

Abstract
ATP11C, a member of the P4-ATPase flippase, translocates phosphatidylserine from the outer to the inner plasma membrane leaflet, and maintains the asymmetric distribution of phosphatidylserine in the living cell. We present the crystal structures of a human plasma membrane flippase, ATP11C–CDC50A complex, in a stabilized E2P conformation. The structure revealed a deep longitudinal crevice along transmembrane helices continuing from the cell surface to the phospholipid occlusion site in the middle of the membrane. We observed that the extension of the crevice on the exoplasmic side is open, and the complex is therefore in an outward-open E2P state, similar to a recently reported cryo-EM structure of yeast flippase Drs2p–Cdc50p complex. We noted extra densities, most likely bound phosphatidylserines, in the crevice and in its extension to the extracellular side. One was close to the phosphatidylserine occlusion site as previously reported for the human ATP8A1–CDC50A complex, and the other in a cavity at the surface of the exoplasmic leaflet of the bilayer. Substitutions in either of the binding sites or along the path between them impaired specific ATPase and transport activities. These results provide evidence that the observed crevice is the conduit along that phosphatidylserine traverses from the outer leaflet to its occlusion site in the membrane and suggest that the exoplasmic cavity is important for phospholipid recognition. They also yield insights into how phosphatidylserine is incorporated from the outer leaflet of the plasma membrane into the transmembrane.
Keywords: Apoptosis, ATPase, crystal structure, phospholipid, plasma membrane, flippase, P-type ATPase
Phospholipids are asymmetrically distributed between the outer and inner leaflets in the plasma membrane of eukaryotic cells. Aminophospholipids such as phosphatidylserine (PtdSer) and phosphatidylethanolamine (PtdEtn) are confined to the inner leaflet, whereas phosphatidylcholine (PtdCho) and sphingomyelin are enriched in the outer leaflet (1). The asymmetric distribution of phospholipids is widely conserved in eukaryotes, being tuned for barrier functions and various signal transductions on the plasma membrane (2). On occasion, this phospholipid asymmetry in the plasma membrane is disrupted, exposing PtdSer on the cell surface. Cells undergoing apoptosis expose PtdSer as an “eat me” signal to phagocytes (3, 4). Activated platelets also display PtdSer as a scaffold for clotting enzyme reactions (5). The amphipathic nature of phospholipids prevents their spontaneous flip-flop movement across the lipid bilayer in most cases, and the translocation of phospholipids therefore needs membrane proteins to overcome the energetic barrier required for phospholipid translocation (2).
Although scramblases mediate nonspecific and bidirectional movement of phospholipids between inner and outer leaflets (6), flippases exhibit ATP-driven, directional and up-hill translocation of phospholipids from the outer to the inner leaflets against their concentration gradient across the membrane bilayer (7–9). Different from other members of cation-transporting P-type ATPases (10–13), type IV P-type ATPase (P4-ATPase) comprises a subfamily of P-type ATPases that transports phospholipids (14). Among all 14 members of P4-ATPases in humans, ATP11A and ATP11C work as aminophospholipid-specific flippases at the plasma membrane (3, 4). They require an accessary subunit, CDC50A, for their correct localization and functional expression on the plasma membrane (15–17). In fact, cells lacking ATP11A and ATP11C, or CDC50A almost completely lose flippase activity for PtdSer and PtdEtn at the plasma membrane, resulting in failure to maintain the asymmetric accumulation of PtdSer in the inner leaflet. Their inactivation causes PtdSer exposure on the cell surface. In apoptotic cells, ATP11A and ATP11C are subjected to a caspase-mediated proteolysis and irreversible inactivation. It has been proposed that calcium ions likely also inhibit the flippase activity for calcium-dependent PtdSer exposure in activated platelets or lymphocytes (18). Regulation of aminophospholipid asymmetry by flippases is physiologically important; ATP11A-deficient mice are embryo lethal (19), and ATP11C-deficient mice display pleiotropic phenotypes such as B-cell lymphopenia (20, 21), cholestasis (22), mild anemia (23), and dystocia. Recently, mutations in the ATP11C gene were identified in patients who suffered from anemia (24).
The directional translocation of specific phospholipids mediated by P4-ATPases is achieved by the cyclic conversion of enzyme conformations, E1, E2, and their autophosphorylated forms E1P and E2P, similar to the Post-Albers–type reaction scheme (14, 25) for cation-transporting P-type ATPases. However, despite the huge size of phospholipids relative to inorganic cations, the mechanism by which P4-flippases translocate phospholipids across the membrane, the so-called “giant substrate problem” (26, 27), has remained elusive until recently and yet is of considerable interest. Now cryo-EM structures of flippases yeast Drs2p—cdc50p (28) and human ATP8A1—CDC50A complexes (29) have greatly advanced our understanding of the underlying molecular mechanism. In the Drs2p structure, there is a membrane crevice, which is most likely the conduit for the translocated phospholipid, although no phospholipids were found there. Several key reaction intermediate structures, including the PtdSer-occluded form, are described in a cryo-EM analysis of human ATP8A1. Unexpectedly, the exoplasmic gate is substantially closed in the E2P state of ATP8A1. Here, we describe a crystal structure of a bona fide human plasma membrane flippase ATP11C—CDC50A complex in the outward-open E2P conformation, analyzed to 3.9 Å resolution. In the crystal structure, we found two extra electron densities in the putative lipid translocation conduit, most likely bound PtdSer. Together with functional analysis, our data provide consolidating evidence for a PtdSer pathway from the outer leaflet to the mid-membrane–binding site.
Results and discussion
Overall structure of the outward-open conformation
Human ATP11C and CDC50A were expressed using the BacMam system (Fig. S1) (see “Experimental procedures”) (30). Purified and deglycosylated proteins were mixed with dioleoylphosphatidylcholine (DOPC) (31), and crystallized in the presence of phosphate analog (32) beryllium fluoride (BeFx) and dioleoylphosphatidylserine (DOPS). Crystals were harvested in the presence of excess DOPS, which was key to preservation of crystal quality. X-ray diffraction data from more than 1,500 individual crystals were merged, and the structure was determined by molecular replacement using the atomic model of the E2BeF state of ATP8A1 (29) as a search model, at a resolution of 3.9 Å with acceptable statistics of Rwork/Rfree = 27.9/34.7 (Fig. S1, Table 1). As seen in most of the other crystallized P-type ATPases, molecular packing occurs as type I crystals in which complexes are embedded in the lipid bilayer (33). The asymmetric unit of the crystal consists of four protomers of ATP11C—CDC50A. Due to different crystal contacts, the appearance of the electron density map differs significantly for each protomer (Fig. S2). Despite limited resolution, however, well-ordered regions, especially the transmembrane (TM) region of protomers A and B were visible at side chain level (Fig. S3). In contrast, protomers C and D showed poor electron densities, which may relate to the relatively large gap between Rwork and Rfree values (6.9%). The overall molecular conformations of the four protomers are essentially the same, although structural variations in some of the loop structures exist (Fig. S2). We therefore focus on the well-ordered protomer A in what follows.
Table 1.
Data collection and refinement statistics
| ATP11 C E2P | |
|---|---|
| Data collection | |
| Number of crystals | 1,588 |
| Resolution (Å)1 | 4.7 × 4.2 × 3.9 (4.9–4.7a, 4.4–4.2b, 4.0–3.9c)2 |
| Space group | P 212121 |
| Cell dimensions | |
| a, b, c (Å) | 100.46, 232.82, 498.89 |
| α, β, γ (˚) | 90, 90, 90 |
| Rmerge | 1.141 (–)3 |
| Rpim | 0.0419 (–)3 |
| I/σI | 15.88 (6.00a, 1.82b, 0.21c) |
| CC1/2 | 0.92 (0.993a, 0.964b, 0.864c) |
| Completeness (%) | 71.32 (81.84a, 35.29b, 11.64c) |
| Redundancy | 776.9 (788.8a, 759.4b, 698.5c) |
| Refinement | |
| Resolution (Å) | 50–3.9 (4.0–3.9) |
| No. of reflections | 82,706,647 (7,342,288) |
| Rwork/Rfree (%) | 27.9/34.8 (26.8/32.6) |
| Wilson B-factor (Å2) | 98.19 |
| No. of atoms | 45,171 |
| Protein | 44,727 |
| Ligands | 444 |
| Average B-factor | 163.19 |
| Protein (Å2) | 163.10 |
| Ligands (Å2) | 172.65 |
| Root mean square deviation | |
| Bond lengths (Å) | 0.007 |
| Bond angles (˚) | 1.35 |
1 The diffraction data are anisotropic. The resolution limits given are for the a*, b*, and c* axes, respectively.
2 Statistics for the indicated resolution shells (a, b, and c) are shown in parentheses.
3 Statistics for the highest-resolution shell are not given due to the large number of merged crystals and their strong anisotropy.
Like other members of the well-characterized, cation-transporting P-type ATPases (11–13), the uphill translocation of aminophospholipids by ATP11C is achieved according to the Post-Albers type reaction scheme (14, 25) (Fig. 1A). The outward-open E2P conformation captures PtdSer or PtdEtn on the outer leaflet and induces dephosphorylation of E2P, thus PtdSer- or PtdEtn-dependent ATP hydrolysis can be detected (Fig. 1B), similar to the inward transport of K+ by Na+,K+-ATPase and H+,K+-ATPase. As we included BeFx and PtdSer for the crystallization, the molecular conformation (34) was expected to be the outward-open E2P state in which PtdSer is bound to the conduit facing toward the exoplasmic side (Fig. 1A). In fact, the overall structure of the ATP11C—CDC50A complex (Fig. 1C) is very close to the corresponding E2P structures of recently reported flippases yeast Drs2p—Cdc50p (28) and human ATP8A1—CDC50A (29) complexes. The sequence identities of each catalytic subunit are 35.3 and 36.2%, respectively (Fig. S4, for sequence alignment) (35). The molecular conformation is also defined by the relative orientations of the cytoplasmic domains (36) (actuator (A), phosphorylation (P), and nucleotide-binding (N) domains), and the arrangement of the 10 TM helices of the catalytic subunit ATP11C. The phosphate analog BeFx likely forms a covalent bond to the invariant aspartate in the 409DKTG signature sequence, which is covered by the 179DGE(S/T) motif located on the surface of the A domain to prevent spontaneous dephosphorylation by the bulk water (Fig. S5). In this conformation, the N domain is segregated from the P domain, and is expected to be relatively flexible compared with the other two domains due to the lack of intimate inter-domain interactions, and this is consistent with its poor electron density (Figs. S2 and S3). The relative orientation of the A and P domains in ATP11C is close to those observed in the Drs2p and ATP8A1 E2P states, and clearly different from that in the ATP8A1 E2-Pi transition state (Fig. S5), indicating that the present ATP11C structure adopts an E2P state. Arrangement of TM3–TM10 helices of ATP11C and those of Drs2p and ATP8A1 are very similar. However, conformation of TM1 and TM2 in these flippases vary (Fig. S5), which results in distinct structures of the exoplasmic gate (Fig. S6) as discussed later. The location of TM1 and TM2 in the ATP11C E2P state is close to that in the Drs2p E2P state, but clearly different from that in the ATP8A1 E2P state (Fig. S5).
Figure 1.
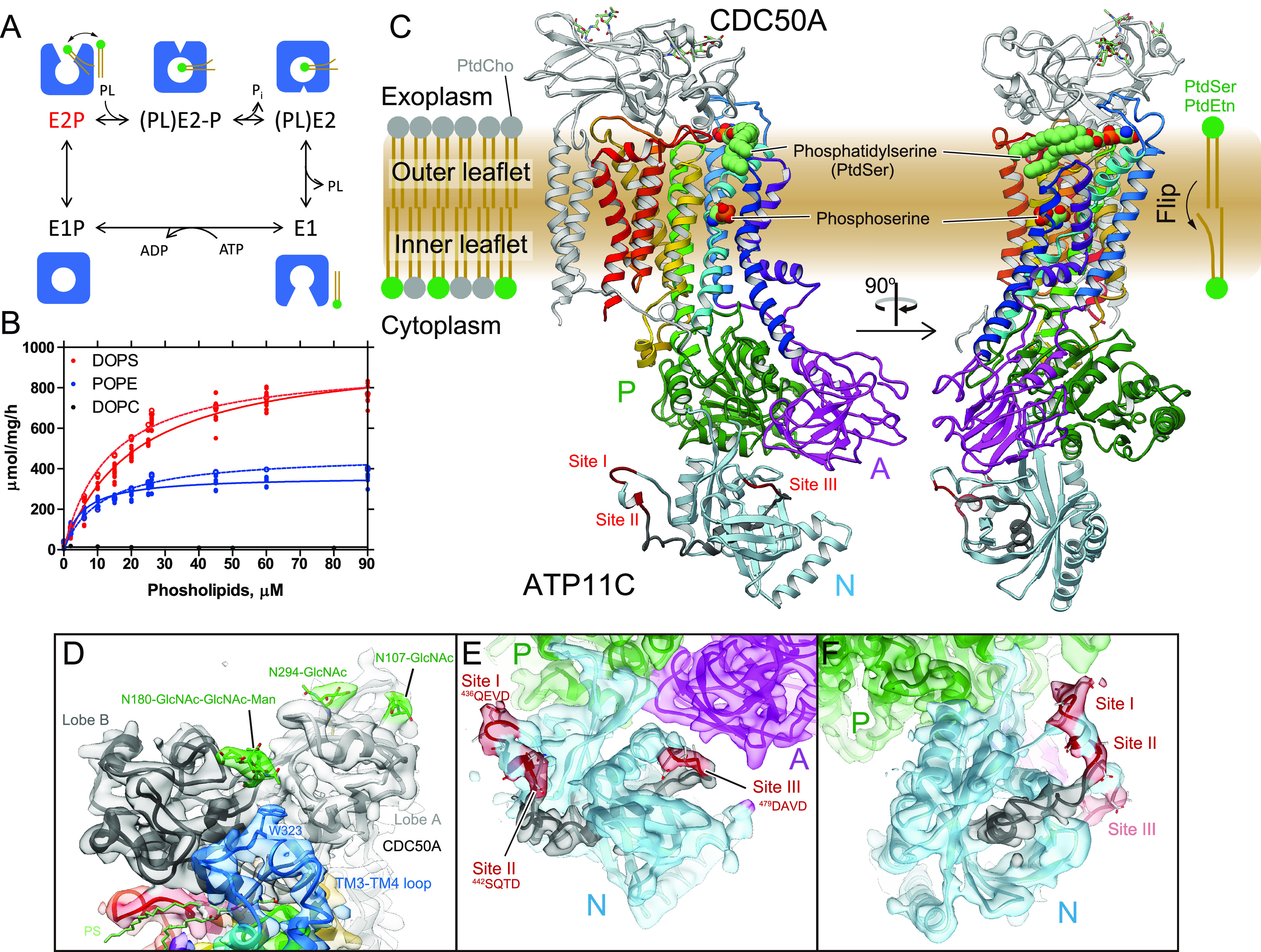
Crystal structure of ATP11C-CDC50A complex. A, reaction scheme of phospholipid (PL) translocation coupled with ATP hydrolysis. Cartoons represent molecular conformations of ATP11C—CDC50A complex (inward- or outward-, and open or occluded states). B, PL-dependent ATPase activity by the purified ATP11C—CDC50A complex. Specific ATPase activities of ATP11c_cryst—CDCQW complex (here referred to as “WT,” see “Experimental procedures”) in the presence of DOPS (red), POPE (blue), or DOPC (black) were plotted as a function of their concentrations (closed symbols and lines). Dotted lines indicate those of unmodified ATP11C—CDC50A complex. C, overall structure of the outward-open E2P state of the ATP11C—CDC50A complex in ribbon representation. Color of the ATP11C gradually changes from the N terminus (purple) to the C terminus (red). CDC50A subunit is shown in gray ribbon, and attached N-linked glycans are displayed as green sticks. A DOPS molecule (PtdSer) and its hydrophilic phosphoserine part are shown as spheres in the exoplasmic cavity and the occlusion site in the middle of the TM domain, respectively. Three cytoplasmic domains are indicated with different colors, and caspase recognition sequences at the N domain surface are indicated in red. Gray ribbon in the N domain thus indicates the region that is removed after the caspase cleavage. D, closed view of the exoplasmic region of CDC50A subunit. Surface represents electron density map (1.5σ). Lobe A (light gray) and lobe B (dark gray) are shown in different colors, and TM3-4 loop is shown in blue with the Trp-323 side chain fitted into the map. N-Linked glycans (green) are highlighted. E and F, close-up of the N domain (light blue) from two different viewpoints. Surface shows electron density map at 1.0σ contour level. Three caspase recognition sites (I, II, and III) and the region between them (Gly-466–Asn-477) are indicated in red and gray, respectively.
The CDC50A subunit consists of 2 TM helices with a long N-terminal tail and a short C terminus on the cytoplasmic side, and a large exoplasmic domain in which four N-linked (Asn-107, Asn-180, Asn-190, and Asn-294) and one O-linked (Ser-292) glycosylation site are located. We extensively investigated various combinations of glycosylation site mutants to improve protein expression and crystal quality, and found that the N190Q/S292W double mutant produced the best crystals, without affecting its PtdSer- or PtdEtn-dependent ATPase activities relative to the unmodified enzyme (Fig. 1B). Because the samples were treated with endoglycosidase to remove excess glycans during purification, an acetylglucosamine (GlcNAc) moiety is retained on each of the three remaining N-linked glycosylation sites. Interestingly, peptide:N-glycosidase treatment, which truncates all glycans including core GlcNAc, produced tiny crystals, suggesting that these GlcNAcs contribute to rather stable crystal packing. We modeled one GlcNAc moiety for each of the two sites (Asn-107, Asn-294), and three glycans at a position close to Asn180 (Asn-180—GlcNAc—GlcNAc—Man). A GlcNAc at the position of Asn-294 in protomer B likely makes a crystal contact with CDC50A subunit in the neighboring symmetry-related protomer A (Fig. S2), consistent with the tiny crystal formed using peptide:N-glycosidase–treated samples. The three glycans at Asn-180 fit into a groove formed between two lobes (lobe A and B) of CDC50A and Trp-323 (in the connecting loop between TM3 and TM4 of ATP11C and conserved in mammalian flippases that require CDC50A), like a wedge, thereby keeping them together and sterically protecting them from the endoglycosidase during purification (Fig. 1D).
The electron density map of the N domain allowed visualization of its overall folding at Cα level, and made it possible to assign the location of the three caspase-recognition sites (Fig. 1, E and F). Inactivation of ATP11A or ATP11C by effector caspases requires cleavage of multiple caspase sites located on the N domain, and a single site cleavage is not enough for full inactivation of the flippase (4, 18). There are three caspase-recognition sites in ATP11C (sites I–III, Fig. 1, Fig. S4), and these are located on loops at both ends of an α-helix–containing region. Site I and site II are very close to each other at the N-terminal side of the helix, and site III is at the C-terminal side. This region, especially the α-helical part between the caspase-recognition sites, is located at the center of the N domain scaffold, and seems to function like a bolt that holds together the surrounding α-helix and several β-sheets, and thus is obviously important for N domain folding. Interaction between this helix and surrounding N domain segments may be sufficient to keep its fold even if one of the caspase sites is cleaved. Cleavage at both ends of the helix must lead to irreversible unfolding of the N domain that is required for the ATP binding. Note, all three sites are exposed on the same side of the N domain with a distance of around 35∼40 Å between two regions (sites I, II, and site III). Such geometry of cleavage sites suggests that instantaneous two-site cleavage may occur by a caspase-3 dimer (37) upon apoptotic signal transduction, rather than single, independent cleavages, given that the distance between the catalytic center of the caspase-3 dimer is around 40 Å.
Outward-open conformation
According to the reaction scheme of P4-flippase (Fig. 1A), the outward-open E2P state, mimicked by the BeFx binding, is a reaction state responsible for the incorporation of PtdSer from the outer leaflet to the transport occlusion site. A structural requirement for the outward-open conformation is therefore the transient formation of a conduit that physically allows the translocation of the phospholipid head group from the outer leaflet to the occlusion site in the middle of the membrane. We found a longitudinal crevice in the peripheral region of TM in ATP11C (Fig. 2A, Fig. S6). The crevice is composed of TM2, TM4, and TM6 helices and runs along TM4, and is continuous from the exoplasmic surface of the lipid bilayer to the middle of the membrane (Fig. 4B). Thus, the unwound part of TM4 (356PVSM motif), which has been implicated in the lipid transport (27), is exposed to the hydrophobic bulk lipid. Comparison with other flippase structures in the corresponding reaction state reveals that the crevice in ATP11C is continuous and wider than that in the Drs2p E2P activated form (28) (see Fig. S6, E and F, for the comparison). In the ATP8A1 E2P state, a membrane crevice is not formed at all (Fig. S6) (29) due to the distinct arrangement of TM1 and TM2 in ATP8A1 compared with that in ATP11C (Fig. S5). The difference may be related to its longer C-terminal regulatory domain in ATP8A1 compared with that in ATP11C (∼38 amino acids) (38). The exoplasmic side of the crevice is closed in the PtdSer-occluded E2-Pi transition state of ATP8A1, and clearly different from that in the ATP11C E2P state (Fig. S6). In ATP11C, TM2 is kinked at Pro-94, an amino acid residue conserved in all mammalian P4-ATPases (Fig. 2B, Fig. S5), so that the exoplasmic side of TM2 departs from the central axis of the crevice in this region. This structural feature enables the formation of the wide and continuous crevice from the exoplasmic side to the PtdSer occlusion site near the unwound part of TM4. Substitution of Pro-94 for alanine significantly reduces apparent affinities for both PtdSer and PtdEtn (Fig. 2C), indicating that the TM2 kink is key for developing a crevice structure that is wide enough for passage of a phospholipid head group. We conclude that the observed longitudinal crevice is in fact the outward facing conduit that enables phospholipid translocation.
Figure 2.
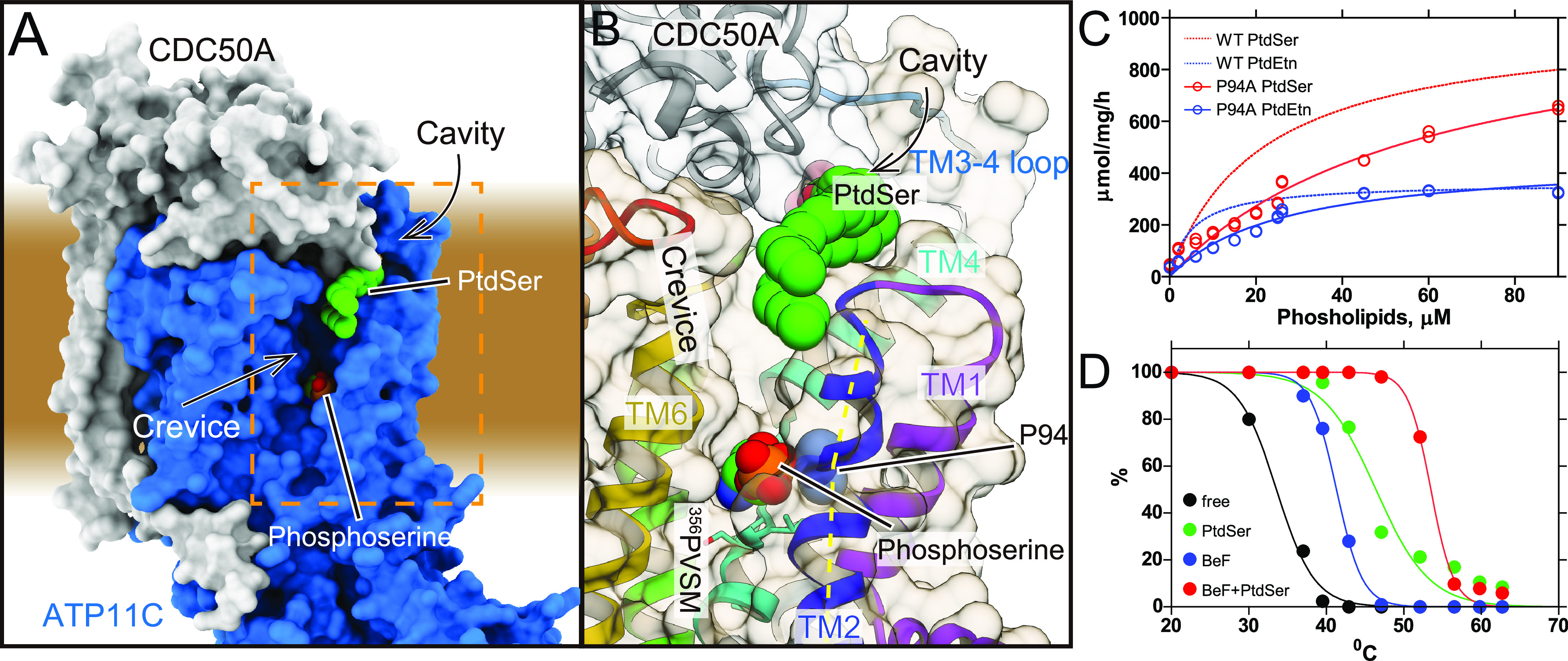
The crevice in the TM region. A, surface representation of the ATP11C—CDC50A complex shows the crevice in the TM region. Green spheres with CPK coloring represent phosphoserine at the occlusion site in the crevice, and PtdSer bound to the exoplasmic cavity. Surfaces of the atomic model of ATP11C and CDC50A are shown in blue and gray, respectively. B, close-up view of the membrane crevice indicated as a dotted box in A. The crevice is mostly composed of TM2, TM4, and TM6. Pro-94 makes a kink at the exoplasmic side of TM2 (yellow dotted lines), which exposes the unwound region of TM4 (PVSM, shown in sticks) to the lipid bilayer phase. C, PtdSer- or PtdEtn-dependent ATPase activities of WT (same as in Fig. 1) and P94A mutant as indicated in the figure. D, thermal stabilities of purified ATP11C—CDC50A complex determined by FSEC (see “Experimental procedures”). Peak values in the FSEC analysis were plotted as a function of treatment temperature in the absence (free) or presence of indicated substrates.
Figure 4.
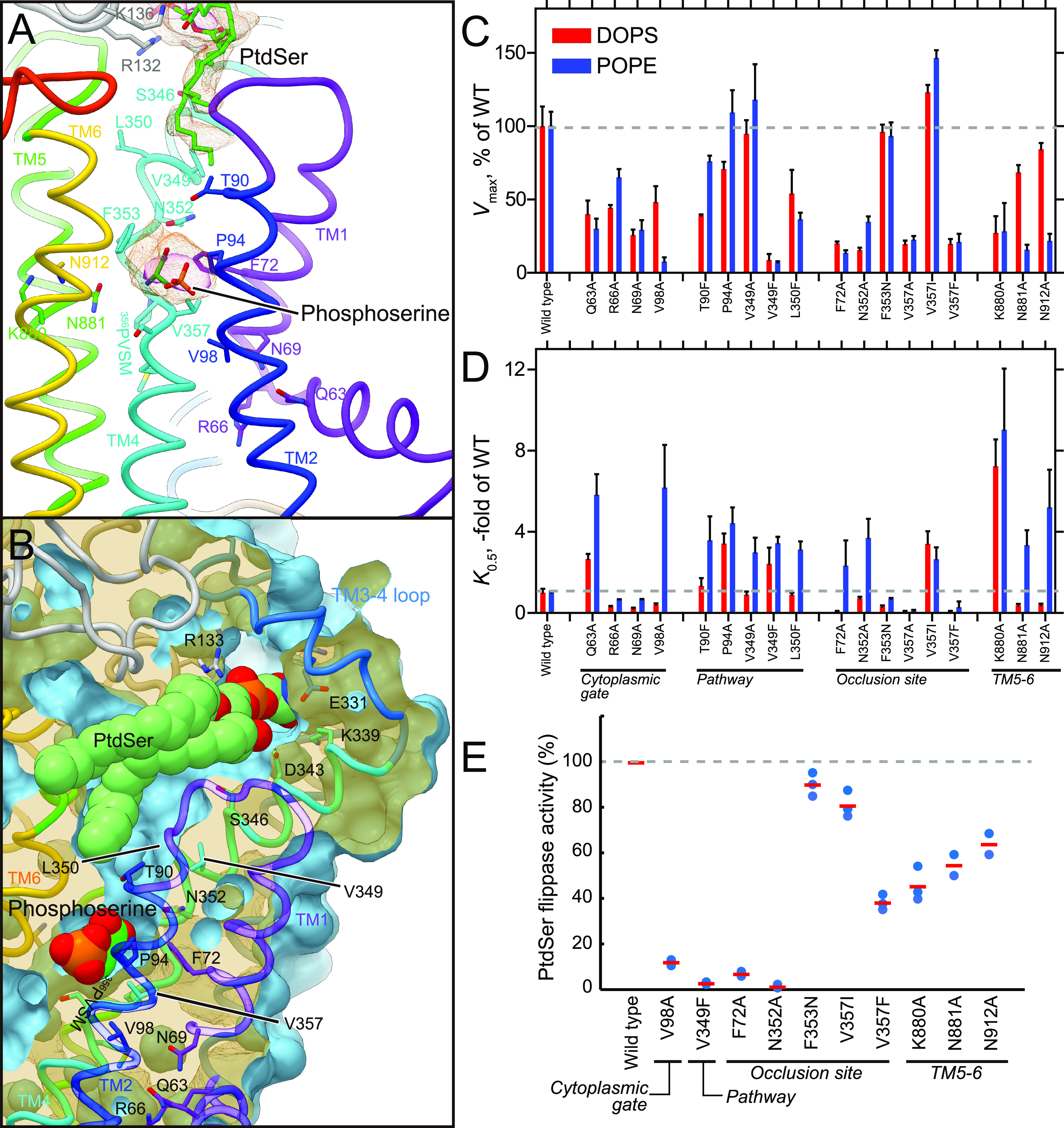
Transmembrane PtdSer occlusion site. A, lateral view of the PtdSer occlusion site in detail. Orange mesh and transparent surface represents the 2Fo − Fc electron density maps around the PtdSer molecule with contour levels of 1.0σ and 2.0σ, respectively. Phospho-l-serine (stick, green with CPK) is modeled according to the observed electron density. B, phospholipid conduit along with TM4. Surface of the atomic model (light blue) is shown with superimposed ribbon model. Only the surface model is clipped by the different plane at the position where TM4 is located, so as to show how the conduit runs along TM4. Its clipped surface is seen as transparent wheat color. The figure is drawn from the TM1 and TM2 viewpoint, with exoplasmic side-up. C and D, Vmax and K0.5 for the indicated mutants determined from their PtdSer- or PtdEtn-dependent ATPase activities, as described in Fig. 3. E, the PtdSer flippase activity. ATP11A-ATP11C knock-out (DKO) T-lymphoma cells expressing WT ATP11C or the indicated mutants were incubated with 1 μm NBD-PS for 3 min. Experiments were performed two or three times, and flippase activity for NBD-PS is shown as a percentage to that of WT ATP11C. Horizontal red bars denote averages.
Electron densities in the crevice and its extension
Well-ordered crystals were generated in the presence of both BeFx and DOPS. The thermal stability (39) of the purified ATP11C—CDC50A complex in the presence of both BeFx and PtdSer (Tm = 53.5 °C) is markedly higher than those in the absence (Tm = 33.6 °C), or presence of either BeFx (Tm = 41.3 °C) or PtdSer alone (Tm = 46.4 °C) (Fig. 2D), indicating that the BeFx-bound form binds PtdSer, and the enzyme likely accumulates in a distinct PtdSer-bound, but not occluded, E2P form in the crystal. This conclusion is supported by the fact that an excess of PtdSer is required for preservation of the crystals when harvested for X-ray diffraction studies (“Experimental procedures”). Because the expected conformation of E2P is an outward-open state, excess PtdSer would favor its accumulation and stabilize the enzyme conformation. These observations qualitatively indicate that added PtdSer is bound to the enzyme in the crystal, and persuaded us to model two PtdSer molecules in the structure (Figs. 1 and 3-5).
Figure 3.
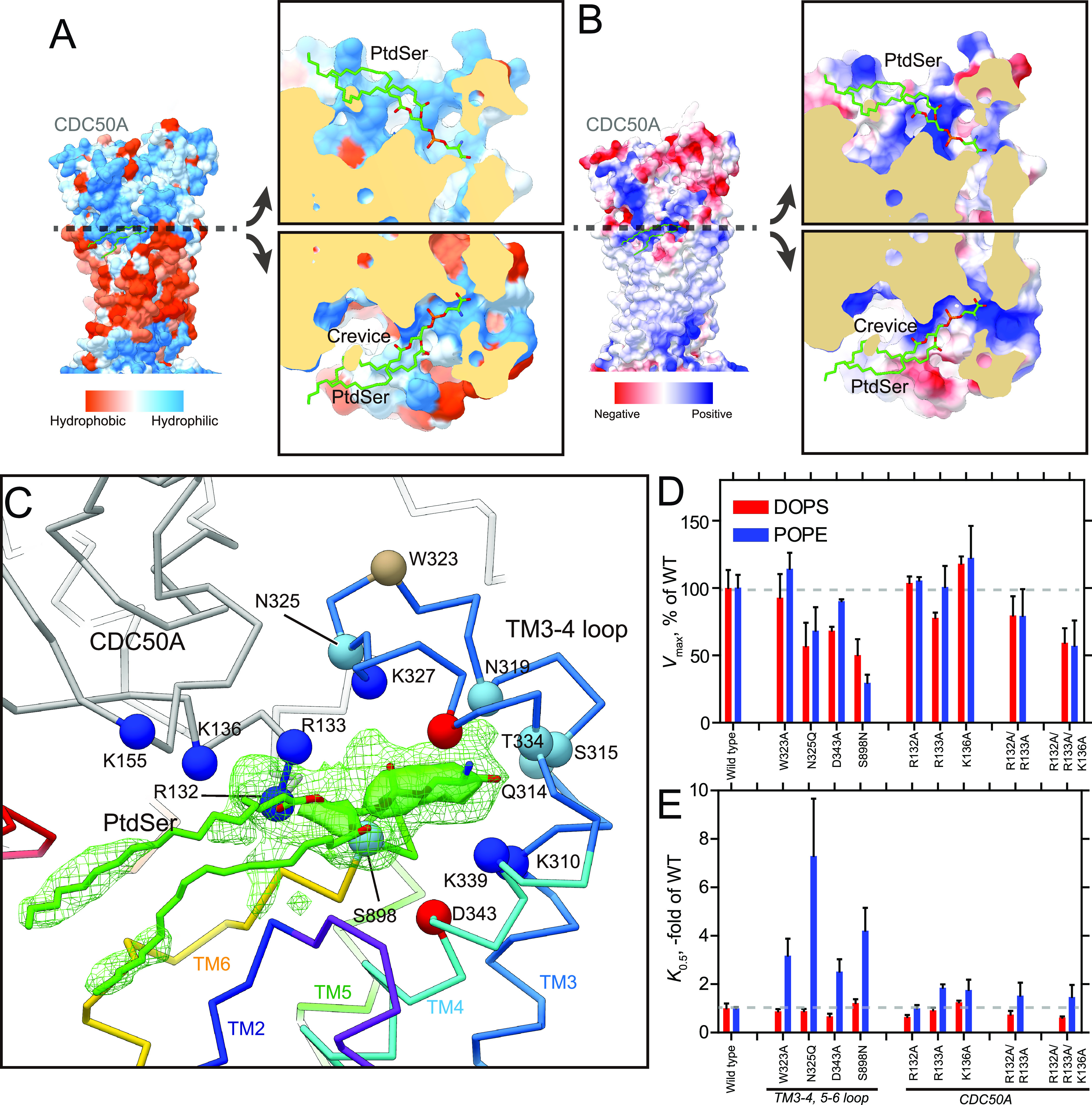
Exoplasmic cavity. A and B, surface representations of the atomic model of ATP11C—CDC50A according to their hydrophobicity (A) or Coulombic surface potentials (B). Each model was sliced at the plane where the phosphate moiety of PtdSer is located, along with the membrane plane, and opened up to show the surface of the exoplasmic cavity interior. Color codes as indicated in the figures. C, PtdSer binding cavity in detail. Models were shown as lines (color codes as described in the legend to Fig. 1), in which positions of Cα for the hydrophilic amino acids likely facing to the PtdSer-bound cavity are shown as spheres. Color of Cα spheres are as follows: blue, basic; red, acidic; light blue, neutral and hydrophilic; wheat, hydrophobic. Green mesh and transparent surface represent the 2Fo − Fc electron density maps around the PtdSer molecule with contour levels of 1.0σ and 2.0σ, respectively. The figure was drawn from the viewpoint parallel to the membrane plane. See Fig. 5 for the electron densities around the exoplasmic cavity. D and E, the Vmax and apparent affinity for PtdSer or PtdEtn derived from the ATPase activities of the indicated mutants. The ATPase activity per mg of purified protein was determined and analyzed in the presence of varying concentrations of PtdSer or PtdEtn (representatives shown in Fig. S7). The Vmax is shown as a percentage of the WT value (D). The apparent affinities for phospholipids are expressed as the concentration giving half-maximum activation (K0.5), and plotted as x-fold of WT values (E).
Although the analyzed resolution is limited, we determined two extra densities for each of the protomers in the asymmetric unit after the initial MR phasing using a homology model of ATP11C based on the PtdSer-free ATP8A1 E2P structure (29); one is located near the unwound part of TM4, and the other in the cavity structure composed of the TM3-4 loop and the CDC50A exoplasmic domain around the outer leaflet surface. These densities are observed, although to different degrees, in the same positions for most protomers (except poorly ordered protomer D). We tentatively modeled a PtdSer head group in the mid-membrane region and a whole PtdSer in the extra density at the surface location in protomer A, but without PtdSer in other protomers due to their relatively weaker densities, and further proceeded with the refinement. Composite omit maps and Fo − Fc maps show significant densities at these positions (Fig. 5), excluding the possibility that the observed extra densities are due to model bias. However, the specific molecular species and detailed conformation could not be explicitly determined in the 3.9 Å resolution electron density map. We cannot rule out the following possibilities, that the species are detergent, another type of phospholipid like PtdCho, or a number of smaller molecules present in the crystal solution. The density around the 356PVSM motif in the unwound part of TM4 is observed at a position close to the PtdSer head group occluded in the cryo-EM structure of the ATP8A1 E2-Pi state (29), supporting its density as the head group of PtdSer in ATP11C (Figs. 4 and 5). The rest of the PtdSer may be disordered in the bulk lipid phase. At the cavity site, the electron density seems to encompass not only the PtdSer head group but also other parts, including its acyl chains (Figs. 3 and 5). The assignment of PtdSer molecules in the density map is further supported by the functional studies described below.
Figure 5.
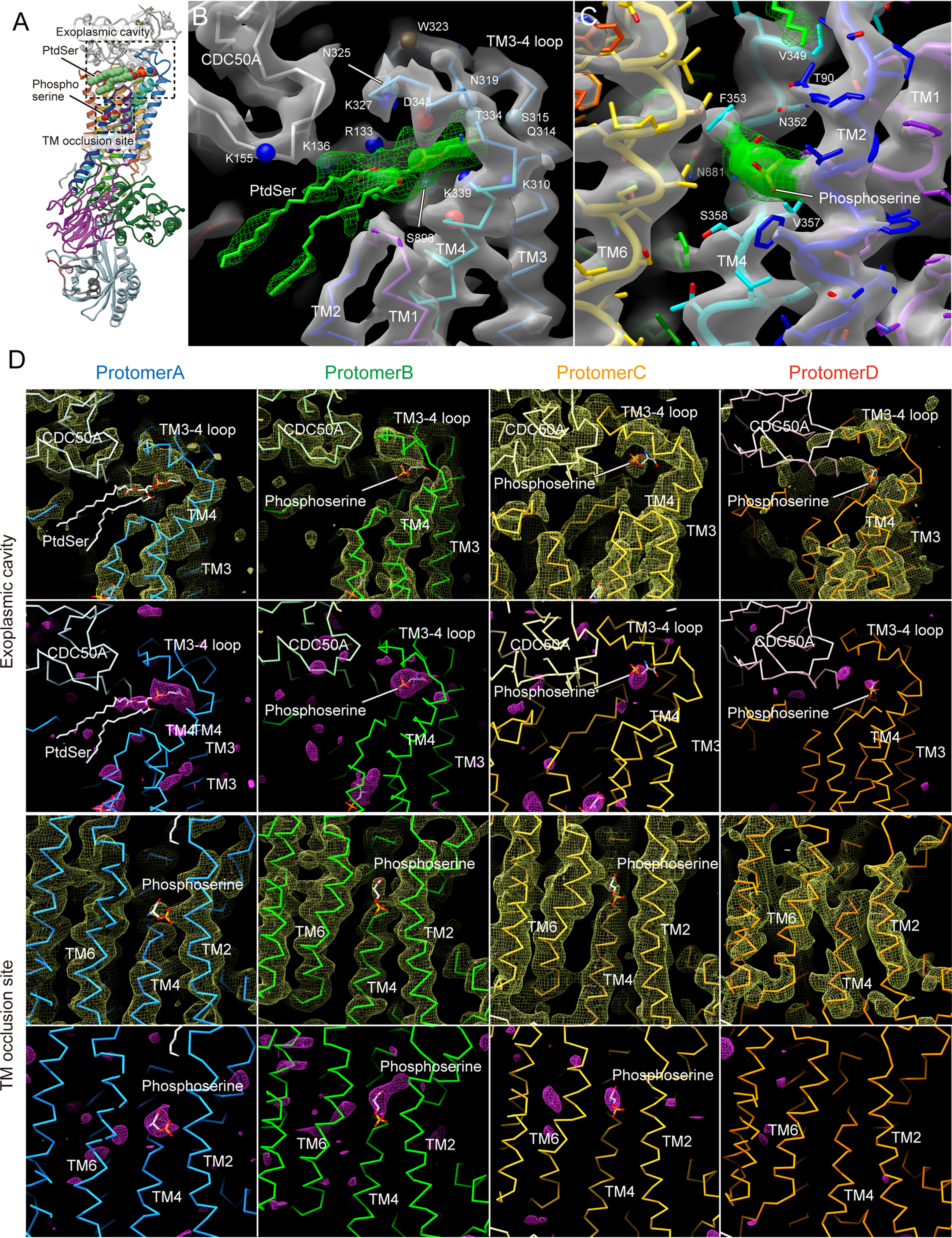
Electron density maps around PtdSer. A, whole enzyme structure as described in the legend to Fig. 1. B and C, close-up view of PtdSer-binding sites indicated as dotted boxes in A. Transparent surface and mesh represent the 2Fo − Fc electron density maps for protein (gray) or around the PtdSer molecule (green) with contour levels of 2.0σ (surface) and 1.0σ (mesh), respectively. Chain trace of protomer A is shown in B as in Fig. 3C. Ribbon models with amino acid residues in stick representations are shown in C. D, composite-omit maps (first and third rows, 2σ, yellow mesh) and Fo − Fc maps (second and fourth rows, 4σ, magenta mesh) around PtdSer (stick representation) in the exoplasmic cavity of protomer A, and phosphoserine (sticks) in the indicated positions of other protomers, except for the TM occlusion site of protomer D are shown with chain trace models of the indicated protomers. Note the Fo − Fc map was drawn using a model in which bound PtdSer and phosphoserine had been removed. For clarity, PtdSer and phosphoserine in protomer A are displayed as in the original model. Because other protomers do not contain bound substrates, phosphoserine molecules are displayed for these protomers according to the Fo − Fc density map.
PtdSer binding at the exoplasmic cavity
There is a cavity at the exoplasmic side of the conduit (Fig. 3), which is formed by the TM3-4 loop of ATP11C and the exoplasmic domain of CDC50A around the surface of the membrane outer leaflet. This cavity is connected to the crevice observed in the TM region (Fig. 3, Fig. S6). Unexpectedly, we found an extra density at this position, and one PtdSer is tentatively modeled here as described above (Figs. 3C and 5). The head group of the PtdSer is accommodated deep in the cavity, the surface of which is highly hydrophilic and electro-positive because of the many basic amino acids from both ATP11C and CDC50A arranged here (Fig. 3, A and B, and Fig. S4). A cluster of basic amino acids in this region is also observed in the cryo-EM structure of ATP8A1 (29). This positively-charged, and hydrophilic cavity would be favorable for attracting the negatively-charged phosphate group of a phospholipid. The hydrocarbon tails of the bound PtdSer lie along the membrane plane, projecting toward the membrane lipid phase. Single replacements of the positively-charged amino acids located at the entrance of the cavity produced only moderate effects on PtdSer- or PtdEtn-dependent ATPase activities, probably due to compensation by the remaining basic amino acids. This interpretation is supported by the ∼50% reduction in the Vmax of the triple mutant (R132ACDC/R133ACDC/K136ACDC) relative to that of WT (Fig. 3D, Fig. S7). Besides these positively-charged amino acids, mutations in other hydrophilic amino acids in this cavity (N325Q/D343A in ATP11C) produce a large reduction in apparent affinity for PtdEtn (Fig. 3E). Ser-898 located in the TM5-6 loop is likely facing to the cavity (Fig. 3C), and its mutation significantly impaired ATPase activity compared with WT (Fig. 3, D and E). Interaction between the TM3-4 loop and CDC50A may also be important, because the apparent affinity for PtdEtn is also reduced in the alanine replacement of Trp-323, the residue of which does not face to the PtdSer but tethers the TM3-4 loop to the exoplasmic domain of CDC50A (Figs. 1D and 3C). It is notable that the stabilities of all mutants used in this study were evaluated by size-exclusion chromatography analysis after purification, and only mutants that gave a sufficiently symmetrical peak were used for ATPase measurement. This means that the reductions in either Vmax or apparent affinities for the above mutants are unlikely to be due to reduced structural integrity. The data therefore support the conclusion that the observed electron density at this position is likely bound PtdSer.
The TM3-4 loop of ATP11C is 6 or 5 amino acids longer than that of ATP8A1 and Drs2p, respectively, and less conserved than other parts of the protein (Fig. S4). However, the hydrophilic and electro-positive nature of this cavity is essentially the same, and may be conserved across P4-ATPases. Interestingly, some mutations in the exoplasmic cavity had differing effects on the apparent affinities for PtdSer and PtdEtn (Fig. 3, D and E), compatible with an intimate association with these transported ligands. This observation is consistent with a previously reported “two-gate” model, derived from chimeric studies on yeast flippase Dnf1, which shows that amino acids in the TM3-4 loop contribute to ligand specificity (26). Such similarity between human and yeast flippases suggests conservation of this binding site.
We then asked what the function of such a PtdSer-binding site could be? Judging from its position at the meeting of membrane and medium, it could be a priming site, where the initial incorporation of translocated phospholipids from the membrane outer leaflet takes place, the beginning of the transport pathway. Our mutagenesis study, however, does not straightforwardly support this hypothesis because mutations in and around the cavity produced rather modest effects on their PtdSer-dependent ATPase activity. However, this can be attributed to the likely multiple interactions between the PtdSer head group and surrounding hydrophilic amino acids. The limited resolution of the present structure does not allow the side chains of amino acids in the cavity to be explicitly determined, yet, considering the hydrophilic nature of the cavity and ligand (Fig. 3A), multiple hydrogen bonds and/or electrostatic interactions can be anticipated. The triple mutant of CDC50A (R132ACDC/R133ACDC/K136ACDC) has severe consequences, and single mutations are likely compensated for by other hydrophilic amino acids. Alternatively, this site may be a low affinity regulatory site, such that low affinity lipid binding at the cavity and conduit entrance is inhibitory because it restricts access. The hypothesis may be related to the inhibition of PtdSer-dependent ATPase activity in the presence of high concentration of PtdSer (27).
PtdSer occlusion site
As described above, there is an extra density at the canonical TM occlusion site in the middle of the membrane (Fig. 4), close to where PtdSer is occluded in the ATP8A1 E2-Pi transition state. We could only model part of a PtdSer (phospho-l-serine moiety) molecule in the observed electron density (Fig. 4A), the other part including the acyl chains are likely disordered in the bulk lipid phase. Like other flippases as well as cation-transporting P-type ATPases, the conserved proline (Pro-356) in the 356PVSM sequence (corresponding to a PISL motif in most of the other P4-ATPases) causes a characteristic unwinding at the middle of TM4, enabling accommodation of a PtdSer head group (Fig. 5). The conduit ends at Val-357 in the 356PVSM motif (Fig. 4B), which has been implicated as a gating residue to the cytoplasmic inner leaflet (27), similar to the glutamate residue in the corresponding PEGL motif of P2-type ATPases (40, 41). Hydrophobic Val-98 (corresponding to Ile-115 in ATP8A2) supports the gating residue Val-357 on its cytoplasmic side, and these two hydrophobic amino acids form a tight seal, the cytoplasmic gate, which prevents penetration of the PtdSer head group to the cytoplasmic side in the outward-facing E2P state. In fact, mutation of these amino acids severely impairs PtdSer- or PtdEtn-dependent ATPase activity as well as PtdSer transport activity in the plasma membrane (Fig. 4, C–E), consistent with previous predictions for other flippases (26). Mutation of the hydrophilic amino acids located at the cytoplasmic side of TM1 (Q63A, R66A, N69A (corresponds to Asn-220 in yeast flippase Dnf1)) (42) also showed reduced Vmax values for ATPase activity relative to WT. These amino acids may contribute to the rigidity of the cytoplasmic gate, or are actually part of the conduit to the inner leaflet when the cytoplasmic gate opens. Val-357 in ATP11C corresponds to Ile-357 in ATP8A1, Ile-364 in ATP8A2, and Ile-508 in Drs2p, thus of almost all the P4-ATPases only ATP11A and ATP11C have a valine residue in this position (methionine in ATP8B3). In the case of ATP11C, mutation V357I produces a remarkable reduction in apparent affinity for PtdSer and PtdEtn, whereas the Vmax is slightly higher than that of WT (Fig. 4C). In fact, the transport activity of V357I is comparable with that of WT (Fig. 4E). In contrast, the ATPase activities of V357A and V357F, and the transport activity of V357F are significantly reduced compared with those of WT. Evidently, a specific size is important for gating residue Val-357. Mutagenesis studies also reveal the important contribution of the conserved residues around the occlusion site (Phe-72, Asn-352) and TM5-6 (Lys-880, Asn-881, and Asn-912); all alanine mutants showed significant reductions in either Vmax or apparent affinity for PtdSer and/or PtdEtn determined through ATPase activity profiles, and transport activities as well, in good agreement with previous studies of ATP8A2 (27, 43). Interestingly, Phe-343 is not conserved among P4-ATPases, despite its close position to the phospholipid head group. In other PtdSer-transporting P4-ATPases, including ATP8A1, ATP8A2, and Drs2p, Phe-343 in ATP11C is replaced with asparagine (Fig. S4), and the hydrophilic residue actually contributes to PtdSer coordination in the ATP8A1 E2-Pi structure (29). The F354N mutant in ATP11C showed significantly higher affinity for PtdSer and PtdEtn relative to WT, whereas keeping its Vmax of ATPase activity and PtdSer transport activity comparable with WT level. Therefore, a hydrophilic or smaller side chain in this position is favorable for the accommodation of the aminophospholipid head group in the occlusion site. It can be speculated that ATP11C has Val-357 and Phe-354 instead of the most conserved isoleucine and asparagine of other PtdSer-dependent flippases, to fine-tune the PtdSer and PtdEtn transport activity relevant to a plasma membrane flippase. As an important note, mutations Vl98A, F72A, N881A, and N912A increases apparent affinity for PtdSer, but lower that for PtdEtn, relative to the WT (Fig. 4D), therefore these mutants discriminate between PtdSer and PtdEtn.
The transport mechanism of ATP11C and P4-flippases
The present crystal structure with bound PtdSer provides structural and functional evidence for the transport model based on the recently reported cryo-EM structures in which a TM crevice is proposed to be a transport conduit for the phospholipids. In addition, a newly determined potential PtdSer-binding site at the exoplasmic cavity suggests a novel model regarding incorporation of phospholipids from the outer leaflet of the bilayer into the protein (Fig. 6).
Figure 6.

A phospholipid transport model for ATP11C. Schematic drawing of the PtdSer incorporation mechanism suggested from the present study of the ATP11C—CDC50A complex. In the outward-open E2P state (present structure), phospholipid enters from the surface of outer leaflet to the exoplasmic cavity by changing its orientation. Trapped phospholipid head group diffuses along the membrane crevice with its hydrophobic tails extending out to the hydrophobic core of the bilayer. Close-up view of the membrane crevice indicated as a dotted box is shown on the right. Phospholipid head group traverses from positively-charged exoplasmic cavity to the occlusion site near Val-357 along with TM4.
So far two structures are available as outward-facing conformations of P4-flippase. The E2P state of Drs2p shows a membrane crevice, which is likely the conduit for the phospholipid, but without bound phospholipid (28) (Fig. S6B). PtdSer is occluded in the ATP8A1 E2-Pi state, and as benefits an occlusion state, the exoplasmic gate is substantially closed (Fig. S6D). Therefore, the present crystal structure of the E2P state of ATP11C, in which PtdSer is bound to the crevice, which is wide enough along its length for a phospholipid to traverse (Fig. S6, A and E), resolves the uncertainly and provides structural evidence that the observed membrane crevice is indeed the phospholipid conduit. The crevice is formed between TM2 and TM6, and along TM4, in good agreement with the previously proposed model for ATP8A2 (27) but different from others (26, 44). Mutagenesis studies of ATP11C provide further support for the conduit's role. Val-349 in TM4 projects into the conduit (Fig. 4, A and B). Its replacement with bulky phenylalanine (V349F) severely impairs PtdSer- and PtdEtn-dependent ATPase activity as well as PtdSer transport activity relative to WT, in contrast to the more moderate effects of an alanine substitution (Fig. 4, C–E). In addition, phenylalanine mutations of residues close to Val-349 (T90F in TM2 and L350F in TM4) also significantly lower ATPase activity relative to the WT. These amino acid residues are located between, and distant from, the two substrate-binding sites and likely bulky substitutions impede diffusion of the phospholipid head group from the exoplasmic leaflet to the occlusion site.
We then asked how phospholipids are incorporated into the conduit. The newly determined putative PtdSer-binding site at the exoplasmic cavity may be one of the entrance sites for phospholipid from the outer leaflet. The electro-positive and hydrophilic nature of the cavity likely attracts the head group of the phospholipid from the outer leaflet layer of the membrane. The exoplasmic cavity connects to the longitudinal crevice along TM4 (Fig. 2, Fig. S6). Therefore, once the phospholipid head group is incorporated into the exoplasmic cavity, it may diffuse along the crevice, whereas keeping its hydrocarbon chains largely projecting toward the hydrophobic bulk lipid. We cannot exclude, however, the possibility that the exoplasmic PtdSer binding is irrelevant, because the affinity is too low or the site too short-lived. The crystals were harvested in the presence of high concentrations of PtdSer, and most of the single mutations in the cavity region had only modest effects on ATPase activity compared with those in the occlusion site or residues between the two PtdSer-binding sites.
Translocation of the phospholipid across the two leaflets is energetically extremely unfavorable due to the amphiphilic nature of phospholipids (45), and the rate of spontaneous phospholipid flip-flop is of the order of several hours to several days. In our envisaged translocation mechanism, the most energy-consuming step may be moving the hydrophilic head group from the outer membrane surface to the transport conduit. In this step the phospholipid head group needs to disconnect from the polar interactions formed with neighboring phospholipids, surrounding membrane proteins as well as water molecules, and also needs to change its orientation ∼90° from a vertical to horizontal orientation in the lipid bilayer. Our structure and transport model could answer the fundamental question in the translocation mechanism of flippase: how does a phospholipid reach the transport conduit from the outer leaflet? The hydrophilic surface of the exoplasmic cavity interior provides an environment similar to the water-facing membrane surface and lowers the energetic barrier required for acquisition of the phospholipid head group. Many membrane transport proteins seem to employ a common strategy for the translocation of their specific substrates; the environment of the substrate-binding site of the protein mimics that of the location from where the substrate comes (46, 47). Flippases are not an exception, and this strategy is applied in the most energy-consuming step in the sequence of the lipid flipping process by the ATP11C—CDC50A complex.
Experimental procedures
Protein expression and purification
Human ATP11C (NCBI: XM_005262405.1)(4) was subcloned into a hand-made vector as described previously (13). Both of the N-terminal 7 amino acids (ΔN7) and the C-terminal 38 amino acids (ΔC38) of hATP11C were truncated, and the FLAG epitope tag (DYKDDDDK), His6 tag, the enhanced green fluorescence protein (EGFP) followed by a tobacco etch virus (TEV) protease recognition sequence were attached to the N-terminal of the deletion mutant (ATP11C_cryst). Human CDC50A cDNA (NCBI: NM_018247.3) was subcloned into the vector independently. The N190Q/S292W double mutation was introduced into the construct to regulate its glycosylation status (CDC50A_QW). The former mutation is simply aimed to prevent N-linked glycosylation. The latter is to eliminate O-linked glycosylation at Ser-292 and at the same time to increase the efficiency of N-linked glycosylation at Asn-294 (48), because these residues appeared to be glycosylated alternatively. The heterodimer composed of ATP11C_cryst and CDC50A_QW was successfully expressed in the plasma membrane using baculovirus-mediated transduction of mammalian Expi293 cells (Thermo) for 48 h at 31.5 °C as described previously (30, 49). We confirmed that the effect of deletion and glycosylation-deficient mutations are negligible as seen in their ATPase activities (Fig. 1B). The harvested cells were directly solubilized with 1.5% (w/v) n-decyl β-d-maltoside in a lysis buffer containing 40 mm MES/Tris (pH 6.5), 200 mm NaCl, 2 mm Mg(CH3COO)2, 1 mm ATP, 1 mm DTT, 0.1 mm EGTA, and protease inhibitor mixture (Roche) on ice for 20 min. After removing the insoluble material by ultracentrifugation (200,000 × g for 1 h), the supernatant was mixed with anti-FLAG M2 affinity resin (Sigma-Aldrich) for 1 h at 4 °C. The resin was washed with 20 column volumes of buffer consisting of 20 mm MES/Tris (pH 6.5), 200 mm NaCl, 5% (v/v) glycerol, 1 mm Mg(CH3COO)2, 0.1 mm ATP, 0.1 mm EGTA, and 0.03% octaethylene glycol monododecyl ether (C12E8, Nikko Chemical). FLAG-EGFP–tagged ATP11C was eluted with 0.2 mg/ml of FLAG peptide (Sigma-Aldrich) in the wash buffer. Eluted proteins were incubated with TEV protease and maltose-binding protein fusion endoglycosidase (EndoHf, New England Biolabs) overnight at 4 °C. Released affinity tags containing FLAG-EGFP were removed from the mixture by a nickel-nitrilotriacetic acid resin (Qiagen). The nonabsorbed fractions were concentrated and subjected to a size-exclusion column chromatograph using a Superose 6 Increase column (GE Healthcare), equilibrated in a buffer comprising 20 mm MES/Tris (pH 6.5), 1% (v/v) glycerol, 50 mm NaCl, 5 mm MgCl2, and 0.03% C12E8. Peak fractions were collected and concentrated to 10 mg/ml. See Fig. S1 for the purity of the sample at each step. The concentrated ATP11C samples were mixed with 1 mm ADP, 0.5 mm BeSO4, 1.5 mm NaF, and 0.1 mg/ml of DOPS, and added to the glass tubes in which a layer of dried DOPC had formed, in a lipid-to-protein ratio of 0.2. C12E8 was added to the glass tubes in a protein-to-detergent ratio of 0.5 to 2.0, and the mixture was incubated overnight at 4 °C in a shaker mixer operated at 120 rpm (31). After removing the insoluble material by ultracentrifugation, lipidated samples were subjected to crystallization. Note that the effect of truncation of both terminal amino acids of ATP11C, the double mutation introduced to CDC50A and deglycosylation of CDC50A on the PtdSer- and PtdEtn-dependent ATPase activity were negligible compared with WT without endoglycosidase treatment.
Gene editing for CDC50A
The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas (CRISPR-associated) system with pX330 vector (Addgene) was used to edit the CDC50A gene in HEK293S GnT1− cells as described (4). The sgRNA sequences for human CDC50A were as follows; 5′-CACCGGGCAACGTGTTTATGTATTA-3′ and 5′-AAACTAATACATAAACACGTTGCCC-3′. Target protospacer sequences are underlined.
Crystallization and data collection
Crystals were obtained by vapor diffusion at 20 °C. The lipidated (10 mg/ml) protein sample obtained either from the Expi293 or CDC-KO cells, containing 1 mm ADP, 0.5 mm BeSO4, 1.5 mm NaF, and 0.1 mg/ml of DOPS was mixed with reservoir solution containing 10% (v/v) glycerol, 14–17% PEG4000, 0.4 m MgSO4, and 2 to 5 mm β-mercaptoethanol. Crystals made using protein samples purified from Expi293 cells grown in a thin plate-like shape with the dimensions of 800 × 500 × 50 μm in 2 weeks. In contrast, crystals from CDC-KO cells grew as small crystals usually less than 50 μm with a polyhedron shape. These crystals were picked up with LithoLoops (Protein Wave Corporation), and flash frozen in liquid nitrogen. The crystals were harvested in the presence of 10% (v/v) glycerol, 14–17% PEG4000, 0.4 m MgSO4, 4 mg/ml of DOPS, 20 mm MES/Tris (pH 6.5), 50 mm NaCl, 5 mm MgCl2, 5 mm β-mercaptoethanol, 2% C12E8, 1 mm ADP, 0.5 mm BeSO4, and 1.5 mm NaF. Note, when crystals were harvested in the absence of DOPS, a small percentage of crystals gave X-ray diffraction better than 4 Å. In the presence of 4 mg/ml of DOPS, however, the number of well-diffracting crystals increased to ∼30%. Despite the different crystal morphologies of the crystals obtained from Expi293 and CDC50A KO cells (Fig. S1), the crystals show the same space group (P212121) and unit cell size (a = 100.5 Å, b = 232.8 Å, c = 492.9 Å, α = β = γ = 90°).
X-ray diffraction data were collected at the SPring-8 beamline BL32XU, BL41XU, and BL45XU. For the large plate-like crystals obtained from Expi293 cells, X-ray diffraction data were collected by the helical scan method (50), or by irradiating with a micro-focus beam directed perpendicularly to the c-axis with the crystals on a 90° bent LithoLoop (Fig. S1). Full data sets could not be collected from CDC-KO cells as they were too small, nor could well-diffracting crystals be determined from their morphology alone. Therefore, multiple crystals were mounted on a ϕ1 μm LithoLoop, and the raster scan was performed to identify well-diffracted crystals. After selecting target crystals, 10° small-wedge data were collected from each individual crystal (Fig. S1). In total 1,588 crystals were used for the data collection, some of which was performed automatically using the ZOO system (51).
Structural determination and analysis
All the diffraction data from the individual 1,588 well-diffracting crystals were processed and merged using the automatic data processing system KAMO (52) with XDS (53). Structure factors were subjected to anisotropy correction using the UCLA MBI Diffraction Anisotropy server (54) (RRID:SCR_018722). A homology model of the ATP11C E2P state was made using a UCSF Chimera (58) based on the cryo-EM structure of the E2P state of ATP8A1 in which no PtdSer is bound (PDB ID 6K7L). Large gaps in the sequence alignment and disordered regions in the ATP8A1 structure are not included in the homology model. The structure was determined by molecular replacement with PHASER, using the homology model as a search model. After the initial phasing, regions that were missing in the model and that showed largely different conformations including TM1 and -2 were manually built, and followed by further iterative model building using COOT (55). Refmac5 (56) and Phenix (57) were used for refinement. The final crystallographic model of BeFx-bound human ATP11C at 3.9 Å resolution, refined to Rwork and Rfree of 0.29 and 0.36, was deposited in the PDB with accession code PDB code 6LKN. Figures were prepared using UCSF Chimera (58) and PYMOL (RRID:SCR_000305).
Activity assay using recombinant proteins
To measure the ATPase activity, a FLAG-EGFP tag connected by the TEV cleavage site to the N-terminal tail of ATP11C_cryst was used to estimate its expression level by fluorescence size-exclusion column chromatography (FSEC) (59). The original and additional mutant complexes of ATP11C_cryst and the CDC50A_QW (here referred to as “WT”) were expressed using the BacMam system and purified in a smaller batch format as described above except for TEV protease digestion and endoglycosidase treatment. The purified proteins (the purity of samples used for the ATPase measurement was comparable with lane 4 of SDS-PAGE in Fig. S1) were subjected to an ATPase activity assay as described previously (47). Briefly, partially purified ATP11C (WT or mutants) was suspended in buffer comprising 40 mm HEPES, 2 mm MgCl2, 2 mm ATP, 2% glycerol, 100 mm NaCl, 0.03 mg/ml of C12E8 (pH 7.0 adjusted by Tris) and the indicated concentrations of phospholipids (DOPS or POPE, dissolved as 10 mg/ml of stock in 2% C12E8), in 96-well–plates. We added 1 mm BeSO4 and 3 mm NaF (BeF3−) for every mutant as a blank. Reactions were initiated by incubating the samples at 37 °C using a thermal cycler, and maintained for 1 h. Reactions were terminated, and the amount of released Pi was determined colorimetrically using a microplate reader (TECAN). Samples used for the ATPase measurement were analyzed by FSEC with Trp fluorescence (excitation 280 nm, emission 320 nm) monitoring, and peak fluorescence values were determined. The peak values of samples were compared with that of a fully purified sample used for the crystallization whose protein concentration was accurately determined by UV absorption, and protein concentrations for each measured sample were estimated. The phospholipid concentration-dependent ATPase activities were plotted, and data fitted as described previously (43) to estimate apparent affinities (K0.5) and Vmax using PRISM4 software. For all measurements, data were duplicated at 12 different phospholipid concentrations for a single measurement, and at least three independent measurements were conducted for each mutant. The K0.5 and Vmax values plotted in Figs. 3 and 4 are mean ± S.D. estimated from at least three independent measurements, with representative ones shown in Fig. S5. Note the PtdSer- or PtdEtn-dependent specific ATPase activities of ATP11C_cryst—CDC50A_QW complex were almost the same as those of WT (Fig. 1A). We therefore refer to ATP11C_cryst—CDC50A_QW as WT in the activity assay for simplicity.
Flippase assay
Flippase activity was determined as described (60). In brief, ATP11A—ATP11C double-deficient WR19L mutant (DKO) cells were transformed with retroviruses carrying cDNA for human FLAG-tagged CDC50A and EGFP-tagged WT ATP11C or mutants. The stable transformants were then subjected to cell sorting for EGFP with FACSAriaII (BD Biosciences), cells at the same EGFP-intensity were sorted and expanded. The amounts of EGFP-tagged proteins and their localization to the plasma membrane were verified by Western blotting and a confocal microscope (FV-1000D; Olympus), respectively. For the flippase assay, DKO cells and their transformants expressing WT ATP11C or mutants were incubated with 1 μm NBD-PS (1-oleoyl-2-{6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3-phosphoserine for 3 min at 20 °C in Hanks' balanced salt solution containing 1 mm MgCl2 and 2 mm CaCl2. The cells were collected by centrifugation, resuspended in Hanks' balanced salt solution containing 5 mg/ml of fatty acid-free BSA to extract nonincorporated lipids, and analyzed by FACSCanto II (BD Biosciences).
Thermal stability
Purified samples were incubated at the indicated temperatures for 10 min in the presence of 40 mm HEPES, 100 mm NaCl, 2 mm MgCl2 (free) with 1 mm BeSO4, 3 mm NaF (BeF), and/or 0.1 mg/ml of DOPS (PtdSer). After incubation, samples were cooled on ice and denatured aggregates were removed using a membrane filter (pore size 0.22 μm), and the resulting filtrates were analyzed by size-exclusion column chromatography using Superose 6 Increase 10/150 GL (GE Healthcare). Peak values at the retention time for the complex were plotted as a function of incubation temperature, and their Tm values estimated.
Data availability
Atomic coordination and structure factors for the structures reported in this work have been deposited in the Protein Data Bank under accession number 6LKN. All remaining data are contained within the article.
Supplementary Material
Acknowledgments
We thank M. Taniguchi for technical assistance, Dr. T. Nishizawa for sharing unpublished results of the ATP8A1 structure, Dr. K. Yamashita for valuable comments on data processing, and Dr. D. McIntosh for improving the manuscript.
This article contains supporting information.
Author contributions—H. N., K. I., K. S., K. H., and K. A. data curation; H. N., K. I., K. S., and K. A. formal analysis; H. N., K. I., K. S., and K. A. investigation; H. N., K. S., K. H., and K. A. methodology; K. I., K. H., and K. A. validation; K. S. resources; K. S. and K. A. writing-review and editing; Y. F., S. N., and K. A. conceptualization; Y. F., S. N., and K. A. supervision; Y. F., S. N., and K. A. funding acquisition; K. A. visualization; K. A. writing-original draft; K. A. project administration
Funding and additional information—This work was supported by Grants-in-Aid for the Scientific Research Grant 17H03653 (to K. A.), grants from the Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) from the Japan Agency for Medical Research and Development (AMED), Takeda Science Foundation, Uehara Science Foundation, The Naito Foundation (to K. A.), the Core Research for Evolutional Science and Technology from JST Grant JPMJCR14M4 (to S. N. and K. A.), Grants-in-Aid for Scientific Research (S), the Japan New Energy and Industrial Technology Development Organization (NEDO), and the Japan Agency for Medical Research and Development (AMED) (to Y. F.). This work is a part of projects 2018B2703 and 2019B2707 at SPring-8. This work was supported in part by the Platform Project for Supporting Drug Discovery and Life Science Research (BINDS) from AMED under Grant number JP18am0101070 (to K. A.).
Conflict of interest—Y. F. is a director of CeSPIA Inc.
- PtdSer
- phosphatidylserine
- PtdEtn
- phosphatidylethanolamine
- PtdCho
- phosphatidylcholine
- DOPC
- dioleoylphosphatidylcholine
- BeFx
- beryllium fluoride
- DOPS
- dioleoylphosphatidylserine
- TM
- transmembrane
- EGFP
- enhanced green fluorescence protein
- TEV
- tobacco etch virus
- FSEC
- fluorescence size-exclusion column chromatography
- NBD-PS
- (1-oleoyl-2-{6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-gylcero-3-phosphoserine.
References
- 1. Leventis P. A., and Grinstein S. (2010) The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427 10.1146/annurev.biophys.093008.131234 [DOI] [PubMed] [Google Scholar]
- 2. Pomorski T., and Menon A. K. (2006) Lipid flippases and their biological functions. Cell. Mol. Life Sci. 63, 2908–2921 10.1007/s00018-006-6167-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Segawa K., and Nagata S. (2015) An apoptotic “eat me” signal: phosphatidylserine exposure. Trends Cell Biol. 25, 639–650 10.1016/j.tcb.2015.08.003 [DOI] [PubMed] [Google Scholar]
- 4. Segawa K., Kurata S., Yanagihashi Y., Brummelkamp T. R., Matsuda F., and Nagata S. (2014) Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168 10.1126/science.1252809 [DOI] [PubMed] [Google Scholar]
- 5. Bevers E. M., Wiedmer T., Comfurius P., Zhao J., Smeets E. F., Schlegel R. A., Schroit A. J., Weiss H. J., Williamson P., Zwaal R. F., and Sims P. J. (1995) The complex of phosphatidylinositol 4,5-bisphosphate and calcium ions is not responsible for Ca2+-induced loss of phospholipid asymmetry in the human erythrocyte: a study in Scott syndrome, a disorder of calcium-induced phospholipid scrambling. Blood 86, 1983–1991 10.1182/blood.V86.5.1983.bloodjournal8651983 [DOI] [PubMed] [Google Scholar]
- 6. Williamson P. (2015) Phospholipid scramblases supplementary issue: cellular anatomy of lipid traffic. Lipid Insights 8s1, LPI.S31785–44 10.4137/LPI.S31785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang X., Halleck M. S., Schlegel R. A., and Williamson P. (1996) A subfamily of P-type ATPases with aminophospholipid transporting activity. Science 272, 1495–1497 10.1126/science.272.5267.1495 [DOI] [PubMed] [Google Scholar]
- 8. Coleman J. A., Kwok M. C. M., and Molday R. S. (2009) Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine Flippase in photoreceptor disc membranes. J. Biol. Chem. 284, 32670–32679 10.1074/jbc.M109.047415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shin H.-W., and Takatsu H. (2019) Substrates of P4-ATPases: beyond aminophospholipids (phosphatidylserine and phosphatidylethanolamine). FASEB J. 33, 3087–3096 10.1096/fj.201801873R [DOI] [PubMed] [Google Scholar]
- 10. Palmgren M. G., and Axelsen K. B. (1998) Evolution of P-type ATPases. Biochim. Biophys. Acta 1365, 37–45 10.1016/S0005-2728(98)00041-3 [DOI] [PubMed] [Google Scholar]
- 11. Toyoshima C., Nakasako M., Nomura H., and Ogawa H. (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405, 647–655 10.1038/35015017 [DOI] [PubMed] [Google Scholar]
- 12. Morth J. P., Pedersen B. P., Toustrup-Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., and Nissen P. (2007) Crystal structure of the sodium–potassium pump. Nature 450, 1043–1049 10.1038/nature06419 [DOI] [PubMed] [Google Scholar]
- 13. Abe K., Irie K., Nakanishi H., Suzuki H., and Fujiyoshi Y. (2018) Crystal structures of the gastric proton pump. Nature 556, 214–229 10.1038/s41586-018-0003-8 [DOI] [PubMed] [Google Scholar]
- 14. Andersen J. P., Vestergaard A. L., Mikkelsen S. A., Mogensen L. S., Chalat M., and Molday R. S. (2016) P4-ATPases as phospholipid flippases-structure, function, and enigmas. Front. Physiol. 7, e275 10.3389/fphys.2016.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saito K., Fujimura-Kamada K., Furuta N., Kato U., Umeda M., and Tanaka K. (2004) Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 3418–3432 10.1091/mbc.e03-11-0829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bryde S., Hennrich H., Verhulst P. M., Devaux P. F., Lenoir G., and Holthuis J. C. (2010) CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J. Biol. Chem. 285, 40562–40572 10.1074/jbc.M110.139543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Segawa K., Kurata S., and Nagata S. (2018) The CDC50A extracellular domain is required for forming a functional complex with and chaperoning phospholipid flippases to the plasma membrane. J. Biol. Chem. 293, 2172–2182 10.1074/jbc.RA117.000289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Segawa K., Kurata S., and Nagata S. (2016) Human type IV P-type ATPases that work as plasma membrane phospholipid flippases and their regulation by caspase and calcium. J. Biol. Chem. 291, 762–772 10.1074/jbc.M115.690727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perez-Garcia V., Fineberg E., Wilson R., Murray A., Mazzeo C. I., Tudor C., Sienerth A., White J. K., Tuck E., Ryder E. J., Gleeson D., Siragher E., Wardle-Jones H., Staudt N., Wali N., et al. (2018) Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 555, 463–468 10.1038/nature26002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yabas M., Teh C. E., Frankenreiter S., Lal D., Roots C. M., Whittle B., Andrews D. T., Zhang Y., Teoh N. C., Sprent J., Tze L. E., Kucharska E. M., Kofler J., Farell G. C., Bröer S., et al. (2011) ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat. Immunol. 12, 441–449 10.1038/ni.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siggs O. M., Arnold C. N., Huber C., Pirie E., Xia Y., Lin P., Nemazee D., and Beutler B. (2011) The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat. Immunol. 12, 434–440 10.1038/ni.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siggs O. M., Schnabl B., Webb B., and Beutler B. (2011) X-linked cholestasis in mouse due to mutations of the P4-ATPase ATP11C. Proc. Natl. Acad. Sci. U.S.A. 108, 7890–7895 10.1073/pnas.1104631108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yabas M., Coupland L. A., Cromer D., Winterberg M., Teoh N. C., D'Rozario J., Kirk K., Bröer S., Parish C. R., and Enders A. (2014) Mice deficient in the putative phospholipid flippase ATP11C exhibit altered erythrocyte shape, anemia, and reduced erythrocyte life span. J. Biol. Chem. 289, 19531–19537 10.1074/jbc.C114.570267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arashiki N., Takakuwa Y., Mohandas N., Hale J., Yoshida K., Ogura H., Utsugisawa T., Ohga S., Miyano S., Ogawa S., Kojima S., and Kanno H. (2016) ATP11C is a major flippase in human erythrocytes and its defect causes congenital hemolytic anemia. Haematologica 101, 559–565 10.3324/haematol.2016.142273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coleman J. A., Vestergaard A. L., Molday R. S., Vilsen B., and Andersen J. P. (2012) Critical role of a transmembrane lysine in aminophospholipid transport by mammalian photoreceptor P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. U.S.A. 109, 1449–1454 10.1073/pnas.1108862109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baldridge R. D., and Graham T. R. (2013) Two-gate mechanism for phospholipid selection and transport by type IV P-type ATPases. Proc. Natl. Acad. Sci. U.S.A. 110, E358–E367 10.1073/pnas.1216948110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vestergaard A. L., Coleman J. A., Lemmin T., Mikkelsen S. A., Molday L. L., Vilsen B., Molday R. S., Dal Peraro M., and Andersen J. P. (2014) Critical roles of isoleucine-364 and adjacent residues in a hydrophobic gate control of phospholipid transport by the mammalian P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. U.S.A. 111, E1334–E1343 10.1073/pnas.1321165111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Timcenko M., Lyons J. A., Januliene D., Ulstrup J. J., Dieudonné T., Montigny C., Ash M. R., Karlsen J. L., Boesen T., Kühlbrandt W., Lenoir G., Moeller A., and Nissen P. (2019) Structure and autoregulation of a P4-ATPase lipid flippase. Nature 571, 366–370 10.1038/s41586-019-1344-7 [DOI] [PubMed] [Google Scholar]
- 29. Hiraizumi M., Yamashita K., Nishizawa T., and Nureki O. (2019) Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science 365, 1149–1155 10.1126/science.aay3353 [DOI] [PubMed] [Google Scholar]
- 30. Dukkipati A., Park H. H., Waghray D., Fischer S., and Garcia K. C. (2008) BacMam system for high-level expression of recombinant soluble and membrane glycoproteins for structural studies. Protein Expr. Purif. 62, 160–170 10.1016/j.pep.2008.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gourdon P., Andersen J. L., Hein K. L., Bublitz M., Pedersen B. P., Liu X.-Y., Yatime L., Nyblom M., Nielsen T. T., Olesen C., Vuust Møller J., Nissen P., and Morth J. P. (2011) HiLiDe-systematic approach to membrane protein crystallization in lipid and detergent. Cryst. Growth Des. 11, 2098–2106 10.1021/cg101360d [DOI] [Google Scholar]
- 32. Danko S., Yamasaki K., Daiho T., Suzuki H., and Toyoshima C. (2001) Organization of cytoplasmic domains of sarcoplasmic reticulum Ca2+-ATPase in E1P and E1ATP states: a limited proteolysis study. FEBS Lett. 505, 129–135 10.1016/S0014-5793(01)02801-0 [DOI] [PubMed] [Google Scholar]
- 33. Norimatsu Y., Hasegawa K., Shimizu N., and Toyoshima C. (2017) Protein-phospholipid interplay revealed with crystals of a calcium pump. Nature 545, 193–198 10.1038/nature22357 [DOI] [PubMed] [Google Scholar]
- 34. Danko S., Daiho T., Yamasaki K., Kamidochi M., Suzuki H., and Toyoshima C. (2001) ADP-insensitive phosphoenzyme intermediate of sarcoplasmic reticulum Ca2+-ATPase has a compact conformation resistant to proteinase K, V8 protease, and trypsin. FEBS Lett. 489, 277–282 10.1016/S0014-5793(01)02111-1 [DOI] [PubMed] [Google Scholar]
- 35. Katoh K., Misawa K., Kuma K.-I., and Miyata T. (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acid Res. 30, 3059–3066 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Toyoshima C., Norimatsu Y., Iwasawa S., Tsuda T., and Ogawa H. (2007) How processing of aspartylphosphate is coupled to lumenal gating of the ion pathway in the calcium pump. Proc. Natl. Acad. Sci. U.S.A. 104, 19831–19836 10.1073/pnas.0709978104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. MacKenzie S. H., and Clark A. C. (2012) Death by caspase dimerization. Adv Exp. Med. Biol. 747, 55–73 10.1007/978-1-4614-3229-6_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takatsu H., Takayama M., Naito T., Takada N., Tsumagari K., Ishihama Y., Nakayama K., and Shin H. W. (2017) Phospholipid flippase ATP11C is endocytosed and downregulated following Ca2+-mediated protein kinase C activation. Nat. Commun. 8, 1423 10.1038/s41467-017-01338-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hattori M., Hibbs R. E., and Gouaux E. (2012) A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening. Structure 20, 1293–1299 10.1016/j.str.2012.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inesi G., Lewis D., Toyoshima C., Hirata A., and De Meis L. (2008) Conformational fluctuations of the Ca2+-ATPase in the native membrane environment: effects of pH, temperature, catalytic substrates, and thapsigargin. J. Biol. Chem. 283, 1189–1196 10.1074/jbc.M707189200 [DOI] [PubMed] [Google Scholar]
- 41. Tsunekawa N., Ogawa H., Tsueda J., Akiba T., and Toyoshima C. (2018) Mechanism of the E2 to E1 transition in Ca2+ pump revealed by crystal structures of gating residue mutants. Proc. Natl. Acad. Sci. U.S.A. 115, 12722–12727 10.1073/pnas.1815472115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roland B. P., and Graham T. R. (2016) Directed evolution of a sphingomyelin flippase reveals mechanism of substrate backbone discrimination by a P4-ATPase. Proc. Natl. Acad. Sci. U.S.A. 113, E4460–E4466 10.1073/pnas.1525730113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mikkelsen S. A., Mogensen L. S., Vilsen B., Molday R. S., Vestergaard A. L., and Andersen J. P. (2019) Asparagine 905 of the mammalian phospholipid flippase ATP8A2 is essential for lipid substrate–induced activation of ATP8A2 dephosphorylation. J. Biol. Chem. 294, 5970–5979 10.1074/jbc.RA118.007240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jensen M. S., Costa S. R., Duelli A. S., Andersen P. A., Poulsen L. R., Stanchev L. D., Gourdon P., Palmgren M., Günther Pomorski T., and López-Marqués R. L. (2017) Phospholipid flipping involves a central cavity in P4 ATPase. Sci. Rep 7, 17621 10.1038/s41598-017-17742-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pomorski T. G., and Menon A. K. (2016) Lipid somersaults: uncovering the mechanisms of protein-mediated lipid flipping. Prog. Lipid Res. 64, 69–84 10.1016/j.plipres.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Doyle D. A., Morais Cabral J., Pfuetzner R. A., Kuo A., Gulbis J. M., Cohen S. L., Chait B. T., and MacKinnon R. (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77 10.1126/science.280.5360.69 [DOI] [PubMed] [Google Scholar]
- 47. Yamamoto K., Dubey V., Irie K., Nakanishi H., Khandelia H., Fujiyoshi Y., and Abe K. (2019) A single K+-binding site in the crystal structure of the gastric proton pump. eLife 8, e47701 10.7554/eLife.47701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murray A. N., Chen W., Antonopoulos A., Hanson S. R., Wiseman R. L., Dell A., Haslam S. M., Powers D. L., Powers E. T., and Kelly J. W. (2015) Enhanced aromatic sequons increase oligosaccharyltransferase glycosylation efficiency and glycan homogeneity. Chem. Biol. 22, 1052–1062 10.1016/j.chembiol.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Goehring A., Lee C. H., Wang K. H., Michel J. C., Claxton D. P., Baconguis I., Althoff T., Fischer S., Garcia K. C., and Gouaux E. (2014) Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 9, 2574–2585 10.1038/nprot.2014.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Flot D., Mairs T., Giraud T., Guijarro M., Lesourd M., Rey V., van Brussel D., Morawe C., Borel C., Hignette O., Chavanne J., Nurizzo D., McSweeney S., and Mitchell E. (2010) The ID23-2 structural biology microfocus beamline at the ESRF. J. Synchrotron Radiat. 17, 107–118 10.1107/S0909049509041168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hirata K., Yamashita K., Ueno G., Kawano Y., Hasegawa K., Kumasaka T., and Yamamoto M. (2019) ZOO: an automatic data-collection system for high-throughput structure analysis in protein microcrystallography. Acta Crystallogr. Sect. D Struct. Biol. 75, 138–150 10.1107/S2059798318017795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamashita K., Hirata K., and Yamamoto M. (2018) KAMO: towards automated data processing for microcrystals. Acta Crystallogr. Sect. D Struct. Biol. 74, 441–449 10.1107/S2059798318004576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kabsch W. (2010) XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strong M., Sawaya M. R., Wang S., Phillips M., Cascio D., and Eisenberg D. (2006) Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 8060–8065 10.1073/pnas.0602606103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 56. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 59. Kawate T., and Gouaux E. (2006) Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681 10.1016/j.str.2006.01.013 [DOI] [PubMed] [Google Scholar]
- 60. Segawa K., Yanagihashi Y., Yamada K., Suzuki C., Uchiyama Y., and Nagata S. (2018) Phospholipid flippases enable precursor B cells to flee engulfment by macrophages. Proc. Natl. Acad. Sci. U.S.A. 115, 12212–12217 10.1073/pnas.1814323115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordination and structure factors for the structures reported in this work have been deposited in the Protein Data Bank under accession number 6LKN. All remaining data are contained within the article.