

Abstract
Poly(A)-specific ribonuclease (PARN) is a 3′-exoribonuclease that plays an important role in regulating the stability and maturation of RNAs. Recently, PARN has been found to regulate the maturation of the human telomerase RNA component (hTR), a noncoding RNA required for telomere elongation. Specifically, PARN cleaves the 3′-end of immature, polyadenylated hTR to form the mature, nonpolyadenylated template. Despite PARN's critical role in mediating telomere maintenance, little is known about how PARN's function is regulated by post-translational modifications. In this study, using shRNA- and CRISPR/Cas9-mediated gene silencing and knockout approaches, along with 3′-exoribonuclease activity assays and additional biochemical methods, we examined whether PARN is post-translationally modified by acetylation and what effect acetylation has on PARN's activity. We found PARN is primarily acetylated by the acetyltransferase p300 at Lys-566 and deacetylated by sirtuin1 (SIRT1). We also revealed how acetylation of PARN can decrease its enzymatic activity both in vitro, using a synthetic RNA probe, and in vivo, by quantifying endogenous levels of adenylated hTR. Furthermore, we also found that SIRT1 can regulate levels of adenylated hTR through PARN. The findings of our study uncover a mechanism by which PARN acetylation and deacetylation regulate its enzymatic activity as well as levels of mature hTR. Thus, PARN's acetylation status may play a role in regulating telomere length.
Keywords: post-translational modification (PTM), protein acetylation, p300, sirtuin1 (SIRT1), polyadenylation, RNA processing, human telomerase RNA component (hTR), poly(A)-specific ribonuclease (PARN), genome maintenance, noncoding RNA, acetylation, sirtuin 1 (SIRT1)
Poly(A)-specific ribonuclease (PARN) is an RNA-processing enzyme involved in many biological processes, such as telomere maintenance and DNA damage response (1, 2). PARN is a 3′-exoribonuclease that cleaves RNA bases at the 3′-end, one nucleotide at a time (3–6). PARN has the highest substrate affinity for adenosines, conferring on it the title of a deadenylase; however, PARN is also efficient at cleaving other bases (6). PARN's deadenylase activity allows it to remove poly(A) tails at the 3′-end of both coding and noncoding RNAs (3–5, 7). PARN's deadenylation of cytoplasmic mRNAs (e.g. TP53 mRNA) has a destabilizing effect (2, 5, 8). However, PARN's deadenylation of noncoding RNAs has been shown to promote their stability (9). PARN also mediates the maturation of noncoding RNAs by cleaving excess adenosine and nonadenosine bases at the 3′-end (1, 10). PARN can regulate the stability and maturation of a variety of noncoding RNAs, such as microRNAs, Y RNAs, small Cajal body RNAs (scaRNAs), small nucleolar RNAs (snoRNAs), 18S rRNA, and the human telomerase RNA component (hTR) (1, 10–17).
PARN's regulation of the human telomerase RNA component has been extensively studied, as it has been linked to several telomere-associated diseases. Telomeres are repetitive DNA sequences found at the ends of DNA that protect it from replicative damage (18, 19). The telomerase ribonucleoprotein complex required for telomere elongation includes the telomerase reverse transcriptase enzyme as well as the telomerase RNA component (20). HTR is a noncoding RNA with an H/ACA domain, transcribed from the TERC gene (21). HTR is essential for telomere elongation, as it is the template that telomerase reverse transcriptase uses to add TTAGGG repeats at the ends of chromosomes (20). Clinical studies in patients suffering from telomere-associated diseases, such as dyskeratosis congenita, Hoyeraal–Hreidarsson syndrome, idiopathic pulmonary fibrosis, and bone marrow failure, have demonstrated that genetic variants that result in the loss of PARN or enzymatically inactivating PARN mutations are linked to disease onset (1, 22–26). PARN is required to process the immature, polyadenylated hTR into its mature, 451-base form. Thus, lack of catalytically active PARN results in an accumulation of immature, polyadenylated hTR that is degraded via the nuclear RNA exosome. Due to decreased levels of mature hTR, there is insufficient template for telomere elongation in PARN-deficient cells. Therefore, loss of PARN function results in excessive telomere shortening via its regulation of hTR (1, 27–30).
Although PARN's activity is important in regulating global RNA stability and telomere maintenance, thus far little is known about how PARN's function is regulated in cells. Post-translational modifications (PTMs) can dynamically regulate the function, expression, localization, and interactions of proteins. Examples of common and reversible PTMs include acetylation, methylation, and phosphorylation (31). To date, only one of PARN's amino acid residues has been convincingly shown to be modified by phosphorylation. MAPKAP kinase-2 (MK2) phosphorylates PARN at serine 557, which prevents it from binding a negative regulator of PARN, nuclear cap–binding protein (NCBP1/CBP80), and regulates cell cycle progression (32, 33). However, several high-throughput MS screenings suggest that PARN may be post-translationally modified by other modifications, such as Nε-amino lysine acetylation, although it has never been validated (34, 35). Nε-Lysine acetylation is dynamically regulated by lysine (K) acetyltransferases (KATs), which catalyze protein acetylation, and lysine deacetylases (HDACs), which reverse this reaction (36, 37). Acetylation of histone proteins is known to mainly regulate transcription; however, the role of acetylation in regulating the function of nonhistone proteins extends to many other functions (38).
Acetylation of RNA-processing enzymes plays an important role in regulating RNA turnover (39). An example of an RNA-editing enzyme regulated by acetylation is the nuclear poly(A)-binding protein (PABP1). PABP1 is required for poly(A) tail elongation after transcription as well as export of poly(A) mRNA from the nucleus to the cytoplasm for protein synthesis. When PABP1 is deacetylated by sirtuin1 (SIRT1), a class III HDAC, it deactivates PABP1's poly(A)-binding ability, preventing poly(A) mRNA export and translation (40). Furthermore, acetylation of the 3′-exoribonuclease CCR4-associated factor 1 (CAF1) by CBP/p300 decreases levels of polyadenylated mRNAs, which destabilizes mRNAs globally (41). These examples highlight how lysine acetylation plays a critical role in regulating the function of RNA-processing enzymes.
Because little is known about PARN's post-translational regulation, in this study, we investigated whether PARN protein is post-translationally modified by lysine acetylation and its functional relevance. We report that PARN is acetylated primarily by p300 at lysine 566, and the class III HDAC, SIRT1, is responsible for reversing PARN's acetylation. Furthermore, we also found that acetylation of PARN decreases its enzymatic activity both in vitro, using a synthetic 5′-fluorescein RNA probe, and in vivo, by measuring endogenous levels of polyadenylated hTR. Our results also elucidate a mechanism by which SIRT1 regulates levels of polyadenylated hTR through PARN.
Results
PARN is primarily acetylated by p300
Inspection of multiple high-throughput MS data obtained from various human cell lines revealed that the PARN protein is potentially acetylated (34, 35). However, biochemical data confirming PARN's lysine acetylation are lacking. To determine whether PARN is acetylated and which acetyltransferases catalyze the acetylation of PARN, V5-tagged PARN was co-transfected with various KATs in 293T cells. Immunoprecipitation of the V5 tag and immunoblotting with anti-acetyllysine (AcK) antibody revealed that PARN can be acetylated by the lysine acetyltransferases: ortholog of Drosophila males absent on the first (MOF), p300/CBP-associated factor (PCAF), and p300 (Fig. 1A). To determine whether PARN is also naturally acetylated in cells, endogenous PARN immunoprecipitation experiments were performed in A549 cells that express high protein levels of PARN. Because PARN is expressed in the nucleolus, nucleus, and cytoplasm (15, 33, 42, 43), subcellular fractionation was performed before testing endogenous PARN acetylation to assess whether PARN is acetylated in a specific subcellular organelle. The results demonstrate that PARN is endogenously acetylated in both the nuclear and cytoplasmic fraction of A549 cells (Fig. 1B).
Figure 1.
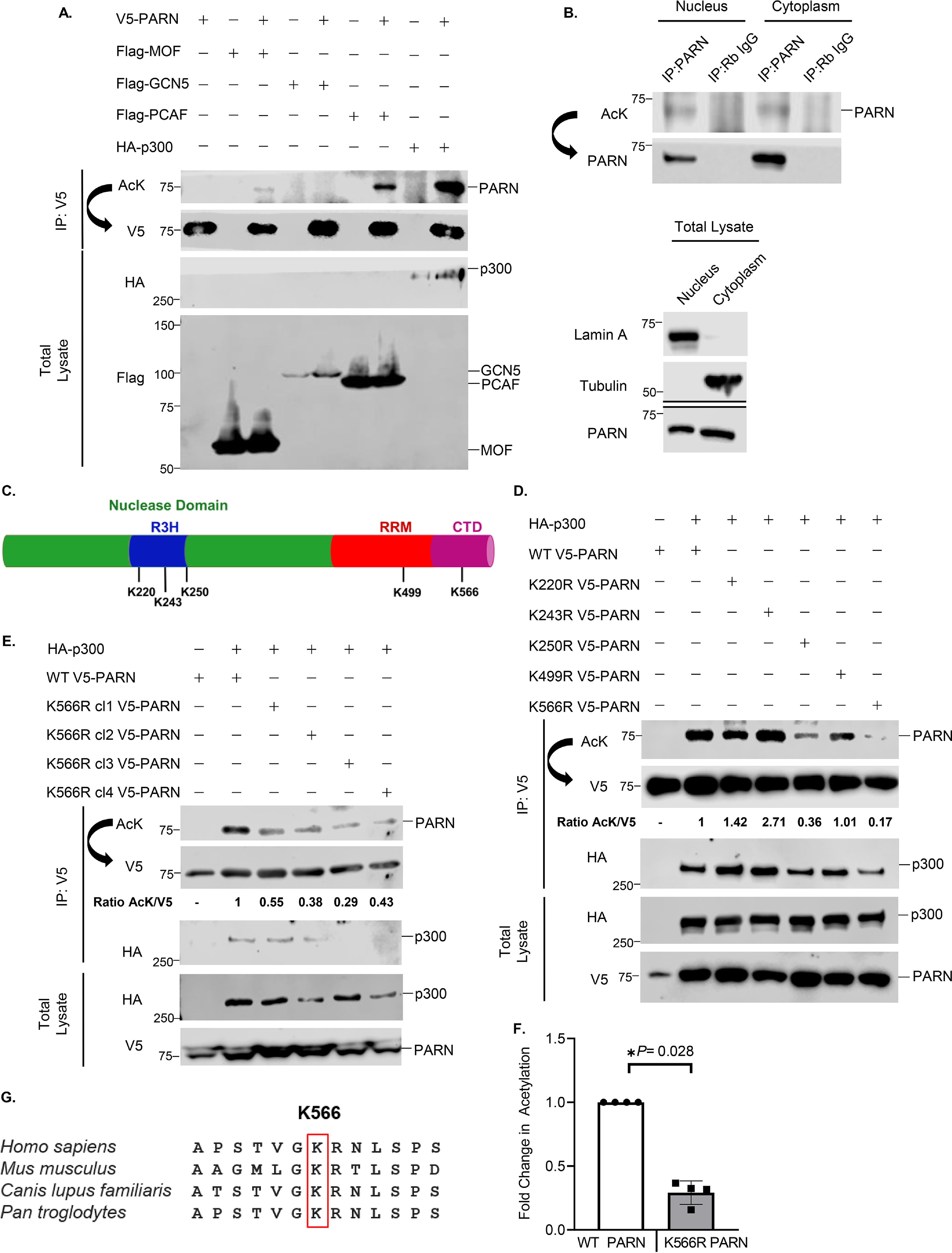
PARN is primarily acetylated by p300. A, Western blot analysis of exogenous PARN acetylation by different lysine acetyltransferases. 293T cells were transfected for 48 h with V5-tagged PARN and four different lysine acetyltransferases. Protein extracts were immunoprecipitated (IP) with anti-V5 antibody and immunoblotted with a nonspecific anti-AcK antibody. Membranes were stripped under low-stringency conditions and reblotted using anti-V5 antibody. B, Western blotting of endogenous PARN acetylation in different subcellular organelles. A549 cells underwent subcellular fractionation and were immunoprecipitated with anti-PARN antibody. Western blotting of total lysate was also performed to monitor subcellular fractionation by using lamin A (a nuclear marker) and tubulin (a cytoplasmic marker). Western blotting of endogenous PARN in each subcellular organelle was also run in parallel. C, diagram of PARN protein domains. The nuclease domain, nucleic acid–binding domain (R3H), RNA-binding domain (RRM), and C-terminal domain (CTD) are indicated. The specified lysine residues were potentially acetylated in high-throughput MS data deposited on PhosphositePlus. D, Western blotting of lysine-to-arginine mutant PARN constructs assessing acetylated residues. Each potentially acetylated lysine residue indicated in C was mutated to arginine using site-directed mutagenesis. 293T cells were co-transfected with HA-tagged p300 and V5-tagged PARN variants to detect changes in acetylation as described in A. The ratio of AcK to V5 was quantified and normalized to WT PARN to determine the effect of the lysine-to-arginine mutations on PARN's acetylation. E, Western blot analysis confirming that K566R is PARN's acetylated residue. Four different K566R plasmids obtained from different mutant clones were used to confirm whether the K566R PARN mutant decreased PARN acetylation by p300. The ratio of AcK to V5 was quantified and normalized to WT PARN. F, quantification of four independent Western blots comparing the acetylation of WT PARN and K566R PARN. HA-tagged p300 was co-transfected with either v5-tagged WT PARN or v5-tagged K566R cl1 PARN in 293T cells as described in A. Within each Western blot, the AcK/V5 ratio was quantified and normalized to levels of WT PARN. The normalized signals from each biological replicate are displayed as individual data points (n = 4). Data are presented as the mean ± S.D. (error bars). For statistical analysis, an unpaired, two-sided Wilcoxon–Mann–Whitney test was performed (*, p < 0.05). G, comparison of PARN's primary sequence between different species. The red box outlines Lys-566, the highly conserved amino acid that is acetylated in PARN.
To identify which PARN residue is acetylated, putatively acetylated lysine residues identified in high-throughput MS studies (34) were individually mutated to arginine (Fig. 1C). Because arginine structurally resembles an unacetylated lysine but cannot be acetylated, lysine-to-arginine (K→R) mutations were used to help identify acetylated residues. Because p300 has the highest detectable acetylation levels of PARN, p300 was co-transfected with WT PARN and PARN mutants to determine acetylation sites. The K566R PARN mutant had a consistent decrease in acetylation compared with the other residues (Fig. 1D). To further verify that Lys-566 is an acetylated residue, the experiment was repeated using four plasmids expressing K566R PARN obtained from different clones. All K566R mutants had a decrease in p300-induced acetylation of PARN (Fig. 1E), supporting the finding that Lys-566 is one of PARN's acetylated residues. In fact, Western blotting quantification of acetylated WT and K566R PARN confirms that K566R PARN has a statistically significant reduction in acetylation (Fig. 1F). Interestingly, PARN's Lys-566 residue is also highly conserved across different species (Fig. 1G). We also found that in addition to acetylating PARN, p300, PCAF, and MOF co-immunoprecipitated with PARN (Fig. 1 (D and E) and Figs. S1C and S2A). Overall, these data validate that PARN is acetylated and that lysine 566 is a critical acetylated residue.
PARN is deacetylated by SIRT1
To determine which lysine deacetylase enzyme reverses PARN's acetylation, cells were treated with different HDAC inhibitors. Trichostatin A (TSA) is an inhibitor of class I and II HDACs, whereas nicotinamide inhibits the class III deacetylases, the sirtuins (SIRTs) (44, 45). Nicotinamide, but not TSA, enhanced PARN's acetylation in the presence of p300 and MOF (Fig. 2A and Fig. S1A). Furthermore, nicotinamide treatment also enhanced PCAF-induced PARN acetylation (Fig. 2B). This strongly suggests that the sirtuins are responsible for deacetylating PARN. Given that many deacetylases and their substrates interact, we screened for which sirtuin potentially regulates PARN by performing co-immunoprecipitation experiments between V5-tagged PARN and FLAG-tagged SIRT1–7. The results show that V5-PARN interacts strongly with SIRT1 and modestly with SIRT3, SIRT5, SIRT6, and SIRT7 (Fig. 3A). Further investigation of the sirtuins revealed that SIRT1 reverses PARN acetylation by p300, whereas SIRT3, SIRT5, and SIRT7 decrease PARN's acetylation but to a lesser degree (Fig. 3B). SIRT1 also prevents MOF- and PCAF-induced PARN acetylation (Figs. S1 (B and C) and S2). Consistent with the role of SIRT1 as the deacetylase enzyme that regulates PARN's acetylation, treatment with the SIRT1-specific inhibitor, EX-527 (46), modestly increased the level of p300-acetylated PARN (Fig. 2A).
Figure 2.

Nicotinamide treatment enhances PARN's acetylation. A, Western blotting analysis of PARN acetylation after treatmnent with lysine deacetylase inhibitors. V5-tagged PARN and HA-tagged p300 were co-transfected in 293T cells for 48 h, and subsequently, cells were treated with different lysine deacetylase inhibitors for 18 h. TSA is an inhibitor of class I and II lysine deacetylases, whereas nicotinamide inhibits the class III deacetylases, called sirtuins. EX-527 is a specific SIRT1 inhibitor. Protein extracts were immunoprecipitated (IP) with anti-V5 antibody and immunoblotted with a nonspecific anti-AcK antibody. Membranes were stripped under low-stringency conditions and reblotted using the anti-V5 antibody. The ratio of AcK to V5 was quantified and normalized to WT PARN. B, Western blotting of PARN acetylation by different lysine acetyltransferases after nicotinamide treatment. 293T cells were transfected for 48 h with V5-tagged PARN and four different lysine acetyltransferases. Additionally, cells were treated with DMSO or nicotinamide for 4 h, immunoprecipitated, and immunoblotted as described in A. For each lysine acetyltransferase, the ratio of AcK to V5 was quantified and normalized to WT PARN.
Figure 3.

PARN is deacetylated by SIRT1. A, Western blotting of interaction between PARN and SIRT1–7. V5-PARN was co-transfected with FLAG-tagged SIRT1-7. Immunoprecipitation (IP) using anti-FLAG antibody and Western blot analysis using anti-V5 antibody were used to assess interaction. B, Western blotting analysis of sirtuins that deacetylate p300-acetylated PARN. Sirtuins that were found to interact with PARN in A were co-transfected with V5-tagged PARN and HA-tagged p300 in 293T cells for 48 h. Protein extracts were immunoprecipitated with anti-V5 antibody and immunoblotted with a nonspecific anti-AcK antibody. Membranes were stripped under low-stringency conditions and reblotted using the anti-V5 antibody. The ratio of AcK to V5 was quantified and normalized to WT PARN. C, Western blotting analysis of endogenous PARN acetylation in SIRT1 WT (control) and SIRT1 KO HeLa cells. Cells were immunoprecipitated with anti-PARN antibody and immunoblotted with anti-acetyllysine antibody. Membranes were stripped under low-stringency conditions and reblotted using the anti-PARN antibody. The ratio of AcK to PARN was quantified and normalized to HeLa control cells. Western blotting of total lysate was also performed, and the ratio of PARN/vinculin in SIRT1 WT and SIRT1 KO HeLa cells was quantified.
To confirm that SIRT1 plays a role in deacetylating PARN endogenously, SIRT1 WT and SIRT1 knockout (KO) HeLa cells were immunoprecipitated with anti-PARN antibody and immunoblotted with anti-acetyllysine antibody. The results of this experiment demonstrate that PARN is also endogenously acetylated in HeLa cells, and PARN's acetylation is enhanced in SIRT1 KO cells (Fig. 3C). In addition to increasing PARN's acetylation, SIRT1 KO also increased PARN protein levels in HCT116, HeLa, and U2OS cells (Fig. 3C and Fig. S3A), whereas PARN transcript levels were unaffected (Fig. S3B). These results indicate that SIRT1 does not transcriptionally regulate PARN, and the increase of PARN protein in SIRT1 KO cells is likely due to post-translational regulation of PARN. In all, these data indicate that SIRT1 is the key enzyme responsible for deacetylating PARN.
Acetylation of PARN decreases its enzymatic activity
Because we established that PARN is modified by acetylation, we wanted to determine whether acetylation of PARN affects its enzymatic activity. To enrich for hypoacetylated PARN and p300-acetylated PARN, the following steps were performed. First, 293T cells were co-transfected with V5-tagged WT PARN and either HA-tagged control or HA-tagged p300 plasmids, as ectopic p300 expression can dramatically increase PARN acetylation. Then PARN was immunoprecipitated with anti-V5 antibody and eluted with V5 peptide. This allowed us to purify hypoacetylated PARN with no detectable acetylation (PARN) and hyperacetylated PARN with enriched acetylation (Ac-PARN). The protein concentrations were quantified and normalized by comparison with a panel of BSA standards (Fig. 4A), and Western blotting was performed to confirm PARN's acetylation status (Fig. 4B). An in vitro exoribonuclease activity assay was performed using a 5′-fluorescein–tagged RNA probe (Fig. 4C). Because PARN can only cleave the 3′-end, the length of the RNA probe can be used to assess PARN's enzymatic activity. When PARN is active, it shortens RNA length, causing the RNA to migrate lower in a denaturing polyacrylamide urea gel. Furthermore, the loss of the first nine adenosine residues can be labeled as PARN's deadenylase activity, whereas the cleavage of the entire RNA probe is indicative of PARN's overall exoribonuclease activity (Fig. 4D). Serial dilution experiments of PARN and Ac-PARN show that at lower concentrations, Ac-PARN has more bands at A9 compared with PARN, corresponding to a decrease in PARN's deadenylase activity (Fig. 4E). Time course experiments demonstrate that acetylation decreases both PARN's deadenylase and exoribonuclease activity, especially when it comes to cleaving cytosine and uracil residues (Fig. 4, F and G). Together, these data demonstrate that acetylation of PARN reduces its enzymatic activity in vitro.
Figure 4.
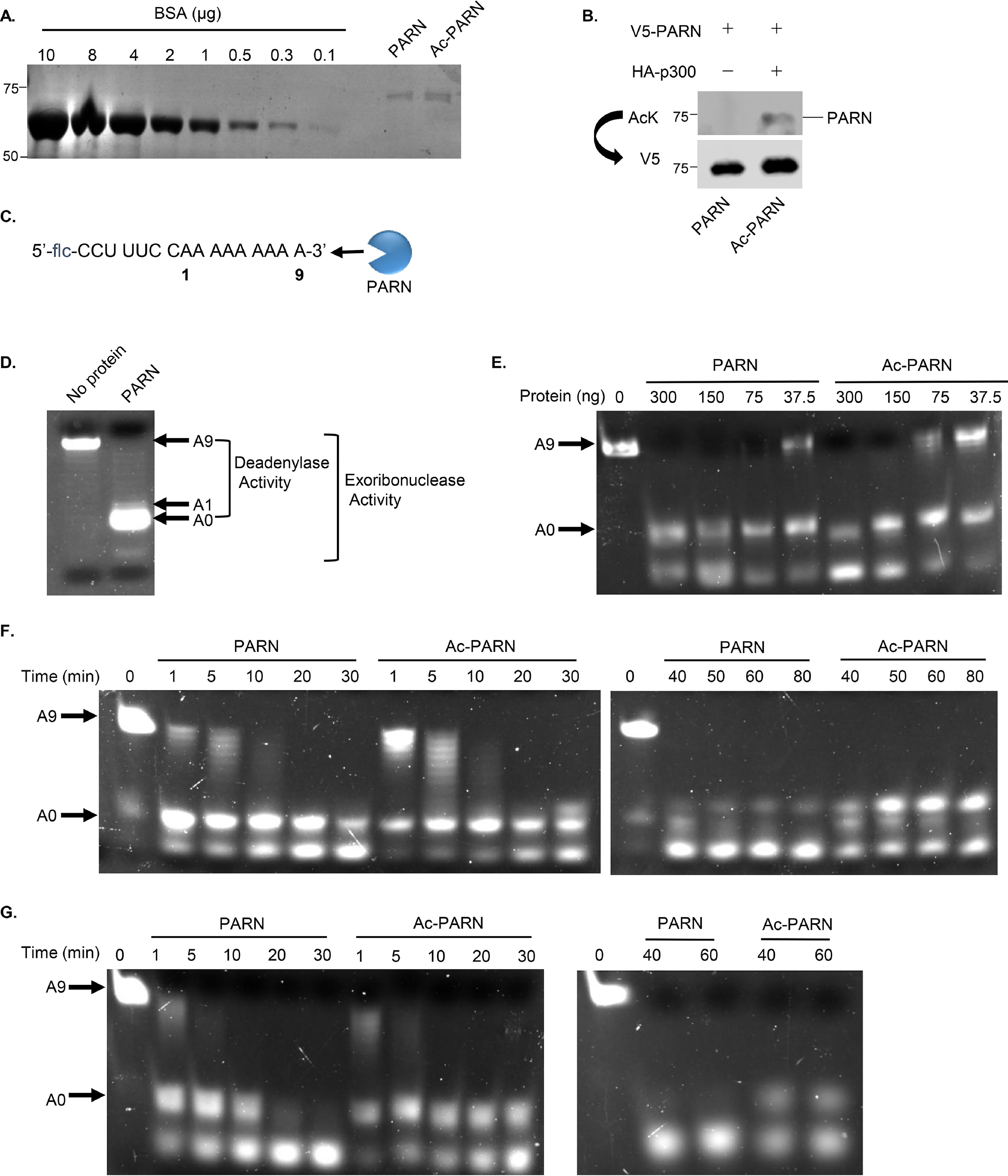
Acetylation of PARN decreases its enzymatic activity. A, Coomassie Blue staining of purified WT PARN (PARN) and p300-acetylated WT PARN (Ac-PARN) was used to quantify purified protein levels. B, Western blotting confirming that only Ac-PARN is acetylated. C, RNA probe used as substrate to assess PARN's activity. The RNA contains a 5′-fluorescein (flc) group and nine adenosine repeats on the 3′-end. An illustration of PARN demonstrates the 3′→5′ direction of its exoribonuclease activity. D, a representative in vitro 3′-exoribonuclease activity assay demonstrates how RNA probe length and migration are used to assess PARN's activity. A negative control sample without protein and a sample containing purified PARN were incubated with 5′-flc RNA at 30 °C for 1 h. After the reaction was stopped, samples were loaded onto a 20% polyacrylamide/50% urea/TBE gel. A9, the full-length RNA probe shown in C; A0, the fully deadenylated RNA probe. E, in vitro activity assay comparing the enzymatic activity of PARN and Ac-PARN serial dilutions. Different concentrations of PARN and Ac-PARN were incubated with 5′-flc RNA and processed as described in D. The same reaction mix without any protein was used as a control (0 ng). F and G, in vitro activity time course assay comparing the enzymatic activity of PARN and Ac-PARN. 5′-flc RNA probe was incubated with 75 ng (F) and 300 ng (G) of purified PARN and Ac-PARN at 30 °C over the indicated time. After the reaction, the samples were processed as described in D. The same reaction mix without any protein was used as a control (0 min).
Reducing PARN's acetylation rescues its enzymatic activity
Given that acetylation decreases PARN's exoribonuclease activity and the discovery that the K566R PARN mutation decreases acetylation, the effect of Ac-K566R PARN on enzymatic activity was investigated. To perform these experiments, hypoacetylated PARN and hyperacetylated PARN were purified as described previously, whereas Ac-K566R PARN was enriched by co-transfecting HA-tagged p300 and V5-tagged K566R PARN in 293T cells. The proteins were purified, normalized, and probed to confirm acetylation status (Fig. 5A and Fig. S4A) before in vitro exoribonuclease assays were performed. Our results show that Ac-PARN still has lower enzymatic activity compared with PARN. However, Ac-K566R PARN has enzymatic activity that is similar to PARN, as shown in serial dilution (Fig. 5B and Fig. S4B) and time course experiments using different concentrations of purified proteins (Fig. 5C and Fig. S4C). The in vitro activity assay from the 30-min time point (Fig. 5C) was adapted to a histogram, which emphasizes how reducing acetylation with the K566R mutant results in RNA length comparable with that of hypoacetylated PARN (Fig. 5D). Overall, these data illustrate that reducing PARN's acetylation restores its enzymatic activity.
Figure 5.
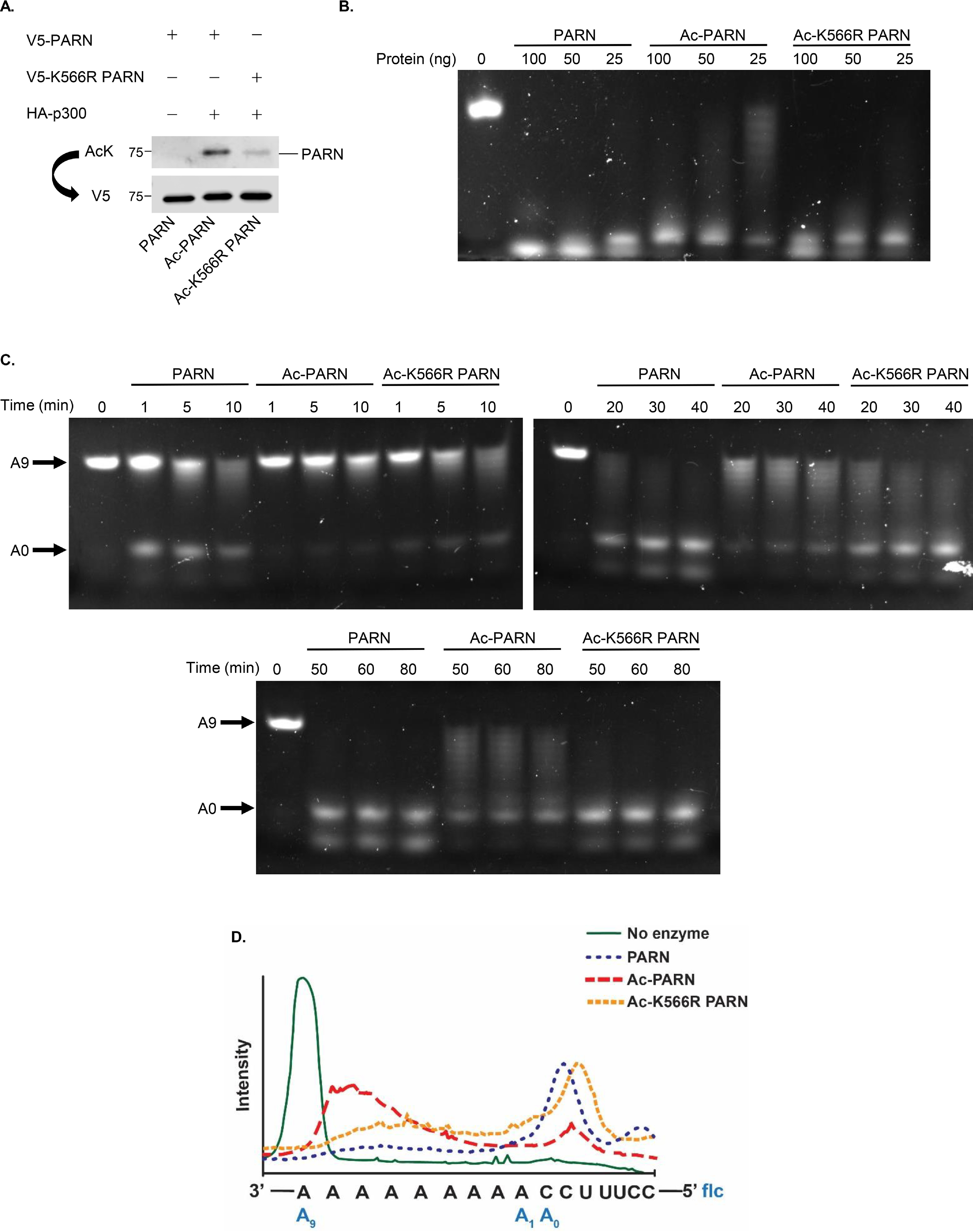
Reducing PARN's acetylation rescues its enzymatic activity. A, Western blot analysis confirming the acetylation status and normalization of purified WT PARN (PARN), p300-acetylated WT PARN (Ac-PARN), and p300-acetylated K566R PARN (Ac-K566R PARN). B, in vitro activity assay comparing the enzymatic activity of PARN, Ac-PARN, and Ac-K566R PARN serial dilutions. Serial dilutions of PARN, Ac-PARN, and Ac-K566R PARN were incubated with 5′-flc RNA at 30 °C for 1 h. After the reactions were stopped, samples were loaded onto a 20% polyacrylamide/50% urea/TBE gel. The same reaction mix without any protein was used as a control (0 ng). C, in vitro activity time course assay comparing the enzymatic activities of PARN, Ac-PARN, and Ac-K566R PARN. 25 ng of purified PARN, Ac-PARN, and Ac-K566R PARN were incubated with 5′-fluorescein RNA at 30 °C over the indicated time. Then samples were processed as described in B. The same reaction mix without any protein was used as a control (0 min). D, histogram of the 30-min time point of the in vitro activity assay shown in C demonstrates how reducing acetylation via Ac-K566R PARN restores PARN's activity similar to WT PARN levels. The histogram was generated using ImageJ and Adobe Illustrator.
PARN acetylation regulates levels of adenylated hTR
Because we found that acetylation affects PARN's activity in vitro, using a synthetic RNA probe, we further tested the effect of PARN acetylation in vivo by using an endogenous RNA target. Given that PARN is known to deadenylate and cleave the excess 3′-end of the hTR to form a mature template, levels of polyadenylated hTR were used to indicate relative enzymatic activity of acetylated PARN (1, 27–30). PARN KO cells were generated in 293FT cells to remove the expression of endogenous PARN protein (Fig. 6A and Fig. S5). RT-qPCRs of 293FT cells show that PARN KO increases levels of adenylated hTR while decreasing total levels of hTR (Fig. 6B), which is consistent with the results from other studies (25, 30). When PARN is depleted, the poly(A) tail of hTR cannot be cleaved, resulting in the accumulation of adenylated hTR (1). However, because adenylated hTR is prone to degradation via the nuclear RNA exosome, there is a decrease in total hTR levels (Fig. 6B) (1, 27, 28). Ectopic expression of WT and K566R PARN in PARN KO cells partially rescued hTR levels (Fig. 6, A and B). To test how PARN acetylation affects levels of endogenous hTR adenylation, PARN KO cells were co-transfected with HA-tagged p300 as well as WT PARN (Ac-PARN) or K566R PARN (Ac-K566R PARN) (Fig. 6, C and D). After confirming PARN protein levels were equally expressed (Fig. 6C), RT-qPCR was performed to examine levels of adenylated hTR transcripts (Fig. 6D). Interestingly, K566R PARN, which has low levels of acetylation, significantly decreases levels of adenylated hTR compared with WT PARN (Fig. 6D), which is hyperacetylated. Lower levels of adenylated hTR in K566R-transfected cells indicate that K566R PARN, with reduced acetylation, has increased deadenylase activity compared with hyperacetylated WT PARN. This is consistent with our in vitro findings that acetylation decreases PARN's enzymatic activity, and K566R PARN has higher activity compared with Ac-PARN. In sum, these data support the role of acetylation in decreasing PARN's enzymatic activity and increasing levels of adenylated hTR.
Figure 6.

PARN acetylation regulates levels of adenylated hTR. A, Western blotting of 293FT control and PARN KO cells, induced with CRISPR-Cas9. Cells were transfected for 48 h with control plasmid, v5-tagged WT PARN or v5-tagged K566R PARN to rescue PARN protein expression. B, RT-qPCR of adenylated and total hTR in 293FT control and PARN KO cells (n = 4) confirms that PARN regulates hTR. Cells were transfected as described in A. Each symbol (▴, Δ, ▪ and □) represents an independent biological replicate (n = 4). C, Western blot analysis demonstrates that levels of Ac-PARN and Ac-K566R PARN are even. 293FT PARN KO cells were transfected with either HA-tagged p300 and WT PARN (Ac-PARN) or HA-tagged p300 and K566R PARN (Ac-K566R PARN). D, RT-qPCR comparing levels of adenylated hTR in PARN KO cells expressing Ac-PARN and Ac-K566R PARN (n = 4). PARN KO cells were transfected as described in C. Data are presented as the mean ± S.D. (error bars). All statistical analyses were performed using unpaired, two-sided Wilcoxon–Mann–Whitney tests (*, p < 0.05; ns, not significant).
SIRT1 regulates levels of adenylated hTR through PARN
Given our findings that SIRT1 is responsible for deacetylating PARN, and PARN acetylation plays an important role in regulating levels of adenylated hTR, we then investigated whether SIRT1 can regulate hTR through PARN. Considering that SIRT1 KO and SIRT1 inhibition increase levels of acetylated PARN (Figs. 2 and 3C and Fig. S1A) and acetylation decreases PARN's enzymatic activity (Figs. 4–6), we hypothesized that disruption of SIRT1 would result in an accumulation of adenylated hTR. Because HCT116 and HeLa cells express high levels of hTR, they were used in SIRT1 KO and SIRT1 inhibition experiments (Fig. 7). As predicted, both SIRT1 KO (Fig. 7A) and SIRT1 inhibition using EX-527 (Fig. 7B) significantly increased levels of adenylated hTR. Next, we wanted to establish whether SIRT1 regulates hTR independently of PARN or through a PARN-dependent mechanism. To assess whether SIRT1 regulates adenylation of hTR through PARN, SIRT1 was stably knocked down using lentiviral transduction in PARN WT and PARN KO cells (Fig. 7C). As expected, SIRT1 knockdown (KD) significantly increased adenylated hTR in PARN WT cells. However, in PARN KO cells, SIRT1 KD could not alter levels of adenylated hTR (Fig. 7D). These results suggest that SIRT1 regulates levels of adenylated hTR through PARN.
Figure 7.

SIRT1 regulates levels of adenylated hTR through PARN. A, RT-qPCR of HCT116 (n = 4) and HeLa cells (n = 4) reveals that SIRT1 KO increases levels of adenylated hTR. B, RT-qPCR of HCT116 cells (n = 4) shows that SIRT1 inhibition increases levels of adenylated hTR. HCT116 cells were treated with DMSO or EX-527, a SIRT1-specific inhibitor, for 18 h (n = 4). C, Western blotting confirming stable SIRT1 knockdown in 293FT control and PARN KO cells. 293FT control and PARN KO cells were transduced with lentivirus containing either pLKO.1 control or pLKO.1 shSIRT1 to stably knock down SIRT1. The ratio of SIRT1 to β-actin was quantified to confirm successful knockdown. D, RT-qPCR of adenylated hTR demonstrates that SIRT1 regulates hTR through PARN. 293FT control and PARN KO cells described in C were used to test levels of adenylated hTR. Each symbol (▴, Δ, ■, and □) represents an independent biological replicate (n = 4). Data are presented as the mean ± S.D. (error bars). All statistical analyses were performed using unpaired, two-sided Wilcoxon–Mann–Whitney tests (*, p < 0.05; ns, not significant).
In addition to hTR, we also tested whether SIRT1 could regulate the adenylation of PARN's other noncoding RNA targets. snoRNAs and scaRNAs have been reported to be deadenylated by PARN (17, 26, 42). Thus, we tested whether SIRT1 could regulate the adenylation of snoRNAs and scaRNAs. Consistent with our findings that SIRT1 regulates PARN's activity, SIRT1 KO increased levels of adenylated snoRNA10 (Fig. S6), scaRNA8, and scaRNA13 (Fig. S7). These results further solidify our conclusion that SIRT1 can regulate the adenylation of PARN's targets.
Discussion
Acetylation and deacetylation of histone proteins have been extensively studied and are well-known to regulate transcription. However, over the past 2 decades, partly due to advances in MS, many nonhistone proteins have also been found to undergo acetylation and deacetylation (37, 38). Our results reveal that both endogenously and exogenously expressed PARN can be post-translationally modified by acetylation in vivo, which further supports that acetylation and deacetylation extend beyond histone protiens. Moreover, we demonstrate a mechanism in which acetylation by p300 and deacetylation by SIRT1 play an important role in regulating PARN's 3′-exoribonuclease activity, thereby mediating the maturation of the telomerase RNA component. These data further solidify the notion that the consequences of protein acetylation are much broader than transcription regulation.
PARN can regulate telomere maintenance by processing immature, polyadenylated hTR into the mature, 451-base form that serves as a template for telomere elongation (1). Given our finding that SIRT1 regulates hTR through PARN, it is interesting to note that just like PARN and hTR, SIRT1 has been reported to regulate telomere length. For example, some studies examined the role of SIRT1 on telomere length by using mouse embryonic fibroblasts (MEFs) from SIRT1+/+, SIRT1−/−, and SIRT1 gain-of-function (SIRT1super) mice. These previous studies found that SIRT1super MEFs had longer telomeres than SIRT1+/+ MEFs, whereas SIRT−/− MEFs had shorter telomere lengths compared with SIRT1+/+ MEFs. These results demonstrate that SIRT1 is important in regulating telomere length, just like PARN and hTR. Furthermore, they also found the effect of SIRT1super on telomere length was absent in mice with concomitant loss of TERC (the gene that encodes hTR), which strongly suggests the effect of SIRT1super on telomere length depends on telomerase activity (47, 48). However, the mechanism by which SIRT1 regulates telomere length in a telomerase-dependent manner has not been examined prior to this study. Instead, other studies have evaluated how SIRT1 regulates telomere integrity by epigenetically maintaining chromatin structure (49). Specifically, SIRT1 transcriptionally regulates the telomerase reverse transcriptase enzyme and proteins in the telomere-protecting shelterin complex (50–52). It is also important to note that a recent study demonstrated how telomere dysfunction can regulate SIRT1 protein expression in mice, which suggests that there may be a dual regulatory mechanism between telomeres and SIRT1 (53). Results from these previous studies that highlight the role of SIRT1 in regulating telomere length are consistent with our conclusion that SIRT1 regulates the telomerase RNA component through PARN.
Given that hTR regulates telomere length and our finding that SIRT1 regulates hTR, we examined the published literature to determine whether SIRT1 and hTR have similar biological roles. Interestingly, both SIRT1 and hTR are involved in aging and senescence, processes that are also associated with telomere length (54–60). In fact, TERC−/− mice have shortened lifespans, signs of pulmonary aging, and induced senescence when compared with TERC+/+ mice (61, 62). Furthermore, knockdown of hTR in human induced pluripotent stem cells results in cellular senescence (63). SIRT1 also regulates cellular senescence, as SIRT1 inhibition has been shown to induce cellular senescence, similar to the effect of hTR ablation (64–66). Absence of SIRT1 also reduces lifespan in zebrafish, whereas expression of SIRT1 in the hypothalamus of mice is related to increased lifespan and a delay in aging-associated phenotypes (67, 68). Additionally, mesenchymal stem cells from aged rats had lower SIRT1 mRNA and protein levels as well as decreased telomerase activity (52). This is consistent with the finding that SIRT1 gene expression is positively correlated with telomere length in humans (69, 70). Based on the analysis of the literature and our experimental findings, we hypothesize that low levels of SIRT1 in aging would result in an accumulation of acetylated PARN with lower enzymatic activity. Increased levels of acetylated PARN with lower activity would result in elevated levels of immature, polyadenylated hTR (illustrated in Fig. 8), causing low telomerase activity and telomere shortening. Future experiments are necessary to examine the role of PARN in SIRT1-related aging.
Figure 8.

Proposed working model for how PARN is regulated by acetylation/deacetylation. Based on our experimental findings, we propose that PARN acetylation regulates its enzymatic activity, which in turn mediates levels of immature, adenylated hTR. When PARN is acetylated by p300 at lysine 566, it decreases PARN's enzymatic activity, resulting in elevated levels of adenylated hTR. However, when SIRT1 deacetylates PARN, it restores PARN's enzymatic activity, allowing PARN to deadenylate hTR.
PARN deadenylates the poly(A) tail of various noncoding RNAs, including snoRNAs and scaRNAs, which is required for their maturation and enables them to direct RNA modifications with the help of auxiliary proteins (17, 24, 26, 42, 71–73). Our finding that SIRT1 can also regulate the adenylation of PARN's snoRNA and scaRNA targets further validates that SIRT1 can regulate the adenylation of noncoding RNA targets through PARN.
Our results show that lysine Lys-566 is one of PARN's main acetylated residues, as mutating lysine 566 to arginine (K566R) significantly reduced PARN's acetylation. Interestingly, K566R is reported to be a naturally occurring PARN mutation in humans that is not associated with disease (74). In fact, our data demonstrate that the K566R mutation restores PARN's enzymatic activity by reducing its acetylation, suggesting that K566R PARN may have a protective effect, as PARN's activity is required for telomere maintenance.
Based on our findings, PARN is endogenously acetylated in some cell lines but not in others. Therefore, PARN's acetylation status is likely cell type–dependent and may be induced under biological conditions that were not tested in this study.
Our study also revealed that SIRT1 KO results in an increase both in PARN's acetylation and overall protein levels. Given that SIRT1 does not transcriptionally alter PARN's gene expression, we hypothesize that SIRT1 post-translationally regulates PARN's protein levels through acetylation. There are many reports on how acetylation can stabilize proteins by protecting them from ubiquitination and preventing their proteasomal degradation (75–78). Interestingly, lysine 566 was found to be putatively ubiquitinated in different human cancer cell lines (79). Therefore, it is possible that acetylation of PARN could prevent its ubiquitination, resulting in elevated protein but not transcript levels. It is important to note that although SIRT1 KO increases PARN protein levels, this protein also has elevated acetylation compared with SIRT1 WT cells. Because acetylation can reduce PARN's enzymatic activity, this explains why SIRT1 KO cells have increased levels of immature, adenylated hTR despite their elevated PARN protein levels.
Although SIRT1 consistently abolished PARN's acetylation, based on our studies, we cannot confirm whether this effect is direct or indirect. Results from previous studies have shown that SIRT1 binds to and regulates the activity of p300 (80). SIRT1 can also bind and deacetylate MOF, which inhibits its enzymatic activity and promotes its degradation (76). Therefore, overexpression of SIRT1 could result in the deacetylation of PARN by changing the activities of KATs that regulate PARN. Similarly, ablation of SIRT1 could increase acetylation, as it can no longer inhibit KAT activity. It is important to note that regardless of whether the effect of SIRT1 on PARN is direct or indirect, it does not alter the chief conclusion of our paper, that SIRT1 can regulate levels of adenylated hTR through PARN.
Based on our findings, we propose a new mechanism by which acetylation of PARN regulates levels of adenylated hTR in cells (summarized in Fig. 8). We propose that when PARN is hypoacetylated, it has high enzymatic activity. However, when PARN is acetylated by p300 at Lys-566, it decreases PARN's enzymatic activity, as shown through in vitro exoribonuclease activity assays. Additionally, reducing PARN's acetylation with the K566R mutation increases PARN's enzymatic activity, similar to nonacetylated PARN, and lowers levels of adenylated hTR compared with acetylated WT PARN. This confirms that reducing acetylation increases PARN's 3′-exoribonuclease activity. Furthermore, because SIRT1 deacetylates PARN, SIRT1 KO and inhibition result in an increase in acetylated PARN. SIRT1 KO, KD, and inhibition also elevate levels of immature, polyadenylated hTR. Our finding that PARN depletion could inhibit the effect of SIRT1 KD on polyadenylated hTR strongly suggests that PARN plays an important role in SIRT1-mediated hTR regulation. Overall, our findings elucidate a new mechanism through which SIRT1 can regulate levels of polyadenylated hTR by deacetylating PARN.
Experimental procedures
Cell culture and transient transfection
293T, 293FT, HCT116 (p53 WT), U2OS, and HeLa cells were obtained from ATCC, cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin-streptomycin, and incubated at 37 °C, with 5% CO2 in sterile conditions.
For transient transfection experiments, cells were seeded overnight and transfected using Lipofectamine 2000 (Thermo Fisher Scientific, 11668019). FLAG-MOF (76), FLAG-PCAF (81), FLAG-GCN5 (82), HA-p300 (83), FLAG SIRT1–7 (84), and Myc-SIRT1(64) plasmids have been described previously.
Drug treatments
Different lysine deacetylase inhibitors were used to treat cells for specified times. 100 ng/ml TSA was used to broadly inhibit class I and II HDACs. 10 mm nicotinamide was used to widely inhibit class III HDACs, the sirtuins. To specifically inhibit SIRT1, cells were incubated with 1 μm EX-527 for 18 h.
Preparation and transduction of shRNA
To generate stable knockdown cell lines, lentivirus was prepared by transfecting 293FT cells with pLKO.1 control or pLKO.1 encoding shRNA targeting SIRT1 (shSIRT1, GTACCGGCATGAAGTGCCTCAGATATTACTCGAGTAATATCTGAGGCACTTCATGTTTTTTG) along with third-generation packaging vectors. After 48 h, the supernatant was collected, filtered (0.22 μm), and used to transduce cells. 48 h post-transduction, cells were selected for puromycin resistance and used for subsequent experiments.
Site-directed mutagenesis
The pLX304 plasmid encoding human PARN was purchased from DNASU (HsCD00438869). Plasmids expressing K→R mutants (K220R, K243R, K250R, K499R, and K566R) were generated via site-directed mutagenesis using pLX304 PARN template, dNTPs, primers (listed in Table S1), and PfuUltra High-Fidelity DNA Polymerase (Agilent Technologies, NC9751972). The resulting PCR product was incubated with DPNI (New England Biolabs, R0176S) for 2 h at 37 °C to remove nonmutated template. The digested product was transformed in DH5α competent bacteria (37 °C, overnight) and cultured in Luria–Bertani medium with ampicillin (37 °C, overnight), and plasmids were recovered using a miniprep kit (Promega, A1222). Mutations were confirmed by DNA sequencing.
Generation of CRISPR-induced knockout cells
To generate PARN and SIRT1 KO cells, specific guide RNAs (listed in Table S2) were cloned into pSpCas9(BB)-2A-Puro (PX459) V2.0 and lentiCRISPR v2, respectively, as described previously (85, 86). pSpCas9(BB)-2A-Puro (PX459) V2.0 and LentiCRISPR v2 were gifts from Feng Zhang (Addgene plasmid 62988; RRID:Addgene_62988) and (Addgene plasmid 52961; RRID:Addgene_52961), respectively.
PX459 plasmid containing PARN guide RNA was transfected into 293FT cells using Lipofectamine 2000 for 48 h (Thermo Fisher Scientific, 11668019). LentiCRISPR v2 plasmid containing SIRT1 guide RNA was used to generate lentivirus targeting SIRT1. The virus was used to transduce HCT116, U2OS and HeLa cells. All cells were selected with puromycin and underwent monoclonal expansion. Western blotting was performed to assess KO efficiency.
Western blotting
Cells were harvested and lysed using NETN lysis buffer (20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 100 mm NaCl, 0.5% NP-40, and protease inhibitor) and centrifuged at 12,000 × g for 10 min at 4 °C to extract protein. Protein concentrations were quantified using Protein Assay Dye (Bio-Rad, 5000006; Eppendorf, BioSpectrometer). Protein samples were boiled at 95 °C for 10 min and resolved by SDS-PAGE. Proteins were transferred to nitrocellulose membranes (Bio-Rad, TransBlot Turbo), blocked in 5% milk for 1 h, and incubated with primary antibody (Table S3) diluted in 5% milk overnight. Then membranes were incubated in secondary antibody (Table S3) for 1 h at room temperature and detected using a chemiluminescence detection kit (Thermo Fisher Scientific, PI34580, PI34076, and PI34095).
Immunoprecipitation
For immunoprecipitation assays, cells were lysed using NETN lysis buffer (20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 100 mm NaCl, 0.5% NP-40, and protease inhibitor). Samples were centrifuged at 12,000 × g for 10 min at 4 °C to extract protein. 20 µl of total lysate was saved for Western blot analysis. The remainder of the protein was precleared with protein-agarose A/G (depending on the primary antibody) for 2 h, rotating at 4 °C. After preclearing, samples were centrifuged at 5,000 × g for 1 min at 4 °C and incubated with antibody-cross-linked agarose A/G overnight (Table S3), rotating at 4 °C. The next day, the beads were washed five times with NETN lysis buffer. After the last wash, all supernatants were aspirated, and 20 µl of 2× protein-loading buffer was added to beads. The protein samples were boiled and resolved by SDS-PAGE, followed by Western blotting.
In vitro 3′-exoribonuclease activity assay
To purify PARN for the exoribonuclease activity assay, 293T cells were co-transfected with plasmids expressing 1) HA-tagged control and WT PARN, 2) HA-tagged p300 and WT PARN, and 3) HA-tagged p300 and K566R PARN. After 48 h, protein was extracted using radioimmune precipitation assay buffer (20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 300 mm NaCl, 0.5% NP-40, 0.1% SDS, and protease inhibitor) and immunoprecipitated with anti-V5-agarose G cross-linked beads overnight, rotating at 4 °C. The next day, the beads were washed five times with radioimmune precipitation assay buffer, and V5-tagged PARN was eluted by incubating beads with 100 µg/µl V5 peptide (Sigma–Aldrich; V7754) for 2 h, rotating at 4 °C. Post-elution, protein samples were purified and concentrated using centrifugal filters (EMD Millipore; UFC501096). Purified protein samples were aliquoted and frozen at −80 °C. Protein samples were quantified and normalized via Coomassie Blue staining with BSA standards (Research Products International, A30075) and Western blotting. After normalizing protein concentrations, gel-based exoribonuclease activity assays were performed using a synthetic RNA probe (5′-fluorescein-CCU UUC CAA AAA AAA A-3′) as described previously (87).
RT-qPCR
RNA was extracted using a high pure RNA isolation kit (Roche Applied Science, 11828665001). 1 µg of RNA was converted to cDNA with the qScript™ Flex cDNA Synthesis Kit (QuantaBio, 101414-112) using either oligo-dT20 primers to test adenylated transcripts or random primers to test total transcript levels. The synthesized cDNA, primers (listed in Table S4), and BrightGreen (Abm, MasterMix-R) were used to perform qPCR (Applied Biosystems, QuantStudio3). Gene expressions were normalized to the housekeeping gene, GAPDH. All RT-qPCR results were analyzed using 2−ΔΔCT.
Statistical analyses
All data are shown as the mean and S.D. of four biological replicates, unless otherwise specified. Biological replicates are shown as data points in the respective figures. For RT-qPCR experiments, each biological replicate is the average of three technical replicates. For statistical analyses between two groups, an unpaired, two-sided, nonparametric t test, the Wilcoxon–Mann–Whitney test, was performed using GraphPad Prism. Exact p values are indicated for each experiment. p values <0.05 were considered statistically significant (*).
Data availability
All representative data are contained within the article. Raw data are available upon request from Eden Dejene (edendejene@gwu.edu) or Edward Seto (seto@gwu.edu).
Supplementary Material
Acknowledgments
We thank Dr. Sebastiaan Winkler (University of Nottingham, UK) for sharing the protocol for gel-based fluorescence detection of 3′-exoribonuclease activity.
This article contains supporting information.
Author contributions—E. A. D., and E. S. conceptualization; E. A. D., H. L., and E. S. resources; E. A. D., Y. L., Z. S., H. L., and E. S. data curation; E. A. D. formal analysis; E.A.D., Y. L., and E. S. supervision;E. A. D., and E. S. funding acquisition; E. A. D. validation; E. A. D., and Z. S. investigation; E. A. D., visualization; E. A. D., and Y. L. methodology; E. A. D. writing-original draft; E. A. D. and E. S. project administration; E. A. D., Y. L., and E. S. writing-review and editing.
Funding and additional information—This work was supported by National Institutes of Health Grants 3R01CA187040-05S1 (to E. A. D.) and R01CA187040 and R01CA169210 (to E. S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- PARN
- poly(A)-specific ribonuclease
- scaRNA
- small Cajal body RNA
- snoRNA
- small nucleolar RNA
- hTR
- human telomerase RNA component
- PTM
- post-translational modification
- KAT
- lysine acetyltransferase
- HDAC
- histone deacetylase
- TSA
- trichostatin A
- SIRT
- sirtuin
- KO
- knockout
- AcK
- acetyllysine
- qPCR
- quantitative PCR
- KD
- knockdown
- MEF
- mouse embryo fibroblast
- NP-40
- Nonidet P-40
- flc
- fluorescein
- CBP
- cAMP response element-binding protein.
References
- 1. Moon D. H., Segal M., Boyraz B., Guinan E., Hofmann I., Cahan P., Tai A. K., and Agarwal S. (2015) Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat. Genet. 47, 1482–1488 10.1038/ng.3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang X., Devany E., Murphy M. R., Glazman G., Persaud M., and Kleiman F. E. (2015) PARN deadenylase is involved in miRNA-dependent degradation of TP53 mRNA in mammalian cells. Nucleic Acids Res. 43, 10925–10938 10.1093/nar/gkv959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aström J., Aström A., and Virtanen A. (1991) In vitro deadenylation of mammalian mRNA by a HeLa cell 3′ exonuclease. EMBO J. 10, 3067–3071 10.1002/j.1460-2075.1991.tb07858.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aström J., Aström A., and Virtanen A. (1992) Properties of a HeLa cell 3′ exonuclease specific for degrading poly(A) tails of mammalian mRNA. J. Biol. Chem. 267, 18154–18159 [PubMed] [Google Scholar]
- 5. Körner C. G., and Wahle E. (1997) Poly(A) tail shortening by a mammalian poly(A)-specific 3′-exoribonuclease. J. Biol. Chem. 272, 10448–10456 10.1074/jbc.272.16.10448 [DOI] [PubMed] [Google Scholar]
- 6. Henriksson N., Nilsson P., Wu M., Song H., and Virtanen A. (2010) Recognition of adenosine residues by the active site of poly(A)-specific ribonuclease. J. Biol. Chem. 285, 163–170 10.1074/jbc.M109.043893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Copeland P. R., and Wormington M. (2001) The mechanism and regulation of deadenylation: identification and characterization of Xenopus PARN. RNA 7, 875–886 10.1017/s1355838201010020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Devany E., Zhang X., Park J. Y., Tian B., and Kleiman F. E. (2013) Positive and negative feedback loops in the p53 and mRNA 3′ processing pathways. Proc. Natl. Acad. Sci. U. S. A. 110, 3351–3356 10.1073/pnas.1212533110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Houseley J., LaCava J., and Tollervey D. (2006) RNA-quality control by the exosome. Nat. Rev. Mol. Cell Biol. 7, 529–539 10.1038/nrm1964 [DOI] [PubMed] [Google Scholar]
- 10. Montellese C., Montel-Lehry N., Henras A. K., Kutay U., Gleizes P.-E., and O'Donohue M.-F. (2017) Poly(A)-specific ribonuclease is a nuclear ribosome biogenesis factor involved in human 18S rRNA maturation. Nucleic Acids Res. 45, 6822–6836 10.1093/nar/gkx253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoda M., Cifuentes D., Izumi N., Sakaguchi Y., Suzuki T., Giraldez A. J., and Tomari Y. (2013) Poly(A)-specific ribonuclease mediates 3′-end trimming of Argonaute2-cleaved precursor microRNAs. Cell. Rep. 5, 715–726 10.1016/j.celrep.2013.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katoh T., Hojo H., and Suzuki T. (2015) Destabilization of microRNAs in human cells by 3′ deadenylation mediated by PARN and CUGBP1. Nucleic Acids Res. 43, 7521–7534 10.1093/nar/gkv669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee D., Park D., Park J. H., Kim J. H., and Shin C. (2019) Poly(A)-specific ribonuclease sculpts the 3′ ends of microRNAs. RNA 25, 388–405 10.1261/rna.069633.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shukla S., Bjerke G. A., Muhlrad D., Yi R., and Parker R. (2019) The RNase PARN controls the levels of specific miRNAs that contribute to p53 regulation. Mol. Cell 73, 1204–1216.e4 10.1016/j.molcel.2019.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishikawa H., Yoshikawa H., Izumikawa K., Miura Y., Taoka M., Nobe Y., Yamauchi Y., Nakayama H., Simpson R. J., Isobe T., and Takahashi N. (2017) Poly(A)-specific ribonuclease regulates the processing of small-subunit rRNAs in human cells. Nucleic Acids Res. 45, 3437–3447 10.1093/nar/gkw1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shukla S., and Parker R. (2017) PARN modulates Y RNA stability and its 3′-end formation. Mol. Cell Biol. 37, e00264–17 10.1128/mcb.00264-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berndt H., Harnisch C., Rammelt C., Stöhr N., Zirkel A., Dohm J. C., Himmelbauer H., Tavanez J. P., Hüttelmaier S., and Wahle E. (2012) Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA 18, 958–972 10.1261/rna.032292.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berendes H. D., and Meyer G. F. (1968) A specific chromosome element, the telomere of Drosophila polytene chromosomes. Chromosoma 25, 184–197 10.1007/BF00327177 [DOI] [PubMed] [Google Scholar]
- 19. Greider C. W. (1991) Telomeres. Curr. Opin. Cell Biol. 3, 444–451 10.1016/0955-0674(91)90072-7 [DOI] [PubMed] [Google Scholar]
- 20. Beattie T. L., Zhou W., Robinson M. O., and Harrington L. (1998) Reconstitution of human telomerase activity in vitro. Curr. Biol. 8, 177–180 10.1016/S0960-9822(98)70067-3 [DOI] [PubMed] [Google Scholar]
- 21. Mitchell J. R., Cheng J., and Collins K. (1999) A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol. Cell Biol. 19, 567–576 10.1128/mcb.19.1.567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tummala H., Walne A., Collopy L., Cardoso S., de la Fuente J., Lawson S., Powell J., Cooper N., Foster A., Mohammed S., Plagnol V., Vulliamy T., and Dokal I. (2015) Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J. Clin. Invest. 125, 2151–2160 10.1172/JCI78963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burris A. M., Ballew B. J., Kentosh J. B., Turner C. E., Norton S. A., NCI DCEG Cancer Genomics Research Laboratory, NCI DCEG Cancer Sequencing Working Group, Giri N., Alter B. P., Nellan A., Gamper C., Hartman K. R., and Savage S. A. (2016) Hoyeraal-Hreidarsson syndrome due to PARN mutations: fourteen years of follow-up. Pediatr. Neurol. 56, 62–68.e1 10.1016/j.pediatrneurol.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dodson L. M., Baldan A., Nissbeck M., Gunja S. M. R., Bonnen P. E., Aubert G., Birchansky S., Virtanen A., and Bertuch A. A. (2019) From incomplete penetrance with normal telomere length to severe disease and telomere shortening in a family with monoallelic and biallelic PARN pathogenic variants. Hum. Mutat. 40, 2414–2429 10.1002/humu.23898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benyelles M., Episkopou H., O'Donohue M. F., Kermasson L., Frange P., Poulain F., Burcu Belen F., Polat M., Bole-Feysot C., Langa-Vives F., Gleizes P. E., de Villartay J. P., Callebaut I., Decottignies A., and Revy P. (2019) Impaired telomere integrity and rRNA biogenesis in PARN-deficient patients and knock-out models. EMBO Mol. Med. 11, e10201 10.15252/emmm.201810201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dhanraj S., Gunja S. M., Deveau A. P., Nissbeck M., Boonyawat B., Coombs A. J., Renieri A., Mucciolo M., Marozza A., Buoni S., Turner L., Li H., Jarrar A., Sabanayagam M., Kirby M., et al. (2015) Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN). J. Med. Genet. 52, 738–748 10.1136/jmedgenet-2015-103292 [DOI] [PubMed] [Google Scholar]
- 27. Shukla S., Schmidt J. C., Goldfarb K. C., Cech T. R., and Parker R. (2016) Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nat. Struct. Mol. Biol. 23, 286–292 10.1038/nsmb.3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tseng C. K., Wang H. F., Burns A. M., Schroeder M. R., Gaspari M., and Baumann P. (2015) Human telomerase RNA processing and quality control. Cell. Rep. 13, 2232–2243 10.1016/j.celrep.2015.10.075 [DOI] [PubMed] [Google Scholar]
- 29. Nguyen D., Grenier St-Sauveur V., Bergeron D., Dupuis-Sandoval F., Scott M. S., and Bachand F. (2015) A polyadenylation-dependent 3′ end maturation pathway is required for the synthesis of the human telomerase RNA. Cell. Rep. 13, 2244–2257 10.1016/j.celrep.2015.11.003 [DOI] [PubMed] [Google Scholar]
- 30. Roake C. M., Chen L., Chakravarthy A. L., Ferrell J. E. Jr., Raffa G. D., and Artandi S. E. (2019) Disruption of telomerase RNA maturation kinetics precipitates disease. Mol. Cell 74, 688–700.e3 10.1016/j.molcel.2019.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spoel S. H. (2018) Orchestrating the proteome with post-translational modifications. J. Exp. Bot. 69, 4499–4503 10.1093/jxb/ery295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reinhardt H. C., Hasskamp P., Schmedding I., Morandell S., van Vugt M. A., Wang X., Linding R., Ong S. E., Weaver D., Carr S. A., and Yaffe M. B. (2010) DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell. 40, 34–49 10.1016/j.molcel.2010.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duan T.-L., He G.-J., Hu L.-D., and Yan Y.-B. (2019) The intrinsically disordered C-terminal domain triggers nucleolar localization and function switch of PARN in response to DNA damage. Cells 8, 836 10.3390/cells8080836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schölz C., Weinert B. T., Wagner S. A., Beli P., Miyake Y., Qi J., Jensen L. J., Streicher W., McCarthy A. R., Westwood N. J., Lain S., Cox J., Matthias P., Mann M., Bradner J. E., et al. (2015) Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 33, 415–423 10.1038/nbt.3130 [DOI] [PubMed] [Google Scholar]
- 36. Yang X. J., and Seto E. (2008) The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 10.1038/nrm2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Drazic A., Myklebust L. M., Ree R., and Arnesen T. (2016) The world of protein acetylation. Biochim. Biophys. Acta 1864, 1372–1401 10.1016/j.bbapap.2016.06.007 [DOI] [PubMed] [Google Scholar]
- 38. Allfrey V. G., Faulkner R., and Mirsky A. E. (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 51, 786–794 10.1073/pnas.51.5.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shimazu T., Horinouchi S., and Yoshida M. (2007) Multiple histone deacetylases and the CREB-binding protein regulate pre-mRNA 3′-end processing. J. Biol. Chem. 282, 4470–4478 10.1074/jbc.M609745200 [DOI] [PubMed] [Google Scholar]
- 40. Shan P., Fan G., Sun L., Liu J., Wang W., Hu C., Zhang X., Zhai Q., Song X., Cao L., Cui Y., Zhang S., and Wang C. (2017) SIRT1 functions as a negative regulator of eukaryotic poly(A)RNA transport. Curr. Biol. 27, 2271–2284.e5 10.1016/j.cub.2017.06.040 [DOI] [PubMed] [Google Scholar]
- 41. Sharma S., Poetz F., Bruer M., Ly-Hartig T. B., Schott J., Séraphin B., and Stoecklin G. (2016) Acetylation-dependent control of global poly(A) RNA degradation by CBP/p300 and HDAC1/2. Mol. Cell 63, 927–938 10.1016/j.molcel.2016.08.030 [DOI] [PubMed] [Google Scholar]
- 42. Son A., Park J. E., and Kim V. N. (2018) PARN and TOE1 constitute a 3′ end maturation module for nuclear non-coding RNAs. Cell. Rep. 23, 888–898 10.1016/j.celrep.2018.03.089 [DOI] [PubMed] [Google Scholar]
- 43. Kim J. H., and Richter J. D. (2006) Opposing polymerase-deadenylase activities regulate cytoplasmic polyadenylation. Mol. Cell 24, 173–183 10.1016/j.molcel.2006.08.016 [DOI] [PubMed] [Google Scholar]
- 44. Yoshida M., Kijima M., Akita M., and Beppu T. (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 265, 17174–17179 [PubMed] [Google Scholar]
- 45. Chuang D. M., Leng Y., Marinova Z., Kim H. J., and Chiu C. T. (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32, 591–601 10.1016/j.tins.2009.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gertz M., Fischer F., Nguyen G. T., Lakshminarasimhan M., Schutkowski M., Weyand M., and Steegborn C. (2013) Ex-527 inhibits sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. U. S. A. 110, E2772–E2781 10.1073/pnas.1303628110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Palacios J. A., Herranz D., De Bonis M. L., Velasco S., Serrano M., and Blasco M. A. (2010) SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 191, 1299–1313 10.1083/jcb.201005160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Bonis M. L., Ortega S., and Blasco M. A. (2014) SIRT1 is necessary for proficient telomere elongation and genomic stability of induced pluripotent stem cells. Stem Cell Reports 2, 690–706 10.1016/j.stemcr.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. El Ramy R., Magroun N., Messadecq N., Gauthier L. R., Boussin F. D., Kolthur-Seetharam U., Schreiber V., McBurney M. W., Sassone-Corsi P., and Dantzer F. (2009) Functional interplay between Parp-1 and SirT1 in genome integrity and chromatin-based processes. Cell Mol. Life Sci. 66, 3219–3234 10.1007/s00018-009-0105-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen J., Zhang B., Wong N., Lo A. W., To K. F., Chan A. W., Ng M. H., Ho C. Y., Cheng S. H., Lai P. B., Yu J., Ng H. K., Ling M. T., Huang A. L., Cai X. F., et al. (2011) Sirtuin 1 is upregulated in a subset of hepatocellular carcinomas where it is essential for telomere maintenance and tumor cell growth. Cancer Res. 71, 4138–4149 10.1158/0008-5472.CAN-10-4274 [DOI] [PubMed] [Google Scholar]
- 51. Yamashita S., Ogawa K., Ikei T., Udono M., Fujiki T., and Katakura Y. (2012) SIRT1 prevents replicative senescence of normal human umbilical cord fibroblast through potentiating the transcription of human telomerase reverse transcriptase gene. Biochem. Biophys. Res. Commun. 417, 630–634 10.1016/j.bbrc.2011.12.021 [DOI] [PubMed] [Google Scholar]
- 52. Chen H., Liu X., Zhu W., Chen H., Hu X., Jiang Z., Xu Y., Wang L., Zhou Y., Chen P., Zhang N., Hu D., Zhang L., Wang Y., Xu Q., et al. (2014) SIRT1 ameliorates age-related senescence of mesenchymal stem cells via modulating telomere shelterin. Front. Aging Neurosci. 6, 103 10.3389/fnagi.2014.00103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Amano H., Chaudhury A., Rodriguez-Aguayo C., Lu L., Akhanov V., Catic A., Popov Y. V., Verdin E., Johnson H., Stossi F., Sinclair D. A., Nakamaru-Ogiso E., Lopez-Berestein G., Chang J. T., Neilson J. R., et al. (2019) Telomere dysfunction induces sirtuin repression that drives telomere-dependent disease. Cell. Metab. 29, 1274–1290.e9 10.1016/j.cmet.2019.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fitzpatrick A. L., Kronmal R. A., Gardner J. P., Psaty B. M., Jenny N. S., Tracy R. P., Walston J., Kimura M., and Aviv A. (2007) Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am. J. Epidemiol. 165, 14–21 10.1093/aje/kwj346 [DOI] [PubMed] [Google Scholar]
- 55. Rudolph K. L., Chang S., Lee H. W., Blasco M., Gottlieb G. J., Greider C., and DePinho R. A. (1999) Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 96, 701–712 10.1016/S0092-8674(00)80580-2 [DOI] [PubMed] [Google Scholar]
- 56. Cherif H., Tarry J. L., Ozanne S. E., and Hales C. N. (2003) Ageing and telomeres: a study into organ- and gender-specific telomere shortening. Nucleic Acids Res. 31, 1576–1583 10.1093/nar/gkg208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hastings R., Li N. C., Lacy P. S., Patel H., Herbert K. E., Stanley A. G., and Williams B. (2004) Rapid telomere attrition in cardiac tissue of the ageing Wistar rat. Exp. Gerontol. 39, 855–857 10.1016/j.exger.2004.02.003 [DOI] [PubMed] [Google Scholar]
- 58. Harley C. B., Futcher A. B., and Greider C. W. (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- 59. Bodnar A. G., Ouellette M., Frolkis M., Holt S. E., Chiu C. P., Morin G. B., Harley C. B., Shay J. W., Lichtsteiner S., and Wright W. E. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352 10.1126/science.279.5349.349 [DOI] [PubMed] [Google Scholar]
- 60. van Deursen J. M. (2014) The role of senescent cells in ageing. Nature 509, 439–446 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Giorgio M., Stendardo M., Migliaccio E., and Pelicci P. G. (2016) P66SHC deletion improves fertility and progeric phenotype of late-generation TERC-deficient mice but not their short lifespan. Aging Cell 15, 446–454 10.1111/acel.12448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen R., Zhang K., Chen H., Zhao X., Wang J., Li L., Cong Y., Ju Z., Xu D., Williams B. R., Jia J., and Liu J. P. (2015) Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J. Biol. Chem. 290, 30813–30829 10.1074/jbc.M115.681619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jose S. S., Tidu F., Burilova P., Kepak T., Bendickova K., and Fric J. (2018) The telomerase complex directly controls hematopoietic stem cell differentiation and senescence in an induced pluripotent stem cell model of telomeropathy. Front. Genet. 9, 345 10.3389/fgene.2018.00345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Langley E., Pearson M., Faretta M., Bauer U. M., Frye R. A., Minucci S., Pelicci P. G., and Kouzarides T. (2002) Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 21, 2383–2396 10.1093/emboj/21.10.2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ota H., Akishita M., Eto M., Iijima K., Kaneki M., and Ouchi Y. (2007) Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell. Cardiol. 43, 571–579 10.1016/j.yjmcc.2007.08.008 [DOI] [PubMed] [Google Scholar]
- 66. Wan Y. Z., Gao P., Zhou S., Zhang Z. Q., Hao D. L., Lian L. S., Li Y. J., Chen H. Z., and Liu D. P. (2014) SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell 13, 890–899 10.1111/acel.12247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kim D. H., Jung I. H., Kim D. H., and Park S. W. (2019) Knockout of longevity gene Sirt1 in zebrafish leads to oxidative injury, chronic inflammation, and reduced life span. PLoS ONE 14, e0220581 10.1371/journal.pone.0220581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Satoh A., Brace C. S., Rensing N., Cliften P., Wozniak D. F., Herzog E. D., Yamada K. A., and Imai S. (2013) Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 18, 416–430 10.1016/j.cmet.2013.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Valerio D., Luddi A., De Leo V., Labella D., Longobardi S., and Piomboni P. (2018) SA1/SA2 cohesion proteins and SIRT1-NAD+ deacetylase modulate telomere homeostasis in cumulus cells and are eligible biomarkers of ovarian aging. Hum. Reprod. 33, 887–894 10.1093/humrep/dey035 [DOI] [PubMed] [Google Scholar]
- 70. Kim S., Bi X., Czarny-Ratajczak M., Dai J., Welsh D. A., Myers L., Welsch M. A., Cherry K. E., Arnold J., Poon L. W., and Jazwinski S. M. (2012) Telomere maintenance genes SIRT1 and XRCC6 impact age-related decline in telomere length but only SIRT1 is associated with human longevity. Biogerontology 13, 119–131 10.1007/s10522-011-9360-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tycowski K. T., You Z. H., Graham P. J., and Steitz J. A. (1998) Modification of U6 spliceosomal RNA is guided by other small RNAs. Mol. Cell 2, 629–638 10.1016/S1097-2765(00)80161-6 [DOI] [PubMed] [Google Scholar]
- 72. Ganot P., Jády B. E., Bortolin M. L., Darzacq X., and Kiss T. (1999) Nucleolar factors direct the 2′-O-ribose methylation and pseudouridylation of U6 spliceosomal RNA. Mol. Cell Biol. 19, 6906–6917 10.1128/mcb.19.10.6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kiss T. (2001) Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 20, 3617–3622 10.1093/emboj/20.14.3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Petrovski S., Todd J. L., Durheim M. T., Wang Q., Chien J. W., Kelly F. L., Frankel C., Mebane C. M., Ren Z., Bridgers J., Urban T. J., Malone C. D., Finlen Copeland A., Brinkley C., Allen A. S., et al. (2017) An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 196, 82–93 10.1164/rccm.201610-2088OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grönroos E., Hellman U., Heldin C. H., and Ericsson J. (2002) Control of Smad7 stability by competition between acetylation and ubiquitination. Mol. Cell. 10, 483–493 10.1016/S1097-2765(02)00639-1 [DOI] [PubMed] [Google Scholar]
- 76. Peng L., Ling H., Yuan Z., Fang B., Bloom G., Fukasawa K., Koomen J., Chen J., Lane W. S., and Seto E. (2012) SIRT1 negatively regulates the activities, functions, and protein levels of hMOF and TIP60. Mol. Cell Biol. 32, 2823–2836 10.1128/MCB.00496-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li H., Wittwer T., Weber A., Schneider H., Moreno R., Maine G. N., Kracht M., Schmitz M. L., and Burstein E. (2012) Regulation of NF-κB activity by competition between RelA acetylation and ubiquitination. Oncogene 31, 611–623 10.1038/onc.2011.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ling H., Peng L., Wang J., Rahhal R., and Seto E. (2018) Histone deacetylase SIRT1 targets Plk2 to regulate centriole duplication. Cell Rep. 25, 2851–2865.e3 10.1016/j.celrep.2018.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Akimov V., Barrio-Hernandez I., Hansen S. V. F., Hallenborg P., Pedersen A. K., Bekker-Jensen D. B., Puglia M., Christensen S. D. K., Vanselow J. T., Nielsen M. M., Kratchmarova I., Kelstrup C. D., Olsen J. V., and Blagoev B. (2018) UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 25, 631–640 10.1038/s41594-018-0084-y [DOI] [PubMed] [Google Scholar]
- 80. Bouras T., Fu M., Sauve A. A., Wang F., Quong A. A., Perkins N. D., Hay R. T., Gu W., and Pestell R. G. (2005) SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 280, 10264–10276 10.1074/jbc.M408748200 [DOI] [PubMed] [Google Scholar]
- 81. Yang X.-J., Ogryzko V. V., Nishikawa J.-I., Howard B. H., and Nakatani Y. (1996) A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382, 319–324 10.1038/382319a0 [DOI] [PubMed] [Google Scholar]
- 82. Mao X., Gluck N., Li D., Maine G. N., Li H., Zaidi I. W., Repaka A., Mayo M. W., and Burstein E. (2009) GCN5 is a required cofactor for a ubiquitin ligase that targets NF- B/RelA. Genes Dev. 23, 849–861 10.1101/gad.1748409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Aizawa H., Hu S.-C., Bobb K., Balakrishnan K., Ince G., Gurevich I., Cowan M., and Ghosh A. (2004) Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303, 197–202 10.1126/science.1089845 [DOI] [PubMed] [Google Scholar]
- 84. Michishita E., Park J. Y., Burneskis J. M., Barrett J. C., and Horikawa I. (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635 10.1091/mbc.e05-01-0033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Maryati M., Kaur I., Jadhav G. P., Olotu-Umoren L., Oveh B., Hashmi L., Fischer P. M., and Winkler G. S. (2014) A fluorescence-based assay suitable for quantitative analysis of deadenylase enzyme activity. Nucleic Acids Res. 42, e30 10.1093/nar/gkt972 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All representative data are contained within the article. Raw data are available upon request from Eden Dejene (edendejene@gwu.edu) or Edward Seto (seto@gwu.edu).