

Abstract
Type 1 cGMP-dependent protein kinases (PKGs) play important roles in human cardiovascular physiology, regulating vascular tone and smooth-muscle cell phenotype. A mutation in the human PRKG1 gene encoding cGMP-dependent protein kinase 1 (PKG1) leads to thoracic aortic aneurysms and dissections. The mutation causes an arginine-to-glutamine (RQ) substitution within the first cGMP-binding pocket in PKG1. This substitution disrupts cGMP binding to the pocket, but it also unexpectedly causes PKG1 to have high activity in the absence of cGMP via an unknown mechanism. Here, we identified the molecular mechanism whereby the RQ mutation increases basal kinase activity in the human PKG1α and PKG1β isoforms. Although we found that the RQ substitution (R177Q in PKG1α and R192Q in PKG1β) increases PKG1α and PKG1β autophosphorylation in vitro, we did not detect increased autophosphorylation of the PKG1α or PKG1β RQ variant isolated from transiently transfected 293T cells, indicating that increased basal activity of the RQ variants in cells was not driven by PKG1 autophosphorylation. Replacement of Arg-177 in PKG1α with alanine or methionine also increased basal activity. PKG1 exists as a parallel homodimer linked by an N-terminal leucine zipper, and we show that the WT chain in WT-RQ heterodimers partly reduces basal activity of the RQ chain. Using hydrogen/deuterium-exchange MS, we found that the RQ substitution causes PKG1β to adopt an active conformation in the absence of cGMP, similar to that of cGMP-bound WT enzyme. We conclude that the RQ substitution in PKG1 increases its basal activity by disrupting the formation of an inactive conformation.
Keywords: protein kinase G (PKG), cyclic GMP (cGMP), mutant, mutagenesis, autophosphorylation, enzyme structure, hydrogen/deuterium exchange, thoracic aortic aneurysm and dissection (TAAD), kinase signaling, cardiovascular system, hydrogen exchange mass spectrometry
The type 1 cGMP-dependent protein kinases play key roles in the cardiovascular system, including modulation of vascular tone, inhibition of thrombosis, and protection from cardiac hypertrophy/fibrosis. (1). At the cellular level, protein kinase G (PKG) regulates intracellular calcium concentrations and the contractile machinery's sensitivity to calcium; PKG also controls gene transcription and apoptosis (2). The kinases are downstream effectors of the natriuretic peptide/nitric oxide (NO)-guanylate cyclase signaling pathways and are the targets of drugs that raise intracellular cGMP levels by releasing NO, directly activating guanylate cyclase, or inhibiting cGMP breakdown (3).
Mammalian cells express two PKG1 isoforms, PKG1α and PKG1β, which are produced from alternative transcriptional start utilization and/or alternate splicing from the same gene; they differ in their first ∼100 amino acids (1). PKG1 is a single-chain kinase with an N-terminal regulatory domain and a C-terminal catalytic domain (Fig. 1A). The regulatory domain can be further divided into functional subdomains. Located at the very N terminus are isoform-specific leucine/isoleucine zipper (LZ) domains, which mediate homodimerization and target the PKG1 isoforms to specific substrates (4–10). The LZ domains are followed by isoform-specific autoinhibitory (AI) domains, which contain pseudosubstrate sequences and inhibit kinase activity by binding within the catalytic cleft to block substrate access. The AI domains also contain autophosphorylation sites that can activate the kinases in the absence of cGMP, i.e. autophosphorylation of S65 in PKG1α and S80 in PKG1β leads to cGMP-independent kinase activation (11, 12). Whereas the N termini are unique, PKG1α and PKG1β have identical cyclic nucleotide binding (CNB-A and CNB-B) and catalytic domains. PKG is activated by a cGMP-induced conformational change in the regulatory domain, which pulls the pseudosubstrate sequence from the catalytic cleft (13). The cGMP-induced exposure of the catalytic cleft in PKG1α has been previously shown using hydrogen/deuterium exchange MS (H/DX-MS) (13).
Figure 1.

In vitro kinase activity of PKG1α R177Q and PKG1β R192Q. A, domain organization of PKG1 highlighting the locations of autophosphorylation sites and the activating RQ mutation. The regulatory domain contains leucine zipper (LZ), autoinhibitory (AI), and cyclic nucleotide binding (CNB-A and CNB-B) subdomains. B and C, kinase assays were performed using purified PKG1α and PKG1β as described in Experimental procedures. Reactions were stopped after 1.5 min (B) or 5 min (C). Data are from three independent protein preparations, with each point representing the average of three kinase reactions for each preparation. Bars show means ± S.D., n = 3.
In humans, a mutation in the gene for PKG1 causes familial thoracic aortic aneurysms and dissections (14). The mutation changes a conserved arginine residue in CNB-A to glutamine and causes a high basal kinase activity in the absence of cGMP (R177Q in PKG1α and R192Q in PKG1β) (14). A knock-in mouse heterozygous for the same mutation develops age-dependent aortic dilatation and aortic media degeneration with elastin fiber breaks (15). Whereas the mutation causes an ∼32,000-fold lower cGMP affinity in CNB-A, the mutant kinase has the same Vmax as the WT enzyme under saturating cGMP conditions (14). Exactly how the RQ mutation leads to PKG1 activation is unknown. In the current study, we describe some unique biochemical properties of the mutant kinase, and we used H/DX-MS to compare the conformations of WT and R192Q PKG1β in the absence and presence of cGMP. We found that the mutant enzyme adopts an active conformation in the absence of cGMP that resembles the conformation of the cGMP-bound WT enzyme.
Results
PKG1α R177Q and PKG1β R192Q kinase activity in vitro
We originally found that PKG1α with an R177Q mutation (RQ-PKG1α) caused the kinase to be 90–95% active in the absence of cGMP (14). PKG1β is produced as a splice variant from the same gene, but despite having the same CNB-A and CNB-B domains as PKG1α, the amount of cGMP required to half-maximally activate PKG1β is ∼3.5-fold higher than that of PKG1α (11). Because the RQ mutation is in CNB-A, we checked to see how the mutation affected PKG1β activity. The N terminus of PKG1β is 15 amino acids longer than that of PKG1α, and R192Q PKG1β (RQ-PKG1β) is directly analogous to RQ-PKG1α (Fig. 1A). We found that both RQ-PKG1α and RQ-PKG1β had high basal activities compared with those of the WT enzymes (Fig. 1B), but the basal activity of RQ-PKG1β was consistently higher than that of RQ-PKG1α (88.4% ± 5.3% versus 57.5% ± 8.5% of maximal activity in the presence of cGMP). The basal activity of RQ-PKG1α was lower than we previously reported (14). In our previous study, we measured kinase activity at 10-min time points versus 1.5 min for the assays shown in Fig. 1B (under the conditions used, the assays are linear up to 11 min). At longer time points, activity might be partially driven by PKG1 autophosphorylation (shown below), because autophosphorylation can increase basal kinase activity in the absence of cGMP (12, 16, 17). At a 5-min time point, basal RQ-PKG1β activity reached 97.8% ± 6.1% versus 75.8% ± 20% of maximal activity for RQ-PKG1α (Fig. 1C). Thus, at longer time points, there is an apparent increase in basal activity in the RQ mutants, which may be driven by autophosphorylation.
Higher autophosphorylation of RQ-PKG1 in vitro but not in cells
We next compared autophosphorylation of WT and mutant kinases. Autophosphorylation of S65 in PKG1α and S80 in PKG1β leads to cGMP-independent kinase activation in vitro (12, 16, 17). First, we examined the ability of WT and mutant PKG1α and PKG1β to autophosphorylate in vitro. Purified kinases were incubated in the same buffer used for kinase assays (without peptide substrate) in the presence and absence of 10 μm cGMP, and phosphate incorporation was analyzed by SDS-PAGE/autoradiography. We found that RQ-PKG1α and RQ-PKG1β had higher rates of autophosphorylation than the respective WT enzymes (Fig. 2A and B). It should be noted that the amount of 32PO4 incorporation by autophosphorylation cannot be directly compared between PKG1α and PKG1β, as 32PO4–γ-ATP-specific activity differed between reactions. Second, to determine whether differential autophosphorylation occurred in cells, we incubated transiently transfected 293T cells with [32P]orthophosphate, and some cells were treated with a final concentration of 250 μm 8-(4-chlorophenylthio)-guanosine–3′5′-cyclic monophophosphate (8-pCPT–cGMP), as indicated. The kinases were isolated under the same conditions used when purifying them for in vitro assays, and 32PO4 incorporation was analyzed by SDS-PAGE/autoradiography. We found equivalent amounts of 32PO4 incorporated in the WT and mutant enzymes (Fig. 2C and D), indicating the mutant kinases were not purified in a more highly autophosphorylated state. The observed 32PO4 incorporation likely represents phosphorylation of the activation loop in the catalytic domain, which is necessary for catalytic activity (18). Again, the amount of 32PO4 incorporation into PKG1α versus PKG1β cannot be directly compared, as experiments were performed at separate times.
Figure 2.

The RQ mutation causes higher autophosphorylation in vitro but not in cells. A, in vitro PKG1α autophosphorylation. Purified PKG1α was incubated for 5 min with 32PO4–γ-ATP under reaction conditions identical to those used for in vitro kinase assays (in the absence of peptide substrate). Phosphate incorporation was determined by SDS-PAGE/autoradiography. Equal loading of the kinase is shown by Western blotting with an anti-Flag antibody. B, performed as in panel A, except using purified PKG1β. C, 293T cells were transfected with expression vectors for Flag-tagged WT and R177Q PKG1α. Six hours posttransfection, cells were incubated with 32PO4 for three hours, and then some cells were treated with 8-pCPT–cGMP for one hour. Upper, PKG was isolated by anti-FLAG immunoprecipitation, and phosphate incorporation was analyzed by SDS-PAGE/autoradiography. Lower, equal PKG amounts were determined by anti-Flag Western blots. D, autophosphorylation of PKG1β in 293T cells performed as in panel C.
Phosphorylation of VASP in intact cells
We next compared WT and RQ mutant kinase activity in intact cells, using vasodilator-stimulated phosphoprotein (VASP) phosphorylation as a readout (19). We cotransfected 293T cells with expression vectors for Myc-tagged VASP and WT or RQ-PKG1α or RQ-PKG1β, and 24 h posttransfection, some wells were treated with a final concentration of 100 μm 8-pCPT–cGMP for one hour. VASP phosphorylation was determined by SDS-PAGE/immunoblotting to detect a gel shift induced by Ser239 phosphorylation (20). 8-pCPT–cGMP treatment of cells transfected with WT PKG1α caused a complete upward shift in VASP migration (Fig. 3A, compare lanes 3 and 4). However, in cells transfected with RQ-PKG1α, most VASP migrated at the shifted position in the absence of cGMP (Fig. 3A, compare lanes 3 and 5), and no further change in migration occurred after treatment with 8-pCPT–cGMP (Fig. 3A, compare lanes 5 and 6). Similar results were obtained when VASP was cotransfected with WT and RQ-PKG1β (Fig. 3B). Thus, the RQ-PKGs had an increased basal activity in cells under conditions where autophosphorylation did not differ from that of the WT enzymes (Fig. 2C and D).
Figure 3.

The RQ mutation increases PKG kinase activity in intact cells. A, 293T cells were cotransfected with expression vectors for Myc-tagged VASP and either WT or R177Q PKG1α. At 24 h posttransfection, some cells were treated with 100 μm 8-pCPT–cGMP for one hour. Cell lysates were analyzed by SDS-PAGE/immunoblotting using antibodies recognizing Myc-epitope (upper) or PKG1 (lower). B, experiment was performed as in panel A, but VASP was cotransfected with WT or R192Q PKG1β. The gel shift of VASP indicates S239 phosphorylation.
Effect of interchain interactions on RQ mutant activity
In the initial report describing RQ-PKG1α, we observed that in WT/RQ heterodimers, the WT chain appeared to inhibit the activity of the mutant chain (14). This observation was based on the assumption that equal transfection of WT and mutant Flag-tagged constructs would produce a combination of WT/WT, WT/RQ, and RQ/RQ dimers in a proportion of 1:2:1; however, the exact proportion of each dimer in the preparations was not known. To more carefully assess the role of interchain contacts in regulating RQ-PKG activity, we developed a novel experimental approach using a kinase-dead PKG. In this method, Flag-tagged dead PKG is used to purify active, untagged PKG (Fig. 4A). Under these conditions, the Flag-purified kinase is a mixture of inactive dead/dead homodimers and dead/active heterodimers. Importantly, the kinase activity measured is that of active chains dimerized to Flag-tagged dead chains. This approach enabled us to compare the effect of the Flag-tagged dead chain, with or without the RQ mutation, on the activity of the untagged, active chain (interchain effect) without having to account for how the mutation affects the activity of its own kinase domain (intrachain effect). We found that basal kinase activity for both RQ-PKG1α and RQ-PKG1β was lower when the dead chain contained WT CNB-A than with a dead chain with RQ mutant CNB-A (Fig. 4B and C). For PKG1α, basal activity increased from 44.5 to 77.4% of cGMP-stimulated maximal activity when the dead chain contained the RQ mutation versus no CNB-A mutation. For PKG1β, the activity increased from 72.2 to 96.4%. These results clearly demonstrate that the two chains do not act independently and highlight a critical role that interchain contacts play in mediating PKG1 activity. These results also suggest that in humans carrying one copy of the mutant PRKG1 gene, the pathological consequences of the mutation are most likely reduced by expression of functionally normal PKG1 from the WT gene.
Figure 4.

Interchain communication regulates RQ-PKG1 activity. A, PKG1 domain map showing dimerized active and dead PKG chains. RQ indicates the site of the activating mutation in CNB-A, and DA indicates mutation of the catalytic aspartic acid to alanine, which causes a loss of kinase activity. B and C, 293T cells were cotransfected with Flag-tagged dead PKG1, with or without the RQ mutation, and untagged active RQ-mutant PKG1. In vitro kinase reactions were stopped at 1.5 min to measure the activity of untagged PKG1α R177Q (B) or PKG1β R192Q (C). Data are from three independent protein preparations, with each point representing the average from three kinase reactions for each preparation. Bars show means ± S.D., n = 3. **, p > 0.01 by two-tailed Student's t test.
Effects of cyclic nucleotide analog inhibitors on RQ PKG1 activity
We have recently shown that RQ-PKG1 basal activity can be inhibited by the small peptide inhibitor DT-2, which targets the catalytic cleft (15). Because the RQ mutation is within CNB-A of the regulatory domain, we tested whether cyclic nucleotide analog inhibitors could reduce basal activity of the mutant enzymes. We found that 8-(4-chlorophenylthio)guanosine–3′5′-cyclic monophophorothioate, Rp-isomer (Rp-8-pCPT–cGMPS), showed a slight agonist activity toward RQ-PKG1α but had a negligible effect on RQ-PKG1β (Fig. 5A and B). The drug did not inhibit the activity of either kinase at concentrations of up to 300 μm. 8-(4-Chlorophenylthio)-β-phenyl-1,N2-ethenoguanosine–3′5′-cyclic monophophorothioate, Rp-isomer (Rp-8-pCPT-PET–cGMPS), showed partial agonist activity toward both RQ-PKG1α and RQ-PKG1β, but at concentrations of ≥100 μm, it inhibited basal activity of both mutant enzymes (Fig. 5C and D). Interestingly, we found that Rp-8-pCPT-PET–cGMPS also inhibited the isolated PKG1 catalytic domain (Fig. 5E), indicating that the inhibition seen in the full-length kinases was not because of binding within the cyclic nucleotide binding pockets. We conclude that the cyclic nucleotide-based inhibitors do not inhibit RQ mutant PKG1α/β basal activity by binding to CNB-A or -B.
Figure 5.

Effects of cyclic nucleotide analog inhibitors on RQ PKG1 activity. In vitro basal kinase activity of purified R177Q PKG1α or R192Q PKG1β measured in the presence of increasing concentrations of Rp-8-pCPT–cGMPS (A and B) or Rp-8-pCPT-PET–cGMPS (C and D). E, In vitro kinase activity of isolated PKG1 catalytic domain (CD) measured in the presence of increasing Rp-8-pCPT-PET-cGMPS concentrations. For all reactions, basal activity in the absence of inhibitor was set at 100%. Data are the average from three experiments using three independent protein preparations. Bars show means ± S.D., n = 3.
Effect of alternate amino acid substitutions at R177 on basal kinase activity of PKG1α
At the present time, there are no published crystal structures for PKG1 in the inhibited state. However, there are a number of structures for the closely related cAMP-dependent protein kinase (PKA) in the inhibited state. Using the PKA RIα/Cα holoenzyme crystal structure (PDB entry 2QCS) as a template, we used SWISS-Modeler to build a model of inactive PKG1α (21). Examination of the model shows that Arg177 makes a number of contacts with remote, nonsequential residues located in CNB-A (Fig. 6A). The Arg177 hydrophobic arm appears to pack between the side chain of Ile131 and the backbone region of Gly137. These residues are conserved in PKA RIα, suggesting that hydrophobic packing interactions play a general role in stabilizing the inactive state of PKG and PKA. In addition, the charged guanido group of Arg177 appears to be neutralized by interactions with the backbone carbonyl groups of Leu139 and Gly167.
Figure 6.

Mutation of R177 to Ala, Met, or Gln leads to increased basal kinase activity. A, molecular model of inactive PKG1α showing putative packing of PKGα R177 in an inactive conformation. B, in silico-predicted destabilizing effects of mutations at R177 using the DUET server (ΔΔG in kcal/mol). C, kinase assays comparing basal to maximum cGMP-stimulated activation of WT and mutant PKG1α. Data are from triplicate reactions from a single protein preparation. The experiment was performed twice with similar results.
Next, we used the DUET server to predict the degree to which different R177 mutations would destabilize interactions with contact residues and disrupt the ability of PKG1 to adopt an inhibited conformation (22). The DUET server combines the approaches of two previous in silico prediction algorithms (mCSM and SDM) to more accurately predict the destabilizing effect of mutations on proteins (23, 24). For example, we chose alanine because it should not produce novel contacts but would lose the hydrophobic interactions with Ile131 and Gly137 and the polar contacts with the Leu139 and Gly167 backbone carbonyl groups. DUET predicted that Met would be less and Gln more destabilizing than Ala at position 177 (Fig. 6B). From the structure shown in Fig. 6A, it is evident that replacing Arg177 with Gln would place Gln's polar side chain (specifically the carboxyl group) in the hydrophobic environment that normally packs around the third methylene group in Arg177's hydrophobic arm.
We tested how the three mutations affect PKG1α activity in vitro and found that all of these substitutions caused an increase in basal activity compared with WT enzyme (Fig. 6C). Each reaction is normalized to the maximum activity seen in the presence of 10 μm cGMP in a 1.5-min reaction to minimize autophosphorylation effects. When comparing our assay results to the predicted destabilizing effects of the mutants, we found that, as predicted, Gln was the most destabilizing; however, contrary to DUET analysis, Met was twice as effective as Ala in increasing basal activity.
Deuterium exchange analysis shows that RQ-PKG1β is in an active conformation
To probe how the RQ mutation affects the conformation of PKG1, we performed H/DX-MS on WT and RQ mutant PKG1β. Aliquots of purified proteins were incubated with or without cGMP and subsequently incubated with buffered D2O. At specific time points, H/D exchange was stopped by adding ice-cold acidic quench buffer. Incorporated deuterons were localized by proteolysis, HPLC fractionation of peptides, and MS. The H/D exchange data were then analyzed with respect to a PKG1β structural model (Fig. 7A and B).
Figure 7.
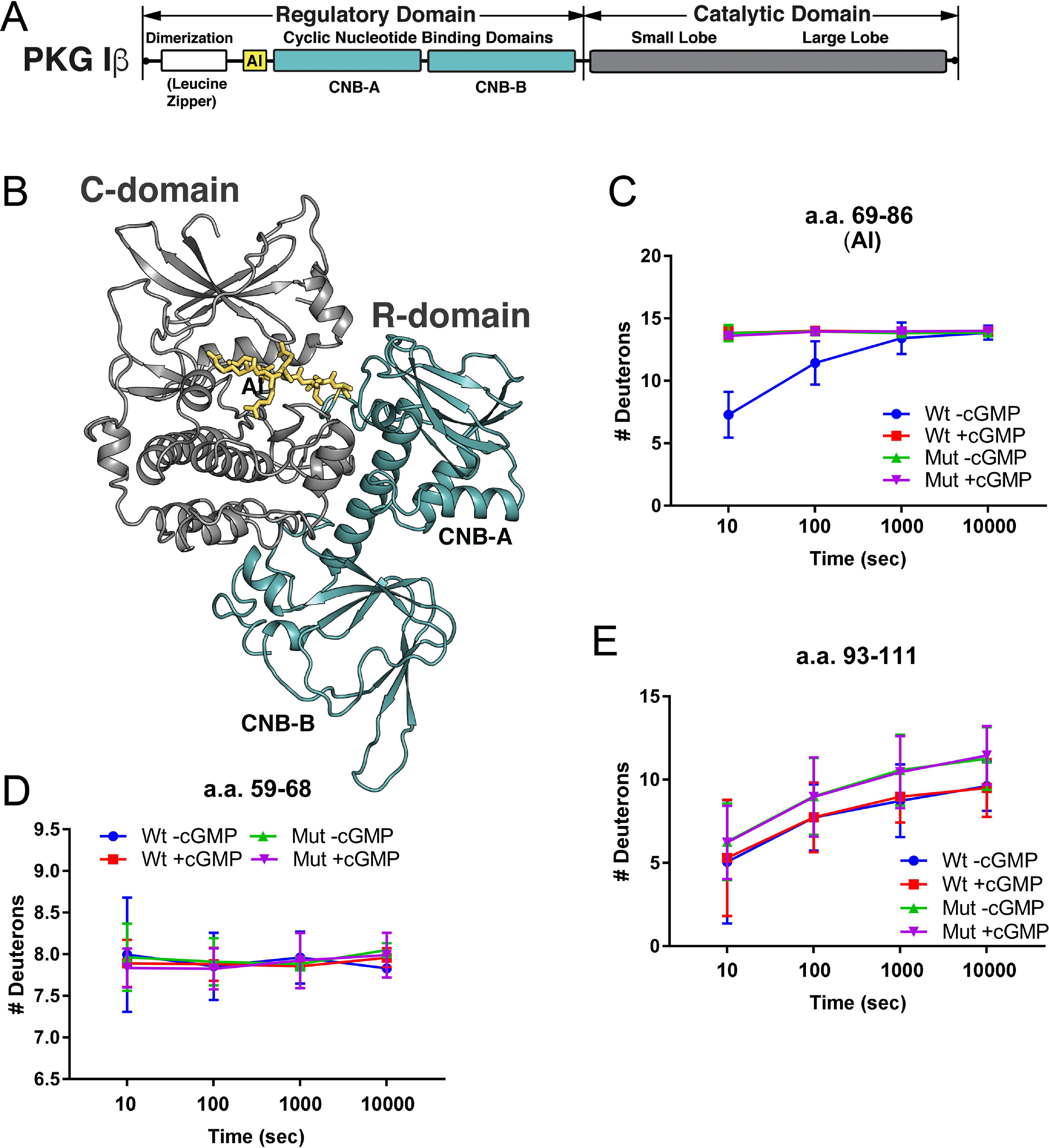
H/D exchange in inhibitory loop residues. A, PKG1β domain organization colored according to the molecular model shown in panel B. LZ, leucine zipper; AI, autoinhibitory loop; CNB-A and CNB-B, cyclic nucleotide binding pockets; and catalytic, the catalytic domain. (B) Molecular model of PKG1β in an inhibited conformation with the regulatory domain CNBs colored teal, catalytic domain in gray, and amino acids 69–86 of the linker/autoinhibitory loop colored yellow. (C) Time-dependent H/D exchange in residues 69–86 (AI), which bind within the catalytic cleft. (D) Time-dependent H/D exchange in residues 59–68, which are N terminal to the autoinhibitory loop. (E) Time-dependent H/D exchange in residues 93–111, which are C terminal to the autoinhibitory loop. Wt, WT PKGIβ; Mut, R192Q PKGIβ. Graphs show the number of deuterons incorporated into the peptides as a function of time. H/D exchange data are averages from two independent H/D exchange reactions performed with two separate protein preparations.
We first examined peptides within the regulatory domain (Fig. 7B, shown in yellow and teal, starting with the autoinhibitory loop, AI). The addition of cGMP to WT PKG1β caused increased solvent exposure in a peptide comprising the autoinhibitory loop (residues 69–86, shown in yellow); however, in RQ-PKG1β, even in the absence of cGMP this peptide had levels of deuterium incorporation equal to that of the WT in the presence of cGMP, and H/D exchange was not increased in the presence of cGMP (Fig. 7C). In contrast to the autoinhibitory loop peptide, deuterium incorporation in a peptide (aa 59–68) just N terminal to the autoinhibitory loop showed identical H/D exchange behavior between WT and mutant PKG, and cGMP had no effect (Fig. 7D). In addition, a peptide (aa 93–111) just C terminal to the loop binding the catalytic cleft showed a trend toward higher H/D exchange in mutant PKG1β, but H/D exchange was not altered by cGMP in either the WT or mutant protein (Fig. 7E). Thus, cGMP induces increased solvent exposure of the autoinhibitory loop in WT PKG1β, but the autoinhibitory loop of RQ-PKG1β shows high solvent exposure even in the absence of cGMP, and exposure is not further increased in the presence of cGMP.
Next, we examined peptides in the catalytic domain (Fig. 8, shown in gray). Alverdi et al. have previously used H/D exchange to examine cGMP-induced conformational changes in PKG1α and observed that cGMP caused increased H/D exchange within peptides flanking the catalytic cleft (13). We found a similar phenomenon in WT PKG1β; in the presence of cGMP, H/D exchange increased in peptides comprising amino acids 521–533 (shown in green), 547–556 (shown in red), and 565–583 (shown in blue), all of which flank the catalytic cleft (Fig. 8). However, in RQ-PKG1β, in the absence of cGMP, H/D exchange within these peptides resembled H/D exchange in the WT enzyme in the presence of cGMP, and H/D exchange of the mutant enzyme did not increase when cGMP was added. Taken together, our data demonstrate that the RQ mutation causes PKG1 to adopt an active conformation in the absence of cGMP, similar to the WT, cGMP-bound kinase.
Figure 8.
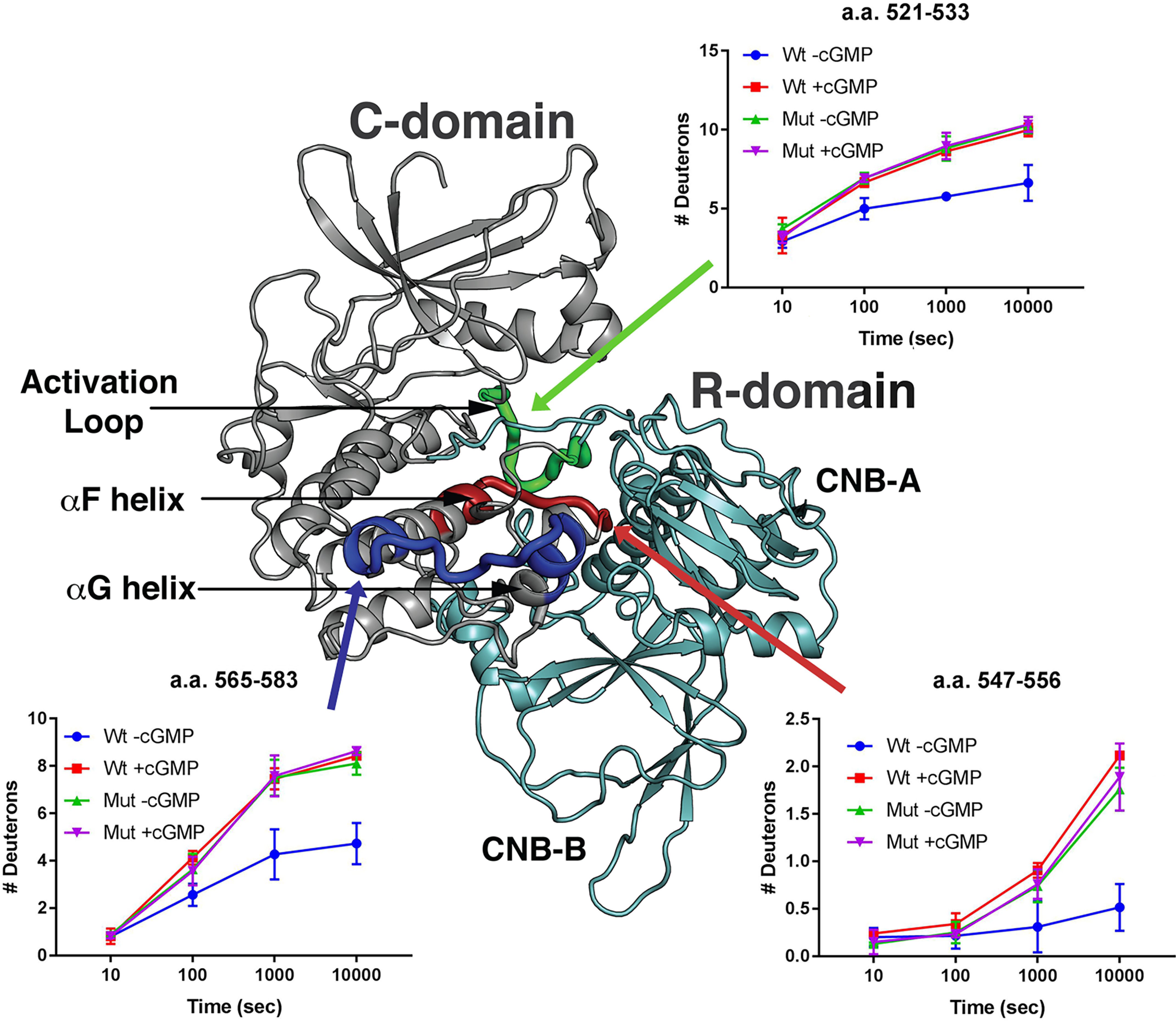
H/D exchange in the catalytic cleft of WT and R192Q PKG1β. H/D exchange profiles of peptides from WT (Wt) and R192Q (Mut) PKG1β in the presence or absence of cGMP are shown mapped to a molecular model of inactive PKG1β. The regulatory domain is colored teal and the catalytic domain is mainly colored gray, with the following exceptions: the region containing amino acids 521–533 is colored green; 547–556 is colored red; and 565–583 is colored dark blue. Graphs show the number of deuterons incorporated into the peptides as a function of time. H/D exchange data are the averages from two independent H/D exchange reactions performed with separate protein preparations.
Discussion
In this study, we probed how the RQ-PKG1 mutation associated with familial thoracic aortic aneurysms and aortic dissections leads to increased basal kinase activity. In the original report describing mutant PKG1α, we were surprised to find that the mutation led to increased basal activity with normal cGMP-stimulated maximal activity, as the mutation disrupted cGMP binding to CNB-A (14). In our current study, we found that the mutation also increased basal activity of PKG1β. In fact, the mutation appeared to increase basal activity of RQ-PKG1β more than that of RQ-PKG1α. This difference is most likely driven by the unique N termini, which cause isoform-specific cGMP affinities and activation constants (25, 26).
De Jong et al. first reported PKG1 autophosphorylation in 1977 (27). Subsequently, Aitken et al. found that PKG1α underwent autophosphorylation on multiple sites in vitro, and that autophosphorylation at these sites occurred at different rates (16). Notably, whereas phosphorylation of Thr59 occurs relatively quickly, phosphorylation at Ser65 is much slower; however, autophosphorylation at Ser65 leads to cGMP-independent kinase activation, whereas modification of Thr59 does not (11). Similar results were found for PKG1β, where autophosphorylation of Ser64 occurs quickly but has no effect on basal kinase activity, and autophosphorylation at Ser80 occurs slowly, leading to cGMP-independent activity (12).
Although we found that the RQ mutation caused both PKG1α and PKG1β to have higher rates of autophosphorylation in vitro than the WT enzymes (with or without cGMP), we did not detect increased autophosphorylation of mutant PKG in intact cells. The reason why PKG1α and PKG1β autophosphorylate in vitro and not in cells is not known. Preliminary experiments suggest that it is because of the relatively higher amount of Mg2+ relative to the ATP concentration used in in vitro reactions (D. Casteel, unpublished results). In addition, it is unknown why the mutant enzymes undergo more rapid autophosphorylation in vitro than the WT enzymes; the mutation may release the enzyme's phosphotransfer activity without reducing the catalytic cleft's access to the autoinhibitory loop (where the autophosphorylation sites are located). Consistent with our results indicating a lack of autophosphorylation in intact cells, Vallur et al. could not detect PKG1α/PKG1β autophosphorylation in a number of cell types using phospho-specific antibodies (28). It has been hypothesized that PKG1 autophosphorylation leads to sustained PKG1 signaling after cellular cGMP levels fall (29); however, our findings, combined with those of Vallur et al., cast doubt on the physiological significance of PKG1 autophosphorylation.
As mentioned, we previously demonstrated that the activity of RQ-PKG1α could be inhibited by DT-2 in vitro (15). Another class of PKG inhibitors consists of modified cGMP analogs in which one of the phosphate oxygen atoms is replaced with sulfur, i.e. Rp-phosphorothioate cGMP analogs (30). These inhibitors bind within the cGMP-binding pockets and prevent activation of WT enzyme by blocking cGMP access. We tested two of these inhibitors for their ability to inhibit RQ-PKG1α and RQ-PKG1β and found that both inhibitors showed partial agonist activity toward the mutant kinases. Our findings are consistent with partial agonist activities of various Rp-phosphorothioate cGMP analogs seen by others with WT PKG1α (31, 32). Interestingly, at higher concentrations, Rp-pCPT-PET–cGMPS inhibited both RQ-PKG1α and RQ-PKG1β, but we found that it also inhibited the isolated PKG1 catalytic domain. Whereas the exact mechanism of inhibition of the catalytic domain remains unknown, it is possible that Rp-pCPT-PET-cGMPS acts as an ATP analog inhibitor.
We used a novel method to demonstrate the role of interchain communication in modulating activity of the mutant kinase. In WT/RQ heterodimers, the activity of the mutant chain is clearly inhibited by the WT chain. How interchain interactions control PKG1 activity has not been thoroughly studied; however, two reports have provided some preliminary insights. Based on the crystal structure of PKG1α's cyclic nucleotide binding pockets, Osborne et al. identified a hydrophobic interface that formed between the two chains of a PKG1 dimer (33). When the hydrophobic residues (and a neighboring asparagine) were mutated to alanines, the basal activity of the kinase increased, suggesting that the interchain interaction stabilized the inactive conformation of the enzyme (33). In another study, Kim et al. identified a cGMP-mediated interface between the two chains that maintained the kinase in the active conformation (34). Consistent with this, when N189 in the interface was changed to alanine, the activation constant (Ka) for cGMP in full-length/dimeric PKG1β increased from 140 nm to 2.5 μm, whereas the mutations had only a slight effect on the Ka for cGMP in truncated/monomeric PKG1β. It should be noted that the Ka for cGMP was already increased in the truncated PKG1β (1.5 μm); thus, the effect of the interface mutation might have been masked by an apparently decreased cGMP affinity in the monomeric enzyme. The new method described in this work should be useful in further studies examining how interchain communication regulates PKG1 activity.
Our H/D exchange analysis demonstrated that the RQ mutation causes PKG1β to adopt a conformation that resembles the active cGMP-bound WT protein, even in the absence of cGMP. Specifically, in WT PKG1β we observed increased H/D exchange in peptides comprising the autoinhibitory loop and the catalytic cleft in the presence, compared with the absence, of cGMP. In the mutant enzyme, in the absence of cGMP, the autoinhibitory loop and catalytic cleft peptides showed H/D exchange behavior that mirrored H/D exchange in cGMP-bound WT, and adding cGMP did not lead to a further increase in H/D exchange. This result was a bit surprising, because RQ-PKG1β is not 100% active (i.e. it can still be stimulated by cGMP). However, RQ-PKG1β is 88% active and the H/D exchange analysis may not be sensitive enough to detect a 10–12% increase in deuteration upon cGMP binding. In addition, PKG1β's inhibited conformation may be stabilized by Mg2+/ATP, which is present in the kinase assays but not in the H/D exchange reactions. In PKA, the affinity between RIα/regulatory and Cα/catalytic subunits is 125 nm in the absence of Mg2+/ATP, but the affinity decreases to <0.05 nm in the presence of Mg2+/ATP (35). It would be interesting to perform H/D exchange experiments on full-length WT and RQ mutant PKG in the presence of Mg2+ and a nonhydrolyzable ATP analog to see if these ligands partially stabilize the inactive conformation and decrease H/D exchange in peptides within the autoinhibitory loop and catalytic cleft.
In conclusion, using H/D exchange MS, we found that the RQ mutation causes PKG1β to adopt an active conformation in the absence of cGMP. A dynamic equilibrium exists between inactive and active conformations of PKG1, and cyclic nucleotide binding normally stabilizes an active conformation of the WT enzyme, as previously discussed for PKA (36). Because mutations other than RQ at position 177 of PKG1α also led to activation, the mechanism does not appear to involve glutamine making unique contacts that stabilize an active conformation; rather, activation results from the destabilization of an inactive conformation. In comparison, single-amino-acid substitutions in small RAS GTPases and G protein-coupled receptors also lead to constitutive activity by shifting the proteins' conformation to an active state (37–39). Whereas high levels of RQ-PKG1 autophosphorylation might drive some of the increased activity seen in vitro, autophosphorylation does not appear to occur in cells and, thus, does not contribute to increased signaling of the mutant kinase in vivo. The fact that the inactive WT chain in WT/RQ heterodimers reduces the activity of the mutant chain suggests that interventions reducing cGMP levels in patients carrying RQ mutant PKG1 reduces the pathological effects of the mutation.
Experimental procedures
Materials
Anti-Flag affinity gel, anti-Flag M2 antibodies, and Flag peptide were from Sigma. Monoclonal mouse anti-Myc-epitope antibody was from Santa Cruz Biotechnology. Horseradish peroxidase-conjugated goat anti-mouse antibodies were from Jackson ImmunoResearch Laboratories. Kemptide was from Anaspec. Radioisotopes were from Perkin Elmer. Protease inhibitor mixture was from Calbiochem. Cell culture medium was from Cellgro, and fetal bovine serum was from Cellgro and Atlanta Biologicals. Tissue culture plates were from Fisher Scientific. KOD Hot Start DNA polymerase was from Novagen. Restriction enzymes and T4 DNA ligase were from New England Biolabs. Lipofectamine 2000 was from Life Technologies. 8-pCPT–cGMP, Rp-8-pCPT–cGMPS, and Rp-8-pCPT-PET–cGMPS were from Biolog. General supplies and chemicals were from Sigma and Fisher Scientific.
Expression plasmids
Flag-tagged expression constructs for PKG1α, PKG1β, PKG1β-CD, and RQ-PKG1α have been described previously (14, 40). RQ-PKG1β was created by swapping the PKG1β N terminus for the PKG1α N terminus in the mutant construct using an internal NcoI site. Myc-tagged human VASP was constructed by PCR using VSV-tagged VASP as a template (41) with the following primers: 5′-GCTGAATTCGCCGCCATGGCTGCCATCCGGAAGAAAC-3 (sense) and 5′-GCTCTCGAGTCACAAGACAAGGCACCC-3′ (antisense). The PCR product was digested with EcoRI and XhoI and ligated into a vector that put an in-frame Myc tag at the N terminus. Additional PKG1α mutations were generated by overlapping extension PCR using high-fidelity KOD Hot Start DNA polymerase (42). All constructs that underwent a PCR step were sequenced to ensure the presence of the desired mutation and the absence of PCR-generated errors.
Protein expression and purification
For kinase assays, 293T cells were plated into 6-well cluster dishes such that they were 90–95% confluent the next day. For each protein preparation, three wells were transfected with 2 μg Flag-tagged expression construct per well using Lipofectamine 2000. The next day (16–20 h later), cells were scraped in PBS (10 mm Na2HPO4, 2 mm KH2PO4, 137 mm NaCl, 2.7 mm KCl, pH 7.4), pelleted by centrifugation, and snap frozen in a dry ice-ethanol bath. The pellets were stored at −80 °C until needed. Cells were lysed in 400 μl ice-cold buffer A (PBS, 0.1% NP-40) with 1× protease inhibitor mixture (Calbiochem), and lysates were cleared by centrifugation (16,000 × g, 10 min at 4 °C). Cleared lysates were incubated with 20 μl anti-Flag affinity gel for one hour at 4 °C with constant mixing. Beads were washed twice with 200 μl buffer A, twice with 200 μl PBS containing a total of 500 mm NaCl, and twice with 200 μl PBS. Bound protein was eluted by incubating the beads 4 times with 10 μl elution buffer (PBS with 100 μg/ml Flag peptide). For each elution step, the beads were incubated with buffer for 5 min on ice. The four eluates for each protein were pooled.
For H/DX-MS analysis, 293T cells were plated into eight 10-cm tissue culture dishes, and the next day four dishes each were transfected with 12 μg Flag-tagged expression construct encoding WT or RQ PKG1β using Lipofectamine 2000. Cells were harvested as described above, each pellet was lysed in 400 μl ice-cold buffer A (PBS, 0.1% NP-40) with 1× protease inhibitor mixture (Calbiochem), and lysates were cleared by centrifugation (16,000 × g, 10 min at 4 °C). Cleared lysates were incubated with 80 μl anti-Flag affinity gel for one hour at 4 °C with constant mixing. Beads were washed twice with 500 μl buffer A, twice with 500 μl PBS containing a total of 500 mm NaCl, and twice with 500 μl 10 mm Tris (pH 7.2) with 150 mm NaCl. Bound proteins were eluted by incubating the beads 4 times with 40 μl elution buffer (10 mm Tris [pH 7.2], 150 mm NaCl and 200 μg/ml Flag peptide). For each elution step, the beads were incubated with buffer for 5 min on ice. The four eluates for each protein were pooled and dialyzed overnight at 4 °C in 10 mm Tris (pH 7.2), 150 mm NaCl. The proteins were concentrated to ∼2 µg/μl using a 10,000-molecular-weight-cutoff Microcon device (Millipore).
Kinase assays
Flag-tagged purified WT and mutant PKG1 proteins were diluted to ∼1 ng/μl in KPEM buffer (10 mm potassium phosphate [pH 7.0], 1 mm EDTA, and 25 mm mercaptoethanol) with 0.1% BSA. To avoid possible oxidation-induced activation of PKG1α, assays were performed within one hour of purification. Diluted kinase (10 μl) was added to 5 μl 3× kinase reaction mix (120 mm HEPES [pH 7.0], 24 μg Kemptide, 30 mm MgCl2, 180 μm ATP, 1.8 μCi 32PO4–γ-ATP) with or without cGMP at a final concentration of 10 μm. In some reactions, the indicated amounts of inhibitors were added to reaction mixes. Reactions were incubated at 30 °C and stopped at 1.5, 2, or 5 min by spotting on P81 phosphocellulose paper. Unincorporated 32PO4–γ-ATP was removed by washing 4 times in 2 liters of 0.452% o-phosphoric acid. 32PO4 incorporation was measured by liquid scintillation counting. Curve fitting for kinase reactions with inhibitors was done by nonparametric analysis using Graphpad Prism. For Rp-8-pCPT–cGMPS, which only showed agonist activity in the full-length RQ mutants, we used log (agonist) versus response (stimulation). Because the assays using Rp-8-pCPT-PET–cGMPS with full-length kinases showed a partial agonist and then inhibitory response, we used the bell-shaped curve. Finally, for assays examining the effect of Rp-8-pCPT–cGMPS on PKGI catalytic domain activity, we used log (agonist) versus response (inhibition).
Autophosphorylation assays
For in vitro autophosphorylation reactions, purified WT and RQ mutant PKG1α and PKG1β were incubated under reaction conditions identical to those used for kinases assays, except that Kemptide was omitted. Reaction mixtures were incubated at 30 °C for 1.5 min and were stopped by adding SDS sample buffer. Reactants were separated by SDS-PAGE, and proteins were transferred to Immobilon P. Phosphate incorporation was determined by autoradiography. Equal loading was determined by Western blotting with anti-Flag antibodies.
For autophosphorylation studies in intact cells, 293T cells were seeded into 6-well cluster dishes the day before transfection, such that they would be 80–90% confluent at the time of transfection. The cells were transfected with expression vectors for Flag-tagged WT and RQ mutant PKG1α or PKG1β using Lipofectamine 2000 (Life Technologies). Four hours posttransfection, cells were placed in phosphate-free DMEM supplemented with 100 μCi [32P]orthophosphate per well. Three hours later, a final concentration of 250 μM 8-pCPT–cGMP (Biolog) was added to the appropriate wells for one hour. The cells were harvested by scraping in buffer A supplemented with 1× protease inhibitor mixture (Calbiochem). Cleared lysates were incubated with anti-Flag M2 affinity gel (Sigma) for one hour at 4 °C. The beads were washed, and phosphorylation levels in bound proteins were determined by SDS-PAGE/autoradiography.
Molecular modeling of inactive PKG1β
The model of PKG1β in its inactive state was generated using the Swiss Model server (21). Briefly, the primary sequence of PKG1β was used as the input, with the PKA in structure 2QCS used as the template (43). This PKA structure contains the catalytic domain bound by the regulatory domain and represents the inactive conformation, and each PKG1β domain was modeled independently. To create the full model, the server-generated structures were aligned with the PKA regulatory and catalytic chains in 2QCS using PyMol (Schrödinger, LLC).
Hydrogen/deuterium exchange analysis
Peptide fragment optimization was performed by incubating 2 μg purified WT or RQ-PKG1β in quench buffer containing different final concentrations of guanidium HCl (0.5, 1, 2, and 4 M). We found that 2 M guanidium gave the best peptide coverage (>90%). Before performing the H/D exchange reactions, aliquots of WT and RQ-PKG1β were incubated with 20 μM cGMP on ice for 1 h. Exchange reactions were initiated by adding 30 μl PKG to 90 μl buffered D2O (12.5 mm Tris, pH 7.2, and 150 mm NaCl). At the appropriate time points, H/D exchange was terminated by adding 24 μl of the exchange reaction to 36 μl ice-cold quench buffer (3.6 M guanidium HCl, 0.8% formic acid, and 16.6% glycerol). Undeuterated samples were prepared by incubating 6 μl PKG for 10 s with 18 μl buffered H2O (as described above), followed by the addition of 36 μl quench buffer. Fully deuterated reference samples were prepared by incubating 6 μl PKG with 18 μl D2O–0.8% formic acid at room temperature overnight and then adding quench buffer. All samples were split into two 30-μl aliquots, frozen on dry ice, and stored at −80°C until analysis by proteolysis/MS. H/D exchange experiments were performed twice using two independent protein preparations for both WT and RQ-PKG1β. Tables S1 and S2 contain H/D exchange data for all of the manually confirmed peptides identified in the first H/DX-MS experiment.
MS, quantification of deuterium incorporation, corrections for back exchange, and data analysis were performed as described previously (44). Briefly, samples were quickly thawed at 4 °C, proteolyzed by passing through a pepsin column, and collected on a C18 reverse-phase column at 0 °C. Peptides were eluted using a linear gradient of 0.046% (v/v) TFA, 6.4% (v/v) acetonitrile to 0.03% (v/v) TFA, and 38.4% (v/v) acetonitrile over a 30-min run. Eluted peptides were analyzed using an Orbitrap Elite mass spectrometer (ThermoFisher) and identified using Proteome Discoverer software (ThermoFisher). The centroids of the isotopic envelopes of nondeuterated, time course-deuterated, and fully deuterated peptides were measured using DXMS Explorer software (Sierra Analytics). Corrections for back exchange were made by measuring deuterium loss in peptides derived from the fully deuterated samples.
Data availability
All of the relevant data are contained within the article.
Supplementary Material
This article contains supporting information.
Author contributions—M. H. C., S. A., T. H., A. T., S. L., and D. E. C. investigation; C. K., R. B. P., and D. E. C. formal analysis; C. K. and D. E. C. visualization; R. B. P. resources; R. B. P. funding acquisition; R. B. P. and D. E. C. writing-review and editing; D. E. C. conceptualization; D. E. C. data curation; D. E. C. supervision; D. E. C. validation; D. E. C. methodology; D. E. C. writing-original draft; D. E. C. project administration.
Funding and additional information—This work was supported by the National Institutes of Health grants R01-HL132141 (to R. B. P.), R01-GM090161 (to C. K.), and 1U19AI117905, R01-GM020501 (to S. L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare no conflicts of interest with the content of this article.
- PKG
- protein kinase G
- AI
- autoinhibitory
- H/D
- hydrogen-deuterium
- H/DX-MS
- hydrogen/deuterium exchange MS
- Rp-8-pCPT-PET–cGMPS
- 8-(4-chlorophenylthio)-β-phenyl-1,N2-ethenoguanosine–3′5′-cyclic monophophorothioate, Rp-isomer
- 8-pCPT–cGMP
- 8-(4-Chlorophenylthio)-guanosine–3′5′-cyclic monophophosphate
- Rp-8-pCPT–cGMPS
- 8-(4-chlorophenylthio)guanosine–3′5′-cyclic monophophorothioate, Rp-isomer.
References
- 1. Hofmann F., Bernhard D., Lukowski R., and Weinmeister P. (2009) cGMP regulated protein kinases (cGK). Handb. Exp. Pharmacol. 191, 137–162 10.1007/978-3-540-68964-5_8 [DOI] [PubMed] [Google Scholar]
- 2. Francis S. H., Busch J. L., Corbin J. D., and Sibley D. (2010) cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62, 525–563 10.1124/pr.110.002907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Friebe A., Sandner P., and Schmidtko A. (2017) Meeting report of the 8th International Conference on cGMP “cGMP: generators, effectors, and therapeutic implications” at Bamberg, Germany, from June 23 to 25, 2017. Naunyn. Schmiedebergs Arch. Pharmacol. 390, 1177–1188 10.1007/s00210-017-1429-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Casteel D. E., Zhuang S., Gudi T., Tang J., Vuica M., Desiderio S., and Pilz R. B. (2002) cGMP-dependent protein kinase I beta physically and functionally interacts with the transcriptional regulator TFII-I. J. Biol. Chem. 277, 32003–32014 10.1074/jbc.M112332200 [DOI] [PubMed] [Google Scholar]
- 5. Kato M., Blanton R., Wang G. R., Judson T. J., Abe Y., Myoishi M., Karas R. H., and Mendelsohn M. E. (2012) Direct binding and regulation of RhoA protein by cyclic GMP-dependent protein kinase Ialpha. J. Biol. Chem. 287, 41342–41351 10.1074/jbc.M112.421040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schlossmann J., Ammendola A., Ashman K., Zong X., Huber A., Neubauer G., Wang G. X., Allescher H. D., Korth M., Wilm M., Hofmann F., and Ruth P. (2000) Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature 404, 197–201 10.1038/35004606 [DOI] [PubMed] [Google Scholar]
- 7. Surks H. K., Mochizuki N., Kasai Y., Georgescu S. P., Tang K. M., Ito M., Lincoln T. M., and Mendelsohn M. E. (1999) Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 286, 1583–1587 10.1126/science.286.5444.1583 [DOI] [PubMed] [Google Scholar]
- 8. Tang K. M., Wang G-R., Lu P., Karas R. H., Aronovitz M., Heximer S. P., Kaltenbronn K. M., Blumer K. J., Siderovski D. P., Zhu Y., Mendelsohn M. E., Tang M., and Wang G. (2003) Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 9, 1506–1512 10.1038/nm958 [DOI] [PubMed] [Google Scholar]
- 9. Yuasa K., Michibata H., Omori K., and Yanaka N. (1999) A novel interaction of cGMP-dependent protein kinase I with troponin T. J. Biol. Chem. 274, 37429–37434 10.1074/jbc.274.52.37429 [DOI] [PubMed] [Google Scholar]
- 10. Yuasa K., Omori K., and Yanaka N. (2000) Binding and phosphorylation of a novel male germ cell-specific cGMP-dependent protein kinase-anchoring protein by cGMP-dependent protein kinase Ialpha. J. Biol. Chem. 275, 4897–4905 10.1074/jbc.275.7.4897 [DOI] [PubMed] [Google Scholar]
- 11. Busch J. L., Bessay E. P., Francis S. H., and Corbin J. D. (2002) A conserved serine juxtaposed to the pseudosubstrate site of type I cGMP-dependent protein kinase contributes strongly to autoinhibition and lower cGMP affinity. J. Biol. Chem. 277, 34048–34054 10.1074/jbc.M202761200 [DOI] [PubMed] [Google Scholar]
- 12. Smith J. A., Francis S. H., Walsh K. A., Kumar S., and Corbin J. D. (1996) Autophosphorylation of type Ibeta cGMP-dependent protein kinase increases basal catalytic activity and enhances allosteric activation by cGMP or cAMP. J. Biol. Chem. 271, 20756–20762 10.1074/jbc.271.34.20756 [DOI] [PubMed] [Google Scholar]
- 13. Alverdi V., Mazon H., Versluis C., Hemrika W., Esposito G., van den Heuvel R., Scholten A., and Heck A. J. (2008) cGMP-binding prepares PKG for substrate binding by disclosing the C-terminal domain. J. Mol. Biol. 375, 1380–1393 10.1016/j.jmb.2007.11.053 [DOI] [PubMed] [Google Scholar]
- 14. Guo D. C., Regalado E., Casteel D. E., Santos-Cortez R. L., Gong L., Kim J. J., Dyack S., Horne S. G., Chang G., Jondeau G., Boileau C., Coselli J. S., Li Z., Leal S. M., Shendure J., National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project. (2013) Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet. 93, 398–404 10.1016/j.ajhg.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwaerzer G. K., Kalyanaraman H., Casteel D. E., Dalton N. D., Gu Y., Lee S., Zhuang S., Wahwah N., Schilling J. M., Patel H. H., Zhang Q., Makino A., Milewicz D. M., Peterson K. L., Boss G. R., et al. (2019) Aortic pathology from protein kinase G activation is prevented by an antioxidant vitamin B12 analog. Nat. Commun. 10, 3533 10.1038/s41467-019-11389-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aitken A., Hemmings B. A., and Hofmann F. (1984) Identification of the residues on cyclic GMP-dependent protein kinase that are autophosphorylated in the presence of cyclic AMP and cyclic GMP. Biochim. Biophys. Acta 790, 219–225 10.1016/0167-4838(84)90025-6 [DOI] [PubMed] [Google Scholar]
- 17. Francis S. H., Smith J. A., Colbran J. L., Grimes K., Walsh K. A., Kumar S., and Corbin J. D. (1996) Arginine 75 in the pseudosubstrate sequence of type Ibeta cGMP-dependent protein kinase is critical for autoinhibition, although autophosphorylated serine 63 is outside this sequence. J. Biol. Chem. 271, 20748–20755 10.1074/jbc.271.34.20748 [DOI] [PubMed] [Google Scholar]
- 18. Feil R., Bigl M., Ruth P., and Hofmann F. (1993) Expression of cGMP-dependent protein kinase in Escherichia coli. Mol. Cell Biochem. 127–128, 71–80 10.1007/BF01076758 [DOI] [PubMed] [Google Scholar]
- 19. Halbrugge M., Friedrich C., Eigenthaler M., Schanzenbacher P., and Walter U. (1990) Stoichiometric and reversible phosphorylation of a 46-kDa protein in human platelets in response to cGMP- and cAMP-elevating vasodilators. J. Biol. Chem. 265, 3088–3093 [PubMed] [Google Scholar]
- 20. Butt E., Abel K., Krieger M., Palm D., Hoppe V., Hoppe J., and Walter U. (1994) cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J. Biol. Chem. 269, 14509–14517 [PubMed] [Google Scholar]
- 21. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 10.1093/nar/gku340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pires D. E., Ascher D. B., and Blundell T. L. (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42, W314–W319 10.1093/nar/gku411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pires D. E., Ascher D. B., and Blundell T. L. (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342 10.1093/bioinformatics/btt691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Worth C. L., Preissner R., and Blundell T. L. (2011) SDM–a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 39, W215–W222 10.1093/nar/gkr363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruth P., Landgraf W., Keilbach A., May B., Egleme C., and Hofmann F. (1991) The activation of expressed cGMP-dependent protein kinase isozymes I alpha and I beta is determined by the different amino-termini. Eur. J. Biochem. 202, 1339–1344 10.1111/j.1432-1033.1991.tb16509.x [DOI] [PubMed] [Google Scholar]
- 26. Ruth P., Pfeifer A., Kamm S., Klatt P., Dostmann W. R., and Hofmann F. (1997) Identification of the amino acid sequences responsible for high affinity activation of cGMP kinase Ialpha. J. Biol. Chem. 272, 10522–10528 10.1074/jbc.272.16.10522 [DOI] [PubMed] [Google Scholar]
- 27. de Jonge H. R., and Rosen O. M. (1977) Self-phosphorylation of cyclic guanosine 3':5'-monophosphate-dependent protein kinase from bovine lung. Effect of cyclic adenosine 3':5'-monophosphate, cyclic guanosine 3':5'-monophosphate and histone. J. Biol. Chem. 252, 2780–2783 [PubMed] [Google Scholar]
- 28. Vallur R., Kalbacher H., and Feil R. (2014) Catalytic activity of cGMP-dependent protein kinase type I in intact cells is independent of N-terminal autophosphorylation. PLoS ONE 9, e98946 10.1371/journal.pone.0098946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Francis S. H., Morris G. Z., and Corbin J. D. (2008) Molecular mechanisms that could contribute to prolonged effectiveness of PDE5 inhibitors to improve erectile function. Int. J. Impot. Res. 20, 333–342 10.1038/ijir.2008.4 [DOI] [PubMed] [Google Scholar]
- 30. Wolfertstetter S., Huettner J. P., and Schlossmann J. (2013) cGMP-dependent protein kinase inhibitors in health and disease. Pharmaceuticals 6, 269–286 10.3390/ph6020269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valtcheva N., Nestorov P., Beck A., Russwurm M., Hillenbrand M., Weinmeister P., and Feil R. (2009) The commonly used cGMP-dependent protein kinase type I (cGKI) inhibitor Rp-8-Br-PET-cGMPS can activate cGKI in vitro and in intact cells. J. Biol. Chem. 284, 556–562 10.1074/jbc.M806161200 [DOI] [PubMed] [Google Scholar]
- 32. Taylor M. S., Okwuchukwuasanya C., Nickl C. K., Tegge W., Brayden J. E., and Dostmann W. R. (2004) Inhibition of cGMP-dependent protein kinase by the cell-permeable peptide DT-2 reveals a novel mechanism of vasoregulation. Mol. Pharmacol. 65, 1111–1119 10.1124/mol.65.5.1111 [DOI] [PubMed] [Google Scholar]
- 33. Osborne B. W., Wu J., McFarland C. J., Nickl C. K., Sankaran B., Casteel D. E., Woods V. L. Jr., Kornev A. P., Taylor S. S., and Dostmann W. R. (2011) Crystal structure of cGMP-dependent protein kinase reveals novel site of interchain communication. Structure 19, 1317–1327 10.1016/j.str.2011.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim J. J., Lorenz R., Arold S. T., Reger A. S., Sankaran B., Casteel D. E., Herberg F. W., and Kim C. (2016) Crystal structure of PKG I:cGMP complex reveals a cGMP-mediated dimeric interface that facilitates cGMP-induced activation. Structure 24, 710–720 10.1016/j.str.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herberg F. W., Dostmann W. R., Zorn M., Davis S. J., and Taylor S. S. (1994) Crosstalk between domains in the regulatory subunit of cAMP-dependent protein kinase: influence of amino terminus on cAMP binding and holoenzyme formation. Biochemistry 33, 7485–7494 10.1021/bi00189a057 [DOI] [PubMed] [Google Scholar]
- 36. Badireddy S., Yunfeng G., Ritchie M., Akamine P., Wu J., Kim C. W., Taylor S. S., Qingsong L., Swaminathan K., and Anand G. S. (2011) Cyclic AMP analog blocks kinase activation by stabilizing inactive conformation: conformational selection highlights a new concept in allosteric inhibitor design. Mol. Cell. Proteomics 10, M110.004390 10.1074/mcp.M110.004390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simanshu D. K., Nissley D. V., and McCormick F. (2017) RAS proteins and their regulators in human disease. Cell 170, 17–33 10.1016/j.cell.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Han X. (2014) Constitutively active chemokine CXC receptors. Adv. Pharmacol. 70, 265–301 10.1016/B978-0-12-417197-8.00009-2 [DOI] [PubMed] [Google Scholar]
- 39. Malik R. U., Ritt M., DeVree B. T., Neubig R. R., Sunahara R. K., and Sivaramakrishnan S. (2013) Detection of G protein-selective G protein-coupled receptor (GPCR) conformations in live cells. J. Biol. Chem. 288, 17167–17178 10.1074/jbc.M113.464065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schwappacher R., Rangaswami H., Su-Yuo J., Hassad A., Spitler R., and Casteel D. E. (2013) cGMP-dependent protein kinase Ibeta regulates breast cancer cell migration and invasion via interaction with the actin/myosin-associated protein caldesmon. J. Cell Sci. 126, 1626–1636 10.1242/jcs.118190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhuang S., Nguyen G. T., Chen Y., Gudi T., Eigenthaler M., Jarchau T., Walter U., Boss G. R., and Pilz R. B. (2004) Vasodilator-stimulated phosphoprotein activation of serum-response element-dependent transcription occurs downstream of RhoA and is inhibited by cGMP-dependent protein kinase phosphorylation. J. Biol. Chem. 279, 10397–10407 10.1074/jbc.M313048200 [DOI] [PubMed] [Google Scholar]
- 42. Higuchi R., Krummel B., and Saiki R. K. (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16, 7351–7367 10.1093/nar/16.15.7351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim C., Cheng C. Y., Saldanha S. A., and Taylor S. S. (2007) PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043 10.1016/j.cell.2007.07.018 [DOI] [PubMed] [Google Scholar]
- 44. Marsh J. J., Guan H. S., Li S., Chiles P. G., Tran D., and Morris T. A. (2013) Structural insights into fibrinogen dynamics using amide hydrogen/deuterium exchange mass spectrometry. Biochemistry 52, 5491–5502 10.1021/bi4007995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the relevant data are contained within the article.