

Abstract
Opioid Use Disorder (OUD) is a debilitating neuropsychiatric condition characterized by compulsive opioid use, dependence, and repeated relapse after periods of abstinence. Given the high risk of developing OUD following prescription opioid use, the continued need for opioid-induced analgesia, and the limitations of current OUD treatments, it is necessary to develop novel, non-opioid based treatments for OUD and decrease abuse potential of prescription opioids. Recent evidence suggests that negative allosteric modulation (NAM) of the M5 muscarinic acetylcholine receptor (M5 mAChR) may provide an alternative therapeutic approach for the treatment of OUD. Previous studies demonstrated localization of M5 mAChR expression within the mesocorticolimbic reward circuitry and that the selective M5 NAM ML375 attenuates both cocaine and alcohol self-administration in rats. In the present study, the effects of ML375 were evaluated in rats self-administering the μ-opioid agonists oxycodone or remifentanil on a progressive ratio (PR) schedule or on cue-reactivity (a rodent model of relapse) in the absence of oxycodone following 72 hours of abstinence. ML375 reduced the PR break point for oxycodone and remifentanil self-administration and attenuated cue-elicited responding. Importantly, ML375 did not affect sucrose pellet-maintained responding on a PR schedule or opioid-induced antinociception using the hot-plate and tail-flick assays. We also confirm expression of M5 mAChR mRNA in the ventral tegmental area and show that this is primarily on dopamine (tyrosine hydroxylase mRNA-positive) neurons. Taken together, these findings suggest that selective functional antagonism of the M5 mAChR may represent a novel, non-opioid based treatment for OUD.
Keywords: antinociception, opioid self-administration, M5 muscarinic, ML375, negative allosteric modulator, oxycodone
Graphical Abstract
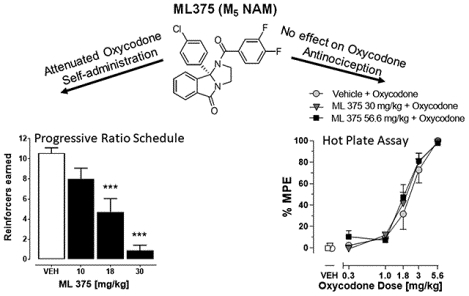
Prescription opioid analgesics are effective pain medications that work through selective activation of μ-opioid receptors and represent one of the most commonly prescribed drug classes worldwide.1–3 However, use of opioid analgesics is also associated with high risks of misuse, dependence, and overdose due to their strong rewarding effects, which have resulted in a global epidemic of opioid use disorder (OUD).2–7 OUD is a debilitating, neuropsychiatric disorder characterized by compulsive opioid use, dependence, and repeated relapse after periods of abstinence.8 Within the United States alone, it is estimated that over 4% of the adult population misuse prescription opioids and ~85% of individuals with OUD are considered to have prescription pain reliever use disorder.3,5,8 In addition, almost two-thirds of all estimated drug-related overdose deaths involve opioids, with nearly half of those attributed to prescription pain medications.9 Current FDA-approved drugs for the treatment of opioid dependence involve both opioid maintenance and detoxification strategies that are also linked with high rates of relapse, abuse potential, and adverse events, including respiratory depression and overdose.10 To date, there is no FDA-approved medication for the prevention of OUD.
For decades, the primary goal has been to identify ways to maintain the desirable analgesic effects of μ-opioid receptor agonists, while lessening their abuse liability through development of novel ligands with preferential efficacy at various opioid receptor subtypes and/or biased μ-opioid receptor signaling. Unfortunately, these approaches have yet to translate into novel therapeutics.9 In general, the reinforcing effects of μ-opioid receptor agonists are attributed to acute and long-term pathophysiological adaptations in the canonical mesocorticolimbic reward circuitry that persist into abstinence and contribute to the high rates of drug craving and relapse.11 The mesocorticolimbic reward circuitry is represented by midbrain dopamine (DA) cell bodies located in the ventral tegmental area (VTA) and their respective projection targets, including the nucleus accumbens (NAc) and prefrontal cortex.11–13 Acutely, μ-opioid receptor agonists selectively hyperpolarize μ-opioid receptor-expressing γ-aminobutyric acid (GABA) interneurons in the VTA resulting in reduced tonic GABA inhibition of VTA DA cell firing and subsequent increases in DA release in the NAc.14,15 Thus, there remains a critical unmet need to develop novel, non-opioid-based treatments for OUD that target the mesocorticolimbic reward circuitry in order to help reduce the rates of relapse and overdose, as well as prevent initial misuse in opioid-naïve patients requiring prescription opioid analgesics.
Recent attention has focused on the M5 muscarinic acetylcholine receptor (M5 mAChR) in motivated behaviors, including drug self-administration, and targeting this receptor may represent an alternative strategy for the reduction and/or blockade of the reinforcing effects of μ-opioid receptor agonists. Of the five mAChR subtypes (M1-M5) activated by acetylcholine (ACh), the M5 mAChR is the only subtype expressed in the ventral midbrain, including the ventral tegmental area [VTA], with sparse expression across other brain regions.16,17 Based on this localization, it is speculated that M5 may provide an important control of midbrain DA neuronal activity under normal conditions and after exposure to drugs of abuse, such as μ-opioid receptor agonists and stimulants. Consistent with this hypothesis, increased DA cell firing in the VTA and extracellular DA release in the NAc were absent following electrical stimulation of brainstem cholinergic projection neurons to the VTA in mice expressing a functionally inactive M5 mAChR (M5 knockout [KO] mice).18–21 Moreover, both the reinforcing effects and strength of cocaine as well as opioid place preference were significantly reduced in the M5 KO mice.22–24 Additionally, severity of naloxone-induced morphine withdrawal symptoms were also reduced in the M5 KO mice.24 In contrast, the acute analgesic effects of morphine and the development of tolerance to these effects remained unaltered in the M5 KO mice relative to the wildtype control mice.24 Unfortunately, until recently there has been a lack of sufficiently selective M5 tool compounds due in part to the fact that the ACh binding site is highly conserved across the five mAChR subtypes, making it difficult to develop orthosteric ligands with high subtype selectivity. Over the last decade, we have made tremendous progress in discovering subtype-selective mAChR allosteric modulators that do not target the orthosteric binding site of ACh, but instead act at more topographically distinct allosteric sites to provide unprecedented selectivity for the M5 mAChR.25 This strategy has led to the discovery of the first systemically active M5 negative allosteric modulator (NAM) ML37526–28 with suitable pharmacokinetic properties for investigating the role of M5 receptors in preclinical models of addiction. In previous studies, functional antagonism of the M5 mAChR subtype by ML375 attenuated cocaine and ethanol self-administration in rats at doses that did not affect non-drug related behaviors or general motor function.27,28 In the present study, we investigated whether functional inhibition of M5 mAChRs in rats would provide an alternative approach to block the reinforcing effects of μ-opioid receptor analgesics, i.e., oxycodone and remifentanil, and attenuate “cue-reactivity” (a model of drug seeking/relapse), without altering the analgesic effects of oxycodone. Currently there are no M5-specific antibodies and selective M5 radioligands available to assess the distribution of this receptor within the mesocorticolimbic reward circuit. To gain insights into the possible site(s) of actions of ML375, we therefore used fluorescent in situ hybridization to determine the expression of M5 mAChR mRNA in dopaminergic and GABAergic neurons of the VTA.
Here, ML375 dose-dependently attenuated the reinforcing strength of oxycodone and remifentanil as well as cue-mediated responding in the absence of oxycodone, at doses that did not affect sucrose pellet-maintained responding. Importantly, at the doses tested, ML375 did not alter the analgesic effects of oxycodone, nor alter plasma or brain exposure. Lastly, fluorescent in situ hybridization studies confirmed that M5 mRNA is primarily expressed in dopaminergic neurons within the VTA. Our findings suggest selective M5 NAMs could provide a novel therapeutic approach for attenuating the reinforcing effects μ-opioid receptor agonists without altering their analgesic properties.
RESULTS AND DISCUSSION
ML375 attenuated remifentanil self-administration
Under an FR10 schedule of reinforcement, rats exhibited a typical biphasic dose-response curve (see Figure 1A,B). Compared to saline self-administration, rats earned more infusions at 1 and 3, but not 10 μg/kg/infusion of remifentanil (F(3,31)=8.52, p< 0.001), and exhibited higher response rates at 3 mg/kg/infusion of remifentanil (F(3,29)=5.36, p< 0.01). Administration of 30 mg/kg ML375 reduced the number of reinforcers earned at 3 mg/kg/infusion (Fdose (2,48)=2.76, p=0.07; Ftreatment (1,48)=9.8771, p=0.01; F dose x treatment (2,48)=0.68, n.s.) with a trend towards reducing the response rate (Fdose (2,48)=2.69, p=0.08; Ftreatment (1,48)=6.38, p=0.05; F dose x treatment (3,58)=0.54, n.s.), but did not alter reinforcers earned (t14=0.28, n.s.) and response rate (t10=1.38, n.s.) when saline was self-administered.
Figure 1. ML375 attenuated remifentanil self-administration.
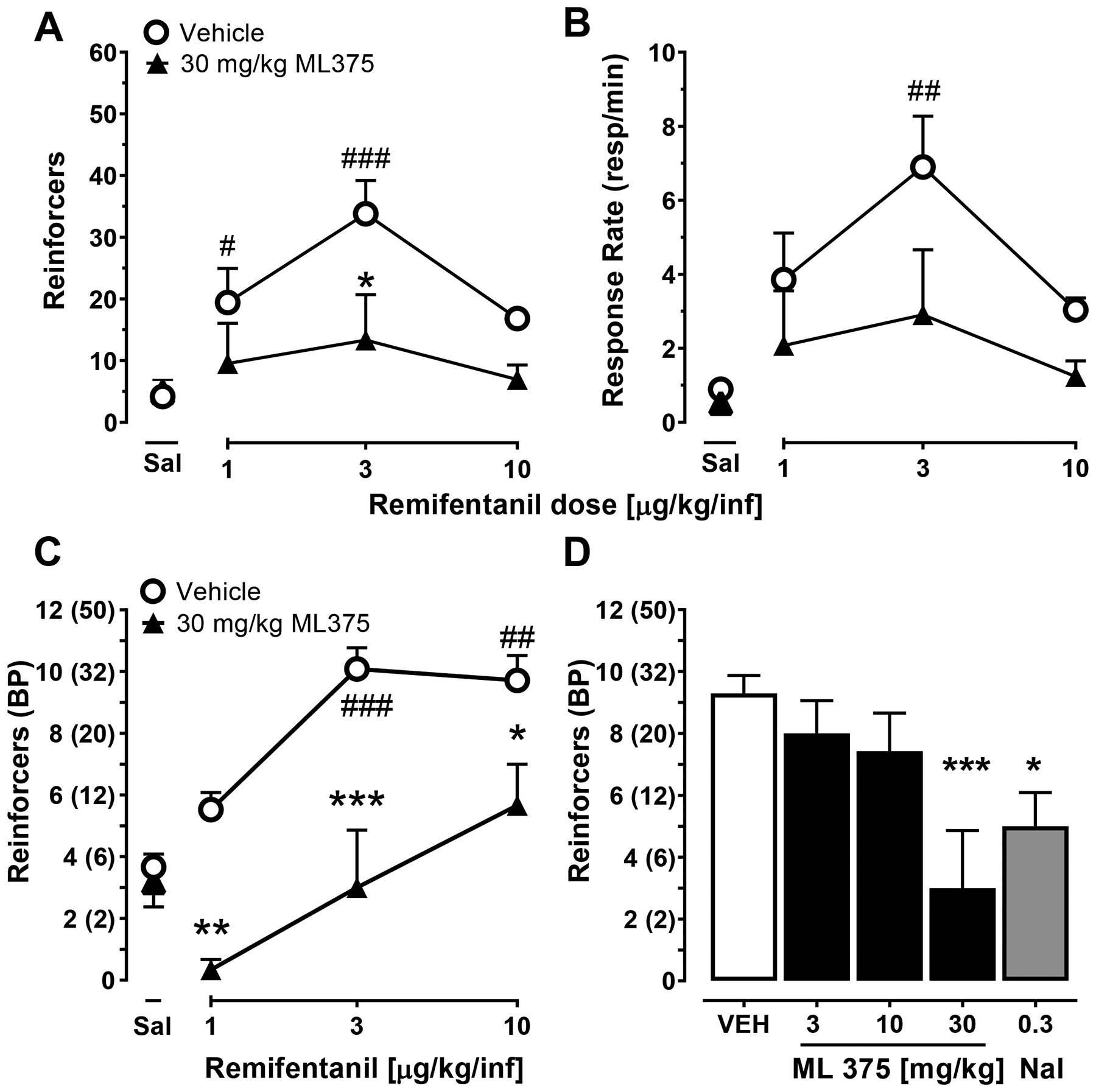
ML375 dose-dependently attenuates remifentanil self-administration maintained under a fixed (A,B) and progressive (C,D) ratio schedule of reinforcement in rats. ML375 reduced number of reinforcers/session (A) and response rates (responses/min, [B]) under a FR 10 schedule of reinforcement. (C) 30 mg/kg ML375 attenuates the number of reinforcers earned across a range of oxycodone doses. (D) ML375 dose-dependently reduces the number of reinforcers earned on a single dose of remifentanil (3 μg/kg/injection). Values represent the mean ± S.E.M. of 8–9 (A), 6–9 (B), 6 (C), and 6–13 (D) animals/group. * p<0.05, ** p<0.01; *** p<0.001 vs. respective vehicle-treated groups; # p<0.05; ## p<0.01; ### p<0.001 vs. saline self-administration. BP, breakpoint; Sal, saline.
Under a progressive ratio (PR) schedule of reinforcement, levels of responding at remifentanil doses of 3 and 10 μg/kg/infusion were greater than when saline was self-administered (F(3,15)=29.9, p<0.001; see Figure 1C). A single dose of ML375 (30 mg/kg) decreased the number of reinforcers earned at all three doses of remifentanil (Fdose (2,20)=11.6, p<0.001; Ftreatment (1,10)=35.3, p<0.001; Fdose x treatment (2,20)=1.1, n.s.), but did not alter saline self-administration (t10=0.56, n.s.).
Figure 1D shows the effects of ML375 and the opioid antagonist naltrexone on remifentanil self-administration at a dose of remifentanil (3 μg/kg/injection) that maintains peak levels of responding under both the FR and PR schedules of reinforcement (Figure 1A,C). Both ML375 (30 mg/kg) and naltrexone (0.3 mg/kg) decreased the number of reinforcers earned (Fdose (4,34)=5.57, p<0.01) compared to when vehicle was administered prior to self-administration sessions.
ML375 attenuated oxycodone self-administration
To determine the dose range of ML375 that could be effective in altering oxycodone self-administration, the effects of ML375 (10–30 mg/kg, i.p.) on oxycodone (56 μg/kg/infusion) were determined under a progressive ratio (PR) schedule of reinforcement. This dose of oxycodone has previously been shown to be at or near the peak of the oxycodone dose-response curve under a PR schedule, and on the descending limb under a FR schedule of reinforcement.29–34 ML375 (18 and 30 mg/kg) and the FDA-approved nonselective μ-opioid receptor antagonist naltrexone (0.3 mg/kg) decreased the number of reinforcers earned (Ftreatment (4,24)=20.1, p<0.0001) compared to vehicle-treated animals (p<0.001); ML375 (30 mg/kg) also reduced reinforcers earned compared to naltrexone (p<0.01) (see Figure 2A).
Figure 2. ML375 attenuated oxycodone self-administration under a progressive ratio schedule of reinforcement.
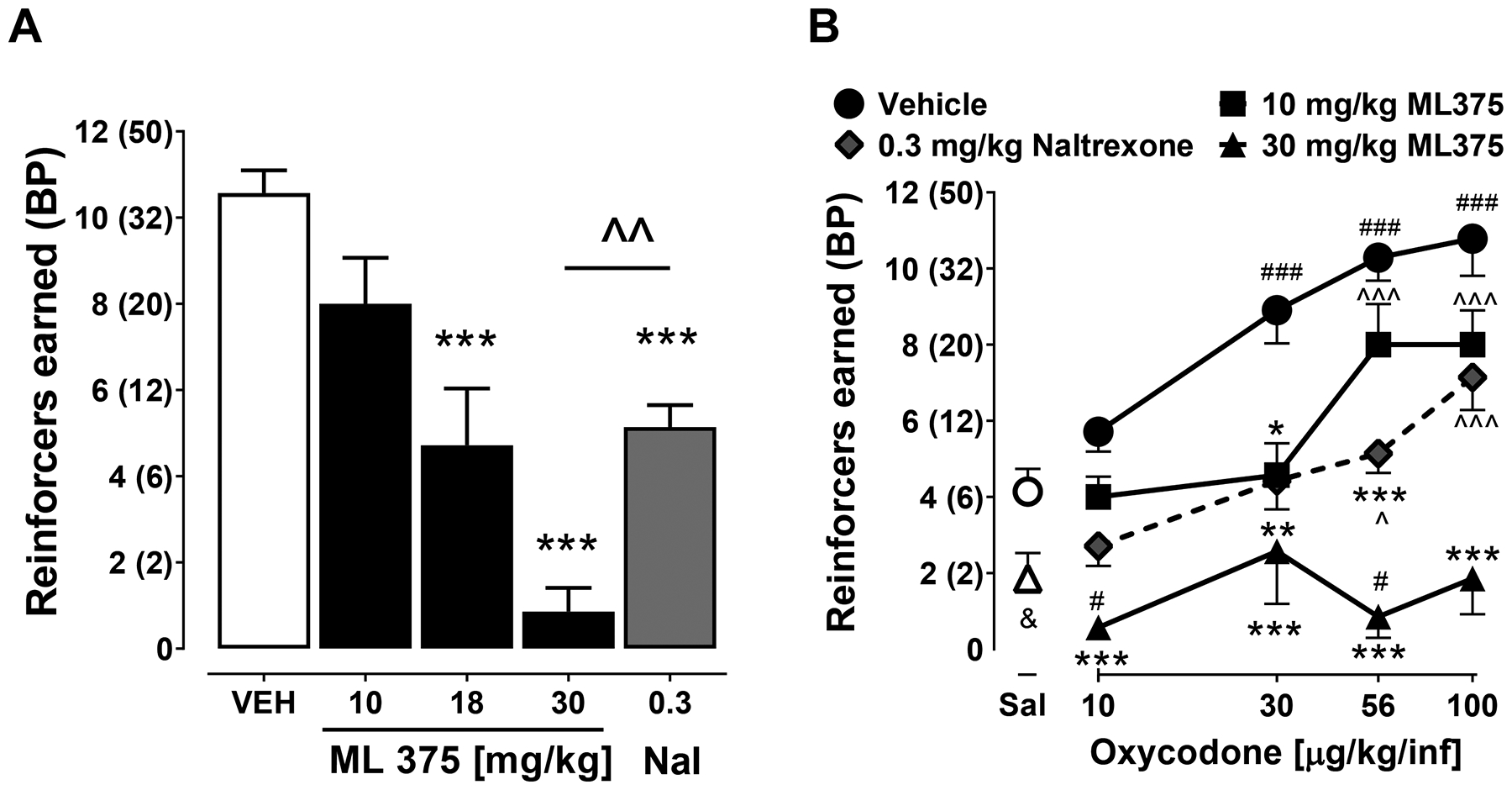
ML375 dose-dependently reduces the number of reinforcers earned when oxycodone is self-administered (A) at 56 μg/kg/infusion or (B) across a dose range of 10–100 μg/kg/infusion. Values represent the mean ± S.E.M of 7 animals/group (A, B); * p<0.05, ** p<0.01, *** p<0.001 vs. respective vehicle-treated groups, ^ p<0.05, ^^ p<0.01, ^^^ p<0.001 vs. respective groups treated with 30 mg/kg ML375 i.p.; # p<0.05, ### p<0.001 vs. vehicle-treated saline self-administration group; & p<0.05, t-test between 30 mg/kg ML375 treatment and vehicle on saline self-administration. BP, breakpoint.
Next, to determine whether the effects of ML375 would be surmounted by increasing doses of oxycodone, we examined 10 and 30 mg/kg ML375 or 0.3 mg/kg naltrexone across a full dose response function of oxycodone (10–100 μg/kg/infusion). As shown in Figure 2B, ML375 and naltrexone decreased the number of reinforcers earned (Fdose (3,18)=11.2, p<0.001; Ftreatment (3,18)=62.3, p<0.0001; F dose x treatment (9,54)=2.42, p< 0.05). Compared to vehicle administration, 30 mg/kg ML375 decreased the number of reinforcers earned across all four doses of oxycodone and 10 mg/kg ML375 decreased the number of reinforcers earned when 30 μg/kg/infusion oxycodone was self-administered. In comparison, naltrexone (0.3 mg/kg) decreased the number of reinforcers earned when 30 and 56 μg/kg/infusion oxycodone were self-administered (all p<0.05). Rats treated with 30 mg/kg ML375 earned fewer reinforcers than rats treated with 10 mg/kg ML375 or 0.3 mg/kg naltrexone at the 56 and 100 μg/kg/infusion doses of oxycodone. Lastly, to directly compare the profile of the most effective dose of ML375 (30 mg/kg) to responding when saline was self-administered, a separate two-way repeated measures ANOVA compared effects of 30 mg/kg ML375 and vehicle administration on oxycodone and saline self-administration. Multiple comparisons following main effects (Fdose (4,24)=7.02, p<0.001; Ftreatment (1,6)=140.3, p<0.001; F dose x treatment (4,24)=8.7, p= 0.001) showed that compared to vehicle administration, ML375 (30 mg/kg) did not alter lever pressing for saline (p>0.05). Only administration of 30 mg/kg ML375 resulted in lower active lever presses in response to 10 and 56 μg/kg/infusion of oxycodone than vehicle administration prior to saline self-administration (p<0.05). Due to concern of Type II statistical error, we also conducted a paired, two-tailed t-test to specifically compare effects of 30 mg/kg ML375 on saline self-administration (t6=2.73, p<0.05). Lastly, as expected, oxycodone engendered a dose-dependent increase in responding. A significantly greater number of reinforcers were earned when unit doses of 30, 56, and 100 μg/kg/infusion oxycodone were available compared to saline (p<0.001). Importantly, this suggests that the effects of ML375 (30 mg/kg) are not surmountable by increasing oxycodone doses up to 100 μg/kg/infusion and that 30 mg/kg ML375 completely abolished the reinforcing strength of oxycodone across a broad range of doses.
ML375 attenuated oxycodone-associated cue reactivity after 72 hours of abstinence
Figure 3 shows the dose effects of ML375 on oxycodone-associated cue-induced reactivity. Rats were counterbalanced in different treatment groups based on oxycodone self-administration prior to the test session (see Supplemental Figure S1). During cue-reactivity test session, pretreatment with ML375 reduced active lever responses in comparison to vehicle pretreatment (Ftreatment (3,44)=9.02, p<0.001; Flever(1,44)= 41.8, p<0.001; F lever x treatment (3,44)=4.15, p< 0.05). Compared to vehicle treatment, both 10 and 30 mg/kg ML375 (p<0.05), but not naltrexone (p>0.05), decreased cue reactivity-induced responding on the active lever. Finally, ML375 did not alter inactive lever responses compared to the vehicle-treated group (p>0.05; see Supplemental Figure S1).
Figure 3. ML375 attenuated responding maintained by oxycodone-associated cues following ~72 hours of abstinence.
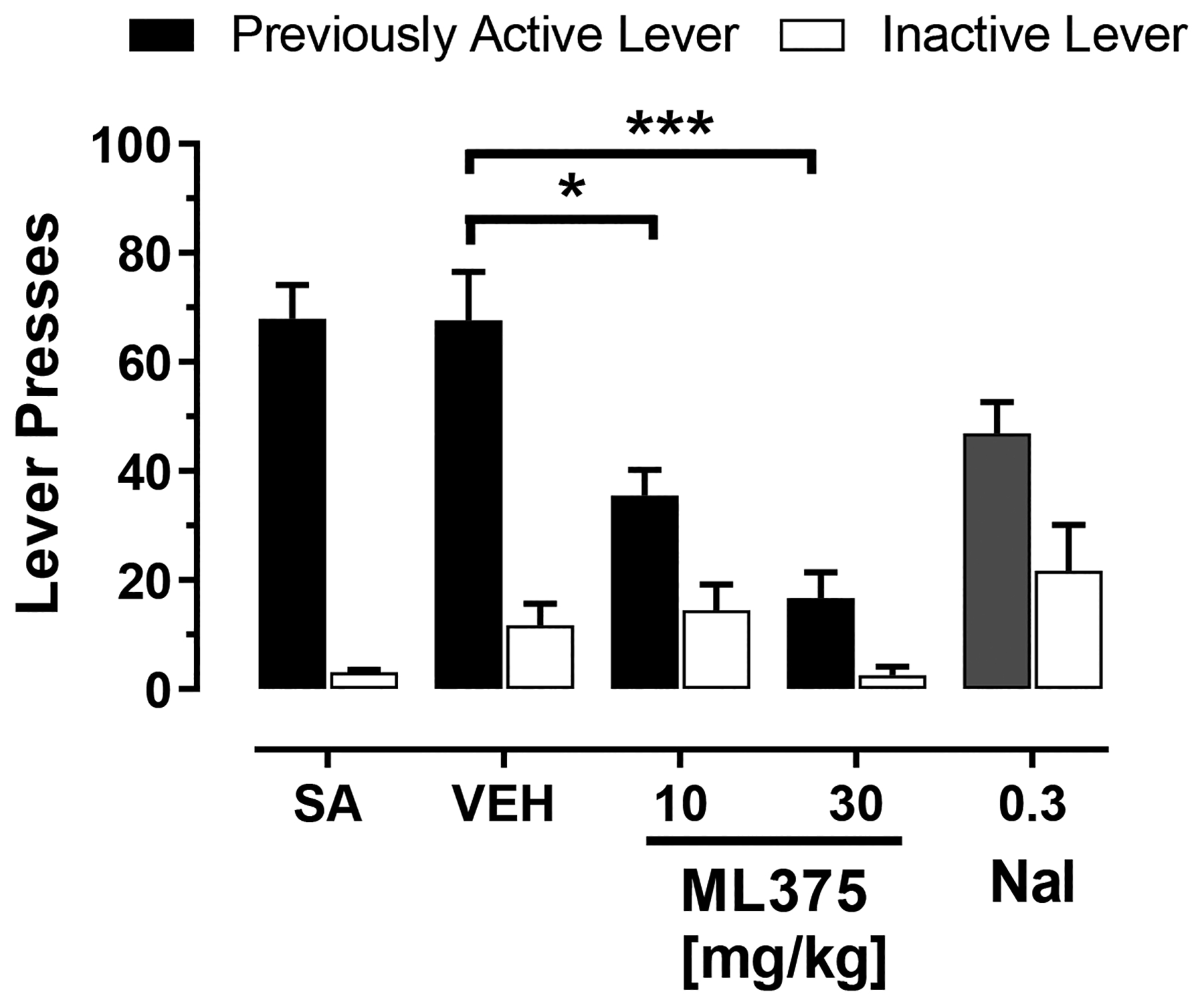
ML375 attenuates responding on the previously active lever (solid bars) but not inactive lever (open bars) following ~72 hrs of abstinence from stable self-administration of 0.56 μg/kg/infusion oxycodone. Values represent the mean ± S.E.M 6–8 animals/group for the cue-reactivity groups, * p<0.05, *** p<0.001 vs. respective vehicle-treated groups. The solid and open bars show the 3-day mean of active and inactive lever presses (total of 27 rats), respectively, during oxycodone self-administration (SA) preceding forced abstinence. Nal, naloxone.
ML375 did not affect the acute antinociceptive effects of oxycodone
Administration of ML375 alone did not change the nociceptive response in the hot plate test when compared to vehicle treatment (F(2,16)=0.32, n.s.; Figure 4A). Oxycodone dose-dependently increased antinociception as measured by an increase in percent of maximum possible response (%MPE; ED50=2.40 mg/kg) and this oxycodone effect was not altered by co-administration of 30 or 56.6 mg/k ML375 (FML375 dose (2,90)=0.72, n.s., Foxycodone dose (4,90)=98.4, p < 0.001, and FML375 x oxycodone dose (8,90)=0.40, n.s.).
Figure 4. ML375 does not alter the antinociceptive effects of oxycodone in the hot plate and tail flick assays.
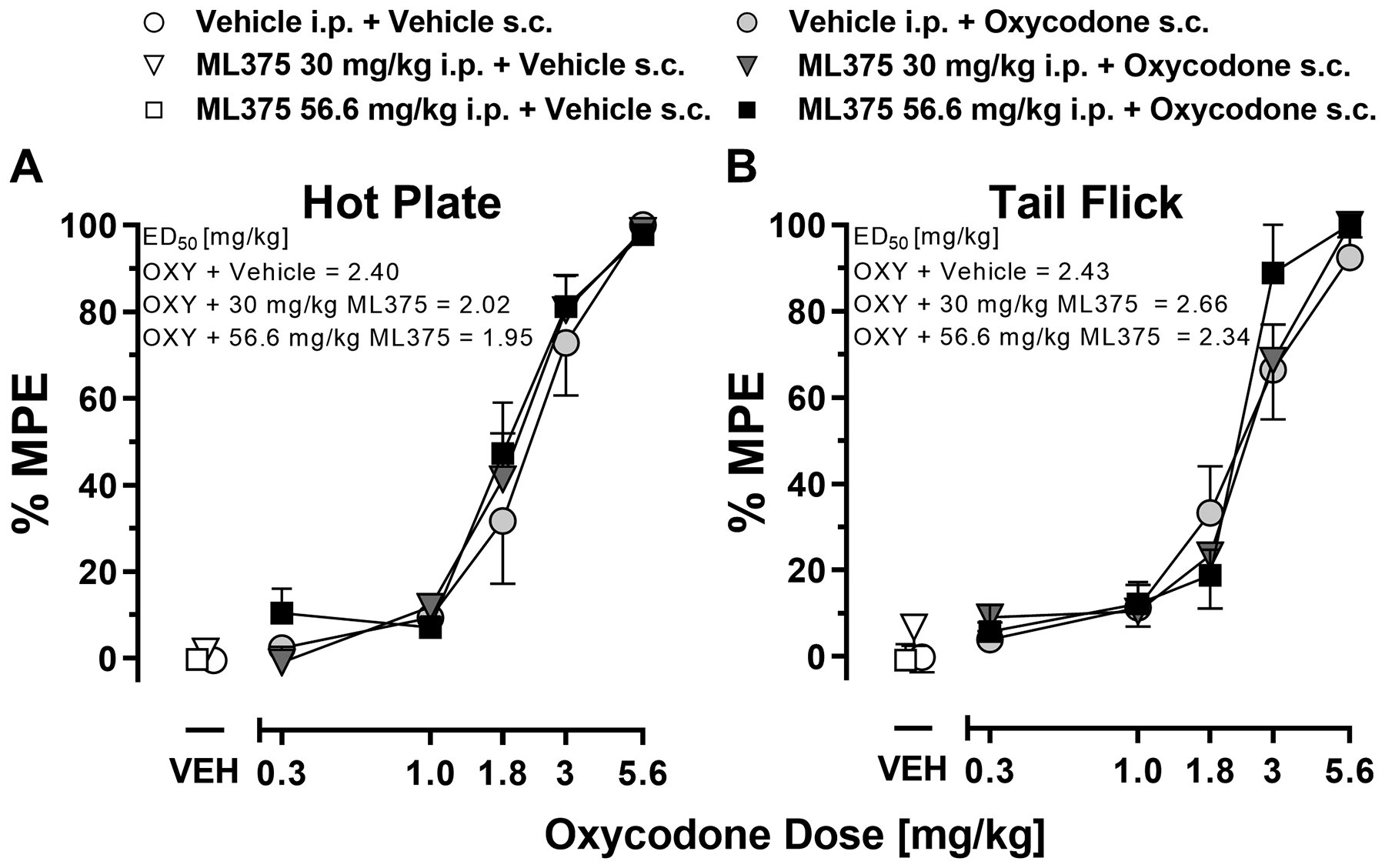
Oxycodone dose-dependently increased latency for rats to withdraw their hindpaw from hot plate (A) or to remove their tail from a hot water bath (B). Data are presented as % maximum possible effect (MPE) reflecting a change from baseline and represent the mean ± S.E.M. of 6–8 animals/group (hot plate) and 7–9 animals/group (tail flick). Compared to vehicle-treated animals, ML375 (30 and 56.6 mg/kg, i.p.) administration did not alter the % MPE of oxycodone-treated animals.
In the tail flick test (Figure 4B), ML375 treatment alone – as compared to vehicle - did not change the latency to withdraw the tail from a 55 °C water bath (F(2,20)=1.16, n.s.). The dose-dependent anti-nociceptive effects of oxycodone (ED50=2.43 mg/kg) where not altered by co-administration of ML375 (30–56.6 mg/kg) (FML375 dose (2,100)=0.74, n.s., Foxycodone dose (4,100)=82.9, p < 0.001, and FML375 x oxycodone dose (8,100) = 0.30, n.s.). These studies confirm that within a dose range that attenuated oxycodone self-administration, ML375 did not affect oxycodone-induced antinociception as determined in the hotplate and tail flick assays, two well-characterized rodent models of supraspinal and spinal mediated nociception. These data suggest that M5 mAChR modulation may selectively affect circuits underlying drug-related motivation and reinforcement versus antinociception.
Lack of drug-drug interactions between ML375 and oxycodone
To rule out the possibility that the observed behavioral interactions between ML375 and oxycodone were caused by a pharmacokinetic drug-drug interaction, resulting in altered exposure of one or both compounds, we confirmed that brain and plasma concentrations remained unaltered for the top doses of both ML375 and oxycodone when given alone or in combination. As shown in Supplementary Table S1, combined administration of oxycodone and ML375 resulted in brain and plasma concentrations that were similar to those observed following single administration of each compound alone.
M5 mRNA is expressed in dopaminergic neurons in the ventral tegmental area
In four rats, a total of 2942 cells expressing message for M5, tyrosine hydroxylase (TH) mRNA-positive neurons (a positive marker for DA neurons), the vesicular GABA transporter (vGAT;, a marker of GABAergic cells), or any combination thereof were analyzed in two ventral midbrain regions, specifically the VTA and substantia nigra pars compacta (SNc). While negative control sections through the midbrain DA cell groups were devoid of discrete fluorescent staining in each channel (Figure 5E,H), labeling for M5, TH, and vGAT was seen in these ventral midbrain nuclei (Figure 5A–D). The mean cell density (number of cells /mm2) for each cell type in the VTA is shown in Supplementary Table 2. Subjectively, the labeling intensity of M5 mRNA appeared to decrease in a lateral to medial gradient (Figure 5A), i.e. from SNc towards the midline nuclei of the VTA. This labeling pattern was remarkably similar to the distribution of TH mRNA-positive neurons, in the same regions (see Figure 5A–B). In the VTA, a small fraction of M5 cells were single labeled, while the majority of M5 cells (~75%) co-expressed TH (see Figures 5C,D and 6A). About half of the TH-labelled cells in the VTA expressed M5, compared to 80% of TH-labelled cells in the SNc (see Figure 6B and Supplemental Figure S2B). These numbers are in agreement with earlier data showing a high degree of colocalization of the D2 dopamine receptor, another marker for midbrain DA neurons, and M5 in the SNc of rats.16 Less than 15% of cells expressing vGAT in the VTA and SNc expressed M5 (see Figures 5C,D, 6C and Supplemental Figure S2C). In both the shell and core sub-regions of the NAc, as well as in the dorsal striatum, intense labeling for vGAT mRNA, but very sporadic M5 and no TH mRNA labeling were seen (Figure 5,6).
Figure 5. Distribution of M5 message in the ventral midbrain of rats as revealed by fluorescent in situ hybridization histochemistry.
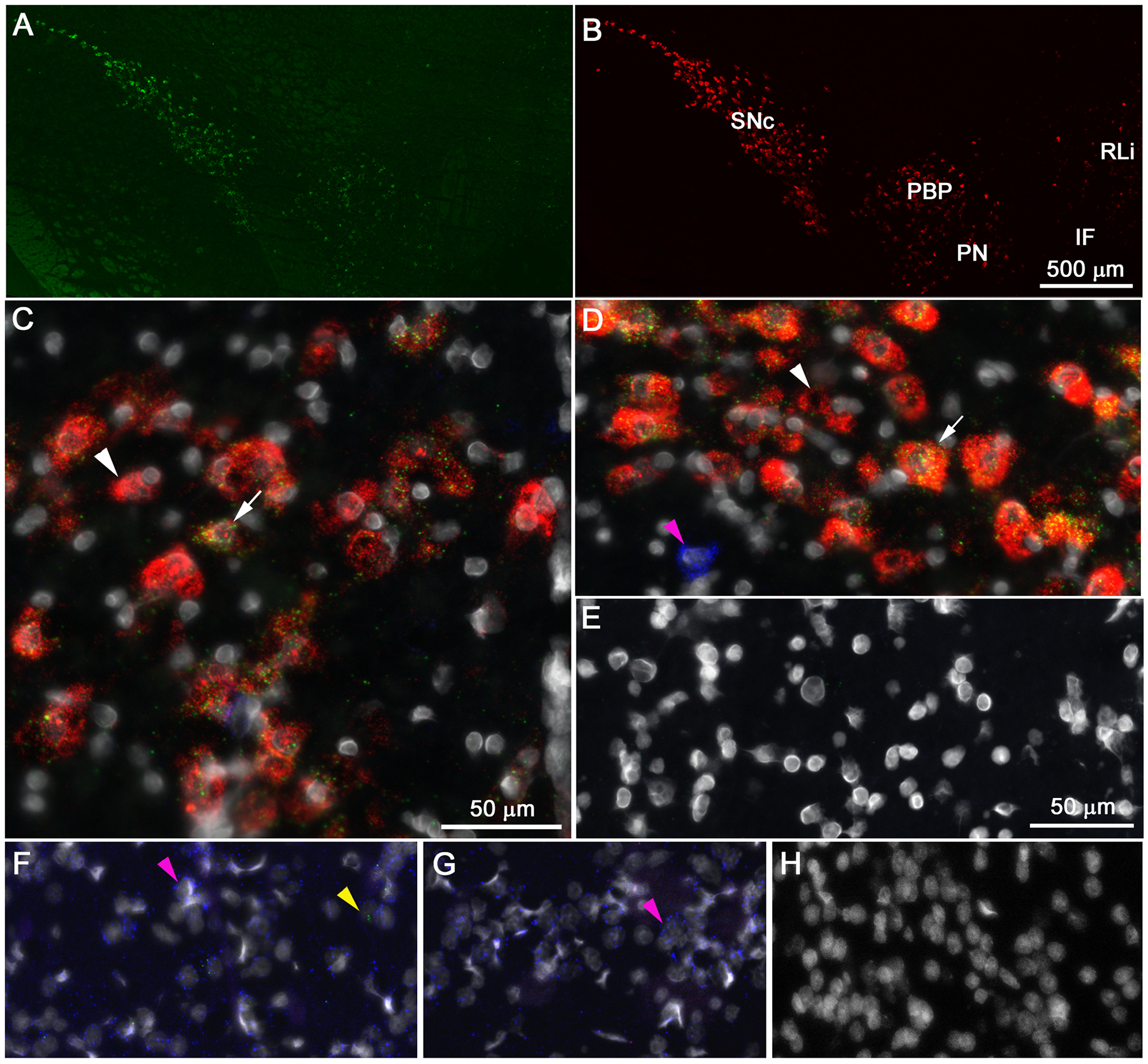
Low power photomicrographs show similar distribution of M5 (green [A]) and TH (red [B]) mRNA in the midbrain with higher expression of both mRNA species in the substantia nigra pars compacta (SNc) than in the ventral tegmental area (VTA). Panels C - G show high power photomicrographs of triple labeling for M5 (green), TH (red), the vesicular GABA transporter (vGAT [blue]) and DAPI stained nuclei (white) in the parabrachial pigmented nucleus (PBP) of the VTA, C) and SNc (D). Noticeable fluorescent signals are absent in the negative control sections through the VTA (E) and nucleus accumbens (NAc [H]). In the SNc (D), and to a lesser degree in the PBP (C), cells co-expressing M5 and TH (white arrow) are more abundant than single labeled TH cells (white arrowhead). Panels F – H show triple-labeled sections through the shell and core (F) region of the NAc, the dorsomedial striatum (G) as well as a negative control section from the NAc (H). Most cells in the NAc and CP express vGAT (magenta arrowhead), whereas M5 puncta (yellow arrowhead) are only sporadically encountered in the NAS and not at all in the CP. Scale bar in panel B applies also to panel A and scale bar in panel E applies to panels D – H.
Figure 6. Co-expression of M5, TH, or vGAT transcripts in the ventral tegmental area.
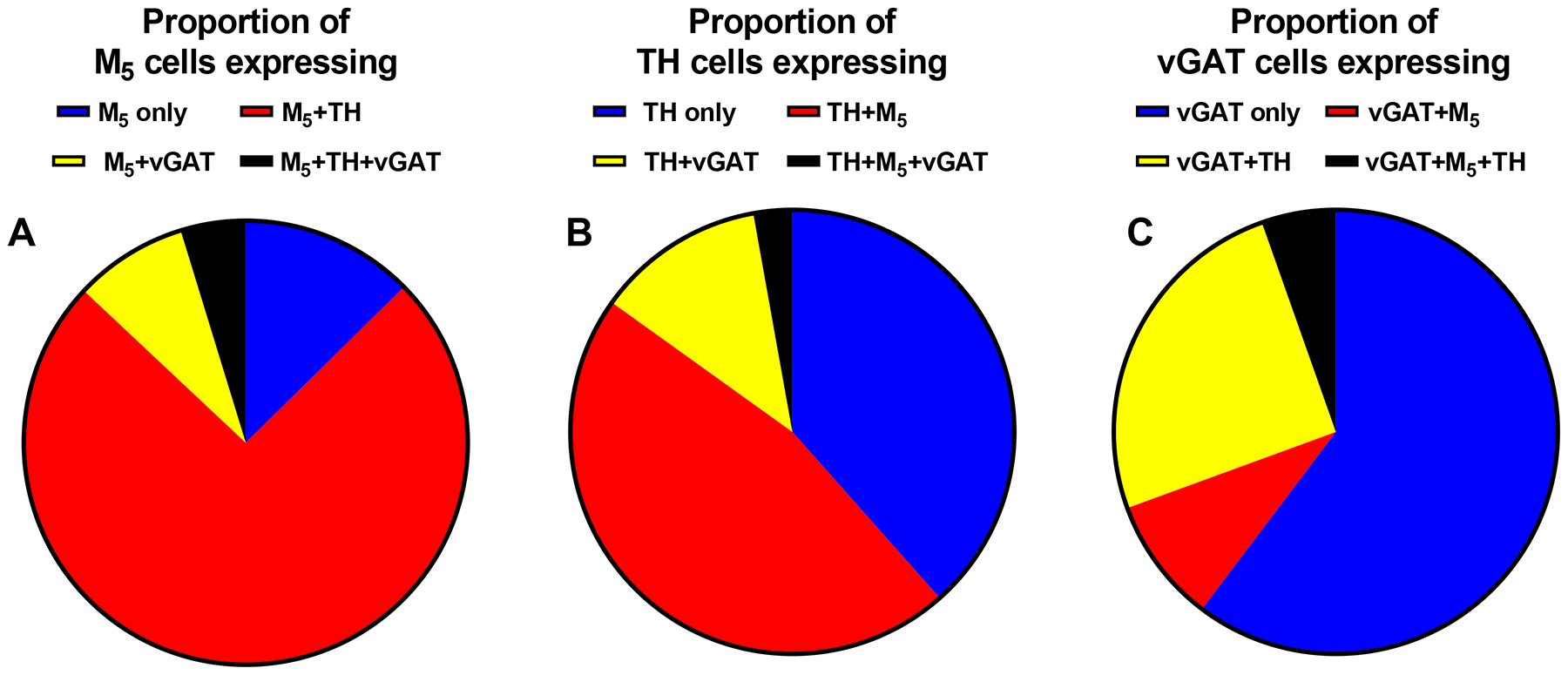
Proportion of M5, (A) TH (B), or vGAT (C) cells in the ventral tegmental area (VTA) that are single labeled or double labeled for TH or vGAT (A), M5 or vGAT (B), or M5 or TH (C) as well as cells triple labeled for these mRNA species (A – C). Data are means of 4 animals per group.
Early anatomical studies have reported that cholinergic neurons located in the laterodorsal and pedunculopontine tegmental nuclei project to the ventral midbrain forming excitatory synapses with VTA DA cells that primarily innervate the NAc.35,36 Local administration of nonselective cholinergic agonists and antagonists into the ventral midbrain elicit and reduce DA release, respectively in the striatum and NAc.37–39 In the absence of either selective M5 antibodies or radioligands for detection of the M5 mAChR receptor protein itself, our fluorescent in situ hybridization studies provided the first demonstration that a large percentage of the dopaminergic neurons in the VTA express M5 mRNA. While the specific site(s) of action for the observed effects of ML375 remains unknown, the present anatomical findings support the possible interpretation that direct inhibition of M5 mAChRs located on DA neurons in the VTA, the only mAChR subtype expressed in this region,16,17 may account for the attenuation of reinforcing effects of opioids through a reduction in opioid-induced increases in DA release in the NAc. In contrast, the limited co-localization of M5 mRNA and vGAT in the VTA suggest that the behavioral effects of ML375 are less likely to be the result of direct M5-mediated action on GABAergic interneurons within the VTA, which become disinhibited by μ-opioid receptor agonists.14,15,40 While such interpretations are possible, previous studies in the nigrostriatal circuitry using the M5 positive allosteric modulator VU0238429 have demonstrated that the role of M5 mAChRs in regulating ventral midbrain dopaminergic circuitry may be multifaceted. For example, selective activation of M5 mAChRs expressed within the nigrostriatal DA pathway results in opposing physiological outcomes depending on the location of the receptor.41 In particular, activation of somatodendritic M5 mAChRs receptors by VU0238429 expressed on SNc neurons increased DA neuron firing, while activation of M5 receptors in the dorsal striatum induced an inhibition in DA release.41 Differential effects of M5 mAChR activation is in accordance with studies by Berizzi et al28, who reported that injections of ML375 into the dorsolateral, but not dorsomedial striatum reduced ethanol self-administration. Lastly, the limited co-localization of vGAT and TH within the VTA is of particular interest. While previous studies have shown co-release of GABA and DA within the VTA, this was through non-canonical pathways that did not require vGAT.42 Co-localization of vGAT and TH has been shown to occur within a small subpopulation of neurons in the arcuate nucleus;43–45 however, this has not previously been shown within the VTA. Further exploration is still needed to understand the physiological consequences of this medio-lateral gradient of M5 mAChR distribution, to understand efferent projections of these M5-containing DA neurons, as well as to determine if co-localization of M5 and TH is altered following opioid self-administration. Current physiologic and neurochemical studies are now focused on evaluating the relative contribution(s) of the selective M5 NAM ML375 on different populations of M5 mAChRs within the mesolimbic DA circuitry in opioid-naïve and opioid-experienced animals.
ML375 did not affect food-maintained responding on a PR schedule or spontaneous locomotor exploration
In order to confirm that a reduction in opiate self-administration or cue-reactivity are not attributed to a reduction in motivation or sedation, we established that ML375 did not alter sucrose pellet-maintained responding or spontaneous locomotor activity across the same dose range (Supplementary Figure S3). In rats trained to self-administer sucrose pellets under a PR schedule of reinforcement, ML375 did not alter the reinforcing strength at doses that attenuated oxycodone-related behaviors (t6=1.44, n.s.; Supplementary Figure S3A). Importantly these rats had a history of oxycodone self-administration (from Figure 2). In addition, ML375 (10 and 30 mg/kg) did not alter the time course of locomotor exploration (F dose (2,108)=0.52, n.s., Ftime (11,108)=27.0, p < 0.001, and Fdose x time (22,108)=0.0.67, n.s. [Supplementary Figure S3B]) and total ambulation counts in an open field (F (2,9)=0.12, n.s.; Supplementary Figure 3B insert).
In summary, functional antagonism of the M5 mAChR produced robust attenuation of the reinforcing strength of oxycodone and remifentanil as well as oxycodone cue-elicited responding in a model of drug craving/relapse in rats. The observed efficacy of ML375 in these preclinical models was achieved in a dose range that had no effect on non-drug maintained responding or spontaneous locomotion. Importantly, ML375 also had no effect on oxycodone-induced antinociception, nor altered exposure levels of oxycodone when given in combination. Moreover, the in situ hybridization studies confirmed that the M5 mAChR subtype is predominately expressed on DA-containing neurons within the VTA supporting the hypothesis that actions of ML375 may be mediated through modulation of mesolimbic dopaminergic signaling. Given the lack of success in developing new opioid-based drugs that maintain analgesic effects while reducing and/or eliminating abuse liability, the present findings support further development of selective M5 NAMs as an alternative non-opioid based medication approach for OUD.
Here, we showed that the selective M5 NAM ML375 decreased the reinforcing effects and the relative reinforcing strength of the μ-opioid receptor agonists, thereby extending previous studies demonstrating a reduction in the reinforcing effects of other substances of abuse including cocaine and ethanol.27,28 ML375 attenuated effects of both remifentanil, a short-acting opiate, and oxycodone, one of the most widely misused prescription μ-opioid receptor analgesics contributing to the opioid epidemic.46 Notably, ML375 produced a robust dose-dependent attenuation of the reinforcing strength of oxycodone, with a complete attenuation of responding at the 30 mg/kg dose, such that responding was lower than when saline was substituted for oxycodone. Furthermore, the magnitude of the reductions in remifentanil and oxycodone self-administration by ML375 were greater than those seen with the FDA-approved μ-opioid receptor antagonist naltrexone at the single dose tested (Figure 1B; Supplementary Figure S1D). While only a single dose of naltrexone was examined, this dose was selected based on a previous report that demonstrated a decrease or a downward shift in remifentanil self-administration in rats maintained under a FR schedule.47
We also demonstrated the potential for selective negative allosteric modulation of M5 mAChRs to block cue-induced reactivity of responding following oxycodone self-administration in a cue-reactivity rodent model of drug craving/relapse.29,48 Classic preclinical models of relapse involve extinguishing responding in the absence of drug-related cues following stable self-administration and then examining the ability of novel therapeutics to block cue-induced reinstatement of responding. However, in an effort to increase validity to clinical populations, we employed 72 hours of forced abstinence and separation from the drug-related environment followed by assessment of the effects of the context and cues alone to maintain responding. Under the cue condition, pretreatment of ML375 attenuated levels of responding to those observed when ML375 was administered prior to sessions when rats self-administered saline for multiple sessions under the PR schedule and drug-cues were still present (Figure 2). An alternative interpretation could be that ML375 facilitates the rate of extinguishing non-drug reinforced responding (e.g. extinction learning) that, if substantiated, could be beneficial as an aid to cognitive behavioral therapy strategies.49 Future studies to explore the effects of M5 mAChR NAMs on aspects of learning and memory under baseline and following opioid self-administration paradigms are warranted.
As previously mentioned, one of the primary objectives of the opioid research community over the last two decades has been to optimize the analgesic properties of μ-opioid receptor agonists, while diminishing their abuse liability.50 The vast majority of these efforts have focused on the development of novel ligands for the μ-opioid receptors themselves that exhibit biased signaling towards less addicting properties. However, to date these biased signaling approaches for developing better and safer μ-opioid receptor agonists have failed to yield any novel therapeutics that are devoid of μ-opioid-mediated abuse liability. Moreover, these strategies have not resulted in compounds for treating patients with existing OUD. Recent efforts are examining the utility of developing non-opioid based mechanisms to treat aspects of OUD.29,51 Such non-opioid based therapeutic approaches have great potential for selective modulation of reward-related circuitry without altering antinociception, as reported in the present studies as well as with DA D3 receptor antagonists.30,31,51 Together, our findings suggest that development of selective M5 NAMs could provide a novel non-opioid therapeutic approach for the prevention of opioid relapse, but may also provide a unique preventative therapy for blocking the initiation of opioid misuse that leads to OUD.
Current efforts are ongoing to optimize M5 mAChR NAMs with more suitable properties to further investigate the therapeutic potential of this mechanism for the treatment of different stages of OUD. In particular, a M5 mAChR NAM with a shorter elimination half-life is needed to conduct repeated dosing studies. While simple schedules of reinforcement and antinociception were employed in the present studies, future studies using more optimized M5 NAMs will investigate this novel mechanism under other schedules of reinforcement to model additional aspects of OUD including behavioral choice studies and repeated dosing paradigms.52–55 Additionally, future studies will evaluate these optimized M5 NAMs in models of acute and chronic pain that may model underlying factors contributing to OUD, including potential antinociception in oxycodone-naïve and oxycodone-experienced male and female rodents.56,57
METHODS
Drugs
Remifentanil hydrochloride, oxycodone hydrochloride, and naltrexone hydrochloride were obtained from (Sigma-Aldrich, St. Louis, Missouri) and were dissolved in saline (remifentanil, oxycodone) or sterile water (naltrexone). ML375 was synthesized in house as described previously58 and formulated as a macrosuspension in 5% dimethylsulfoxide (Sigma) and 20% (w/v) hydroxypropyl-β-cyclodextrin (Trappsol, CTD, Inc., Alachua, Florida) and administered intraperitoneally (i.p.) in a volume of 2–4 mL/kg.
Subjects
Adult male Sprague-Dawley rats (Envigo, Indianapolis, Indiana) were housed in groups of two or three under a 12/12-h light/dark cycle with water and standard rodent chow (Harlan Teklad, Madison, WI) provided ad libitum. Behavioral studies were conducted during the second half of the 12-h light phase. All experiments were approved by and conducted in accordance with the Vanderbilt University’s Institutional Animal Care and Use Committee and followed the guidelines set forth by the National Research Council’s Guide for Care and Use of Laboratory Animals.59
Opioid self-administration
Self-administration procedures have been described previously27,60 (see Supplemental Materials for additional details). Briefly, rats (260 – 300 g) were implanted with a chronic indwelling jugular vein catheter that was connected to a vascular access button. Following recovery, rats with no prior behavioral history began training for self-administration studies. Rats were connected to an external infusion pump for drug delivery and placed in operant chambers.
Sessions started with extension of two levers, a priming infusion of the dose of drug available for that session, and illumination of a houselight. Rats were trained over the course of 5–10 sessions to respond under a fixed-ratio 10 (FR10; remifentanil) or fixed-ratio 3 schedule (FR3; oxycodone) on the active lever resulting in the retraction of both levers, illumination of the stimulus light above the active lever, and infusion of remifentanil or oxycodone over the course of 4–6 seconds. The stimulus light remained on for 10 sec following completion of each FR after which the light turned off and levers were extended again.
To evaluate the ability of ML375 to attenuate the reinforcing strength of oxycodone, following stable self-administration of 56 μg/kg/infusion oxycodone under a FR3 schedule, a progressive ratio (PR) schedule was implemented such that each subsequent ratio increased according to the following equation; ratio = [5 × e(R × 0.2)] – 5,27,61 (see Supplemental Materials for more detail). Rats self-administered one dose of oxycodone until rates of responding stabilized before a test session was implemented. Order of opioid and ML375 doses were randomized.
To evaluate the ability of ML375 to affect “cue-reactivity”, a model of relapse and drug seeking, separate groups of rats were trained to reliably self-administer 56 μg/kg/infusion of oxycodone under a FR3 schedule for a minimum of 7 days of responding with >10 infusions and a 3-day stability criteria with <25% variability in number of infusions and no upward or downward trends evident. Animals were then returned to their home cage and self-administration studies were suspended for 72 h. Following 72 h of experimenter-forced abstinence, animals were injected 15 min (vehicle or ML375 [3–30 mg/kg]) or 20 min (vehicle or naltrexone [0.3 mg/kg]) before being placed in the operant chamber. During this “cue-reactivity” session rats were exposed to all cues associated with oxycodone self-administration under an FR1 schedule. However, each response on the previously active lever resulted in illumination of the light above the lever, noise of the infusion pump, yet no fluid being delivered through the catheter.29,48 Following completion of cue-reactivity studies, a subset of seven rats was gradually food-restricted to 85% free feeding weight to assess, if ML375 affected sucrose pellet-maintained responding in opioid-exposed rats. Once the rats showed stable responding on the same PR schedule (FR3), effects of vehicle or 30 mg/kg ML 375 were determined.
Oxycodone-induced antinociception
The effects of ML375 on oxycodone-induced anti-nociception were assessed in the hot plate and tail flick tests (See Supplemental Methods for details). Animals were tested before compound administration (baseline) and then administered vehicle or ML375 [30 – 56.6 mg/kg, i.p.]. 15 minutes later animals were administered (vehicle or oxycodone [0.3 – 5.6 mg/kg, s.c.]). Anti-nociception was evaluated 30 minutes after oxycodone administration. Data are expressed as % maximum possible effect (%MPE) which was calculated as follows: %MPE = [(test latency − baseline latency)/(cutoff time − baseline latency)]*100.
Drug-drug interactions
To test for a potential pharmacokinetic drug-drug interaction, rats were injected with ML375 (56.6 mg/kg, i.p.) or vehicle followed 15 min later by administration of oxycodone (5.6 mg/kg, s.c.) or vehicle. After an additional 30 min, animals were sacrificed and trunk blood and brains were collected. Methods for bioanalysis of in vivo plasma and brain samples can be found in Supplemental Methods.
Fluorescent in situ hybridization
A version of fluorescent in situ hybridization called RNAscope (Advanced Cell Diagnostics [ACD], Hayward, CA) was used to simultaneously visualize transcripts for M5 (Chrm5) in combination with tyrosine hydroxylase (TH) and vesicular γ-aminobutyric acid (GABA) transporter (vGAT, Slc32a1), markers of dopaminergic and GABAergic cells, respectively.
Four rats, 6–8 weeks of age, were decapitated under isoflurane anesthesia and their brains were rapidly extracted and flash frozen on dry ice, and stored at −80 °C. 16-μm-thick sections through the ventral midbrain and the striatal complex were cut on a cryostat. Sections were kept frozen until they were fixated in 4% paraformaldehyde and prepared for flourescent in situ hybridization. All subsequent steps used the RNAscope multiplex fluorescent kit v1 and followed the manufacturer’s protocol (https://acdbio.com/technical-support/user-manuals) as outlined by Ghamari-Langroudi et al. (2015).62 Sections through the VTA labeled with three probe sets of bacterial RNA (ACD) served as a negative control for tissue autofluorescence and for non-specific labeling due to assay reagents and procedures. DAPI (4’,6-diamidino-2-phenylindole) staining of nuclei was used to identify cells.
Image acquisition and analysis
5–6 sections/rat were imaged throughout the VTA, including a negative control, using a Nikon STORM microscope used in wide field mode. First, negative control sections were thresholded using Fiji63 followed by identical adjustments of the remaining sections. Subsequently, in each color channel the number of labeled puncta overlying and/or adjacent to DAPI-stained nuclei was counted in 50 VTA cells in the negative control section. Cells were considered labeled for a given RNA species if their puncta count was equal to or greater than the mean + 3 x standard deviation of the puncta count/cell of the negative control.
Cells were counted throughout the VTA, including the parabrachial pigmented nucleus (PBP), paranigral nucleus (PN), interfascicular nucleus (IF), rostral linear nucleus (RLi), and the A10 ventral rostral (A10vr) and A10 dorsal caudal regions as defined by Phillipson et al. (1979)64 and Hökfelt et al., (1984).65 The substantia nigra pars compacta (SNc) served as a control region where M5 expression had been assessed before;16 methods and results are included in the supplemental material. The number of single labeled (M5, TH or vGAT), double labeled (M5/TH, M5/vGAT, or TH/vGAT), and triple labeled (M5/TH/vGAT) cells was determined and expressed as number of cells/mm2. Additionally, for each mRNA species, the proportion of M5, TH, and vGAT cells that were single, double and triple labeled was calculated.
Data analysis
For all self-administration studies, the number of reinforcers per session or response rates (lever presses when a reinforcer was available) were examined. Data (mean ± SEM) were plotted as a function of remifentanil or oxycodone self-administration dose, or as a function of ML375 or naltrexone dose when administered prior to opioid self-administration. One-way ANOVAs were conducted to examine main effects of treatment (ML375 or naltrexone) on a single dose of remifentanil or oxycodone self-administration; two-way ANOVAs were conducted to examine effects of treatment (ML375 or naltrexone) across a dose response curve for opioid self-administration. For remifentanil self-administration studies, non-repeated ANOVAs were conducted (n=6–9 rats/treatment; each rat was tested with at least two conditions to be included); for oxycodone self-administration studies, repeated measures ANOVAs were applied (n=7/treatment). For cue-reactivity tests, a non-repeated two-way ANOVA was conducted to assess between-group effects of treatment (ML375 or naltrexone) on active and inactive lever responding. For hot plate and tail flick assays, a one-way ANOVA was conducted to compare effects of ML375 in combination with vehicle with vehicle + vehicle treatment. The dose response curves for oxycodone in the presence or absence of ML375 were analyzed by two-way ANOVA. Further, a nonlinear regression analysis for each dose response curve was conducted to determine ED50 values for each treatment condition. Significant main effects were followed by Bonferroni’s post hoc test comparing each test condition with respective vehicle-treated conditions (one-way ANOVAs) or multiple comparisons to all treatment conditions (two-way ANOVAs). All statistics and graphical representations were conducted using GraphPad Prism version 7.02. Repeated measures ANOVAs did not assume sphericity and applied the Geisser-Greenhouse correction method.
Supplementary Material
ACKNOWLEDGEMENTS
We thank William Hudson Robb, Rebecca Sale, Nathan Iyer, Richard Fu and Edith Duncan for technical assistance and Dr. Ariel Y. Deutch for helpful discussion during the preparation of this manuscript.
FUNDING AND DISCLOSURE
This work was supported by the National Institutes of Health National Institute on Drug Abuse Grants [DA037207(CKJ/CWL), DA042129(RWG)]. Data analysis was performed in part through the use of the Vanderbilt Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126).
Financial Disclosures: M.B., T.M.B., C.W.L., and C.K.J. received research/salary support from AstraZeneca, Lundbeck, Ono and/or Bristol Myers Squibb Pharmaceuticals. T.M.B., C.K.J., C.W.L. are inventors on multiple composition of matter patents protecting allosteric modulators of GPCRs. The remaining authors declare no competing financial interests.
ABBREVIATIONS
- ACh
acetylcholine
- BP
breakpoint
- DA
dopamine
- DAPI
4’,6-diamidino-2-phenylindole
- HPLC
high-performance liquid chromatography
- LCMS
liquid chromatography-coupled mass spectroscopy
- mAChR
muscarinic acetylcholine receptor
- MPE
maximal possible effect
- NAM
negative allosteric modulator
- NAc
nucleus accumbens
- OUD
opioid use disorder
- OXY
oxycodone
- SA
self-administration
- SNc
substantia nigra pars compacta
- VTA
ventral tegmental area
Footnotes
Supporting Information: Supplemental Tables and Figures are included providing additional methodology and data pertaining to behavior, pharmacokinetic analysis, and in-situ hybridization histochemistry.
REFERENCES
- (1).Skolnick P; Volkow ND Re-Energizing the Development of Pain Therapeutics in Light of the Opioid Epidemic. Neuron 2016, 92 (2), 294–297. 10.1016/j.neuron.2016.09.051. [DOI] [PubMed] [Google Scholar]
- (2).Degenhardt L; Whiteford H; Hall WD The Global Burden of Disease Projects: What Have We Learned about Illicit Drug Use and Dependence and Their Contribution to the Global Burden of Disease? Drug Alcohol Rev. 2014, 33 (1), 4–12. 10.1111/dar.12088. [DOI] [PubMed] [Google Scholar]
- (3).Volkow ND; McLellan AT Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. N. Engl. J. Med 2016, 374 (13), 1253–1263. 10.1056/NEJMra1507771. [DOI] [PubMed] [Google Scholar]
- (4).Chen LH; Hedegaard H; Warner M Drug-Poisoning Deaths Involving Opioid Analgesics: United States, 1999–2011. NCHS Data Brief 2014, No. 166, 1–8. [PubMed] [Google Scholar]
- (5).Compton WM; Jones CM; Baldwin GT Relationship between Nonmedical Prescription-Opioid Use and Heroin Use. N. Engl. J. Med 2016, 374 (2), 154–163. 10.1056/NEJMra1508490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cicero TJ; Ellis MS; Harney J Shifting Patterns of Prescription Opioid and Heroin Abuse in the United States The New England journal of medicine. United States: October 2015, pp 1789–1790. 10.1056/NEJMc1505541. [DOI] [PubMed] [Google Scholar]
- (7).Shei A; Rice JB; Kirson NY; Bodnar K; Birnbaum HG; Holly P; Ben-Joseph R Sources of Prescription Opioids among Diagnosed Opioid Abusers. Curr. Med. Res. Opin 2015, 31 (4), 779–784. 10.1185/03007995.2015.1016607. [DOI] [PubMed] [Google Scholar]
- (8).Substance Abuse and Mental Health Services; Administration. Key Substance Use and Mental Health Indicators in the United States: Results from the 2016 National Survey on Drug Use and Health (HHS Publication No. SMA 17–5044, NSDUH Series H-52). 2017. 10.1088/1741-2560/7/4/046002. [DOI] [Google Scholar]
- (9).Machelska H; Celik MO Advances in Achieving Opioid Analgesia Without Side Effects. Front. Pharmacol 2018, 9, 1388 10.3389/fphar.2018.01388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Stotts AL; Dodrill CL; Kosten TR Opioid Dependence Treatment: Options in Pharmacotherapy. Expert Opin. Pharmacother 2009, 10 (11), 1727–1740. 10.1517/14656560903037168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nestler EJ Historical Review: Molecular and Cellular Mechanisms of Opiate and Cocaine Addiction. Trends Pharmacol. Sci 2004, 25 (4), 210–218. 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
- (12).Fallon JH Collateralization of Monoamine Neurons: Mesotelencephalic Dopamine Projections to Caudate, Septum, and Frontal Cortex. J. Neurosci 1981, 1 (12), 1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Takada M; Hattori T Organization of Ventral Tegmental Area Cells Projecting to the Occipital Cortex and Forebrain in the Rat. Brain Res. 1987, 418 (1), 27–33. [DOI] [PubMed] [Google Scholar]
- (14).Gysling K; Wang RY Morphine-Induced Activation of A10 Dopamine Neurons in the Rat. Brain Res. 1983, 277 (1), 119–127. [DOI] [PubMed] [Google Scholar]
- (15).Johnson SW; North RA Opioids Excite Dopamine Neurons by Hyperpolarization of Local Interneurons. J. Neurosci 1992, 12 (2), 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Weiner DM; Levey AI; Brann MR Expression of Muscarinic Acetylcholine and Dopamine Receptor MRNAs in Rat Basal Ganglia. Proc. Natl. Acad. Sci. U. S. A 1990, 87 (18), 7050–7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vilaro MT; Palacios JM; Mengod G Localization of M5 Muscarinic Receptor MRNA in Rat Brain Examined by in Situ Hybridization Histochemistry. Neurosci. Lett 1990, 114 (2), 154–159. [DOI] [PubMed] [Google Scholar]
- (18).Forster GL; Yeomans JS; Takeuchi J; Blaha CD M5 Muscarinic Receptors Are Required for Prolonged Accumbal Dopamine Release after Electrical Stimulation of the Pons in Mice. J. Neurosci 2002, 22 (1), RC190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yeomans JS; Takeuchi J; Baptista M; Flynn DD; Lepik K; Nobrega J; Fulton J; Ralph MR Brain-Stimulation Reward Thresholds Raised by an Antisense Oligonucleotide for the M5 Muscarinic Receptor Infused near Dopamine Cells. J. Neurosci 2000, 20 (23), 8861–8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yeomans J; Forster G; Blaha C M5 Muscarinic Receptors Are Needed for Slow Activation of Dopamine Neurons and for Rewarding Brain Stimulation. Life Sci. 2001, 68 (22–23), 2449–2456. [DOI] [PubMed] [Google Scholar]
- (21).Wang H; Ng K; Hayes D; Gao X; Forster G; Blaha C; Yeomans J Decreased Amphetamine-Induced Locomotion and Improved Latent Inhibition in Mice Mutant for the M5 Muscarinic Receptor Gene Found in the Human 15q Schizophrenia Region. Neuropsychopharmacology 2004, 29 (12), 2126–2139. 10.1038/sj.npp.1300502. [DOI] [PubMed] [Google Scholar]
- (22).Fink-Jensen A; Fedorova I; Wortwein G; Woldbye DPD; Rasmussen T; Thomsen M; Bolwig TG; Knitowski KM; McKinzie DL; Yamada M; et al. Role for M5 Muscarinic Acetylcholine Receptors in Cocaine Addiction. J. Neurosci. Res 2003, 74 (1), 91–96. 10.1002/jnr.10728. [DOI] [PubMed] [Google Scholar]
- (23).Thomsen M; Woldbye DPD; Wortwein G; Fink-Jensen A; Wess J; Caine SB Reduced Cocaine Self-Administration in Muscarinic M5 Acetylcholine Receptor-Deficient Mice. J. Neurosci 2005, 25 (36), 8141–8149. 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Basile AS; Fedorova I; Zapata A; Liu X; Shippenberg T; Duttaroy A; Yamada M; Wess J Deletion of the M5 Muscarinic Acetylcholine Receptor Attenuates Morphine Reinforcement and Withdrawal but Not Morphine Analgesia. Proc. Natl. Acad. Sci. U. S. A 2002, 99 (17), 11452–11457. 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bender AM; Garrison AT; Lindsley CW The Muscarinic Acetylcholine Receptor M5: Therapeutic Implications and Allosteric Modulation. ACS Chem. Neurosci 2018. 10.1021/acschemneuro.8b00481. [DOI] [PubMed] [Google Scholar]
- (26).Gentry PR; Kokubo M; Bridges TM; Noetzel MJ; Cho HP; Lamsal A; Smith E; Chase P; Hodder PS; Niswender CM; et al. Development of a Highly Potent, Novel M5 Positive Allosteric Modulator (PAM) Demonstrating CNS Exposure: 1-((1H-Indazol-5-Yl)Sulfoneyl)-N-Ethyl-N-(2-(Trifluoromethyl)Benzyl)Piperidine-4- Carboxamide (ML380). J. Med. Chem 2014, 57 (18), 7804–7810. 10.1021/jm500995y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gunter BW; Gould RW; Bubser M; Mcgowan KM; Lindsley CW; Jones CK Selective Inhibition of M5muscarinic Acetylcholine Receptors Attenuates Cocaine Self-Administration in Rats. Addict. Biol 2017. 10.1111/adb.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Berizzi AE; Perry CJ; Shackleford DM; Lindsley CW; Jones CK; Chen NA; Sexton PM; Christopoulos A; Langmead CJ; Lawrence AJ Muscarinic M5 Receptors Modulate Ethanol Seeking in Rats. Neuropsychopharmacology 2018, 43 (7), 1510–1517. 10.1038/s41386-017-0007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Neelakantan H; Holliday ED; Fox RG; Stutz SJ; Comer SD; Haney M; Anastasio NC; Moeller FG; Cunningham KA Lorcaserin Suppresses Oxycodone Self-Administration and Relapse Vulnerability in Rats. ACS Chem. Neurosci 2017, 8 (5), 1065–1073. 10.1021/acschemneuro.6b00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).You ZB; Gao JT; Bi GH; He Y; Boateng C; Cao J; Gardner EL; Newman AH; Xi ZX The Novel Dopamine D3 Receptor Antagonists/Partial Agonists CAB2–015 and BAK4–54 Inhibit Oxycodone-Taking and Oxycodone-Seeking Behavior in Rats. Neuropharmacology 2017, 126, 190–199. 10.1016/j.neuropharm.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).You Z-B; Bi G-H; Galaj E; Kumar V; Cao J; Gadiano A; Rais R; Slusher BS; Gardner EL; Xi Z-X; et al. Dopamine D3R Antagonist VK4–116 Attenuates Oxycodone Self-Administration and Reinstatement without Compromising Its Antinociceptive Effects. Neuropsychopharmacology 2018, 116 (July), 1–10. 10.1038/s41386-018-0284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Beardsley PM; Aceto MD; Cook CD; Bowman ER; Newman JL; Harris LS Discriminative Stimulus, Reinforcing, Physical Dependence, and Antinociceptive Effects of Oxycodone in Mice, Rats, and Rhesus Monkeys. Exp. Clin. Psychopharmacol 2004, 12 (3), 163–172. 10.1016/j.foodchem.2016.09.132. [DOI] [PubMed] [Google Scholar]
- (33).Townsend EA; Naylor JE; Negus SS; Edwards SR; Qureshi HN; McLendon HW; McCurdy CR; Kapanda CN; do Carmo JM; da Silva FS; et al. Effects of Nalfurafine on the Reinforcing, Thermal Antinociceptive, and Respiratory-Depressant Effects of Oxycodone: Modeling an Abuse-Deterrent Opioid Analgesic in Rats. Psychopharmacology (Berl). 2017, 234 (17), 2597–2605. 10.1007/s00213-017-4652-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gauvin D V; McComb, M.; Code, R.; Dalton, J. A.; Baird, T. J. Abuse Liability Assessment of Hydrocodone under Current Draft Regulatory Guidelines. J. Pharmacol. Toxicol. Methods 2015, 75, 118–129. 10.1016/j.vascn.2015.05.003. [DOI] [PubMed] [Google Scholar]
- (35).Omelchenko N; Sesack SR Cholinergic Axons in the Rat Ventral Tegmental Area Synapse Preferentially onto Mesoaccumbens Dopamine Neurons. J. Comp. Neurol 2006, 494 (6), 863–875. 10.1002/cne.20852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mesulam MM; Mufson EJ; Wainer BH; Levey AI Central Cholinergic Pathways in the Rat: An Overview Based on an Alternative Nomenclature (Ch1-Ch6). Neuroscience 1983, 10 (4), 1185–1201. [DOI] [PubMed] [Google Scholar]
- (37).Gronier B; Perry KW; Rasmussen K Activation of the Mesocorticolimbic Dopaminergic System by Stimulation of Muscarinic Cholinergic Receptors in the Ventral Tegmental Area. Psychopharmacology (Berl). 2000, 147 (4), 347–355. [DOI] [PubMed] [Google Scholar]
- (38).Blaha CD; Allen LF; Das S; Inglis WL; Latimer MP; Vincent SR; Winn P Modulation of Dopamine Efflux in the Nucleus Accumbens after Cholinergic Stimulation of the Ventral Tegmental Area in Intact, Pedunculopontine Tegmental Nucleus-Lesioned, and Laterodorsal Tegmental Nucleus-Lesioned Rats. J. Neurosci 1996, 16 (2), 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lester DB; Miller AD; Pate TD; Blaha CD Midbrain Acetylcholine and Glutamate Receptors Modulate Accumbal Dopamine Release. Neuroreport 2008, 19 (9), 991–995. 10.1097/WNR.0b013e3283036e5e. [DOI] [PubMed] [Google Scholar]
- (40).Di Chiara G; Imperato A Drugs Abused by Humans Preferentially Increase Synaptic Dopamine Concentrations in the Mesolimbic System of Freely Moving Rats. Proc. Natl. Acad. Sci. U. S. A 1988, 85 (14), 5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Foster DJ; Gentry PR; Lizardi-Ortiz JE; Bridges TM; Wood MR; Niswender CM; Sulzer D; Lindsley CW; Xiang Z; Conn PJ M5 Receptor Activation Produces Opposing Physiological Outcomes in Dopamine Neurons Depending on the Receptor’s Location. J. Neurosci 2014, 34 (9), 3253–3262. 10.1523/JNEUROSCI.4896-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tritsch NX; Ding JB; Sabatini BL Dopaminergic Neurons Inhibit Striatal Output through Non-Canonical Release of GABA. Nature 2012, 490 (7419), 262–266. 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Marshall CJ; Desroziers E; McLennan T; Campbell RE Defining Subpopulations of Arcuate Nucleus GABA Neurons in Male, Female, and Prenatally Androgenized Female Mice. Neuroendocrinology 2017, 105 (2), 157–169. 10.1159/000452105. [DOI] [PubMed] [Google Scholar]
- (44).Zhang X; van den Pol AN Dopamine/Tyrosine Hydroxylase Neurons of the Hypothalamic Arcuate Nucleus Release GABA, Communicate with Dopaminergic and Other Arcuate Neurons, and Respond to Dynorphin, Met-Enkephalin, and Oxytocin. J. Neurosci 2015, 35 (45), 14966–14982. 10.1523/JNEUROSCI.0293-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Everilta BJ; Hokfelfi T; Wuc J-Y; Goldsteina M Coexistence of Tyrosine Hydroxylase-Like and Gamma-Aminobutyric Acid-Like Immunoreactivities in Neurons of the Arcuate Nucleus. Neuroendocrinology 1984, 39, 189–191. [DOI] [PubMed] [Google Scholar]
- (46).Rosenblum A; Parrino M; Schnoll SH; Fong C; Maxwell C; Cleland CM; Magura S; Haddox JD Prescription Opioid Abuse among Enrollees into Methadone Maintenance Treatment. Drug Alcohol Depend. 2007, 90 (1), 64–71. 10.1016/j.drugalcdep.2007.02.012. [DOI] [PubMed] [Google Scholar]
- (47).Tanda G; Mereu M; Hiranita T; Quarterman JC; Coggiano M; Katz JL Lack of Specific Involvement of (+)-Naloxone and (+)-Naltrexone on the Reinforcing and Neurochemical Effects of Cocaine and Opioids. Neuropsychopharmacology 2016, 41 (11), 2772–2781. 10.1038/npp.2016.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dimet AL; Cisneros IE; Fox RG; Stutz SJ; Anastasio NC; Cunningham KA; Dineley KT A Protocol for Measuring Cue Reactivity in a Rat Model of Cocaine Use Disorder. J. Vis. Exp 2018, No. 136 10.3791/55864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Nic Dhonnchadha BA; Kantak KM Cognitive Enhancers for Facilitating Drug Cue Extinction: Insights from Animal Models. Pharmacol. Biochem. Behav 2011, 99 (2), 229–244. 10.1016/j.pbb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Austin Zamarripa C; Edwards SR; Qureshi HN; Yi JN; Blough BE; Freeman KB The G-Protein Biased Mu-Opioid Agonist, TRV130, Produces Reinforcing and Antinociceptive Effects That Are Comparable to Oxycodone in Rats. Drug Alcohol Depend. 2018, 192, 158–162. 10.1016/j.drugalcdep.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Jordan CJ; Humburg B; Rice M; Bi G-H; You Z-B; Shaik AB; Cao J; Bonifazi A; Gadiano A; Rais R; et al. The Highly Selective Dopamine D3R Antagonist, RVK4–40, Attenuates Oxycodone Reward and Augments Analgesia in Rodents. Neuropharmacology 2019. 10.1016/j.neuropharm.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mello NK; Negus SS Preclinical Evaluation of Pharmacotherapies for Treatment of Cocaine and Opioid Abuse Using Drug Self-Administration Procedures. Neuropsychopharmacology 1996, 14 (6), 375–424. 10.1016/0893-133X(95)00274-H. [DOI] [PubMed] [Google Scholar]
- (53).Haney M; Spealman R Controversies in Translational Research: Drug Self-Administration. Psychopharmacology (Berl). 2008, 199 (3), 403–419. 10.1007/s00213-008-1079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Czoty PW; Stoops WW; Rush CR Evaluation of the “Pipeline” for Development of Medications for Cocaine Use Disorder: A Review of Translational Preclinical, Human Laboratory, and Clinical Trial Research. Pharmacol. Rev 2016, 68 (3), 533–562. 10.1124/pr.115.011668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Banks ML; Townsend EA; Negus SS Testing the 10 Most Wanted: A Preclinical Algorithm to Screen Candidate Opioid Use Disorder Medications. Neuropsychopharmacology 2019, 44 (6), 1011–1012. 10.1038/s41386-019-0336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Whiteside GT; Pomonis JD; Kennedy JD An Industry Perspective on the Role and Utility of Animal Models of Pain in Drug Discovery. Neurosci. Lett 2013, 557 Pt A, 65–72. 10.1016/j.neulet.2013.08.033. [DOI] [PubMed] [Google Scholar]
- (57).Negus SS; Bilsky EJ; Do Carmo GP; Stevenson GW Rationale and Methods for Assessment of Pain-Depressed Behavior in Preclinical Assays of Pain and Analgesia. Methods Mol. Biol 2010, 617, 79–91. 10.1007/978-1-60327-323-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Gentry PR; Kokubo M; Bridges TM; Kett NR; Harp JM; Cho HP; Smith E; Chase P; Hodder PS; Niswender CM; et al. Discovery of the First M5-Selective and CNS Penetrant Negative Allosteric Modulator (NAM) of a Muscarinic Acetylcholine Receptor: (S)-9b-(4-Chlorophenyl)-1-(3,4-Difluorobenzoyl)-2,3-Dihydro-1 H -Imidazo[2,1- a ]Isoindol-5(9b H)-One (ML375). J. Med. Chem 2013, 56 (22), 9351–9355. 10.1021/jm4013246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Council NR Guide for the Care and Use of Laboratory Animals: Eighth Edition, 8th Editio.; The National Academies Press: Washington, DC, 2011. 10.17226/12910. [DOI] [Google Scholar]
- (60).Gould RW; Amato RJ; Bubser M; Joffe ME; Nedelcovych MT; Thompson AD; Nickols HH; Yuh JP; Zhan X; Felts AS; et al. Partial MGlu 5 Negative Allosteric Modulators Attenuate Cocaine-Mediated Behaviors and Lack Psychotomimetic-Like Effects. Neuropsychopharmacology 2016, 41 (4). 10.1038/npp.2015.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Richardson NR; Roberts DC Progressive Ratio Schedules in Drug Self-Administration Studies in Rats: A Method to Evaluate Reinforcing Efficacy. J. Neurosci. Methods 1996, 66 (1), 1–11. [DOI] [PubMed] [Google Scholar]
- (62).Ghamari-Langroudi M; Digby GJ; Sebag JA; Millhauser GL; Palomino R; Matthews R; Gillyard T; Panaro BL; Tough IR; Cox HM; et al. G-Protein-Independent Coupling of MC4R to Kir7.1 in Hypothalamic Neurons. Nature 2015, 520 (7545), 94–98. 10.1038/nature14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Schindelin J; Arganda-Carreras I; Frise E; Kaynig V; Longair M; Pietzsch T; Preibisch S; Rueden C; Saalfeld S; Schmid B; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9 (7), 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Phillipson O The Cytoarchitecture of the Interfascicular Nucleus and the Ventral Tegmental Area of Tsai in the Rat. J Comp. Neurol 1979, No. 187, 85–98. [DOI] [PubMed] [Google Scholar]
- (65).Hokfelt T; Martensson R; Bjorklund A; Kleinau S; Goldstein M No Title. In Handbook of Chemical Neuroanatomy, Classsical Neurotransmitters in the CNS; Bjorklund A, Hokfelt T, Eds.; Elsevier, Amsterdam, 1984; pp 277–379. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.