

Abstract
The activation of the Wnt/β-catenin signaling pathway is critical for skeletal development but surprisingly little is known about the requirements for the specific frizzled receptors that recognize Wnt ligands. To define the contributions of individual frizzled proteins to osteoblast function, we profiled the expression of all 10 mammalian receptors during calvarial osteoblast differentiation. Expression of Fzd4 was highly up-regulated during in vitro differentiation and therefore targeted for further study. Mice lacking Fzd4 in mature osteoblasts had normal cortical bone structure but reduced cortical tissue mineral density and also exhibited an impairment in femoral trabecular bone acquisition that was secondary to a defect in the mineralization process. Consistent with this observation, matrix mineralization, markers of osteoblastic differentiation, and the ability of Wnt3a to stimulate the accumulation of β-catenin were reduced in cultures of calvarial osteoblasts deficient for Fzd4. Interestingly, Fzd4-deficienct osteoblasts exhibited an increase in the expression of Fzd8 both in vitro and in vivo, which suggest that the two receptors may exhibit overlapping functions. Indeed, ablating a single Fzd8 allele in osteoblast-specific Fzd4 mutants produced a more severe effect on bone acquisition. Taken together, our data indicate that Fzd4 is required for normal bone development and mineralization despite compensation from Fzd8.
Keywords: Osteoblast, Wnt signaling, Frizzled-4, Frizzled-8, Bone
Introduction
Acquisition of peak bone mass and skeletal remodeling are controlled by a constellation of local growth factors, systemic hormones, and their cognate receptors and signaling networks (Karsenty, 2000; Mundy et al., 1995). Over the last two decades, Wnt/β-catenin signaling has emerged as a central regulator of bone metabolism, with an influence on nearly all facets of skeletal physiology (Regard et al., 2012). In this pathway, the stability and nuclear localization of the transcription factor β-catenin are regulated by a proteolytic mechanism (reviewed in (MacDonald et al., 2012; Niehrs, 2012)). In the absence of Wnt ligands, cytosolic β-catenin is phosphorylated and targeted for proteasomal degradation by glycogen synthase kinase 3, a component of a destruction complex that also contains casein kinase Iα, Axin, and Adenomatosis polyposis coli. The binding of Wnts to a receptor complex containing a frizzled (Fzd) receptor and low-density lipoprotein-related protein-5 (LRP5) or LRP6 co-receptor leads to the recruitment of the destruction complex to the cell surface and its inactivation. This allows β-catenin to accumulate and subsequently translocate to the nucleus, where it trans-activates genes under the control of Lymphoid enhancer-binding factor (LEF)/T cell factor (TCF) that influence cellular proliferation and differentiation. Combinations of Wnt ligands, frizzled receptors, and co-receptors also activate planar cell polarity and Wnt-calcium pathways (Niehrs, 2012).
The importance of Wnt/β-catenin signaling in skeletal physiology was first revealed by the observation that mutations in Wnt3a impact axial skeletal development in the mouse (Greco et al., 1996) and subsequently by the identification of causative mutations in LRP5 in the autosomal recessive disorder osteoporosis-pseudoglioma (OPPG), a condition that results in severe osteoporosis, fractures, and skeletal deformity (Gong et al., 2001). Genetic alterations in Wnt signaling components are now known to underlie a large number of human bone diseases, ranging from a recessive form of osteogenesis imperfecta resulting from mutations in WNT1 (Fahiminiya et al., 2013; Keupp et al., 2013; Laine et al., 2013) to the extremely high bone mass found in patients with sclerosteosis and van Buchem disease, owing to alterations in the SOST gene that encodes sclerostin (Balemans et al., 2001; Balemans et al., 2002; Brunkow et al., 2001), a secreted inhibitor of Wnt/β-catenin signaling (Li et al., 2005; Semenov et al., 2005). The development of genetic mouse models have confirmed the importance of this pathway in skeletal physiology and elaborated on the impact of Wnt signaling on bone cell function. For example, disrupting the expression of Lrp5 specifically in osteoblasts recapitulates in mice the low bone mass that is evident in OPPG (Cui et al., 2011; Joeng et al., 2011; Riddle et al., 2013), while manipulating the expression of β-catenin revealed the transcription factor is required for commitment to the osteoblast lineage, full osteoblast maturation, and proper coupling of osteoblasts and osteoclasts (Glass et al., 2005; Hill et al., 2005; Holmen et al., 2005).
While recent studies have started to assign specific functions to individual Wnt ligands (Laine et al., 2013; Zheng et al., 2012), surprisingly little is known about the functions of individual Fzd receptors. Named for the irregular patterning of hair and disoriented ommatidia of the compound eye in Drosophila mutants (Adler, 1992), Fzd receptors bear structural similarity to G-protein coupled receptors (Vinson et al., 1989). Seven-transmembrane domains are linked by three intracellular loops and followed by a carboxy-terminal tail, which act to facilitate interactions with signaling partners (Umbhauer et al., 2000; Wong et al., 2003). In the extracellular space, Wnt ligands “pinch” a 120-amino acid cysteine-rich domain (CRD) at the amino-terminal end of the receptor (Janda et al., 2012). In mammals, 10 Fzd genes have been identified and each receptor has the capacity to interact with a number of individual Wnt ligands, but the binding affinity and functional outcome vary among ligand:receptor pairs (Hsieh et al., 1999; Rulifson et al., 2000; Wang et al., 2016; Ye et al., 2011). Nearly all ten receptors have been assigned developmental functions and, in many cases, partial functional redundancy has been observed among closely related receptors (Liu et al., 2012; Wang et al., 2006; Ye et al., 2011; Yu et al., 2012).
In this study, we investigated the function of Fzd4 during osteoblast differentiation and in vivo bone acquisition. We demonstrate that Fzd4 expression increases during osteoblast maturation and contributes to the stabilization of β-catenin after Wnt ligand stimulation. Moreover, the selective ablation of Fzd4 expression in mature osteoblasts impairs trabecular bone acquisition in the distal femur despite a compensatory increase in the expression of Fzd8. Complementary in vitro and in vivo data indicated the expression of Fzd4 by the osteoblast is required for normal matrix mineralization.
Materials and Methods
Animal models-
The Institutional Animal Care and Use Committee of the Johns Hopkins University approved all procedures using mice. Fzd4−/−, Fzd8−/−, Ndp−/y, and Fzd4flox mice have been described previously (Wang et al., 2001; Ye et al., 2009; Ye et al., 2011). To generate osteoblast-specific Fzd4 knockouts, Fzd4flox mice were crossed with Ocn-Cre mice, in which Cre recombinase expression is driven by the human osteocalcin promoter (Zhang et al., 2002). Fzd4flox mice were originally maintained on a mixed background of C57BL/6 and 129, but were backcrossed onto the C57BL/6 background (6 generations) using C57BL/6 mice originally obtained from The Jackson Laboratories.
Skeletal Phenotyping-
High-resolution images of the mouse femur, L5 vertebra, and skull were acquired using a desktop microtomographic imaging system (Skyscan 1172, Bruker) in accordance with the recommendations of the American Society for Bone and Mineral Research (ASBMR) (Bouxsein et al., 2010). Bones were scanned with an isotropic voxel size of 10 μm at 50 keV and 200 μA using a 0.5-mm aluminum filter. Trabecular bone parameters in the distal femur were assessed in a region of interest 500 μm proximal to the growth plate and extending for 2 mm. Cortical bone structure was assessed in a 500 μm region of interest centered on the mid-diaphysis. In the spine, trabecular bone parameters were assessed between the cranial and caudal growth plates. Tissue mineral density was assessed by scanning phantoms of known density with bone samples. Dynamic bone formation was assessed at 12 weeks of age by 2 injections of calcein separated by 7 days. After embedding in methylmethacrylate, 3 μm were cut with a Microm microtome. Static histomorphometric indices were measured at standardized sites under the growth plate using a semiautomatic method (Osteoplan II, Kontron) in compliance with the guidelines of the nomenclature committee of the ASBMR (Dempster et al., 2013). Serum bone turnover markers P1NP and CTX were measured by ELISA (IDS, AC-33F1 and AC-06F1). Femurs analyzed by three-point bending were harvested and immediately stored at −80 °C in PBS-soaked gauze. Femurs were thawed, then scanned with microCT before performing mechanical testing. Femurs were oriented on standard fixtures and a monotonic displacement ramp of 0.1 mm/s was applied until failure, with force and displacement acquired digitally. The force-displacement curves were converted to stress-strain using microCT-based geometry and analyzed using a custom GNU Octave script.
LacZ-staining-
Fzd4+/− neonates were harvested on ice to remove the skin and organs, then fixed for 8 hr at 4°C in 0.2% glutaraldehyde containing 5 mM EGTA, 10 mM MgCl2, and 100 mM NaH2PO4 (pH 7.3). Samples were then stained overnight at 4°C in X-gal solution containing 20 mM Tris-HCl (pH 7.5) 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and 1 mg/ml X-gal. After post-fixation in 4% paraformaldehyde (PFA) at 4°C overnight, samples were cleared using increasing concentrations of glycerol (20%, 50%, and 80%) in 1% potassium hydroxide. Stained samples were placed in 100% glycerol for imaging using a digital camera and macro lens (Fujifilm XT-1, 60 mm f2.4). For samples from 3 week old Fzd4+/− mice, tissues were fixed in 4% PFA at 4°C for 1 hour followed by staining in X-gal solution for 24–48 hours at 32°C. Stained sections were fixed in 4% PFA overnight at 4°C before embedding in OCT and cutting 10μm sections for analysis. Samples from Fzd4+/+ mice were processed in parallel as a negative control. Positive LacZ staining was confirmed by immunofluorescent staining using a Fzd4 antibody from Santa Cruz Biotechnology (sc135108).
Primary Osteoblast Culture-
Mouse osteoblasts were isolated from the calvaria of 1- to 3-day-old neonates by serial digestion in 1.8 mg/ml collagenase (Worthington Biochemical). For in vitro deletion of Fzd4, osteoblasts isolated from Fzd4flox mice were infected with adenovirus encoding Cre recombinase or GFP (Vector Biolabs). A multiplicity of infection of 100 was used in all experiments, and gene deletion was confirmed by quantitative PCR (qPCR). Osteoblast differentiation was induced by supplementing αMEM containing 10% serum with 10 mM β-glycerol phosphate and 50 μg/ml ascorbic acid. Differentiation was confirmed by Alkaline Phosphatase and Alizarin red S staining for mineralization according to standard techniques. Cellular proliferation was assessed by flow cytometry (FACSCalibur) after labeling osteoblast cultures with 10 μM BrdU for 24 hours and staining with anti-BrdU-APC and 7-amino-actinomycin D (BD Biosciences). Recombinant mouse Wnt3a was obtained from R&D Systems. Pyrophosphate levels were quantified using a commercially available kit (Sigma-Aldrich, MAK168).
Gene Expression and Western Blotting-
Total RNA was extracted from bone tissue (flushed free of marrow) or osteoblasts cultures using Trizol (Invitrogen). The iScript cDNA synthesis system (Bio-rad) was used to reverse transcribe 1 μg of RNA. Two microliters of cDNA was then subjected to PCR amplification using iQ SYBR Green Supermix (Bio-rad). Primer sequences were obtained from PrimerBank (http://pga.mgh.harvard.edu/primerbank/index.html). Reactions were normalized to endogenous b-actin reference transcript. Western blot analyses were carried out according to standard technique using primary antibodies obtained from Cell Signal (β-catenin, 2698; Actin, 3700), Santa Cruz Biotechnology (Fzd4, sc135108), and Novus Biologicals (Fzd8, NBP1–00833).
Statistics-
Sample sizes are provided in figure legends. In vitro studies were repeating using at least 2 independently isolated osteoblast cultures. Results are presented as mean ± SEM. Statistical analyses were performed using unpaired, two-tailed Student’s t test or One-way ANOVA (for comparison of bone parameters in Fzd4;Fzd8 mutants). A p-value less than 0.05 was considered significant. In all figures, * p < 0.05.
Results
Expression of Fzd4 is up-regulated during osteoblast differentiation.
Mammalian genomes contain 10 Fzd genes that can participate in the propagation of Wnt signaling (Wang et al., 2016). As a first step in characterizing the function of individual Fzds during osteoblast differentiation, we profiled the expression of all 10 genes by qPCR during the in vitro differentiation of calvarial osteoblasts indexed by the mRNA levels of Bglap2 (Figure 1A). Expression of Fzd1, Fzd2, Fzd3, Fzd5, Fzd6, and Fzd7 were all detectable by qPCR (Cycle counts ranging from 22–24), but mRNA levels did not change during in vitro differentiation (data not shown). Expression of Fzd10 was not detected by qPCR (data not shown). By contrast, the expression of Fzd4, Fzd8, and Fzd9 were increased at at least one time-point during the differentiation process (Figure 1B). Most notably, the expression of Fzd4 exhibited a consistent increase as osteoblasts matured such that mRNA levels at day 14 were more than 7-fold higher than those at day 0 and were accompanied by an increase in Fzd4 protein levels (Figure 1C).
Figure 1. Fzd4 is highly expressed by osteoblasts.
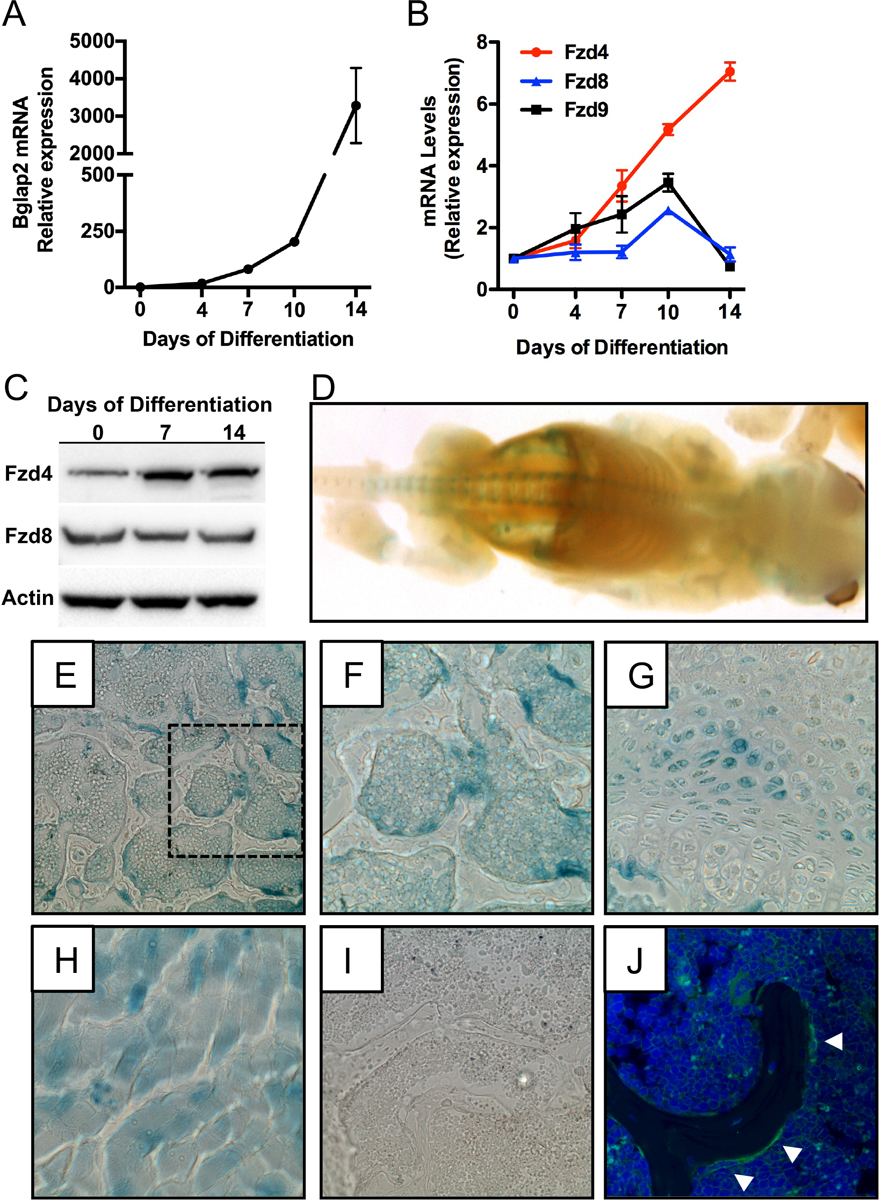
(A and B) qPCR analysis of Bglap2 (A) and Fzd4, Fzd8, Fzd9 mRNA levels in primary osteoblasts cultured under osteogenic conditions for up to 14 days. (C) Western blot analysis of Fzd4 and Fzd8 protein levels in primary osteoblasts. (D) Whole mount X-gal staining of newborn (P0) Fzd4+/− mice that contain a lacZ reporter gene under the control of the endogenous promoter. (E-G) X-gal staining in the trabecular bone compartment (E and F) and growth plate (G) in the distal femur of 3 week old Fzd4+/− mice. 10X original magnification. Panel F shows a 20X magnification of the boxed section of panel E. (H and I) X-gal staining of skeletal muscle (H) from Fzd4+/− mice and distal femur (I) of Fzd4+/+ mice were used as a positive and negative control for staining, respectively). (J). Immunofluorescent staining for Fzd4 in the distal femur of a 3 week old mouse. White arrowheads indicated Fzd4 expressing osteoblasts.
To confirm that Fzd4 was also expressed by osteoblasts in vivo, we performed X-gal staining in newborn and 3 week old Fzd4+/− mice that incorporate a lacZ reporter beginning at the initiator methionine of the endogenous Fzd4 gene (Wang et al., 2001). In whole mounts of newborn mice, weak X-gal staining was evident in the calvaria, rib cage, and developing long bones of the limbs (Figure 1D) while stronger staining was detected in the vertebral column. In the older mice, prominent X-gal staining was evident in cuboidal osteoblasts located on trabecular bone surfaces (Figure 1E and F), marrow elements, and a subset of reserve chondrocytes in the distal femur (Figure 1G). X-gal staining of skeletal muscle (Figure 1H) served as a positive control (Wang et al., 2001), while Fzd4+/+ mice, which exhibited no staining, served as a negative control (Figure 1I). Immunofluorescent staining for Fzd4 exhibited a similar expression pattern in vivo (Figure 1J). Thus, Fzd4 is well expressed by osteoblasts both in vitro and in vivo and exhibits the most dramatic change in expression among the 10 receptors during calvarial osteoblast differentiation.
Osteoblastic expression of Fzd4 is required for normal bone acquisition.
Fzd4−/− mice develop kyphosis, ataxia, and myocyte atrophy (Wang et al., 2001), suggestive of a role for this Fzd receptor in musculoskeletal physiology. However, the presence of additional complicating phenotypes required the development of a tissue-specific loss of function model to investigate the role of Fzd4 in osteoblasts in vivo. Osteoblast-specific mutants (referred to hereafter as ΔFzd4) were generated by crossing Fzd4flox mice (Ye et al., 2009) with mice expressing the Ocn-Cre transgene (Zhang et al., 2002) and were born at the expected Mendelian ratios. Compared to controls littermates, ΔFzd4 mice exhibited a 60% decrease in the expression Fzd4 in the femur (Figure 2A) and significant decreases in the expression of markers of activated Wnt/β-catenin signaling (Figure 2B).
Figure 2. Fzd4 expression in osteoblasts is required from normal bone acquisition.
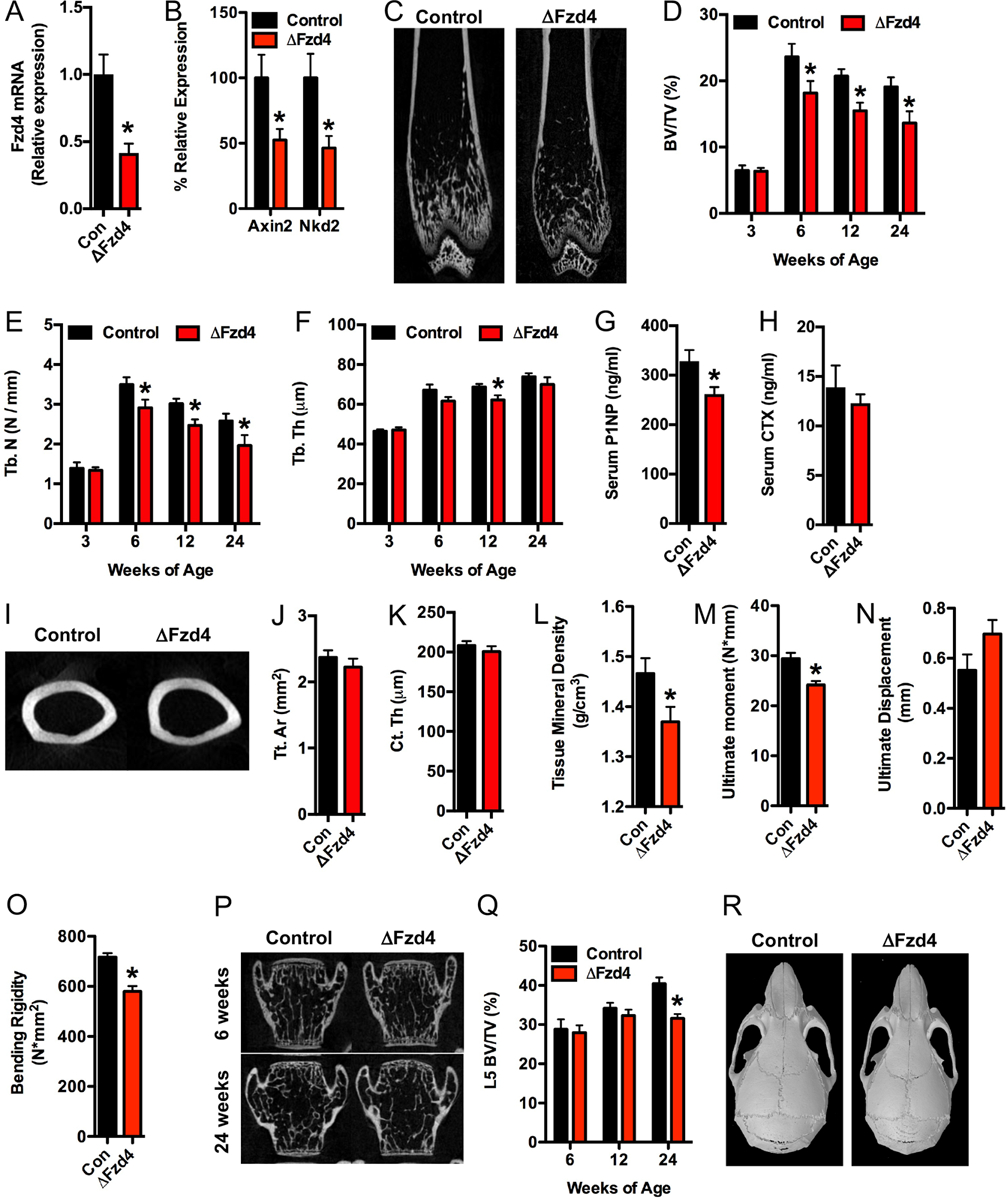
(A and B) qPCR analysis of Fzd4 (A) and Axin2 and Nkd2 (B) mRNA levels in the femur bone of control (Fzd4flox) and ΔFzd (Fzd4flox; Ocn-Cretg/+) mice (n=7–10 mice/genotype). (C) Representative microCT images of the distal femur of 6 week old control and ΔFzd4 mice. (D-F) Quantification of trabecular bone volume per tissue volume (D, BV/TV), trabecular number (E, Tb.N) and trabecular thickness (F, Tb.Th) in the distal femur (n=7–10 mice/genotype). (G and H) Serum measurements of the bone turnover markers P1NP (G) and CTX (H) in 12 week old mice (n=6–10mice/genotype). (I) Representative microCT images of the femoral mid-diaphysis of 24week old control and ΔFzd4 mice. (J-L) Quantification of tissue area (J, Tt.Ar), cortical thickness (K, Ct. Th) and tissue mineral density (L) at the mid-diaphysis (n=5–10mice/genotype). (M-O) Quantification of ultimate moment (M), ultimate displacement (N) and bending rigidity (O) by 3pt bend test of 24 week old femurs (n=8–10 mice/genotype). (P) Representative microCT images of the L5 vertebrae of 6 week old and 24 week old control and ΔFzd4 mice. (Q) Quantification of L5 trabecular bone volume per tissue volume (n=8–10 mice/genotype). (R) Computer reconstructions of skull morphology from microCT images of 6 week old control and ΔFzd4 mice. All results are expressed as mean ± SEM. *, p<0.05.
MicroCT analysis revealed normal trabecular bone structure in the distal femur of male ΔFzd4 mice at 3 weeks of age (Figure 2D). However, the mutants failed to achieve the peak in trabecular bone volume evident at 6 weeks of age in control littermates and maintained significantly lower bone volume through 24 weeks of age (Figure 2C and D) primarily due to reductions in trabecular number (Figure 2E). Mutant mice also exhibited a very modest decrease in trabecular thickness at 12 weeks of age (Figure 2F). Histomorphometric analyses performed in the trabecular bone compartment under the femoral growth plate of 12 week old mice confirmed the decrease in bone volume (Table I). This analysis suggested that the reduction in bone volume was secondary to a defect in matrix deposition and bone mineralization as osteoblasts numbers per bone perimeter were significantly increased but osteoid thickness and the mineral apposition rate were significantly decreased in ΔFzd4 mice relative to controls. In further support of this idea, serum levels of P1NP were significantly reduced in the mutants (Figure 2G). Indices of bone resorption including the numbers of osteoclasts per bone surface (Table I) and serum CTX levels (Figure 2H) were not affected by the loss of Fzd4 function in osteoblasts.
Table I.
Static and Dynamic Histomorphometry in the Distal Femur
| Bone parameter‡ | Control (n = 10) |
ΔFzd4 (n = 11) |
|---|---|---|
| Bone Structure | ||
| Bone volume/tissue volume (BV/TV; %) | 13.17 ± 0.82 | 10.74 ± 0.68* |
| Trabecular Thickness (μm) | 32.76 ± 1.81 | 29.35 ± 1.68 |
| Trabecular Spacing (μm) | 212.24 ± 11.47 | 247.73 ± 11.47* |
| Bone Formation | ||
| Osteoid surface/bone surface (OS/BS; %) | 2.29 ± 0.85 | 2.17 ± 0.39 |
| Osteoid thickness (O.Th; μm) | 2.87 ± 0.36 | 1.73 ± 0.19* |
| Osteoblast surface/bone surface (Ob.S/BS; %) | 0.82 ± 0.30 | 1.89 ± 0.57 |
| Osteoblast number/bone perimeter (NOb/BPm; no./mm) | 48.80 ± 14.18 | 130.72 ± 32.51* |
| Bone Erosion | ||
| Erosion surface/bone surface (ES/BS; %) | 2.40 ± 0.48 | 2.16 ± 0.42 |
| Osteoclast surface (Oc.S/BS; %) | 2.17 ± 0.46 | 1.90 ± 0.37 |
| Osteoclast number/bone perimeter (NOc/BPm; no./mm) | 80.10 ± 16.00 | 75.45 ± 14.29 |
| Bone Dynamics | ||
| Mineral apposition rate (MAR; μm/day) | 1.48 ± 0.07 | 1.07 ± 0.12* |
| Mineralizing surface/bone surface (MS/BS; %) | 9.25 ± 0.86 | 8.44 ± 1.07 |
| Bone formation rate/bone surface (BFR/BS; mm3/cm2/yr) | 51.36 ± 6.32 | 29.76 ± 3.23* |
Values are shown as mean ± SEM
p < 0.05
Cortical bone structure at the mid-diaphysis of the femur was comparable in control and ΔFzd4 mice (Figure 2I–K), but differences in signal intensity in microCT scans and the reduction in mineral apposition rate evident in trabecular bone and lead us to question whether the cortical bone of the mutants is hypo-mineralized. Consistent with this hypothesis, analysis of tissue mineral density at the mid-diaphysis revealed a significant decrease in the ΔFzd4 mice relative to controls (Figure 2L). Mechanical testing by three point bending (Figure 2M–O) revealed that the decrease in mineral density in the mutants was sufficient to alter bone strength as the ultimate moment (Figure 2M) and bending rigidity (Figure 2O) were significantly decreased in ΔFzd4 mice.
Despite the strong staining for the Fzd4-lacZ reporter in the vertebral column of newborn mice, the effect of Fzd4 loss of function on bone structure in the vertebrae was age-dependent. The mutants exhibited normal trabecular bone volume through 12 weeks of age, but exhibited a significant reduction at 24 weeks (Figure 2P and Q). Skull morphology was comparable in the control and ΔFzd4 mice (Figure 2R). When taken together, these data suggest that Fzd4 signaling is required for normal osteoblast function particularly during the acquisition of trabecular bone volume in long bones and the mineralization of cortical bone.
Loss Fzd4 impairs osteoblast differentiation and Wnt signaling in vitro.
To further characterize the function of Fzd4 in osteoblasts, we assessed differentiation and mineral production in calvarial osteoblasts isolated from Fzd4flox mice infected with adenoviral constructs directing the expression of Cre recombinase (ΔFzd4) or green fluorescent protein (control). Cultures of ΔFzd4 osteoblasts exhibited a 65% reduction in Fzd4 mRNA relative to control cultures (Figure 3A). Disrupting the expression of Fzd4 induced a modest but statistically significant increase in cellular proliferation indexed by BrdU incorporation (Figure 3B), but had the greatest effect on late stage osteoblast differentiation. Staining for alkaline phosphatase activity was equivalent between control and ΔFzd4 osteoblasts after 14 days of differentiation, but matrix mineralization was dramatically reduced (Figure 3C and D), which is compatible with the reduced mineral apposition rate observed in vivo (Table I). qPCR analysis also revealed significant reductions in the mRNA levels of genes encoding Runx2, Osterix (Sp7), Osteocalcin (Bglap2) and Osteoprotegerin (Tnfrsf11b) (Figure 3E and F). By contrast, the expression of inhibitors of mineralization including Ank, Enpp1, and Spp1 (Figure 3G) and the abundance of pyrophosphate (Figure 3H) were increased in the ΔFzd4 osteoblasts relative to controls, which may explain why calcium deposition was so dramatically affected.
Figure 3. Fzd4 is required for normal osteoblast differentiation and Wnt signaling in vitro.
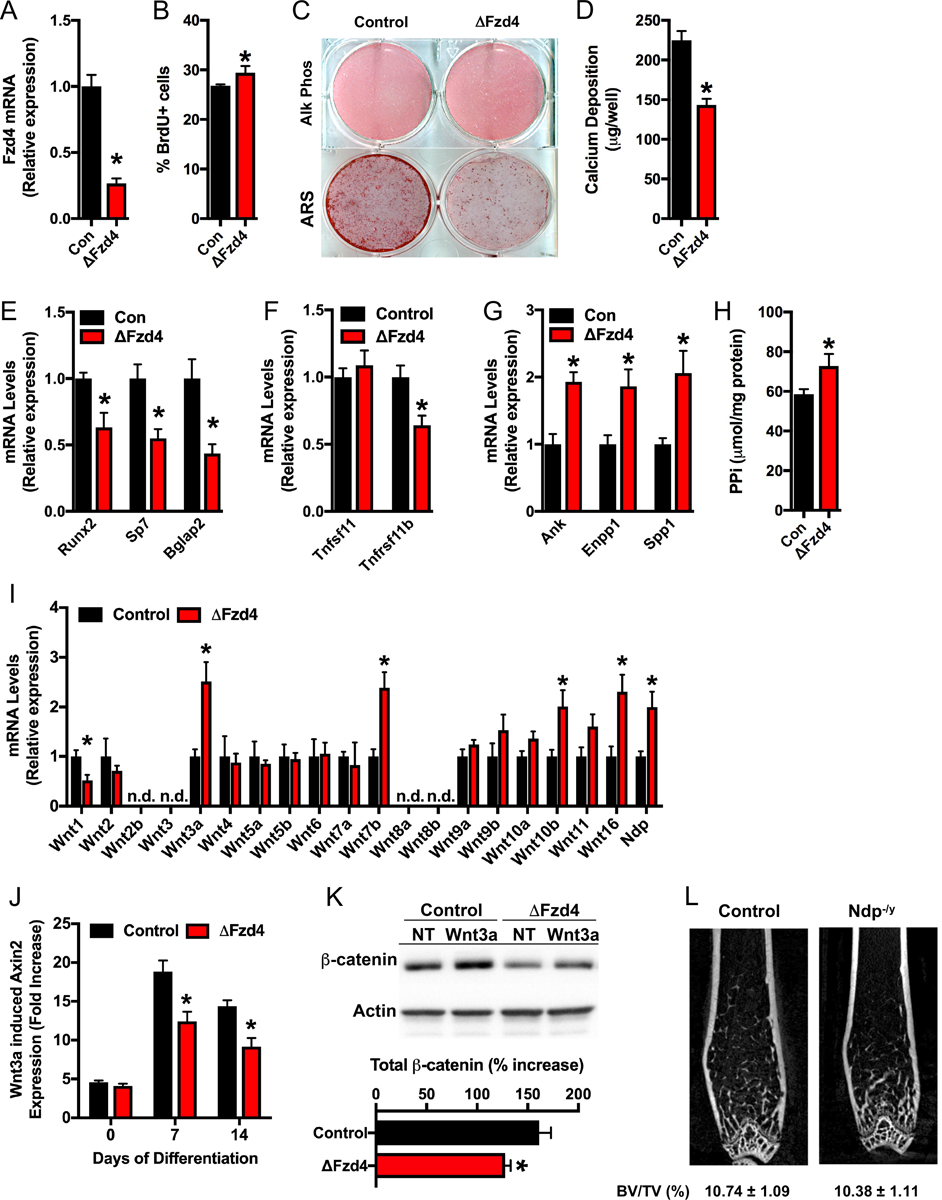
(A) qPCR analysis of Fzd4 mRNA levels in control and ΔFzd4 osteoblasts. (B) Assessment of osteoblast proliferation by flow cytometric analysis of BrdU incorporation. (C) Alkaline phosphatase and Alizarin red S (ARS) staining of cultures of control and ΔFzd4 osteoblasts cultured for 14 days under osteogenic conditions. (D) Quantification of calcium deposition via the extraction of ARS stain. (E and F) qPCR analysis of osteogenic markers (E) and regulators of osteoclastogenesis (F). (G) qPCR analysis of inhibitors of matrix mineralization. (H) Quantification of pyrophosphate (PPi) in cultures of control and ΔFzd4 osteoblasts. (I) qPCR analysis of Wnt ligand mRNA levels in control and ΔFzd4 osteoblasts (n.d., not detected). (J) Fold increase in Axin2 mRNA levels after Wnt3a (100ng/ml) treatment of differentiating control and ΔFzd4 osteoblasts. (K) Western blot analysis and quantification of β-catenin protein levels in control and ΔFzd4 osteoblasts left untreated (NT) or treated with Wnt3a. (L) Representative microCT images of the distal femur of 12 week old control and Ndp−/y mice and quantification of trabecular bone volume (n=9–11mice/genotype). All results are expressed as mean ± SEM. *, p<0.05.
As a first step in determining the signaling mechanism by which Fzd4 impacts osteoblast differentiation, we profiled the expression of Wnt ligands in control and ΔFzd4 osteoblasts (Figure 3I). The expression levels of Wnt3a, Wnt7b, Wnt10b, and Wnt16 were significantly upregulated in ΔFzd4 osteoblasts, which suggests that the receptor likely functions in the activation of canonical Wnt/β-catenin signaling. Indeed, differentiating ΔFzd4 osteoblasts exhibited an impairment in both the induction of Axin2, a β-catenin target gene (Jho et al., 2002), and the accumulation of β-catenin protein after Wnt3a stimulation (Figure 3J and K). The reduction in baseline β-catenin levels in ΔFzd4 osteoblasts could be due to an insensitivity of Fzd4 mutant osteoblasts to endogenous Wnt production. mRNA levels of Ndp, which encodes the Norrin protein that binds Fzd4 and Lrp5 to stimulate β-catenin signaling (Ye et al., 2009), were also up-regulated in ΔFzd4 osteoblasts when compared to controls (Figure 3I). However, bone architecture in the distal femur of Ndp−/y mice was comparable to controls, indicating that this ligand is unlikely to play a role in bone development. Thus, Fzd4 appears to regulate osteoblast differentiation and matrix mineralization by facilitating Wnt/β-catenin signaling.
Fzd4 deficiency leads to a compensatory increase in Fzd8 expression.
Pairs of Fzd receptors have been demonstrated to exhibit functional overlap during the development of a number of tissues (Wang et al., 2006; Ye et al., 2011; Yu et al., 2012). To determine if other Fzds compensate for the loss of Fzd4 function in osteoblasts, we examined the expression of other receptors in control and ΔFzd4 osteoblasts cultured in vitro. Among the 10 Fzd receptors, the expression of Fzd8 was significantly up-regulated in ΔFzd4 osteoblasts (Figure 4A). Since a similar increase in Fzd8 expression was also evident in the femur of ΔFzd4 mice (Figure 4B), we crossed ΔFzd4 mice with Fzd8+/− mice (Ye et al., 2011) to generate mice that lacked Fzd4 in osteoblasts and one allele of Fzd8 globally (ΔFzd4; Fzd8+/−) and control littermates. In support of the idea that Fzd8 acts to compensate for the lack of Fzd4 signaling, trabecular bone volume (Figure 4C–E) in the distal femur of 8 week old male ΔFzd4; Fzd8+/− mice was reduced to a greater degree than that in ΔFzd4 littermates, even though bone volume was not significantly affected in Fzd8+/− mice. More strikingly, trabecular bone volume in the L5 vertebrae of ΔFzd4; Fzd8+/− was significantly reduced in 8 week old mice even though there was no effect of eliminating the expression of Fzd4 alone at this timepoint (Figure 4F–H). Taken together, these data indicate that the lack of Fzd4 signaling in the osteoblast is partially compensated for by an up-regulation of Fzd8 expression and suggest that functions of the two receptors are at least partially redundant in the osteoblast.
Figure 4. Fzd8 compensated for the loss of Fzd4 function in osteoblasts.
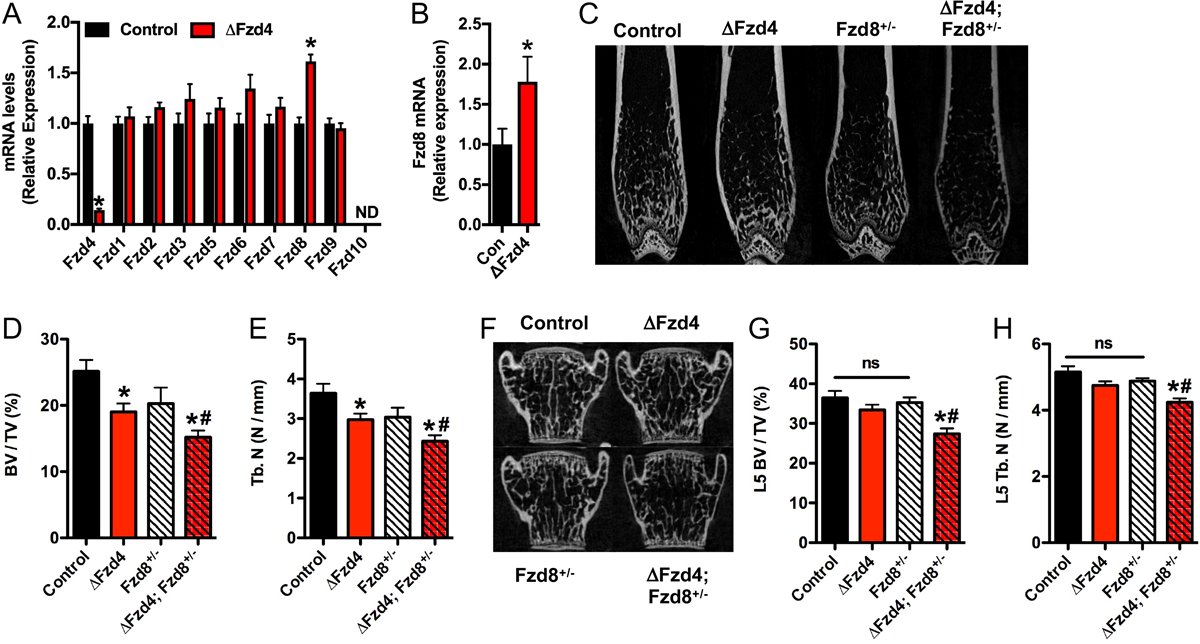
(A) qPCR analysis of Fzd receptor expression in control and ΔFzd4 osteoblasts cultured in vitro. (B) qPCR analysis of Fzd8 mRNA levels in the femur of 6 week old control and ΔFzd4 mice (n=7–8 mice/genotype). (C) Representative microCT images of the distal femur of mutant mice. (D and E) Quantification of trabecular bone volume per tissue volume (D, BV/TV) and trabecular number (E, Tb.N) in the distal femur of 8 week old mice (n=7–11 mice/genotype). (E) Representative microCT images of the L5 vertebrae. (G and H) Quantification of trabecular bone volume per tissue volume (G, BV/TV) and trabecular number (H, Tb.N) in the L5 vertebrae of 8 week old mice (n=7–11 mice/genotype). All results are expressed as mean ± SEM. *, p<0.05 vs control mice. # p<0.05 vs ΔFzd4 mice. One-Way ANOVA was used to compare bone structural parameters across the 4 groups.
Discussion
The anabolic effects of Wnt/β-catenin signaling in the osteoblast are well-established, but the identity of the seven-transmembrane Fzd receptors required for the initiation of signaling remains relatively unknown. In this study, we profiled the expression of all 10 mammalian Fzds during in vitro osteoblast differentiation and found that Fzd4 exhibited the most dramatic increase in expression. Fzd4 was also highly expressed by the osteoblast in vivo and genetic disruption of Fzd4 expression in mature osteoblasts impaired trabecular bone acquisition and reduced tissue mineral density in the cortical bone compartment. To the best of our knowledge this is the first report on a tissue-specific knockout of a Fzd receptor in the osteoblast and the first to show an effect of Fzd receptor ablation on Wnt/β-catenin signaling in bone.
Our analyses of bone structure and bone cell function indicate that the reduction in bone volume evident in the mutant mice is due to Fzd4’s action in the late stage of osteoblast differentiation. Osteoid thickness and the mineralization rate were reduced in ΔFzd4 mice, while matrix mineralization was reduced in vitro in cultures of mutant osteoblasts. Osteoblast numbers were actually increased in vivo and may represent a compensatory mechanism to overcome the defect in matrix deposition and mineralization. Additionally, disrupting the expression of Fzd4 in vitro only reduced responsivity to Wnt3a when osteoblasts had been induced to differentiate. These data suggest that another Fzd receptor must mediate the effects of Wnt/β-catenin signaling on osteoblast proliferation and that different receptors temporally control osteoblast function. Along these same lines, Wnt/β-catenin signaling is known to control osteoclast development (Glass et al., 2005; Holmen et al., 2005), but bone resorption was normal in the the Fzd4 mutants.
Consistent with the idea that Fzd4 regulates Wnt/β-catenin in mature osteoblasts, the skeletal phenotype of Fzd4 mutants corresponds well with the phenotype of mouse mutants in which Lrp5 expression was ablated in the same cell population (Lrp5flox; Ocn-Cre) (Riddle et al., 2013). In these Lrp5 mutants, we reported that trabecular bone volume was initially normal but that the mice developed a reduction in bone volume with increasing age that was secondary to impaired osteoblast activity. Moreover, calvarial osteoblasts deficient for Lrp5 exhibited normal alkaline phosphatase activity but greatly impaired matrix mineralization, a phenotype nearly identical to what we report here for Fzd4 mutant osteoblasts. While a direct interaction between Fzd4 and Lrp5 was not explored in our study, the two mediators of Wnt signaling coordinate vascular development in the retina (Xu et al., 2004; Ye et al., 2009) and activate β-catenin signaling in HEK293 cells (Mikels et al., 2006). Additionally, Fzd4 expression was found to be increased in an RNAseq analysis of Lrp5 deficient osteoblasts (Ayturk et al., 2013).
Two previous studies have utilized global knockout mice to explore the requirements for specific Fzds during bone development. In humans, the FZD9 gene is located in a region of chromosome 7 that is deleted in Williams-Beuren Syndrome (OMIM 194050), a neurodevelopmental disorder that also presents with reduced bone mineral density (Cherniske et al., 2004). Albers and colleagues (Albers et al., 2011) demonstrated that Fzd9−/− mice develop osteopenia in association with reduced bone formation and impaired in vitro osteoblast differentiation indicating that this receptor is important for osteoblast function. However, Fzd9-deficient osteoblasts exhibited normal activation of β-catenin signaling after Wnt3a treatment, which suggests that Fzd9 regulates osteoblast function via a different mechanism. Fzd9−/− mice also exhibit abnormalities in lymphocyte development (Ranheim et al., 2005) that could indirectly effect bone mass in vivo (Ponzetti et al., 2019). Albers and colleagues (Albers et al., 2013) also reported that Fzd8−/− mice develop age-related osteopenia, but in the case of this model, bone formation is normal and the defect was reported to be inherent to the osteoclast.
As noted above, a number of Fzd receptors exhibit functional overlap (Wang et al., 2006; Ye et al., 2011; Yu et al., 2012). In light of our data, the lack of an effect of Fzd8 deficiency on osteoblast function in the Albers study (Albers et al., 2013) could be related to a primacy for and functional redundancy by Fzd4. In Fzd4 mutant mice and cultured osteoblasts, we found that the expression of Fzd8 was increased. In turn, disrupting only a single allele of Fzd8 in the context of Fzd4 deficiency was sufficient to further decrease trabecular bone volume in the femur and induce osteopenia in the spine at a younger age. A similar interaction between Fzd4 and Fzd8 has also been described in the developing kidney. In this tissue, Ye et al (Ye et al., 2011) showed that abolishing Fzd4 expression results in a ~15% reduction in kidney length, while kidney length was normal in Fzd8−/− mice. Fzd4−/−; Fzd8−/− double mutants exhibited postnatal lethality with severe hypoplasia of the kidney (Ye et al., 2011). Additional studies will be necessary to fully clarify the molecular mechanisms responsible for the apparent redundancy between Fzd4 and Fzd8 since reporter assays suggest the ability of the receptors to transduce signals from individual Wnt ligands is not identical (Yu et al., 2012). The differential effects we observed in the femur and spine, with trabecular bone in the femur being more sensitive to the ablation of Fzd4, could be related to the relative expression of each receptor at these skeletal sites or the expression of specific ligands or antagonists.
In summary, our studies demonstrate a key role for Fzd4 in the propagation of Wnt/β-catenin signaling in the osteoblast and normal skeletal homeostasis. Even though therapeutic strategies designed to enhance the activity of Wnt/β-catenin signaling are becoming available in the clinic, we believe functional genetic and molecular interrogation of the large number of Wnt signaling components present in bone may still aid in the development of new small-molecule therapies that increase bone mass and strength. Indeed, the G protein coupled receptor family, of which Fzd receptors form a sub-class (Foord et al., 2005), are targeted by greater than 30% of all available therapeutics (Wise et al., 2002). Our studies highlight the potential value in targeting Fzd4.
Acknowledgments
We are grateful to Drs. Jeremy Nathans and Marie-Claude Faugere for kindly providing mice and assisting with skeletal histomorphometric analyses, respectively.
Funding
Work in the authors’ laboratories is supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (DK099134, R.C.R) and the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development (BX003724, R.C.R).
Footnotes
Conflict of Interest Statement
Dr. Williams maintains an SRA with Janssen and is a Surrozen stockholder and member of the Scientific Advisory Board. All other authors declare no conflicts of interest.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Adler PN (1992). The genetic control of tissue polarity in Drosophila. Bioessays, 14(11), 735–741. [DOI] [PubMed] [Google Scholar]
- Albers J, Keller J, Baranowsky A, Beil FT, Catala-Lehnen P, Schulze J, Amling M, & Schinke T (2013). Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J Cell Biol, 200(4), 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers J, Schulze J, Beil FT, Gebauer M, Baranowsky A, Keller J, Marshall RP, Wintges K, Friedrich FW, Priemel M, Schilling AF, Rueger JM, Cornils K, Fehse B, Streichert T, Sauter G, Jakob F, Insogna KL, Pober B, Knobeloch KP, Francke U, Amling M, & Schinke T (2011). Control of bone formation by the serpentine receptor Frizzled-9. J Cell Biol, 192(6), 1057–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayturk UM, Jacobsen CM, Christodoulou DC, Gorham J, Seidman JG, Seidman CE, Robling AG, & Warman ML (2013). An RNA-seq protocol to identify mRNA expression changes in mouse diaphyseal bone: applications in mice with bone property altering Lrp5 mutations. J Bone Miner Res, 28(10), 2081–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, & Van Hul W (2001). Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hu Mol Genet, 10(5), 537–543. [DOI] [PubMed] [Google Scholar]
- Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, & Van Hul W (2002). Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet, 39(2), 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, & Muller R (2010). Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res, 25(7), 1468–1486. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, & Mulligan J (2001). Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. AM J Hum Genet, 68(3), 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniske EM, Carpenter TO, Klaiman C, Young E, Bregman J, Insogna K, Schultz RT, & Pober BR (2004). Multisystem study of 20 older adults with Williams syndrome. Am J Med Genet A, 131(3), 255–264. [DOI] [PubMed] [Google Scholar]
- Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, Zhong Z, Matthes S, Jacobsen CM, Conlon RA, Brommage R, Liu Q, Mseeh F, Powell DR, Yang QM, Zambrowicz B, Gerrits H, Gossen JA, He X, Bader M, Williams BO, Warman ML, & Robling AG (2011). Lrp5 functions in bone to regulate bone mass. Nat Med, 17(6), 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, & Parfitt AM (2013). Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res, 28(1), 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, & Rauch F (2013). Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet, 50(5), 345–348. [DOI] [PubMed] [Google Scholar]
- Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, Davenport AP, Spedding M, & Harmar AJ (2005). International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev, 57(2), 279–288. [DOI] [PubMed] [Google Scholar]
- Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, & Karsenty G (2005). Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell, 8(5), 751–764. [DOI] [PubMed] [Google Scholar]
- Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, & Warman ML (2001). LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell, 107(4), 513–523. [DOI] [PubMed] [Google Scholar]
- Greco TL, Takada S, Newhouse MM, McMahon JA, McMahon AP, & Camper SA (1996). Analysis of the vestigial tail mutation demonstrates that Wnt-3a gene dosage regulates mouse axial development. Genes Dev, 10(3), 313–324. [DOI] [PubMed] [Google Scholar]
- Hill TP, Spater D, Taketo MM, Birchmeier W, & Hartmann C (2005). Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell, 8(5), 727–738. [DOI] [PubMed] [Google Scholar]
- Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, & Williams BO (2005). Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem, 280(22), 21162–21168. [DOI] [PubMed] [Google Scholar]
- Hsieh JC, Rattner A, Smallwood PM, & Nathans J (1999). Biochemical characterization of Wnt-frizzled interactions using a soluble, biologically active vertebrate Wnt protein. Proc Natl Acad Sci U S A, 96(7), 3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda CY, Waghray D, Levin AM, Thomas C, & Garcia KC (2012). Structural basis of Wnt recognition by Frizzled. Science, 337(6090), 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, & Costantini F (2002). Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol, 22(4), 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joeng KS, Schumacher CA, Zylstra-Diegel CR, Long F, & Williams BO (2011). Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo. Dev Biol, 359(2), 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G (2000). Bone formation and factors affecting this process. Matrix Biol, 19(2), 85–89. [DOI] [PubMed] [Google Scholar]
- Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, Fischer B, Yigit G, Janda CY, Becker J, Breer S, Altunoglu U, Grunhagen J, Krawitz P, Hecht J, Schinke T, Makareeva E, Lausch E, Cankaya T, Caparros-Martin JA, Lapunzina P, Temtamy S, Aglan M, Zabel B, Eysel P, Koerber F, Leikin S, Garcia KC, Netzer C, Schonau E, Ruiz-Perez VL, Mundlos S, Amling M, Kornak U, Marini J, & Wollnik B (2013). Mutations in WNT1 cause different forms of bone fragility. AM J Hum Genet, 92(4), 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M, Lu JT, Pekkinen M, Wessman M, Heino TJ, Nieminen-Pihala V, Aronen M, Laine T, Kroger H, Cole WG, Lehesjoki AE, Nevarez L, Krakow D, Curry CJ, Cohn DH, Gibbs RA, Lee BH, & Makitie O (2013). WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med, 368(19), 1809–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, & Wu D (2005). Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem, 280(20), 19883–19887. [DOI] [PubMed] [Google Scholar]
- Liu C, Bakeri H, Li T, & Swaroop A (2012). Regulation of retinal progenitor expansion by Frizzled receptors: implications for microphthalmia and retinal coloboma. Hu Mol Genet, 21(8), 1848–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, & He X (2012). Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb Perspect Biol, 4(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikels AJ, & Nusse R (2006). Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol, 4(4), e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy GR, Boyce B, Hughes D, Wright K, Bonewald L, Dallas S, Harris S, Ghosh-Choudhury N, Chen D, Dunstan C, & et al. (1995). The effects of cytokines and growth factors on osteoblastic cells. Bone, 17(2 Suppl), 71S–75S. [DOI] [PubMed] [Google Scholar]
- Niehrs C (2012). The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol, 13(12), 767–779. [DOI] [PubMed] [Google Scholar]
- Ponzetti M, & Rucci N (2019). Updates on Osteoimmunology: What’s New on the Cross-Talk Between Bone and Immune System. Front Endocrinol (Lausanne), 10, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranheim EA, Kwan HC, Reya T, Wang YK, Weissman IL, & Francke U (2005). Frizzled 9 knock-out mice have abnormal B-cell development. Blood, 105(6), 2487–2494. [DOI] [PubMed] [Google Scholar]
- Regard JB, Zhong Z, Williams BO, & Yang Y (2012). Wnt signaling in bone development and disease: making stronger bone with Wnts. Cold Spring Harb Perspect Biol, 4(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle RC, Diegel CR, Leslie JM, Van Koevering KK, Faugere MC, Clemens TL, & Williams BO (2013). Lrp5 and Lrp6 Exert Overlapping Functions in Osteoblasts during Postnatal Bone Acquisition. PLoS One, 8(5), e63323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulifson EJ, Wu CH, & Nusse R (2000). Pathway specificity by the bifunctional receptor frizzled is determined by affinity for wingless. Mol Cell, 6(1), 117–126. [PubMed] [Google Scholar]
- Semenov M, Tamai K, & He X (2005). SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem, 280(29), 26770–26775. [DOI] [PubMed] [Google Scholar]
- Umbhauer M, Djiane A, Goisset C, Penzo-Mendez A, Riou JF, Boucaut JC, & Shi DL (2000). The C-terminal cytoplasmic Lys-thr-X-X-X-Trp motif in frizzled receptors mediates Wnt/beta-catenin signalling. EMBO J, 19(18), 4944–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson CR, Conover S, & Adler PN (1989). A Drosophila tissue polarity locus encodes a protein containing seven potential transmembrane domains. Nature, 338(6212), 263–264. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chang H, Rattner A, & Nathans J (2016). Frizzled Receptors in Development and Disease. Curr Top Dev Biol, 117, 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo N, & Nathans J (2006). The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci, 26(8), 2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huso D, Cahill H, Ryugo D, & Nathans J (2001). Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J Neurosci, 21(13), 4761–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise A, Gearing K, & Rees S (2002). Target validation of G-protein coupled receptors. Drug Discov Today, 7(4), 235–246. [DOI] [PubMed] [Google Scholar]
- Wong HC, Bourdelas A, Krauss A, Lee HJ, Shao Y, Wu D, Mlodzik M, Shi DL, & Zheng J (2003). Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol Cell, 12(5), 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, & Nathans J (2004). Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell, 116(6), 883–895. [DOI] [PubMed] [Google Scholar]
- Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, Peachey NS, & Nathans J (2009). Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell, 139(2), 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Wang Y, Rattner A, & Nathans J (2011). Genetic mosaic analysis reveals a major role for frizzled 4 and frizzled 8 in controlling ureteric growth in the developing kidney. Development, 138(6), 1161–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Ye X, Guo N, & Nathans J (2012). Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: evidence for a network of interacting genes. Development, 139(23), 4383–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, & Clemens TL (2002). Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem, 277(46), 44005–44012. [DOI] [PubMed] [Google Scholar]
- Zheng HF, Tobias JH, Duncan E, Evans DM, Eriksson J, Paternoster L, Yerges-Armstrong LM, Lehtimaki T, Bergstrom U, Kahonen M, Leo PJ, Raitakari O, Laaksonen M, Nicholson GC, Viikari J, Ladouceur M, Lyytikainen LP, Medina-Gomez C, Rivadeneira F, Prince RL, Sievanen H, Leslie WD, Mellstrom D, Eisman JA, Moverare-Skrtic S, Goltzman D, Hanley DA, Jones G, St Pourcain B, Xiao Y, Timpson NJ, Smith GD, Reid IR, Ring SM, Sambrook PN, Karlsson M, Dennison EM, Kemp JP, Danoy P, Sayers A, Wilson SG, Nethander M, McCloskey E, Vandenput L, Eastell R, Liu J, Spector T, Mitchell BD, Streeten EA, Brommage R, Pettersson-Kymmer U, Brown MA, Ohlsson C, Richards JB, & Lorentzon M (2012). WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet, 8(7), e1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]