

Abstract
With the trend of moving molecular tests from clinical laboratories to on-site testing, there is a need for nucleic acid based diagnostic tools combining the sensitivity, specificity and flexibility of established diagnostics with the ease, cost effectiveness and speed of isothermal amplification and detection methods. A promising new nucleic acid detection method is Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated nuclease (Cas)-based sensing. In this method Cas effector proteins are used as highly specific sequence recognition elements that can be combined with many different read-out methods for on-site point-of-care testing. This review covers the technical aspects of integrating CRISPR/Cas technology in miniaturized sensors for analysis on-site. We start with a short introduction to CRISPR/Cas systems and the different effector proteins and continue with reviewing the recent developments of integrating CRISPR sensing in miniaturized sensors for point-of-care applications. Finally, we discuss the challenges of point-of-care CRISPR sensing and describe future research perspectives.
Keywords: Point-of-care, CRISPR/Cas, DNA sensing, Genomic diagnostic tools, CRISPR sensing, Diagnosis
Graphical abstract

Highlights
-
•
Rapid and reliable detection of nucleic acid is critical in many different fields.
-
•
CRISPR/Cas effector protein complexes could be used for specific DNA detection in so-called point-of-care testing.
-
•
A short and comprehensive introduction to CRISPR sensing is given.
-
•
We review recent developments of integrating CRISPR sensing in Point-of-care devices.
-
•
The challenges of (point-of-care) CRISPR sensing are discussed.
1. Introduction
The access to rapid and reliable detection methods of nucleic acids is critical in many different fields, such as life sciences, environmental monitoring, biotechnology and maybe most importantly health care. Sensing of pathogens on the basis of their genetic information, for instance by monitoring circulating cell-free DNA/RNA particles related to various types of diseases makes early diagnosis and treatment possible. However, this depends on in methods that allow the detection of ultralow concentrations of nucleic acids with high sensitivity and specificity. The currently ongoing COVID-19 outbreak, affecting millions of people around the globe, again shows the importance of early and specific infectious disease detection for patient and health care in general, and for risk prevention of further spread.
Established genomic diagnostic tools, such as polymer chain reaction (PCR), are superior in terms of specific amplification of genomic sequences but come with several drawbacks in terms of usability and costs (Scheler et al., 2014). Most of the established nucleic acid sensing methods need complex and bulky instrumentation, as well as trained personnel to operate them. The assays are costly to perform due to multiple (manual) steps that are both time and material consuming.
Isothermal amplification methods have been developed (e.g. LAMP and RPA) that are faster than PCR and can be operated at constant temperature, eliminating the need for sophisticated equipment like thermocyclers (Martzy et al., 2019). However, these new advantages come with tradeoffs in terms of sensitivity and specificity. Even after optimization, single-nucleotide polymorphisms (SNPs) cannot always be discriminated (Zanoli and Spoto, 2013), while these small nucleotide changes will be crucial in both pathogen and disease detection.
Therefore, there is a need for nucleic acid based diagnostic tools combining the sensitivity, specificity and flexibility of established genomic diagnostics, with the ease, cost effectiveness and speed of isothermal amplification methods. Such a newly developed diagnostic tool will be perfect for point-of-care (POC) testing. Point-of-care testing is defined as a (medical) diagnostic tool near or even at the point-of-care, so bringing testing on site (Kost et al., 2008). This on-site testing is accomplished by integrating assays in (trans)portable devices. In ideal point-of-care testing, the steps one needs to perform from raw sample to understandable result should be minimized, allowing unskilled operators to perform analysis. This trend asks for total-analysis devices, integrating sample pre-treatment, target recognition and signal acquisition (sensing) in a single device. These total analysis systems could comprise entire microfluidic chips, paper-based sensors or even single tube reactions, as long as they are accompanied by easy protocols and accessible read-out methods.
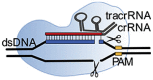
A promising alternative to established genomic diagnostic tools that fits the specifications for POC sensing is Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated nuclease (Cas)-based sensing. The origin of CRISPR based sensing lies in the discovery of the collateral cleavage activity by effector protein Cas13a. When this effector protein is associated with a CRISPR RNA (crRNA), this crRNA can specifically complement with a target sequence, inducing enzymatic cleavage of both the targeted sequence and untargeted collateral cleavage of all single stranded RNA (ssRNA) in its neighborhood. (Abudayyeh et al., 2016). This discovery in 2016 was quickly followed by the first proof that this trans-cleavage can be used in a biosensor (East-Seletsky et al., 2016). The first comprehensive and applicable CRISPR (bio)sensing system developed was SHERLOCK (Gootenberg et al., 2017), relying on the collateral trans-cleavage of quenched fluorescent nucleotides after target binding (Fig. 1 ), enabling signal amplification up to 10,000 times the original concentration of the target sequence. Over the years, the versatility of Cas effectors from different families, including Cas9, Cas12, Cas13 and Cas14, has proven to work for nucleotide sensing with high sensitivity and selectivity (down to zeptomolar (10−21 M) concentrations and single nucleotide resolution).
Fig. 1.

Schematic representation of SHERLOCK's detection mechanism using Cas13a effector proteins to detect single stranded RNA.
Overall, CRISPR based (bio)sensing seems to be an excellent candidate to fulfill the needs of ultrasensitive nucleotide detection. Nowadays, a small number of review papers exists concerning CRISPR sensing, for example Zhou et al., summarizing applications of the CRISPR/Cas system to the recognition and detection of nucleotides and summarizing the applications to test pathogens ( L. Zhou et al., 2018). Later work of Li et al. in 2019 provides a detailed classification of CRISPR/Cas biosensing systems and proposes their future utility ( Li et al., 2019a). In 2020 Aman et al. covered recent advances in the field of CRISPR-based biosensing technologies and highlighted insights into their potential use in multiple applications (Aman et al., 2020). While possible POC applications were discussed in these review papers, no review papers have as yet focused on the technical aspects of integrating CRISPR/Cas technology in miniaturized sensors for analysis at the point-of-care. The field of CRISPR sensing might be at the early stage of development, but the potential of this sensing method for incorporation in POC devices has been already repeatedly shown and new publications showing its versatility are appearing rapidly. In this paper we will introduce the CRISPR/Cas systems and the different effector proteins, review the recent developments, discuss the challenges of POC CRISPR sensing and describe future research perspectives.
2. Classification of CRISPR/Cas systems
Three decades ago, the first CRISPR/Cas systems were described in bacterial genomes (Ishino et al., 1987; Van Soolingen et al., 1993). It took many years to show that CRISPR and Cas genes were part of the adaptive immune system of bacteria, used as a defense mechanism against foreign nucleotides (Bolotin et al., 2005; Mojica et al., 2005; Pourcel et al., 2005). CRISPR/Cas effector proteins exist in a wide variety, varying in size, structure, composition, and target. The relatively new and active field of CRISPR research is still developing new effector proteins and (genetically) optimizing current systems for different applications.
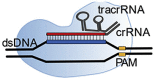
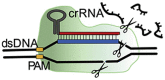
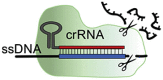
CRISPR/Cas systems can be divided into two classes, each consisting of several types and subtypes (Makarova et al., 2020). CRISPR/Cas class 1 systems comprise complexes of multiple effector proteins, where each protein performs a single function in the CRISPR process. Class 2 CRISPR/Cas systems are characterized by one single effector protein that has multiple functions within the CRISPR process. Class 2 systems are most used in bioengineering and CRISPR diagnostics due to their simplicity in combination with their high efficiency. Therefore, the focus of this review will be on class 2 CRISPR/Cas systems. Class 2 systems can be subdivided in 3 types commonly used in CRISPR sensing: Type II, V and VI. An overview of these types, the corresponding signature effector proteins and properties can be found in Table 1 .
Table 1.
Overview of properties of CRISPR types corresponding to class 2 CRISPR systems commonly used in CRISPR based bioengineering and sensing.
|
Type |
II |
V |
VI |
||
|---|---|---|---|---|---|
| Effector protein | (sp)Cas9 | (sp)dCas9 | (fn)Cas12a | Cas14a/b | Cas13a/b |
 |
 |
 |
 |
 |
|
| Spacer length | 18-24 nt | 18-24 nt | 18-25 nt | 20-40 nt | 22-30 nt |
| PAM required? | Yes | Yes | Yes | No | No, PFS |
| PAM sequence | NGG | NGG | (T)TTN | -a | non-G |
| Cleavage | blunt-end cut | – | single staggered cut | single (staggereda) cut | Many cleavage sites |
| Target type | dsDNA | – | DNA (ds/ssb) | DNA (ss/dsa) | RNA (ss) |
| Collateral cleavage? | No | No | DNA (ss) | DNA (ss) | RNA (ss) |
T-rich PAM sequences, like TTTA could allow dsDNA cleavage.
ssDNA can also be detected by Cas12, however less specific.
Class 2 CRISPR/Cas systems are characterized by a single multidomain protein that associates with an RNA sequence to form a ribonucleoprotein (RNP) surveillance complex. This RNA sequence is called guide RNA (gRNA). gRNA consists of a customizable component called the crRNA, that defines the specificity and selectivity towards target DNA, and a non-coding RNA part containing two-dimensional structures. This non-coding part facilitates association between gRNA and the effector protein by extensive hydrogen bond contacts and aromatic stacking between gRNA and the effector protein (Chen and Doudna, 2017; Slaymaker et al., 2019). The interaction between the gRNA and the protein induces structural changes to the effector protein, as confirmed by crystallography (Nishimasu et al., 2014; Slaymaker et al., 2019; Yamano et al., 2016). These structural changes ‘activate’ the effector protein and induce the formation of the RNP surveillance complex, which scans nucleic acids and targets sequences complementary to its crRNA for enzymatic degradation.
2.1. Type II effector proteins
A signature family from the type II proteins are the Cas9 effector proteins. Fig. 2 shows the DNA recognition process of a Cas9-crRNAs ribonucleotide complex. Type II CRISPR/Cas effector proteins, like Cas9, use their customizable crRNA, called spacer (red), to target protospacer sequences (blue) on double stranded DNA (dsDNA). These protospacer sequences are complementary to a spacer sequence (red), created on the customizable crRNA. Besides the crRNA, Cas9 effector proteins have an extra crRNA, called transactivating crRNA (tracrRNA). TracrRNA (green) is essential for target sequence recognition and target sequence cleavage, as well as the association between the crRNAs and Cas effector protein (Chen and Doudna, 2017). This two component crRNA system was simplified by Jinek et al. to a single component system by the development of a single-guide RNA (sgRNA) that combines both crRNAs (Jinek et al., 2012).
Fig. 2.

Schematic of Cas9 effector protein (light blue) during the dsDNA recognition process (black). In the case of Cas9 effector proteins, two crRNAs are associated by nucleotide hybridization. The tracrRNA (green) is crucial in the association between crRNA and effector protein, creating a RNP complex. The other crRNA has the spacer sequence (red), which could complement with a target sequences' protospacer (blue). The RNP complex can only unwind dsDNA next to a so-called PAM sequence (yellow). Without PAM sequence recognition by the RNP, no complementation between spacer and target sequence is possible. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The crRNA sequence is customizable: by changing the spacer sequence present on the crRNA one can change the genomic target sequence. However, sequence possibilities are limited by the effector protein specific protospacer adjacent motif (PAM) sequence (yellow, Fig. 3 ). This PAM sequence can be found upstream (from 5’ -> 3’) of the protospacer on the dsDNA. PAM sequence recognition by the RNP complex originates from the task of CRISPR proteins in the adaptive immune system of bacteria. The purpose of the CRISPR system is to protect bacteria from invading bacteriophages, viruses. If the bacterium survives an infection, class 1 CRISPR effector proteins, together with Cas9 trim part of the viral DNA and add this to a so-called CRISPR array. In this way the bacterium creates a library of previous infections to fight the viral infection in the future (Fig. 2A). This CRISPR array, is transcribed and used for the formation of Cas9 RNP complexes, that screen for complementary (viral) sequences (Fig. 2B). However, the CRISPR array contains the same sequence as the viral DNA. This is where the PAM sequence comes in: since Cas9 is involved in the protospacer recruitment, this protospacer sequence is always followed by a Cas9 specific PAM sequence. Only if this PAM sequence is present next to the target sequence, cleavage will take place. This is only the case for the protospacer present on the viral DNA and not for the spacer on the CRISPR array (Mali et al., 2013).
Fig. 3.

Schematic overview of CRISPR adaptive immune system of bacteria. A) Bacteriophages can insert their genome in the cell of the bacterium, resulting in replication of the virus and eventually cell death of the bacterium. However, if a bacterium survives an infection, part of the viral genome can be inserted in the CRISPR array, a library of all previous infections. The protospacer sequences are cut-out specifically by CRISPR class 1 proteins in combination with Cas9 upon viral DNA recognition. This protospacer is inserted in the CRISPR array, resulting in an embedded memory of the virus. B) if the virus returns, the CRISPR array is transcribed in a long pre-crRNA sequence, containing spacer sequences complementary to the viral DNA. The tracrRNA complements with part of the crRNA, resulting in recruitment of Cas9 effector proteins, trimming of the pre-crRNA and formation of a RNP complex. Because the sequence of the CRISPR array and viral DNA are identical, it is important to distinguish between the array and viral DNA. Upon complementation between the RNP and a DNA sequence, the DNA sequence will be destroyed. To avoid cleavage of the CRISPR array, the PAM sequence is of importance. Only sequences followed by a specific PAM sequence will be recognized and will be cleaved by the RNP.
So, by the PAM sequence, which is a nucleotide combination of typically 2–4 bases which is not present in the genome of the bacterium, bacteria can distinguish their own DNA from foreign DNA. Besides this function, the PAM sequence also accelerates the search process for viral DNA in the bacterium cell. Cas9 only has to unwind the DNA containing a PAM sequence. By unwinding the DNA, complementation between spacer and protospacer can take place, initiating the enzymatic cleavage properties of the effector protein, in this example Cas9. This cleavage results in blunt-end double strand breaks in the target dsDNA (Fig. 3). Besides enzymatic cleavage, the Cas9 effector protein possesses excellent sequence recognition. The enzymatically deactivated Cas9 effector protein (dCas9) follows the same target recognition process but lacks the ability to perform blunt-end strand breaks.
The PAM sequence is dependent on the type of Cas effector protein and the species it originates from. PAM sequence recognition does not take place via the crRNA, but by the effector protein itself. In the case of the commonly used spCas9 from Streptococcus pyogenes, the PAM sequences following the protospacer is 5′-NGG-3’ (where N could be any type of nucleotide). Depending on the type of Cas9, non-specific DNA cleavage occurs due to PAM region mismatches.
2.2. Type V effector proteins
The type V effector proteins Cas12 and Cas14 exert two types of enzymatic cleavage reactions: Besides the target sequence cleavage, similar to Cas9, they also induce untargeted collateral cleavage of single stranded (ssDNA) upon protospacer-target recognition (Harrington et al., 2018; Makarova et al., 2020). Cas12 and Cas14 are capable of protospacer recognition on both ssDNA and dsDNA, where Cas12 has more specificity towards dsDNA and Cas14 more towards ssDNA.
Commonly used Cas12 subtypes in CRISPR engineering and sensing are Cas12a and Cas12b. Cas12b has not been studied thoroughly due to the high temperature requirements of the protein (48 °C, compared to the 37 °C for Cas12a). Nevertheless, genetic engineering has optimized Cas12b for use at lower temperatures (Strecker et al., 2019). Cas12a/b requires a unique, specific PAM sequence, just like Cas9, to enable dsDNA unwinding. The PAM sequence of Cas12, 5’-(T)TTN-3’ (where N could be any nucleotide base), can be found upstream (from 5’-> 3′) of the protospacer sequence. This PAM sequence is less crucial when sensing ssDNA, and it has been shown that enzymatic cleavage activity can still be possible with mutated PAM sequences on the ssDNA target sequence (Chen et al., 2018). While subtype Cas12a is more specific, with less off-target cutting compared to Cas9, the on-target cleavage efficiency of Cas12a is also relatively low (Kim et al., 2016; Kleinstiver et al., 2016). The high on-target specificity is lost when sensing ssDNA, making the system less suitable for distinguishing very similar ssDNA sequences (Kim et al., 2016; Kleinstiver et al., 2016).
The Cas14 family, composed of Cas14a, Cas14b and Cas14c, was recently discovered and is a ssDNA nuclease, functioning PAM-independent (Harrington et al., 2018). However, dsDNA recognition can also be triggered by a T-rich PAM sequence (Karvelis et al., n.d.). While Cas12a has problems with the discrimination of very similar ssDNA sequences even in the presence of a PAM sequence, Cas14a showed recognition and discrimination of ssDNA sequences with only one nucleotide difference. This improved specificity raises the possibility to use Cas14a for the detection of ssDNA SNPs without PAM sequence requirements.
2.3. Type VI effector proteins
While subtype II and V target ssDNA and/or dsDNA, type VI effector proteins specifically target RNA sequences. Effector proteins from the Cas13 family form RNP complexes with a single guide RNA sequence, consisting of a hairpin loop for association and a 28–30 nt spacer for target recognition. Similar to type V effector proteins, two types of enzymatic cleavage reactions can be performed upon target recognition: in addition to the programmable RNAse activity towards the target sequence, non-specific cleavage of nearby RNA sequences is initiated (Abudayyeh et al., 2017; Omar O. Abudayyeh et al., 2016). The targeted cleavage of Cas13 is not in the complementary protospacer, but further downstream, allowing multiple targeted cleavage steps since the protospacer is not damaged by the enzymatic reaction (Terns, 2018). The Cas13 effector proteins do not require a PAM sequence, but have a protospacer flanking site (PFS) where a guanine base directly following the protospacer has a negative effect on complementation between crRNA and target ssRNA, while any other nucleotide does not influence the enzymatic activity (Omar O Abudayyeh et al., 2016). Commonly used subtypes of the Cas13 family in biotechnology and sensing sciences are Cas13a and Cas13b, where Cas13b is thought to be more robust and stable in cell engineering (Konermann et al., 2018).
3. CRISPR/Cas sensing in POC sensors
POC sensing is a rapidly fast-developing field in clinical diagnostics and is expected to define the future of diagnostics in health care. POC sensors such as the handheld glucose sensors for diabetes patients are widely known, but rapid development now also allows for sensors for the detection of proteins, small (inorganic) molecules and nucleic acids. According to the World Health Organization (WHO) POC testing needs to follow the ASSURED guidelines (Affordable, Sensitive, Specific, User-friendly, Robust and rapid, Equipment-free, Deliverable to all people who need the test) (Kosack et al., 2017). According to these guidelines, it should be possible for a non-specialist to conduct and interpret the test in a variety of settings, including low resource communities. CRISPR/Cas effector proteins were previously used in nucleotide sensing applications and showed, in combination with (s)gRNA, that they can specifically target nucleic acids with high efficiency and could therefore be used in combination with POC devices for highly effective nucleic acid detectors.
As mentioned in the introduction, the first CRISPR sensing methods were designed using Cas13a effector proteins (previously known as C2c2), which exerts two distinct RNAse activities. Besides the cleavage of the target sequence, similar to Cas9 effector proteins, a second active site was found that, upon target binding, shows untargeted ssRNA degradation activity (East-Seletsky et al., 2016). By varying the amount of fluorophore quencher-labelled reporter RNA added to RNP-RNA complexes, East-Seletsky et al. were able to measure an increase in fluorescence with increasing target RNA concentrations (East-Seletsky et al., 2016). They showed that the collateral cleavage activity could lead to enormous signal amplification. In their experimental setup, 10 pM of a target RNA is predicted to result in only around 0.02% of the Cas13a–crRNA complex bound to target RNA. However, 25–50% of the fluorophore quencher-labelled reporter RNA was degraded, suggesting a massive turnover capacity of at least 10,000 turnovers per bound target RNA. This shows the potential to measure ultralow concentrations by signal amplification, rather than (isothermal) nucleic acid target amplification.
By the use of T7 promoter it was shown by Gootenberg et al. that Cas13a effector proteins could also be used for indirect (ds)DNA sensing by transcribing the DNA amplification products to RNA, enabling the detection of DNA by SHERLOCK (Gootenberg et al., 2017). With the discovery of the collateral cleavage activity of Cas12a, a DNA targeting effector protein, the possibility for dsDNA sensing without the need of an amplification step was introduced. Similar to Cas13a, multiple turnovers for nonspecific cleavage were observed for the nonspecific ssDNA substrate, enabling signal amplification. The discovery of Cas12a enabled the development of DNA detection assays like DETECTR (Chen et al., 2018) and HOLMES (Li et al., 2018). While a great step forward, these assays, that still employ a nucleic acid amplification step in addition to the signal amplification obtained by the effector proteins to further reduce the limit of detection.
In the next sections we will discuss CRISPR POC sensing methods based on their read-out mechanism: Fluorescence, colorimetric or electronic. Table 2 gives an overview of all current POC CRISPR sensing methods described in this review paper.
Table 2.
Overview of major characteristics of classified POC CRISPR nucleotide sensing applications.
| Type | Method | sensor type | Effector | Ampli-fication | Quanti-tative | LODa | Multi-plex | Read-out | Time | Sample type | Target Type | disease/pathogen | Refs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II | NASBACC | Toehold switch | Cas9 | NASBA | Semi | 1 fM | No | Colorimetric | 3 h | Raw | DNA | Zika virus | Pardee et al. (2016) |
| CASFLA | LFA | (d)Cas9 | PCR/RPA | Yes | 82 aM | No | Colorimetric | 1–1.5 h | Raw | DNA | African Swine Fever | Wang et al., 2020 | |
| Cas9nAR | LFA | Cas9 | SDA | Semi | 100 copies | No | Colorimetric | ~3 h | Pre-treated | DNA | Salmonella typhimurium and Escherichia coli | Wang et al., 2020 | |
| - | LFA | Cas9 | - | Semi | 1 nM | No | Colorimetric | < 30 min | Pre-treated | DNA | Various pathogens | Hu et al. (2020) | |
| CRISPR-chip | Field-effect transistor | Cas9 | – | Yes | 2.3 fM | No | Potentiometry | 15 min | Pre-treated | DNA | Duchenne muscular dystrophy | Hajian et al. (2019) | |
| – | Micro array | dCas9 | – | Yes | 10 CFU/ml | No | Fluorescence | 30 min | Raw | DNA | MRSA bacterium | Guk et al. (2017) | |
| – | Nanoelectrokinetic chip | dCas9 | – | No | – | No | Fluorescence | <1 h | Pre-treated | DNA | HIV | Lee et al. (2018) | |
| – | SMR sensor | dCas9 | (rt-)RPA | Yes | ~0.6 fM | No | Change in R.I. | ~20 min | Raw | DNA/RNA | Tick born disease | Koo et al., 2018a | |
| – | Nanopore sensors | dCas9 | – | No | Single copy | No | Resistive pulse | 1–2 h | Synthetic | DNA | – | Yang et al. (2018) | |
| – | Nanopore sensors | dCas9 | – | No | Single copy | No | Resistive pulse | <1h | Synthetic | DNA | – | Weckman et al. (2019) | |
| V | SCAN | Nanopore sensors | Cas12a | - | No | 10 nM | No | Resistive pulse | 1 h | Synthetic | DNA | HIV | Nouri et al. (2020) |
| – | One-pot reaction | Cas12a | LAMP | Yes | 0.05% GM DNA | No | Fluorescence | 45 min | Pre-treated | DNA | GMOs | Wu et al. (2020a) | |
| ULC | One-pot reaction | Cas12a | Uracil-LAMP | Yes | Single copy | No | Fluorescence | <1 h | Pre-treated | DNA | Citrus greening disease | Qian et al. (2019) | |
| – | One-pot reaction | Cas12a | LAMP | Yes | ~1 fM | No | Fluorescence | <1 h | Raw | DNA | African Swine Fever | Qian et al. (2020) | |
| – | Portable microplatereader | Cas12a | – | Semi | 1 pM/0.2 nM | Yes | Fluorescence | 1 h | Pre-treated | DNA | African Swine Fever | He et al. (2020) | |
| DAMR | 3D printer inset/tube | Cas12a | RPA | Yes | 10-100 copies | Yes | Fluorescence | <1 h | Raw | DNA | HPV | Yin et al. (2020) | |
| IMPACT | PDMS micropillar chip | Cas12a | – | Semi | 0.1–10 nM | No | Fluorescence | ~10 min | Raw | DNA | African Swine Fever | Hass et al. (2020) | |
| CORDS | LFA | Cas12a | RAA | No | 10 fM | No | Fluorescence/Colorimetric | ~1h | Pre-treated | DNA | African Swine Fever | Bai et al. (2019) | |
| - | LFA | Cas12a | RPA | No | 10 pM | No | Fluorescence/Colorimetric | 15–60 min | Raw | RNA | SARS-CoV-2 | Curti et al. (2020) | |
| DETECTR | LFA | Cas12a | RT-LAMP | No | 20 aM | No | Fluorescence/Colorimetric | 30–40 min | Pre-treated | RNA | SARS-CoV-2 | Broughton et al. (2020) | |
| AIOD-CRISPR | One-pot reaction | Cas12a | RPA | Semi | ~1 copy | No | Fluorescence/Colorimetric | <1 h | Pre-treated | DNA | SARS-Cov-2/HIV | Ding et al. (2020) | |
| V | CIA-LFB | LFA | Cas12a | LAMP | No | 3.4 aM | No | Colorimetric | 30min–1 h | Raw | DNA | P.aeruginosa | Mukama et al. (2020a) |
| CIA-LFB | LFA | Cas12a | PCR/LAMP | Semi | 3.1 aM | No | Colorimetric | <1 h | Raw | DNA | HPV | Mukama et al. (2020b) | |
| – | LFA | Cas12a | RPA | Semi | 0.24 fM | No | Colorimetric | ~3 h | Raw | DNA | HPV | Tsou et al. (2019) | |
| MAV-chip | Platinum nanoreporter | Cas12a | – | Yes | 10pM | Yes | Colorimetric | ~1 h | Pre-treated | DNA | Liver cancer | Shao et al. (2019) | |
| CRISPR-μPAD | ssDNA-anchored hydrogel on paper fluidics | Cas12a | – | Yes | 400 pM | No | Colorimetric | 1–4h | Synthetic | DNA/RNA | Ebola virus | English et al. (2019) | |
| RT-RPA | 11 aM | No | Electronic | ||||||||||
| E-CRISPR | Electrochemical chip | Cas12a | – | Yes | 1 pM/0.2 nM | No | Electrochemical | ~1 h | Raw | DNA | HPV/Parvovirus | Dai et al. (2019) | |
| – | Electrochemical chip | Cas12a | – | Yes | 30 pM/100 pM* | No | Electrochemical | ~1 h | Pre-treated/Raw* | DNA | HPV | Zhang et al., 2020 | |
| HOLMESv2 | One-pot reaction | Cas12b | LAMP | Yes | 10aM/1pM* | No | Fluorescence | <1h | Pre-treated/Raw* | DNA/RNA | Several viruses and DNA methylation | L. Li et al. (2019) | |
| CasDetec | One-pot reaction | Cas12b | RT-RAA | Semi | 1x104 copies/mL | No | Fluorescence/Colorimetric | <1 h | Pre-treated | DNA | SARS-CoV-2 | Guo et al. (2020a) | |
| - | AuNP colorimetric | Cas12a | LAMP | Yes | 50 aM | No | Colorimetric | - | Synthetic | DNA | Hepatitis B | R. Zhou et al. (2020) | |
| VI | – | Fluorescence reader with paper fluidics | Cas13a | – | Yes | 3.7 nM | No | Fluorescence | 20 min | Synthetic | RNA | – | Katzmeier et al. (2019) |
| – | LFA | Cas13a | RT RPA | No | 90 aM–9 aM | No | Fluorescence/Colorimetric | <2 h | Raw | RNA | Zika/dengue virus | Myhrvold et al. (2018) | |
| SHERLOCKv2 | LFA | Cas13a | RPA | No | 20–200 aM | No | Fluorescence/Colorimetric | <1 h | Pre-treated | RNA | SARS-CoV-2 | F. Zhang et al. (2020) | |
| SHERLOCKv2 | LFA | Cas13a | RT-RPA | No | 0.5–2.3 aM | No | Colorimetric | <2 h | Raw | DNA | BK polyomavirus and cytomegalovirus | Kaminski et al. (2020) | |
| – | LFA | Cas13a | RPA | No | 10 copies | No | Colorimetric | ~1 h | Pre-treated | DNA/RNA | White Spot Syndrome Virus | Sullivan et al. (2019) | |
| – | LFA | Cas13a | RPA | No | ~1.x105 copies/ml | No | Colorimetric | 1–2 h | Pre-treated | RNA | Porcine reproductive and respiratory syndrome virus | ||
| PECL-CRISPR | Electrochemiluminescence | Cas13a | EXPAR | Yes | 1 fM | No | Electrochemiluminescence | < 2 h | Pre-treated | miRNA | Various tumor cells | Zhou et al., 2020 | |
| Multi | – | AuNP colorimetric | Cas12a/Cas13a | RPA/PCR | Semi | 200 copies | No | Colorimetric | >20 min | Pre-treated | DNA/RNA | Various pathogenic bacteria | Yuan et al. (2020) |
| E-DNA | Electrochemical chip | Cas9/Cas12a | – | Yes | ~10 fM- 100 fM | No | Electrochemical | <1 h | Raw | DNA | Parvovirus | Xu et al. (2020) |
All values are taken from the original publications.
3.1. Fluorescence chips and portable read-out systems
The first CRISPR sensing systems were based on an increase in fluorescence upon target sequence recognition by the RNP complex. A major advantage of fluorescence-based sensing is its background-free sensing, drastically increasing the signal-to-noise ratio compared to other optical techniques. The miniaturization of fluorometers enables performance of established fluorescence based CRISPR sensing methods, like SHERLOCK, HOLMES and DETECTR on site. However, this established fluorescence based CRISPR sensing techniques include numerous manual (pipetting), and/or nucleotide amplification steps, besides there need of large volumes of (expensive) chemicals. This compromises the user-friendliness and cost effectiveness of these established systems for POC applications. In this section dedicated POC sensors based on CRISPR sensing will be reviewed that reduce the number of manual steps, relatively easy and cheap to perform.
Katzmeier et al. developed a pocket-sized fluorescence detector for less than €15 that enables fluorescence read-out of CRISPR/Cas13a assays on paper (Katzmeier et al., 2019). Their detection method consists of a portable detector unit, which can be customized to fluorophores of interest by the use of plastic filter foils. The filter paper, on which the CRISPR sensing takes place, can be placed in a cartridge, which ensures perfect positioning of the sample for sensing. The individual CRISPR consumables are freeze dried on filter paper, allowing long time storage at room temperature. Addition of target-RNA solution resulted in resuspension and, in the presence of target-RNA, activation of the collateral cleavage of fluorophore quencher-labelled reporter RNA. A fast increase in fluorescence signal allows detection of RNA concentrations around 3 nM within 20 min. The assay performance on filter paper is less efficient compared to in-solution assays like SHERLOCK, HOLMES and DETECTR, but remained in the sub-nanomolar to nanomolar range (Gootenberg et al., 2017).
Another fully automated, fluorescence based CRISPR/Cas13a sensing system was presented by Qin et al. They reported a in field-testing POC kit for Ebola RNA detection, which could be used in combination with a custom-made table-top integrated fluorometer. Within 5 min, concentrations of purified Ebola RNA as low as 10 pM could be sensed amplification-free. The microfluidic chip consists of a pneumatic and fluidic layer, allowing fully automated fluid handling and fast reaction times. The integrated fluorometer has a XYZ translation stage, which can be used to move between detection reservoirs for rapid multiplexing. In their original design, 24 assays could be run in parallel within 30 min. Due to the absence of an amplification step, no expensive temperature regulation is needed and the price of 24 assays was calculated to be ~$6 USD (Qin et al., 2019).
He et al. developed an amplification-free POC system for rapid and accurate virus detection. The African Swine Fever Virus (ASFV) is a DNA containing virus and can be sensed with CRISPR/Cas12a effector proteins. Due to the high specificity and selectivity of the Cas12a-crRNA RNP complex, differentiation between closely related viruses was possible, and a detection limit of 1 pM could be reached within 2 h for pre-treated DNAs. By increasing incubation time, improved detection limits of 100 fM could be reached. For bodily fluids of infected animals, the LOD increases due to autofluorescence. The POC system consists of a disposable cartridge in combination with a custom-made fluorometer. The cartridge could hold up to eighty individual samples and was simply fabricated by punching pre-cured polydimethyl siloxane (PDMS) using a biopsy punch and bonding this PDMS slab to a glass slide. By the use of a parabolic mirror all individual detection chambers could be measured (He et al., 2020). Downside of this method is the need to individually add consumables to the chambers at the right time, requiring trained personnel.
A fluorescent array based CRISPR sensing technique was developed by Guk et al. in 2017. An NTA-modified glass surface was functionalized with Histidine6 tagged dCas9/sgRNA complexes specific for the MRSA bacterium. Spotting of genomic DNA of the MRSA bacterium resulted in hybridization between the DNA and the sgRNA of the dCas9 effector protein. This binding was visualized by fluorescently labeling the DNA, where the fluorescence intensity could be related to the spotted DNA sequence concentration on the glass surface with a LOD of 10 cfu/ml (Guk et al., 2017). This visualization method forms a direct downside of this technique: while the spotting opens possibilities towards multiplex sensing, it is a labor-intensive process. Furthermore, the intercalating dye used is carcinogenic and washing the slides with this dye could be potentially dangerous. Incorporating this sensor in a closed (microfluidic) and automated system however could improve the usability of the technology in the POC field.
As an alternative, Lee et al. developed a nano-electrokinetic chip where DNA and DNA-dCas9 complexes were separated via electrophoresis (Lee et al., 2018). A charge selective junction in the chip formed a concentration boundary layer, creating an electric field gradient, and therefore a spatial gradient in electrophoretic force which works against the drag force from the flow in the channel. Since DNA, dCas9 and DNA-dCas9 complexes have different electrophoretic mobilities, the authors could concentrate either the DNA-dCas9 complexes at a certain position in the chip (to pre-concentrate a specific DNA sequence before analysis) or both free DNA and DNA-dCas9 complexes (for direct analysis by the use of fluorescent intercalating dyes (one vs two lines). The spacer region on the crRNA can be designed to adjust the specificity and selectivity of the assay, down to distinguishing SNPs. A downside of this system is that it can only be used for relatively short DNA fragments (50–100 bp), as large DNA fragments (700–1000 bp) will not show enough mobility difference on the addition of the relatively small dCas9. The major issue with this platform is however its limited compatibility with physiological conditions. Cells, debris, or even ions in solution might interfere with the working principle, making (integrated) DNA extraction necessary when measuring real samples, increasing the complexity of the device (Sonker et al., 2017).
3.2. Colorimetric read-out systems
Colorimetric sensors are optical sensors that measure a change in color occurring in the presence of certain analytes. Colorimetric sensors are widespread in POC applications due to the high accessibility, ease of use and low costs. Also, in CRISPR sensing the use of colorimetric sensors is popular, due to direct visualization of target sequences of interest and easy interpretation of results.
3.2.1. Lateral flow assays
Lateral flow assays (LFAs) are known for their wide applicability in protein and antibody sensing. Lately, LFAs have been adapted for nucleic acid testing, nucleic acid analysis and gene identification (Corstjens et al., 2001; Mao et al., 2009). By using tagged primers, the amplicons of interest could be sandwiched between membrane and gold nanoparticle (AuNP) probes for capture and signal read-out. However, proper distinguishing between primer-dimers, incorrect amplicons due to the low temperature amplification, and the amplicons of interest is very difficult, leading to false-positives (Crannell et al., 2016; Li et al., 2017). Nucleic acid hybridization on LFAs is more specific, but requires single stranded amplicons and PCR reaction which need sophisticated thermocyclers, making this method unsuitable for POC applications (Xu et al., 2014).
The major advantage of LFAs is that they are easy to interpret as their results are typically Boolean and can be read out by the naked eye. However, LFAs lack accuracy and efficiency, which makes these conventional paper-based platforms not suitable for the more quantitative analysis which is desired in clinical applications (Kasetsirikul et al., 2020). The ability of CRISPR/Cas complexes to discriminate between primer-dimers, non-target amplicons and amplicons of interest could make them the missing link in the current LFA field. Furthermore, both ssDNA, dsDNA and ssRNA can be detected by choosing different Cas effector proteins.
Various LFA platforms were adapted to incorporate Cas effector proteins as target sequence recognition element. In most of these platforms, a commercially available universal test strip, the HybriDetect-Universal Lateral Flow Assay Kit, was used. This dipstick was originally designed for qualitative or even quantitative rapid testing of proteins, antibodies or gene amplicons, but has been adapted to function in several LFA based CRISPR sensing methods (Bai et al., 2019; Chang et al., 2019; Gootenberg et al., 2018; Kaminski et al., 2020; Mukama et al., 2020b; Sullivan et al., 2019; Tsou et al., 2019). This platform offers a Streptavidine line as well as an antibody line that can capture anti-FITC coated AuNPs (Fig. 4 ). By dual labeling of single-stranded reporter nucleotides on both 5′ end and 3’ end with Biotin and FITC, the intensity of the test line could be a measure for the amount of collateral cleavage performed by the type V and VI Cas effector proteins. By calibration the collateral cleavage activity can be related to the target sequence concentration present in the original sample. It was shown that in this way femtomolar (10−15 M) concentrations could be measured within 1 h without target sequence amplification, which could be lowered to a LOD of attomolar (10−18 M) or even zeptomolar (10−21 M) concentrations by isothermal amplification. While read-out of LFAs is simple and does not need sophisticated machinery, these assays require multiple manual pipetting steps prior to the LFA step. Ideally, these steps could be minimized by incorporating Cas sequence recognition and cleavage on the paper membrane, resulting a single dip of the assay in the sample of interest prior to read-out.
Fig. 4.

A) Type V/VI based LFA, adapted from (Bai et al., 2019) with permission from Creative Commons Attribution License (CC BY) B) Type II based LFA, adapted from with permission from (Wang et al., 2020). Copyright 2020 American Chemical Society.
While most LFA based CRISPR sensors make use of type V and VI effector proteins, Wang et al. are the only team who developed an LFA assay based on type II effector proteins, which they called the CRISPR/dCas9-mediated lateral flow nucleic acid assay (CASLFA) method (Wang et al., 2020) (Fig. 4B). In their paper two CASFLA strategies were developed. In both cases the dCas9 proteins are immobilized on the paper membrane by the associated target dsDNA. For this purpose, biotin containing primers are introduced to the target sequence by PCR or isothermal amplification, to enable interaction between the target sequence and a streptavidin coated test line. Prior to application of the amplicons to the paper membrane, a dCas9 reaction is performed, forming RNP-target sequence complexes that could interact with the test line. The two CASFLA methods differ in the way the AuNPs (for visualization) interact with the RNP complex. The first strategy is based on the DNA-unwinding-based hybridization. Major disadvantage of this method is the need for customized AuNP-DNA probes for different application purposes, since the target sequence defines the AuNP-DNA probe sequences. Therefore, a second method was developed where the three-dimensional aptamer structure of the crRNA was probed with AuNP-ssDNA conjugates. This universal detection technique is independent of the target sequence and can be applied to all sequences, as long as the non-coding crRNA is preserved. Concentrations down to 0.8 fM could be sensed by combining the second CASLFA method with PCR. The need for an amplification step for both biotin incorporation and sample amplification is a downside of this technique. However, due to the cheap and easy-to-fabricate LFAs this technique could be promising.
Another Cas9 based LFA assay was developed by Wang et al. This assay is based on the sequence -specific cleavage activity of Cas9 (Wang et al., 2020). Similar to the previously discussed work by X. Wang et al. an amplification step is needed to incorporate reactive groups for binding to the lateral flow strip and the microspheres for visualization during primer annealing. Since Cas9 does not provide collateral cleavage upon target recognition, no signal amplification takes place, resulting in a relatively higher LOD. Hu et al. reported an Horse-radish peroxidase (HRP) based signal amplification step for Cas9 based LFAs. In their work they showed simple and fast freezing-based dual labeling of AuNPs with both DNA and HRP proteins. The HRP protein can catalyzed a color reaction, resulting in test lines being better visual even at 1–10 nM concentrations of target sequence (Hu et al., 2020).
3.2.2. Other colorimetric based read-out sensors
Besides the use of LFAs, other methods have been developed to visualize the results of CRISPR detection of nucleic acids in a POC device. These sensing methods can roughly be divided in methods that do or do not require prior target amplification. First, we will discuss the methods that require some sort of target amplification prior to their sensing technique.
For pathogen and disease related nucleotide sensing, being able to discriminate on the single nucleotide level can be extremely important. An example application where sensing with SNP resolution is important, is the detection of the Zika virus pathogen: the American and African Zika strain have only a single nucleotide difference. Pardee et al. developed a paper-based POC method based on so-called “toehold switches” to distinguish American and African Zika strains (Fig. 5 A). Toehold switches are regulators that enable precise control over gene expression (Green et al., 2014). Upon binding of a “trigger” sequence, the toehold switch can activate gene expression. In the case of the paper-based POC method, the toehold switch, once activated, induces the translation of an enzyme that converts a yellow substrate to a purple product. This color change can easily be observed by eye.
Fig. 5.

A) NASBACC reaction. During amplification trigger sequences are incorporated that act as activators for the toehold switches. If PAM sequences are present, cleavage will take place by RNP Cas9 complex, resulting in a loss of trigger sequences. Reproduced with permission from (Pardee et al., 2016). Copyright 2020 Elsevier. B) Schematic of RCA-based isothermal reaction, resulting in ssDNA sequences with a three-dimensional structure that facilitate dCas9 binding, inducing HRP fusion and activation. Reproduced with permission from (Wang et al., 2020). Copyright 2020 American Chemical Society C) Schematic representation of the Cas12a RNP and Cas13a RNP posing target-dependent trans-cleavage activity, resulting in a degradation of linker DNA, inhibiting AuNP aggregation. Reproduced with permission from (Yuan et al., 2020). Copyright 2020 American Chemical Society.
The trigger sequence is key in this colorimetric detection method and is artificially introduced in the nucleotide sequences of the Zika viruses during isothermal amplification. Cas9 was added after amplification and could distinguish between American and African Zika by the single nucleotide difference in the PAM sequence. The American Zika strain could trigger Cas9's cleavage activity, resulting in a loss of the trigger sequences by enzymatic cleavage. The PAM sequence defines which different sequences could be used as target substrates, so the RNP complex can only detect complementary sequences that include a PAM sequence. Therefore, only African and not American Zika viruses could induce the color change from yellow to purple by trigger sequence binding to the toehold switch. The individual components of the reaction could be freeze-dried, enabling easy incorporation into a paper-based sensor. This single nucleotide resolution CRISPR sensing method was called NASBACC and is, based on the isothermal amplification in combination with the paper-based platform, a low-cost sensor to be used in low resource settings (Pardee et al., 2016).
Qiu et al. developed a colorimetric detection system comprising of isothermal amplification, detection and reporting based on rolling circle amplification (RCA) in a single reaction tube (Wang et al., 2020). The RCA step allows the dsDNA targeting dCas9 to sense microRNA (miRNA) sequences with single nucleotide precision in clinical serum samples (Fig. 5B). miRNA are small non-coding RNA sequences that function in RNA silencing and post-transcriptional regulation of genes, and altered miRNA expression has been related to multiple diseases. RCA is an isothermal unidirectional amplification method that, different from other amplification techniques, continuously adds single nucleotides to a primer, which results in a long ssDNA molecule containing tens to hundreds of tandem repeats, complementary to a template sequence. The long ssDNA sequence that is formed is self-complementary, forming dsDNA secondary structures. These dsDNA structures are targeted by the spacer sequence of the dCas9-RNP complex. Instead of ‘regular’ dCas9 effector proteins, dCas9-split horseradish peroxidase (HRP) fusion enzymes are used. These effector proteins have half of an HRP enzyme attached. When two effector proteins are in close proximity due to the three-dimensional structures of the dsDNA structures, fusion of the attached HRP enzymes can take place, resulting in an active enzyme. The HRP activity can be followed by a simple color reaction. The current system, as presented in the published work of Qiu et al. requires less than 4 h for femtomolar detection levels. By integrating the system in a microfluidic device, optimizing protein concentrations and assay steps we believe that this total reaction time can be reduced, and the procedure could be simplified, enabling POC detection.
Yuan et al. adapted a AuNP-based colorimetric assay by incorporating Cas12a and Cas13a effector proteins for target amplification-free CRISPR sensing (Yuan et al., 2020). Traditional AuNP colorimetric assays are based on hybridization between target nucleotides and nucleotides present on the AuNPs. The AuNPs are typically hybridized with (at least) two different sequences that correspond to different parts of the target sequence. When multiple AuNPs hybridize with the same target sequence, particle aggregation is initiated. Due to the electromagnetic properties of the AuNPs, this aggregation will initiate a color change from red to purple, which can easily be observed by the naked eye. However, faint color changes of low concentration of target sequence result in low signal-to-noise ratios. Furthermore, for each new target sequence, new AuNPs must be conjugated with different DNA probes. To circumvent this, Yuan et al. designed a CRISPR sensing technique where the RNP complex acts as a target recognition element and the AuNPs are used for signal amplification (Fig. 5C). In the case of target recognition, Cas12a/13a initiates collateral cleavage of all ssDNA/ssRNA, allowing degradation of the sequence needed for AuNP aggregation. Upon addition of AuNPs, no aggregation will take place, indicating the presence of the target DNA. The absorbance at 520 nm could be used to find a linear correlation between target sequence concentrations and red color shift of the solution. The LOD of this detection system was as low as 500 fM, in the same order of magnitude as established fluorescent CRISPR sensing methods (Gootenberg et al., 2017) (Li et al., 2019). Zhou et al. used a similar detection technique based on AuNPs. However, instead of only a red to blue color shift as a read-out, two reaction tubes where used for orthogonal color readout. This method allows quantification in a larger concentration range than usual (Zhou et al., 2020).
Shao et al. proposed a visual and stand-alone SNP detection method based on CRISPR/Cas12a cleavage (Shao et al., 2019). By their volumetric bar-chart chip, multiple cancer mutations could be identified and quantified in serum. The sensing mechanism is based on platinum nanoparticles (PtNPs), which are tethered to magnetic beads by ssDNA. The microfluidic chip consists of a top slide and a bottom slide (Fig. 6 ). On the bottom slide three different chambers are present, that can be connected by moving the top slide (‘SlipChip concept'). Target sequences are added to the first chamber. In the presence of target DNA, Cas12a will degrade the ssDNA tether, resulting in free PtNPs in solution. By the sliding mechanism the free nanoparticles are transferred to the second chamber, while a magnet retains the magnetic beads in the first chamber. If no target sequence is present in the first chamber, the PtNPs are retained in the first chamber. In the second chamber, the PtNPs catalyze the reaction of H2O2 to O2, forming an overpressure that pushes detection-ink into the readout channel, over a certain distance. The amount of PtNPs is related to the amount of O2 produced and in this way the distance over which the ink travels can be related to the amount of target sequence present in the first chamber. The platform can easily be adapted for multiplexing by increasing the amount of parallel reaction chambers. Although only Cas12a was used as the sensing enzyme, other Cas family proteins could also be applied in their platform by adjusting the tether between the PtNP and the magnetic bead. The detection limit was consistent with established methods (10 pM) (Gootenberg et al., 2017).
Fig. 6.

Working principle of the volumetric bar-chart chip as developed by Shao et al. a) The modified CRISPR/Cas12a detection system based on the PtNPs and magnetic beads. Upon target sequence binding by Cas12a, the ssDNA tether will be cleaved and PtNPs and magnetic beads can be separated. b) General overview of the ‘SlipChip’ design and operation. c) Cross-section view of the chip in the state of assay/reagent loading, sliding, and readout. Reproduced with permission from (Koo et al., 2018a). Copyright 2019 American Chemical Society.
dCas9 was also utilized in a single microring resonator (SMR) biosensor, enabling real-time detection of both DNA and RNA samples (Koo et al., 2018a). A SMR is a set of waveguides of which one is a closed loop. Light of a specific wavelength is coupled into the waveguide and propagates in it by total internal reflection. However, the light can interfere with a small region outside the waveguide by its so-called evanescent field. Changes in refractive index in this region affect the wavelength of the light within the waveguide. Because of the ring structure, the light passes the region of small changes in refractive index multiple times, enhancing the wavelength shift induced. In this example, the ring was functionalized with ssDNA/ssRNA to capture the target sequences, and binding of the target sequence induces the change in the refractive index, which is sensed. dCas9 is added to the assay to induce a three times larger change in refractive index and in this way ‘amplify’ the signal. With this method, genetically very similar tick-borne diseases could be distinguished in serum samples at the single molecule level within 20 min. The detection sensitivity of around 0.6 aM is about 100 times lower than that of conventional RT-PCR assays (Koo et al., 2018a).
3.3. Electronic read-out systems
Several sensor types have been created by researchers so far, in which no target-amplification is needed to measure at a very low LOD. These sensors are a graphene-based field effect transistor (gFET) (Hajian et al., 2019), nanopore sensors (Weckman et al., 2019; Yang et al., 2018), electrochemical sensors (Bruch et al., 2019; Dai et al., 2019; Xu et al., 2020; D. Zhang et al., 2020) and a conductivity sensor combined with a DNA gel (English et al., 2019).
3.3.1. Binding-dependent CRISPR sensors
The graphene-based field effect transistor (gFET) and the nanopore sensors do not use the cleavage of an effector protein, but rather the binding of dCas9-crRNA complexes.
The gFET was the first sensor to employ electronic readout in CRISPR sensing. Using dCas9, Haijan et al. use a gFET to obtain an electronic signal when a specific DNA sequence is present (Hajian et al., 2019). Their sensor is based on the principle that the proximity of a charged DNA molecule to the graphene will reduce the resistance of the system, leading to a higher current response. This proximity is achieved by the immobilizing dCas9 on the graphene, which then selectively binds target DNA and keeps it close to the graphene surface. The dCas9 is bound to the graphene surface via a PBA linker comprised of a planar pyrene ring system that electrostatically interacts with the graphene and a carboxylate group that covalently couples to the dCas9 (Fig. 7 A). Subsequently the rest of the surface is blocked to prevent the non-specific adsorption of charged molecules. A drop of the sample is placed on the device, left to incubate, after which it is washed and the conductivity of the system is measured via two platinum electrodes on the graphene. They report a LOD of 15 fM and a total measurement time of 15 min (Hajian et al., 2019).
Fig. 7.

An overview of the mechanisms in the different electronic sensors described in the literature. A) gFET (Hajian et al., 2019), B) nanopore sensor, adjusted from (Yang et al., 2018), C) different types of electrochemical sensors with MB as an electrochemical tag, based on targeted (II) and collateral cleavage (I & III) (Dai et al., 2019; Xu et al., 2020; D. Zhang et al., 2020) D) collateral cleavage based sensor with a glucose oxidase electrochemical tag. Adapted from (Bruch et al., 2019), according to Creative Commons Attribution License (CC BY 4.0) E) collateral cleavage of DNA-gel based sensor with combined colorimetric and amperometric readout (English et al., 2019).
The nanopore sensors exploit the binding of dCas9 effector proteins to DNA and are based on the characteristic blockade signal of DNA-Cas9 complexes translocating through a nanopore (Fig. 7B). This translocation results in a dip in the ionic current through the pore that could be analyzed and interpreted. Using nanopores as a sensing strategy enables detection of dsDNA without unzipping or temperature changes and also opens up opportunities for multiplexing. Yang et al. showed that the binding position of the dCas9 effector protein could be deduced from the ionic current spike, which was later also confirmed by Weckman et al. (Weckman et al., 2019; Yang et al., 2018). The latter authors designed multiple crRNA sequences for dCas9 that could all bind to the same DNA sequence, creating ‘barcodes’ in the ionic current traces specific for the different DNA sequences. In this way they were able to identify two types of DNA target sequences in a mixture of ‘background’ DNA, demonstrating the specificity and selectivity of the dCas9 effector protein in combination with nanopore sensing.
While it is possible to observe the difference between the translocation signal of the dCas9 and the DNA, the measurement method is still dependent on a single signal. While this means that a single copy can be detected, it also means that for samples with low amounts of the specific DNA sequence, the time needed for a measurement is very long, as the chance of the target DNA strand appearing in the proximity of the pore is not very high. While nanopore sensing could be useful for identifying DNA, it is low throughput, and therefore a very slow method for low LOD measurements. A more recent publication by Nouri et al. potentially solved this measurement time issue of low concentrations of target DNA exploiteding the trans-cleavage of Cas12a and measuring the occurrence of reporter DNA fragments (Nouri et al., 2020).
3.3.2. Electrochemical sensors
For electrochemical sensors different strategies have been employed. In several systems a reporter methylene blue (MB) is placed on an electrode surface via DNA as an electrochemical signaling tag, measuring directly at the cleavage site (Fig. 7C). In a second system, a catalyst is placed on the surface, while the compound to be oxidized is provided in the solution, which is then measured with an electrode located at some distance in their device (Fig. 7D). Another method uses an electrochemical reaction with Ru(phen)2(dppz) 2+-DNA complexes to create a red luminescence indicating the presence of cleaved RNA after amplification.
There are systems using MB reported that are very closely related in their methods, with a few key differences. Dai et al. exploited the trans-cleavage activity of Cas12a to obtain a high signal without the need for DNA amplification via an electrochemical sensor (Dai et al., 2019). In their “E-CRISPR” sensor a gold measurement electrode (measuring 33 by 8 mm) is used, which is functionalized with a thiolated-ssDNA-MB conjugate (Fig. 7C). For their measurement, a (10 min preincubated) 20–30 μL drop of sample is deposited onto the electrode area. In case of the target DNA being present, the Cas12a complex is activated and will cut the single stranded DNA immobilized on the surface, which releases the MB from the surface. After washing, a square wave voltammetry measurement is performed, in which the reduction of MB will cause a peak in the current at a certain potential. The removal of MB from the surface via cleavage decreases the current peak after the incubation step as compared to before. This current difference indicates the presence of the target DNA and an experimental LOD of 50 pM was obtained (Dai et al., 2019).
In a later paper by the same group, Xu et al. used the cleavage activity of Cas9 to enhance the performance of an E-DNA sensor, previously reported by Fan et al., which measures hybridization of DNA via proximity of an electrochemical signaling tag to a gold surface (Fan et al., 2003). In this sensor, they used single stranded hairpin DNA with a MB electrochemical signaling tag that was immobilized on gold electrodes. In the standard sensor, without CRISPR-enhancement, the distance between the signaling tag and electrode upon binding of target ssDNA to the hairpin will result in an increase of the electron tunneling distance via the unfolding of the hairpin, measurable as a decrease in the current. The addition of the Cas9 effector protein will cause the electrochemical signaling probe to be released from the surface via cleavage upon recognition of PAM and target sequence (Fig. 7C–III). This specific cleavage of Cas9 lowers the LOD of the “normal” sensor by at least two orders of magnitude to 100 fM concentration of target DNA (Xu et al., 2020). In this second publication, the sensor was not (yet) incorporated in a POC device, but can be applied to a similar electrode as the system of Dai et al. Using Cas12a, a larger signal was obtained and the LOD could be reduced to 10 fM. The increase in sensitivity compared to the first sensor is caused using a DNA-hairpin, which brings the MB closer to the surface and increases the current response. However, the fact that there is no collateral cleavage, but only cutting of the target DNA, means that another step could be made towards increasing the sensitivity of the system.
Zhang et al. used the same approach as Xu et al. to increase the sensitivity of the system by Dai et al., with MB molecules brought close to a gold surface by hairpin DNA (Zhang et al., 2020) (Fig. 7C–II). The hairpin in their system could bring the MB closer to the surface than the dsDNA, increasing the response, and caused less problems with steric hindrance than the system of Dai et al.. Where Xu et al. cleaved the target DNA directly on the surface, Zhang et al. used the same method as Dai et al. where the target DNA was cleaved in solution by Cas12a, and used the subsequent collateral cleavage of the reporter hairpin DNA on the surface to get a signal response. The replacement of the dsDNA by hairpin DNA allowed them to increase their current response and reduce the LOD to 30 pM.
A more indirect measurement method is used in the device created by Bruch et al., where glucose oxidase is bound to the surface of a microfluidic chip via an RNA strand. The authors use the collateral cleavage activity of Cas12a to cut the reporter RNA, which then either makes it impossible for the glucose oxidase to be bound to the channel surface, or releases it from the channel surface (for the off-chip and on-chip version of their device respectively). After a washing step, depending on the presence and concentration of the target DNA, the glucose oxidase will oxidize the glucose in the channel solution, producing D-glucono-δ-lactone and H2O2. The H2O2 is then amperometrically detected in the downstream electrochemical cell. In case of the presence of target DNA, the current will be lower than in a control measurement (Fig. 7d). Off-chip cleavage provided a signal with a LOD of 18, 10, and 2 pM for cleavage times of 1 h, 3 h and 24 h respectively, with a slightly different dynamic range up to 1000 pM. On-chip cleavage could reach a LOD of 2.2 nM with hourly solution changes (Bruch et al., 2019). The authors report that their device has lower material costs than the devices mentioned above and requires less hands-on time for taking the measurement, but this comes at the cost of the need for more sophisticated equipment for sample handling.
Zhou et al. used Cas13a to detect the presence of miRNA from tumor cells (Zhou et al., 2020). The presence of the miRNA which induces collateral cleavage of an EXPAR specific pre-trigger primer. In its uncleaved state the pre-trigger primer is not able to bind an EXPAR template, however upon trans-cleavage by the RNP, the primer can hybridize with the amplification template, initiating the formation of large amounts of dsDNA via an EXPAR amplification step. This dsDNA formation, which acts as an signal amplification step, can be visualized by addition of Ru(phen)2(dppz)2+, which produces a luminescence signal upon interaction by the major groove of dsDNA. Under optimized conditions, a limit of detection down to 1 fM could be reached. Their sensing method was translated to a paper-based bipolar electrode electrochemiluminescence (pBPE-ECL) biosensing platform, consisting of a simple paper based device, on which an electrochemical reaction was driven by a portable DC voltage supply. Their luminescence approach makes specific detection possible without the need of extensive washing procedures, which improves reproducibility and simplifies the experimental procedure, which is of great interest in POC sensing (T. Zhou et al., 2020).
3.3.3. DNA-gel based sensing
Another approach was described by English et al., who discovered that one could use CRISPR mediated cleavage of DNA hydrogels as a sensing method (English et al., 2019). This was then applied to two possible systems for electronic readout.
The first system was a DNA-gel fuse. A conductive carbon black nanoparticle loaded DNA-gel was cured on top of two interdigitated electrodes to connect them. If the DNA is cut by the RNP-target DNA complex, the connection between the electrodes will be interrupted, increasing the resistance of the system (English et al., 2019). The gel acts as a fuse between the electrodes. For measurements, the electrode pad is placed in an Eppendorf tube which also contains the solution with the RNP complex. The measurement is however not quantitative and takes 3–12 h, depending on the concentration of target DNA (English et al., 2019). To obtain a faster and more quantitative result, the authors created a paper-based POC device, in which they combined optical and electronic read-out (English et al., 2019). They inserted a DNA cross-linked polyacrylamide gel precursor, that together with ssDNA crosslinks to form a hydrogel gel in the paper channels. The extent of gel formation depends inversely on the extent of degradation of the ssDNA in the gel. By degrading the crosslinker via trans-cleavage by Cas12a, the channel is less obstructed by the gel, and the level of flow of a colored buffer that was added afterwards can therefore be linked to the amount of dsDNA that complemented with the Cas12a effector protein (Fig. 7E). With the visual output, a concentration down to 400 pM could be detected (English et al., 2019). To reduce the user error from the visual output, an electric readout was achieved by including two electrodes that sandwiched the channel. These electrodes recorded the conductance, which depends on the number of electrolyte ions in the channel introduced by the buffer. The conductance change could be directly linked to the penetration length of the buffer and with this method a similar LOD could be obtained. This device needs a pre-incubation time of 4 h and a 2-min readout.
4. Challenges in POC CRISPR sensing systems
The field of CRISPR sensing is premature: With only half a decade of research there are still many challenges to overcome. In this section the most common and (in our opinion) limiting challenges are discussed that are faced in the implementation of CRISPR sensing, both in general and specifically in POC applications.
4.1. Sequence limitations
The sequences one can detect using CRISPR/Cas effector proteins are limited, which is a general problem in CRISPR sensing. CRISPR effector proteins are guided by crRNA and are able to recognize target sequences on DNA and RNA that are complementary to the crRNA. The spacer sequence, present on the crRNA, can be tailored to the target sequence. However, as discussed above for (d)Cas9 and Cas12a/b, an effector specific PAM sequence is crucial to unwind the dsDNA strands and allow complementation between crRNA and the target sequence. Depending on the Cas effector protein family, mismatches between spacer and target sequence are tolerated. In general, the overall mismatch tolerance depends on the number of mismatches and the position of the mismatch compared to the PAM sequence, where distant mismatches are generally more tolerated. It was shown for Cas9 that less than 5% of crRNA sequences with two or more mismatches to the target sequence is functional in target binding (Anderson et al., 2015) and mismatches near the PAM region are less tolerated (Pattanayak et al., 2013). However, at a high abundance of RNP complexes, mismatches are more tolerated and even mismatches in the PAM region result in enzymatic target cleavage by Cas9 (Pattanayak et al., 2013). Furthermore, the sequence and length of the gRNA seems to influence the mismatch tolerance, varying dramatically between gRNAs (Hsu et al., 2013; Ran et al., 2013). Compared to Cas9, Cas12a has a lower mismatch tolerance and greater specificity (Kim et al., 2016; Kleinstiver et al., 2016). This lower tolerance can be explained by the fact that changes in the three-dimensional structure of the effector protein require more base pair matching between spacer and target DNA for Cas12 effector proteins compared to Cas9 (Strohkendl et al., 2018). When designing CRISPR assays where high specificity is crucial, the choice of effector protein and sensing region on the target sequence needs to be carefully selected. to avoid background signals of non-specific (although probably low) background signal.
When discriminating DNA sequences on the single nucleotide level, one must carefully design the spacer sequence in such a way that this SNP is present in a region where the effector protein is sensitive to single base pair mismatches (generally close to the PAM region) (Lei et al., 2017; Li et al., 2018; Yamano et al., 2016). Another possibility would be introducing SNPs in the PAM region itself to distinguish between pathogen genotypes, as was introduced in the NASBACC method (Pardee et al., 2016). A downside of this method is the need of SNPs of interest in a sequence that can act as a PAM sequence for a certain effector protein, which is not always possible. Selection of an effector protein from a different species or different family could offer a solution, since each effector protein has a specific PAM sequence. However, in the case of short target sequences it might be impossible to find a PAM sequence matching one of the effector proteins.
To avoid PAM related sequence limitations with dsDNA sensing, the PAM sequence can be introduced during an amplification step as was shown by Li et al. in their HOLMES approach (L. Li et al., 2019; Li et al., 2018). In their approach target recognition is only dependent on the complementation between the target sequence and the crRNA's spacer sequence.
Completely PAM-free sensing is enabled by effector proteins from the Cas14 and Cas13 family. Cas14 only requires a thymine-rich PAM sequence for dsDNA recognition, but ssDNA recognition does not require a specific sequence adjacent to the target sequence. In the case of Cas13 effector proteins a PFS is needed. Here it negatively affects the target recognition when the first base following the protospacer is a guanine (Abudayyeh et al., 2016). These limitations can generally be overcome by shifting the target sequence in such a way that the first base following the protospacer will be any base except a guanine (Gootenberg et al., 2018).
4.2. Standardization
Standardization is the key to effective POC CRISPR sensing. Both procedures and individual tests have to be standardized in order to be sure that every user will obtain the same results. While standardization is a general issue in POC testing, it becomes more important in the case of protein-based sensors. Salt levels, temperature, pH and/or the abundance of reaction inhibitors might interfere in the assembly between (s)gRNA and effector protein and the cleavage activity of the effector proteins, inducing a change in the generated signal (Dai et al., 2019; Mekler et al., 2016; Sundaresan et al., 2017). These issues can be solved by providing a test dilution series for an on-site calibration or spike-in samples with known concentrations, which however compromises the ease of testing. Ideally, CRISPR sensing can be standardized like pH paper, using a color chart to compare and interpret the result.
4.3. Quantitative analysis
For molecular diagnosis, quantitative analysis is of great importance. For multiple diseases, the concentration level of nucleic acids found in liquid biopsies varies with progression of the disease and quantification may be quantitative needed for diagnostics. Similar to standardization, quantification of the results is difficult to establish without on-site calibration. Established CRISPR based sensing systems like HOLMES, DETECTR and SHERLOCK are unfortunately not able to quantify the amount of nucleic acids and are only qualitative. HOLMESv2 is able to quantify the nucleic acid target, but only when combined with a real-time PCR reaction, which requires bulky machinery. Here, measuring multiple samples at the same time, as in multiplex analysis, would allow for on-board calibration, where known target concentrations can be measured simultaneously under the same conditions, cancelling out factors that influence the performance of the sensor and affect measurement results.
4.4. Multiplexing
Besides the need for calibration, gaining precise and conclusive information by genomic diagnosis requires detection of several target sequences simultaneously, so-called multiplexing (Dincer et al., 2017). This multiplexed genomic sensing in a single reaction volume is challenging due to limitations in signal acquisition strategies, interference between the multiple detection steps and possible cross reactions, compromising both the sensitivity and specificity. Major issue with single reaction volume multiplexing is the non-specific cleavage of the same type of nucleotide as being sensed. This results in a signal bias for the first sequence to be bound to an RNP complex. This binding initiates non-specific collateral cleavage of all ssDNA/ssRNA sequences, possibly destroying the other targets. This issue is only present for Cas13 (ssRNA detection and cleavage), Cas14 (ssDNA detection and cleavage) and in some cases for Cas12 (ssDNA detection and cleavage).
So far only two multiplexed biosensing systems were published based on CRISPR/Cas systems (Gootenberg et al., 2018; Shao et al., 2019). Li et al. describe three different strategies for in vitro based multiplexed CRISPR sensing: Cas effector-based, signal transduction-based and separation-based (Y. Li et al., 2019b). Gootenberg et al. achieved Cas effector-based multiplex sensing of four different genomic targets by using four different Cas effector proteins with cutting preferences to different nucleotide sequences (Gootenberg et al., 2018). This multiplexed approach is limited by the amount of different Cas effector proteins with different nucleotide preferences, which is currently limited by biotechnology.
Signal transduction-based multiplexing requires physically separated signal transducers, that, by immobilization of Cas effectors with different spacer sequences, could sense different target sequences. Interference between RNP and target sequence can be measured by for example electrochemistry (Hajian et al., 2019; W. Zhou et al., 2018). By simultaneous read-out of multiple sensors, multiplex measurements can be achieved. A major downside of this method is that collateral cleavage actitivity cannot be used, decreasing (relative) signal output.
The last, and in our opinion most promising multiplex method is the separation-based method. By physical separation Shao et al. achieved multiplex CRISPR sensing by separating Cas effector proteins in different microfluidic channels. In this way each channel is used to test a different sequence by changing the spacer sequence on the crRNA (Shao et al., 2019). This microfluidic approach, using which spatial separation can be achieved, is not limited by the current status of the CRISPR biotechnology or advanced transducers for read-out. By increasing the number of microfluidic channels, relatively easy to both produce and use POC tests may be developed for highly multiplexed nucleotide sensing.
4.5. Target amplification
To lower the LOD, many of the published CRISPR biosensing methods require nucleic acid amplification prior to CRISPR sensing. Standard amplification methods, such as PCR, are not suitable for incorporation in POC devices due to the need for a bulky thermocycler in combination with the cycling of multiple temperature steps from 60 °C to 95 °C which typically leads to long reaction times (≥90 min).
To allow CRISPR sensing in remote areas without trained personnel, multiple techniques based on isothermal amplification can be used that are more straightforward to use and can be incorporated in small handheld devices. Table 3 shows an overview of the isothermal amplification techniques that have been applied in combination with CRISPR sensing.
Table 3.
An overview of isothermal amplification methods commonly used in combination with CRISPR sensing.
| Name | Description | Typical time | Typical temperature | Refs |
|---|---|---|---|---|
| NASBA | Nucleic acid sequence-based amplification is a method used to amplify RNA. | ~30 min | 41 °C | Pardee et al. (2016) |
| LAMP | Loop-mediated isothermal amplification is a single tube technique for the amplification of DNA. It uses 4–6 primers, which form loop structures to facilitate subsequent rounds of amplification. | 15–60 min | ~65 °C | (Li et al., 2019; Mukama et al., 2020a; Qian et al., 2020) |
| RCA | Rolling circle amplification starts from a circular DNA template and a short DNA or RNA primer to form a long single stranded molecule. | ~120 min | 20–37 °C | Wang et al., 2020 |
| RPA | Recombinase polymerase amplification is a low temperature DNA and RNA amplification technique. | 5–60 min | 37–42 °C | (Y. Chang et al., 2019; English et al., 2019; Kellner et al., 2019; Khan et al., 2019; Sullivan et al., 2019; X. Wang et al., 2020; Williams et al., 2019) |
| SDA | Strand Displacement Amplification (SDA) employs a restriction endonuclease, which is capable of nicking of its recognition site, and a DNA exonuclease deficient polymerase, which is capable of initiating synthesis at a nick. | 90 min | 37–55 °C | Zhou et al., 2018 |
The most often employed isothermal amplification methods are RPA and LAMP, which operate between 37 and 42 °C (could also be used at room temperature, although this influences the reaction kinetics), and ~65 °C respectively. RPA and LAMP offer fast amplification times (from ~15 to 60 min for LAMP and 5–60 min for RPA depending on the initial nucleotide concentrations) which is of great interest for POC applications. A major advantage of RPA over LAMP is that it requires much lower temperatures during amplification. However, lower operation temperatures also have drawbacks, such as non-specific amplification products and the formation of primer dimers (Zaghloul and El-Shahat, 2014). By the introduction of CRISPR-complexes, which are highly specific to their target sequence, these unwanted products do not interfere with the sensing process and therefore do not influence the end result.
When combining isothermal amplification and CRISPR sensing in a single POC device, the operation temperature of the isothermal amplification should be in the same range as the operation temperature of the RNP complex. Different effector proteins have different temperature optima. The activity of Cas12a for example has an optimum temperature around 28 °C, while Cas12b has an optimal activity around 48 °C. The high temperature tolerance of Cas12b was exploited in HOLMESv2, where both LAMP and CRISPR sensing was incorporated in a one-pot reaction at 55 °C (L. Li et al., 2019).
As an alternative to matching the temperatures of amplification and sensing, one can physically separate both reactions. To enable one-pot CRISPR reactions dehydrated CRISPR may for example be attached to the lid of a reaction tube for enhanced temperature stability (Wu et al., 2020b) (Qian et al., 2019). Wu et al. showed in 2020 their design of a lid where the Cas12a solution was separated from the reaction solution by a sealed film (Wu et al., 2020a). After the LAMP amplification step, the lid could be tightened, allowing a needle to puncture the sealed film (Fig. 8 A). In their most recent publication these authors developed a Poly(methyl methacrylate) chip to physically separate the Cas12a solution and LAMP reaction products (Wu et al., 2020b). After LAMP reaction, the operator can move the liquid from the reaction to the detection chamber by shaking the chip (Fig. 8B).
Fig. 8.

One-pot reactions by physically separating CRISPR from LAMP isothermal amplification reactions. A) Special designed reaction tube. This lid consists of two components, where the bottom part separates Cas effector proteins from the tube by a sealed film. The top part of the lid consists of a needed, that could puncture film to start the CRISPR sensing process. Taken from (Wu et al., 2020a) with permission. Copyright 2020 Elsevier. B) Poly(methyl methacrylate) chip where a reaction tube could be connected to the chip via a connection structure. LAMP amplification and heating takes place in the tube, after which the mixture can be moved to the Cas effector solution for signal read-out by fluorescence. Taken from [69] with permission. Copyright 2020 Elsevier.
Yin et al. developed an alternative method for separating target amplification and CRISPR reactions. In their dynamic aqueous multiphase reaction (DAMR) system they took advantage of density differences of sucrose concentrations (Yin et al., 2020). By phase separation, a two-phase system was established. In the high-density bottom layer, the RPA reaction was initiated. The amplified nucleotides could diffuse to the low-density top-phase where the Cas12a RNP could complement and initiate collateral cleavage activity of quenched fluorescent probe sequences. In this way incompatible reactions could be performed in one pot. The system was evaluated in reaction tubes and multiplex three-dimensional printed inserts for multi-well plates. It was found that 10–100 copies of Human Papilloma Virus DNA spiked in untreated human plasma could be sensed in less than 1 h, showing the high tolerance of the DAMR system to inhibitors.
5. Conclusion and future outlook
While many CRISPR sensing applications were published in the last four years, the CRISPR nucleic acid detection field is still very much in its infancy. As discussed in this review paper, there are still many challenges before CRISPR sensing can replace established biomolecular nucleic acid sensing techniques such as PCR.
In this review, we specifically focused on the POC detection of nucleic acids. It was shown that CRISPR sensing may provide rapid diagnostics, but assay times are still influenced by the need for target amplification. Therefore, type V and VI effector proteins have the highest potential in POC applications as they provide signal amplification by the untargeted collateral cleavage activity of reporter nucleic acids. This method has been used for fluorescent, colorimetric and electronic read-out systems and could detect nucleic acids at as low as femtomolar concentrations in a diversity of sample matrixes. Besides DNA and RNA sensing, detection of proteins and small (inorganic) molecules was also enabled by CRISPR-based sensing (Dai et al., 2019; Liang et al., 2019; Xiong et al., 2020).
In the current COVID-19 outbreak, a number of CRISPR-based POC sensing method have been adapted or newly developed (Broughton et al., 2020; Curti et al., 2020; Ding et al., 2020; Guo et al., 2020b; Kellner et al., 2019; Wang et al., 2020 ; Zhang et al., 2020). This shows the enormous potential and versatility of CRISPR-based POC sensing in the detection and qualitative analysis of infectious pathogens. Most publications state that their CRISPR sensing method can be adapted and reconfigured within days to detect other viruses, arming the medical diagnostic world for future pandemics.
Future work will need to focus on portable microfluidic-based POC cartridges, carrying all needed reagents on-board, either in-solution or lyophilized. At the same time, laboratory based quantitative testing devices are needed, such as those based on microchips. The focus will mainly be on multiplex sensing, allowing on-board quality checks and calibration, while measuring for multiple sequences simultaneously. Furthermore, we expect a trend with (isothermal) amplification being replaced by (genetically modified) Cas effector proteins with collateral cleavage activity for signal amplification purposes.
CRediT authorship contribution statement
Jeanne E. van Dongen: Writing - original draft, Writing - review & editing. Johanna T.W. Berendsen: Writing - original draft, Writing - review & editing. Renske D.M. Steenbergen: Writing - review & editing. Rob M.F. Wolthuis: Writing - review & editing. Jan C.T. Eijkel: Writing - review & editing, Supervision. Loes I. Segerink: Writing - review & editing, Supervision.
Declaration of competing interest
RDMS is a minority stockholder of Self-screen B.V., a spin-off company of VUmc.
Acknowledgments
The Weijerhorst Foundation and the Cancer Center Amsterdam areacknowledged for financial support. We thank Daniel Warmerdam for helpful insights and support.
References
- Abudayyeh O.O., Gootenberg J.S., Essletzbichler P., Han S., Joung J., Belanto J.J., Verdine V., Cox D.B.T., Kellner M.J., Regev A., Lander E.S., Voytas D.F., Ting A.Y., Zhang F. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh Omar O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B.T., Shmakov S., Makarova K.S., Semenova E., Minakhin L., Severinov K., Regev A., Lander E.S., Koonin E.V., Zhang F. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;80(353) doi: 10.1126/science.aaf5573. aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman R., Mahas A., Mahfouz M. Nucleic acid detection using CRISPR/cas bio-sensing technologies. ACS Synth. Biol. 2020 doi: 10.1021/acssynbio.9b00507. [DOI] [PubMed] [Google Scholar]
- Anderson E.M., Haupt A., Schiel J.A., Chou E., Machado H.B., Strezoska Ž., Lenger S., McClelland S., Birmingham A., Vermeulen A., Smith A.V.B. Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity. J. Biotechnol. 2015;211:56–65. doi: 10.1016/j.jbiotec.2015.06.427. [DOI] [PubMed] [Google Scholar]
- Bai J., Lin H., Li H., Zhou Y., Liu J., Zhong G., Wu L., Jiang W., Du H., Yang J., Xie Q., Huang L. Cas12a-Based on-site and rapid nucleic acid detection of african swine fever. Front. Microbiol. 2019;10:2830. doi: 10.3389/fmicb.2019.02830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin A., Quinquis B., Sorokin A., Dusko Ehrlich S. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- Broughton J.P., Deng X., Yu G., Fasching C.L., Servellita V., Singh J., Miao X., Streithorst J.A., Granados A., Sotomayor-Gonzalez A., Zorn K., Gopez A., Hsu E., Gu W., Miller S., Pan C.-Y., Guevara H., Wadford D.A., Chen J.S., Chiu C.Y. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruch R., Baaske J., Chatelle C., Meirich M., Madlener S., Weber W., Dincer C., Urban G.A. CRISPR/Cas13a-Powered electrochemical microfluidic biosensor for nucleic acid amplification-free miRNA diagnostics. Adv. Mater. 2019;31:1905311. doi: 10.1002/adma.201905311. [DOI] [PubMed] [Google Scholar]
- Chang Y., Deng Y., Li T., Wang J., Wang T., Tan F., Li X., Tian K. Visual detection of porcine reproductive and respiratory syndrome virus using CRISPR-Cas13a. Transbound. Emerg. Dis. 2019;67:564–571. doi: 10.1111/tbed.13368. [DOI] [PubMed] [Google Scholar]
- Chen J.S., Doudna J.A. The chemistry of Cas9 and its CRISPR colleagues. Nat. Rev. Chem. 2017;1 doi: 10.1038/s41570-017-0078. [DOI] [Google Scholar]
- Chen J.S., Ma E., Harrington L.B., Da Costa M., Tian X., Palefsky J.M., Doudna J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science (80-. ) 2018;360:436–439. doi: 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corstjens P., Zuiderwijk M., Brink A., Li S., Feindt H., Niedbala R.S., Tanke H. Use of up-converting phosphor reporters in lateral-flow assays to detect specific nucleic acid sequences: a rapid, sensitive DNA test to identify human papillomavirus type 16 infection. Clin. Chem. 2001;47:1885–1893. doi: 10.1093/clinchem/47.10.1885. [DOI] [PubMed] [Google Scholar]
- Crannell Z., Castellanos-Gonzalez A., Nair G., Mejia R., White A.C., Richards-Kortum R. Multiplexed recombinase polymerase amplification assay to detect intestinal Protozoa. Anal. Chem. 2016;88:1610–1616. doi: 10.1021/acs.analchem.5b03267. [DOI] [PubMed] [Google Scholar]
- Curti L., Pereyra-Bonnet F., Gimenez C. bioRxiv; 2020. An Ultrasensitive, Rapid, and Portable Coronavirus SARS-CoV-2 Sequence Detection Method Based on CRISPR-Cas12; p. 971127. 2020.02.29. [DOI] [Google Scholar]
- Dai Y., Somoza R.A., Wang L., Welter J.F., Li Y., Caplan A.I., Liu C.C. Exploring the trans-cleavage activity of CRISPR-cas12a (cpf1) for the development of a universal electrochemical biosensor. Angew. Chem. Int. Ed. 2019;58:17399–17405. doi: 10.1002/anie.201910772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincer C., Bruch R., Kling A., Dittrich P.S., Urban G.A. Multiplexed point-of-care testing – xPOCT. Trends Biotechnol. 2017;35:728–742. doi: 10.1016/j.tibtech.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X., Yin K., Li Z., Liu C. bioRxiv; 2020. All-in-One Dual CRISPR-Cas12a (AIOD-CRISPR) Assay: A Case for Rapid, Ultrasensitive and Visual Detection of Novel Coronavirus SARS-CoV-2 and HIV Virus. 2020.03.19.998724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A., O'Connell M.R., Knight S.C., Burstein D., Cate J.H.D., Tjian R., Doudna J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–273. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English M.A., Soenksen L.R., Gayet R.V., De Puig H., Angenent-Mari N.M., Mao A.S., Nguyen P.Q., Collins J.J. Programmable CRISPR-responsive smart materials. Science. 2019;365:780–785. doi: 10.1126/science.aaw5122. 80. [DOI] [PubMed] [Google Scholar]
- Fan C., Plaxco K.W., Heeger A.J. Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of DNA. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9134–9137. doi: 10.1073/pnas.1633515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a and Csm6. Science. 2018;360:439–444. doi: 10.1126/science.aaq0179. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A., Myhrvold C., Bhattacharyya R.P., Livny J., Regev A., Koonin E.V., Hung D.T., Sabeti P.C., Collins J.J., Zhang F. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A.A., Silver P.A., Collins J.J., Yin P. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014;159:925–939. doi: 10.1016/j.cell.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guk K., Keem J.O., Hwang S.G., Kim H., Kang T., Lim E.K., Jung J. A facile, rapid and sensitive detection of MRSA using a CRISPR-mediated DNA FISH method, antibody-like dCas9/sgRNA complex. Biosens. Bioelectron. 2017;95:67–71. doi: 10.1016/j.bios.2017.04.016. [DOI] [PubMed] [Google Scholar]
- Guo L., Sun X., Wang X., Liang C., Jiang H., Gao Q., Dai M., Qu B., Fang S., Mao Y., Chen Y., Feng G., Gu Q., Wang L., Wang R.R., Zhou Q., Li W. bioRxiv; 2020. SARS-CoV-2 Detection with CRISPR Diagnostics. 2020.04.10.023358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Sun X., Wang X., Liang C., Jiang H., Gao Q., Dai M., Qu B., Fang S., Mao Y., Chen Y., Feng G., Gu Q., Wang R.R., Zhou Q., Li W. SARS-CoV-2 detection with CRISPR diagnostics. Cell Discov. 2020 doi: 10.1038/s41421-020-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajian R., Balderston S., Tran T., deBoer T., Etienne J., Sandhu M., Wauford N.A., Chung J.Y., Nokes J., Athaiya M., Paredes J., Peytavi R., Goldsmith B., Murthy N., Conboy I.M., Aran K. Detection of unamplified target genes via CRISPR–Cas9 immobilized on a graphene field-effect transistor. Nat. Biomed. Eng. 2019;3:427–437. doi: 10.1038/s41551-019-0371-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington L.B., Burstein D., Chen J.S., Paez-Espino D., Ma E., Witte I.P., Cofsky J.C., Kyrpides N.C., Banfield J.F., Doudna J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018;362:839–842. doi: 10.1126/science.aav4294. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass K.N., Bao M., He Q., Park M., Qin P., Du K. bioRxiv; 2020. Integrated Micropillar Polydimethylsiloxane Accurate CRISPR Detection (IMPACT) System for Rapid Viral DNA Sensing. 2020.03.17.994137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q., Yu D., Bao M., Korensky G., Chen J., Shin M., Kim J., Park M., Qin P., Du K. High-throughput and all-solution phase African Swine Fever Virus (ASFV) detection using CRISPR-Cas12a and fluorescence based point-of-care system. Biosens. Bioelectron. 2020;154 doi: 10.1016/j.bios.2020.112068. [DOI] [PubMed] [Google Scholar]
- Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., Cradick T.J., Marraffini L.A., Bao G., Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M., Yuan C., Tian T., Wang X., Sun J., Xiong E., Zhou X. Single-step, salt-aging-free, and thiol-free freezing construction of AuNP-based bioprobes for advancing CRISPR-based diagnostics. J. Am. Chem. Soc. 2020;142:35. doi: 10.1021/jacs.0c00217. [DOI] [PubMed] [Google Scholar]
- Ishino Y., Shinagawa H., Makino K., Amemura M., Nakatura A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isoenzyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987;169:5429–5433. doi: 10.1128/jb.169.12.5429-5433.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;(337):816–821. doi: 10.1126/science.1225829. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski M.M., Alcantar M.A., Lape I.T., Greensmith R., Huske A.C., Valeri J.A., Marty F.M., Klämbt V., Azzi J., Akalin E., Riella L.V., Collins J.J. A CRISPR-based assay for the detection of opportunistic infections post-transplantation and for the monitoring of transplant rejection. Nat. Biomed. Eng. 2020;4:601–609. doi: 10.1038/s41551-020-0546-5. [DOI] [PubMed] [Google Scholar]
- Karvelis T., Bigelyte G., Young J.K., Hou Z., Zedaveinyte R., Pociute K., Silanskas A., Venclovas Č., Siksnys V. PAM recognition by miniature CRISPR-Cas14 triggers programmable double-stranded DNA cleavage. n.d. [DOI] [PMC free article] [PubMed]
- Kasetsirikul S., Shiddiky M.J.A., Nguyen N.T. Challenges and perspectives in the development of paper-based lateral flow assays. Microfluid. Nanofluidics. 2020 doi: 10.1007/s10404-020-2321-z. [DOI] [Google Scholar]
- Katzmeier F., Aufinger L., Dupin A., Quintero J., Lenz M., Bauer L., Klumpe S., Sherpa D., Dürr B., Honemann M., Styazhkin I., Simmel F.C., Heymann M. A low-cost fluorescence reader for in vitro transcription and nucleic acid detection with Cas13a. PloS One. 2019;14 doi: 10.1371/journal.pone.0220091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner M.J., Koob J.G., Gootenberg J.S., Abudayyeh O.O., Zhang F. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 2019;14:2986–3012. doi: 10.1038/s41596-019-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan H., Khan A., Liu Yufeng, Wang S., Bibi S., Xu H., Liu Yuan, Durrani S., Jin L., He N., Xiong T. CRISPR-Cas13a mediated nanosystem for attomolar detection of canine parvovirus type 2. Chin. Chem. Lett. 2019;30:2201–2204. doi: 10.1016/j.cclet.2019.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Kim J., Hur J.K., Been K.W., Yoon S.H., Kim J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016;34:863–868. doi: 10.1038/nbt.3609. [DOI] [PubMed] [Google Scholar]
- Kleinstiver B.P., Tsai S.Q., Prew M.S., Nguyen N.T., Welch M.M., Lopez J.M., McCaw Z.R., Aryee M.J., Joung J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016;34:869–874. doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S., Lotfy P., Brideau N.J., Oki J., Shokhirev M.N., Hsu P.D. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell. 2018;173:665–676. doi: 10.1016/j.cell.2018.02.033. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B., Kim D. eun, Kweon J., Jin C.E., Kim S.H., Kim Y., Shin Y. CRISPR/dCas9-mediated biosensor for detection of tick-borne diseases. Sensor. Actuator. B Chem. 2018;273:316–321. doi: 10.1016/j.snb.2018.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosack C.S., Page A.L., Klatser P.R. A guide to aid the selection of diagnostic tests. Bull. World Health Organ. 2017;95:639–645. doi: 10.2471/BLT.16.187468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kost G.J., Tran N.K., Louie R.F. Encyclopedia of Analytical Chemistry. John Wiley & Sons, Ltd; Chichester, UK: 2008. Point-of-Care testing: principles, practice, and critical-emergency-disaster medicine. [DOI] [Google Scholar]
- Lee H., Choi J., Jeong E., Baek S., Kim H.C., Chae J.H., Koh Y., Seo S.W., Kim J.S., Kim S.J. DCas9-mediated nanoelectrokinetic direct detection of target gene for liquid biopsy. Nano Lett. 2018;18:7642–7650. doi: 10.1021/acs.nanolett.8b03224. [DOI] [PubMed] [Google Scholar]
- Lei C., Li S.Y., Liu J.K., Zheng X., Zhao G.P., Jin W. The CCTL (Cpf1-assisted Cutting and Taq DNA ligase-assisted Ligation) method for efficient editing of large DNA constructs in vitro. Nucleic Acids Res. 2017;45 doi: 10.1093/nar/gkx018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Li S., Wu N., Wu J., Wang G., Zhao G., Wang J. HOLMESv2: a CRISPR-cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 2019;8:2228–2237. doi: 10.1021/acssynbio.9b00209. [DOI] [PubMed] [Google Scholar]
- Li S., Gu Y., Lyu Y., Jiang Y., Liu P. Integrated graphene oxide purification-lateral flow test strips (iGOP-LFTS) for direct detection of PCR products with enhanced sensitivity and specificity. Anal. Chem. 2017;89:12137–12144. doi: 10.1021/acs.analchem.7b02769. [DOI] [PubMed] [Google Scholar]
- Li S.Y., Cheng Q.X., Li X.Y., Zhang Z.L., Gao S., Cao R.B., Zhao G.P., Wang J., Wang J.M. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018;4:20. doi: 10.1038/s41421-018-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Li S., Wang J., Liu G. CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol. 2019;37:730–743. doi: 10.1016/j.tibtech.2018.12.005. [DOI] [PubMed] [Google Scholar]
- Li Y., Liu L., Liu G. CRISPR/Cas multiplexed biosensing: a challenge or an insurmountable obstacle? Trends Biotechnol. 2019;37:792–795. doi: 10.1016/j.tibtech.2019.04.012. [DOI] [PubMed] [Google Scholar]
- Liang M., Li Z., Wang W., Liu J., Liu L., Zhu G., Karthik L., Wang M., Wang K.F., Wang Z., Yu Jing, Shuai Y., Yu Jiaming, Zhang L., Yang Z., Li C., Zhang Q., Shi T., Zhou L., Xie F., Dai H., Liu X., Zhang J., Liu G., Zhuo Y., Zhang B., Liu C., Li S., Xia X., Tong Y., Liu Y., Alterovitz G., Tan G.Y., Zhang L.X. A CRISPR-Cas12a-derived biosensing platform for the highly sensitive detection of diverse small molecules. Nat. Commun. 2019;10:1–9. doi: 10.1038/s41467-019-11648-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova K.S., Wolf Y.I., Iranzo J., Shmakov S.A., Alkhnbashi O.S., Brouns S.J.J., Charpentier E., Cheng D., Haft D.H., Horvath P., Moineau S., Mojica F.J.M., Scott D., Shah S.A., Siksnys V., Terns M.P., Venclovas Č., White M.F., Yakunin A.F., Yan W., Zhang F., Garrett R.A., Backofen R., van der Oost J., Barrangou R., Koonin E.V. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020 doi: 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Esvelt K.M., Church G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods. 2013 doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X., Ma Y., Zhang A., Zhang L., Zeng L., Liu G. Disposable nucleic acid biosensors based on gold nanoparticle probes and lateral flow strip. Anal. Chem. 2009;81:1660–1668. doi: 10.1021/ac8024653. [DOI] [PubMed] [Google Scholar]
- Martzy R., Kolm C., Krska R., Mach R.L., Farnleitner A.H., Reischer G.H. Challenges and perspectives in the application of isothermal DNA amplification methods for food and water analysis. Anal. Bioanal. Chem. 2019;411:1695–1702. doi: 10.1007/s00216-018-1553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekler V., Minakhin L., Semenova E., Kuznedelov K., Severinov K. Kinetics of the CRISPR-Cas9 effector complex assembly and the role of 3′-terminal segment of guide RNA. Nucleic Acids Res. 2016;44:2837–2845. doi: 10.1093/nar/gkw138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica F.J.M., Díez-Villaseñor C., García-Martínez J., Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- Mukama O., Wu J., Li Z., Liang Q., Yi Z., Lu X., Liu Yujie, Liu Yumei, Hussain M., Makafe G.G., Liu J., Xu N., Zeng L. An ultrasensitive and specific point-of-care CRISPR/Cas12 based lateral flow biosensor for the rapid detection of nucleic acids. Biosens. Bioelectron. 2020;112143 doi: 10.1016/j.bios.2020.112143. [DOI] [PubMed] [Google Scholar]
- Mukama O., Yuan T., He Z., Li Z., Habimana J. de D., Hussain M., Li W., Yi Z., Liang Q., Zeng L. A high fidelity CRISPR/Cas12a based lateral flow biosensor for the detection of HPV16 and HPV18. Sensor. Actuator. B Chem. 2020;316:128119. doi: 10.1016/j.snb.2020.128119. [DOI] [Google Scholar]
- Myhrvold C., Freije C.A., Gootenberg J.S., Abudayyeh O.O., Metsky H.C., Durbin A.F., Kellner M.J., Tan A.L., Paul L.M., Parham L.A., Garcia K.F., Barnes K.G., Chak B., Mondini A., Nogueira M.L., Isern S., Michael S.F., Lorenzana I., Yozwiak N.L., MacInnis B.L., Bosch I., Gehrke L., Zhang F., Sabeti P.C. Field-deployable viral diagnostics using CRISPR-Cas13. Science. 2018;360:444–448. doi: 10.1126/science.aas8836. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouri R., Jiang Y., Lian X.L., Guan W. Sequence-specific recognition of HIV-1 DNA with solid-state CRISPR-cas12a-assisted nanopores (SCAN) ACS Sens. 2020;5:1273–1280. doi: 10.1021/acssensors.0c00497. [DOI] [PubMed] [Google Scholar]
- Pardee K., Green A.A., Takahashi M.K., Braff D., Lambert G., Lee J.W., Ferrante T., Ma D., Donghia N., Fan M., Daringer N.M., Bosch I., Dudley D.M., O'Connor D.H., Gehrke L., Collins J.J. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell. 2016;165:1255–1266. doi: 10.1016/j.cell.2016.04.059. [DOI] [PubMed] [Google Scholar]
- Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C., Salvignol G., Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- Qian C., Wang R., Wu H., Zhang F., Wu J., Wang L. Uracil-mediated new photospacer-adjacent motif of Cas12a to realize visualized DNA detection at the single-copy level free from contamination. Anal. Chem. 2019;91:11362–11366. doi: 10.1021/acs.analchem.9b02554. [DOI] [PubMed] [Google Scholar]
- Qian C., Wu H., Shi Y., Wu J., Chen H. Dehydrated CRISPR-mediated DNA analysis for visualized animal-borne virus sensing in the unprocessed blood sample. Sensor. Actuator. B Chem. 2020;305 doi: 10.1016/j.snb.2019.127440. [DOI] [Google Scholar]
- Qin P., Park M., Alfson K.J., Tamhankar M., Carrion R., Patterson J.L., Griffiths A., He Q., Yildiz A., Mathies R., Du K. Rapid and fully microfluidic Ebola virus detection with CRISPR-cas13a. ACS Sens. 2019;4:1048–1054. doi: 10.1021/acssensors.9b00239. [DOI] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., Zhang F. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheler O., Glynn B., Kurg A. Nucleic acid detection technologies and marker molecules in bacterial diagnostics. Expert Rev. Mol. Diagn. 2014 doi: 10.1586/14737159.2014.908710. [DOI] [PubMed] [Google Scholar]
- Shao N., Han X., Song Y., Zhang P., Qin L. CRISPR-Cas12a coupled with platinum nanoreporter for visual quantification of SNVs on a volumetric bar-chart chip. Anal. Chem. 2019;91:12384–12391. doi: 10.1021/acs.analchem.9b02925. [DOI] [PubMed] [Google Scholar]
- Slaymaker I.M., Mesa P., Kellner M.J., Kannan S., Brignole E., Koob J., Feliciano P.R., Stella S., Abudayyeh O.O., Gootenberg J.S., Strecker J., Montoya G., Zhang F. High-resolution structure of Cas13b and biochemical characterization of RNA targeting and cleavage. Cell Rep. 2019;26:3741–3751. doi: 10.1016/j.celrep.2019.02.094. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonker M., Sahore V., Woolley A.T. Recent advances in microfluidic sample preparation and separation techniques for molecular biomarker analysis: a critical review. Anal. Chim. Acta. 2017 doi: 10.1016/j.aca.2017.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J., Jones S., Koopal B., Schmid-Burgk J., Zetsche B., Gao L., Makarova K.S., Koonin E.V., Zhang F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019;10 doi: 10.1038/s41467-018-08224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohkendl I., Saifuddin F.A., Rybarski J.R., Finkelstein I.J., Russell R. Kinetic basis for DNA target specificity of CRISPR-cas12a. Mol. Cell. 2018;71:816–824. doi: 10.1016/j.molcel.2018.06.043. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan T.J., Dhar A.K., Cruz-Flores R., Bodnar A.G. Rapid, CRISPR-based, field-deployable detection of white spot syndrome virus in shrimp. Sci. Rep. 2019;9:1–7. doi: 10.1038/s41598-019-56170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan R., Parameshwaran H.P., Yogesha S.D., Keilbarth M.W., Rajan R. RNA-independent DNA cleavage activities of Cas9 and Cas12a. Cell Rep. 2017;21:3728–3739. doi: 10.1016/j.celrep.2017.11.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terns M.P. CRISPR-based technologies: impact of RNA-targeting systems. Mol. Cell. 2018 doi: 10.1016/j.molcel.2018.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou J.H., Leng Q., Jiang F. A CRISPR test for detection of circulating nuclei acids. Transl. Oncol. 2019;12:1566–1573. doi: 10.1016/j.tranon.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Soolingen D., De Haas P.E.W., Hermans P.W.M., Groenen P.M.A., Van Embden J.D.A. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis. J. Clin. Microbiol. 1993;31:1987–1995. doi: 10.1128/jcm.31.8.1987-1995.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Shen X., Wang T., Chen P., Qi N., Yin B.-C., Ye B.-C. A lateral flow strip combined with Cas9 nickase-triggered amplification reaction for dual food-borne pathogen detection. Biosens. Bioelectron. 2020;112364 doi: 10.1016/j.bios.2020.112364. [DOI] [PubMed] [Google Scholar]
- Wang R., Zhao X., Chen X., Qiu X., Qing G., Zhang H., Zhang L., Hu X., He Z., Zhong D., Wang Y., Luo Y. Rolling circular amplification (RCA)-Assisted CRISPR/Cas9 cleavage (RACE) for highly specific detection of multiple extracellular vesicle MicroRNAs. Anal. Chem. 2020;92:2176–2185. doi: 10.1021/acs.analchem.9b04814. [DOI] [PubMed] [Google Scholar]
- Wang X., Xiong E., Tian T., Cheng M., Lin W., Wang H., Zhang G., Sun J., Zhou X. Clustered regularly interspaced short palindromic repeats/cas9-mediated lateral flow nucleic acid assay. ACS Nano. 2020;14:2497–2508. doi: 10.1021/acsnano.0c00022. [DOI] [PubMed] [Google Scholar]
- Weckman N.E., Ermann N., Gutierrez R., Chen K., Graham J., Tivony R., Heron A., Keyser U.F. Multiplexed DNA identification using site specific dCas9 barcodes and nanopore sensing. ACS Sens. 2019;4:2065–2072. doi: 10.1021/acssensors.9b00686. [DOI] [PubMed] [Google Scholar]
- Williams M.A., O'Grady J., Ball B., Carlsson J., de Eyto E., McGinnity P., Jennings E., Regan F., Parle-McDermott A. The application of CRISPR-Cas for single species identification from environmental DNA. Mol. Ecol. Resour. 2019;19:1106–1114. doi: 10.1111/1755-0998.13045. [DOI] [PubMed] [Google Scholar]
- Wu H., He J. song, Zhang F., Ping J., Wu J. Contamination-free visual detection of CaMV35S promoter amplicon using CRISPR/Cas12a coupled with a designed reaction vessel: rapid, specific and sensitive. Anal. Chim. Acta. 2020;1096:130–137. doi: 10.1016/j.aca.2019.10.042. [DOI] [PubMed] [Google Scholar]
- Wu H., Qian C., Wu C., Wang Z., Wang D., Ye Z., Ping J., Wu J., Ji F. End-point dual specific detection of nucleic acids using CRISPR/Cas12a based portable biosensor. Biosens. Bioelectron. 2020 doi: 10.1016/j.bios.2020.112153. [DOI] [PubMed] [Google Scholar]
- Xiong Ying, Zhang J., Yang Z., Mou Q., Ma Y., Xiong Yonghua, Lu Y. Functional DNA regulated CRISPR-cas12a sensors for point-of-care diagnostics of non-nucleic-acid targets. J. Am. Chem. Soc. 2020;142:207–213. doi: 10.1021/jacs.9b09211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Jin T., Dai Y., Liu C.C. Surpassing the detection limit and accuracy of the electrochemical DNA sensor through the application of CRISPR Cas systems. Biosens. Bioelectron. 2020;155:112100. doi: 10.1016/j.bios.2020.112100. [DOI] [PubMed] [Google Scholar]
- Xu Y., Liu Y., Wu Y., Xia X., Liao Y., Li Q. Fluorescent probe-based lateral flow assay for multiplex nucleic acid detection. Anal. Chem. 2014;86:5611–5614. doi: 10.1021/ac5010458. [DOI] [PubMed] [Google Scholar]
- Yamano T., Nishimasu H., Zetsche B., Hirano H., Slaymaker I.M., Li Y., Fedorova I., Nakane T., Makarova K.S., Koonin E.V., Ishitani R., Zhang F., Nureki O. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165:949–962. doi: 10.1016/j.cell.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Restrepo-Pérez L., Bengtson M., Heerema S.J., Birnie A., Van Der Torre J., Dekker C. Detection of CRISPR-dCas9 on DNA with solid-state nanopores. Nano Lett. 2018;18:6469–6474. doi: 10.1021/acs.nanolett.8b02968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin K., Ding X., Li Z., Zhao H., Cooper K., Liu C. bioRxiv; 2020. Dynamic Aqueous Multiphase Reaction System for Simple, Sensitive and Quantitative One- Pot CRISPR-Cas12a Based Molecular Diagnosis Kun Yin; pp. 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C., Tian T., Sun J., Hu M., Wang Xusheng, Xiong E., Cheng M., Bao Y., Lin W., Jiang J., Yang C., Chen Q., Zhang H., Wang H., Wang Xiran, Deng X., Liao X., Liu Y., Wang Z., Zhang G., Zhou X. Universal and naked-eye gene detection platform based on CRISPR/Cas12a/13a system. Anal. Chem. 2020 doi: 10.1021/acs.analchem.9b05597. [DOI] [PubMed] [Google Scholar]
- Zaghloul H., El-Shahat M. Recombinase polymerase amplification as a promising tool in hepatitis C virus diagnosis. World J. Hepatol. 2014 doi: 10.4254/wjh.v6.i12.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoli L.M., Spoto G. Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors. 2013;3:18–43. doi: 10.3390/bios3010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Yan Y., Que H., Yang T., Cheng X., Ding S., Zhang X., Cheng W. CRISPR/Cas12a-Mediated interfacial cleaving of hairpin DNA reporter for electrochemical nucleic acid sensing. ACS Sens. 2020;5:557–562. doi: 10.1021/acssensors.9b02461. [DOI] [PubMed] [Google Scholar]
- Zhang F., Abudayyeh O.O., Gootenberg J.S., Sciences C., Mathers L. A protocol for detection of COVID-19 using CRISPR diagnostics. Bioarchive. 2020:1–8. https://www.broadinstitute.org/files/publications/special/COVID-19%20detection%20(updated).pdf [Google Scholar]
- Zhou L., Peng R., Zhang R., Li J. The applications of CRISPR/Cas system in molecular detection. J. Cell Mol. Med. 2018 doi: 10.1111/jcmm.13925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R., Li Y., Dong T., Tang Y., Li F. A sequence-specific plasmonic loop-mediated isothermal amplification (LAMP) assay with orthogonal color readouts enabled by CRISPR Cas12a. Chem. Commun. 2020 doi: 10.1039/d0cc00397b. [DOI] [PubMed] [Google Scholar]
- Zhou T., Huang R., Huang M., Shen J., Shan Y., Xing D. CRISPR/Cas13a powered portable electrochemiluminescence chip for ultrasensitive and specific MiRNA detection. Adv. Sci. 2020;1903661:1–10. doi: 10.1002/advs.201903661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Hu L., Ying L., Zhao Z., Chu P.K., Yu X.F. A CRISPR–Cas9-triggered strand displacement amplification method for ultrasensitive DNA detection. Nat. Commun. 2018;9:1–11. doi: 10.1038/s41467-018-07324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]