at the Dartmouth conference in the summer of 1956 (12). Since then, AI has had cycles of success as well as so-called “AI winters” (13). Recently, AI has significantly advanced and gained increasing interest in a wide range of fields, including healthcare (14), engineering (15), and transportation (16). This increased focus on AI applications has been fueled by the growing availability of big data in healthcare and the rapid advancement of numerous analytical techniques (10).
Machine learning is a popular AI technique (Fig. 1) whereby computers can accurately adapt or modify their actions (e.g., making predictions). Machine learning algorithms can be classified into two major categories: supervised learning and unsupervised learning (17). In supervised learning, the algorithm uses generalizations to respond appropriately to a set of training examples. Training examples are input-output data that are provided in the dataset to be learned. Because the output data here are known to be the correct responses (or correct answers), they are termed as “targets.” The machine learning model eventually aims to predict an output that is closer to the target.
Fig. 1.

Schematic showing the relationship between AI, machine learning, and artificial neural networks (left), and a number of applications of artificial neural networks in pharmaceutical sciences (right)
Examples of supervised machine learning methods include regression analysis, support vector machines (SVMs), random forests (RF), and ANNs. Unsupervised learning is based on feature extraction methods in which no examples are provided (17), such as principal component analysis (PCA). Some supervised machine learning models may also support unsupervised machine learning models such as SVMs and ANNs (18). Table I shows a comparison of several machine learning methods commonly used in pharmaceutical research. Linear regression, ANNs, KNN, SVM, DT, and RF are common machine learning methods used in pharmaceutical sciences; PCA is considered as an unsupervised dimensionality reduction technique usually integrated into computing transformation of unlabeled data to find a lower-dimensional set of axes (12). Although PCT may be considered as a statistical technique used to analyze multidimensional data, it is usually incorporated as a preprocessing tool in machine learning (19).
Table I.
Comparison of Different Machine Learning Methods Commonly Used in Pharmaceutical Research* (12,17,25–33)
| Machine learning method | Learning algorithm | Machine learning model | Learning problem | Dataset size | Advantages | Disadvantages/limitations |
|---|---|---|---|---|---|---|
| Linear regression | Supervised | Parametric | Regression | Varies (25) | Easy implementation | Applicable only for linear modeling |
| Artificial Neural Networks (ANNs) | Supervised and unsupervised | Parametric | Classification and regression | Large | Modeling complex nonlinear relationships |
Overfitting/underfitting Relatively slow training time |
| K-Nearest Neighbor (KNN) | Supervised | Nonparametric | Classification and regression | Large (dependent on noise level in data) | Simple and easy to implement with single pre-defined parameter (i.e., the number of nearest neighbors) | Intolerant of noise |
| Support vector machine (SVM) | Supervised and unsupervised | Nonparametric | Classification and regression | Small |
Able to represent complex functions Offer resistant to overfitting |
Relatively slow training time High complexity of the model Long computing time |
| Decision tree (DT) | Supervised | Nonparametric | Classification and regression | small |
Easier than RF Can deal with noisy and missing data Fast |
Unstable. Its performance can be affected by slight variations in the training data |
| Random forest (RF) | Supervised | Nonparametric | Classification and regression | large | Similar to decision trees in addition to its capability to overcome overfitting | Complex |
| Principal Component Analysis (PCA) | Unsupervised | Feature extraction | Classification and regression | Large | Reduces the dimensionality of multivariate data while maintaining the relevant information in the original dataset | It assumes Gaussian distribution of data which might limit their use if data are not normally distributed such as gene expression data |
| LightGBM** | Supervised | Nonparametric | Classification and regression | Large |
Fast training speed High efficiency and accuracy |
Sensitive to overfitting |
*Note that this table presents a general comparison of the different machine learning methods commonly used in drug research and development. Different methods may have relative variations compared with other methods
**LightGBM is an emerging machine learning method recently been implemented in pharmaceutical sciences
In addition, there are other machine learning methods used in pharmaceutical sciences such as the fuzzy logic algorithm. In this method, reasoning with logical expressions is used to describe membership in fuzzy sets (12). This method has the advantage of eliminating the need for expert knowledge regarding the system, considers the noise in the data, and produces easily interpretable predictions (20). Fuzzy logic algorithm provided good prediction models for analyzing gene expression data (20). Additionally, genetic algorithm (GA) is a population-based method commonly used as an optimization technique. This algorithm also offers the advantage of modeling nonlinear relationships. In pharmaceutical research, GA is mainly used in quantitative structure-activity relationship (QSAR) studies as a feature selection tool (21,22). Recently, there is an emergence of several novel machine learning applications in pharmaceutical settings using non-conventional machine learning techniques such as light gradient boosting machine (lightGBM). This method has offered numerous useful features as compared to the other classic machine learning methods as shown in Table I.
Furthermore, an emerging machine learning technique is the transfer learning. Transfer learning is based on reusing a pre-trained model in order to build a new, improved model to address the intended target (23). In transfer learning, a relatively large dataset size of the original model is an important determinant for optimum transfer learning performance. Important recent progress of using transfer learning has been achieved in the field of pharmaceutical sciences (24) as will be discussed in a following section.
Moreover, machine learning models can be classified into two categories: parametric and nonparametric models (Table I). Parametric models summarize data with a set of constant number of parameters (regardless of the number of training examples), whereas nonparametric models are dependent on the number of parameters and therefore on the number of training examples (12). Table I summarizes the common parametric and nonparametric machine learning methods encountered in different drug research and development studies. Note that each of these machine learning methods may have further subtypes, and a general comparison among these models can be unfair. For example, although certain machine learning methods may require large datasets, an optimum dataset size is usually lacking. The reader is encouraged to refer to the cited references for details. Additionally, no machine learning method is generally considered superior to all others, and each problem (classification or regression) should be addressed individually.
Artificial Neural Networks
ANNs are biologically inspired computational models that mimic the brain’s ability to learn by example (Fig. 2). Our brains consist of billions of processing units called neurons. These neurons are fully interconnected through an enormous number of synapses that connect one neuron to another (34). A biological neuron consists of a cell body that contains a nucleus and controls cell activities, dendrites, which compose the fine threads among neurons and carry the information to the cell, and axons, which consist of one long thread that transports information to the next cell (34).
Fig. 2.
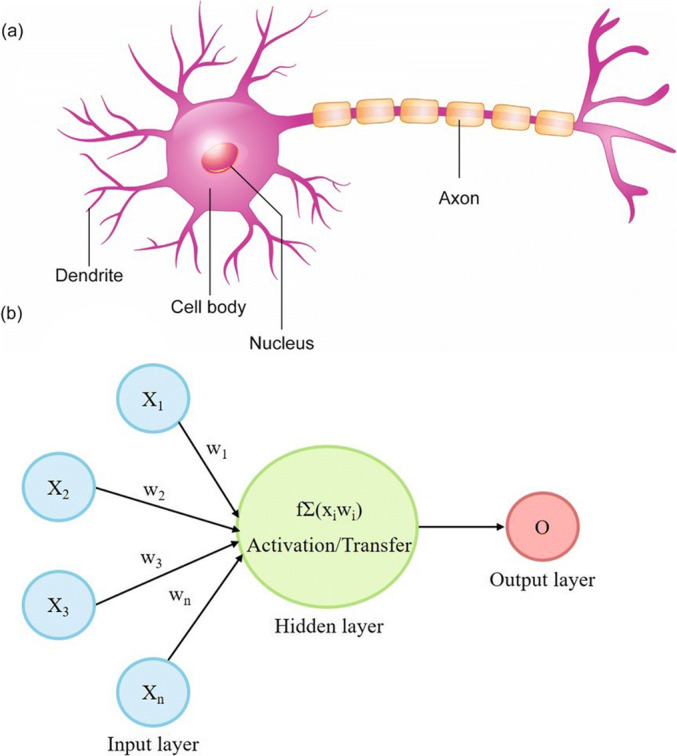
Schematic of a typical biological neuron (a), and an artificial neural network (b)
Similar to human neurons, ANNs consist of artificial neurons or processing elements (PEs) that are connected via coefficients (weights) (34). A typical ANN (Fig. 2) consists of three main structural components: input, hidden, and output layers. The first layer of an artificial neuron is the input layer, which corresponds to the dendrites of the biological neuron and transfers information to the next layer. The following layer is the hidden layer, which is the middle layer between the input layer and the output layer. The hidden layer connects these two layers through certain coefficients (weights). Each hidden layer consists of a number of neurons (also called nodes). The choice of the number of neurons in the hidden layer of ANNs is generally achieved by a trial-and-error approach (35). Although there is no definite number of neurons to be used, using too few neurons in the hidden layer may result in a reduction in the ANN learning ability, whereas too many neurons in the hidden layer may result in the memorization or overfitting of the training data, ultimately decreasing the generalization ability of the ANN. Thus, the number of hidden neurons in the neural network that will give the highest correlation coefficient (r2) and lowest error (i.e., the minimum difference between observed and predicted values) should be selected as the optimal ANN. The final layer of an artificial neuron is the output layer, which consists of the outputs (targets). Moreover, by examining the magnitude of the ANN connection weights, ANNs can provide quantitative estimates of the relative importance of the input variables for the output in question (36). Figure 2 illustrates a schematic representation of a typical biological neuron (a) and an ANN (b).
The process of designing a neural network that can learn to ultimately solve a problem occurs through iterative use of examples with known answers (targets). This process is called learning or training. The learning/training process as illustrated in Fig. 2 starts with receiving signals (inputs) from the input layer. These inputs are multiplied by connection weights and summed in the hidden layer. The results are then sent to the output layer through a transfer function. Several activation functions are available including identity, logistic, tanh, and exponential functions (17,37). The sigmoid function is a commonly used activation function in pharmaceutical research. During neural network learning, a process called “error back-propagation” is usually implemented (38). In back-propagation, the weights are adjusted to minimize the error between the calculated (predicted) output and the observed (target) output.
ANNs are particularly powerful in modeling nonlinear relationships and can make highly accurate predictions due to their ability to analyze complex data primarily based on generalization and pattern recognition (39,40). Nevertheless, some challenges with using ANNs can be encountered, such as trapping at local minima, controlling noise, and overfitting/underfitting. To avoid local minima and control noise, a time-invariant noise algorithm (TINA) can be implemented (41). In addition, there are various ways to overcome overfitting/underfitting problems, including splitting the data into training and validation sets (42). This technique can reduce overfitting. Moreover, stopping the training process at the right point can also prevent both overfitting and underfitting (17). Figure 3 illustrates the optimum stopping point for ANN training.
Fig. 3.
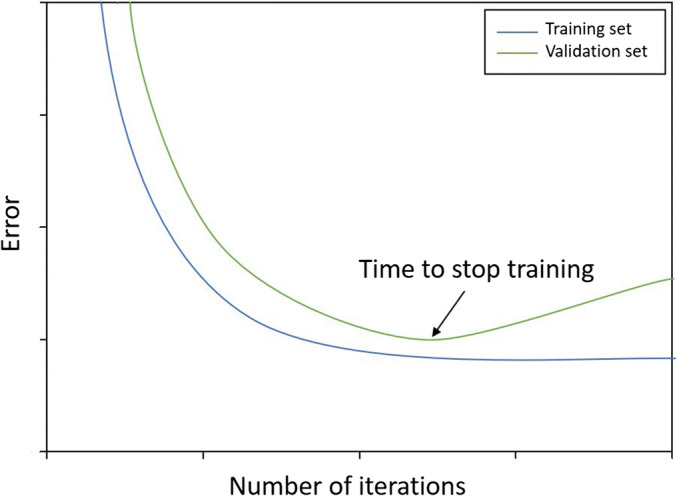
An illustration of the effect of overfitting/underfitting of the data on the training and validation error curves showing the optimum point where the training/learning process of ANNs should stop
From ANNs to Deep Learning
DL is a machine learning technique that is also a representation learning method (43). The state-of-the-art of DL methods includes recent advances in neural networks. The major difference between ANNs and DL is that DL includes larger numbers of hidden layers (usually more than three), and each layer comprises many more nodes. Therefore, DL uses multiple levels of representations that can ultimately learn very complex functions. Generally, DL requires very large training sets, which may limit the use of such methods. There are different types of neural network architectures in DL, including convolutional neural networks (CNNs), recurrent neural networks (RNNs), and fully connected feed-forward networks, which have been comprehensively discussed elsewhere (44). DL has become very popular and has gained interest in diverse research areas of pharmaceutical research such as in pharmaceutical formulation development (45), drug discovery (46), and drug repurposing (47). Their predictability and generalization performance are generally better than that of other machine learning methods, such as SVMs and RFs (45). This can be due to the improvements in algorithms, computers, and the availability of large datasets. Specific DL applications in pharmaceutical sciences is an interesting topic for future reviews.
APPLICATIONS OF MACHINE LEARNING IN PHARMACEUTICAL SCIENCES
Machine learning has been utilized in different pharmaceutical applications from the early stages of drug discovery to the late phases of drug development. The following sections present three major areas of pharmaceutical sciences that have witnessed a considerable use of ANNs together with a number of other machine learning methods. These studies can be categorized into drug design and discovery, preformulation, and formulation studies.
Machine Learning in Drug Design and Discovery
Drug discovery accounts for a significant share of the machine learning applications in pharmaceutical sciences, mainly due to the use of high-throughput screening, combinatorial chemistry, and computer-aided drug design (45,48). One of the early areas in which ANNs were applied is QSAR studies (49–51). The QSAR approach correlates the physicochemical properties of a compound with the corresponding chemical or biological activities (52,53). The most commonly used physicochemical properties in QSAR studies include molecular weight, partition coefficient (logP), and hydrogen bonding capacity. Because QSAR studies usually involve complex and nonlinear characteristics, ANNs were among the best available QSAR modeling tools. Additionally, due to their usefulness and success, the importance of neural networks has continued to grow in drug discovery with the rapid rise of QSAR studies based on ANNs (54). Table II summarizes several input-output data used to build various machine learning models in different QSAR studies.
Table II.
Summarization of Input-Output Data Used to Build Various Pharmaceutical Machine Learning Models in Different QSAR Studies
| Machine learning method* | Learning algorithm | Dataset size | Inputs/descriptors | Output(s)/purpose | Reference |
|---|---|---|---|---|---|
| ANN Linear regression | Supervised | 30 |
5 molecular descriptors: - water–accessible surface area - polar surface area - maximal electrostatic potentials - ovality - hydrophobicity (logP) |
Prediction of tumor specificity of chemotherapeutic agents | (55) |
|
ANN SVM DT RF |
Supervised | 89 |
10 molecular descriptors related to: - hydrophobicity - electronic features - topological features - protein-inhibitor interactions |
Prediction of activity of HIV inhibitors | (56) |
|
RBFNN KNN SVM RF |
Unsupervised and supervised for RBFNN model and the other models, respectively |
Twodatasets: 128 (Phenol dataset) 105 (ROCK dataset) |
- For phenol datasets: 6 molecular descriptors (related 2D and 3D descriptors such as log P). - For ROCK datasets: 6 molecular descriptors (related 2D descriptors such as ring count) |
Prediction of the biological activity of various phenols and Rho kinase (ROCK) inhibitors. | (57) |
|
ANN Linear partial least squares (linear statistical method) |
Supervised | 36 |
4 molecular descriptors: - minimum bond dissociation enthalpy - electron transfer enthalpy - proton affinity - hydration energy |
Prediction of antioxidant activity of flavonoids | (58) |
|
RF ANN |
Supervised | 91 |
166 molecular descriptors including: - structure - topology - molecular connectivity index - geometric descriptors |
Prediction of the carcinogenicity of polycyclic aromatic hydrocarbons | (59) |
| ANN | Supervised | 33 | 6 molecular descriptors (related 2D and 3D descriptors) | Prediction of anti-malarial activity of imidazolopiperazine compounds | (60) |
|
ANN SVM |
Supervised | 639 |
341 molecular descriptors related to: - simple constitutional - topological indices - electrotopological state indices - charge-based - hydrogen-bonding descriptors |
Prediction of nephrotoxicity of traditional Chinese medicines ingredients | (61) |
*The top-ranked machine learning methods in each of these studies demonstrated better predictive ability than the other machine learning methods tested. ANN artificial neural network, SVM support vector machine, DT decision tree, RF random forest, KNN K-nearest neighbor, RBFNN radial basis function neural network
Machine Learning in Pharmaceutical Preformulation
Preformulation is the stage of drug development in which the physicochemical properties of a drug substance are assessed. Determining the physicochemical properties of a drug substance is very important because it governs various parameters, such as its solubility, stability, interaction with excipients, and ultimately, bioavailability (62).
Determining the aqueous solubility of a new drug substance is an essential first step in preformulation. Any drug to be absorbed must possess a certain degree of water solubility. This is true for oral, parenteral, ophthalmic, topical, and other routes of administration. Various solubilization techniques are used to improve the aqueous solubilities of drug substances, such as using surfactant, complexation, salt formation, using hydrotropes, or forming cocrystals (36,63,64).
The in silico prediction of the aqueous solubility of drug substances has gained significant interest using different computational approaches, such as molecular dynamics simulations (65) and machine learning techniques (36). For example, Damiati et al. (2017) developed a machine learning application using ANNs to predict the solubility enhancement effect of several hydrotrope molecules. The input data consisted of experimental data together with 10 physicochemical properties (used as descriptors) related to 10 hydrotrope molecules at different hydrotrope concentrations. The physicochemical properties included logP, melting point, and hydrogen bonding capacity. The developed ANN model was subsequently used to predict the solubility enhancement of another 16 potential hydrotrope molecules from an external dataset. The trained model was also able to identify new prospective hydrotropes for the drug molecule. In addition to providing accurate predictions, by determination of the connection weights, the developed ANN was able to provide a quantitative assessment of the relative importance of various physicochemical properties that are required for a good hydrotrope (36). The reported use of ANNs in the prediction of solubility enhancements for drug substances and their successful use in other solubility applications in various research areas (66,67) are encouraging for further exploration of their potential uses in more pharmaceutical preformulation research.
Moreover, based on the pharmacokinetic profile of a drug substance, a suitable pharmaceutical formulation can be designed. As stated earlier, important progress has been achieved in utilizing the emerging machine learning technique of transfer learning in pharmaceutical settings. Ye et al. (2019) developed an integrated transfer learning and multitask learning approach for the prediction of four pharmacokinetic-related properties, namely oral bioavailability (BA), plasma protein binding rate (PPBR), apparent volume of distribution at steady-state (VDss), and elimination half-life (HL). Eight molecular descriptors have been used for 1104 approved drug molecules. Descriptors included molecular weight, hydrogen bond donor count, hydrogen bond acceptor count, rotatable bond count, topological polar surface area, heavy atom count, complexity, and covalently bonded unit count. The developed model showed good performance and generalization ability compared to other conventional machine learning techniques including partial least-squares regression (PLSR), SVM, ANNs, RF, and KNN (24). In preformulation studies, transfer learning is a promising machine learning approach for further exploration.
Machine Learning in Pharmaceutical Formulations
Another stage of drug development is the formulation of pure drug substances into drug products to be administered by patients. ANNs have gained significant interest in this area and became the most popular machine learning tool in pharmaceutical formulation prediction (45). Table III summarizes numerous pharmaceutical researches that have been performed utilizing ANNs (as the only method used or as an approach that outperformed other machine learning methods) in the area of pharmaceutical formulation development in the past 20 years. This table compares these studies from different machine learning aspects including the diverse input-output data used, amount of data (dataset sizes), input variables, and purpose(s). Notably, a large number of these studies have utilized ANNs for the development and optimization of formulations and the prediction of formulation- and process-related factors associated with different parameters, such as drug dissolution and release. Additionally, the optimization of formulations (including the optimization of ingredients and/or operating conditions) using machine learning tools—particularly ANNs—has provided considerable success and displayed great promise for future applications that usually require fast and efficient manufacturing.
Table III.
Summarization of Input-Output Data Used to Build Various ANN Models in Different Pharmaceutical Formulation Studies
| Dataset size | Inputs/variables | Output(s) | Purpose | Reference |
|---|---|---|---|---|
| 125 |
19 variables related to: - the composition of the formulations - the processing conditions |
- Time taken for 10% of the drug to be released - Time taken for 90% of the drug to be released |
Prediction of the most important formulation and processing variables contributing to the in vitro dissolution of sustained-release (SR) minitablets | (70) |
|
Two datasets: 154 (for synthetic samples) 169 (for pharmaceutical samples) |
- 5 principle components for synthetic samples - 6 principle components for pharmaceutical samples |
Concentrations of 3 vitamins in synthetic and pharmaceutical samples | Prediction of vitamins in synthetic and pharmaceutical samples | (71) |
| 30 |
3 input variables: - acid concentration - acid solution to chitin ratio - reaction time |
Percentage production yield of glucosamine | Prediction of glucosamine production yield from chitin under various reaction conditions | (72) |
| 180 |
4 input variables related to different formula ingredients: - Methocel® K100M - xanthan gum - Carbopol® 974P - Surelease® |
In vitro dissolution time profiles at six different sampling times | Development and optimization of sustained-release salbutamol sulfate formulation | (73) |
| 300 |
5 input variables related to 5 active ingredients and excipients (three physical–chemical properties of active ingredients in addition to two formulation factors): - solubility - mean particle size - specific surface area - the weight ratios of microcrystalline cellulose - the weight ratios of magnesium stearate |
Tablet tensile strength and disintegration time before and after accelerated test | Prediction of responses to differences in quantities of excipients and physical–chemical properties of active ingredients in tablets | (74) |
| 327 |
6 input variables related to 14 active ingredients: - melting point - solubility - specific surface area - mean particle size - size distribution - contents of APIs |
- Tablet tensile strength - Disintegration time |
Prediction of the contribution of different physicochemical properties of APIs to tablet properties | (75) |
| 15 |
3 formulation factors: - weight ratio of drug to lipid - the concentration of polymer - the concentration of surfactant |
- Drug loading efficiency - Mean particle size |
Optimization of controlled-release nanoparticle formulation | (76) |
| 45 |
3 input variables: - chitosan (Cs) concentration - potasodium tripolyphosphate (TPP) concentration - mass ratio of Cs and TPP |
- Nanoparticle size - Percentage yield |
Optimization of formulation parameters of chitosan-tripolyphosphate nanoparticles | (77) |
| 43 |
7 input variables: - alginate percentage - concentration of CaCl2 solution in the emulsion - percentage of Tween™ 85 in the emulsion - percentage of Tween™ 85 in the receptor bath - flow rates of alginate - flow rates of emulsion - frequency of vibration |
- Shape - Oil content - Oil distribution |
Optimization of encapsulation of active pharmaceutical ingredients (API) for efficient delivery of hydrophobic compounds | (78) |
| 20 |
3 input variables: - the amounts of drug (pilocarpine hydrochloride) - the amounts of bile salt (sodium deoxycholate) - the amounts of water |
Entrapment efficiency | Optimization of ocular formulation of flexible nano-liposomes containing pilocarpine hydrochloride | (79) |
| 16 |
3 input variables: - amount of oil - amount of surfactant - amount of co-surfactant |
Minimal globule size | Optimization of self-emulsifying drug delivery system | (80) |
| 8 |
2 formulation variables: - ratio of carrier to coating - type of solubilizing agent |
Amount of API resealed in 10 min and 30 min | Development of a new liquisolid formulation | (81) |
| 160 | 160 NIR and Raman spectral data of each of intact tablets | Dissolution of the tablets | Prediction of the in vitro dissolution of pharmaceutical tablets | (82) |
| 29 |
4 formulation and process variables: - microcrystalline cellulose concentration - sodium starch glycolate concentration - spheronization time - extrusion speed |
- Drug release (at 15 min, 30 min, 45 min, and 60 min) - Aspect ratio - Yield |
Prediction of the effects of formulation and process variables on drug release | (83) |
| 144 | Amino acid composition of each monoclonal antibody and different formulation conditions (i.e., pH and salt concentrations) |
- Melting temperature - Aggregation onset - Temperature - Interaction parameter |
Prediction of biophysical properties of therapeutic monoclonal antibodies | (84) |
| 32 |
4 input variables: - concentration of shell material - concentration of core material - type of shell material - type of core material |
- Tensile strength - Brittleness index |
Prediction of powder compact ability of tablets using core/shell technique | (85) |
| 646 |
24 variables related to: - formulation (including molecular weight, melting point, hydrogen bonding for both drug and polymer) - experimental conditions (including temperature, relative humidity, and storage time) |
Stability results | Prediction of the physical stability of solid dispersions | (86) |
Recently, non-traditional machine learning techniques have been utilized in the development of in silico predictive models in pharmaceutical formulation. LightGBM has recently shown high potential predictive ability compared to conventional machine learning methods in pharmaceutical formulation researches. Zhao et al. (2019) compared lightGBM, RF, and DL for the prediction of complexation free energy between cyclodextrins (CDs) and guest molecules with a dataset consisting of 3000 data points. Over 30 numerous descriptors related to the guest molecule, CD, and experimental conditions have been implemented in designing the machine learning models. LightGBM showed better prediction performance compared to the other models including RF and DL (33). Gao et al. (2020) also implemented the lightGBM method for prediction of complexation performance of 341 drugs/phospholipid complex formulations described by over 40 molecular descriptors related to the properties of the drugs, solvents, and experimental conditions. Compared with other conventional machine learning techniques such as SVM and DT, lightGBM model showed the best predictive performance for predicting drug/phospholipid complexation (68). Also, in 2020, He and co-workers used lightGBM to predict the particle size and polydispersity index (PDI) of nanocrystals prepared by different methods. The dataset consisted of 910 experimental size data and 341 PDI data under various conditions and using various API-, stabilizer-, and nanocrystal preparation-related descriptors. The prediction performance of lightGBM was better than that obtained from several classic machine learning methods including deep neural networks (DNN), SVM, and DT for both size and PDI datasets (69). In all these lightGBM studies, it has been proved that lightGBM is a powerful and promising machine learning technique that can be further explored in the future for various pharmaceutical applications not only for its ability to provide accurate predictions but also due to its capability to provide an informative assessment of the importance of the input descriptors.
CURRENT AND FUTURE PROSPECTS
Benefits, Risks, and Efforts
In terms of applying AI and machine learning technologies in common day-to-day pharma needs, a number of aspects are to be considered including the benefits, risks, and efforts.
The benefits of machine learning applications in pharmaceutical sciences are evident. This is true for both the classic machine learning tools such as ANNs as well as for the newly emerging tools such as lightGBM. Accelerating advances across the entire spectrum of the development of drug substances and drug products by dramatically reducing the timeline in unnecessary attempts is a substantial benefit of AI in pharmaceutical settings. This may not only allow for improving outcomes in less time, but it also can help to find more efficient solutions in order to sustain manufacturing efficiency and rapid throughput. In addition, depending upon the therapeutic class, the problem of high drug attrition rates (87) can be reduced. Thus, the high costs associated with drug research and development processes can be significantly reduced if performed in silico using data digitalization and reduced extensive laboratory testing.
For instance, considering a real pharmaceutical problem in which substantial efforts are needed is the problem of low aqueous solubilities of drugs. It is estimated that approximately 90% of drug candidates in research and development pipelines are poorly water-soluble (88). Considering that only small quantities (< 50 mg) of a drug substance exist in early preformulation (62), determining the baseline solubility and subsequently the optimum solubilization technique for each drug substance may require extensive screening and laboratory work, as well as substantial resources. If well-trained and well-validated machine learning models can be incorporated in such settings, only drug candidates that show positive results in silico may then undergo laboratory testing. Thus, successful drug candidates can ultimately reach the intended patient in less time and with less material waste.
Based on the type of data, there is an important advantage of machine learning is that no restrictions are encountered while implementing machine learning algorithms. Different types of data, including binary classification, multiple classes, and continuous data all can be modeled and analyzed by machine learning. Moreover, machine learning models may be used individually or in combination. Compared with traditional statistical models, a number of machine learning technologies (e.g., ANNs) offer the advantage of modeling complex and nonlinear relationships that are frequently encountered in pharmaceutical sciences. Traditional models are usually used to find inference about relationships in the data, whereas machine learning models are designed to model complex relationships which can ultimately produce accurate predictions. For example, the nature of the solubilization effect using hydrotropes is complex, nonlinear, and do not follow a constant pattern (36). Traditional statistical tool would not be able to provide accurate predictions for the solubilizing effect of these systems, whereas machine learning models not only were able to produce highly accurate predictions, but also proved to be powerful tools that can provide useful insights into the relative importance of the different input features in determining the outputs by interrogation of the connection weights. In addition, the machine learning approach also provided valuable insights that eventually lead to the identification of new prospective solubilizing compounds (36).
The quality of data is one of the challenges that must be considered when using AI and machine learning in the pharmaceutical sciences. Quality encompasses the consistency, reliability, accuracy, availability, and accessibility of the data. The dataset size also should be considered. Small dataset size can be modeled using simple machine learning methods; if the dataset size is large and more complex to be modeled using simple machine learning methods, the advanced ANN models based on DL approach can offer a potential solution. Other challenges that must also be considered include the training/learning time, underfitting, and overfitting. Therefore, the risk of applying unreliable machine learning models can be eliminated if these challenges were appropriately considered, and well-trained and well-validated machine learning models were carefully designed. Hence, digitalizing pharmaceutical data using AI may require domain experience and the ability to train algorithms; each machine learning method should be implemented “task specifically.”
AI and Pharma Industry
The pharmaceutical industry would greatly benefit from the use of AI and machine learning, due to its wide range of applications as discussed in this review. From proof of concept to product evaluation and marketing, AI can be applied to nearly every aspect of drug development.
With the long-standing figures of an average of $2.6 billion and over 10 years to develop new medicines (89), AI can offer a substantial investment to hasten and improve this process. In the last 10 years, there is a remarkable growing number of pharmaceutical companies and startups using AI in drug research and development. A number of pharma companies either collaborated with or acquired AI technologies such as Novartis and Pfizer with IBM Watson (90). Mak and Pichika (2019) provided a comprehensive list of AI and pharmaceutical companies and the corresponding collaboration areas in drug development such as drug repurposing, personalized medicine, and drug discovery (91). Other areas where pharma companies have been actively investigating in AI applications include process automation, robotic manufacturing, and targeted marketing (90). Investing in data management and AI power can sustain manufacturing efficiency and rapid throughput of data digitalization which is powered by advancing algorithms as well as the availability of the diverse, complex, and large amount of data. AI may, therefore, improve decision-making and eventually create new and better medicines. Nonetheless, it has been reported that AI has not yet influenced the pharma industry significantly due to several reasons/challenges suggested by Henstock (2019) including data management (e.g., managing diversity and large amount of data), finding solutions for a large number of problems, insufficient skillsets, shifting towards alternatives to traditional scientific approaches, and lack of investments. To overcome these challenges, the author also suggested internal investment in data management and AI talent (90).
Mary and co-workers in 2019 conducted a survey-based study to clarify and understand the adoption and effect of AI in pharmaceutical and biotechnology companies. Across 217 organizations, a number of important AI activities have been identified including the use of AI for patient selection and recruitment for clinical trials, in addition to identification of medicinal products data gathering. Major factors for not utilizing AI technology have been identified including lack of skilled staff, safety, regulatory, and compliance concerns, and budget constraints (92).
Regulatory and Recommendation Insights
In terms of regulatory and recommendation insights, Food and Drug Administration (FDA) recently published a discussion paper “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) - Discussion Paper and Request for Feedback” which discusses the current approach made to subject software as a medical device driven by AI and machine learning to a premarket review in order to ensure safety and effectiveness. Several types of changes/modifications which may have an impact on users (including patients, healthcare professionals, and others) using these softwares have been reviewed. For example, changes/modifications related to re-changing the inputs, training with new data sets, and change in the AI/ML architecture. To ensure lifecycle safety and effectiveness from its premarket development to postmarked performance, FDA also proposed a total product lifecycle regulatory approach for AI/ML-Based SaMD (TPLC) to acquire evaluation and monitoring of a software product (93).
Beyond Traditional Applications
The growing success of machine learning technologies, particularly ANNs, in many pharmaceutical settings showed great potentials for the development of beyond traditional machine learning applications. This trend has already begun in areas such as drug and gene delivery. Therapeutic agents are often transported into the cell using special transporter systems such as cell-penetrating peptides (CPPs). The efficiency of CPPs is usually investigated and screened based on extensive laboratory work, which has recently been successfully performed in silico using ANNs. The developed CPPs/ANN model provided highly accurate predictions and informative assessments for 13 different input features (94). Additionally, drug repurposing also can highly benefit from these technologies (95).
At present, although the first AI-designed drug has not reached the market yet, there is an ongoing race to find a treatment for the current COVID-19 pandemic. AI plays an important role in the ongoing efforts by identifying potential molecules that could be used as anti-COVID-19 drugs. For example, Benevolent AI (96,97) reported the use of machine learning to identify drugs for COVID-19 in which clinical trials are already underway.
CONCLUSIONS
Digitalizing pharmaceutical sciences is a very promising area in which numerous AI and machine learning technologies can be discovered and effectively employed. The growing success of machine learning technologies in many pharmaceutical settings shows great potentials for the development of beyond traditional AI applications. In practice, the choice of the machine learning method to be implemented may depend on various factors, including the type of the data and the size of the dataset. Therefore, the choice of which machine learning method should be implemented can be considered task-specific. With a sufficient amount of carefully curated data, building high-value applications using advancing AI algorithms may become a common practice that has the potential to solve many challenges in drug research and development. It is likely that AI will flourish a new era of digital pharmaceutical sciences with efficient, fast, and economical solutions.
Compliance with Ethical Standards
Competing Interests
The author declares that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Laney D. 3D data management: controlling data volume, velocity and variety. META Group Res Note. 2001;6(70):1. [Google Scholar]
- 2.Chen M, Mao S, Liu Y. Big data: a survey. Mobile Netw Appl. 2014;19(2):171–209. [Google Scholar]
- 3.Heidari S, Alborzi M, Radfar R, Afsharkazemi MA, Rajabzadeh GA. Big data clustering with varied density based on MapReduce. J Big Data. 2019;6(1):77. [Google Scholar]
- 4.Javornik M, Nadoh N, Lange D. Data is the new oil. Towards user-centric transport in Europe: Springer; 2019. p. 295–308.
- 5.Zeng ML. Smart data for digital humanities. J Data Inf Sci. 2017;2(1):1–12. [Google Scholar]
- 6.Hashem IAT, Yaqoob I, Anuar NB, Mokhtar S, Gani A, Khan SU. The rise of “big data” on cloud computing: review and open research issues. Inf Syst. 2015;47:98–115. [Google Scholar]
- 7.Ware M, Mabe M. The STM report: an overview of scientific and scholarly journal publishing. 2015.
- 8.PubMed. Database resources of the national center for biotechnology information. Bethesda (MD): U.S. National Library of Medicine. Available from: https://www.ncbi.nlm.nih.gov/pubmed/?term=pharmaceutical+sciences. Data accessed August 17, 2019.
- 9.Wakabayashi G, Sasaki A, Nishizuka S, Furukawa T, Kitajima M. Our initial experience with robotic hepato-biliary-pancreatic surgery. J Hepato-Biliary-Pancreat Sci. 2011;18(4):481–487. doi: 10.1007/s00534-011-0388-3. [DOI] [PubMed] [Google Scholar]
- 10.Jiang F, Jiang Y, Zhi H, Dong Y, Li H, Ma S, Wang Y, Dong Q, Shen H, Wang Y. Artificial intelligence in healthcare: past, present and future. Stroke Vasc Neurol. 2017;2(4):230–243. doi: 10.1136/svn-2017-000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCulloch WS, Pitts W. A logical calculus of the ideas immanent in nervous activity. Bull Math Biophysics. 1943;5(4):115–133. [PubMed] [Google Scholar]
- 12.Russell SJ, Norvig P. Artificial intelligence: a modern approach. 3rd ed: Pearson Education Limited; 2016.
- 13.Fast E, Horvitz E. Long-term trends in the public perception of artificial intelligence. Thirty-First AAAI Conference on Artificial Intelligence 2017.
- 14.Esteva A, Robicquet A, Ramsundar B, Kuleshov V, DePristo M, Chou K, Cui C, Corrado G, Thrun S, Dean J. A guide to deep learning in healthcare. Nat Med. 2019;25(1):24–29. doi: 10.1038/s41591-018-0316-z. [DOI] [PubMed] [Google Scholar]
- 15.Frank MR, Wang D, Cebrian M, Rahwan I. The evolution of citation graphs in artificial intelligence research. Nat Mach Intell. 2019;1(2):79–85. [Google Scholar]
- 16.Zhao J, Liang B, Chen Q. The key technology toward the self-driving car. Int J Intell Unmanned Syst. 2018;6(1):2–20. [Google Scholar]
- 17.Marsland S Machine learning: an algorithmic perspective. 2nd ed: CRC Press; 2015.
- 18.Lo Y-C, Rensi SE, Torng W, Altman RB. Machine learning in chemoinformatics and drug discovery. Drug Discov Today. 2018;23(8):1538–1546. doi: 10.1016/j.drudis.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kachrimanis K, Rontogianni M, Malamataris S. Simultaneous quantitative analysis of mebendazole polymorphs A–C in powder mixtures by DRIFTS spectroscopy and ANN modeling. J Pharm Biomed Anal. 2010;51(3):512–520. doi: 10.1016/j.jpba.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Woolf PJ, Wang Y. A fuzzy logic approach to analyzing gene expression data. Physiol Genomics. 2000;3(1):9–15. doi: 10.1152/physiolgenomics.2000.3.1.9. [DOI] [PubMed] [Google Scholar]
- 21.Serra A, Önlü S, Festa P, Fortino V, Greco D. MaNGA: a novel multi-niche multi-objective genetic algorithm for QSAR modelling. Bioinformatics. 2020;36(1):145–153. doi: 10.1093/bioinformatics/btz521. [DOI] [PubMed] [Google Scholar]
- 22.De P, Bhattacharyya D, Roy K. Exploration of nitroimidazoles as radiosensitizers: application of multilayered feature selection approach in QSAR modeling. Struct Chem. 2020:1–13.
- 23.Li X, Fourches D. Inductive transfer learning for molecular activity prediction: next-gen QSAR models with MolPMoFiT. J Cheminformatics. 2020;12:1–15. doi: 10.1186/s13321-020-00430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye Z, Yang Y, Li X, Cao D, Ouyang D. An integrated transfer learning and multitask learning approach for pharmacokinetic parameter prediction. Mol Pharm. 2018;16(2):533–541. doi: 10.1021/acs.molpharmaceut.8b00816. [DOI] [PubMed] [Google Scholar]
- 25.Knofczynski GT, Mundfrom D. Sample sizes when using multiple linear regression for prediction. Educ Psychol Meas. 2008;68(3):431–442. [Google Scholar]
- 26.Ougiaroglou S, Evangelidis G, editors. A simple noise-tolerant abstraction algorithm for fast k-nn classification. International Conference on Hybrid Artificial Intelligence Systems; 2012: Springer.
- 27.Zhao Y, Zhang Y. Comparison of decision tree methods for finding active objects. Adv Space Res. 2008;41(12):1955–1959. [Google Scholar]
- 28.Mogensen UB, Ishwaran H, Gerds TA. Evaluating random forests for survival analysis using prediction error curves. J Stat Softw. 2012;50(11):1–23. doi: 10.18637/jss.v050.i11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bengio Y, Delalleau O, Simard C. Decision trees do not generalize to new variations. Comput Intell. 2010;26(4):449–467. [Google Scholar]
- 30.Yao F, Coquery J, Lê Cao K-A. Independent principal component analysis for biologically meaningful dimension reduction of large biological data sets. BMC Bioinformatics. 2012;13(1):24. doi: 10.1186/1471-2105-13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali J, Khan R, Ahmad N, Maqsood I. Random forests and decision trees. Int J Comp Sci Issues (IJCSI) 2012;9(5):272. [Google Scholar]
- 32.Gertrudes J, Maltarollo VG, Silva R, Oliveira PR, Honorio KM, Da Silva A. Machine learning techniques and drug design. Curr Med Chem. 2012;19(25):4289–4297. doi: 10.2174/092986712802884259. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Q, Ye Z, Su Y, Ouyang D. Predicting complexation performance between cyclodextrins and guest molecules by integrated machine learning and molecular modeling techniques. Acta Pharm Sin B. 2019;9(6):1241–1252. doi: 10.1016/j.apsb.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agatonovic-Kustrin S, Beresford R. Basic concepts of artificial neural network (ANN) modeling and its application in pharmaceutical research. J Pharm Biomed Anal. 2000;22(5):717–727. doi: 10.1016/s0731-7085(99)00272-1. [DOI] [PubMed] [Google Scholar]
- 35.Shin-ike K. A two phase method for determining the number of neurons in the hidden layer of a 3-layer neural network. Proc SICE Ann Conf. 2010;2010:238–242. [Google Scholar]
- 36.Damiati SA, Martini LG, Smith NW, Lawrence MJ, Barlow DJ. Application of machine learning in prediction of hydrotrope-enhanced solubilisation of indomethacin. Int J Pharm. 2017;530(1–2):99–106. doi: 10.1016/j.ijpharm.2017.07.048. [DOI] [PubMed] [Google Scholar]
- 37.Statistica®. Help documentations. 2017 TIBCO Software Inc. (17JUL2018).
- 38.Rumelhart DE, Hinton GE, Williams RJ. Learning representations by back-propagating errors. Cogn Model. 1988;5(3):1. [Google Scholar]
- 39.Sutariya V, Groshev A, Sadana P, Bhatia D, Pathak Y. Artificial neural network in drug delivery and pharmaceutical research. Open Bioinform J. 2013;7:49–62. [Google Scholar]
- 40.Krogh A. What are artificial neural networks? Nat Biotechnol. 2008;26(2):195–197. doi: 10.1038/nbt1386. [DOI] [PubMed] [Google Scholar]
- 41.Burton RM, Jr, Mpitsos GJ. Event-dependent control of noise enhances learning in neural networks. Neural Netw. 1992;5(4):627–637. [Google Scholar]
- 42.Nazir J, Barlow DJ, Lawrence MJ, Richardson CJ, Shrubb I. Artificial neural network prediction of aerosol deposition in human lungs. Pharm Res. 2002;19(8):1130–1136. doi: 10.1023/a:1019889907976. [DOI] [PubMed] [Google Scholar]
- 43.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 44.Chen H, Engkvist O, Wang Y, Olivecrona M, Blaschke T. The rise of deep learning in drug discovery. Drug Discov Today. 2018;23(6):1241–1250. doi: 10.1016/j.drudis.2018.01.039. [DOI] [PubMed] [Google Scholar]
- 45.Yang Y, Ye Z, Su Y, Zhao Q, Li X, Ouyang D. Deep learning for in vitro prediction of pharmaceutical formulations. Acta Pharm Sin B. 2019;9(1):177–185. doi: 10.1016/j.apsb.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep neural nets as a method for quantitative structure–activity relationships. J Chem Inf Model. 2015;55(2):263–274. doi: 10.1021/ci500747n. [DOI] [PubMed] [Google Scholar]
- 47.Aliper A, Plis S, Artemov A, Ulloa A, Mamoshina P, Zhavoronkov A. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharm. 2016;13(7):2524–2530. doi: 10.1021/acs.molpharmaceut.6b00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan HS, Shan H, Dahoun T, Vogel H, Yuan S. Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci. 2019;40:801. doi: 10.1016/j.tips.2019.07.013. [DOI] [PubMed] [Google Scholar]
- 49.Aoyama T, Suzuki Y, Ichikawa H. Neural networks applied to structure-activity relationships. J Med Chem. 1990;33(3):905–908. doi: 10.1021/jm00165a004. [DOI] [PubMed] [Google Scholar]
- 50.Hirst JD, Sternberg MJ. Prediction of structural and functional features of protein and nucleic acid sequences by artificial neural networks. Biochemistry. 1992;31(32):7211–7218. doi: 10.1021/bi00147a001. [DOI] [PubMed] [Google Scholar]
- 51.Aoyama T, Ichikawa H. Basic operating characteristics of neural networks when applied to structure-activity studies. Chem Pharm Bull. 1991;39(2):358–366. [Google Scholar]
- 52.Liu G, Yang X, Zhong H. Molecular design of flotation collectors: a recent progress. Adv Colloid Interf Sci. 2017;246:181–195. doi: 10.1016/j.cis.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 53.Hansch C, Maloney PP, Fujita T, Muir RM. Correlation of biological activity of phenoxyacetic acids with Hammett substituent constants and partition coefficients. Nature. 1962;194(4824):178–180. [Google Scholar]
- 54.Niculescu SP. Artificial neural networks and genetic algorithms in QSAR. J Mol Struct THEOCHEM. 2003;622(1–2):71–83. [Google Scholar]
- 55.Uesawa Y, Mohri K, Kawase M, Ishihara M, Sakagami H. Quantitative structure–activity relationship (QSAR) analysis of tumor-specificity of 1, 2, 3, 4-tetrahydroisoquinoline derivatives. Anticancer Res. 2011;31(12):4231–4238. [PubMed] [Google Scholar]
- 56.Hdoufane I, Bjij I, Soliman M, Tadjer A, Villemin D, Bogdanov J, Cherqaoui D. In silico SAR studies of HIV-1 inhibitors. Pharmaceuticals. 2018;11(3):69. doi: 10.3390/ph11030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheikhpour R, Sarram M, Rezaeian M, Sheikhpour E. QSAR modelling using combined simple competitive learning networks and RBF neural networks. SAR QSAR Environ Res. 2018;29(4):257–276. doi: 10.1080/1062936X.2018.1424030. [DOI] [PubMed] [Google Scholar]
- 58.Žuvela P, David J, Yang X, Huang D, Wong MW. Non-linear quantitative structure–activity relationships modelling, mechanistic study and in-silico design of flavonoids as potent antioxidants. Int J Mol Sci. 2019;20(9):2328. doi: 10.3390/ijms20092328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li N, Qi J, Wang P, Zhang X, Zhang T, Li H. Quantitative structure–activity relationship (QSAR) study of carcinogenicity of polycyclic aromatic hydrocarbons (PAHs) in atmospheric particulate matter by random forest (RF) Anal Methods. 2019;11(13):1816–1821. [Google Scholar]
- 60.Yousefinejad S, Mahboubifar M, Eskandari R. Quantitative structure–activity relationship to predict the anti-malarial activity in a set of new imidazolopiperazines based on artificial neural networks. Malar J. 2019;18(1):310. doi: 10.1186/s12936-019-2941-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun Y, Shi S, Li Y, Wang Q. Development of quantitative structure-activity relationship models to predict potential nephrotoxic ingredients in traditional Chinese medicines. Food Chem Toxicol. 2019;128:163–170. doi: 10.1016/j.fct.2019.03.056. [DOI] [PubMed] [Google Scholar]
- 62.Gaisford S, Saunders M. Essentials of pharmaceutical preformulation: John Wiley & Sons, Ltd; 2013.
- 63.Babu NJ, Nangia A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst Growth Des. 2011;11(7):2662–2679. [Google Scholar]
- 64.Yalkowsky SH. Techniques of solubilization of drugs: PharmaMed Press; 1981.
- 65.Hossain S, Kabedev A, Parrow A, Bergström C, Larsson P. Molecular simulation as a computational pharmaceutics tool to predict drug solubility, solubilization processes and partitioning. Eur J Pharm Biopharm. 2019;137:46–55. doi: 10.1016/j.ejpb.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen G, Luo X, Zhang H, Fu K, Liang Z, Rongwong W, Tontiwachwuthikul P, Idem R. Artificial neural network models for the prediction of CO2 solubility in aqueous amine solutions. Int J Greenhouse Gas Control. 2015;39:174–184. [Google Scholar]
- 67.Meesattham S, Charoensiritanasin P, Ongwattanakul S, Liang Z, Tontiwachwuthikul P, Sema T. Predictions of equilibrium solubility and mass transfer coefficient for CO2 absorption into aqueous solutions of 4-diethylamino-2-butanol using artificial neural networks. Petroleum. 2018;(in press).
- 68.Gao H, Ye Z, Dong J, Gao H, Yu H, Li H, et al. Predicting drug/phospholipid complexation by the lightGBM method. Chem Phys Lett. 2020;747:137354. [Google Scholar]
- 69.He Y, Ye Z, Liu X, Wei Z, Qiu F, Li H-F, et al. Can machine learning predict drug nanocrystals? J Control Release. 2020. [DOI] [PubMed]
- 70.Leane MM, Cumming I, Corrigan OI. The use of artificial neural networks for the selection of the most appropriate formulation and processing variables in order to predict the in vitro dissolution of sustained release minitablets. AAPS PharmSciTech. 2003;4(2):129–140. doi: 10.1208/pt040226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barthus RC, Mazo LH, Poppi RJ. Simultaneous determination of vitamins C, B6 and PP in pharmaceutics using differential pulse voltammetry with a glassy carbon electrode and multivariate calibration tools. J Pharm Biomed Anal. 2005;38(1):94–99. doi: 10.1016/j.jpba.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 72.Valizadeh H, Pourmahmood M, Mojarrad JS, Nemati M, Zakeri-Milani P. Application of artificial intelligent tools to modeling of glucosamine preparation from exoskeleton of shrimp. Drug Dev Ind Pharm. 2009;35(4):396–407. doi: 10.1080/03639040802422088. [DOI] [PubMed] [Google Scholar]
- 73.Chaibva F, Burton M, Walker RB. Optimization of salbutamol sulfate dissolution from sustained release matrix formulations using an artificial neural network. Pharmaceutics. 2010;2(2):182–198. doi: 10.3390/pharmaceutics2020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takagaki K, Arai H, Takayama K. Creation of a tablet database containing several active ingredients and prediction of their pharmaceutical characteristics based on ensemble artificial neural networks. J Pharm Sci. 2010;99(10):4201–4214. doi: 10.1002/jps.22135. [DOI] [PubMed] [Google Scholar]
- 75.Onuki Y, Kawai S, Arai H, Maeda J, Takagaki K, Takayama K. Contribution of the physicochemical properties of active pharmaceutical ingredients to tablet properties identified by ensemble artificial neural networks and Kohonen’s self-organizing maps. J Pharm Sci. 2012;101(7):2372–2381. doi: 10.1002/jps.23134. [DOI] [PubMed] [Google Scholar]
- 76.Li Y, Abbaspour MR, Grootendorst PV, Rauth AM, Wu XY. Optimization of controlled release nanoparticle formulation of verapamil hydrochloride using artificial neural networks with genetic algorithm and response surface methodology. Eur J Pharm Biopharm. 2015;94:170–179. doi: 10.1016/j.ejpb.2015.04.028. [DOI] [PubMed] [Google Scholar]
- 77.Hashad RA, Ishak RA, Fahmy S, Mansour S, Geneidi AS. Chitosan-tripolyphosphate nanoparticles: optimization of formulation parameters for improving process yield at a novel pH using artificial neural networks. Int J Biol Macromol. 2016;86:50–58. doi: 10.1016/j.ijbiomac.2016.01.042. [DOI] [PubMed] [Google Scholar]
- 78.Rodríguez-Dorado R, Landín M, Altai A, Russo P, Aquino RP, Del Gaudio P. A novel method for the production of core-shell microparticles by inverse gelation optimized with artificial intelligent tools. Int J Pharm. 2018;538(1–2):97–104. doi: 10.1016/j.ijpharm.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 79.Zhao F, Lu J, Jin X, Wang Z, Sun Y, Gao D, Li X, Liu R. Comparison of response surface methodology and artificial neural network to optimize novel ophthalmic flexible nano-liposomes: characterization, evaluation, in vivo pharmacokinetics and molecular dynamics simulation. Colloids Surf B: Biointerfaces. 2018;172:288–297. doi: 10.1016/j.colsurfb.2018.08.046. [DOI] [PubMed] [Google Scholar]
- 80.Parikh KJ, Sawant KK. Comparative study for optimization of pharmaceutical self-emulsifying pre-concentrate by design of experiment and artificial neural network. AAPS PharmSciTech. 2018;19(7):3311–3321. doi: 10.1208/s12249-018-1173-2. [DOI] [PubMed] [Google Scholar]
- 81.Barmpalexis P, Grypioti A, Eleftheriadis GK, Fatouros DG. Development of a new aprepitant liquisolid formulation with the aid of artificial neural networks and genetic programming. AAPS PharmSciTech. 2018;19(2):741–752. doi: 10.1208/s12249-017-0893-z. [DOI] [PubMed] [Google Scholar]
- 82.Nagy B, Petra D, Galata DL, Démuth B, Borbás E, Marosi G, et al. Application of artificial neural networks for process analytical technology-based dissolution testing. Int J Pharm. 2019;567:118464. doi: 10.1016/j.ijpharm.2019.118464. [DOI] [PubMed] [Google Scholar]
- 83.Manda A, Walker RB, Khamanga SM. An artificial neural network approach to predict the effects of formulation and process variables on prednisone release from a multipartite system. Pharmaceutics. 2019;11(3):109. doi: 10.3390/pharmaceutics11030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gentiluomo L, Roessner D, Augustijn D, Svilenov H, Kulakova A, Mahapatra S, Winter G, Streicher W, Rinnan Å, Peters GHJ, Harris P, Frieß W. Application of interpretable artificial neural networks to early monoclonal antibodies development. Eur J Pharm Biopharm. 2019;141:81–89. doi: 10.1016/j.ejpb.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 85.Lou H, Chung JI, Kiang Y-H, Xiao L-Y, Hageman MJ. The application of machine learning algorithms in understanding the effect of core/shell technique on improving powder compactability. Int J Pharm. 2019;555:368–379. doi: 10.1016/j.ijpharm.2018.11.039. [DOI] [PubMed] [Google Scholar]
- 86.Han R, Xiong H, Ye Z, Yang Y, Huang T, Jing Q, et al. Predicting physical stability of solid dispersions by machine learning techniques. J Control Release. 2019;311:16–25. doi: 10.1016/j.jconrel.2019.08.030. [DOI] [PubMed] [Google Scholar]
- 87.Hutchinson L, Kirk R. High drug attrition rates—where are we going wrong? Nature Reviews Clinical Oncology. 2011;8(4):189–90. [DOI] [PubMed]
- 88.Kalepu S, Nekkanti V, editors. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm Sin B; 2015. [DOI] [PMC free article] [PubMed]
- 89.Mohs RC, Greig NH. Drug discovery and development: Role of basic biological research. Alzheimer's & Dementia: Translational Research & Clinical Interventions. 2017;3(4):651–7. [DOI] [PMC free article] [PubMed]
- 90.Henstock PV. Artificial intelligence for pharma: time for internal investment. Trends Pharmacol Sci. 2019;40(8):543–546. doi: 10.1016/j.tips.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 91.Mak K-K, Pichika MR. Artificial intelligence in drug development: present status and future prospects. Drug Discov Today. 2019;24(3):773–780. doi: 10.1016/j.drudis.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 92.Lamberti MJ, Wilkinson M, Donzanti BA, Wohlhieter GE, Parikh S, Wilkins RG, Getz K. A study on the application and use of artificial intelligence to support drug development. Clin Ther. 2019;41(8):1414–1426. doi: 10.1016/j.clinthera.2019.05.018. [DOI] [PubMed] [Google Scholar]
- 93.US Food and Drug Administration. Proposed regulatory framework for modifications to artificial intelligence/machine learning (AI/ML)-based software as a medical device (SAMD)—discussion paper and request for feedback. 2019 Accessed 08 June 2020. Available from: https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-software-medical-device.
- 94.Damiati SA, Alaofi AL, Dhar P, Alhakamy NA. Novel machine learning application for prediction of membrane insertion potential of cell-penetrating peptides. Int J Pharm. 2019;567:118453. doi: 10.1016/j.ijpharm.2019.118453. [DOI] [PubMed] [Google Scholar]
- 95.Paranjpe MD, Taubes A, Sirota M. Insights into computational drug repurposing for neurodegenerative disease. Trends Pharmacol Sci. 2019;40(8):565–576. doi: 10.1016/j.tips.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Richardson PJ, Corbellino M, Stebbing J. Baricitinib for COVID-19: a suitable treatment?–Authors’ reply. Lancet Infect Dis. 2020. [DOI] [PMC free article] [PubMed]
- 97.McCall B. COVID-19 and artificial intelligence: protecting health-care workers and curbing the spread. Lancet Digital Health. 2020;2(4):e166–e1e7. doi: 10.1016/S2589-7500(20)30054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]