

Abstract
CYP2D metabolizes many drugs that act within the brain, and variable expression of CYP2D in the brain may alter local drug and metabolite levels sufficiently to affect behavioral responses. Transgenic mice that express human CYP2D6 (TG) were compared to wild type mice (WT). Following selective inhibition of human CYP2D6 in TG brain, we demonstrated in vivo that human CYP2D6 in the brain was sufficient to alter a drug-induced behavioral response. After a 4-h pre-treatment with intracerebroventricular (i.c.v.) propranolol, CYP2D activity in vivo and in vitro was reduced in TG brain, whereas CYP2D activity in vivo, but not in vitro, was reduced in WT brain. After a 24-h pre-treatment with i.c.v. propranolol, CYP2D activity in vivo and in vitro was reduced in TG brain, whereas CYP2D activity in vivo and in vitro was not changed in WT brain. These results indicate that i.c.v. propranolol irreversibly inhibited human CYP2D6 in TG brain but not mouse CYP2D in TG and WT brain. Pre-treatments with propranolol did not change liver CYP2D activity in vivo or in vitro. Furthermore, 24-h pre-treatment with i.c.v. propranolol resulted in a significant decrease of the haloperidol-induced catalepsy response in TG, but not in WT, without changing serum haloperidol levels in either mouse line. These studies reveal a new tool to selectively and irreversibly inhibit human CYP2D6 in TG brain and indicate that human CYP2D6 has a functional role within the brain sufficient to impact the central nervous system response from peripherally administered drugs.
Keywords: CYP2D6, Drug metabolism, Neurotoxicology, Haloperidol, Propranolol
Introduction
Most therapeutic drugs are metabolized by cytochrome P450 enzymes (CYPs) [1]. CYPs are highly expressed in liver, where a majority of drug metabolism occurs, altering systemic
drug and metabolite levels [2]. However, plasma drug levels do not always correlate with therapeutic effect, especially for centrally acting drugs [3, 4]. Some CYPs, including CYP2D (referred to as CYP2D6 for human and herein as CYP2D for all other species), are also expressed and enzymatically active in the brain and other extrahepatic organs, though usually at lower levels compared to liver. Unlike hepatic CYP2D [5], CYP2Ds in the brain can be readily induced by drugs [6-8]. The human CYP2D6 gene is highly polymorphic; some people have no CYP2D6 enzyme activity, while some have ultrarapid metabolism due to multiple copies of the CYP2D6 gene [9]. Thus, for CYP2D6 in the brain, these genetic and environmental influences can result in variation between people that may alter brain drug and metabolite levels, and consequently alter drug response. In humans, genetic variation in CYP2D6 is associated with differing personality traits [10-12]. Moreover, it has been associated with differences in resting brain perfusion rates [13], brain activity during a cognitive task [14], and neurocognition as assessed with a systematic battery of cognitive tests [10], suggesting a role for CYP2D6 in the brain. Of note, CYP2D6 metabolizes a large number of clinical substrates that act in the brain, such as antipsychotics (e.g. haloperidol), antidepressants, and opioids [1]. In addition, endogenous ligands, such as trace amines (e.g., tyramine to dopamine [15] and 5-methoxytryptamine to serotonin [16]) and neurosteroids (e.g., progesterone [17]), are metabolized by CYP2D6. CYP2D6 can also metabolize neurotoxins, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [18], which can induce Parkinson-like symptoms [19]. Thus, variable CYP2D6-mediated drug metabolism specifically within the brain may alter brain drug and metabolite levels and have a resulting impact on the therapeutic or neurotoxic effects [20]. In rat brain, a role has been demonstrated for CYP2D-mediated metabolism in drug consequences [21-23]. However, it is unclear if human CYP2D6 can function within the brain and contribute meaningfully to central drug metabolism and response.
A useful method to study the role of CYP2D in brain drug metabolism and response involves the central administration (with no impact on hepatic CYP2D) of pharmacological inhibitors to animals. Propranolol is an in vivo mechanism-based inhibitor (MBI) of liver CYP2D in humans [24], rats [25], and mice [26], and an in vitro MBI of CYP2D in liver microsomes of humans [26, 27], rats [25, 28], and mice [26]. An MBI is a substrate that is metabolized to a reactive intermediate, which in turn covalently and irreversibly binds to the enzyme, rendering it inactive [26]. MBIs result in inhibition that is relatively long lasting, with activity returning following new enzyme synthesis [29]. Selective and irreversible inhibition of CYP2D in rat brain, without inhibiting CYP2D in rat liver, was demonstrated in vivo using 24-h pre-treatment with intracerebroventricular (i.c.v.) propranolol before subcutaneous (s.c.) administration of the CYP2D-substrate haloperidol [21]. Catalepsy is an acute response to first generation anti-psychotics, including haloperidol, and is used to predict the likelihood of a drug (e.g., an antipsychotic) causing extrapyramidal side effects in humans [30]. Rats that were given 24-h pre-treatment with i.c.v. propranolol prior to haloperidol administration had reduced catalepsy [21].
As a translational step towards studying CYP2D6 in vivo in human brain, CYP2D6-transgenic mice (TG), which express human CYP2D6 in addition to mouse CYP2D isozymes [31], were compared to wild-type C57BL/6J mice (WT) [26]. Propranolol is an MBI of liver CYP2D6 (in humans and TG) and of mouse liver CYP2D (in TG and WT) [26], and thus we expected that propranolol administered into the mouse brain would also act as an MBI of both CYP2D6 (in TG brain) and mouse CYP2D (in TG and WT brain). Initial experiments revealed that i.c.v. propranolol acts as an MBI of CYP2D6 in TG brain, but not of mouse CYP2D in WT brain (see 9results). Although unexpected, this provided a novel strategy to study human CYP2D6 in the brain alone (leaving mouse CYP2Ds in the brain and liver, and CYP2D6 in the liver unaffected) in a live animal. This provided a model for evaluating the role of CYP2D6 in vivo in the mammalian brain. While studies in humans have associated genetic variation in CYP2D6 with differences in neurobiological function, this CYP2D6 transgenic mouse model could provide a method for directly testing the roles of CYP2D6 in the brain. Using this model, we demonstrated that human CYP2D6 in the brain is active in vivo and in vitro; furthermore, we demonstrated that there is sufficient CYP2D6 activity in the brain to alter haloperidol-induced catalepsy in mice in vivo. These data indicate that, within this mouse model, human CYP2D6 expressed in the brain is functional in vivo and contributes to drug response. In addition, this model provides a novel tool to understand better the role that CYP2D6 in brain plays in drug response, neurotoxicity, and endogenous neurotransmitter metabolism.
Methods
Animals and Propranolol Pre-treatment
Male TG [31] and WT (Charles River, Saint-Constant, Canada) mice aged 8 to 12 weeks, were housed in groups of 3 to 4 under a 12-h light/12-h dark cycle with water and chow supplied ad libitum; all procedures were conducted in the light phase. Experiments were performed in accordance with the NIH guidelines for the care and use of laboratory animals, and with approval of the University of Toronto Animal Care Committee. The derivation of these TG mice has previously been described [31]. Briefly, TG were produced by microinjecting fertilized FVB/N mouse eggs with the CYP2D6 gene (Genbank accession number BX247885, PAC clone RP4-669P10). The insert contained the complete human CYP2D6 gene sequence (exon 1–9), including the 5′- and 3′-flanking sequences, as well as the pseudogenes CYP2D7P1 and CYP2D8P1. TG founders were mated with C57BL/6J mice (WT). After successive matings, confirmed homozygous TGs were established. Both polymerase chain reaction and Southern blot analyses were used to confirm the incorporation of the full length CYP2D6 gene. Within our own breeding program, TG homozygosity was confirmed by crosses with WT. All TG used were homozygous [26] and genotyped prior to use according to previously published methods [31]. TG and WT were randomly assigned to i.c.v. pre-treatment with 2 μL of either 80 μg propranolol hydrochloride (~ 2.67 mg/kg for an average 30 g mouse; Sigma-Aldrich Canada, Oakville, Canada) in vehicle (sterile 20% w/v 2-hydroxypropyl-β-cyclodextrin, Sigma-Aldrich Canada, Oakville, Canada) or vehicle alone, at either 4-h or 24-h before experimental testing (n = 11–12 per mouse line per pre-treatment). Pre-treatment solutions were injected i.c.v. via a guide cannula at bregma coordinates of dorsal-ventral – 2.3 mm, lateral-medial – 1.0 mm, and anterior-posterior – 0.9 mm over 2 min, and the injector was left in place for another minute.
DOR Formation from DEX In Vivo
The probe drug dextromethorphan (DEX) undergoes CYP2D-specific O-demethylation to dextrorphan (DOR) [32]. After a 4-h or 24-h pre-treatment with i.c.v. propranolol or vehicle, 30 mg/kg DEX hydrobromide (Sigma-Aldrich Canada, Oakville, Canada) in sterile saline was injected intraperitoneally (i.p.). A saphenous vein blood sample was collected 30 min post-DEX injection, samples were then spun at 5,000g for 10 min, and serum was collected to measure drug levels. Immediately following blood draw, mice were sacrificed, and brains and livers were dissected. For brain drug levels, a half-brain minus the cerebellum was homogenized in 0.25 M Na2CO3 (1:5 w/v), centrifuged at 12,500g for 20 min, and the supernatant was assayed. The ratio of DOR/DEX, assessed from brain tissue and from serum, was used as an index of in vivo CYP2D activity.
DOR Formation In Vitro
The formation rate of the metabolite DOR during in vitro incubations of brain membranes and liver microsomes, derived from in vivo drug pre-treated mice, was used as an index of CYP2D activity [21]. Assay conditions were optimized for linear DOR formation [21, 26]. In vitro brain membrane incubations were performed the same day as sacrifice to prevent loss of CYP2D activity from the freezing of brain tissues [33], and in vitro liver microsome incubations were performed at a later date using liver tissues stored at − 80 °C [34]. Brain cerebellar membranes and liver microsomes were prepared as previously described [33, 34]. Briefly, mouse livers were thawed on ice and homogenized in 1.15% KCl buffer, centrifuged at 9000g for 20 min, followed by the supernatant being centrifuged at 100,000g for 90 min. The resultant microsomal pellets were resuspended in a volume of 1.15% KCl. Whole individual cerebella (approximately 300–400 μg) were thawed on ice and homogenized in artificial cerebrospinal fluid (ACSF), centrifuged at 1,700g for 5 min, followed by the supernatant being centrifuged at 100,000g for 90 min. The resultant membrane pellets were resuspended in 150 μL volume of ACSF. Cerebellum was used due to high CYP2D expression in rodents [7, 31] and humans [35]. For brain, freshly prepared membranes from individual whole cerebella were incubated with 50 μM DEX (approximate Vmax) [26] and 1 mM NADPH in ACSF (pH 7.4) for 90 min at 37 °C under 95% O2/5% CO2 in a final volume of 1 mL. DOR tartrate (5 ng DOR, Sigma-Aldrich Canada, Oakville, Canada) was added to each stopped incubate immediately before extraction to ensure that DOR levels were above the limit of detection. The spiked 5 ng DOR was subsequently subtracted from the DOR measured in each sample to calculate the amount of enzymatically formed DOR. For liver, microsomes (50 μg) were incubated with 5 μM DEX (approximate Km) [26] and 1 mM NADPH in 100 mM potassium phosphate buffer (pH 7.4) for 10 min at 37 °C in a final volume of 0.5 mL. Reactions were stopped with an equal volume of hexane-butanol (95:5 v/v). A variety of incubation controls (e.g., buffer + NADPH, buffer + NADPH + DEX, buffer + NADPH + fresh membranes/microsomes, buffer + NADPH + DEX + denatured membranes/microsomes) were tested, and no DOR formation was detected, confirming that the DOR identified from the brain and liver incubations was enzymatically formed.
Haloperidol-Induced Catalepsy
Catalepsy in mice was measured as the latency to remove their forepaws from a surface raised 9 cm above the cage floor, with a maximum cut-off of 420 s [21]. To determine dose and time frame for catalepsy testing, TG and WT were given s.c. haloperidol (0.05, 0.1, or 0.2 mg/kg; Omega, Montreal, Canada). Catalepsy was measured at baseline (immediately prior to the haloperidol injection), and at 15, 30, 45, 60, 90, 120, 150, and 180 min post-haloperidol injection.
To test the impact of inhibiting CYP2D6 in brain on catalepsy response, TG and WT (n = 10 per group) were given 24-h pre-treatment with i.c.v. propranolol or vehicle prior to 0.1 mg/kg s.c. haloperidol, and catalepsy was measured 120 min post-haloperidol injection. To confirm that pre-treatments with i.c.v. propranolol had no effect on serum haloperidol levels, saphenous vein blood samples were collected 60 min and 180 min post-haloperidol injection. Sufficient time separated blood collection and catalepsy measurement to minimize the impact of stress from blood collection on catalepsy testing while also ensuring that the earliest blood draw was after haloperidol Tmax in mice [36]. Mice acted as their own controls, given a pre-treatment of either i.c.v. propranolol or vehicle, and after a two-week washout, given the alternative pre-treatment. For both mouse lines, the order of pre-treatment had no effect on catalepsy (all p > 0.2).
Drug Measurements
DEX and DOR levels in brain and serum samples, and from in vitro incubates, were quantified using HPLC with standard curves for DEX and DOR (5–500 ng/ml) as previously described [21], with a limit of quantification of 0.5 ng for both DEX and DOR. Serum haloperidol levels were quantified using LCMS with standard curves for haloperidol (5–500 ng/ml) as previously described [21], with a limit of quantification of 0.025 ng for haloperidol.
Pharmacokinetic Parameters and Statistical Analyses
The data were analyzed using GraphPad Prism (version 6.0c, La Jolla, California, USA). Mouse line comparisons of brain DOR/DEX ratio and brain DOR formation were analyzed by two-tailed, unpaired t tests. It was expected that i.c.v. propranolol pre-treatment would result in inhibition of CYP2D in mouse brain based on previous similar inhibition work in rats [21-23]. Thus, analyses of the results assessing the impact of i.c.v. propranolol pre-treatment were preplanned as within mouse line analyses. Pre-treatment comparisons of brain DOR/DEX ratio, brain DOR formation rate, serum DOR/DEX ratio, and liver DOR formation rate were analyzed between animal and within mouse line by two-tailed, unpaired t tests. Catalepsy scores from the dose response were first analyzed by two-way repeated measures ANOVA separately for each mouse line (dose × time). This was followed by a two-way ANOVA of catalepsy data at 120 min post-haloperidol injection (dose × mouse line). Post-hoc Bonferroni analyses were then performed. Pre-treatment comparisons of catalepsy data at 120 min post-haloperidol injection and serum haloperidol levels were each analyzed within animal (2-week crossover of pre-treatment) and within mouse line by two-tailed, paired t tests. A value of p < 0.05 was considered statistically significant. Data were provided in the figures as normalized to mean vehicle pre-treatment controls within each mouse line to illustrate the relative impact of pre-treatment with propranolol; all statistical analyses were performed with raw, non-normalized data.
Results
The Brain DOR/DEX Ratio In Vivo was Higher and the Brain DOR Formation Rate In Vitro was Faster in TG Compared to WT
The brain DOR/DEX ratio was significantly higher (243%, Fig. 1a) and DOR formation rate by brain membranes in vitro was significantly faster (137%, Fig. 1b) in TG compared to WT. This is consistent with TG expressing human CYP2D6 in the brain, in addition to mouse CYP2D in the brain, compared with WT that express mouse CYP2D alone.
Fig. 1.

TG had higher brain DOR/DEX ratio in vivo and faster brain DOR formation rate in vitro in brain membranes compared to WT. a Brain DOR/DEX ratio and b brain DOR formation rate are shown for TG and WT. P values are calculated using a two-tailed, unpaired t test
The 4-h Pre-treatment with i.c.v. Propranolol Decreased the Brain DOR/DEX Ratio In Vivo in TG and WT, and Decreased the Brain DOR Formation Rate In Vitro in TG, but Not in WT
After the 4-h pre-treatment with i.c.v. propranolol (versus vehicle) (Fig. 2a), the brain DOR/DEX ratio was significantly decreased in both TG (22% decrease) and in WT (47% decrease) (Fig. 2b). This suggests inhibition of CYP2D in vivo in brains of both mouse lines. After the 4-h pre-treatment with i.c.v. propranolol, the in vitro DOR formation rate was significantly decreased (39%) in brain membranes from TG, but the i.c.v. propranolol pre-treatment had no effect on the brain DOR formation rate in WT (Fig. 2c). This suggests that i.c.v. propranolol pre-treatment in vivo acted as an MBI of the human CYP2D6 in TG brain but acted as a competitive inhibitor of mouse CYP2D in TG and WT brain.
Fig. 2.

The 4-h pre-treatment with i.c.v. propranolol decreased the brain DOR/DEX ratio in vivo in TG and WT, and decreased the DOR formation rate in vitro in brain membranes in TG, but not in WT. a Experimental design. b Brain DOR/DEX ratio, c brain DOR formation rate, d serum DOR/DEX ratio, and e liver DOR formation rate are shown for TG (left side) and WT (right side). The data is illustrated as the mean, relative to the vehicle pre-treatment group within mouse line, plus standard deviation. The brain DOR/DEX data from one TG animal was excluded due to DEX levels being 9-fold above the mean brain DEX level. Data analysis was run on all remaining animals. However, the brain DOR/DEX data from one WT animal was excluded from this figure (value 3.09). P values are calculated using a two-tailed, unpaired t test
As expected, after the 4-h pre-treatment with i.c.v. propranolol (versus vehicle) in vivo, there was no difference in the serum DOR/DEX ratio (Fig. 2d) or in the in vitro liver DOR formation rate (Fig. 2e) in either mouse line. Thus, i.c.v. propranolol pre-treatment in vivo did not result in propranolol entering the systemic system and inhibiting liver CYP2D in vivo (serum DOR/DEX ratio) or in vitro (liver microsome DOR formation) in either TG or WT.
The 24-h Pre-treatment with i.c.v. Propranolol Decreased the Brain DOR/DEX Ratio In Vivo and Decreased the Brain DOR Formation Rate In Vitro in TG, but Not in WT
After the 24-h pre-treatment with i.c.v. propranolol (versus vehicle) (Fig. 3a), the in vivo brain DOR/DEX ratio was significantly decreased (34%) in TG but not in WT (Fig. 3b). The in vitro DOR formation rate was also significantly decreased (37%) in TG but not in WT (Fig. 3c). These data suggest that, following this longer pre-treatment interval, i.c.v. propranolol acted as an MBI of human CYP2D6 in TG brain but not of mouse CYP2D in TG and WT brain.
Fig. 3.

The 24-h pre-treatment with i.c.v. propranolol decreased the brain DOR/DEX ratio in vivo and the DOR formation rate in vitro in brain membranes in TG, but not in WT. a Experimental design. b Brain DOR/DEX ratio, c brain DOR formation rate, d serum DOR/DEX ratio, and e liver DOR formation rate are shown for TG (left side) and WT (right side). The data is illustrated as the mean, relative to the vehicle pre-treatment group within mouse line, plus standard deviation. P values are calculated using a two-tailed, unpaired t test
As expected, after the 24-h pre-treatment with i.c.v. propranolol (versus vehicle) in vivo, there was no difference in the serum DOR/DEX ratio (Fig. 3d) or in the liver in vitro DOR formation rate (Fig. 3e) in either mouse line. Thus, i.c.v. propranolol pre-treatment in vivo did not result in propranolol entering the systemic system and inhibiting liver CYP2D in vivo (serum DOR/DEX ratio) or in vitro (liver microsome DOR formation) in either TG or WT.
Haloperidol-Induced Catalepsy Response Was Higher in TG Compared to WT
Catalepsy was tested following different haloperidol doses and time points post-haloperidol injection to select parameters that allowed for the detection of changes in catalepsy. Catalepsy reached a stable level by 60 min, which was maintained to 180 min post-haloperidol injection (Fig. 4a) as observed previously in rats [21] and mice [37, 38]. There was a main effect of dose and time for TG (F(2, 10) = 27.11, p < 0.001; F(8, 80) = 48.78, p < 0.001) and for WT (F(2,11) = 25.94, p < 0.001; F(8.88) = 11.66, p < 0.001). Catalepsy from 0.1 mg/kg haloperidol was significantly higher than catalepsy from0.05mg/kg and significantly lower than catalepsy from 0.2 mg/kg haloperidol in TG (p < 0.01 and p < 0.01, respectively) and in WT (p < 0.05 and p < 0.001, respectively). At 120 min post-haloperidol catalepsy, there was a main effect of dose (F(2.21) = 10.50, p < 0.01) and mouse line (F(1.21) = 15.35, p < 0.001). Catalepsy at 120 min after 0.1 mg/kg haloperidol was significantly higher after 0.05 mg/kg haloperidol and lower after 0.2 mg/kg haloperidol for TG (p < 0.05 and p < 0.05, respectively); this did not reach significance for WT (p = 0.09 and p = 0.11, respectively). Therefore, subsequent experiments tested catalepsy at 120 min after a 0.1 mg/kg haloperidol injection. We also collected blood at times before and after the catalepsy to ensure no impact on serum haloperidol levels after the i.c.v. propranolol pre-treatments.
Fig. 4.
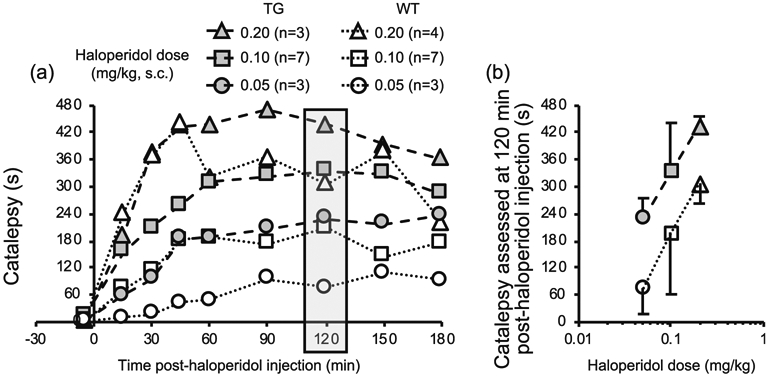
TG experienced longer catalepsy, compared to WT. a Haloperidol-induced catalepsy is assessed. b Catalepsy at 120 min post-haloperidol (shaded in a) is in the linear portion of the dose response curve for each line. The data (b) is illustrated as the mean catalepsy plus (for TG) or minus (for WT) standard deviation. Effect of dose (two-way ANOVA, F2.21 = 10.50, p < 0.001) and mouse line (two-way ANOVA, F1.21 = 15.35, p < 0.001)
TG had significantly higher (67%) catalepsy compared to WT (Fig. 4b). This suggests that a CYP2D-mediated haloperidol metabolite is responsible for catalepsy consistent with data from rats [21].
The 24-h Pre-treatment with i.c.v. Propranolol Decreased Catalepsy in TG, but Not in WT
The 24-h pre-treatment with i.c.v. propranolol (versus vehicle) (Fig. 5a) did not change mean baseline catalepsy response in TG (7 s versus 9 s, respectively) or in WT (3 s versus 3 s, respectively). After the 24-h pre-treatment with i.c.v. propranolol (versus vehicle), catalepsy was significantly decreased (33%) in TG, but not in WT (Fig. 5b). This is consistent with i.c.v. propranolol inhibiting CYP2D-mediated haloperidol metabolism in TG, but not in WT, brain. As expected, after the 24-h pre-treatment with i.c.v. propranolol, there was no difference in serum haloperidol AUC60–180 (Fig. 5c) or serum haloperidol levels at 60 min and 180 min post-haloperidol injection (Online Resource 1a, b) in either TG or WT. This indicates that i.c.v. propranolol pre-treatment in vivo did not result in propranolol entering the systemic system and inhibiting liver CYP2D-mediated haloperidol metabolism in either TG or WT.
Fig. 5.

The 24-h pre-treatment with i.c.v. propranolol decreased the haloperidol-induced catalepsy in TG, but not in WT. a Experimental design. b Mean catalepsy and c serum haloperidol AUC60–180 are shown for TG (left side) and WT (right side). Lines represent data from individual mice crossed by pre-treatment. The data is illustrated as the mean, relative to the vehicle pre-treatment group within mouse line, plus standard deviation. P value is calculated using a two-tailed, paired t test
Summary of Results
A summary of pre-treatment inhibitory effects can be found in Online Resource 2. The 4-h pre-treatment with i.c.v. propranolol decreased the brain DOR/DEX ratio in vivo and decreased the brain DOR formation rate in vitro in TG. Conversely, in WT, the 4-h pre-treatment with i.c.v. propranolol decreased the brain DOR/DEX ratio in vivo but did not decrease the brain DOR formation rate in vitro. The 24-h pretreatment with i.c.v. propranolol decreased the brain DOR/DEX ratio in vivo and decreased the brain DOR formation rate in vitro in TG, and had no effect on either in WT. In vitro inhibition of CYP2D in TG and not in WT after both pre-treatment intervals suggests that propranolol acts as an MBI of human CYP2D6 in TG brain, but not of mouse CYP2D in TG and WT brain. The brain DOR/DEX ratio, brain DOR formation rate, and catalepsy at 120 min were higher in TG compared to WT, consistent with the additional contribution of CYP2D6 in TG versus WT. The 24-h pre-treatment with i.c.v. propranolol (versus vehicle) decreased the catalepsy response in TG, but not in WT, demonstrating that CYP2D6 in the brain is functional in vivo with sufficient enzymatic activity to alter drug response. In all experiments, serum drug levels in vivo and liver enzymatic activity in vitro were unaltered by i.c.v. inhibitor pre-treatments.
Discussion
Herein, we examined the role of human CYP2D6 expressed in mouse brain and determined that it was (1) irreversibly inhibited by propranolol, while mouse CYP2Ds in brain were not, and (2) functional both in vivo and in vitro, at activity levels sufficient to change drug concentrations and drug response. This is the first demonstration of human CYP2D6 in the brain contributing meaningfully to drug metabolism and response in vivo.
Propranolol is an MBI of mouse liver CYP2D and human liver CYP2D6 [26], and thus was expected to act as an MBI of CYP2D in the brain. The 4-h pre-treatment with i.c.v. propranolol was initially chosen primarily for two reasons. First, propranolol should be cleared, as 4-h is greater than 5 half-lives of propranolol [39], reported as ~ 45 min in Swiss Webster mice [40]. Second, there should be minimal new mouse CYP2D protein synthesis in the brain following MBI treatment (while unknown for mouse CYP2D, the human CYP2D6 turnover rate is estimated at ~ 51 h [41, 42]). However, after the 4-h pre-treatment with i.c.v. propranolol, the in vivo brain DOR/DEX ratio was decreased (Fig. 2), suggesting that there is inhibition of CYP2D in the brain in vivo. This is consistent with a reported extended propranolol half-life of ~ 2 h in mouse brain [40]. However, after a 4-h pre-treatment with i.c.v. propranolol, there was no reduction in rate of brain DOR formation in vitro by WT washed brain membranes (propranolol is not present in the in vitro incubations). This suggests that propranolol in vivo is a competitive inhibitor, not an MBI, of mouse CYP2D in the brain. In contrast, after the 4-h pre-treatment with i.c.v. propranolol, there was a reduction in rate of brain DOR formation in vitro by TG washed brain membranes. This suggests that propranolol is an MBI of human CYP2D6 in TG brain. Consistent with this, after the 24-h pre-treatment with i.c.v. propranolol, there was no impact in WT on mouse CYP2D activity in the brain in vivo or in vitro (see WT, Fig. 3), while propranolol inhibited human CYP2D6 in TG brain in vivo and in vitro (see TG, Fig. 3). With both pre-treatment times, in both mouse lines, and for both DEX and haloperidol, i.c.v. propranolol had no effect on serum drug levels in vivo or hepatic activity in vitro, indicating that although it is an MBI for hepatic mouse and human CYP2Ds, propranolol did not leave the brain in sufficient amounts to alter hepatic activity.
Catalepsy is an animal model used to identify antipsychotic drugs that may produce acute extrapyramidal side effects [30]; catalepsy resembles bradykinesia, and other parkinsonian symptoms in humans [37]. In rats, a CYP2D-mediated haloperidol metabolite formed in the brain contributes to haloperidol-induced catalepsy [21]. Here, we demonstrated that catalepsy was more severe in TG than in WT. This is consistent with the additional activity of CYP2D6 in TG brain contributing to more rapid central metabolism of haloperidol and hence greater catalepsy. Pre-treatment with propranolol had no effect on baseline catalepsy response nor on serum haloperidol levels in both TG and WT, consistent with the very small amount injected having no relevant impact and/or being fully cleared within 24-h. The 24-h pre-treatment with i.c.v. propranolol before haloperidol administration reduced catalepsy in TG, but not in WT. This provides additional evidence that CYP2D6 in TG brain has sufficient activity to affect drug-response, and that this model that can be used to assess the role of CYP2D6 in brain on other drugs and toxins. Of note, many other anti-psychotics are also CYP2D6 substrates (e.g., aripiprazole, risperidone [43]) and thus may be similarly affected by CYP2D6 in the brain.
Previously we have shown that a 24-h pre-treatment with intraperitoneal propranolol (20 mg/kg) acted as an MBI of mouse liver CYP2D in TG and WT, increased serum haloperidol, and prolonged catalepsy (a shift in apparent dose as seen in Fig. 4b) [26]. Some propranolol given systemically may have escaped hepatic metabolism and could have crossed the blood brain barrier, potentially inhibiting local activation by CYP2D in the brain. However, on balance, the effect on catalepsy following intraperitoneal propranolol at this dose and timing was an increase, not a decrease, suggesting a predominant effect of inhibiting the hepatic CYP2D enzyme and in turn increasing apparent dose.
We have shown that propranolol acts as an MBI of mouse liver CYP2D [26], but not mouse CYP2D in the brain. This may be due to different complements of mouse CYP2D isoforms expressed in liver and brain. The mRNA of six CYP2D isozymes (CYP2D9, 2D10, 2D11, 2D22, 2D26, 2D40) were found in C57BL/6J mouse liver, but the mRNA of only two (CYP2D10, 2D22) were found in C57BL/6J mouse brain [44]. There is also some evidence that mRNA of other CYP2Ds are expressed in the brain [45-47]; differences between studies in brain isoforms detected may be due to differing mouse strains or techniques used. Interestingly, the cellular and regional expression pattern of mouse brain CYP2D [48] is similar to that of human CYP2D6 in both human [35, 49] and TG [31] brain, with some minor differences (see [48] for a detailed comparison). Organ-specific transcription of CYP2D isoforms has also been seen in rats, where CYP2D4 mRNA is predominantly found in brain, but not in liver [50, 51]. In addition, it is not clear which other CYP2D isoforms are translated and functional in brain, relative to liver. Propranolol metabolism by CYP2D is required before CYP2D can be irreversibly inhibited in a mechanism-based manner [26]. Rat CYP2D1, 2D2, 2D3, and 2D4, and human CYP2D6 can mediate propranolol 4-, 5-, and 7-hydroxylation, and N-desisopropylation to different extents (e.g., CYP2D1 catalyzed neither propranolol 5-hydroxylation nor propranolol 7-hydroxylation) [52]. Likewise, mouse CYP2D22, which is found in the brain, has a different binding affinity, rate of metabolism, inhibition profile, and allosteric behavior compared to human CYP2D6 [53, 54], and possibly metabolizes propranolol differently relative to other mouse CYP2D isoforms. Thus, our data is consistent with enzymatically active CYP2D isoforms expressed in the mouse brain being insensitive to propranolol as an MBI, while sensitive to it as a competitive inhibitor (Figs. 2 and 3).
The wide variation in CYP2D6 expression in brain between humans is due to genetic variation and is also associated with age and exposure to inducers [6, 8, 35, 55, 56]. Variation in CYP2D6 is tissue-specific, whereby human smokers and alcoholics have higher CYP2D6 in the brain, but similar liver CYP2D6, compared to non-smokers and non-alcoholics, respectively [8, 35, 56]. Moreover, chronic nicotine and alcohol exposure in rats and African Green monkeys induces CYP2D in the brain, but not the liver [7, 8, 57]. The role of variable CYP2D in the brain could be explored with this approach of using TG expressing CYP2D6 in the brain in combination with a 24-h pre-treatment with i.c.v. propranolol. The role of CYP2D6 in brain drug response, including therapeutic effects, abuse liability, and toxicity could be examined for the many classes of CYP2D6 substrates, which include antipsychotics, antidepressants, opioids, and psychostimulants [1, 43, 58]. Furthermore, as some pharmaceutical and environmental neurotoxins are among CYP2D6 substrates, this model may be useful for examining the role of CYP2D6 in the brain in susceptibility to and protection from neurotoxicity and neurodegeneration.
Being a CYP2D6 poor metabolizer has been associated with increased risk for Parkinson’s disease (PD) [59-61]. Pro-neurotoxins such as MPTP readily enter the brain, producing dopaminergic neuronal loss that resembles the neurodegeneration seen in patients with PD [62]. These pro-neurotoxins are detoxified by CYP2D6 [18], which is expressed in brain regions affected by PD [48]. Human smokers have an approximately 50% lower relative risk of developing PD compared to age-matched non-smokers [63]. Chronic nicotine administration induces CYP2D in the brain (but not the liver) of rats and African Green monkeys [7, 8, 57]. This suggests that nicotine may be neuroprotective by increasing CYP2D-mediated neurotoxin inactivation in brain. Based on post-mortem studies, individuals with PD were more likely to have the CYP2D6*4 null allele (associated with no detectable CYP2D6 in liver) [1, 55] than individuals without PD [55]. In addition, individuals with PD had 50% lower CYP2D6 levels in the brain compared to age-matched individuals without PD, even after controlling for CYP2D6 genotype [55]. Taken together, this suggests that CYP2D6 in the brain may detoxify neurotoxins, reducing the risk for these disorders. In contrast, individuals with lower CYP2D6 expression in the brain, independent of CYP2D6 genotype, may be at increased risk for PD. Thus, evidence for a functional role of CYP2D6 in the brain in neurotoxicity and neuroprotection could be further assessed using this model of TG mice and propranolol pre-treatment.
This work has a few limitations that should be considered. First, female mice, and the impact of estrous cycle, were not investigated in this initial study. Second, additional behaviors affected by haloperidol administration, such as locomotion, were not investigated. Third, the relative expression of human CYP2D6 compared to mouse CYP2D in TG mouse brain was not characterized (via immunoblotting), as antibodies that detect mouse CYP2D, but not the human CYP2D6, in the brain were unavailable. Lastly, this is a model system using exogenously expressed human CYP2D6 in TG. Thus, we have demonstrated that CYP2D6 expressed in the mouse brain can have a role in drug response in vivo. Despite the similarity in CYP2D6 expression patterns (e.g., in neurons of the frontal cortex, the striatum, and the molecular and granular layer of the cerebellum) in human [35, 49] and TG [31] brains, these data do not necessarily directly reflect expression or function of endogenous CYP2D6 in human brain.
Our current understanding of genetic variation and environmental regulation on the activity of human CYP2D6 in the brain in terms of its impact on drug metabolism and response is still lacking, but also as it relates to brain function and behavior. While associations between genetic variation in human CYP2D6 and personality [10-12], cognition [10, 14], schizophrenia [64], eating disorders [65, 66], and suicidality [67, 68] have been documented, suggesting a role for CYP2D6 in the brain, the mechanisms remain unknown. The transgenic mouse and inhibition approach described here can be used for the first time to directly test the function of CYP2D6 in the brain, independent of the liver.
In conclusion, the 24-h pre-treatment with propranolol is a new tool to selectively and irreversibly inhibit human CYP2D6 in TG brain, providing a novel approach to examine the impact of variation in CYP2D6 in the brain on local drug metabolism and response (using WT as controls where needed). CYP2D6 in the brain may influence the severity of acute extrapyramidal side effects from haloperidol via a catalepsy-causing metabolite formed within the brain. Our findings demonstrate that human CYP2D6 in the brain is functional and that it can meaningfully alter a central response to a drug given peripherally.
Supplementary Material
Acknowledgements
We thank Bin Zhao for his technical assistance with the liquid chromatography–mass spectrometry assay, Fariba Baghai Wadji for her expert support with animal husbandry and procedures, and Qian Zhou for genotyping the transgenic animals.
Funding Information This work was supported by a Canada Research Chair in Pharmacogenomics (R.F.T.); the Canadian Institutes of Health Research (FDN 154294, FRN 132557); the Campbell Family Mental Health Research Institute of the Centre for Addiction and Mental Health (CAMH); and the CAMH Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s12035-020-01896-4) contains supplementary material, which is available to authorized users.
Conflict of Interest R.F.T. has consulted for Quinn Emanuel and Ethismos. All other authors declare no conflict of interest.
Human and Animal Rights and Informed Consent All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Experiments were performed in accordance with the NIH guidelines for the care and use of laboratory animals, and with approval of the University of Toronto Animal Care Committee.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zanger UM, Schwab M (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138(2013):103–141 [DOI] [PubMed] [Google Scholar]
- 2.Wrighton SA, Stevens JC (1992) The human hepatic cytochromes P450 involved in drug metabolism. Crit Rev Toxicol 22(1):1–21 [DOI] [PubMed] [Google Scholar]
- 3.Ding X, Kaminsky LS (2003) Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol 43(1):149–173 [DOI] [PubMed] [Google Scholar]
- 4.Michels R, Marzuk PM (1993) Medical Progress - Progress in psychiatry (1). N Engl J Med 329(8):552–560 [DOI] [PubMed] [Google Scholar]
- 5.Edwards RJ, Price RJ, Watts PS, Renwick AB, Tredger JM, Boobis AR, Lake BG (2003) Induction of cytochrome P450 enzymes in cultured precision-cut human liver slices. Drug Metab Dispos 31(3):282–288 [DOI] [PubMed] [Google Scholar]
- 6.Miksys S, Tyndale RF (2013) Cytochrome P450-mediated drug metabolism in the brain. J Psychiatry Neurosci 38(3):152–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yue J, Miksys S, Hoffmann E, Tyndale RF (2008) Chronic nicotine treatment induces rat CYP2D in the brain but not in the liver: an investigation of induction and time course. J Psychiatry Neurosci 33(1 ):54–63 [PMC free article] [PubMed] [Google Scholar]
- 8.Mann A, Miksys S, Lee A, Mash DC, Tyndale RF (2008) Induction of the drug metabolizing enzyme CYP2D in monkey brain by chronic nicotine treatment. Neuropharmacology 55(7): 1147–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaedigk A (2013) Complexities of CYP2D6 gene analysis and interpretation. Int Rev Psychiatry 25(5):534–553 [DOI] [PubMed] [Google Scholar]
- 10.Peñas-Lledó EM, Dorado P, Pacheco R, González I, Llerena A (2009) Relation between CYP2D6 genotype, personality, neurocognition and overall psychopathology in healthy volunteers. Pharmacogenomics 10(7): 1111–1120 [DOI] [PubMed] [Google Scholar]
- 11.Kirchheiner J, Lang U, Stamm T, Sander T, Gallinat J (2006) Association of CYP2D6 genotypes and personality traits in healthy individuals. J Clin Psychopharmacol 26(4):440–442 [DOI] [PubMed] [Google Scholar]
- 12.Roberts RL, Luty SE, Mulder RT, Joyce PR, Kennedy MA (2004) Association between cytochrome P450 2D6 genotype and harm avoidance. Am J Med Genet B Neuropsychiatr Genet 127(1):90–93 [DOI] [PubMed] [Google Scholar]
- 13.Kirchheiner J, Seeringer A, Godoy AL, Ohmle B, Maier C, Beschoner P, Sim EJ, Viviani R (2011) CYP2D6 in the brain: genotype effects on resting brain perfusion. Mol Psychiatry 16:333–341 [DOI] [PubMed] [Google Scholar]
- 14.Stingl JC, Esslinger C, Tost H, Bilek E, Kirsch P, Ohmle B, Viviani R, Walter H et al. (2012) Genetic variation in CYP2D6 impacts neural activation during cognitive tasks in humans. Neuroimage 59(3):2818–2823 [DOI] [PubMed] [Google Scholar]
- 15.Hiroi T, Imaoka S, Funae Y (1998) Dopamine formation from tyramine by CYP2D6. Biochem Biophys Res Commun 249(3):838–843 [DOI] [PubMed] [Google Scholar]
- 16.Yu AM, Idle JR, Byrd LG, Krausz KW, Küpfer A, Gonzalez FJ (2003) Regeneration of serotonin from 5-methoxytryptamine by polymorphic human CYP2D6. Pharmacogenet Genomics 13(3): 173–181 [DOI] [PubMed] [Google Scholar]
- 17.Hiroi T, Kishimoto W, Chow T, Imaoka S, Igarashi T, Funae Y (2001) Progesterone oxidation by cytochrome P450 2D isoforms in the brain. Endocrinology 142(9):3901–3908 [DOI] [PubMed] [Google Scholar]
- 18.Coleman T, Ellis SW, Martin IJ, Lennard MS, Tucker GT (1996) 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is N-demethylated by cytochromes P450 2D6, 1A2 and 3A4 - implications for susceptibility to Parkinson's disease. J Pharmacol Exp Ther 277(2):685–690 [PubMed] [Google Scholar]
- 19.Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219(4587):979–980 [DOI] [PubMed] [Google Scholar]
- 20.Krishna DR, Klotz U (1994) Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet 26(2):144–160 [DOI] [PubMed] [Google Scholar]
- 21.Miksys S, Wadji FB, Tolledo EC, Remington G, Nobrega JN, Tyndale RF (2017) Rat brain CYP2D enzymatic metabolism alters acute and chronic haloperidol side-effects by different mechanisms. Prog Neuro-Psychopharmacol Biol Psychiatry 78:140–148 [DOI] [PubMed] [Google Scholar]
- 22.McMillan DM, Tyndale RF (2015) Nicotine increases codeine analgesia through the induction of brain CYP2D and central activation of codeine to morphine. Neuropsychopharmacology 40(7):1804–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMillan DM, Miksys S, Tyndale RF (2019) Rat brain CYP2D activity alters in vivo central oxycodone metabolism, levels and resulting analgesia. Addict Biol 24(2):228–238 [DOI] [PubMed] [Google Scholar]
- 24.Rowland K, Yeo WW, Ellis SW, Chadwick IG, Haq I, Lennard MS, Jackson PR, Ramsay LE et al. (1994) Inhibition of CYP2D6 activity by treatment with propranolol and the role of 4-hydroxy propranolol. Br J Clin Pharmacol 38(1):9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneck DW, Pritchard JF (1981) The inhibitory effect of propranolol pretreatment on its own metabolism in the rat. J Pharmacol Exp Ther 218(3):575–581 [PubMed] [Google Scholar]
- 26.Tolledo EC, Miksys S, Gonzales FJ, Tyndale RF (2019) Propranolol is a mechanism-based inhibitor of CYP2D and CYP2D6 in humanized CYP2D6-transgenic mice: effects on activity and drug response. Br J Pharmacol. 10.1111/bph.14884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lennard MS, Jackson PR, Freestone S, Tucker GT, Ramsay LE, Woods HF (1984) The relationship between debrisoquine oxidation phenotype and the pharmacokinetics and pharmacodynamics of propranolol. Br J Clin Pharmacol 17(6):679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masubuchi Y, Fujita S, Chiba M, Kagimoto N, Umeda S, Suzuki T (1991) Impairment of debrisoquine 4-hydroxylase and related monooxygenase activities in the rat following treatment with propranolol. Biochem Pharmacol 41 (6):861–865 [DOI] [PubMed] [Google Scholar]
- 29.McDonald AG, Tipton KF (2011) Enzymes: irreversible inhibition. eLS:1–17 [Google Scholar]
- 30.Hoffman DC, Donovan H (1995) Catalepsy as a rodent model for detecting antipsychotic drugs with extrapyramidal side effect liability. Psychopharmacology 120(2):128–133 [DOI] [PubMed] [Google Scholar]
- 31.Cheng J, Zhen Y, Miksys S, Beyoglu D, Krausz KW, Tyndale RF, Yu A, Idle JR et al. (2013) Potential role of CYP2D6 in the central nervous system. Xenobiotica 43(11):973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmid B, Schmid B, Bircher J, Bircher J, Preisig R, Preisig R, Küpfer A, Kupfer A (1985) Polymorphic dextromethorphan metabolism: co-segregation of oxidative O-demethylation with debrisoquin hydroxylation. Clin Pharmacol Ther 38(6):618–624 [DOI] [PubMed] [Google Scholar]
- 33.Tyndale RF, Li Y, Li N-Y, Messina E, Miksys S, Sellers EM (1999) Characterization of cytochrome P-450 2D1 activity in rat brain: high-affinity kinetics for dextromethorphan. Drug Metab Dispos 27(8):924–930 [PubMed] [Google Scholar]
- 34.Siu ECK, Wildenauer DB, Tyndale RF (2006) Nicotine self-administration in mice is associated with rates of nicotine inactivation by CYP2A5. Psychopharmacology 184(3):401–408 [DOI] [PubMed] [Google Scholar]
- 35.Miksys S, Rao Y, Hoffmann E, Mash DC, Tyndale RF (2002) Regional and cellular expression of CYP2D6 in human brain: higher levels in alcoholics. J Neurochem 82(6):1376–1387 [DOI] [PubMed] [Google Scholar]
- 36.Zetler G, Baumann GH (1985) Pharmacokinetics and effects of haloperidol in the isolated mouse. Pharmacology 31 (6):318–327 [DOI] [PubMed] [Google Scholar]
- 37.Ionov ID, Severtsev NN (2012) Histamine-and haloperidol-induced catalepsy in aged mice: differential responsiveness to L-DOPA. Psychopharmacology 223(2):191–197 [DOI] [PubMed] [Google Scholar]
- 38.Nishchal BS, Rai S, Prabhu MN, Ullal SD, Rajeswari S, Gopalakrishna HN (2014) Effect of Tribulus terrestris on haloperidol-induced catalepsy in mice. Indian J Pharm Sci 76(6): 564–567 [PMC free article] [PubMed] [Google Scholar]
- 39.Ito S (2011) Pharmacokinetics 101. Paediatr Child Health 16(9): 535–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levy A, Ngai S, Finck A, Kawashima K, Spector S (1976) Disposition of propranolol isomers in mice. Eur J Pharmacol 40(1):93–100 [DOI] [PubMed] [Google Scholar]
- 41.Venkatakrishnan K, Obach RS (2005) In vitro-in vivo extrapolation of CYP2D6 inactivation by paroxetine: prediction of nonstationary pharmacokinetics and drug interaction magnitude. Drug Metab Dispos 33(6):845–852 [DOI] [PubMed] [Google Scholar]
- 42.Yang J, Liao M, Shou M, Jamei M, Yeo KR, Tucker GT, Rostami-Hodjegan A (2008) Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab 9(5): 384–394 [DOI] [PubMed] [Google Scholar]
- 43.Zanger UM, Raimundo S, Eichelbaum M (2004) Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedeberg's Arch Pharmacol 369(1):23–37 [DOI] [PubMed] [Google Scholar]
- 44.Renaud HJ, Cui JY, Khan M, Klaassen CD (2011) Tissue distribution and gender-divergent expression of 78 cytochrome P450 mRNAs in mice. Toxicol Sci 124(2):261–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blume N, Leonard J, Xu ZJ, Watanabe O, Remotti H, Fishman J (2000) Characterization of Cyp2d22, a novel cytochrome P450 expressed in mouse mammary cells. Arch Biochem Biophys 381(2):191–204 [DOI] [PubMed] [Google Scholar]
- 46.Choudhary D, Jansson I, Schenkman JB, Sarfarazi M, Stoilov I (2003) Comparative expression profiling of 40 mouse cytochrome P450 genes in embryonic and adult tissues. Arch Biochem Biophys 414(1):91–100 [DOI] [PubMed] [Google Scholar]
- 47.Yamaori S, Jiang R, Maeda C, Ogawa R, Okazaki H, Aramaki H, Watanabe K (2017) Expression levels of 39 Cyp mRNAs in the mouse brain and neuroblastoma cell lines, C-1300N18 and NB2a–strong expression of Cyp1b1. Fund Toxicol Sci 4(5): 195–200 [Google Scholar]
- 48.Miksys S, Cheung C, Gonzalez FJ, Tyndale RF (2005) Human CYP2D6 and mouse CYP2Ds: organ distribution in a humanized mouse model. Drug Metab Dispos 33(10):1495–1502 [DOI] [PubMed] [Google Scholar]
- 49.Siegle I, Fritz P, Eckhardt K, Zanger UM, Eichelbaum M (2001) Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenet Genomics 11 (3):237–245 [DOI] [PubMed] [Google Scholar]
- 50.Komori M (1993) A novel P450 expressed at the high level in rat brain. Biochem Biophys Res Commun 196(2):721–728 [DOI] [PubMed] [Google Scholar]
- 51.Hiroi T, Imaoka S, Chow T, Funae Y (1998) Tissue distributions of CYP2D1, 2D2, 2D3 and 2D4 mRNA in rats detected by RT-PCR. Biochim Biophys Acta 1380(3):305–312 [DOI] [PubMed] [Google Scholar]
- 52.Hiroi T, Chow T, Imaoka S, Funae Y (2002) Catalytic specificity of CYP2D isoforms in rat and human. Drug Metab Dispos 30(9):970–976 [DOI] [PubMed] [Google Scholar]
- 53.McLaughlin LA, Dickmann LJ, Wolf CR, Henderson CJ (2008) Functional expression and comparative characterization of nine murine cytochromes P450 by fluorescent inhibition screening. Drug Metab Dispos 36(7):1322–1331 [DOI] [PubMed] [Google Scholar]
- 54.Yu AM, Haining RL (2006) Expression, purification, and characterization of mouse Cyp2d22. Drug Metab Dispos 34(7):1167–1174 [DOI] [PubMed] [Google Scholar]
- 55.Mann A, Miksys SL, Gaedigk A, Kish SJ, Mash DC, Tyndale RF (2012) The neuroprotective enzyme CYP2D6 increases in the brain with age and is lower in Parkinson’s disease patients. Neurobiol Aging 33(9):2160–2171 [DOI] [PubMed] [Google Scholar]
- 56.Miksys S, Tyndale RF (2004) The unique regulation of brain cytochrome P450 2 (CYP2) family enzymes by drugs and genetics. Drug Metab Rev 36(2):313–333 [DOI] [PubMed] [Google Scholar]
- 57.Miller RT, Miksys S, Hoffmann E, Tyndale RF (2014) Ethanol self-administration and nicotine treatment increase brain levels of CYP2D in African green monkeys. Br J Pharmacol 171(12): 3077–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou S (2016) Cytochrome P450 2D6: structure, function, regulation and polymorphism. CRC Press, Boca Raton, Florida [Google Scholar]
- 59.McCann SJ, Pond SM, James KM, Le Couteur DG (1997) The association between polymorphisms in the cytochrome P-450 2D6 gene and Parkinson's disease: a case-control study and meta-analysis. J Neurol Sci 153(1):50–53 [DOI] [PubMed] [Google Scholar]
- 60.Lu Y, Mo C, Zeng Z, Chen S, Xie Y, Peng Q, He Y, Deng Y et al. (2013) CYP2D6* 4 allele polymorphism increases the risk of Parkinson’s disease: evidence from meta-analysis. PLoS One 8(12):e84413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anwarullah, Aslam M, Badshah M, Abbasi R, Sultan A, Khan K, Ahmad N, von Engelhardt J, (2017). . ,18. (2017) Further evidence for the association of CYP2D6* 4 gene polymorphism with Parkinson’s disease: a case control study. Genes Environ 39 (1): 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Przedborski S (2007) The MPTP model of Parkinson's disease. Informa Healthcare USA, Inc., New York [Google Scholar]
- 63.Alves G, Kurz M, Lie SA, Larsen JP (2004) Cigarette smoking in Parkinson's disease: influence on disease progression. Mov Disord 19(9):1087–1092 [DOI] [PubMed] [Google Scholar]
- 64.Llerena A, Dorado P, Peñas-Lledó EM, Cáceres MC, De la Rubia A (2007) Low frequency of CYP2D6 poor metabolizers among schizophrenia patients. Pharmacogenomics J 7(6):408–410 [DOI] [PubMed] [Google Scholar]
- 65.Peñas-Lledó EM, Dorado P, Agüera Z, Gratacós M, Estivill X, Fernández-Aranda F, Llerena A (2012) CYP2D6 polymorphism in patients with eating disorders. Pharmacogenomics J 12(2): 173–175 [DOI] [PubMed] [Google Scholar]
- 66.Peñas-Lledó EM, Gonzalez I, Dorado P, Perez B, Calzadilla LR, Alvarez M, Eugania G, Naranjo M et al. (2012) Eating disorder symptoms and CYP2D6 variation in Cuban healthy females: a report from the Ibero-American network of pharmacogenetics. Current Pharmacogenomics and Personalized Medicine 10(4):288–292 [Google Scholar]
- 67.Zackrisson AL, Lindblom B, Ahlner J (2010) High frequency of occurrence of CYP2D6 gene duplication/multiduplication indicating ultrarapid metabolism among suicide cases. Clinical Pharmacology & Therapeutics 88(3):354–359 [DOI] [PubMed] [Google Scholar]
- 68.Peñas-Lledó EM, Blasco-Fontecilla H, Dorado P, Vaquero-Lorenzo C, Baca-Garcýa E, Llerena A (2012) CYP2D6 and the severity of suicide attempts. Pharmacogenomics 13(2): 179–184 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.