

Abstract
The assessment of major organ toxicities through in silico predictive models plays a crucial role in drug discovery. Computational tools can predict chemical toxicities using the knowledge gained from experimental studies which drastically reduces the attrition rate of compounds during drug discovery and developmental stages. The purpose of in silico predictions for drug leads and anticipating toxicological endpoints of absorption, distribution, metabolism, excretion and toxicity, clinical adverse impacts and metabolism of pharmaceutically active substances has gained widespread acceptance in academia and pharmaceutical industries. With unrestricted accessibility to powerful biomarkers, researchers have an opportunity to contemplate the most accurate predictive scores to evaluate drug’s adverse impact on various organs.
A multiparametric model involving physico-chemical properties, quantitative structure-activity relationship predictions and docking score was found to be a more reliable predictor for estimating chemical toxicities with potential to reflect atomic-level insights. These in silico models provide informed decisions to carry out in vitro and in vivo studies and subsequently confirms the molecules clues deciphering the cytotoxicity, pharmacokinetics, and pharmacodynamics and organ toxicity properties of compounds. Even though the drugs withdrawn by USFDA at later phases of drug discovery which should have passed all the state-of-the-art experimental approaches and currently acceptable toxicity filters, there is a dire need to interconnect all these molecular key properties to enhance our knowledge and guide in the identification of leads to drug optimization phases. Current computational tools can predict ADMET and organ toxicities based on pharmacophore fingerprint, toxicophores and advanced machine-learning techniques.
Keywords: Organ toxicities, in silico, physico-chemical properties, multiparametric approach
1. Drug discovery process
The drug discovery process was initiated in the year 1806 when a hypnotic agent called morphine was synthesized. However, the first attempt of drug discovery was mostly attributed to the Avogadro’s atomic hypothesis and coal-tar derivatives synthesis in the 1870s. The process of drug discovery starts with the identification of biological target and lead discovery (candidate, synthesis, characterization, high-throughput screening and assays for therapeutic efficacy) followed by lead optimization through pharmacokinetics and pharmacodynamics studies with preclinical and clinical development (phase-1, 2, 3 and 4). The newly synthesized drug should be approved by United States Food and Drug Administration (USFDA) before introducing to the market. This whole process takes around 15–20 years with massive financial investment. The rational drug design includes new drug discovery based on the knowledge of biological target pertain to therapeutic benefit. However, it mainly focuses on the accurate calculations of binding affinity calculations through customized structure-based approaches.
The receptive substances theory introduced by John Langley in 1905 was the turning point in the milestone of the drug discovery process. Paul Ehrlich, the Father of modern chemotherapy, developed synthetic drugs and arsphenamine (Salvarsan) which was later introduced into market by Sachiro Hata. Those were the first rational approaches guided by structure-activity relationship for sleeping sickness and syphilis in the year 1910 (Lednicer 2006). QSAR (Quantitative Structure-Activity Relationship) found its root from toxicology field and relied on chemical structure and experimentally determined toxicity endpoints to develop relationships. After the discovery of QSAR approach by Hansch and Fujita in 1960, structure- and target-based techniques emerged as two approaches for drug discovery and development (Bolten and DeGregorio 2002). Drug discovery relies on disease mechanisms and their understanding which is further carried through target identification, lead compound discovery, and clinical trials (Barab asi et al. 2011; Chen and Butte 2013).
2. Toxicity overview
Toxicity is damage to an organ in humans or animals as a result of chemical or environmental exposure. A crucial step in determining the probable toxicity of a new chemical entity is the indication of access in our human system and outcomes of that compound in human metabolism. The analysis of potential toxicity initiates a series of events: the characterization of the compound and its transformation achieved through metabolism, followed by the recognition of enzymes which will literally be involved in the metabolism, with consideration of exposure and reaction rates (Bugrim et al. 2004).
Different types of toxicities have been investigated, containing blood/cardiovascular toxicity, carcinogenicity, dermal/ocular toxicity, genetic toxicity (germ cells), hepatotoxicity, immunotoxicity, mutagenicity, nephrotoxicity, neurotoxicity, reproductive toxicity and respiratory toxicity (Ekins 2007). The measurements of toxicity can be classified into severe toxicity, subchronic toxicity and chronic toxicity in relation to developmental and reproductive toxicity (DART) or toxicokinetic studies.
3. Organ toxicity in drug discovery
The principles of target organ toxicity include the importance of pharmacokinetics, metabolic stimulation and (key defense instruments, discharge, species variation) and tissue-specific biochemistry. The evaluation of toxicity and safety is required by Contract Research Organization (CRO) law for every new product or therapy offered by the medical equipment, or drug chemical for specific toxicity of different target organs, such as lung, liver, kidney, nervous system, ear, eye, and the male and female reproductive systems. The main goal of this toxicity assessment is to identify the side effects of a substance or product which may harm (toxicity) in humans (Bugrim et al. 2004). Organ system toxicity seeks high-dose chemotherapy in the marrow transplant setting, broadly as an exact reaction on the organ systems of the chemotherapy and radiation therapy.
It is estimated that more than 900 drugs, toxins and herbs entering the market are withdrawn due to reports of liver injury and ~75 of the idiosyncratic drug reactions leading to liver transplantation or death. Therefore, hepatotoxicity (drug-induced hepatic injury) has been distinguished as one among the most common reasons for withdrawal of drugs from the market and cessation of clinical trials (Mehta et al. 2010). In the case of idiosyncratic toxicities, a ‘black box warning’ is given to medications (Park 2013). Nephrotoxicity in the framework of autologous hematopoietic stem cell transplantation (AHSCT) can be derived from many sources, such as chemotherapeutic agents (cisplatin, nitrosoureas, phenylalanine mustard), and nephrotoxic reactions to antibiotics. Pulmonary drug toxicity was noted in the post-transplant environment. Dose limited cardiac toxicity resulting from chemotherapeutic agents causes arrhythmia, tachycardia, bradycardia, atrial flutter, atrial fibrillation, ventricular fibrillation, hypertension, myocardial infarction and congestive heart failure, etc. may occur (Pai and Nahata 2000).
3.1. In vitro model/in vivo model – methods
Identification and interpretation of disease and effects of drugs in specific cells are carried out during preclinical drug development through in vitro studies (Figure 1). These in vitro studies enhance the understanding of the mechanisms of drug- and chemical-induced toxicities. The U.S. National Toxicology Program (NTP) Breakout Group studied acute oral toxicity data of rodents by a series of studies including the bovine corneal opacity test, the skin permeability assays, the EpiDerm™ model for dermal irritation/corrosivity, a neutral red uptake (NRU) assay for systemic toxicity, a primary rat hepatocyte assay for hepatic toxicity. The Breakout Group considered various organ systems i.e., liver, central nervous system, kidney, heart, hematopoietic system, and lung. The xenobiotic effects on the skin, gastrointestinal tract, and eye through the acute toxicity tests were measured and a database created comparing hepatic toxicity using in vitro and in vivo studies. Table 1 describes a brief outline on currently available in vitro toxicity assays.
Figure 1.

Traditional in vitro modeling approach.
Table 1.
Cell viability (cytotoxicity) assays used for in vitro toxicology.
| Sr. No. | Assay | Reference |
|---|---|---|
| 1 | MTT (3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) | (Freimoser et al. 1999) |
| 2 | MTS Cell Proliferation Colorimetric Assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) | (Malich et al. 1997) |
| 3 | ATP (Adenosine triphosphate phosphate) | (Bowen and Kerwin 1956) |
| 4 | NR (Neutral Red) | (Borenfreund et al. 1988) |
| 5 | ELISA (Enzyme-linked immunosorbent assay) | (Yolken 1978) |
| 6 | XTT (2,3-bis-(2-methoxy-4-nitro-65-sulfophenyl)-2H-tetrazolium-5-carboxanilide) | (De Logu et al. 2003) |
| 7 | WSTs (Water-soluble Tetrazolium salts) | (Ukeda et al. 1999) |
Traditional toxicity testing methods provide insights about the chemical safety in reference to humans through in vivo analysis from animal models but these methods are too expensive for the exploration of species differences (Collins et al. 2008). Tables 2 and 3 tabulated various tests performed for the in vivo analysis in a mouse model. The Tox21 program (Tice et al. 2013) is a collaboration between the National Institute of Environmental Health Sciences/National Toxicology Program, the US Environmental Protection Agency/National Center for Computational Toxicology, the National Institutes of Health Chemical Genomics Center (now within the National Center for Advancing Translational Sciences) and the US Food and Drug Administration, which is focused on in vivo toxicity through in vitro testing. The Tox21 program relies on cell-based assays, including nuclear receptors (Huang et al. 2011) and stress response pathways (Shukla et al. 2010), in a quantitative HTS (qHTS) format in triplicate (Attene-Ramos et al. 2013) using 10 K compound library screening. The self-organizing map (SOM) algorithm (Kohonen 2006) is used, to cluster the compound activity profiles or structure fingerprints based on the similarity between the profiles measured by pair-wise Euclidean distance through SOM Toolbox (http://www.cis.hut.fi/projects/somtoolbox/). This clustered model measures toxic potential (toxicity score) of the compounds on the basis of log P-value, sensitivity, specificity, cutoff score through Fisher’s exact test (Huang et al. 2016).
Table 2.
Experimental study of a particular drug in mice model for in vivo analysis.
| No. of | Duration of | Autopsy | |
|---|---|---|---|
| Group | animal | dosing | (Day) |
| Untreated control | 10 | 30 | 31st |
| Vehicle treated Control | 10 | 30 | 31st |
| Drug-Low Dose (40 mg/kg body weight) | 10 | 30 | 31st |
| Drug-Medium Dose (60mg /kg body weight) | 10 | 30 | 31st |
| Drug-High Dose (120 mg/kg body weight) | 10 | 30 | 31st |
Table 3.
Biochemical parameters analysis.
| Sr. No. | Tests | References |
|---|---|---|
| 1 | Total protein | (Lowry et al. 1951) |
| 2 | Lipid peroxidation | (Ohkawa et al. 1979) |
| 3 | Cholesterol | (Zlatkis et al. 1953) |
| 4 | Succinate dehydrogenase | (Beatty et al. 1966) |
| 5 | ATPase | (Quinn and White 1968) |
| 6 | Acid phosphatase | (Bessey et al. 1946) |
| 7 | Alkaline phosphatases | (Bessey et al. 1946) |
| 8 | Total lipids | (Frings et al. 1972) |
| 9 | Catalase activity | (Luck and Peroxidase 1963) |
| 10 | Superoxide dismutase | (Kakkar et al. 1984) |
| 11 | Total glutathione | (Grunert and Phillips 1951) |
| 12 | Glutathione peroxidase | (Paglia and Valentine 1967) |
| 13 | Glutathione reductase | (Mavis and Stellwagen 1968) |
| 14 | DNA | (Giles and Myers 1965) |
| 15 | RNA | (Mezbaum 1939) |
3.2. Currently available modalities
Collections of chemical assets information are important for informing computational toxicologists on probable toxicity alerts for new compounds. In addition, the aspiration of framing structure–toxicity relationships and correlated prediction models venture into medicinal chemistry area of drug discovery. These relationships are then expressed as mathematical and statistical models to explain the mechanisms of chemical toxicity and permit the prediction of detrimental impacts of these chemicals on human health and/or the environment. Such approaches are of high significance which enables prioritization of chemicals for in vitro and in vivo screenings. A vast amount of data are available to computational toxicologists which arises a greater opportunity of open source software for the establishment of computational models (Vashishtha et al. 2002). For example, Pfizer created a Bayesian model for foreseeing cytotoxicity (Engkvist et al. 2003).
Drug transporters, key membrane proteins that transport drugs and endogenous compounds are beginning to be examined for their role in chemical toxicity (Worth and Cronin 2003). Assessment of corrosiveness by intestinal bile acid is also looked out by human apical sodium-subordinate bile acid transporter (ASBt). Since ASBt is the principal component for intestinal bile corrosive reabsorption, this target may be directly involved in colorectal physiology. A separate 3 D-QSAR and Bayesian models were created utilizing 38 ASBt inhibitors (Cronin et al. 2003). Both models showed great consistency in determining whether a drug will be a ASBt inhibitor. FDA approved numerous medications from different classes, for example, the dihydropyridine calcium channel blockers and 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG CoA-reductase) inhibitors were identified to be ASBt inhibitors.
3.2.1. In silico approaches
Toxicological tests for new chemical entities require the regular use of laboratory animals, infrastructure facilities (BSL3 level) and time. The purpose of computational toxicology is to stimulate or simulate the estimation of possibly hazardous substances through in silico models. Various types of models could be constructed by utilizing such information. For instance, computational chemists model particular toxicological endpoints to relate with the chemical structure to speculate possible clues or fragments which may prove undesirable effects in in vitro and in vivo assays (Muster et al. 2008; Merlot 2010).
In recent years, pharmaceutical companies have introduced toxicity testing as well as ADME studies earlier in the drug development process. The goal is to use in silico methods to predict toxicity even before a drug candidate is synthesized. First, toxicity covers a wide range of adverse effects; second, there is a paucity of data concerning, in particular, chronic toxicities, especially in humans; third, the in silico methods currently available are class-specific and/or are of insufficient accuracy. Despite the limitations, pharmaceutical companies are widely engaged in developing in silico toxicity predictive models (Chen 2004).
The drug discovery process has been fueled by the recent advancements in the bioinformatics and chemoinformatics tools. These in silico methods are reciprocal and can be conveniently integrated into the conventional in vitro and in vivo methods to test pharmacological hypotheses. Computer algorithms or high-order simulations can also be useful to predict product failure (Kaznessis et al. 2001; Rose et al. 2002; Garg and Verma 2006). The prime objective of the in silico models is the accurate prediction of ADME properties to guide in vivo pharmacokinetics of a potential drug molecule in humans otherwise it will exist merely as a virtual structure (Piotrowski et al. 2007).
With advanced improvements of scoring functions in molecular docking as well as close prediction by 3 D-QSAR and pharmacophore/toxicophore approaches, these methods can be unified with chemoinformatic and toxicogenomic techniques into a computational toxicology workflow. It is crucial to define a generalized model in which 3 D computational molecular modeling is used to model the most applicable toxicokinetic, metabolic and molecular toxicological restrictions, thereby facilitating the computational toxicology-driven basis of modern risk assessment while implementing an initiation point for prudent viable molecular design (Piotrowski et al. 2007). Figure 1 shows the schematic view of multiparametric modeling to predict toxicity of different systems.
3.2.2. Statistical modeling approaches
Statistical modeling software, for example, Topkat (http://accelrys.com/items/disclosure studio/toxicology), PASS (Reitz 2014), TPS–SVM (DeFina et al. 2013) and Multicase (http://www.multicase.com/) aims to investigate existing information and consequently assemble models, with a less requirement for human mediation and interpret the results.
3.2.3. Quantitative Structure-Activity relationship models and biological assays
Biological and physicochemical properties of a compound are calculated through QSAR models. A SAR is a qualitative association between a chemical substructure and the potential of a chemical containing the substructure to exhibit a certain biological effect. A QSAR is a mathematical model that quantifies the relationship between the chemical’s structure-related properties (descriptors) and usually a biological effect (e.g., toxicological endpoint) (Patlewicz et al. 2003), (Tong et al. 2003). QSAR models have been broadly utilized as a part of the pharmaceutical business essential for lead disclosure and advancement (Aarsland et al. 2009).
To guarantee the best possible utilization of QSAR models, it is vital to perceive their inborn impediments (Zheng et al. 2009). ADVERPred (http://www.way2drug.com/adverpred/) is the web server for the prediction of adverse effects of drugs which generate the SARs based on PASS (Prediction of Activity Spectra for Substances) software with high accuracy. This web service includes the prediction of toxic endpoints related to myocardial infarction, arrhythmia, cardiac failure, severe hepatotoxicity and nephrotoxicity (Ivanov et al. 2017).
4. Development of the current multiparametric model
Computational chemistry, high-throughput screening (HTS), and numerous toxicogenomic technologies are combined to anticipate probable toxicity (Table 4). Figure 2 shows the flow of presenting work, proceeding with target identification and drug selection through USFDA approval and withdrawal status. The next step is the calculations of physico-chemical and ADMET prediction followed by molecular docking and pharmacophore/toxicophore approaches. These all data are analyzed using various statistical methods, for example, linear discriminant analysis (LDA), principal component analysis (PCA), receiver operating characteristic (ROC), etc. On the basis of these results cut off values are determined to select molecules for further testing.
Table 4.
List of probable targets for organ-specific toxicity.
| Sr. No. | Major organ toxicity |
Possible targets |
|---|---|---|
| 1 | Cardio Toxicity | Human B2-adrenergic G protein-coupled receptor, Human cytochrome P450 2D6, human histamine HI receptor, cytochrome P450 3A4 |
| 2 | Hepatotoxicity | Human HMG-COA reductase, Tesis ACE co-crystal structure, human cyclooxygenase-2 |
| 3 | Nephrotoxicity | Human thymidylate synthase, human androgen receptor, aminoglycoside 4’-O-adenylyltransferase ANT(4’)-IIb, human Renin, human methionine aminopeptidase 2, human cathepsin G, recombinant human dihydrofolate reductase, human microsomal P450 1A2, structure of adenylate kinase mutants, human cytochrome P450 2E1, rat mitochondrial P450 24A1 S57D, cytochrome P450 CYP11A1, DNA polymerase beta mutant 5P20, cytosol aminopeptidase, oligomeic turkey beta1-adrenergic g protein-coupled receptor, human methionine aminopeptidase 2, Human cathepsin G, recombinant human dihydrofolate reductase, cytochrome P450 CYP11A1 |
| 4 | Neurotoxicity | Apo human homogentisate dioxygenase, adenylate cydases, human monoamine oxidase B, prostaglandin h2 synthase-1, mammalian cytochrome P450 2B4, catechol o-methyltransferase, UDP-glucuronic acid binding domain, cyclohexanone monooxygenase, amino terminal domain of the NMDA receptor subunit NR2B, human dopamine D3 receptor, Atu4243-GABA receptor, human insulin, binding protein of ABC transporter, human acetylcholinesterase, human microsomal cytochrome P450 (CYP) 2C19 |
Figure 2.

Flow chart of the multiparametric model for the prediction of organ-specific toxicity.
The pros of this toxicity predictor are (1) An ample amount of chemical space to approach chemical library, (2) To determine the restraint for the procurement of screening-level data, (3) Selection of biological assays with emphasis on resources available that could generate predictive bio-activity profiles, (4) To assess the impact of metabolism on the compounds with proven efficiency in assays, (5) Storage and analyzation of predictive signatures based on a bioinformatics approach, and (6) Preceding the prospective chemicals testing strategy to compete with traditional toxicity testing (Dix et al. 2007). This toxicity predictor recognizes the hypothesis which may guide further work or plan but it will not provide any assurance of successful work so there will be chance of failure.
This review attempts to observe and classify the chemical compound which proposed for the drug discovery process. The proposed work includes three filters having ADMET, QSAR and molecular docking to gauze the potency of selected chemical. However, these filters play a crucial role to classify the chemical compound based on algorithms with higher sensitivity and specificity which leads unknown chemical compounds through screening and generates the accountability of prior results.
5. Recent trends in toxicity predictions
Different approaches are being used nowadays for in vivo, in vitro and in silico with safety biomarkers (Amur et al. 2015) in the initial stages of drug discovery and development stages (Blaauboer et al. 2012). The main focus is to reduce the risk associated with the toxicity predictions through in silico (Knowledge-based and pathway analysis) and in vitro assays (microfluid systems and proteomics approaches) (Matheis et al. 2011; Klaeger et al. 2017). The machine learning and artificial intelligence are the most prominent approaches which predict on-target or off-target related activities in a quantitative manner to facilitate the prioritization of suitable candidates in drug development process (Murphy 2011). These strategies offer accurate predictions of drug toxicity for different datasets using a consensus or ranking methods and thereby, promises to reduce the efforts for in vitro and in vivo experimentation for large set of molecules significantly. In addition, system biology and toxicokinetic-toxico-dynamic models with the framework of neural networks are favorable for toxicity testing of new chemical entity (NCE) uptake and elimination rate as well as effect on target on large scale (Hartung et al. 2012; Hartung 2018). The heart, liver, kidney and nervous system-related biomarkers are being accounted for the prediction of adverse drug reactions (ADRs), drug attrition and withdrawal rate (Dixit and Boelsterli 2007; Bussiere et al. 2009; Brennan et al. 2015). detection and prediction is an emerging field which takes into account the cell lines-based activity to forecast the fate of toxicity in millions of compounds (Pognan 2018).
6. The way ahead
For the toxicity testing, tremendous opportunities are arising with the help of multiparametric models including in silico, in vitro and in vivo studies. Advancement in supercomputing will help to decipher insights into the toxicity problem by providing direction which could accelerate new drug discovery and development. This multiparametric modeling is an emerging era of ‘omics’. Toxicokinetics and pharmacokinetics (PK) outcomes can quantitatively relate the outside centralization of a toxicant in nature to the measurements present in the target tissues. Pharmacodynamic (PD) models will complement in predicting dosage and toxicological endpoints.
The development of drug and its clinical significance in both pre- and post-market is closely regulated by FDA provided the strong implementation of guidelines laid down by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) in research activities of drug development and related studies (Dominguez-Urban 1997; Rägo and Santoso 2008; Suke et al. 2015). ICH also recommends the use of bioinformatics, computational genomics and statistical modeling in the earlier stages of drug development to reduce cost and recover lead molecules with these cutting-edge tools and techniques (Hasan et al. 2014; Romano and Tatonetti 2019). One of the primary evaluations is the drug toxicity assessment of lead candidates in the initial stage of drug discovery to reduce animal testing and promote only promising molecules for in vivo testing in animal models (Cragg et al. 1997; Romano and Tatonetti 2019). To promote discovery through computational models, the FDA Center for Drug Evaluation and Research (CDER), Office of Pharmaceutical Science (OPS) supervise the in silico toxicology models for drug saftey related paradigms (Matthews et al. 2000). The expertise rely on on various aspects of drugs viz. genetic toxicity, evolutionary relationship, chemical features, quantitative statistical probabilities, sensitivity, specificity and other measures obtained through computational models (Valerio 2011; Humphreys et al. 2016). Similarly, the ICH guidelines monitors various safety aspects for carcinogenicity, genotoxicity and reprotoxicity testing in animals. It also recommends the carcinogenicity studies and chronic toxicity testing in non-rodents for six months or longer (Lima and Videira 2018). Collectively, the forecasting of lead molecules through computational toxicology models aid in the retrieval of promising molecules to test in in vivo experiments with reduced animal testings (Kavlock et al. 2008).
Organ toxicity is a problem for the drug discovery process and can cause expensive and time-consuming irreversible drawbacks. This can be further expanded by multiparametric models which will reduce the level of toxicity as well as define the pre-toxic effects which play vital roles in drug design. This includes selection of a drug candidate as well as withdrawn drug followed by ADMET and physicochemical parameters prediction. The molecular docking approach is applied to decipher the binding mode analysis and multiparametric analysis can be performed using LDA, PCA and deviation of cutoff through use of ROC curves. Ultimately, the expenditure of time and funds will decrease through this model for any organ-specific toxicity (Figure 3).
Figure 3.
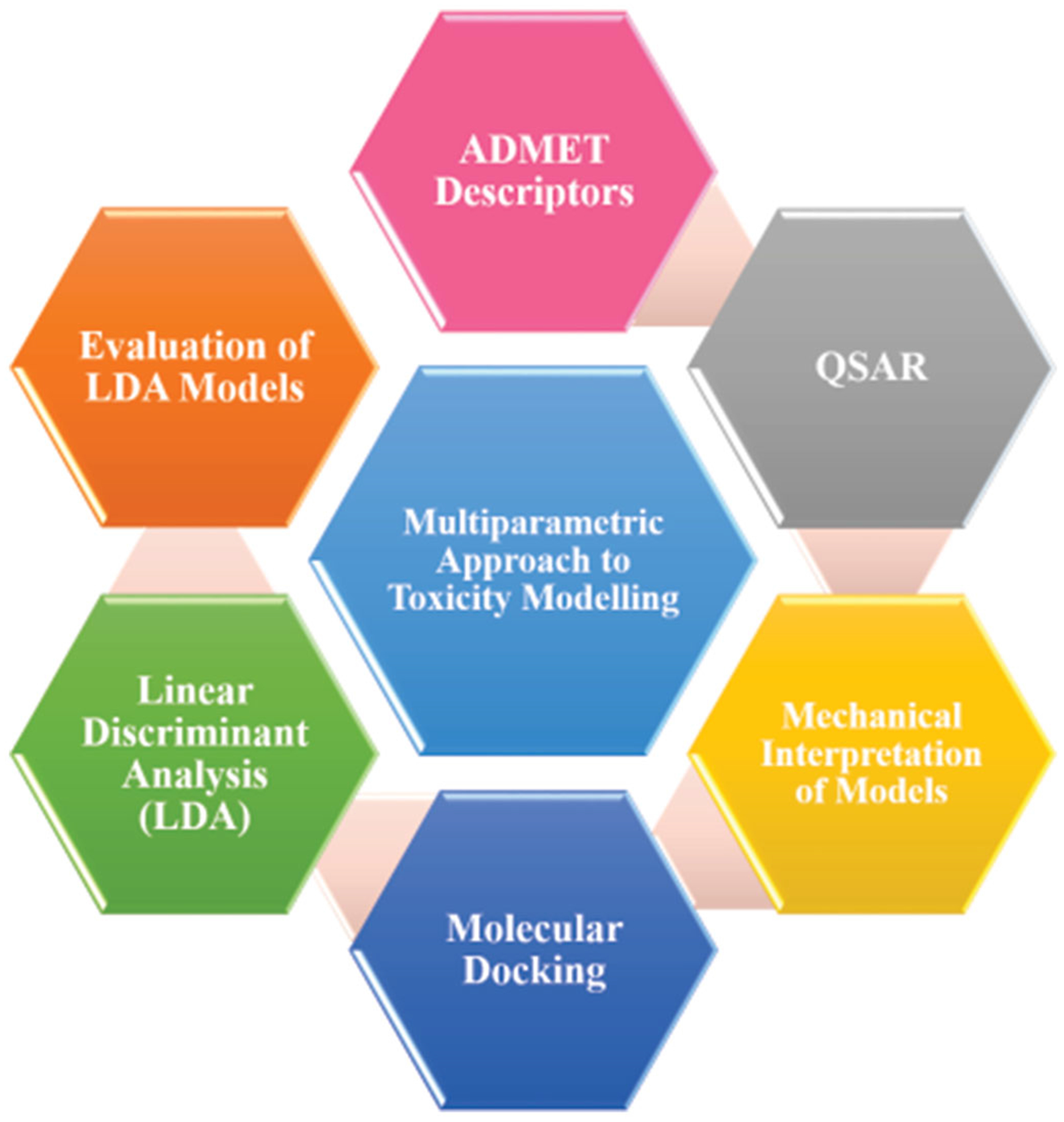
Schematics of the toxicity prediction model.
Abbreviations:
- ADMET
absorption, distribution, metabolism, excretion and toxicity
- QSAR
quantitative structure-activity relationship
- USFDA
United States Food and Drug Administration
- DART
developmental and reproductive toxicity
- AHSCT
autologous hematopoietic stem cell transplantation
- NTP
The U.S. National Toxicology Program
- MTS
Cell Proliferation Colorimetric
- Assay
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NRU
neutral red uptake
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
- NTP
The U.S. National Toxicology Program
- ATP
Adenosine triphosphate phosphate
- NR
Neutral Red
- ELISA
Enzyme-linked immunosorbent assay
- XTT
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
- WSTs
Water-soluble Tetrazolium salts
- SOM
self-organizing map
- ASBt
human apical sodium-subordinate bile acid transporter
- HMG CoA-reductase
3-hydroxy-3-methyl-glutaryl-coenzyme A reductase
- SARs
structure-activity relationships
- HTS
high-throughput screening
- EPA
U.S. Environmental Protection Agency
- PK
pharmacokinetic
- PD
Pharmacodynamic
- LDA
Linear Discriminant Analysis
- PCA
Principal component analysis
- ROC
receiver operating characteristic curve
Footnotes
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Aarsland D, Marsh L, Schrag A. 2009. Neuropsychiatric symptoms in Parkinson’s disease. Mov Disord. 24(15):2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amur S, LaVange L, Zineh I, Buckman -Garner S, Woodcock J. 2015. Biomarker qualification: toward a multiple stakeholder framework for biomarker development, regulatory acceptance, and utilization. Clin Pharmacol Ther. 98(1):34–46. [DOI] [PubMed] [Google Scholar]
- Attene-Ramos MS, Miller N, Huang R, Michael S, Itkin M, Kavlock RJ, Austin CP, Shinn P, Simeonov A, Tice RR, et al. 2013. The Tox21 robotic platform for the assessment of environmental chemicals–from vision to reality. Drug Discov Today. 18(15–16):716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási A-L, Gulbahce N, Loscalzo J. 2011. Network medicine: a network-based approach to human disease. Nat Rev Genet. 12(1):56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty C, Basinger G, Dully C, Bocek R. 1966. Comparison of red and white voluntary skeletal muscles of several species of primates. J Histochem Cytochem. 14(8):590–600. [Google Scholar]
- Bessey OA, Lowry OH, Brock MJ. 1946. A method for the rapid determination of alkaline phosphatase with five cubic millimeters of serum. J Biol Chem. 164:321–329. [PubMed] [Google Scholar]
- Blaauboer BJ, Boekelheide K, Clewell HJ, Daneshian M, Dingemans MM, Goldberg AM, Leist M. 2012. The use of biomarkers of toxicity for integrating in vitro hazard estimates into risk assessment for humans. Altex. 29(4):411–425. [DOI] [PubMed] [Google Scholar]
- Bolten BM, DeGregorio T. 2002. Trends in development cycles. Nat Rev Drug Discov. 1(5):335–336. [DOI] [PubMed] [Google Scholar]
- Borenfreund E, Babich H, Martin-Alguacil N. 1988. Comparisons of two in vitro cytotoxicity assays—the neutral red (NR) and tetrazolium MTT tests. Toxicol In Vitro. 2(1):1–6. [DOI] [PubMed] [Google Scholar]
- Bowen WJ, Kerwin TD. 1956. A simple method for assaying adenosine triphosphate and adenosine diphosphate in mixtures. J Biol Chem. 220(1):9–14. [PubMed] [Google Scholar]
- Brennan FR, Baumann A, Blaich G, de Haan L, Fagg R, Kiessling A, Kronenberg S, Locher M, Milton M, Tibbitts J, et al. 2015. Nonclinical safety testing of biopharmaceuticals–Addressing current challenges of these novel and emerging therapies. Regul Toxicol Pharm. 73(1): 265–275. [DOI] [PubMed] [Google Scholar]
- Bugrim A, Nikolskaya T, Nikolsky Y. 2004. Early prediction of drug metabolism and toxicity: systems biology approach and modeling. Drug Discov Today. 9(3):127–135. [DOI] [PubMed] [Google Scholar]
- Bussiere JL, Martin P, Horner M, Couch J, Flaherty M, Andrews L, Beyer J, Horvath C. 2009. Alternative strategies for toxicity testing of species-specific biopharmaceuticals. Int J Toxicol. 28(3):230–253. [DOI] [PubMed] [Google Scholar]
- Chen C 2004. Humidity in plant tissue culture vessels. Biosyst Eng. 88(2): 231–241. [Google Scholar]
- Chen B, Butte AJ. 2013. Network medicine in disease analysis and therapeutics. Clin Pharmacol Ther. 94(6):627–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Gray GM, Bucher JR. 2008. Transforming environmental health protection. Science (New York, NY). 319(5865):906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg GM, Newman DJ, Snader KM. 1997. Natural products in drug discovery and development. J Nat Prod. 60(1):52–60. [DOI] [PubMed] [Google Scholar]
- Cronin MT, Jaworska JS, Walker JD, Comber MH, Watts CD, Worth AP. 2003. Use of QSARs in international decision-making frameworks to predict health effects of chemical substances. Environ Health Persp. 111(10):1391. p [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Logu A, Borgna R, Uda P, Sanna A, Pellerano ML, Saddi B. 2003. The 2, 3-bis (2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay as rapid colorimetric method for determination of antibiotic susceptibility of clinical Mycobacterium tuberculosis isolates in liquid medium. Clin Lab. 49(7–8):357–365. [PubMed] [Google Scholar]
- DeFina PA, Moser RS, Glenn M, Lichtenstein JD, Fellus J. 2013. Alzheimer’s disease clinical and research update for health care practitioners. J Aging Res. 2013:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix DJ, Houck KA, Martin MT, Richard AM, Setzer RW, Kavlock RJ. 2007. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci. 95(1):5–12. [DOI] [PubMed] [Google Scholar]
- Dixit R, Boelsterli UA. 2007. Healthy animals and animal models of human disease (s) in safety assessment of human pharmaceuticals, including therapeutic antibodies. Drug Discov Today. 12(7–8): 336–342. [DOI] [PubMed] [Google Scholar]
- Dominguez-Urban I 1997. Harmonization in the regulation of pharmaceutical research and human rights: the need to think globally. Cornell Int’l LJ. 30:245. [PubMed] [Google Scholar]
- Ekins S 2007. Computational toxicology: risk assessment for pharmaceutical and environmental chemicals. Hoboken (NJ: ): John Wiley & Sons. [Google Scholar]
- Engkvist O, Wrede P, Rester U. 2003. Prediction of CNS activity of compound libraries using substructure analysis. J Chem Inf Comput Sci. 43(1):155–160. [DOI] [PubMed] [Google Scholar]
- Freimoser FM, Jakob CA, Aebi M, Tuor U. 1999. The MTT [3-(4, 5-dime-thylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] assay is a fast and reliable method for colorimetric determination of fungal cell densities. Appl Environ Microb. 65(8):3727–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frings CS, Fendley TW, Dunn RT, Queen CA. 1972. Improved determination of total serum lipids by the sulfo-phospho-vanillin reaction. Clin Chem. 18(7):673–674. [PubMed] [Google Scholar]
- Garg P, Verma J. 2006. In silico prediction of blood brain barrier permeability: an artificial neural network model. J Chem Inf Model. 46(1): 289–297. [DOI] [PubMed] [Google Scholar]
- Giles KW, Myers A. 1965. An improved diphenylamine method for the estimation of deoxyribonucleic acid. Nature. 206(4979):93–93.14334364 [Google Scholar]
- Grunert R, Phillips P. 1951. A modification of the nitroprusside method of analysis for glutathione. Arch Biochem. 30(2):217–225. [PubMed] [Google Scholar]
- Hartung T 2018. Perspectives on in vitro to in vivo extrapolations. Appl Vitro Tox. 4(4):305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung T, Van Vliet E, Jaworska J, Bonilla L, Skinner N, Thomas R. 2012. Systems toxicology. ALTEX-Altern Anim Ex. 29(2):119–128. [DOI] [PubMed] [Google Scholar]
- Hasan A, Chakrobarty S, Chakrovorty R, Nabi A. 2014. Application of bio-technology and bioinformatics in drug designing and discovery. DOI: 10.1201/b17104-15. [DOI] [Google Scholar]
- Huang R, Xia M, Cho M-H, Sakamuru S, Shinn P, Houck KA, Dix DJ, Judson RS, Witt KL, Kavlock RJ, et al. 2011. Chemical genomics profiling of environmental chemical modulation of human nuclear receptors. Environ Health Persp. 119(8):1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Xia M, Sakamuru S, Zhao J, Shahane SA, Attene-Ramos M, Zhao T, Austin CP, Simeonov A. 2016. Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat Commun. 7(1):10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys WG, Will Y, Guengerich FP. 2016. Toxicology strategies for drug Discovery-Present and future: introduction. Washington, DC: ACS Publications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov SM, Lagunin AA, Rudik AV, Filimonov DA, Poroikov VV. 2017. ADVERPred–Web service for prediction of adverse effects of drugs. J Chem Inf Model. 58(1):8–11. [DOI] [PubMed] [Google Scholar]
- Kakkar P, Das B, Viswanathan PN. 1984. A modified spectrophotometric assay of superoxide dismutase. Indian J Biochem Biophys. 21(2): 130–2. [PubMed] [Google Scholar]
- Kavlock RJ, Ankley G, Blancato J, Breen M, Conolly R, Dix D, Houck K, Hubal E, Judson R, Rabinowitz J, et al. 2008. Computational toxicology—a state of the science mini review. Toxicol Sci. 103(1):14–27. [DOI] [PubMed] [Google Scholar]
- Kaznessis YN, Snow ME, Blankley CJ. 2001. Prediction of blood-brain partitioning using Monte Carlo simulations of molecules in water. J Comput Aid Mol Des. 15(8):697–708. [DOI] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig P-A, Reinecke M, Ruprecht B, Petzoldt S, Meng C, et al. 2017. The target landscape of clinical kinase drugs. Science. 358(6367):eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohonen T 2006. Self-organizing neural projections. Neural Netw. 19(6–7):723–733. [DOI] [PubMed] [Google Scholar]
- Lednicer D 2006. New drug discovery and development. Hoboken (NJ): John Wiley & Sons. [Google Scholar]
- Lima BS, Videira MA. 2018. Toxicology and biodistribution: the clinical value of animal biodistribution studies. Mol Ther. 8:183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J Biol Chem. 193(1):265–275. [PubMed] [Google Scholar]
- Luck H, Peroxidase I. 1963. Of Enzymatic Analysis: New York (NY): Academic Press Inc. [Google Scholar]
- Malich G, Markovic B, Winder C. 1997. The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicology. 124(3):179–192. [DOI] [PubMed] [Google Scholar]
- Matheis K, Laurie D, Andriamandroso C, Arber N, Badimon L, Benain X, Bendjama K, Clavier I, Colman P, Firat H, et al. 2011. A generic operational strategy to qualify translational safety biomarkers. Drug Discov Today. 16(13–14):600–608. [DOI] [PubMed] [Google Scholar]
- Matthews EJ, Benz RD, Contrera JF. 2000. Use of toxicological information in drug design. J Mol Graph Model. 18(6):605–15. [DOI] [PubMed] [Google Scholar]
- Mavis RD, Stellwagen E. 1968. Purification and subunit structure of glutathione reductase from bakers’ yeast. J Biol Chem. 243(4):809–814. [PubMed] [Google Scholar]
- Mehta N, Ozick L, Gbadehan E. 2010. Drug-induced hepatotoxicity. State U New-York Med J. (7):51–57. [Google Scholar]
- Merlot C 2010. Computational toxicology—a tool for early safety evaluation. Drug Discov Today. 15(1–2):16–22. [DOI] [PubMed] [Google Scholar]
- Mezbaum W 1939. Estimation of RNA by orcinol method. J Physiol Chem. 258:117–120. [Google Scholar]
- Murphy RF. 2011. An active role for machine learning in drug development. Nat Chem Biol. 7(6):327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muster W, Breidenbach A, Fischer H, Kirchner S, Müller L, Pähler A. 2008. Computational toxicology in drug development. Drug Discov Today. 13(7–8):303–310. [DOI] [PubMed] [Google Scholar]
- Ohkawa H, Ohishi N, Yagi K. 1979. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 95(2):351–358. [DOI] [PubMed] [Google Scholar]
- Paglia DE, Valentine WN. 1967. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 70(1):158–169. [PubMed] [Google Scholar]
- Pai VB, Nahata MC. 2000. Cardiotoxicity of chemotherapeutic agents: incidence, treatment and prevention. Drug Saf. 22(4):263–302. [DOI] [PubMed] [Google Scholar]
- Park N 2013. Last Updated November 3, 2014 Name: Birnbaum Linda S.. [Google Scholar]
- Patlewicz G, Rodford R, Walker JD. 2003. Quantitative structure-activity relationships for predicting mutagenicity and carcinogenicity. Environ Toxicol Chem. 22(8):1885–1893. [DOI] [PubMed] [Google Scholar]
- Piotrowski PL, Sumpter BG, Malling HV, Wassom JS, Lu PY, Brothers RA, Sega GA, Martin SA, Parang M. 2007. A toxicity evaluation and predictive system based on neural networks and wavelets. J Chem Inf Model. 47(2):676–685. [DOI] [PubMed] [Google Scholar]
- Pognan F 2018. Detection, elimination, mitigation, and prediction of drug-induced liver injury in drug discovery Drug-Induced Liver Toxicity. Berlin (Germany): Springer; p. 21–43. [Google Scholar]
- Quinn P, White I. 1968. Distribution of adenosinetriphosphatase activity in ram and bull spermatozoa. J Reprod Fertil. 15(3):449–452. [DOI] [PubMed] [Google Scholar]
- Rägo L, Santoso B. 2008. Drug regulation: history, present and future. Drug Benefits Risks: Int Textbook Clin Phar. 2:65–77. [Google Scholar]
- Reitz C 2014. Genomic insights into the etiology of Alzheimer’s disease: a review. Advances in genomics & genetics. 4:59–66. [Google Scholar]
- Romano JD, Tatonetti NP. 2019. Informatics and computational methods in natural product drug discovery: a review and perspectives. Front Genet. 10:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose K, Hall LH, Kier LB. 2002. Modeling blood-brain barrier partitioning using the electrotopological state. J Chem Inf Comp Sci. 42(3):651–666. [DOI] [PubMed] [Google Scholar]
- Shukla SJ, Huang R, Austin CP, Xia M. 2010. The future of toxicity testing: a focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov Today. 15(23–24):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suke SG, Kosta P, Negi H. 2015. Role of pharmacovigilance in India: An overview. Online J Public Health Inform. 7(2):e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice RR, Austin CP, Kavlock RJ, Bucher JR. 2013. Improving the human hazard characterization of chemicals: a Tox21 update. Environ Health Persp. 121(7):756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W, Cao X, Harris S, Sun H, Fang H, Fuscoe J, Harris A, Hong H, Xie Q, Perkins R, et al. 2003. ArrayTrack–supporting toxicogenomic research at the US food and drug administration national center for toxicological research. Environ Health Persp. 111(15):1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukeda H, Kawana D, Maeda S, Sawamura M. 1999. Spectrophotometric assay for superoxide dismutase based on the reduction of highly water-soluble tetrazolium salts by xanthine-xanthine oxidase. Biosci Biotech Bioch. 63(3):485–488. [DOI] [PubMed] [Google Scholar]
- Valerio LG. 2011. In silico toxicology models and databases as FDA Critical Path Initiative toolkits. Hum Genomics. 5(3):200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashishtha SC, Hawes EM, McCann DJ, Ghosheh O, Hogg L. 2002. Quaternary ammonium-linked glucuronidation of 1-substituted imidazoles by liver microsomes: interspecies differences and structure-metabolism relationships. Drug Metab Dispos. 30(10): 1070–1076. [DOI] [PubMed] [Google Scholar]
- Worth AP, Cronin MT. 2003. The use of discriminant analysis, logistic regression and classification tree analysis in the development of classification models for human health effects. J Mol Struct. 622(1–2):97–111. [Google Scholar]
- Yolken RH. 1978. ELISA: enzyme-linked immunosorbent assay. Hosp Pract. 13(12):121–127. [DOI] [PubMed] [Google Scholar]
- Zheng X, Ekins S, Raufman J-P, Polli JE. 2009. Computational models for drug inhibition of the human apical sodium-dependent bile acid transporter. Mol Pharmaceut. 6(5):1591–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatkis A, Zak B, Boyle AJ. 1953. A new method for the direct determination of serum cholesterol. Transl Res. 41(3):486–492. [PubMed] [Google Scholar]