

Abstract
Ferroptosis has been reported as a unique form of cell death. However, in recent years, researchers have increasingly challenged the uniqueness of ferroptosis compared to other types of cell death. In this study, we examined whether ferroptosis shares cell death pathways with other types of cell death, especially autophagy, via the autophagic process. Here, we observed that ferroptosis inducers (artesunate and erastin) and autophagy inducers (bortezomib and XIE62–1004) led to autophagosome formation via the endoplasmic reticulum (ER) stress response. Unlike XIE62–1004, artesunate, erastin, and bortezomib, which affect glutathione production or utilization, induced oxidative stress responses - an increase in the levels of heme oxygenase-1 and lipid peroxidation. Oxidative stress responses were attenuated by deletion of autophagy related gene-5 or treatment with autophagy inhibitors (bafilomycin and chloroquine). Our studies provide an overview of common death pathways - the ER stress response-associated autophagic process in ferroptosis and autophagy. We also highlight the role the glutathione redox system plays in the outcome of the autophagic process.
Keywords: ferroptosis, autophagy, glutathione, endoplasmic reticulum stress
Introduction
Ferroptosis is a metabolic stress-related form of cell death [Tarangelo and Dixon, 2018; Tarangelo et al., 2018; Hirschhorn and Stockwell, 2019]. Although this type of cell death was reported as early as the 1950s [de Duve and Beaufy, 1959; Kerr, 1965], the molecular mechanism of ferroptosis was systematically characterized by Stockwell’s group in 2012 [Dixon et al., 2012]. For instance, Maellaro et al. reported that glutathione-depleting agents generate oxidative stress and lipid peroxidation, which leads to liver necrosis [Maellaro et al., 1990]. This report described what we now know as cell death with ferroptotic characteristics that are attributed to cell death mechanisms. Several researchers have reported in recent years that deprivation of cystine by inhibiting the cystine/glutamate antiporter (system xc−) or inhibition of glutathione synthesis/utilization leads to a unique alteration of morphology and ferroptotic cell death [Conrad and Friedmann Angeli, 2015; Dixon et al., 2012; Friedmann Angeli et al., 2014; Lisewski et al., 2014; Xie et al., 2016; _S1_Reference52_S1_Reference52Yang et al., 2014b]. Ferroptosis is considered different from other types of cell death in various ways. For example, ferroptosis does not result in biochemical and morphological changes like uncontrolled release of cell death products into the extracellular space and loss of cell membrane integrity, like what occurs during necrosis [Zong and Thompson, 2006]; formation of double membrane-layered autophagic vacuoles that fuse with a lysosome/vacuole whose hydrolytic enzymes degrade the sequestered organelle, like what occurs during autophagy [Monastyrska and Klionsky, 2006]; or cell shrinkage, plasma membrane blebbing, chromatin condensation, nuclear fragmentation, DNA laddering, and caspase-dependent cleavage of various cellular proteins, like what occurs during apoptosis [He et al., 2009]. Instead, ferroptosis manifests primarily as increased mitochondrial membrane density and mitochondrial shrinkage [Yu et al., 2017]. Although scientists have claimed that this iron-dependent form of programmed cell death is distinct from other types of cell death, recent studies have challenged the uniqueness of ferroptotic cell death [Gao et al., 2016; Hou et al., 2016]. We and other researchers previously reported that ferroptotic agent induces endoplasmic reticulum (ER) stress [Dixon et al., 2014; Hong et al., 2018; Lee et al., 2019; Rahmani et al., 2007]. In this study, we examined whether ferroptosis shares cell death processes with other types of cell death, especially autophagy, via the ER stress response.
Autophagy leads to programmed cell death through the lysosomal degradation of unfolded or misfolded proteins and intracellular structures or organelles [Ding et al., 2007; Rashid et al., 2015]. Autophagy is activated in response to a variety of environmental stressors such as energy depletion, nutritional starvation, and ER stress [Kroemer et al., 2010]. Under ER stress conditions, binding immunoglobulin protein (BiP), which is a major ER chaperone protein, dissociates from the unfolded protein response (UPR) sensors ATF6 (activating transcription factor 6), PERK (PKR-like ER kinase/pancreatic eIF2α kinase), and IRE1 (inositol requiring protein-1) and initiates the UPR signaling pathways [Lee, 2005]. Dissociated BiP translocates from the ER to the cytosol. There, the N-terminal of BiP becomes arginylated through arginyltransferase 1 (ATE1)-encoded Arg-tRNA transferases [Cha-Molstad et al., 2015]. Cytosolic arginylated BiP (R-BiP) activates p62 via binding to its ZZ-type zinc finger domain, promotes p62 aggregates with ubiquitinated proteins, and interacts with microtubule-associated proteins 1A/1B light chain 3B (LC3) on the autophagosome double membrane, which then leads to lysosomal proteolysis [Bjorkoy et al., 2005; Cha-Molstad et al., 2015; Cha-Molstad et al., 2017; Lee, 2005].
Several researchers have observed that ER stress is linked to autophagy, mitochondrial biogenesis, hypoxia signaling, and oxidative stress responses [Bravo et al., 2012; Rashid et al., 2015; Schonenberger and Kovacs, 2015; Verfaillie et al., 2010]. Interestingly, ferroptotic agent also induces ER stress through the UPR and activates ER stress-related response genes such as DR5 (death receptor 5), PUMA (p53 upregulated modulator of apoptosis), ATF4 (activating transcription factor 4), and CHOP (C/EBP (CCAAT‐enhancer‐binding protein)‐homologous protein) [Dixon et al., 2014; Lee et al., 2019; Ohoka et al., 2005; Rahmani et al., 2007; Su and Kilberg, 2008]. In this study, we observed that ferroptotic agent triggers an autophagic cell death process through arginylation of BiP, LC3 lipidation and puncta formation, and autophagosome formation. However, results from studies with the autophagy inducer XIE62–1004 reveal that, unlike ferroptotic agent, XIE62–1004 neither enhances the level of heme oxygenase-1 (HO-1), a marker of oxidative stress, nor alters the intracellular level of glutathione, even though this small molecule induces LC3 lipidation and puncta formation. Our studies suggest that ferroptosis and autophagy share the same ER stress response-triggered autophagic cell death process. However, glutathione production and utilization are probably two of the endogenous effectors involved in regulating the various types of cell death. Reduction of glutathione production and utilization leads to ferroptosis rather than autophagy.
Materials and methods
Cell lines and cell culture conditions
Human colorectal carcinoma HCT116 cells were previously obtained from American Type Culture Collection (ATCC, Manassas, VA). Human melanoma M24met cells were kindly provided by B. M. Mueller (Scripps Research Institute, La Jolla, CA). HCT116 cells and M24met cells were cultured in McCoy’s 5A and Roswell Park Memorial Institute medium‐1640 supplemented with 2 mM glutamine, respectively. Mouse embryonic fibroblasts (MEFs) and their ATG5-deficient (ATG5−/−) cells were obtained from D. Tang (UT Southwestern, Dallas, TX). These cells were cultured in Dulbecco modified Eagle medium. Autophagy-related gene 7 (ATG7)-deficient (ATG7−/−) HCT116 cells were obtained from D. Tang (UT Southwestern, Dallas, TX). All cell lines were maintained with 10% fetal bovine serum and incubated in a humidified atmosphere of 5% CO2 at 37°C.
Chemicals and reagents
ART, ERA, and BOR were purchased from Selleckchem (Houston, TX). Baf.A1 and CQ were purchased from Sigma Aldrich (St. Louis, MO). XIE62–1004 was obtained from Dr. Y.T. Kwon (Seoul National University, Korea).
Cell viability assay
The CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI) was used to determine cell viability. In brief, cells (1 × 104) were seeded in the wells of a 96-well plate. Two days later, cells were treated and CellTiter 96® AQueous One Solution Reagent was added. After 1 hr of incubation at 37°C, the absorbance at 490nm was recorded using an ELISA plate reader and viability was determined.
Western blot analysis and antibodies
Immunoblotting was carried out as previously described [Hong et al., 2018]. The following antibodies were used in this study: anti-CHOP, anti‐BiP, anti‐HO-1, and anti‐LC3, which came from Cell Signaling Technology (Beverly, MA); anti-R-BiP (Cha-Molstad et al., 2015), which came from Dr. Y.T. Kwon (Seoul National University, Korea); and anti‐actin, anti‐rabbit IgG‐horseradish peroxidase (HRP), and anti‐mouse IgG‐HRP, which came from Santa Cruz Biotechnology (Santa Cruz, CA).
Lipid peroxidation assay
The concentration of malondialdehyde (MDA), an end product of lipid peroxidation of polyunsaturated fatty acids, was measured using a Lipid Peroxidation (MDA) Assay Kit (#MAK085, Sigma-Aldrich) according to the manufacturer’s instructions. In brief, cells (6 × 105) were plated onto 70-mm Petri dish. Two days later, cells were treated and harvested. Cells were homogenized on ice in MDA lysis buffer and centrifuged at 13,000 g for 10 min. Supernatant was saved and incubated at 95 °C for 60 min with thiobarbituric acid (TBA) solution. The MDA-TBA adduct was quantified colorimetrically at 532 nm.
ROS assay
Intracellular reactive oxygen species (ROS) level was detected with a microplate reader using a ROS Detection Assay Kit (# K936–100, Biovision, CA) according the manufacturer’s instructions. We utilized H2DCFDA (2′,7′-Dichlorodihydrofluorescein diacetate), a unique cell-permeable fluorogenic probe. In brief, cells (3 × 104) were seeded in the wells of a six-well plate. After three days, cells were treated, washed, stained with H2DCFDA, transferred to microplate, and analyzed with microplate reader.
Measurement of protein concentrations
Protein concentration was determined using a Pierce™ BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA).
Measurement of glutathione concentrations
Total glutathione concentration was detected with a microplate reader at 412 nm using a Glutathione Assay Kit (# CS0260, Sigma-Aldrich, St. Louis, MO) according the manufacturer’s instructions. In brief, cells (3 × 105) were plated onto 60-mm Petri dish. After two days, cells were treated, deproteinized with the 5% 5-Sulfosalicylic Acid Solution, centrifuged to remove the precipitated protein, and then measured glutathione concentrations using a microplate reader.
LC3 imaging
LC3 imaging was carried out as previously described [Song et al., 2018]. Briefly, LC3-GFP-expressing HCT116 cells were grown on coverslips. Cells were fixed in 4% paraformaldehyde for 20 min at room temperature. Nuclei were stained with 40,6-diamidino-2-phenylindole (DAPI; Cell Signaling). Cells were examined with an OlympusFluoview 1000 confocal microscope.
Statistical analysis
All values are represented as mean ± SD. Statistical analysis was performed using one‐way ANOVA analysis as indicated using SigmaPlot software. P values of less than 0.05 were defined as statistically significant. Where indicated, *P < 0.05; **P < 0.01; ***P < 0.001.
Results
Assessment of morphological alterations and viability during treatment with ER stress-inducing agents
To investigate whether the ER stress response plays an important role in the cell death process possibly shared between ferroptosis and autophagy, we chose to study the ferroptotic agents artesunate (ART) and erastin (ERA) and the autophagic agent bortezomib (BOR). These agents are known to induce ER stress [Dixon et al., 2014; Hong et al., 2018] [Lee et al., 2019; Obeng et al., 2006; Yoo et al., 2014]. In the first step, we examined the effects of ART, ERA, or BOR on cell morphology and viability in human colon cancer HCT116 and human melanoma cancer M24met cells. Cells were treated with various doses of the drugs and we observed cell morphology under a light microscope (Figs. 1A and 1C). Data from phase-contrast images showed dose-dependent morphological alterations. The cells became round, shrunken, and detached. However, results from propidium iodide staining showed that not much DNA fragmentation was detected during treatment with the drugs (Figs. 1A and 1C). These results suggest that apoptosis didn’t occur in the cells during treatment with the drugs. Next, cell viability was determined using MTS assay. Our results show decreased cell viability, which was due to increased cytotoxicity during treatment with the drugs (Figs. 1B and 1D).
Figure 1. Evaluation of morphological alterations and cytotoxicity during treatment with artesunate (ART), erastin (ERA), or bortezomib (BOR).
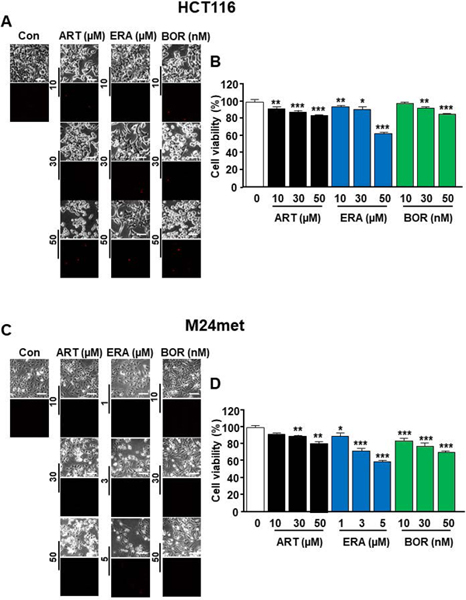
(A, B) HCT116 cells were treated with ART (10–50 μM), ERA (10–50 μM), or BOR (10–50 nM) for 24 h. The cells were stained with propidium iodide. (C, D) M24met cells were treated with ART (10–50 μM), ERA (1–5 μM), or BOR (10–50 nM) for 24 h. The cells were stained with propidium iodide. (A, C) Phase-contrast images or fluorescence images were visualized under a light or fluorescence microscope, respectively. Representative images are shown. (B, D) Cell viability was assessed using MTS assay. The values are indicated as mean ± SD from three independent experiments. p-values: *, 0.05; **, 0.01; ***, 0.001.
Detection of the ER stress response and oxidative stress during treatment with ER stress-inducing agents
Previous studies have shown that oxidative stress and ER stress are strongly connected [Cao and Kaufman, 2014; Malhotra and Kaufman, 2007]. To investigate this connection, we examined the ER stress response and oxidative stress during ART, ERA, and BOR treatments by assessing the ER stress response markers CHOP and BiP, as well as the oxidative-sensor protein HO-1. As shown in Figs. 2A and 2B, the levels of CHOP, BiP, and HO-1 expression were elevated during treatment with the drugs in both HCT116 and M24met cells. Moreover, we observed an increase in ROS levels and lipid peroxidation during treatment with the drugs ( Figs. 2C and 2D). Since ferroptotic agents are related to the glutathione antioxidant system, we investigated the relationship between oxidative stress and intracellular glutathione concentrations. Figure 2E shows that the intracellular levels of glutathione were decreased in a dose-dependent manner during treatments with ERA and BOR. These results suggest a connection between the ER stress response and oxidative stress during treatment with ER stress-inducing agents and involvement of the glutathione antioxidant system in the regulation of defense against oxidative stress.
Figure 2. Assessment of the endoplasmic reticulum stress response, oxidative stress response, lipid peroxidation, and glutathione content during treatment with ART, ERA, or BOR.

(A) HCT116 cells were treated with ART (10–50 μM), ERA (10–50 μM), or BOR (10–50 nM) for 24 h. (B) M24met cells were treated with ART (10–50 μM), ERA (1–5 μM), or BOR (10–50 nM) for 24 h. Whole-cell extracts were analyzed with immunoblotting assay using indicated antibodies. Actin was used as a loading control. Densitometry analysis of the bands from CHOP, BiP, or HO-1 was performed (right panels). The values are indicated as mean ± SD from three independent experiments. p-values: *, 0.05; **, 0.01; ***, 0.001. (C) HCT116 cells were treated with ERA (1–50 μM) or BOR (1–50 nM) for 24 h. Glutathione content of the sample was assayed using a Glutathione Assay Kit. The values are indicated as mean ± SD from three independent experiments. p-values: ***, 0.001. (D) HCT116 cells were treated with ART (50 μM), ERA (50 μM), or BOR (50 nM) for 24 h. Intracellular reactive oxygen species were detected using the fluorescent dye CM-H2DCFDA. The values are indicated as mean ± SD from three independent experiments. p-values: **, 0.01; ***, 0.001. (E) HCT116 cells were treated with ART (10–50 μM) or BOR (10–50 nM) for 24 h. Lipid peroxidation was determined using malondialdehyde (MDA) assay. The values are indicated as mean ± SD from three independent experiments. p-values: ***, 0.001.
Arginylation of BiP and autophagic process during treatment with ART, ERA, or BOR
Several researchers have reported that BiP, which is located in the lumen of the ER, translocates to the cytoplasm during ER stress. During translocation, N-terminal BiP becomes arginylated by ATE1 [Cha-Molstad et al., 2015; Cha-Molstad et al., 2016]. R-BiP associates with p62, an autophagic adaptor, through interaction between its N-terminal arginine and the ZZ domain of p62 [Cha-Molstad et al., 2015; Cha-Molstad et al., 2016]. This interaction induces oligomerization and aggregation of p62 and increases the binding of p62 to the lipidated form of LC3, and then transports p62 to the autophagosome formation site [Cha-Molstad et al., 2015; Cha-Molstad et al., 2016; Shvets et al., 2011]. In this study, we examined whether ER stress agents induce arginylation of BiP during treatment with ART, ERA, or BOR. As shown in Fig. 3A, R-BiP was detected during treatment with the proteasome inhibitor BOR, but not ART. However, R-BiP was detected during treatment with ART or ERA in the presence of the proteasome inhibitor MG132: the amount of R-BiP induced by ART or ERA in the presence of MG132 was significantly larger than that by MG132 alone (Figs. 3A and 3B). These results suggest that R-BiP is a short-lived protein [Shim et al., 2018]. We further examined the autophagic process during treatments with ART, ERA, or BOR. Data from imaging and immunoblotting assays show that all three drugs induced puncta formation and lipidation of LC3 (Figs. 4A and 4B). These results suggest that the ER stress inducers ART, ERA, and BOR trigger autophagosome formation. To confirm ART, ERA, and BOR-induced autophagosome formation and their role in oxidative stress, we employed autophagy related gene 5 (ATG5)-deficient MEFs. Since ATG5 is required for autophagosome formation through involvement of the extension of the phagophoric membrane in autophagic vesicles, autophagosome formation is inhibited in ATG5-deficient (ATG5−/−) cells [Young et al., 2012]. As shown in Fig. 4C, ART, ERA, and BOR-increased lipidation of LC3 (LC3-II) and expression of HO-1 (oxidative stress) were inhibited in ATG5−/− cells. Data from malondialdehyde (MDA) assay confirmed that ERA and BOR-induced lipid peroxidation was suppressed in ATG5−/− cells (Fig. 4D). These results were confirmed using autophagy related gene 7 (ATG7)-deficient HCT116 cells. As shown in Fig. 4E, ART, ERA, and BOR-increased lipidation of LC3 (LC3-II) and expression of HO-1 were inhibited in ATG7−/− cells. These results suggest that autophagosome formation plays an important role in oxidative stress during treatment with ER stress inducers. We further examined these observations with autophagy inhibitors. As shown in Fig. 5A, the amount of the lipidated form of LC3 was further increased during treatment with ART or BOR in the presence of the autophagy inhibitors chloroquine (CQ) and bafilomycin A1 (Baf.A1). In contrast, the levels of ART or BOR-induced HO-1 was decreased in the presence of CQ or Baf.A1. These results suggest that oxidative stress was mediated through the autophagosome and prevented by blocking autophagic flux. Indeed, similar results were observed when the levels of ROS and lipid peroxidation were measured during treatment with ART, ERA, or BOR in the presence/absence of CQ or Baf.A1. The levels of ROS and lipid peroxidation were reduced in the presence of CQ or Baf.A1 during treatment with ART, ERA, or BOR (Figs. 5B and 5C).
Figure 3. Arginylation of BiP during treatment with ART, ERA, or BOR.
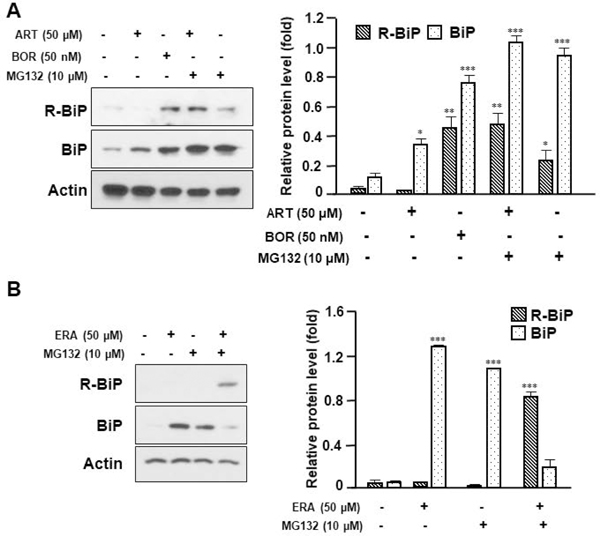
(A) HCT116 cells were treated with ART (50 μM) or BOR (50 nM) in the presence/absence of MG132 (10 μM) for 24 h. Western blotting analysis of BiP and arginylated BiP (R-BiP) was performed after treatment with indicated agents. Actin was used as a loading control. Densitometry analysis of the bands from the R-BiP or BiP was performed (right panel). The values are indicated as mean ± SD from three independent experiments. p-values: *, 0.05; **, 0.01; ***, 0.001. (B) HCT116 cells were treated with ERA (50 μM) in the presence/absence of MG132 (10 μM) for 24 h. Whole-cell extracts were analyzed with immunoblotting assay using indicated antibodies. Actin was used as a loading control. Densitometry analysis of the bands from the R-BiP or BiP was performed (right panel). The values are indicated as mean ± SD from three independent experiments. p-values: ***, 0.001.
Fig 4. Assessment of autophagosome formation, oxidative stress, and lipid peroxidation during treatment with ART, ERA, or BOR.

(A) HCT116 cells expressing the GFP-LC3 gene were treated with ART (10–50 μM), ERA (10–50 μM), or BOR (10–50 nM) for 24 h. LC3 puncta was examined with a confocal microscope. (B) HCT116 cells were treated with ART (10–50 μM), ERA (10–50 μM), or BOR (10–50 nM) for 24 h. Western blotting analysis of LC3-ll levels was performed after treatment with indicated agents. Actin was used as a loading control. Densitometry analysis of the bands from the LC3-II was performed (lower panel). The values are indicated as mean ± SD from three independent experiments. p-values: *, 0.05; **, 0.01; ***, 0.001. (C, D) Wild-type (WT) MEFs or ATG5 knockout (ATG5−/−) MEFs were treated with ART (50 μM), ERA (5 μM), or BOR (50 nM) for 24 h. (C) Whole-cell extracts were analyzed with immunoblotting assay using indicated antibodies. Actin was used as a loading control. Densitometry analysis of the bands from the LC3-II or HO-1 was performed (right panel). The values are indicated as mean ± SD from three independent experiments. p-values: *, 0.05; **, 0.01; ***, 0.001. (D) Lipid peroxidation was determined by malondialdehyde (MDA) assay. The values are indicated as mean ± SD from three independent experiments. p-values: *, 0.05; ***, 0.001. (E) Wild-type (WT) HCT116 or ATG7 knockout (ATG7−/−) HCT116 cells were treated with ART (50 μM), ERA (50 μM), or BOR (50 nM) for 24 h. Whole-cell extracts were analyzed with immunoblotting assay using indicated antibodies. Actin was used as a loading control. Densitometry analysis of the bands from LC3-II or HO-1 was performed (right panel). The values are indicated as mean ± SD from three independent experiments. p-values: **, 0.01; ***, 0.001.
Figure 5. Effect of autophagic process inhibitors on lipid lipidation, oxidative stress, and lipid peroxidation during treatment with ART, ERA, or BOR.

(A, B) HCT116 cells were treated with 50 μM ART (A) or 50 nM BOR (B) in the presence/absence of chloroquine (CQ) (10 μM) or bafilomycin A1 (Baf.A1) (600 nM) for 24 h. Whole-cell extracts were analyzed with immunoblotting assay using indicated antibodies. Actin was used as a loading control. Densitometry analysis of the bands from LC3-II or HO-1 was performed (right panels). The values are indicated as mean ± SD from three independent experiments. p-values: *, 0.05; ***, 0.001. (C) HCT116 cells were treated with ERA (50 μM) or BOR (50 nM) in the presence/absence of CQ (10 μM) or Baf.A1 (600 nM) for 24 h. Intracellular reactive oxygen species was detected using fluorescent dye CM-H2DCFDA. The values are indicated as mean ± SD from three independent experiments. p-values: *, 0.05; **, 0.01; ***, 0.001. (D) HCT116 and M24met cells were treated with ART (50 μM) or BOR (50 nM) in the presence/absence of CQ (10 μM) or Baf.A1 (600 nM) for 24 h. Lipid peroxidation was analyzed using malondialdehyde assay. The values are indicated as mean ± SD from three independent experiments. p-values: **, 0.01; ***, 0.001.
Role of glutathione antioxidant system in the outcome of the autophagic process
To examine the role of the glutathione antioxidant system in the outcome of the autophagic process, we employed the autophagic inducer XIE62–1004, which is a ligand of p62. It binds to the ZZ-domain of p62 and induces autophagy without inducing ER stress and affecting the glutathione antioxidant system. Indeed, lipidation of LC3 and LC3 puncta formation occurred during treatment with XIE62–1004 (Figs. 6A and 6B). However, XIE62–1004 treatment didn’t lead to elevation of BiP and HO-1 expression (Figs. 6A and 6B). Moreover, unlike ERA, XIE62–1004 didn’t affect the level of glutathione (Fig. 6). These results suggest that the glutathione antioxidant system plays an important role in the inhibition of autophagy-associated oxidative stress.
Figure 6. Assessment of endoplasmic reticulum stress response, lipid lipidation, autophagosome formation, oxidative stress, and glutathione content during treatment with the autophagy inducer XIE62-1004.
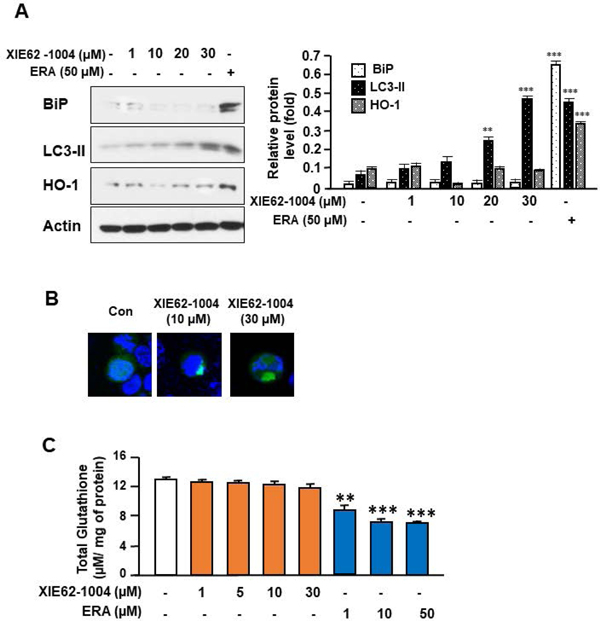
(A) HCT116 cells were treated with XIE62-1004 (1–30 μM) or ERA (50 μM) for 24 h. Whole-cell extracts were analyzed with immunoblotting assay using indicated antibodies. Actin was used as a loading control. Densitometry analysis of the bands from BiP, LC3-II, or HO-1 was performed (right panel). The values are indicated as mean ± SD from three independent experiments. p-values: **, 0.01; ***, 0.001. (B) HCT116 cells expressing the GFP-LC3 gene were treated with XIE62-1004 (10 or 30 μM) for 24 h. LC3 puncta were examined with a confocal microscope. (C) HCT116 cells were treated with XIE62–1004 (1–30 μM) or ERA (1–50 μM) for 24 h. Glutathione content of the sample was assayed using a Glutathione Assay Kit. Error bars represent the mean ± SD from three independent experiments. p-values: **, 0.01; ***, 0.001.
Discussion
Several conclusions can be drawn upon considering the data presented here. First, the network of ART, ERA, and BOR-induced cell death pathways is mediated through the ER stress response. ER stress enhances the level of lipid peroxidation and production of ROS through promotion of the autophagic process during treatment with ART, ERA, or BOR. Moreover, the glutathione redox system plays an important role in the modulation of cell death caused by impaired lipid metabolism [Miess et al., 2018]. The inhibition of glutathione production/utilization promotes autophagic process-induced oxidative death by enhancing lipid peroxidation.
Our data from Figs 2, 4, and 5 clearly demonstrate that ER stress inducers promote ROS production via the autophagic process and subsequently generate oxidative stress-induced responses such as HO-1 gene expression and lipid peroxidation. Several researchers have reported that ROS generation occurs due to the autophagic degradation of the iron storage macromolecule ferritin [Bresgen and Eckl, 2015; Hou et al., 2016]. It is well known that autophagosomes containing ferritin fuse with lysosomes and form autophagolysosomes. Digestion of ferritin in the autophagolysosomes results in the release of ferrous iron (Fe2+) [Asano et al., 2011; Bogdan et al., 2016]. Ferrous iron, a reduced form of iron, reacts with lipid peroxides (ROOH) to produce alkoxy radicals, whereas ferric iron (Fe3+), an oxidized form of iron, reacts more slowly to produce peroxyl radicals [Braughler et al., 1986]. Both radicals can become involved in the propagation of the chain reaction. The end products of these complex metal ion-catalyzed breakdowns of lipid hydroperoxides lead to ferroptosis [Asano et al., 2011; Bogdan et al., 2016]. Nuclear receptor coactivator 4 (NCOA4), a cargo receptor protein, mediates ferritin degradation, and its overexpression increases ferritin degradation and promotes ferroptosis [Mancias et al., 2014; Hou et al., 2016]. Involvement of lysosomes in ferroptosis has been confirmed by recent observations. Several lysosomal inhibitors prevent cell death during treatment with ferroptosis-inducing compounds such as erastin and RSL3 [Torii et al., 2016]. Bafilomycin A1, which disrupts autophagic flux by inhibiting both Ca-P60A/SERCA-dependent autophagosome-lysosome fusion and vacuolar-type H(+)-ATPase-dependent acidification [Mauvezin and Neufeld, 2015], protects cells from ferroptosis. This is probably due to inhibition of ferritinophagic degradation of ferritin [Gao et al., 2016; Gryzik et al., 2017; Hou et al., 2016; Torii et al., 2016; Yang et al., 2014a]. Our data from Fig. 5 also clearly demonstrate that the autophagy inhibitors CQ and Baf.A1 inhibit ROS generation and lipid peroxidation.
Our studies clearly demonstrate that the proteasome inhibitor bortezomib induces oxidative stress (elevation of HO-1 expression, ROS production, and lipid peroxidation) (Fig. 2). Our observations are consistent with those from previous reports [Furfaro et al., 2016; Tibullo et al., 2016]. Bortezomib enhances HO-1 expression in myeloma and neuroblastoma HTLA-230 cells [Furfaro et al., 2016; Tibullo et al., 2016]. Interestingly, bortezomib-induced oxidative stress cannot be prevented because of the reduction of glutathione concentrations [Starheim et al., 2016; Zaal et al., 2017] (Fig. 2E). The role of the glutathione redox system in bortezomib-induced oxidative stress has been previously reported. Cysteine and glutathione supplement abolish bortezomib-induced cytotoxicity in several myeloma cell lines [Starheim et al., 2016; Zaal et al., 2017]. How the proteasome inhibitor bortezomib affects the glutathione redox system remains unanswered. Since the level of glutathione content is decreased during treatment with bortezomib, system xc- cystine/glutamate antiporter and/or glutathione synthetase rather than glutathione peroxidase and transferase may be involved in the biological effects of bortezomib. One possibility is that bortezomib modulates the level of system xc- cystine/glutamate antiporter and/or glutathione synthetase. Obviously, this possibility needs to be further examined to understand the effect of bortezomib on the glutathione redox system.
Acknowledgements
We thank Christine Burr (Department of Surgery, University of Pittsburgh) for her critical review of the manuscript. This study is supported by National Cancer Institute (NCI) R03 CA205267, R03 CA212125, and P30CA047904.
Funding information:National Cancer Institute (NCI) R03 CA205267, R03 CA212125, and P30CA047904.
Abbreviations used in this paper:
- ART
artesunate
- ATE1
arginyltransferase 1
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- ATG5
autophagy-related gene-5
- ATG7
autophagy-related gene 7
- Baf.A1
bafilomycin A1
- BiP
binding immunoglobulin protein
- BOR
bortezomib
- C/EBP
CCAAT-enhancer-binding proteins
- CHOP
CCAAT-enhancer-binding protein homologous protein
- CQ
chloroquine
- DAPI
4’,6-diamidino-2-phenylindole
- DR5
death receptor 5
- eIF2α
eukaryotic initiation factor 2α
- ER
endoplasmic reticulum
- ERA
erastin
- H2DCFDA
2′,7′-Dichlorodihydrofluorescein diacetate
- HO-1
heme oxygenase-1
- HRP
horseradish peroxidase
- IRE1
inositol requiring protein-1
- MDA
malondialdehyde
- MEF
mouse embryo fibroblast
- NCOA4
nuclear receptor coactivator 4
- PERK
PKR-like ER kinase
- PUMA
p53 upregulated modulator of apoptosis
- R-BiP
arginylated BiP
- ROS
reactive oxygen species
- TBA
thiobarbituric acid
- UPR
unfolded protein response
- WT
wild-type
Footnotes
Conflict of interests
The authors declare no competing financial interests.
Data availability statement:
The data sets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
References
- Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K 2011. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol 31:2040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y 2016. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem Sci 41:274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braughler JM, Duncan LA, Chase RL. 1986. The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J Biol Chem 261:10282–9. [PubMed] [Google Scholar]
- Bravo R, Gutierrez T, Paredes F, Gatica D, Rodriguez AE, Pedrozo Z, Chiong M, Parra V, Quest AF, Rothermel BA, Lavandero S 2012. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int J Biochem Cell Biol 44:16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresgen N, Eckl PM. 2015. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 5:808–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao SS, Randal J Kaufman RJ. 2014. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 21: 396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha-Molstad H, Sung KS, Hwang J, Kim KA, Yu JE, Yoo YD, Jang JM, Han DH, Molstad M, Kim JG, Lee YJ, Zakrzewska A, Kim SH, Kim ST, Kim SY, Lee HG, Soung NK, Ahn JS, Ciechanover A, Kim BY, Kwon YT. 2015. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol 17:917–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha-Molstad H, Yu JE, Feng Z, Lee SH, Kim JG, Yang P, Han B, Sung KW, Yoo YD, Hwang J, McGuire T, Shim SM, Song HD, Ganipisetti S, Wang N, Jang JM, Lee MJ, Kim SJ, Lee KH, Hong JT, Ciechanover A, Mook-Jung I, Kim KP, Xie XQ, Kwon YT, Kim BY. 2017. p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat Commun 8:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha-Molstad H, Yu JE, Lee SH, Kim JG, Sung KS, Hwang J, Yoo YD, Lee YJ, Kim ST, Lee DH, Ciechanover A, Kim BY, Kwon YT 2016. Modulation of SQSTM1/p62 activity by N-terminal arginylation of the endoplasmic reticulum chaperone HSPA5/GRP78/BiP. Autophagy 12:426–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Friedmann Angeli JP. 2015. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol Cell Oncol 2:e995047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Duve C, Beaufay H 1959. Tissue fractionation studies. 10. Influence of ischaemia on the state of some bound enzymes in rat liver. Biochem J 73:610–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. 2007. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171:513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR. 2014. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3:e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Radmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Forster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M 2014. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16:1180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furfaro AL, Piras S, Domenicotti C, Fenoglio D, De Luigi A, Salmona M, Moretta L, Marinari UM, Pronzato MA, Traverso N, Nitti M 2016. Role of Nrf2, HO-1 and GSH in Neuroblastoma Cell Resistance to Bortezomib. PLoS One 11:e0152465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X 2016. Ferroptosis is an autophagic cell death process. Cell Res 26:1021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryzik M, Srivastava A, Longhi G, Bertuzzi M, Gianoncelli A, Carmona F, Poli M, Arosio P 2017. Expression and characterization of the ferritin binding domain of Nuclear Receptor Coactivator-4 (NCOA4). Biochim Biophys Acta Gen Subj 1861:2710–2716. [DOI] [PubMed] [Google Scholar]
- He B, Lu N, Zhou Z 2009. Cellular and nuclear degradation during apoptosis. Curr Opin Cell Biol 21:900–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn T, Stockwell BR. 2019. The development of the concept of ferroptosis. Free Radic Biol Med 133:130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Lee DH, Lee YS, Jo MJ, Jeong YA, Kwon WT, Choudry HA, Bartlett DL, Lee YJ. 2017. Molecular crosstalk between ferroptosis and apoptosis: Emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 8:115164–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, Tang D 2016. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12:1425–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF. 1965. A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J Pathol Bacteriol 90:419–35. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B 2010. Autophagy and the integrated stress response. Mol Cell 40:280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS. 2005. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 35:373–81. [DOI] [PubMed] [Google Scholar]
- Lee YS, Lee DH, Jeong SY, Park SH, Oh SC, Park YS, Yu J, Choudry HA, Bartlett DL, Lee YJ. 2019. Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through upregulation of death receptor 5. J Cell Biochem 120:928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisewski AM, Quiros JP, Ng CL, Adikesavan AK, Miura K, Putluri N, Eastman RT, Scanfeld D, Regenbogen SJ, Altenhofen L, Llinas M, Sreekumar A, Long C, Fidock DA, Lichtarge O 2014. Supergenomic network compression and the discovery of EXP1 as a glutathione transferase inhibited by artesunate. Cell 158:916–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maellaro E, Casini AF, Del Bello B, Comporti M 1990. Lipid peroxidation and antioxidant systems in the liver injury produced by glutathione depleting agents. Biochem Pharmacol 39:1513–21. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. 2007. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9:2277–93. [DOI] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. 2014. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, Neufeld TP. 2015. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11:1437–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, Saunders B, Howell M, Downward J, Felsher DW, Peck B, Schulze A 2018. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 37:5435–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monastyrska I, Klionsky DJ. 2006. Autophagy in organelle homeostasis: peroxisome turnover. Mol Aspects Med 27:483–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, Boise LH. 2006. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107:4907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H 2005. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J 24:1243–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani M, Davis EM, Crabtree TR, Habibi JR, Nguyen TK, Dent P, Grant S 2007. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol 27:5499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid HO, Yadav RK, Kim HR, Chae HJ. 2015. ER stress: Autophagy induction, inhibition and selection. Autophagy 11:1956–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonenberger MJ, Kovacs WJ. 2015. Hypoxia signaling pathways: modulators of oxygen-related organelles. Front Cell Dev Biol 3:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim SM, Choi HR, Sung KW, Lee YJ, Kim ST, Kim D, Mun SR, Hwang J, Cha-Molstad H, Ciechanover A, Kim BY, Kwon YT. 2018. The endoplasmic reticulum-residing chaperone BiP is short-lived and metabolized through N-terminal arginylation. Sci Signal 11. [DOI] [PubMed] [Google Scholar]
- Shvets E, Abada A, Weidberg H, Elazar Z 2011. Dissecting the involvement of LC3B and GATE-16 in p62 recruitment into autophagosomes. Autophagy 7:683–8. [DOI] [PubMed] [Google Scholar]
- Song X, Lee DH, Dilly AK, Lee YS, Choudry HA, Kwon YT, Bartlett DL, Lee YJ. 2018. Crosstalk Between Apoptosis and Autophagy Is Regulated by the Arginylated BiP/Beclin-1/p62 Complex. Mol Cancer Res 16:1077–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starheim KK, Holien T, Misund K, Johansson I, Baranowska KA, Sponaas AM, Hella H, Buene G, Waage A, Sundan A, Bjorkoy G 2016. Intracellular glutathione determines bortezomib cytotoxicity in multiple myeloma cells. Blood Cancer J 6:e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N, Kilberg MS. 2008. C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J Biol Chem 283:35106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarangelo A, Dixon S 2018. The p53-p21 pathway inhibits ferroptosis during metabolic stress. Oncotarget 9:24572–24573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, Dixon SJ. 2018. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 22:569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibullo D, Barbagallo I, Giallongo C, Vanella L, Conticello C, Romano A, Saccone S, Godos J, Di Raimondo F, Li Volti G 2016. Heme oxygenase-1 nuclear translocation regulates bortezomibinduced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget 7:28868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, Yamada K 2016. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J 473:769–77. [DOI] [PubMed] [Google Scholar]
- Verfaillie T, Salazar M, Velasco G, Agostinis P 2010. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol 2010:930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D 2016. Ferroptosis: process and function. Cell Death Differ 23:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ND, Tan SH, Ng S, Shi Y, Zhou J, Tan KS, Wong WS, Shen HM. 2014a. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin. J Biol Chem 289:33425–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. 2014b. Regulation of ferroptotic cancer cell death by GPX4. Cell 156:317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JY, Hurwitz BS, Bolyard C, Yu JG, Zhang J, Selvendiran K, Rath KS, He S, Bailey Z, Eaves D, Cripe TP, Parris DS, Caligiuri MA, Yu J, Old M, Kaur B 2014. Bortezomib-induced unfolded protein response increases oncolytic HSV-1 replication resulting in synergistic antitumor effects. Clin Cancer Res 20:3787–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, Kester M, Wang HG. 2012. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 287:12455–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Guo P, Xie X, Wang Y, Chen G 2017. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med 21:648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaal EA, Wu W, Jansen G, Zweegman S, Cloos J, Berkers CR. 2017. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Thompson CB. 2006. Necrotic death as a cell fate. Genes Dev 20:1–15. [DOI] [PubMed] [Google Scholar]