

Abstract
Aortic dissection is a life-threatening aortopathy involving separation of the aortic wall, whose underlying mechanisms are still incompletely understood. Epidemiological evidence suggests that unsaturated fatty acids improve cardiovascular health. Here, using quantitative RT-PCR, histological analyses, magnetic cell sorting and flow cytometry assays, and MS-based lipidomics, we show that the activity of a lipid-metabolizing enzyme, secreted phospholipase A2 group V (sPLA2-V), protects against aortic dissection by endogenously mobilizing vasoprotective lipids. Global and endothelial cell–specific sPLA2-V–deficient mice frequently developed aortic dissection shortly after infusion of angiotensin II (AT-II). We observed that in the AT-II–treated aorta, endothelial sPLA2-V mobilized oleic and linoleic acids, which attenuated endoplasmic reticulum stress, increased the expression of lysyl oxidase, and thereby stabilized the extracellular matrix in the aorta. Of note, dietary supplementation with oleic or linoleic acid reversed the increased susceptibility of sPLA2-V–deficient mice to aortic dissection. These findings reveal an unexplored functional link between sPLA2-driven phospholipid metabolism and aortic stability, possibly contributing to the development of improved diagnostic and/or therapeutic strategies for preventing aortic dissection.
Keywords: aortic dissection, animal model, phospholipase A2, phospholipid metabolism, fatty acid, lipidomics, lysyl oxidase, extracellular matrix, cardiovascular disorder, aortopathy, phospholipase, gene knockout, cardiovascular disease
Aortic dissection is a life-threatening condition that is caused by a tear in the intimal layer of the aorta or bleeding within the aortic wall, leading to the separation of the layers of the aortic wall. Two-thirds of such cases involve dissection of the thoracic ascending aorta, which is highly lethal (10–35%) even if emergency surgery can be performed at specialist centers (1–3). Although a key pathologic feature of aortic dissection is fragility of the aortic medial wall (3), the underlying mechanisms are still incompletely understood. Given that such dissection occurs suddenly without preceding clinical signs and that current treatment strategies are limited mainly to antihypertensive agents, biomarkers that can predict fragility and/or therapeutic targets for stabilization of the aortic wall are needed to improve patient outcomes (1–3).
Although there are only limited opportunities for examining the pathogenesis of aortic dissection in patients who are seen after onset, several recent investigations using animal models have improved our understanding of the pathophysiology of this condition. It has been shown that aortic dissection can be induced by infusion of angiotensin II (AT-II), which raises blood pressure, into mice with an ApoE−/− genetic background, which develop extensive atherosclerosis characterized by an unusual plasma lipid profile (4, 5). However, considering that aortic dissection in humans is not accompanied by typical atherosclerotic plaques in many cases (6), studies of aortic dissection using ApoE−/− mice as a model system might not necessarily reflect all of the potential mechanisms, especially in terms of lipid metabolism.
Two main mechanisms underlying medial fragility of the aortic wall have been proposed: extracellular matrix (ECM) degradation and inflammation, where matrix metalloproteinases (MMPs) and pro-inflammatory cytokines contribute to the onset and severity of aortic dissection (3, 7). In humans, several ECM-related genes, such as TGFB1, ACTA2, and COL3A1, are associated with aortic dissection (3). Mutations in LOX (encoding lysyl oxidase (LOX), which is involved in cross-linking of the ECM proteins elastin and collagen) are also correlated with the condition, dissections of the thoracic ascending aorta being most common in affected patients (3, 8, 9). Indeed, administration of β-aminopropionitrile (BAPN), a LOX inhibitor; genetic deletion of tenascin-C, an ECM glycoprotein; or prolonged treatment with GM-CSF, a pro-inflammatory cytokine, in combination with AT-II infusion is able to induce various degrees of aortic dissection even in the absence of ApoE deletion (7, 10–12). Interestingly, a recent clinical study involving individuals with higher cardiovascular risks showed that the incidences of major cardiovascular events including aortic rupture were lower among those assigned to a Mediterranean diet supplemented with extra-virgin olive oil, which abundantly contains oleic acid (OA) and to a lesser extent linoleic acid (LA), than those assigned to a reduced-fat diet (12), suggesting a potentially beneficial role of these unsaturated fatty acids on cardiovascular health. However, considering that aortic dissection is an uncommon disease with a low incidence (5–30 cases/1 million people/year), the number of patients with this particular disease found in that study (12) might be too low to precisely evaluate the efficacy of the OA/LA-rich diet on the disease conditions.
As well as being supplied exogenously as nutrients, unsaturated fatty acids are largely stored in membrane phospholipids and can be spatiotemporally released by phospholipase A2 (PLA2) enzymes. Among the PLA2 enzymes identified to date, the secreted PLA2 (sPLA2) family comprises the largest subgroup, containing 10 catalytically active isoforms. Individual sPLA2s show distinct tissue distributions and substrate preferences, thereby participating in diverse biological events in response to microenvironmental cues (13). sPLA2s have been implicated in cardiovascular diseases, including atherosclerosis and aortic aneurysm, as the plasma levels of group IIA sPLA2 (sPLA2-IIA), a prototypic inflammatory sPLA2, are correlated with atherosclerotic diseases (14–16), and genetic deletion of several sPLA2s or treatment with varespladib, a pan-sPLA2 inhibitor that broadly inhibits sPLA2s in the group I/II/V/X branch, ameliorates atherosclerosis or aortic aneurysm in mice (17–22), although conflicting evidence also exists (23). Importantly, a phase III clinical trial employing varespladib for treatment of patients with cardiovascular diseases failed to demonstrate efficacy and in fact suggested an increased level of risk (24), raising the possibility that some sPLA2s may have a protective rather than detrimental role in vascular diseases. Thus, the precise roles of sPLA2s and underlying lipid metabolism in aortic diseases remain controversial, and no report has examined the contributions of sPLA2s to aortic dissection.
In our continuing efforts to clarify the biological roles of sPLA2s using various sPLA2-knockout mouse strains in combination with comprehensive lipidomics (25–27), we herein show that mice lacking group V sPLA2 (sPLA2-V), which releases OA and LA preferentially (28, 29), frequently develop dissection in the thoracic ascending aorta, a feature that resembles aortic dissection in humans, shortly after AT-II infusion. sPLA2-V is the major sPLA2 isoform expressed in aortic endothelial cells (ECs), releasing OA and LA in response to AT-II stimulation and protecting against acute dissection of the thoracic ascending aorta in a manner dependent upon LOX-dependent ECM stabilization. We also show that the increased incidence of aortic dissection by endothelial sPLA2-V deficiency could be rescued by dietary supplementation with OA or LA, providing a rationale for the beneficial role of these fatty acids for cardiovascular health.
Results
Expression of sPLA2s in endothelial cells
Quantitative RT-PCR revealed that, among the nearly full set of sPLA2s, Pla2g2d, Pla2g2e, Pla2g3, and Pla2g5 (encoding sPLA2-IID, -IIE, -III, and -V, respectively) were substantially expressed in the aorta of C57BL/6 mice (male, 8–12 weeks old), Pla2g5 showing the highest expression (Fig. 1A). The expression level of Pla2g5 in the aorta was almost equivalent to that in the heart (Fig. 1B), a tissue previously shown to express this sPLA2 most abundantly (30, 31). The expression levels of Pla2g5 in anatomically distinct aortas, including thoracic ascending, thoracic descending, and abdominal aortas, were almost equal (Fig. S1A). Pla2g5 was also expressed in the inferior vena cava, where its expression level was less than half of that in the aortas (Fig. S1A). Immunohistochemical analysis showed that sPLA2-V protein was localized mainly in the EC lining of the aorta (Fig. 1C) as well as in the myocardium (Fig. S1B) in Pla2g5+/+ mice, whereas its staining was absent in Pla2g5−/− mice. To address the aortic expression of sPLA2-V further, we isolated ECs from aortic tissues using anti-CD31 magnetic beads (Fig. 1D). Consistent with the immunohistochemistry (Fig. 1C), Pla2g5 was expressed almost exclusively in the CD31-positive EC fraction (Fig. 1E). In contrast, the expression of Pla2g2d and Pla2g3 in the CD31-positive EC fraction was far lower than that of Pla2g5, and that of Pla2g2e was distributed mainly in the CD31-negative non-EC fraction (Fig. 1E). Thus, sPLA2-V is the major sPLA2 expressed in aortic ECs in mice. As in mouse ECs, PLA2G5 was the most abundant sPLA2 isoform in human ECs (Fig. S1C), where its expression level was higher than that in human vascular smooth muscle cells (VSMCs) and fibroblasts (Fig. S1D).
Figure 1.
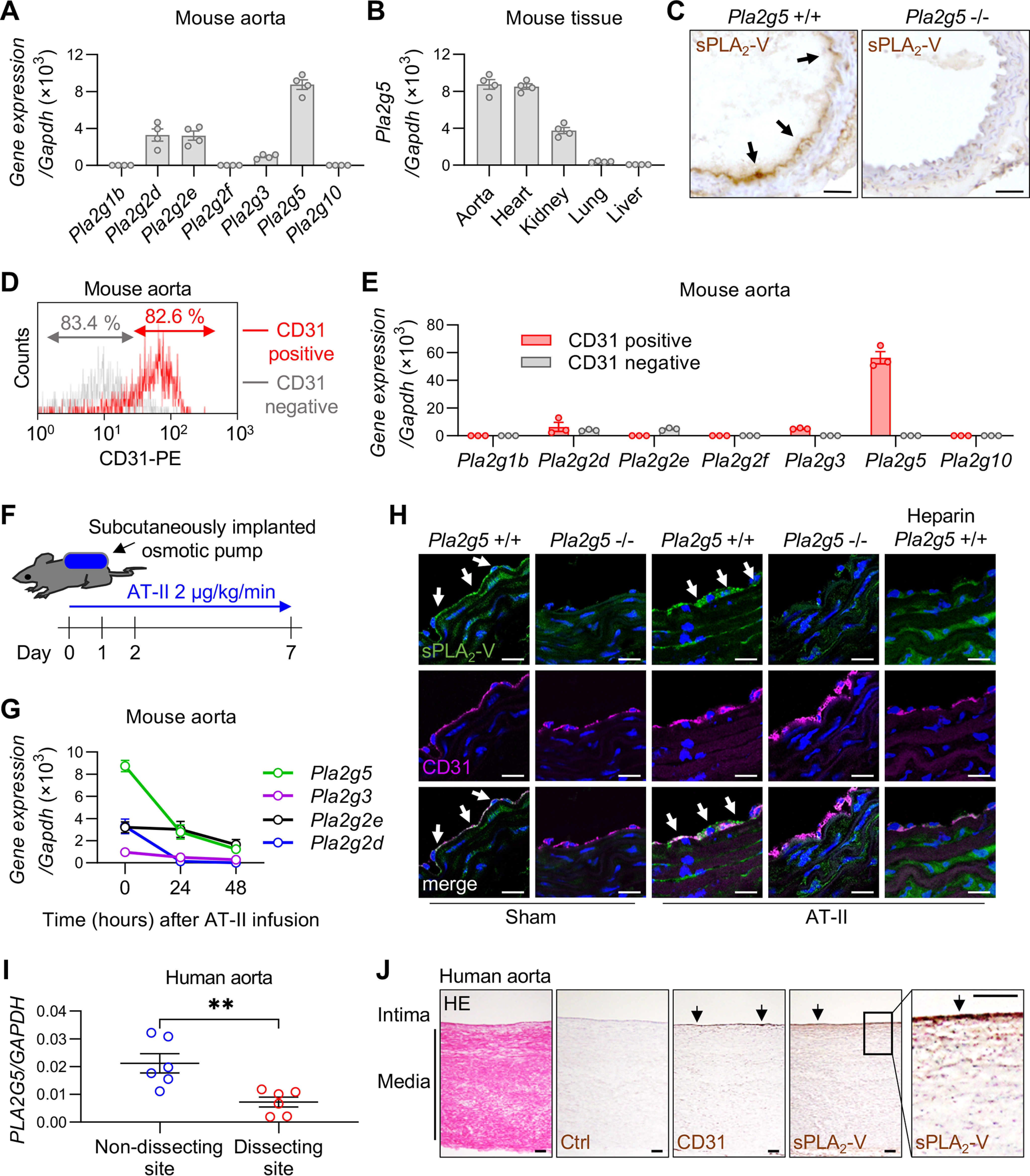
Expression of sPLA2s in the aortas of mice and humans. A, expression of sPLA2 mRNAs relative to Gapdh in the aorta of WT C57BL/6 mice (n = 4). B, expression of Pla2g5 in various tissues of WT mice (n = 4). C, immunohistochemistry of sPLA2-V in the aorta of Pla2g5+/+ and Pla2g5−/− mice. Arrows, positive staining of sPLA2-V. Scale bars, 100 μm. D, flow cytometry of CD31-positive/negative cells from WT mouse aorta. E, expression of sPLA2 mRNAs of CD31 positive/negative cells from WT mouse aorta (n = 3). F, schematic procedure of AT-II infusion into mice using subcutaneously implanted osmotic pumps. G, time course of the expression of sPLA2 mRNAs in WT mouse aorta after AT-II infusion (n = 4). H, immunofluorescence of sPLA2-V (green) and CD31 (magenta) with DAPI (blue) in the aorta of Pla2g5+/+ and Pla2g5−/− mice with or without AT-II infusion for 48 h. The rightmost panels represent the staining of Pla2g5+/+ aorta after AT-II infusion, following perfusion with heparinized saline through the left ventricle before extraction. Arrows indicate positive staining of sPLA2-V. Scale bars, 20 μm. I, expression of PLA2G5 in nondissecting or dissecting site of human aorta (n = 6). **, p < 0.01 by unpaired t test. J, a representative section of human aorta stained with HE and immunohistochemistry of the serial sections with control IgG (Ctrl), anti-CD31 antibody, or anti-sPLA2-V antibody. A boxed area is magnified on the right. Arrows, positive staining. Scale bars, 100 μm. Data are presented as mean ± S.E. (error bars) of the indicated number (n) of biological replicates.
Following AT-II infusion into mice (the procedure is illustrated schematically in Fig. 1F), the aortic expression of Pla2g5 mRNA as well as mRNAs for the other three sPLA2s (Pla2g2d, Pla2g2e, and Pla2g3) declined over 24–48 h (Fig. 1G). Confocal immunofluorescence microscopy confirmed that, prior to AT-II infusion, sPLA2-V protein was distributed mainly in aortic ECs, being colocalized with the signature EC marker CD31, in Pla2g5+/+ mice but not in Pla2g5−/− mice (Fig. 1H). Even after 48 h of AT-II infusion, at which time Pla2g5 mRNA expression was reduced (Fig. 1G), sPLA2-V protein was still present in the EC lining (Fig. 1H). Interestingly, the aortic staining of sPLA2-V was nearly abolished after infusion of heparin into the circulation (Fig. 1H), suggesting that sPLA2-V, a heparin-binding sPLA2 (32), is largely retained on the luminal surface of the aortic endothelium through association with heparan sulfate proteoglycans.
Consistent with the reduced Pla2g5 expression in the aortas of AT-II–infused mice (Fig. 1G), the expression of PLA2G5 in human aortas obtained by curative resection from six patients with aortic dissection, all of whom had undergone aortic replacement, was significantly lower in dissecting sites than in nondissecting sites (Fig. 1I). Immunohistochemical analysis of the nondissecting site of a patient, where the histology of the aortic wall was maintained, showed that sPLA2-V protein was localized mainly in ECs, which were also positive for CD31 in a serial section (Fig. 1J). Precise evaluation of sPLA2-V immunostaining in the dissecting site was difficult because of the collapsed histology. Nonetheless, expression of PLA2G5 was reduced in cultured human ECs after stimulation for 24 h with AT-II in a dose-dependent manner (Fig. S1E). Thus, although sPLA2-V mRNA expression in ECs is decreased after AT-II treatment in both mice and humans, its protein expression is maintained on EC surfaces likely through binding to heparan sulfate proteoglycans.
Increased AT-II–induced thoracic aortic dissection in Pla2g5−/− mice
To clarify the roles of sPLA2s in the pathophysiology of aortic dissection, we studied mice with knockout of the four sPLA2s that had been detected in the aorta. AT-II infusion has commonly been used to trigger aortic dissection in mice; however, these models typically require prolonged exposure to AT-II (>4 weeks) or use of elderly mice (>7 months old) or atherosclerosis-prone ApoE−/− mice, and the dissection takes place in the abdominal or thoracic descending, rather than thoracic ascending, aorta (4, 5, 33). Surprisingly, we found that Pla2g5−/− mice, even at a young age (8–12 weeks old), frequently developed aortic dissection after exposure to AT-II for a shorter period. Thus, when subjected to AT-II infusion only for 7 days, nearly half of Pla2g5−/− mice suffered from aortic dissection, which occurred mainly in the thoracic ascending aorta and often extended to the aortic arch (Fig. 2A). Some of the Pla2g5−/− mice died within 3 days due to rupture of the thoracic aorta, which was confirmed by an immediate autopsy showing intrathoracic hematoma accompanied by a tear of the aortic arch (Fig. 2B). Under the same conditions, only a few WT mice displayed a sign of aortic dissection, and none of them suffered sudden death due to aortic rupture. Histological examination of the lesions revealed dissection of the aortic wall with formation of intramural hematoma (as assessed by hematoxylin and eosin (HE) staining) and disruption of medial elastic fibers (as assessed by Elastica van Gieson (EVG) staining) in Pla2g5−/− mice (Fig. 2C). Thus, on day 7, the incidence of aortic dissection including rupture reached 44% (11 of 25) in Pla2g5−/− mice, whereas few or no aortic lesions were evident in Pla2g2d−/−, Pla2g2e−/−, Pla2g3−/−, or their littermate WT mice (Fig. 2D and Fig. S2 (A–C)). Although hypertension is a main causal factor for aortic dissection and rupture, systolic and diastolic blood pressures as well as heart rates were similarly elevated in both Pla2g5+/+ and Pla2g5−/− mice after 2 days of AT-II infusion (Fig. 2E), indicating that the increased incidence of aortic dissection caused by Pla2g5 deficiency was due to a mechanism other than elevated blood pressure.
Figure 2.
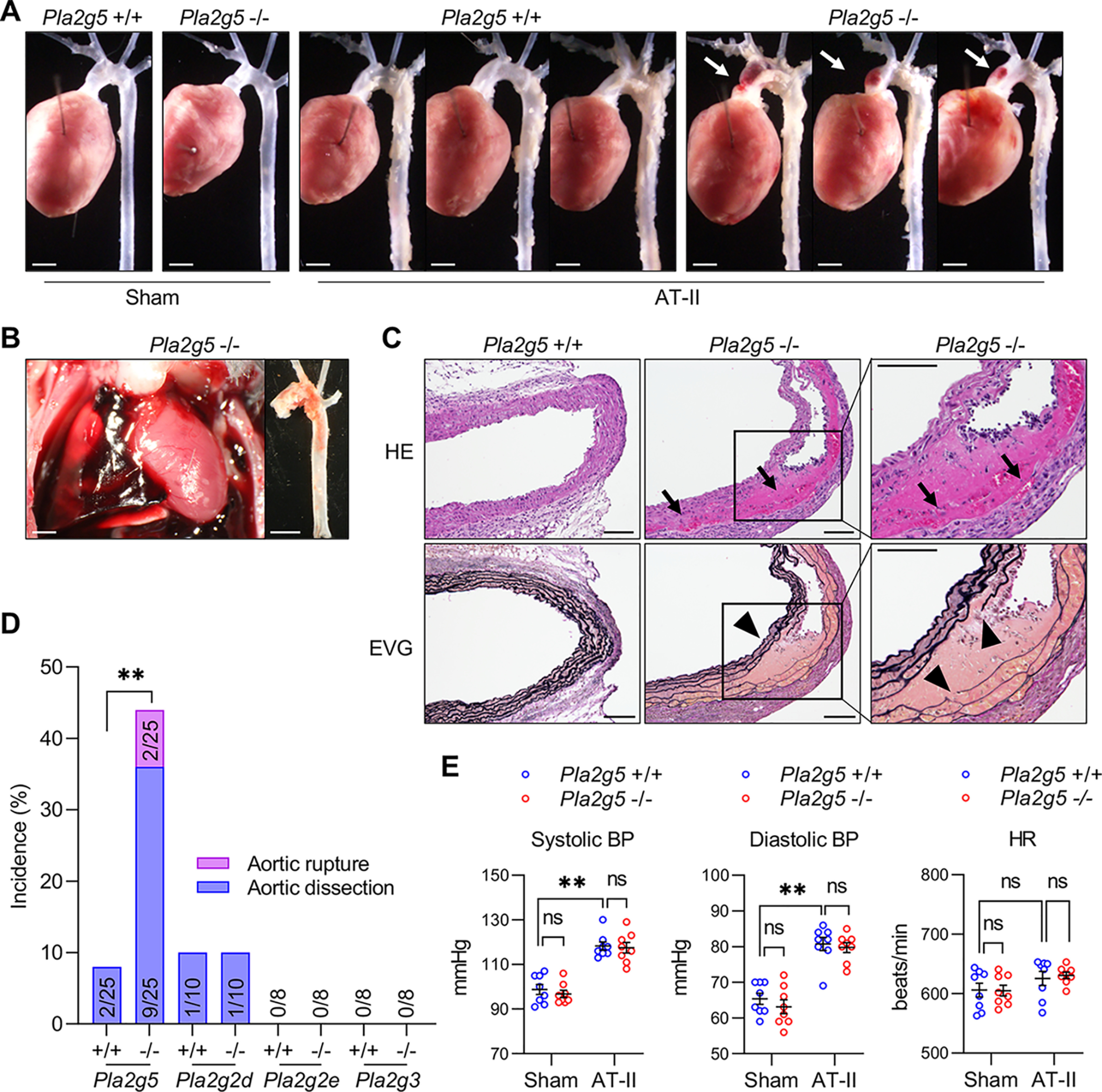
Increased AT-II-induced thoracic aortic dissection in Pla2g5−/− mice. A, representative thoracic aortas of Pla2g5+/+ and Pla2g5−/− mice after 7 days of AT-II infusion or sham control. Arrows indicate aortic dissection with intramural hematoma. Scale bars, 1 mm. B, representative intrathoracic hematoma and ruptured thoracic aorta of a Pla2g5−/− mouse after 3 days of AT-II infusion. Scale bars, 1 mm. C, representative sections of the thoracic aorta of Pla2g5+/+ and Pla2g5−/− mice stained with HE or EVG. Arrows indicate intramural hematoma, and arrowheads indicate dissection of the aortic wall accompanied by elastin fragmentation. Scale bars, 100 μm. Boxes are magnified on the right. D, incidence of thoracic aortic dissection or rupture in Pla2g5−/− (n = 25), Pla2g2d−/− (n = 10), Pla2g2e−/− (n = 8), Pla2g3−/− (n = 8), and littermate WT mice within 7 days of AT-II infusion. **, p < 0.01 by Fisher's exact test. E, systolic blood pressure (BP), diastolic blood pressure, and heart rate (HR) of Pla2g5+/+ and Pla2g5−/− mice with or without AT-II infusion for 2 days (n = 8). **, p < 0.01; ns, not significant by two-way ANOVA with Tukey's multiple-comparison test. Data are presented as mean ± S.E. of the indicated number (n) of biological replicates.
To elucidate the mechanism underlying the increased susceptibility of Pla2g5−/− mice to aortic dissection, we analyzed gene expression in the aortic tissues after 48 h of AT-II infusion, a time point just prior to dissection onset. Quantitative RT-PCR analysis of a panel of genes implicated in aortic stability, vascular remodeling, and inflammation showed that the AT-II–induced up-regulation of Lox mRNA, encoding a prototypic LOX that catalyzes the cross-linking of ECM proteins and thereby strengthens the aortic stability (34), was significantly attenuated in the aorta of Pla2g5−/− mice relative to Pla2g5+/+ mice (Fig. 3A). In contrast, expression of mRNAs for other LOX family members (LOX-like proteins; encoded by Loxl1–4) and vascular remodeling markers (Mmp2, Mmp9, Acta2, Col1a1, Col3a1, and Tgfb1) was similar between Pla2g5+/+ and Pla2g5−/− mice (Fig. 3A). The increased expression of pro-inflammatory cytokines (Il1b, Il6, and Tnf) in response to AT-II challenge was also not affected by Pla2g5 deficiency (Fig. 3A). Kinetic studies showed that the aortic expression of Lox in Pla2g5+/+ mice increased gradually over 48 h during AT-II infusion, whereas this response was significantly attenuated in Pla2g5−/− mice (Fig. 3B). AT-II–induced increases in aortic LOX protein expression (both the pro- and mature forms) (Figs. 3, C and D) and its activity (Fig. 3E) were also reduced in Pla2g5−/− mice relative to Pla2g5+/+ mice. Moreover, elevation of the insoluble/soluble collagen ratio, which reflects an increase of collagen cross-linking by LOX, after AT-II infusion was lower in Pla2g5−/− mice than in Pla2g5+/+ mice (Fig. 3F). After 48 h of AT-II infusion, immunofluorescence analysis using two different anti-LOX antibodies showed that LOX protein was increased mainly in medial VSMC layers beneath the CD31-positive EC layer in Pla2g5+/+ mice, whereas the AT-II–induced LOX staining was attenuated in Pla2g5−/− mice (Fig. 3G and Fig. S2D). These results suggest that Pla2g5 deficiency leads to aortic fragility due, at least in part, to reduced LOX induction in VSMCs and thereby insufficient ECM stability. Moreover, quantitative RT-PCR analysis using the aorta of aortic dissection patients (including samples of both dissecting and nondissecting sites) showed a positive correlation between PLA2G5 and LOX expressions (Fig. 3H), implying human relevance. In contrast, aortic MMP activities as assessed by gelatin zymography were similar between Pla2g5+/+ and Pla2g5−/− mice under both sham and AT-II infusion conditions (Figs. S2, E and F), suggesting that gelatinases, which can promote ECM degradation, did not profoundly affect the underlying phenotype.
Figure 3.
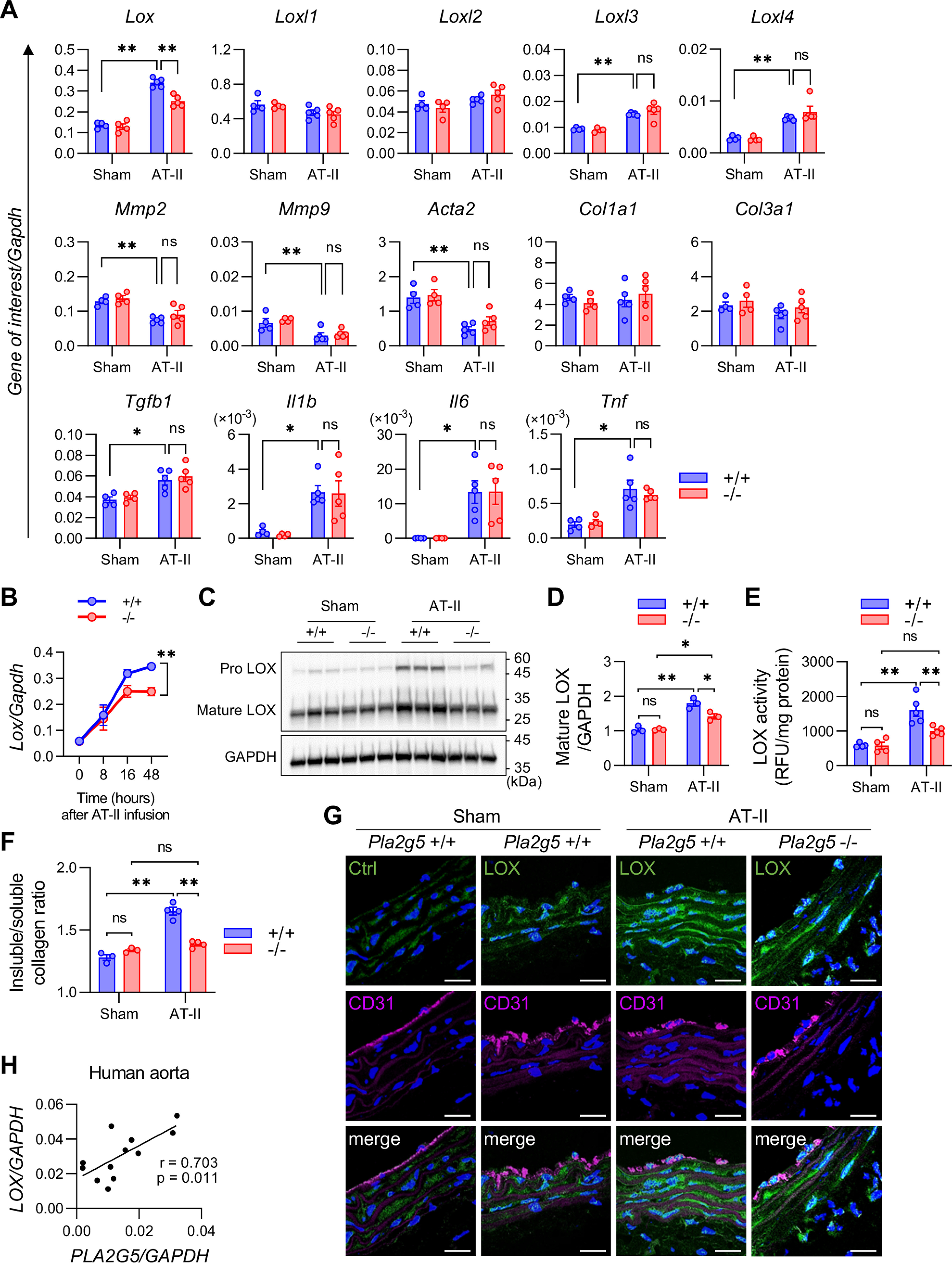
Decreased LOX expression and function in aorta of AT-II–infused Pla2g5−/− mice. A, mRNA expression of LOX family members, vascular remodeling markers, and pro-inflammatory cytokines in the aortas of Pla2g5+/+ and Pla2g5−/− mice with or without AT-II infusion for 48 h (n = 4–5). B, time course of Lox mRNA expression in the aortas of Pla2g5+/+ and Pla2g5−/− mice after AT-II infusion (n = 3–8). Shown are immunoblotting of LOX protein, with GAPDH as an internal control (C), densitometric analysis of mature LOX protein relative to GAPDH (n = 3) (D), LOX activity (n = 4–5) (E), and insoluble/soluble collagen ratio (n = 3–4) (F) in the aortas of Pla2g5+/+ and Pla2g5−/− mice with or without AT-II infusion for 48 h. *, p < 0.05; **, p < 0.01; ns, not significant by two-way ANOVA with Tukey's multiple-comparison test. Data are presented as mean ± S.E. (error bars) of the indicated number (n) of biological replicates (A, B, D, E, and F). G, immunofluorescence of aortas of Pla2g5+/+ and Pla2g5−/− mice with or without AT-II infusion for 48 h using control IgG (Ctrl), anti-LOX antibody (green), and anti-CD31 antibody (magenta) with DAPI (blue). Scale bars, 20 μm. H, correlation of PLA2G5 and LOX expression in human aorta (n = 12). Pearson correlation (r value) and statistical significance (p value) are indicated.
Administration of BAPN, an inhibitor of LOX as well as its homologs LOXL1–4, into mice blocks ECM cross-linking within the vascular wall to trigger aortic dissection (35). Among the LOX members, LOX is the major isoform that is responsible for ∼80% of total LOX activity in aortic VSMCs (36). Indeed, even without AT-II infusion, treatment with BAPN alone led to decreased survival of both Pla2g5+/+ and Pla2g5−/− mice over 2–4 weeks due to aortic rupture (Fig. S3, A–C). Moreover, in the presence of BAPN, AT-II–induced dissection and rupture of the thoracic and abdominal aortas occurred similarly in both genotypes (Fig. S3, D–F), where the systolic and diastolic blood pressures as well as heart rates were similarly elevated in both Pla2g5+/+ and Pla2g5−/− mice after AT-II infusion (Fig. S3G). Thus, the increased aortic instability in Pla2g5−/− mice relative to Pla2g5+/+ mice was bypassed if LOX was pharmacologically inactivated, further supporting the notion that LOX may work downstream of sPLA2-V to facilitate cross-linking of the ECM and thereby contribute to protection from aortic dissection.
Increased aortic dissection in EC-specific Pla2g5-null mice
To ascertain whether sPLA2-V expressed in ECs is indeed responsible for aortic stability, mice carrying a floxed allele of Pla2g5 (Pla2g5flox/flox) were crossed with Tie2-Cre transgenic (Tg) mice to obtain mice lacking sPLA2-V selectively in ECs (Pla2g5flox/flox; Tie2-Cre) (Fig. 4A). Expression of Pla2g5 in the aorta was blunted almost completely in Pla2g5flox/flox; Tie2-Cre mice compared with control Pla2g5flox/flox mice (Fig. 4B), confirming that Cre-mediated recombination efficiently ablated Pla2g5 expression in ECs. Pla2g5 expression was also reduced by about 40% in the heart (Fig. 4B), probably because of its specific ablation in coronary arterial ECs, but not in other cells such as cardiomyocytes (37). Importantly, as in the case of global Pla2g5−/− mice (Fig. 3), Pla2g5flox/flox; Tie2-Cre mice developed dissection of the thoracic aorta more frequently than control Pla2g5flox/flox mice within 7 days after the start of AT-II administration (Fig. 4, C and D). In addition, the induction of LOX at the mRNA (Fig. 4E), protein (Figs. 4, F and G), and activity (Fig. 4H) levels in the aorta after 48 h of AT-II infusion was significantly attenuated in Pla2g5flox/flox; Tie2-Cre mice relative to Pla2g5flox/flox mice. Expression levels of neither other LOX family members, vascular remodeling markers, nor inflammatory markers were affected by Pla2g5 deficiency (Fig. 4E and Fig. S4). After 48 h of AT-II infusion, immunofluorescence analysis showed that LOX protein was increased mainly in medial VSMC layers beneath the CD31-positive EC layer in Pla2g5flox/flox mice, whereas the AT-II–induced LOX staining was attenuated in Pla2g5flox/flox; Tie2-Cre mice (Fig. 4I). Thus, EC-specific deletion of sPLA2-V largely recapitulates its global deficiency. Taken together, these results suggest that sPLA2-V expressed in ECs has a protective role against AT-II–induced aortic dissection at least partly through increased expression of LOX in the aortic tissue.
Figure 4.
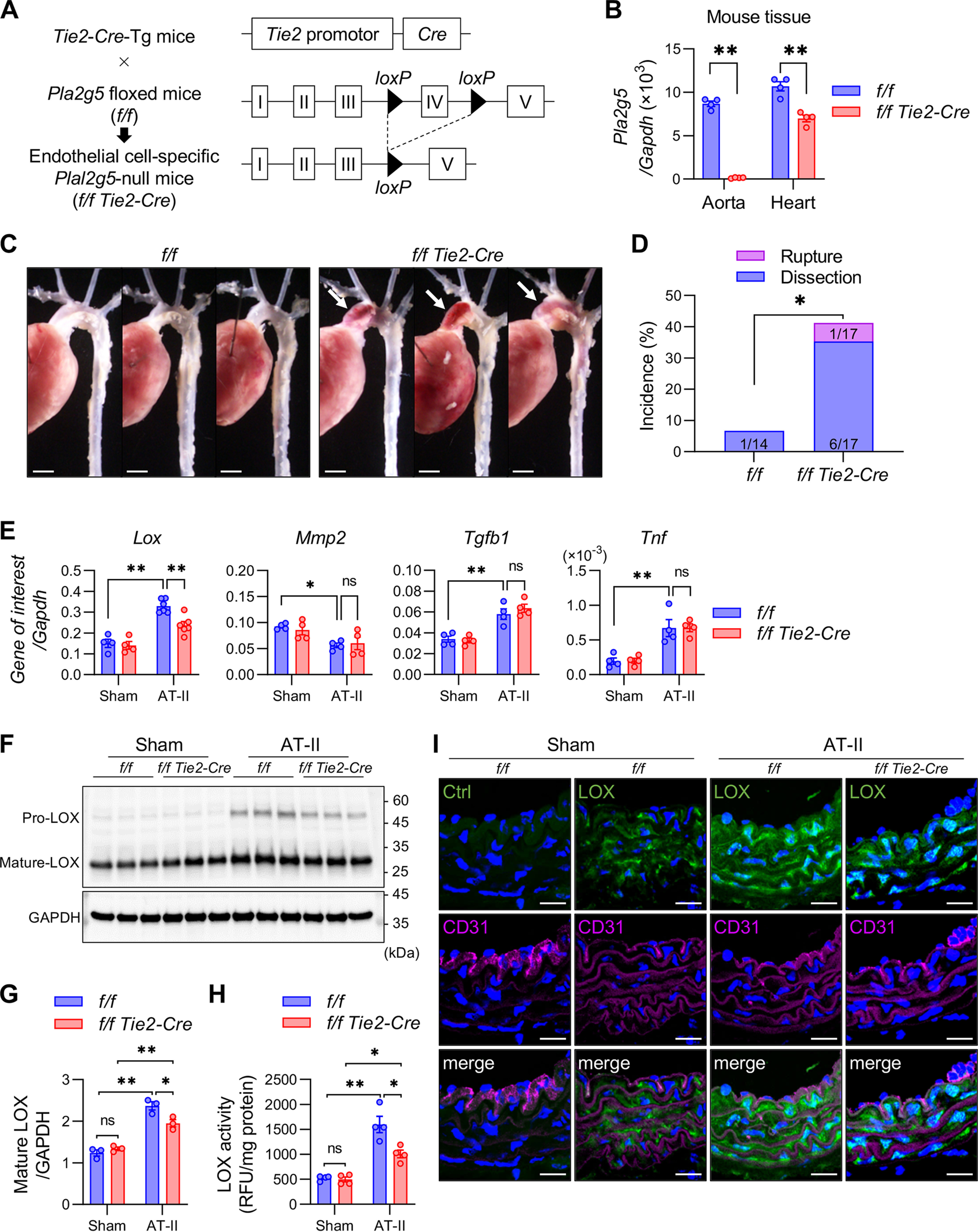
Increased aortic dissection in EC-specific Pla2g5-null mice. A, schematic procedure for generation of EC-specific Pla2g5-null mice. B, expression of Pla2g5 mRNA relative to Gapdh in the aorta and heart of control (f/f) and EC-specific Pla2g5-null (f/f Tie2-Cre) mice (n = 4). C, representative thoracic aortas of control and EC-specific Pla2g5-null mice after 7 days of AT-II infusion. Arrows indicate aortic dissection with intramural hematoma. Scale bars, 1 mm. D, incidence of thoracic aortic dissection or rupture in control (n = 14) and EC-specific Pla2g5-null (n = 17) mice within 7 days of AT-II infusion. E, mRNA expression of Lox, vascular remodeling markers (Mmp2, Acta2, and Tgfb1), and pro-inflammatory cytokine (Tnf) in the aortas of control and EC-specific Pla2g5-null mice with or without AT-II infusion for 48 h (n = 4–6). Shown are immunoblot analysis of LOX protein, with GAPDH as an internal control (F), densitometric analysis of mature LOX protein relative to GAPDH (n = 3) (G), and LOX activity (n = 4) (H) in the aortas of control and EC-specific Pla2g5-null mice with or without AT-II infusion for 48 h. I, immunofluorescence of aortas of control and EC-specific Pla2g5-null mice with or without AT-II infusion for 48 h using control IgG (Ctrl) or anti-LOX antibody (green) and anti-CD31 antibody (magenta) with DAPI (blue). Scale bars, 20 μm. *, p < 0.05; **, p < 0.01; ns, not significant by two-way ANOVA (B, E, G, and H) and by Fisher's exact test (D). Data are presented as mean ± S.E. (error bars) of the indicated number (n) of biological replicates.
Altered lipid profiles in Pla2g5−/− mice after AT-II infusion
Because sPLA2-V is a phospholipase, we reasoned that the aortic protection by sPLA2-V may involve some lipids mobilized by this enzyme. To identify such lipids (fatty acids, lysophospholipids, and their metabolites) that are potentially mobilized by sPLA2-V in mouse aorta, we performed lipidomics analysis by LC coupled with electrospray ionization-tandem MS (ESI-MS/MS) using aortic tissues after 12 h of AT-II infusion, at which time Pla2g5 expression was still high (Fig. 1G) and Lox expression was apparently increased (Fig. 3B) in Pla2g5+/+ mice.
A heat map summary of overall changes in free fatty acids, lysophospholipids, and their metabolites is shown in Fig. 5 (A–C), and quantitative values of the representative metabolites are shown in Fig. 5 (D–G). We found that OA (18:1) and LA (18:2) were markedly elevated in the aorta of Pla2g5+/+ mice following AT-II challenge, whereas these increases occurred only minimally in that of Pla2g5−/− mice (Figs. 5, A and D). Among the highly unsaturated fatty acids (HUFAs), including arachidonic acid (AA; 20:4), eicosapentaenoic acid (EPA; 20:5), and docosahexaenoic acid (DHA; 22:6), which were much less abundant than OA and LA, AA was also significantly reduced in AT-II–infused Pla2g5−/− mice relative to Pla2g5+/+ mice (Fig. 5, A and D). Likewise, several lysophospholipid species, including lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE) and to a lesser extent lysophosphatidic acid (LPA), lysophosphatidylinositol (LPI), and lysophosphatidylserine (LPS), all of which had an unsaturated or monounsaturated fatty acyl chain representing typical PLA2 reaction products, were also elevated in AT-II-treated Pla2g5+/+ mice, whereas these changes of lysophospholipids were substantially lower in Pla2g5−/− mice (Fig. 5, B and E). Although aortic expression of Pla2g4a (encoding cytosolic PLA2α (cPLA2α), a central regulator of AA release) was increased following AT-II infusion, it was not affected by the absence of sPLA2-V (Fig. S5A). These results suggest that sPLA2-V mobilizes OA and LA (and to a much lesser extent AA) from phosphatidylcholine (PC) and phosphatidylethanolamine (PE) in AT-II–stimulated, rather than unstimulated, aortic cells, being apparently consistent with the properties of this enzyme already reported in several other systems (28, 29, 38). Plasma levels of fatty acids and lysophospholipids were not affected by Pla2g5 deficiency regardless of AT-II infusion (Fig. S5, B and C). Among the HUFA metabolites, AT-II treatment increased the aortic production of several AA metabolites and related eicosanoids, among which only prostaglandin E2 (PGE2) was significantly lower in Pla2g5−/− mice than in Pla2g5+/+ mice (Fig. 5, C and F). In addition, lipoxin A4 (LXA4, an anti-inflammatory AA metabolite), as well as several metabolites in the 12/15-lipoxygenase pathway (12- and 15-hyrdroxytetraenoic acids (HETEs)), tended to be increased in AT-II–infused Pla2g5−/− mice relative to Pla2g5+/+ mice (Fig. 5, C and G), with concomitant increases in the expression of Alox12 and Alox15 (encoding 12- and 15-lipoxygenases, respectively) (Fig. S5D), which might reflect a secondary effect of aortic alterations caused by Pla2g5 deficiency.
Figure 5.
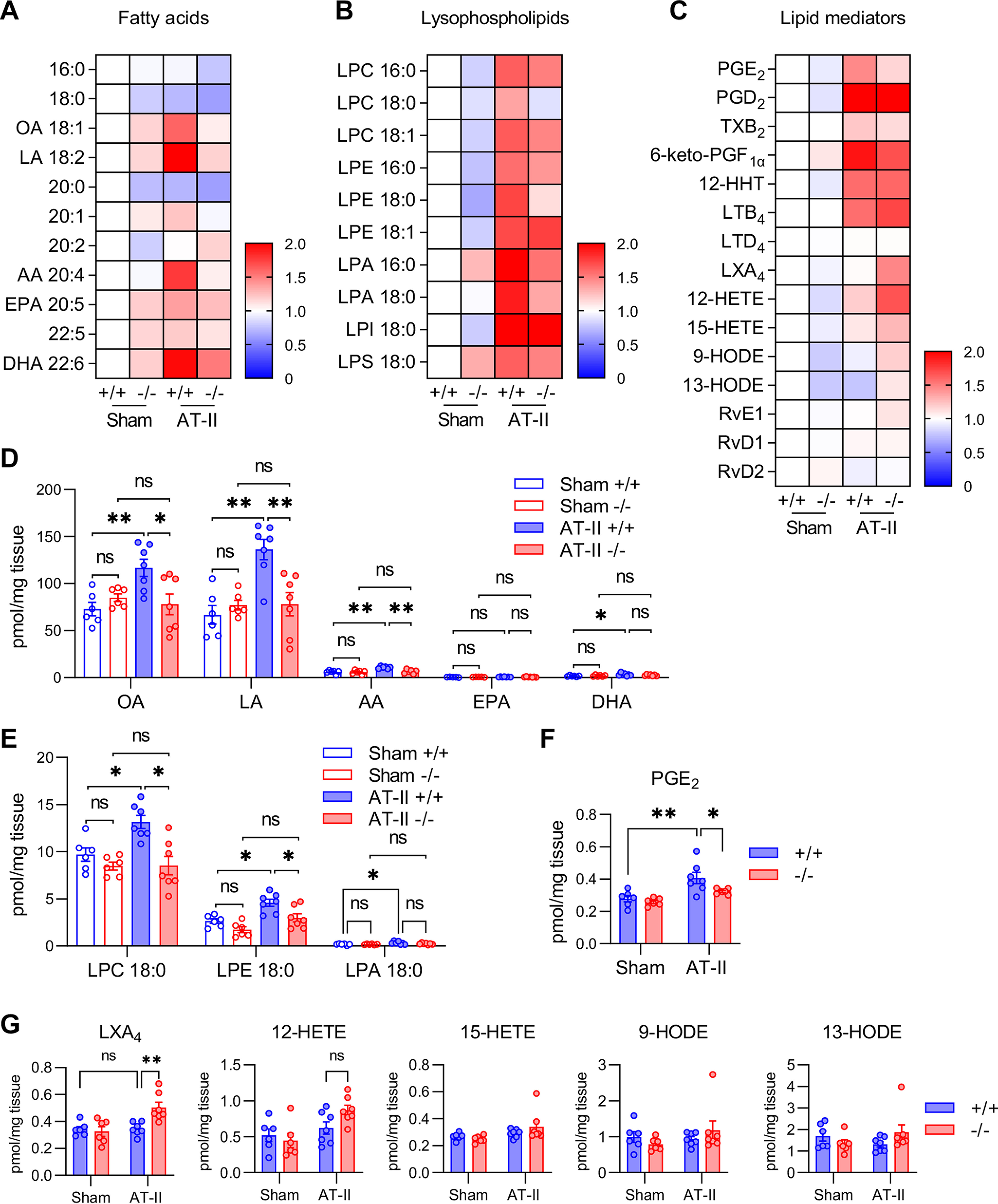
Altered lipid profiles in Pla2g5−/− mice after AT-II infusion. A–C, heat map representation of LC-ESI-MS/MS profiling of fatty acids (A), lysophospholipids (B), and lipid mediators (C) in the aortas of Pla2g5+/+ and Pla2g5−/− mice with or without AT-II infusion for 12 h. -Fold changes relative to sham Pla2g5+/+ are represented by color (n = 6–7). D–G, LC-ESI-MS/MS of unsaturated fatty acids (D), lysophospholipids (E), and HUFA-derived lipid mediators (F and G) presented as pmol/mg of tissue dry weight (n = 6–7). *, p < 0.05; **, p <0.01; ns, not significant by two-way ANOVA followed by Tukey's multiple-comparison test. Data are presented as mean ± S.E. (error bars) of the indicated number (n) of biological replicates.
We also monitored plasma lipoproteins and exosomes, whose phospholipids are potential targets of sPLA2-V, in Pla2g5+/+ and Pla2g5−/− mice. Although sPLA2-V alters the lipoprotein profile by cleaving OA- or LA-containing PC in LDL after its induction in hypertrophic adipocytes during obesity (28), the lipoprotein composition under normal dietary conditions was unaffected by Pla2g5 deficiency regardless of AT-II infusion (Fig. S5E). AT-II infusion increased the protein content and the number of plasma exosomes and decreased their sizes (Fig. S6, A–C). Although sPLA2-V is capable of hydrolyzing microvesicle membranes in vitro (39), plasma exosomes (in terms of protein content, number, and size) did not differ significantly between Pla2g5+/+ and Pla2g5−/− mice regardless of AT-II infusion (Fig. S6, A–C). Specifically, AT-II treatment increased various PE species and some PA species in plasma exosomes (Fig. S6, D–H). However, none of the phospholipid species (PC, PE, PA, PI, and PS) in plasma exosomes was affected by Pla2g5 deficiency with or without AT-II infusion (Figs. S6, D–H). It is thus likely that, in the context of aortic dissection, sPLA2-V retained on the endothelium hydrolyzes phospholipids mainly in membranes of AT-II-activated (or damaged) ECs, rather than those in circulating lipoproteins and exosomes, to release OA and LA.
OA and LA stabilize the aortic wall by up-regulating LOX expression
To address whether sPLA2-V–driven fatty acids or their metabolites could affect LOX expression, we utilized human umbilical vein ECs (HUVECs) and human aortic VSMCs in culture. Consistent with the results of immunohistochemistry (Fig. 3G and Fig. S3A), LOX mRNA was mainly expressed in VSMCs, rather than in ECs, regardless of AT-II stimulation (Fig. 6A). LOX expression in VSMCs was increased ∼4-fold after stimulation for 24 h with TGFβ1, a fibrogenic factor that is known to induce LOX (40) (Fig. 6B), whereas LOX expression in ECs was very low even after stimulation with TGFβ1 (Fig. 6B) or AT-II (Fig. 6A). Importantly, LOX expression in VSMCs was increased after culture with the conditioned medium of AT-II–stimulated but not control ECs, whereas this response was suppressed by a PLA2G5-specific siRNA, which blocked PLA2G5 expression in ECs (Fig. 6, C and D), or by the sPLA2 inhibitor varespladib (Fig. 6E). Although AT-II has been shown to activate cPLA2α (41, 42) and cross-talks between sPLA2-V and cPLA2α in both directions have been reported (43–45), the increased LOX expression in VSMCs after culture with the conditioned medium of AT-II–stimulated ECs was not suppressed by AACOCF3, a cPLA2α inhibitor (Fig. 6F), implying that EC-derived sPLA2-V contributes to LOX induction in VSMCs independently of cPLA2α. Furthermore, in the presence of TGFβ1, OA and LA, two major fatty acids released by sPLA2-V, dose-dependently augmented LOX expression in VSMCs (Fig. 6G) and LOX activity in culture supernatant of VSMCs (Fig. 6H), although these fatty acids alone were ineffective. The LOX-inducing effect of OA or LA was more remarkable when the cells were treated with a suboptimal concentration of TGFβ1 (Fig. 6I). We also found that TGFB1 was increased in ECs, but not in VSMCs, after stimulation for 24 h with AT-II (Fig. 6J and Fig S7 (A and B)). In addition, TGFB1 was also increased in VSMCs after stimulation with TGFβ1 itself (Fig. 6K). It is thus likely that AT-II stimulation of ECs induces TGFβ1, which in turn increases LOX and TGFβ1 expression in VSMCs in paracrine and autocrine manners. However, the expression of TGFB1 induced in both ECs and VSMCs (after stimulation with AT-II or TGFβ1, respectively) was not affected by OA or LA (Fig. 6, J and K).
Figure 6.
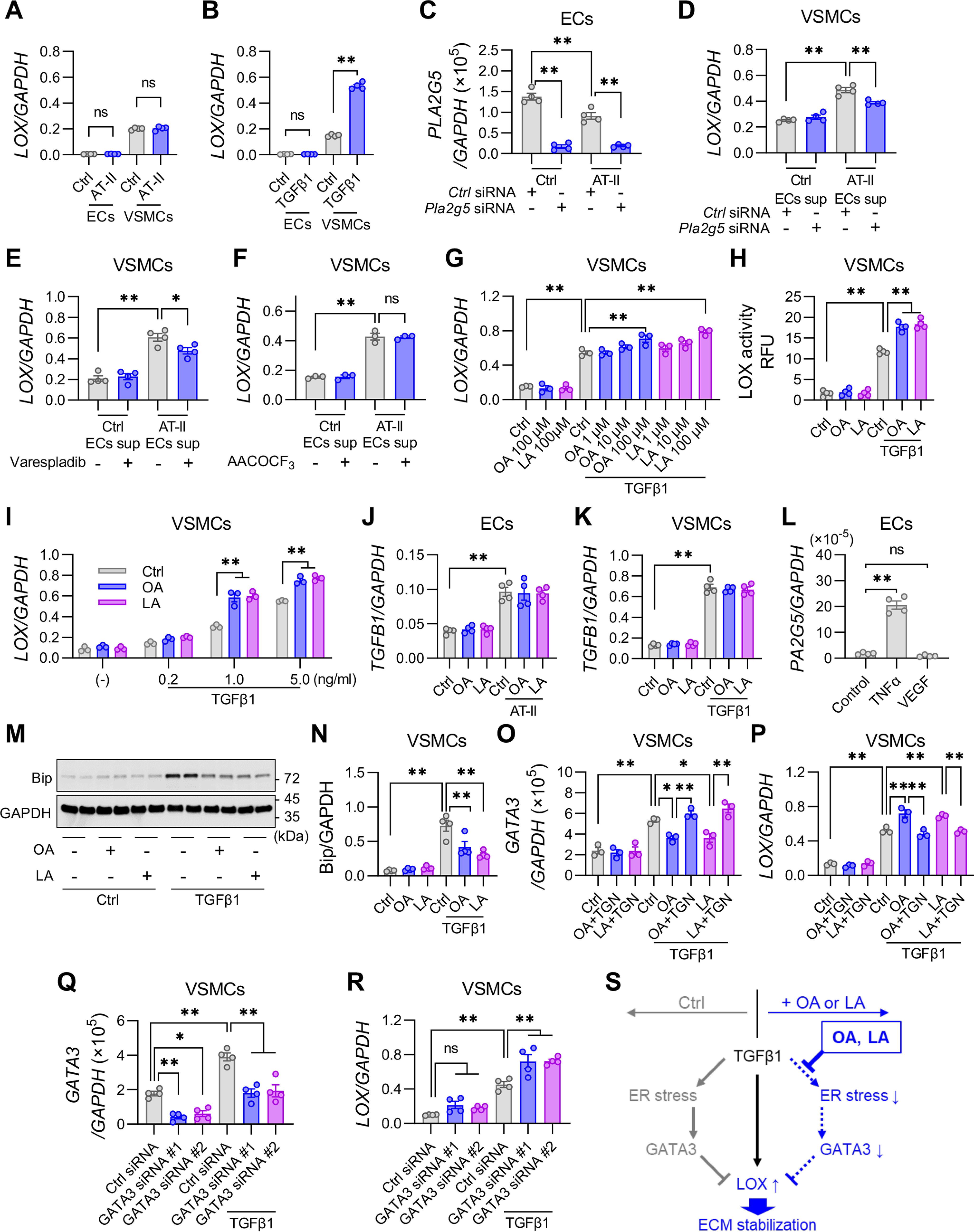
OA and LA attenuate ER stress and increase LOX expression in VSMCs. A, the expression of LOX relative to GAPDH in ECs and VSMCs treated for 24 h with or without AT-II (100 nm) (n = 4). B, the expression of LOX relative to GAPDH in ECs and VSMCs treated for 24 h with or without TGFβ1 (5 ng/ml) (n = 4). C, expression of PLA2G5 in ECs that were pretreated with PLA2G5-specific siRNA or nonsilencing negative control siRNA and then treated for 24 h with or without AT-II (n = 4). D–F, expression of LOX in VSMCs treated for 24 h with the supernatant (sup) of ECs. D, ECs were pretreated with a PLA2G5-specific or control siRNA and then treated for 24 h with or without AT-II (n = 4). E, ECs were treated for 24 h with or without AT-II in the presence or absence of 100 nm varespladib, a pan-sPLA2 inhibitor (n = 4). F, ECs were treated for 24 h with or without AT-II in the presence or absence of 10 μm AACOCF3, a cPLA2α inhibitor (n = 3). G, effect of OA or LA on the expression of LOX relative to GAPDH in VSMCs treated for 24 h with or without TGFβ1 (n = 3). H, LOX activity in supernatant of VSMCs treated for 24 h with or without TGFβ1 in the presence or absence of OA or LA (100 μm) (n = 4). I, expression of LOX in VSMCs treated for 24 h with or without TGFβ1 (0.2–5 ng/ml) in the presence or absence of OA or LA (100 μm) (n = 4). J, expression of TGFB1 relative to GAPDH in ECs treated for 24 h with or without AT-II (100 nm) in the presence or absence of OA or LA (100 μm) (n = 4). K, expression of TGFB1 in VSMCs treated for 24 h with or without TGFβ1 (5 ng/ml) in the presence or absence of OA or LA (100 μm) (n = 4). L, expression of PLA2G5 in ECs treated for 24 h with or without TNFα (10 ng/ml) or VEGF (10 ng/ml) (n = 4). Shown are immunoblot analysis of Bip protein, with GAPDH as an internal control (M), and densitometric analysis of Bip protein relative to GAPDH (n = 4) (N) in VSMCs treated for 24 h with or without TGFβ1 in the presence (+) or absence (−) of OA or LA (100 μm). O and P, effect of OA or LA on the expression of GATA3 (O) or LOX (P) in VSMCs treated for 24 h with or without TGFβ1 in the presence or absence of TGN (10 nm) (n = 3). Q and R, mRNA expression of GATA3 (Q) and LOX (R) in VSMCs that were pretreated with GATA3-specific siRNAs (#1 and #2) or control siRNA and then treated for 24 h with or without TGFβ1 (n = 4). S, a schematic summary of ECM stabilization by OA or LA in VSMCs. Whereas TGFβ1 induces LOX expression through canonical signaling pathways, it also induces ER stress leading to up-regulation of GATA3, which prevents LOX expression as a counter-regulatory arm. OA and LA counteract this process by attenuating TGFβ1-induced ER stress. *, p <0.05; **, p <0.01; ns, not significant by one-way ANOVA followed by Tukey's multiple-comparison test. Data are presented as mean ± S.E. (error bars) of the indicated number (n) of biological replicates.
In contrast to treatment of VSMCs with OA and LA, that with AA, EPA, or DHA decreased, rather than increased, the basal and TGFβ1-induced expression of LOX (Fig. S7C). Of the AA metabolites that were significantly altered in Pla2g5−/− mice (Fig. 5, F and G), PGE2 suppressed both basal and TGFβ1-induced LOX expression, whereas LXA4 had no effect (Fig. S7D). The two major sPLA2-V–driven lysophospholipids, LPC and LPE, had no effect on the basal and TGFβ1-induced LOX expression in VSMCs (Fig. S7, E and F). Collectively, OA and LA, rather than HUFA metabolites and lysophospholipids, have the ability to increase TGFβ1-driven LOX expression in VSMCs. This again argues against the involvement of cPLA2α, which mobilizes AA selectively (41, 42), in the sPLA2-V action in this situation.
In search of other factors that could affect sPLA2-V expression in ECs, we found that PLA2G5 was markedly up-regulated by TNFα, but not by VEGF (Fig. 6L). However, in the process of aortic dissection, the expression of Tnf was elevated (Fig. 3A), whereas that of Pla2g5 was conversely decreased (Fig. 1G), after AT-II treatment. Therefore, it appears that there is no causal relationship between TNFα and sPLA2-V in this disease. Conceivably, TNFα-induced sPLA2-V expression may occur during atherosclerosis, where the expressions of both sPLA2-V and TNFα are increased in the lesions (46).
It has been reported that TGFβ1 increases LOX expression through Smad and MAPK signaling (40). Indeed, phosphorylation of Smad2, Smad3, and p38 MAPK was increased in VSMCs after stimulation with TGFβ1, yet the addition of OA or LA did not influence their phosphorylation levels (Fig. S7G). As an alternative signaling pathway, TGFβ1 has the capacity to induce endoplasmic reticulum (ER) stress (47), and unsaturated fatty acids attenuate ER stress in general (48). Notably, the induction of various ER stress markers, including Bip protein (Fig. 6, M and N) as well as BIP, ATF4, CHOP, ERdj4, EDEM1, and PDIA2 mRNAs (Fig. S7H), after stimulation for 24 h with TGFβ1 was significantly reduced by OA or LA supplementation.
ER stress has been reported to induce the expression of the transcription factor GATA3 (49), which has an inhibitory effect on LOX expression (50). Indeed, expression of GATA3 was induced in VSMCs after stimulation for 24 h with TGFβ1 (Fig. 6O). This TGFβ1-induced expression of GATA3 was reduced by the addition of OA or LA, and this reduction was reversed by the addition of thapsigargin (TGN), another ER stress inducer, although TGN alone did not affect the baseline level of GATA3 expression (Fig. 6O). In the presence of TGFβ1, TGN canceled the LOX-inducing effect of OA and LA (Fig. 6P). Furthermore, GATA3-specific siRNAs, but not a nonsilencing control siRNA, which reduced both the baseline and the TGFβ1-induced GATA3 expression (Fig. 6Q), reciprocally increased the TGFβ1-induced LOX expression (Fig. 6R). These results suggest that suppression of ER stress by OA or LA attenuates the induction of GATA3, thereby promoting the expression of LOX (Fig. 6S).
To address whether the action of OA or LA would be mediated by fatty acid receptors, such as the nuclear receptor PPARγ and the G protein–coupled receptor GPR40/120, we examined the effects of their synthetic agonists (rosiglitazone (RSG) and GW9508, respectively) on LOX expression in VSMCs. Unlike OA and LA (see above), the PPARγ and GPR40/120 agonists failed to increase the basal and TGFβ1-induced expression of LOX (Figs. S7, I and J), suggesting that the action of OA and LA is independent of these fatty acid receptors.
Increased susceptibility of Pla2g5−/− mice to aortic dissection is rescued by OA- or LA-rich diet
Finally, to examine whether exogenous supply of OA and LA could alter the sensitivity of Pla2g5−/− mice to aortic dissection in vivo, Pla2g5−/− mice fed an olive oil–rich diet (containing a high level of OA) or corn oil–rich diet (containing a high level of LA) for 2 weeks were subjected to AT-II infusion. Plasma levels of OA and/or LA, but not other fatty acids such as 16:0 (palmitic acid), 18:0 (stearic acid), 20:4 (AA), 20:5 (EPA), and 22:6 (DHA), were increased equally in both Pla2g5+/+ and Pla2g5−/− mice fed an olive oil–rich or corn oil–rich diet compared with those fed a normal chow diet (Fig. S8A). Although LDL has been reported to down-regulate LOX (51), there were no differences in plasma levels of LDL and high-density lipoprotein, which were increased after feeding of an olive oil– or corn oil–rich diet, between both genotypes (Fig. S8B). Notably, under these conditions, Pla2g5−/− mice fed an olive oil– or corn oil–rich diet were fully protected from aortic dissection (Figs. 7, A and B). Furthermore, although aortic expression of Lox was lower and that of Gata3 was higher in Pla2g5−/− mice than in Pla2g5+/+ mice when a normal chow diet was supplied, the expression of both in Pla2g5−/− mice was fully restored to levels similar to those in Pla2g5+/+ mice after the olive oil– or corn oil–rich diet was supplied (Fig. 7, C and D). In contrast, Pla2g5−/− mice fed a cholesterol-rich, pro-atherogenic Western diet were as susceptible as those fed a chow diet to AT-II–induced aortic dissection (Fig. 7, A and B). Interestingly, aortic expression of Pla2g5 was increased in mice fed an olive oil–rich diet relative to those fed a chow diet (Fig. 7E), implying a feed-forward amplification loop of the sPLA2-V-OA pathway for the aortic stability. These findings collectively suggest that dietary supplementation of OA or LA could rescue the increased susceptibility of Pla2g5−/− mice to aortic dissection. Taken together, we conclude that aortic sPLA2-V preferentially mobilizes OA and LA, which stabilize the extracellular matrix in the aortic wall by augmenting the AT-II–induced up-regulation of LOX, thereby exerting a protective effect against aortic dissection (Fig. 7F).
Figure 7.
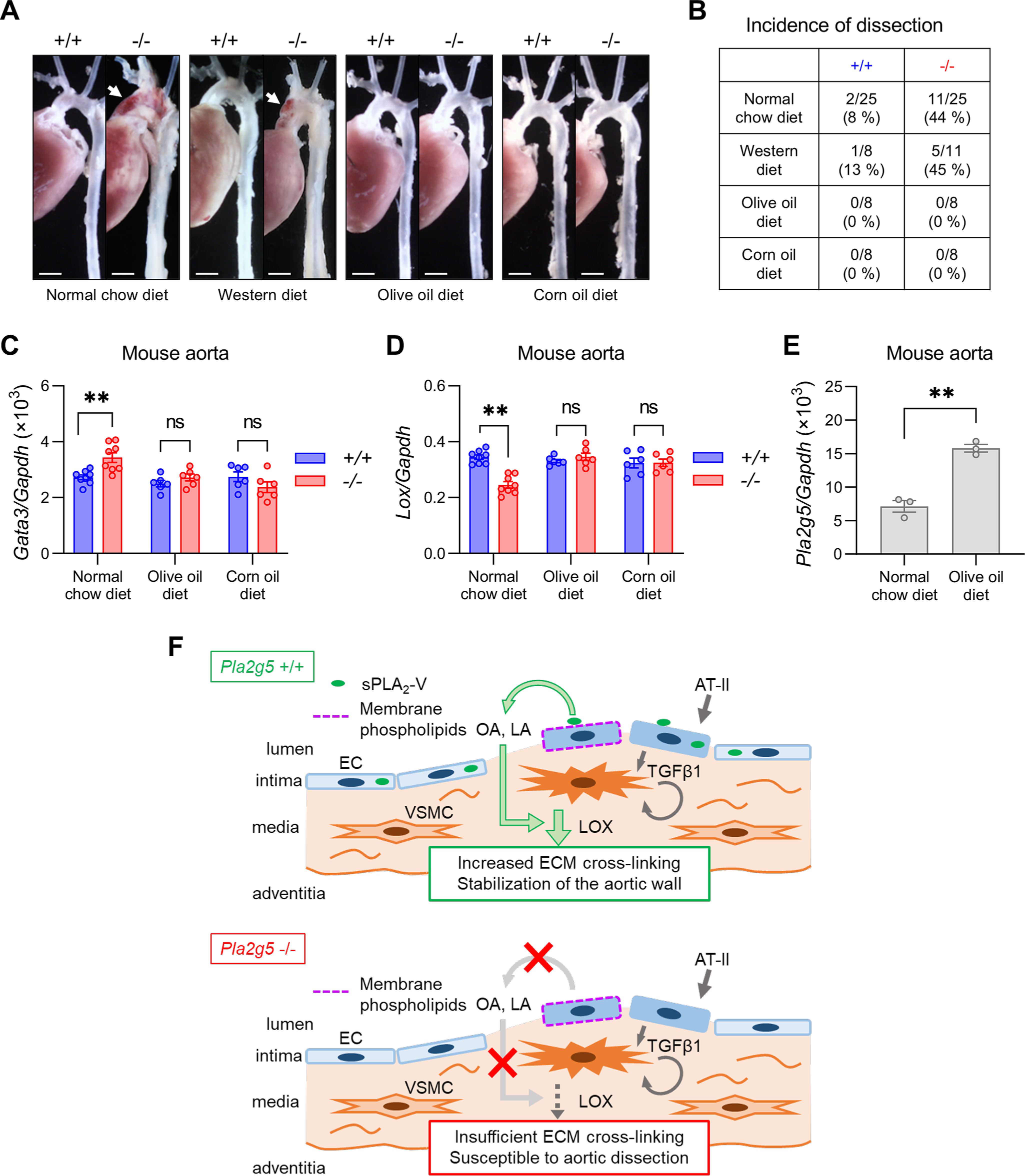
Exogenous supply of OA and LA protects against aortic dissection in Pla2g5−/− mice. A, representative photographs of thoracic aortas in Pla2g5+/+ and Pla2g5−/− mice fed a normal chow diet, a Western diet, an olive oil–rich diet, or a corn oil–rich diet after 7 days of AT-II infusion. Arrows, aortic dissection with intramural hematoma. Scale bars, 1 mm. B, incidence of thoracic aortic dissection or rupture in Pla2g5+/+ and Pla2g5−/− mice fed a normal chow diet (n = 25), a Western diet (n = 8–11), an olive oil–rich diet (n = 8), or a corn oil–rich diet (n = 8) within 7 days of AT-II infusion. C and D, mRNA expression of Lox (C) and Gata3 (D) in the aortas of Pla2g5+/+ and Pla2g5−/− mice after AT-II infusion for 48 h fed normal chow, an olive oil–rich diet, or a corn oil–rich diet (n = 6–8). *, p < 0.05; **, p < 0.01; ns, not significant by two-way ANOVA followed by Tukey's multiple-comparison test. E, Pla2g5 expression in the aorta of Pla2g5+/+ mice fed a normal chow or an olive oil–rich diet (n = 3). **, p < 0.01 by unpaired t test. Data are presented as mean ± S.E. (error bars) of the indicated number (n) of biological replicates. F, schematic diagram of the roles of endothelial sPLA2-V in aortic stabilization. sPLA2-V is a major sPLA2 isoform expressed in aortic ECs and is largely retained on the luminal surface of the aortic endothelium likely through binding to heparan sulfate proteoglycans. Endothelial sPLA2-V acts on membrane phospholipids of AT-II–activated ECs to mobilize OA and LA, which in turn promote AT-II–induced up-regulation of LOX that facilitates ECM cross-linking, thereby stabilizing the aortic wall. Impairment of this sPLA2-V–driven lipid pathway leads to increased susceptibility to aortic dissection.
Discussion
Because of the paucity of preceding clinical signs (except for hypertension) and high mortality associated with aortic dissection and rupture, effective biomarkers that can predict aortic fragility and/or therapeutic targets for stabilization of the aortic wall are needed to mitigate against this life-threatening disease. However, the mechanisms underlying aortic dissection have remained obscure, largely due to the lack of a suitable animal model that mimics the pathology of this disease in humans. It is generally recognized that a Mediterranean diet is beneficial for cardiovascular health (12, 52). Olive oil, a key component of the Mediterranean diet, contains abundant OA, which has been proposed to be associated with reduced risks of cardiovascular disease and mortality (53), although understanding of the beneficial effect of this monounsaturated fatty acid is still incomplete. Herein, as a part of our ongoing attempts to clarify the biological roles of sPLA2s using various sPLA2-knockout mouse strains, we have demonstrated for the first time that OA as well as LA, mobilized endogenously from phospholipids by endothelial sPLA2-V, plays a crucial role in stabilization of the aortic wall, thereby protecting against aortic dissection.
Previous studies have shown that sPLA2-V is expressed in cardiomyocytes, IL-4–driven M2 macrophages, bronchial epithelial cells, and obesity-associated adipocytes, where it plays offensive or protective roles in myocardial infarction, atherosclerosis, asthma, and diet-induced obesity in accordance with disease contexts (17, 19, 28, 29, 43, 54). sPLA2-V is also reportedly expressed in both mouse and human ECs (46, 55), and we have confirmed here that sPLA2-V is indeed distributed almost exclusively in CD31-positive ECs within the aortic tissue, the expression level being far higher than those of other sPLA2s. Thus, in addition to acting as a “metabolic sPLA2” or an “asthmatic (or Th2-prone) sPLA2” (28, 43), sPLA2-V can be regarded as a major “endothelial sPLA2” that controls vascular homeostasis and disease.
Reportedly, several (if not all) sPLA2s, including sPLA2-V, exhibit significant affinity for heparan sulfate proteoglycans on cell surfaces in vitro (32, 56), although in vivo evidence for this event has been lacking. We have demonstrated here that sPLA2-V is retained on the luminal surface of the aortic endothelium and washed out after infusion of heparinized saline into the circulation, providing the first in vivo evidence for the heparin-binding behavior of sPLA2-V. This view is reminiscent of sPLA2-IIA, another heparin-binding sPLA2 that is also released from heparan sulfate proteoglycans on EC surfaces into the circulation in heparinized patients (57). We previously reported that a heparin-binding sPLA2, which is immunoreactive with an anti-sPLA2-IIA antibody, is expressed in HUVECs (58), and based on the expression profile of sPLA2s in ECs both in vivo and in vitro, it is likely that this heparin-binding sPLA2 is attributed to sPLA2-V rather than sPLA2-IIA. Moreover, our results suggest that sPLA2-V retained on aortic ECs mobilizes OA and LA from phospholipids (PC and PE) in AT-II–infused, but not AT-II–untreated, aortic cells in the process of aortic dissection, which also appears to be consistent with in vitro evidence that several sPLA2s, including sPLA2-V, act preferentially on the membranes of activated or damaged, rather than resting, cells (32, 56). In fact, the release of OA and LA by sPLA2-V in aortic tissue is compatible with that in adipose tissue during diet-induced obesity (28) and in lung tissue during asthmatic inflammation (29) and is also consistent with the in vitro finding that it mobilizes OA from cultured cells (38). Thus, beyond the discovery of a novel role of this particular sPLA2 in the aorta, our present study has provided a rationale for the action mode of sPLA2 in an in vivo setting. Although sPLA2-V has the capacity to hydrolyze phospholipids in lipoproteins and microvesicles (e.g. exosomes) (28, 39), it is likely that the OA and LA released by sPLA2-V are derived mainly from the membranes of AT-II–activated ECs, because blood cells and plasma in the aortic lumen were flushed out before sample preparation and because there were no alterations in lipid compositions in plasma lipoproteins and exosomes in PLA2g5−/− mice under the experimental conditions employed here. Nonetheless, the possibility that a minor fraction of these unsaturated fatty acids is liberated from lipoproteins and/or exosomes by endothelial sPLA2-V cannot be fully ruled out and needs further elucidation. It is also possible that certain lipid molecules other than OA and LA would be additionally involved in the sPLA2-V action.
LOX-mediated ECM stabilization plays a protective role against acute aortic dissection (35). Reportedly, LOX expression is promoted by fibrogenic factors such as TGFβ1 (40), whereas this process is counter-regulated by the transcription factor GATA3 (50). We have shown here that OA and LA, which are preferentially released by sPLA2-V, attenuate ER stress, eventually leading to down-regulation of GATA3 and thereby up-regulation of LOX, thus revealing a functional interplay between phospholipid metabolism and ECM stabilization through the action of the endothelial sPLA2. Considering that ECM stabilization is also important for the prevention of atherosclerotic plaque rupture, a major trigger of myocardial infarction (59, 60), the present finding that sPLA2-V contributes to the stabilization of aortic ECM might account, at least partly, for the unfavorable result of the phase III clinical trial of varespladib, a pan-sPLA2 inhibitor, which failed to show efficacy for the treatment of patients with cardiovascular diseases and in fact increased the risk of myocardial infarction (24). Specifically, whereas varespladib prevented atherosclerosis and aneurysm in animal models (19), it would increase the risk of aortic dissection through inhibiting sPLA2-V and thus destabilize the vascular wall. Nonetheless, as varespladib inhibits various sPLA2 isoforms in the group I/II/V/X branch (14–16), we do not rule out the possibility that this agent might have blocked another cardioprotective sPLA2 isoform(s) or even another target(s) unrelated to sPLA2 in the clinical study.
In contrast to our present study, Boyanovsky et al. (21) previously reported that sPLA2-V promotes (rather than prevents) abdominal aortic aneurysm and rupture in response to AT-II infusion in mice with an ApoE−/− genetic background. ApoE−/− mice develop aortic dissection mainly in the supra-renal region of the abdominal aorta, where atherosclerotic lesions with chronic inflammation are evident at the origin of the celiac and mesenteric arteries as early as 6–8 weeks of age, and this may be followed by suprarenal abdominal aortic dissection in response to AT-II infusion (4, 5). Rosengren et al. (46) reported that sPLA2-V is localized in macrophages and VSMCs in the atherosclerotic regions of ApoE- and Ldlr-double knockout mice. Conceivably, under the abnormal lipid profile and vascular inflammation resulting from ApoE or Ldlr deficiency (61, 62), the pro-inflammatory effects of some lipid metabolites driven by sPLA2-V distributed in different cell types might become predominant, masking the protective effect of OA and LA mobilized by endothelial sPLA2-V, although details of the underlying mechanism require further elucidation. Importantly, dissection is generally more common in the ascending aorta than in other aortic regions in humans, probably because of continuous exposure to the systolic jet from the left ventricle (3), which was also observed in our Pla2g5−/− mouse model. In this regard, administration of AT-II to Pla2g5−/− mice, independently of ApoE deletion, can be regarded as a groundbreaking mouse model of aortic dissection, having considerable potential for screening of effective treatments and/or diagnostic markers of this condition. Interestingly, regardless of the different outcomes of aortic rupture, Boyanovsky et al. also reported that sPLA2-V increases the collagen content of atherosclerotic lesions (18), an event that seems to corroborate the LOX-inducing effect of sPLA2-V demonstrated in the present study, again implying the ECM-stabilizing function of sPLA2-V.
We have previously shown that sPLA2-V has an offensive, rather than protective, role in a model of myocardial ischemia-reperfusion injury (43). In that model, the coronary artery is artificially ligated to induce myocardial injury. However, considering the pathological situation, myocardial infarction is induced by the occlusion due to plaque rupture in the coronary artery. The plaque rupture could be suppressed by the sPLA2-V-LOX axis that may stabilize the fibrous cap (extracellular matrix) of the plaque. In the ischemia-reperfusion model, however, the protective effect of the sPLA2-V-LOX axis may be bypassed by the artificial ligation of the coronary artery. Also, in the ischemia-reperfusion model, the contribution of sPLA2-V expressed abundantly in cardiomyocytes and possibly in macrophages to the disease should be taken into account.
Last, it appears that our present findings have some relationships with previous studies describing the potential roles of sPLA2s in cardiovascular homeostasis and diseases. Bernatchez et al. (63) reported that sPLA2-V is expressed in HUVECs and involved in the synthesis of platelet-activating factor (a lysophospholipid-derived mediator) in response to VEGF, although we failed to observe the induction of sPLA2-V by VEGF, and the role of sPLA2-V in platelet-activating factor generation during aortic dissection remains to be elucidated. Consistent with our present study, Sonoki et al. demonstrated that sPLA2-V is induced by TNFα and produces LPC in HUVECs (64) and also showed that an AT-II blocker inhibits TNFα-induced sPLA2-V expression (65). It seems unlikely, however, that there is a functional interplay between sPLA2-V and TNFα in aortic dissection, as described here. It has been shown that sPLA2-IIA, when transgenically overexpressed in macrophages, induces collagen deposition in the atherosclerotic plaque (66), that AT-II induces the expression of sPLA2-IIA in VSMCs (67), and that sPLA2-IIA activates the LOX-mediated collagen pathway in myofibroblasts in the process of cardiac remodeling (68). These findings may indicate some functional redundancy between sPLA2-V and -IIA in the context of cardiovascular disorders, although aortic dissection, arteriosclerosis, and cardiac remodeling are essentially different pathologies, and a direct comparison may not be appropriate.
In summary, our present study has revealed a novel lipid-driven mechanism controlling aortic stability, perturbation of which can be linked to aortic fragility and dissection. Central to this, sPLA2-V plays a novel physiological role as a specific sPLA2 expressed spatiotemporally in aortic ECs by endogenously mobilizing the unsaturated fatty acids OA and LA as critical regulators of aortic LOX expression and thereby ECM stabilization. Our findings provide valuable insights into the functional link between phospholipid metabolism and aortic stabilization, thus contributing to the future development of novel diagnostic and/or therapeutic strategies for aortic dissection. Furthermore, our study might open a potential mechanism underlying the beneficial impact of the olive oil–rich (i.e. OA-rich) Mediterranean diet on cardiovascular health. Because there has been no clinical evidence for the relationship between corn oil–rich (i.e. LA-rich) diet and aortic dissection, the actual effect of corn oil–rich diet on this disease in humans should be carefully investigated in a future study.
Experimental procedures
Mice
Pla2g5−/−, Pla2g2d−/−, and Pla2g2e−/− mice, backcrossed onto a C57BL/6 background for more than 12 generations, were described previously (25, 28, 43). Heterozygous Pla2g3+/− mice (C57BL/6 × 129Sv) were backcrossed to the C57BL/6 background for three generations, and then male and female heterozygotes were intercrossed to obtain Pla2g3−/− mice and littermate Pla2g3+/+ mice (25, 69). Mice expressing Cre recombinase under control of the Tie2 gene promoter (Tie2-Cre transgenic mice) on the C57BL/6 background were purchased from Jackson Laboratory. Mice with a floxed allele in the Pla2g5 gene (Pla2g5flox/flox) (43) were crossed with Tie2-Cre Tg mice to generate EC-specific Pla2g5-null mice (Pla2g5flox/flox; Tie2-CreTg/+) and their littermate controls (Pla2g5flox/flox; Tie2-Cre+/+) (Fig. 4A). In all experiments, only male mice from 8 to 12 weeks of age (weight: 25–28 g) were used. All mice were housed in climate-controlled facilities at 25 °C with a 12-h light-dark cycle, with free access to standard laboratory food (CE2, CLEA) and water. All procedures were performed in accordance with approvals from the institutional animal care and use committees of the University of Yamanashi, the Tokyo Metropolitan Institute of Medical Science, and the University of Tokyo.
Animal models
Acute aortic dissection in mice was induced by subcutaneous infusion of AT-II (A9525, Sigma–Aldrich) for up to 7 days using a micro-osmotic pump (1007D, Alzet). Mice were anesthetized with inhaled isoflurane, and then micro-osmotic pumps were implanted through an incision in the midscapular region under sterile conditions. AT-II was dissolved in PBS and infused at 2 µg/kg/min. Sham animals underwent an identical surgical procedure, except that osmotic pumps containing PBS alone were implanted. As required, BAPN (A3134, Sigma–Aldrich), a LOX inhibitor, was administered for 3 days prior to AT-II treatment and continued for an additional 7 days concurrent with AT-II infusion. BAPN was dissolved in drinking water at a dose of 250 mg/100 ml or 500 mg/100 ml ad libitum. In some experiments, 8-week-old male mice were fed an OA-rich, LA-rich, or Western diet (Research Diets) for 2 weeks. The compositions of these diets are given in Table S1. Mice were observed at least twice a day, and an autopsy was performed immediately if any mice died during AT-II infusion. To harvest aortic tissues, mice were anesthetized with inhaled isoflurane, and then the abdominal and thoracic cavities were opened ventrally. After perfusion with saline through the left ventricle of the heart to remove blood, the aortic tissue was harvested carefully under a stereo microscope (SZ61, Olympus), as described previously (22).
Measurement of blood pressure
Blood pressure in mice was measured by a noninvasive tail-cuff method using a blood pressure device (BP-98A, Softron). Mice went through daily sessions of unrecorded measurements for several days to acclimate them to the procedure. Blood pressure was recorded before and 2 days after AT-II administration. For each mouse, an average of six consecutive measurements were taken.
Quantitative RT-PCR
Total RNA was extracted from tissues or cells using TRIzol reagent (Thermo Fisher Scientific). First-strand cDNA synthesis was performed using a ReverTra Ace qPCR RT Kit (Toyobo). PCRs were carried out using a TaqMan Gene Expression System (Applied Biosystems) on the 7500 Real-Time PCR System (Applied Biosystems). The probe and primer sets used are listed in Table S2.
Histological analysis
Aortas from mice were fixed in 4% (w/v) paraformaldehyde and embedded in paraffin, and then 5-µm-thick serial sections were prepared for HE or EVG staining. Digital images were acquired using a light microscope (BX41, Olympus).
Immunoblotting
Aortic tissues were isolated and homogenized in radioimmune precipitation buffer (Thermo Fisher Scientific) containing protease inhibitor mixture (Thermo Fisher Scientific) using TissueLyser II (Qiagen) with 7-mm stainless beads (Qiagen). The homogenates were centrifuged at 14,000 × g for 30 min at 4 °C, and the supernatants were collected for immunoblotting. Tissue homogenates or cell lysates (10-µg protein equivalents) were subjected to SDS-PAGE on 10% (w/v) gels under reducing conditions. Protein concentrations were determined with a Bradford assay kit (Thermo Fisher Scientific). The separated proteins in the gels were electroblotted onto polyvinylidene difluoride membranes with a semi-dry blotter (Trans-Blot-Turbo, Bio-Rad). After blocking with polyvinylidene difluoride–blocking reagent (NYPBR01, Toyobo), the membranes were probed with primary antibodies at 1:2,000 dilution in Signal Enhancer HIKARI Solution A (02272-74, Nacalai Tesque) at room temperature for 2 h. Primary antibodies used in this study were as follows: rabbit antibody against LOX (ab174316) was from Abcam; rabbit antibody against Bip (#3177), p-Smad2 (#3108), Smad-2 (#5339), p-Smad3 (#9520), Smad3 (#9523), p-p38 MAPK (#4511), p38 MAPK (#8690), p-Akt (#4056), Akt (#9272), and GAPDH (#2118) were from Cell Signaling Technology. Then the membranes were then washed with TBS-T, followed by incubation with horseradish peroxidase–conjugated anti-rabbit IgG (NA934, GE Healthcare) at 1:20,000 dilution in Signal Enhancer HIKARI Solution B (02297-64, Nacalai Tesque) for 1 h and then visualized using ECL Prime system (RPN2236, GE Healthcare). Images were visualized by LAS-4000 (GE Healthcare), and densitometric analysis was carried out using ImageJ software (National Institutes of Health). The whole blots are shown in Fig. S9.
Measurement of soluble/insoluble collagen
The amounts of soluble and insoluble (cross-linked) collagen in aortic tissues were measured using a Sircol collagen assay kit (S1000, S2000, Biocolor) in accordance with the manufacturer's protocol. Briefly, freeze-dried aortic tissues were weighed and incubated in dilute acid/pepsin solution with rotation overnight at 4 °C. The extracts were centrifuged, and the supernatants were collected as the cold acid/pepsin-soluble fraction. The insoluble fraction was extracted by digestion in acid solution at 65 °C for 3 h. The amounts of soluble and insoluble collagen were determined by a colorimetric reaction using Sircol dye (Sirius red dye). Collagen cross-linking was assessed as the ratio of insoluble to soluble collagen.
LOX activity assay
Aortic tissues were isolated and homogenized in 500 µl of 6 m urea in 0.02 m borate buffer (pH 8.0) using TissueLyser II (Qiagen) with 7-mm stainless beads. The homogenates were centrifuged at 14,000 × g for 30 min at 4 °C, and the supernatants were collected for the LOX activity assay. The activity was assessed using a lysyl oxidase activity assay kit (ab112139, Abcam) in accordance with the manufacturer's instructions and expressed as relative fluorescence units/mg of protein.
Gelatin zymography
Gelatinase activity in aortic tissue lysates was determined using a gelatin zymography kit (AK45, Primary Cell) in accordance with the manufacturer's instructions. Briefly, aortic tissues were homogenized in radioimmune precipitation buffer (Thermo Fisher Scientific) using TissueLyser II with 7-mm stainless beads and centrifuged at 14,000 × g for 30 min, and the supernatants were collected for the assay. Total protein (2.5-µg equivalent) was loaded onto gelatin-impregnated polyacrylamide gels and electrophoresed under nonreducing conditions. Then the gels were incubated in the enzymatic reaction buffer for 24 h at 37 °C. After incubation, the gels were stained with Coomassie Brilliant Blue, and then destained in methanol/acetate solution. The gels were scanned, and densitometric analysis was performed using ImageJ software.
Immunohistochemistry
Mouse or human aortic tissues were, snap-frozen in OCT compound and then sliced into 10-µm-thick sections. These sections were fixed with 4% paraformaldehyde for 5 min and then incubated with rabbit anti-sPLA2-V polyclonal antibody (ab23709, Abcam), rabbit anti-LOX mAb (ab174316, Abcam), rabbit anti-LOX polyclonal antibody (ab31238, Abcam), rat anti-mouse CD31 mAb (550247, BD Biosciences), mouse anti-human CD31 mAb (M0823, Agilent), or rabbit anti-human sPLA2-V polyclonal antibody, which did not cross-react with other sPLA2s (70), or control antibody overnight at 4 °C. The sections were then treated with horseradish peroxidase–conjugated secondary antibody (Histofine Simple Stain MAX PO, Nichirei Biosciences) and visualized with diaminobenzidine (Vector Laboratory), followed by counterstaining with hematoxylin. For immunofluorescence, the sections treated with primary antibodies were incubated with secondary antibodies, including goat anti-rabbit IgG conjugated with Alexa Fluor 488 and goat anti-rat IgG conjugated with Alexa Fluor 594 (Thermo Fisher Scientific). Fluorescence images were acquired by a confocal microscope (FV-1000, Olympus).
Magnetic cell sorting (MACS) and flow cytometry analysis
Aortic tissues from five mice were excised, minced in Dulbecco's modified Eagle's medium (DMEM) (Thermo Fisher Scientific), and digested in 1 mg/ml collagenase type II (Worthington) solution with shaking for 30 min at 37 °C. The cell suspensions were then passed through 70-µm nylon cell strainers (Corning), and the cells were washed with MACS buffer (0.5% BSA and 2 mm EDTA in PBS). To isolate ECs, CD31 MicroBeads (130-097-418, Miltenyi Biotec) were used in accordance with the manufacturer's protocol. RNA extracted from the cells was taken for quantitative RT-PCR. For flow cytometry analysis, the cells were stained with phycoerythrin-conjugated anti-mouse CD31 antibody (130-102-608, Miltenyi Biotec) and analyzed using a cell analyzer (EC800, Sony Biotechnology). Dead cells were excluded using the viability dye Zombie Aqua (eBioscience).
LC-ESI-MS/MS
All procedures were performed as described previously (25, 71, 72). In brief, for detection of fatty acid oxygenated metabolites, lipids were extracted by using Sep-Pak C18 cartridges (Waters). Tissues were soaked in 10 volumes of methanol and homogenized with a Polytron homogenizer. After overnight incubation at −20 °C, water was added to the homogenates to give a final methanol concentration of 10% (v/v). The samples in 10% methanol were applied to the cartridges, washed with 10 ml of hexane, eluted with 3 ml of methyl formate, dried under N2 gas, and dissolved in 60% methanol. As internal standards for determination of recovery, 50 pmol of d4-labeled PGE2 (Cayman Chemicals) was added to the samples. The analysis of fatty acid metabolites was performed using a 4000Q-TRAP quadrupole linear ion trap hybrid mass spectrometer (AB Sciex) with reverse-phase LC (LC-30AD; Shimadzu) combined with autosampler (SIL-30ACMP; Shimadzu). The sample was applied to a Kinetex C18 column (2.1 × 150 mm, 1.7-µm particle; Phenomenex) coupled for ESI-MS/MS. The samples injected by the autosampler (10 µl) were directly introduced and separated by a step gradient with mobile phase A (water containing 0.1% acetic acid) and mobile phase B (acetonitrile/methanol = 4:1 (v/v)) at a flow rate of 0.2 ml/min and a column temperature of 45 °C.
For detection of fatty acids, phospholipids, and lysophospholipids, tissues were soaked in 10 volumes of 20 mm Tris-HCl (pH 7.4) and then homogenized with a Polytron homogenizer. Lipids were extracted by the method of Bligh and Dyer (73) and subjected to ESI-MS/MS using a 4000Q-TRAP and LC-30AD with a Kinetex C18 column, as described previously (71). As an internal standard, 50 pmol of d5-labeled EPA (Cayman Chemicals), LPC (17:0), and PE (14:0-14:0) (Avanti Polar Lipids) were added to each sample. The samples were separated by a step gradient with mobile phase A (acetonitrile/methanol/water = 1:1:1 (v/v/v) containing 5 µm phosphoric acid and 1 mm ammonium formate) and mobile phase B (2-propanol containing 5 µm phosphoric acid and 1 mm ammonium formate) at a flow rate of 0.2 ml/min at 50 °C.
Identification was conducted using multiple-reaction monitoring transition and retention times, and quantification was performed based on peak area of the multiple-reaction monitoring transition and the calibration curve obtained using an authentic standard for each lipid (Table S3) (27, 71).
Evaluation of circulating exosomes
Mouse plasma exosomes were isolated using a total exosome isolation kit (4484450, Thermo Fisher Scientific) in accordance with the manufacturer's instructions. Analysis of particle numbers and sizes of plasma exosomes was performed by Theoria Science using a NanoSight nanoparticle-tracking system.
Analyses of plasma lipoproteins
Analysis of mouse plasma lipoproteins was performed by LipoSearch (Skylight Biotech).
Cell culture studies
HUVECs (EA.hy926), obtained from ATCC (CRL-2922), were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 50 µg/ml penicillin/streptomycin (P/S). Human aortic VSMCs (T/G HA-VSMCs), obtained from ATCC (CRL-1999), were cultured in Ham's F-12K medium (Thermo Fisher Scientific) supplemented with 10% FBS, 50 µg/ml P/S, 0.05 mg/ml ascorbic acid, 0.01 mg/ml insulin, 0.01 mg/ml transferrin, 10 ng/ml sodium selenite, and 0.03 mg/ml endothelial cell growth supplement (E2759, Sigma–Aldrich). Human fibroblasts (WI-38), obtained from ATCC (CCL-75), were cultured in DMEM supplemented with 10% FBS and 50 µg/ml P/S. For treatment, the cells were starved in serum-free DMEM containing 0.5% (w/v) fatty acid–free BSA (015-23871, Wako) for 24 h and then treated with or without 5 ng/ml TGFβ1, 10 ng/ml TNFα, or 10 ng/ml VEGF (all from R&D Systems) for 24 h. As required, the cells were treated with 1–100 µm OA, LA, AA, EPA, or DHA, 1 µm PGE2, 1 µm LXA4, 1–100 µm LPC (18:0), or 0.1–10 µm LPE (18:0) (Avanti Polar Lipids); 10 nm TGN, 0.1–10 µm RSG, or 0.1–10 µm GW9508 (GPR40/120 agonist) (all from Cayman Chemicals); 100 nm varespladib (Anthera Pharmaceuticals); or 10 µm AACOCF3 (Cayman Chemicals). Fatty acids were mixed with fatty acid–free BSA at a 3.3:1 molar ratio and then diluted to the required concentration with culture media. Fatty acid-free BSA alone was used as an experimental control. In the gene knockdown experiments, ECs or VSMCs were transfected with Pla2g5 siRNA (assay ID s10597), GATA3 siRNA (assay ID 43432 or 106675, Thermo Fisher Scientific), or negative control siRNA (AM4611, Thermo Fisher Scientific) using Lipofectamine RNAiMAX (13778030, Thermo Fisher Scientific), in accordance with the manufacturer's instructions.
Human samples
Aortic tissues were obtained by curative resection from six patients (Asian, male sex, aged from 58 to 78 years), all of whom underwent aortic graft replacement surgically at the Yamanashi University Hospital (Yamanashi, Japan) from April 16, 2014 to December 31, 2019. The study was approved by the Ethics Committees of Yamanashi University (approval number 1801), with written informed consent from all patients. All studies involving human samples abide by the Declaration of Helsinki principles.
Statistical analysis
Results are expressed as mean ± S.E. Univariate comparison was performed with Fisher's exact test for categorical data. Differences between two groups were assessed by using an unpaired t test. Analysis of more than two groups was carried out either by one-way ANOVA or two-way ANOVA followed by Tukey post hoc comparison test. Survival curves were calculated by the Kaplan–Meier method and compared by the log-rank test. The association between variables was examined using Pearson correlation. GraphPad Prism (version 8.3.1) software was used to conduct statistical analyses.
Data availability
Data supporting the findings of this work are available within the paper and the supporting information. The raw data will be provided by the corresponding author upon request.
Supplementary Material
This article contains supporting information.
Author contributions—K. W. and M. M. conceptualization; K. W. resources; K. W., Y. T., and Y. M. data curation; K. W. formal analysis; K. W. and M. M. funding acquisition; K. W. validation; K. W., Y. T., and Y. M. investigation; K. W. methodology; K. W. writing-original draft; K. K. and M. M. supervision; M. M. writing-review and editing.
Funding and additional information—This work was supported by AMED-CREST Grant JP18gm0710006 and FORCE Grant JP19gm4010005 from the Japan Agency for Medical Research and Development (to M. M.) and Japan Society for the Promotion of Science KAKENHI Grants JP15H05905, JP16H02613, and JP18H05025 (to M. M.) and 25860591, 16K19436, and 19K07502 (to K. W.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- AT-II
- angiotensin II
- AA
- arachidonic acid
- BAPN
- β-aminopropionitrile
- DHA
- docosahexaenoic acid
- ECM
- extracellular matrix
- EC
- endothelial cell
- HUVEC
- human umbilical vein EC
- EPA
- eicosapentaenoic acid
- ER
- endoplasmic reticulum
- EVG
- Elastica van Gieson
- HE
- hematoxylin and eosin
- HETE
- hydroxytetraenoic acid
- HUFA
- highly unsaturated fatty acid
- LA
- linoleic acid
- ESI
- electrospray ionization
- LOX
- lysyl oxidase
- LPA
- lysophosphatidic acid
- LPC
- lysophosphatidylcholine
- LPE
- lysophosphatidylethanolamine
- LPI
- lysophosphatidylinositol
- LPS
- lysophosphatidylserine
- LXA4
- lipoxin A4
- MACS
- magnetic cell sorting
- MMP
- matrix metalloproteinase
- OA
- oleic acid
- PA
- phosphatidic acid
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PI
- phosphatidylinositol
- PS
- phosphatidylserine
- PGE2
- prostaglandin E2
- PLA2
- phospholipase A2
- sPLA2
- secreted PLA2
- sPLA2-V
- group V sPLA2
- sPLA2-IIA
- group IIA sPLA2
- cPLA2α
- cytosolic PLA2α
- TGN
- thapsigargin
- RSG
- rosiglitazone
- VSMC
- vascular smooth muscle cell
- p-
- phosphorylated
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- P/S
- penicillin/streptomycin
- ANOVA
- analysis of variance
- DAPI
- 4′,6-diamidino-2-phenylindole
- MAPK
- mitogen-activated protein kinase
- LDL
- low-density lipoprotein.
References
- 1. Nienaber C. A., and Clough R. E. (2015) Management of acute aortic dissection. Lancet 385, 800–811 10.1016/S0140-6736(14)61005-9 [DOI] [PubMed] [Google Scholar]
- 2. Mussa F. F., Horton J. D., Moridzadeh R., Nicholson J., Trimarchi S., and Eagle K. A. (2016) Acute aortic dissection and intramural hematoma: a systematic review. JAMA 316, 754–763 10.1001/jama.2016.10026 [DOI] [PubMed] [Google Scholar]
- 3. Nienaber C. A., Clough R. E., Sakalihasan N., Suzuki T., Gibbs R., Mussa F., Jenkins M. P., Thompson M. M., Evangelista A., Yeh J. S., Cheshire N., Rosendahl U., and Pepper J. (2016) Aortic dissection. Nat. Rev. Dis. Primers 2, 16053 10.1038/nrdp.2016.71 [DOI] [PubMed] [Google Scholar]
- 4. Saraff K., Babamusta F., Cassis L. A., and Daugherty A. (2003) Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 1621–1626 10.1161/01.ATV.0000085631.76095.64 [DOI] [PubMed] [Google Scholar]
- 5. Trachet B., Aslanidou L., Piersigilli A., Fraga-Silva R. A., Sordet-Dessimoz J., Villanueva-Perez P., Stampanoni M. F. M., Stergiopulos N., and Segers P. (2017) Angiotensin II infusion into ApoE−/− mice: a model for aortic dissection rather than abdominal aortic aneurysm? Cardiovasc. Res. 113, 1230–1242 10.1093/cvr/cvx128 [DOI] [PubMed] [Google Scholar]
- 6. Coady M. A., Rizzo J. A., and Elefteriades J. A. (1999) Pathologic variants of thoracic aortic dissections. Penetrating atherosclerotic ulcers and intramural hematomas. Cardiol. Clin. 17, 637–657 10.1016/S0733-8651(05)70106-5 [DOI] [PubMed] [Google Scholar]
- 7. Son B. K., Sawaki D., Tomida S., Fujita D., Aizawa K., Aoki H., Akishita M., Manabe I., Komuro I., Friedman S. L., Nagai R., and Suzuki T. (2015) Granulocyte macrophage colony-stimulating factor is required for aortic dissection/intramural haematoma. Nat. Commun. 6, 6994 10.1038/ncomms7994 [DOI] [PubMed] [Google Scholar]
- 8. Guo D. C., Regalado E. S., Gong L., Duan X., Santos-Cortez R. L., Arnaud P., Ren Z., Cai B., Hostetler E. M., Moran R., Liang D., Estrera A., Safi H. J., Leal S. M., et al. (2016) LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ. Res. 118, 928–934 10.1161/CIRCRESAHA.115.307130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee V. S., Halabi C. M., Hoffman E. P., Carmichael N., Leshchiner I., Lian C. G., Bierhals A. J., Vuzman D., Mecham R. P., Frank N. Y., and Stitziel N. O. (2016) Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc. Natl. Acad. Sci. U.S.A. 113, 8759–8764 10.1073/pnas.1601442113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kurihara T., Shimizu-Hirota R., Shimoda M., Adachi T., Shimizu H., Weiss S. J., Itoh H., Hori S., Aikawa N., and Okada Y. (2012) Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation 126, 3070–3080 10.1161/CIRCULATIONAHA.112.097097 [DOI] [PubMed] [Google Scholar]
- 11. Kimura T., Shiraishi K., Furusho A., Ito S., Hirakata S., Nishida N., Yoshimura K., Imanaka-Yoshida K., Yoshida T., Ikeda Y., Miyamoto T., Ueno T., Hamano K., Hiroe M., Aonuma K., et al. (2014) Tenascin C protects aorta from acute dissection in mice. Sci. Rep. 4, 4051 10.1038/srep04051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Estruch R., Ros E., Salas-Salvadó J., Covas M. I., Corella D., Arós F., Gómez-Gracia E., Ruiz-Gutiérrez V., Fiol M., Lapetra J., Lamuela-Raventos R. M., Serra-Majem L., Pintó X., Basora J., Muñoz M. A., et al. (2018) Primary prevention of cardiovascular disease with a Mediterranean diet supplemented with extra-virgin olive oil or nuts. N. Engl. J. Med. 378, e34 10.1056/NEJMoa1800389 [DOI] [PubMed] [Google Scholar]
- 13. Murakami M., Sato H., Miki Y., Yamamoto K., and Taketomi Y. (2015) A new era of secreted phospholipase A2. J. Lipid Res. 56, 1248–1261 10.1194/jlr.R058123.1015.1.test [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kugiyama K., Ota Y., Takazoe K., Moriyama Y., Kawano H., Miyao Y., Sakamoto T., Soejima H., Ogawa H., Doi H., Sugiyama S., and Yasue H. (1999) Circulating levels of secretory type II phospholipase A2 predict coronary events in patients with coronary artery disease. Circulation 100, 1280–1284 10.1161/01.cir.100.12.1280 [DOI] [PubMed] [Google Scholar]
- 15. Kugiyama K., Ota Y., Sugiyama S., Kawano H., Doi H., Soejima H., Miyamoto S., Ogawa H., Takazoe K., and Yasue H. (2000) Prognostic value of plasma levels of secretory type II phospholipase A2 in patients with unstable angina pectoris. Am. J. Cardiol. 86, 718–722 10.1016/S0002-9149(00)01069-9 [DOI] [PubMed] [Google Scholar]
- 16. Mallat Z., Steg P. G., Benessiano J., Tanguy M. L., Fox K. A., Collet J. P., Dabbous O. H., Henry P., Carruthers K. F., Dauphin A., Arguelles C. S., Masliah J., Hugel B., Montalescot G., Freyssinet J. M., et al. (2005) Circulating secretory phospholipase A2 activity predicts recurrent events in patients with severe acute coronary syndromes. J. Am. Coll. Cardiol. 46, 1249–1257 10.1016/j.jacc.2005.06.056 [DOI] [PubMed] [Google Scholar]
- 17. Bostrom M. A., Boyanovsky B. B., Jordan C. T., Wadsworth M. P., Taatjes D. J., de Beer F. C., and Webb N. R. (2007) Group V secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Arterioscler. Thromb. Vasc. Biol. 27, 600–606 10.1161/01.ATV.0000257133.60884.44 [DOI] [PubMed] [Google Scholar]
- 18. Boyanovsky B., Zack M., Forrest K., and Webb N. R. (2009) The capacity of group V sPLA2 to increase atherogenicity of ApoE−/− and LDLR−/− mouse LDL in vitro predicts its atherogenic role in vivo. Arterioscler. Thromb. Vasc. Biol. 29, 532–538 10.1161/ATVBAHA.108.183038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fraser H., Hislop C., Christie R. M., Rick H. L., Reidy C. A., Chouinard M. L., Eacho P. I., Gould K. E., and Trias J. (2009) Varespladib (A-002), a secretory phospholipase A2 inhibitor, reduces atherosclerosis and aneurysm formation in ApoE−/− mice. J. Cardiovasc. Pharmacol. 53, 60–65 10.1097/fjc.0b013e318195bfbc [DOI] [PubMed] [Google Scholar]
- 20. Zack M., Boyanovsky B. B., Shridas P., Bailey W., Forrest K., Howatt D. A., Gelb M. H., de Beer F. C., Daugherty A., and Webb N. R. (2011) Group X secretory phospholipase A2 augments angiotensin II-induced inflammatory responses and abdominal aortic aneurysm formation in apoE-deficient mice. Atherosclerosis 214, 58–64 10.1016/j.atherosclerosis.2010.08.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boyanovsky B. B., Bailey W., Dixon L., Shridas P., and Webb N. R. (2012) Group V secretory phospholipase A2 enhances the progression of angiotensin II-induced abdominal aortic aneurysms but confers protection against angiotensin II-induced cardiac fibrosis in apoE-deficient mice. Am. J. Pathol. 181, 1088–1098 10.1016/j.ajpath.2012.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe K., Fujioka D., Saito Y., Nakamura T., Obata J. E., Kawabata K., Watanabe Y., Mishina H., Tamaru S., Hanasaki K., and Kugiyama K. (2012) Group X secretory PLA2 in neutrophils plays a pathogenic role in abdominal aortic aneurysms in mice. Am. J. Physiol. Heart Circ. Physiol. 302, H95–H104 10.1152/ajpheart.00695.2011 [DOI] [PubMed] [Google Scholar]
- 23. Ait-Oufella H., Herbin O., Lahoute C., Coatrieux C., Loyer X., Joffre J., Laurans L., Ramkhelawon B., Blanc-Brude O., Karabina S., Girard C. A., Payré C., Yamamoto K., Binder C. J., Murakami M., et al. (2013) Group X secreted phospholipase A2 limits the development of atherosclerosis in LDL receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 33, 466–473 10.1161/ATVBAHA.112.300309 [DOI] [PubMed] [Google Scholar]
- 24. Nicholls S. J., Kastelein J. J., Schwartz G. G., Bash D., Rosenson R. S., Cavender M. A., Brennan D. M., Koenig W., Jukema J. W., Nambi V., Wright R. S., Menon V., Lincoff A. M., Nissen S. E., and VISTA-16 Investigators (2014) Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA-16 randomized clinical trial. JAMA 311, 252–262 10.1001/jama.2013.282836 [DOI] [PubMed] [Google Scholar]
- 25. Miki Y., Yamamoto K., Taketomi Y., Sato H., Shimo K., Kobayashi T., Ishikawa Y., Ishii T., Nakanishi H., Ikeda K., Taguchi R., Kabashima K., Arita M., Arai H., Lambeau G., et al. (2013) Lymphoid tissue phospholipase A2 group IID resolves contact hypersensitivity by driving antiinflammatory lipid mediators. J. Exp. Med. 210, 1217–1234 10.1084/jem.20121887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taketomi Y., Ueno N., Kojima T., Sato H., Murase R., Yamamoto K., Tanaka S., Sakanaka M., Nakamura M., Nishito Y., Kawana M., Kambe N., Ikeda K., Taguchi R., Nakamizo S., et al. (2013) Mast cell maturation is driven via a group III phospholipase A2-prostaglandin D2-DP1 receptor paracrine axis. Nat. Immunol. 14, 554–563 10.1038/ni.2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamamoto K., Miki Y., Sato M., Taketomi Y., Nishito Y., Taya C., Muramatsu K., Ikeda K., Nakanishi H., Taguchi R., Kambe N., Kabashima K., Lambeau G., Gelb M. H., and Murakami M. (2015) The role of group IIF-secreted phospholipase A2 in epidermal homeostasis and hyperplasia. J. Exp. Med. 212, 1901–1919 10.1084/jem.20141904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sato H., Taketomi Y., Ushida A., Isogai Y., Kojima T., Hirabayashi T., Miki Y., Yamamoto K., Nishito Y., Kobayashi T., Ikeda K., Taguchi R., Hara S., Ida S., Miyamoto Y., et al. (2014) The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell Metab. 20, 119–132 10.1016/j.cmet.2014.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamaguchi M., Samuchiwal S. K., Quehenberger O., Boyce J. A., and Balestrieri B. (2018) Macrophages regulate lung ILC2 activation via Pla2g5-dependent mechanisms. Mucosal Immunol. 11, 615–626 10.1038/mi.2017.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen J., Engle S. J., Seilhamer J. J., and Tischfield J. A. (1994) Cloning and recombinant expression of a novel human low molecular weight Ca2+-dependent phospholipase A2. J. Biol. Chem. 269, 2365–2368 [PubMed] [Google Scholar]
- 31. Sawada H., Murakami M., Enomoto A., Shimbara S., and Kudo I. (1999) Regulation of type V phospholipase A2 expression and function by proinflammatory stimuli. Eur. J. Biochem. 263, 826–835 10.1046/j.1432-1327.1999.00565.x [DOI] [PubMed] [Google Scholar]
- 32. Murakami M., Shimbara S., Kambe T., Kuwata H., Winstead M. V., Tischfield J. A., and Kudo I. (1998) The functions of five distinct mammalian phospholipase A2s in regulating arachidonic acid release. Type IIa and type V secretory phospholipase A2s are functionally redundant and act in concert with cytosolic phospholipase A2. J. Biol. Chem. 273, 14411–14423 10.1074/jbc.273.23.14411 [DOI] [PubMed] [Google Scholar]
- 33. Tieu B. C., Lee C., Sun H., Lejeune W., Recinos A. 3rd, Ju X., Spratt H., Guo D. C., Milewicz D., Tilton R. G., and Brasier A. R. (2009) An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Invest. 119, 3637–3651 10.1172/JCI38308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodriguez C., Martínez-González J., Raposo B., Alcudia J. F., Guadall A., and Badimon L. (2008) Regulation of lysyl oxidase in vascular cells: lysyl oxidase as a new player in cardiovascular diseases. Cardiovasc. Res. 79, 7–13 10.1093/cvr/cvn102 [DOI] [PubMed] [Google Scholar]
- 35. Ren W., Liu Y., Wang X., Jia L., Piao C., Lan F., and Du J. (2016) β-Aminopropionitrile monofumarate induces thoracic aortic dissection in C57BL/6 mice. Sci. Rep. 6, 28149 10.1038/srep28149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mäki J. M., Sormunen R., Lippo S., Kaarteenaho-Wiik R., Soininen R., and Myllyharju J. (2005) Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am. J. Pathol. 167, 927–936 10.1016/S0002-9440(10)61183-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Masuda S., Murakami M., Ishikawa Y., Ishii T., and Kudo I. (2005) Diverse cellular localizations of secretory phospholipase A2 enzymes in several human tissues. Biochim. Biophys. Acta 1736, 200–210 10.1016/j.bbalip.2005.08.014 [DOI] [PubMed] [Google Scholar]
- 38. Balsinde J., Balboa M. A., Yedgar S., and Dennis E. A. (2000) Group V phospholipase A2-mediated oleic acid mobilization in lipopolysaccharide-stimulated P388D1 macrophages. J. Biol. Chem. 275, 4783–4786 10.1074/jbc.275.7.4783 [DOI] [PubMed] [Google Scholar]
- 39. Rousseau M., Belleannee C., Duchez A. C., Cloutier N., Levesque T., Jacques F., Perron J., Nigrovic P. A., Dieude M., Hebert M. J., Gelb M. H., and Boilard E. (2015) Detection and quantification of microparticles from different cellular lineages using flow cytometry. Evaluation of the impact of secreted phospholipase A2 on microparticle assessment. PLoS ONE 10, e0116812 10.1371/journal.pone.0116812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Voloshenyuk T. G., Landesman E. S., Khoutorova E., Hart A. D., and Gardner J. D. (2011) Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine 55, 90–97 10.1016/j.cyto.2011.03.024 [DOI] [PubMed] [Google Scholar]
- 41. Khan N. S., Song C. Y., Jennings B. L., Estes A. M., Fang X. R., Bonventre J. V., and Malik K. U. (2015) Cytosolic phospholipase A2α is critical for angiotensin II-induced hypertension and associated cardiovascular pathophysiology. Hypertension 65, 784–792 10.1161/HYPERTENSIONAHA.114.04803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rao G. N., Lassègue B., Alexander R. W., and Griendling K. K. (1994) Angiotensin II stimulates phosphorylation of high-molecular-mass cytosolic phospholipase A2 in vascular smooth-muscle cells. Biochem. J. 299, 197–201 10.1042/bj2990197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yano T., Fujioka D., Saito Y., Kobayashi T., Nakamura T., Obata J. E., Kawabata K., Watanabe K., Watanabe Y., Mishina H., Tamaru S., and Kugiyama K. (2011) Group V secretory phospholipase A2 plays a pathogenic role in myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 90, 335–343 10.1093/cvr/cvq399 [DOI] [PubMed] [Google Scholar]
- 44. Kikawada E., Bonventre J. V., and Arm J. P. (2007) Group V secretory PLA2 regulates TLR2-dependent eicosanoid generation in mouse mast cells through amplification of ERK and cPLA2α activation. Blood 110, 561–567 10.1182/blood-2006-10-052258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Han W. K., Sapirstein A., Hung C. C., Alessandrini A., and Bonventre J. V. (2003) Cross-talk between cytosolic phospholipase A2α (cPLA2α) and secretory phospholipase A2 (sPLA2) in hydrogen peroxide-induced arachidonic acid release in murine mesangial cells: sPLA2 regulates cPLA2α activity that is responsible for arachidonic acid release. J. Biol. Chem. 278, 24153–24163 10.1074/jbc.M300424200 [DOI] [PubMed] [Google Scholar]
- 46. Rosengren B., Peilot H., Umaerus M., Jönsson-Rylander A. C., Mattsson-Hulten L., Hallberg C., Cronet P., Rodriguez-Lee M., and Hurt-Camejo E. (2006) Secretory phospholipase A2 group V: lesion distribution, activation by arterial proteoglycans, and induction in aorta by a Western diet. Arterioscler. Thromb. Vasc. Biol. 26, 1579–1585 10.1161/01.ATV.0000221231.56617.67 [DOI] [PubMed] [Google Scholar]
- 47. Chen C. A., Chang J. M., Chang E. E., Chen H. C., and Yang Y. L. (2018) Crosstalk between transforming growth factor-β1 and endoplasmic reticulum stress regulates α-smooth muscle cell actin expression in podocytes. Life Sci. 209, 9–14 10.1016/j.lfs.2018.07.050 [DOI] [PubMed] [Google Scholar]
- 48. Ishiyama J., Taguchi R., Akasaka Y., Shibata S., Ito M., Nagasawa M., and Murakami K. (2011) Unsaturated FAs prevent palmitate-induced LOX-1 induction via inhibition of ER stress in macrophages. J. Lipid Res. 52, 299–307 10.1194/jlr.M007104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qiang G., Kong H. W., Fang D., McCann M., Yang X., Du G., Blüher M., Zhu J., and Liew C. W. (2016) The obesity-induced transcriptional regulator TRIP-Br2 mediates visceral fat endoplasmic reticulum stress-induced inflammation. Nat. Commun. 7, 11378 10.1038/ncomms11378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chu I. M., Michalowski A. M., Hoenerhoff M., Szauter K. M., Luger D., Sato M., Flanders K., Oshima A., Csiszar K., and Green J. E. (2012) GATA3 inhibits lysyl oxidase-mediated metastases of human basal triple-negative breast cancer cells. Oncogene 31, 2017–2027 10.1038/onc.2011.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rodríguez C., Raposo B., Martínez-González J., Casaní L., and Badimon L. (2002) Low density lipoproteins downregulate lysyl oxidase in vascular endothelial cells and the arterial wall. Arterioscler. Thromb. Vasc. Biol. 22, 1409–1414 10.1161/01.atv.0000033818.21748.99 [DOI] [PubMed] [Google Scholar]
- 52. Martínez-González M. A., Gea A., and Ruiz-Canela M. (2019) The Mediterranean diet and cardiovascular health. Circ. Res. 124, 779–798 10.1161/CIRCRESAHA.118.313348 [DOI] [PubMed] [Google Scholar]
- 53. Guasch-Ferré M., Hu F. B., Martinez-González M. A., Fitó M., Bulló M., Estruch R., Ros E., Corella D., Recondo J., Gómez-Gracia E., Fiol M., Lapetra J., Serra-Majem L., Muñoz M. A., Pintó X., et al. (2014) Olive oil intake and risk of cardiovascular disease and mortality in the PREDIMED study. BMC Med. 12, 78 10.1186/1741-7015-12-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ohta S., Imamura M., Xing W., Boyce J. A., and Balestrieri B. (2013) Group V secretory phospholipase A2 is involved in macrophage activation and is sufficient for macrophage effector functions in allergic pulmonary inflammation. J. Immunol. 190, 5927–5938 10.4049/jimmunol.1203202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. López-Sagaseta J., Puy C., Tamayo I., Allende M., Cerveró J., Velasco S. E., Esmon C. T., Montes R., and Hermida J. (2012) sPLA2-V inhibits EPCR anticoagulant and antiapoptotic properties by accommodating lysophosphatidylcholine or PAF in the hydrophobic groove. Blood 119, 2914–2921 10.1182/blood-2011-05-353409 [DOI] [PubMed] [Google Scholar]
- 56. Murakami M., Koduri R. S., Enomoto A., Shimbara S., Seki M., Yoshihara K., Singer A., Valentin E., Ghomashchi F., Lambeau G., Gelb M. H., and Kudo I. (2001) Distinct arachidonate-releasing functions of mammalian secreted phospholipase A2s in human embryonic kidney 293 and rat mastocytoma RBL-2H3 cells through heparan sulfate shuttling and external plasma membrane mechanisms. J. Biol. Chem. 276, 10083–10096 10.1074/jbc.M007877200 [DOI] [PubMed] [Google Scholar]
- 57. Nakamura H., Kim D. K., Philbin D. M., Peterson M. B., Debros F., Koski G., and Bonventre J. V. (1995) Heparin-enhanced plasma phospholipase A2 activity and prostacyclin synthesis in patients undergoing cardiac surgery. J. Clin. Invest. 95, 1062–1070 10.1172/JCI117752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murakami M., Kudo I., and Inoue K. (1993) Molecular nature of phospholipases A2 involved in prostaglandin I2 synthesis in human umbilical vein endothelial cells. Possible participation of cytosolic and extracellular type II phospholipases A2. J. Biol. Chem. 268, 839–844 [PubMed] [Google Scholar]
- 59. Ovchinnikova O. A., Folkersen L., Persson J., Lindeman J. H., Ueland T., Aukrust P., Gavrisheva N., Shlyakhto E., Paulsson-Berne G., Hedin U., Olofsson P. S., and Hansson G. K. (2014) The collagen cross-linking enzyme lysyl oxidase is associated with the healing of human atherosclerotic lesions. J. Intern. Med. 276, 525–536 10.1111/joim.12228 [DOI] [PubMed] [Google Scholar]
- 60. Lusis A. J. (2000) Atherosclerosis. Nature 407, 233–241 10.1038/35025203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Getz G. S., and Reardon C. A. (2016) ApoE knockout and knockin mice: the history of their contribution to the understanding of atherogenesis. J. Lipid Res. 57, 758–766 10.1194/jlr.R067249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Getz G. S., and Reardon C. A. (2016) Do the Apoe−/− and Ldlr−/− mice yield the same insight on atherogenesis? Arterioscler. Thromb. Vasc. Biol. 36, 1734–1741 10.1161/ATVBAHA.116.306874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bernatchez P. N., Winstead M. V., Dennis E. A., and Sirois M. G. (2001) VEGF stimulation of endothelial cell PAF synthesis is mediated by group V 14 kDa secretory phospholipase A2. Br. J. Pharmacol. 134, 197–205 10.1038/sj.bjp.0704215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sonoki K., Iwase M., Sasaki N., Ohdo S., Higuchi S., Takata Y., and Iida M. (2008) Secretory PLA2 inhibitor indoxam suppresses LDL modification and associated inflammatory responses in TNFα-stimulated human endothelial cells. Br. J. Pharmacol. 153, 1399–1408 10.1038/bjp.2008.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sonoki K., Iwase M., Ohdo S., Ieiri I., Matsuyama N., Takata Y., and Kitazono T. (2012) Telmisartan and N-acetylcysteine suppress group V secretory phospholipase A2 expression in TNFα-stimulated human endothelial cells and reduce associated atherogenicity. J. Cardiovasc. Pharmacol. 60, 367–374 10.1097/FJC.0b013e3182646ccc [DOI] [PubMed] [Google Scholar]
- 66. Ghesquiere S. A., Gijbels M. J., Anthonsen M., van Gorp P. J., van der Made I., Johansen B., Hofker M. H., and de Winther M. P. (2005) Macrophage-specific overexpression of group IIa sPLA2 increases atherosclerosis and enhances collagen deposition. J. Lipid Res. 46, 201–210 10.1194/jlr.M400253-JLR200 [DOI] [PubMed] [Google Scholar]
- 67. Luchtefeld M., Bandlow N., Tietge U. J., Grote K., Pfeilschifter J., Kaszkin M., Beck S., Drexler H., and Schieffer B. (2007) Angiotensin II type 1-receptor antagonism prevents type IIA secretory phospholipase A2-dependent lipid peroxidation. Atherosclerosis 194, 62–70 10.1016/j.atherosclerosis.2006.09.024 [DOI] [PubMed] [Google Scholar]
- 68. Martin R., Gutierrez B., Cordova C., Roman A. S., Alvarez Y., Hernandez M., Cachofeiro V., and Nieto M. L. (2020) Secreted phospholipase A2-IIA modulates transdifferentiation of cardiac fibroblast through EGFR transactivation: an inflammation-fibrosis link. Cells 9, 396 10.3390/cells9020396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Murase R., Taketomi Y., Miki Y., Nishito Y., Saito M., Fukami K., Yamamoto K., and Murakami M. (2017) Group III phospholipase A2 promotes colitis and colorectal cancer. Sci. Rep. 7, 12261 10.1038/s41598-017-12434-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Degousee N., Ghomashchi F., Stefanski E., Singer A., Smart B. P., Borregaard N., Reithmeier R., Lindsay T. F., Lichtenberger C., Reinisch W., Lambeau G., Arm J., Tischfield J., Gelb M. H., and Rubin B. B. (2002) Groups IV, V, and X phospholipases A2s in human neutrophils: role in eicosanoid production and Gram-negative bacterial phospholipid hydrolysis. J. Biol. Chem. 277, 5061–5073 10.1074/jbc.M109083200 [DOI] [PubMed] [Google Scholar]
- 71. Yamamoto K., Miki Y., Sato H., Murase R., Taketomi Y., and Murakami M. (2017) Secreted phospholipase A2 specificity on natural membrane phospholipids. Methods Enzymol. 583, 101–117 10.1016/bs.mie.2016.09.007 [DOI] [PubMed] [Google Scholar]
- 72. Yamamoto K., Taketomi Y., Isogai Y., Miki Y., Sato H., Masuda S., Nishito Y., Morioka K., Ishimoto Y., Suzuki N., Yokota Y., Hanasaki K., Ishikawa Y., Ishii T., Kobayashi T., et al. (2011) Hair follicular expression and function of group X secreted phospholipase A2 in mouse skin. J. Biol. Chem. 286, 11616–11631 10.1074/jbc.M110.206714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this work are available within the paper and the supporting information. The raw data will be provided by the corresponding author upon request.