

Abstract
Agonist stimulation of G-protein–coupled receptors (GPCRs) typically leads to phosphorylation of GPCRs and binding to multifunctional proteins called β-arrestins (βarrs). The GPCR–βarr interaction critically contributes to GPCR desensitization, endocytosis, and downstream signaling, and GPCR–βarr complex formation can be used as a generic readout of GPCR and βarr activation. Although several methods are currently available to monitor GPCR–βarr interactions, additional sensors to visualize them may expand the toolbox and complement existing methods. We have previously described antibody fragments (FABs) that recognize activated βarr1 upon its interaction with the vasopressin V2 receptor C-terminal phosphopeptide (V2Rpp). Here, we demonstrate that these FABs efficiently report the formation of a GPCR–βarr1 complex for a broad set of chimeric GPCRs harboring the V2R C terminus. We adapted these FABs to an intrabody format by converting them to single-chain variable fragments and used them to monitor the localization and trafficking of βarr1 in live cells. We observed that upon agonist simulation of cells expressing chimeric GPCRs, these intrabodies first translocate to the cell surface, followed by trafficking into intracellular vesicles. The translocation pattern of intrabodies mirrored that of βarr1, and the intrabodies co-localized with βarr1 at the cell surface and in intracellular vesicles. Interestingly, we discovered that intrabody sensors can also report βarr1 recruitment and trafficking for several unmodified GPCRs. Our characterization of intrabody sensors for βarr1 recruitment and trafficking expands currently available approaches to visualize GPCR–βarr1 binding, which may help decipher additional aspects of GPCR signaling and regulation.
Keywords: G-protein–coupled receptors (GPCRs), β-arrestins, cellular signaling, synthetic antibody, intrabody, biased agonism, trafficking, biosensors, cell signaling, protein phosphorylation, signal transduction, bioluminescence resonance energy transfer (BRET), confocal microscopy
G-protein–coupled receptors (GPCRs) recognize a diverse set of ligands and initiate a broad spectrum of downstream signaling responses (1). Upon agonist stimulation, GPCRs couple to three major subfamilies of cellular proteins namely, the heterotrimeric G-proteins, GPCR kinases, and β-arrestins (βarrs) (1). Of these, βarrs are multifunctional adaptor proteins, which play a central role in regulatory and signaling paradigms of GPCRs (2, 3). βarrs are evenly distributed in the cytoplasm under basal condition, and upon agonist stimulation, they typically translocate to the plasma membrane to interact with activated and phosphorylated receptors (4).
Binding of βarrs to GPCRs at the plasma membrane results in termination of G-protein coupling and desensitization of receptors through a steric hindrance-based mechanism (5). Subsequently, βarrs either dissociate from the receptors and relocalize back in the cytoplasm or traffic into endosomal vesicles in complex with the receptors (2, 4). These two different patterns are referred to as “class A” and “class B,” respectively (4). βarrs also contribute in a number of downstream GPCR signaling pathways such as ERK1/2 MAP kinase activation, although strict G-protein independence of such mechanisms are currently being discussed and debated (6–9).
Considering the multifaceted roles of βarrs, understanding the details of their interaction with GPCRs continues to be a frontier area in GPCR research (10). The interaction of βarrs with GPCRs involves two distinct components (11, 12). One is receptor phosphorylation, primarily in the C terminus but also in the intracellular loops, and the other is the intracellular side of receptor transmembrane bundle, referred to as the receptor core (11, 12). There are several assays that are currently used to measure GPCR–βarr interaction, including those based on resonance energy transfer (13–15), enzyme complementation (16), and reporter responses (17, 18). However, developing novel sensors is desirable to expand the currently available toolbox and complement the existing assays.
Previous studies have suggested that receptor phosphorylation is not only sufficient to promote βarr binding, but it can also induce βarr conformations capable of mediating receptor endocytosis and signaling (19–21). These findings raise the possibility that biochemical reagents such as antibodies, which selectively recognize βarr conformation triggered by the interaction of phosphorylated receptor, may serve as sensors for βarr recruitment and trafficking. Here, we develop and characterize intrabody sensors derived from synthetic antibody fragments (FABs) against βarr1 that report the formation of GPCR–βarr1 complexes and allow us to monitor βarr1 trafficking in cellular context.
Results
Synthetic antibody fragments report the formation of β2V2R–βarr1 complex
Agonist-induced receptor phosphorylation is a key determinant for βarr recruitment (11). A phosphopeptide corresponding to the C terminus of the human vasopressin V2 receptor, referred to as V2Rpp, has been used extensively as a surrogate to induce active βarr conformation in vitro (22–25). We have previously generated and characterized a set of synthetic FABs that selectively recognize V2Rpp-bound βarr1 (26). We have also used one of these FABs, referred to as Fab30, to monitor the interaction of βarr1 with a chimeric β2-adrenergic receptor harboring V2R C terminus (referred to as β2V2R) and V2R (25). As the first step toward developing these FABs as potential sensors of GPCR–βarr interaction and trafficking, we first confirmed their ability to report the formation of β2V2R–βarr1 complex in vitro (Fig. 1, A–D). Here, we used lysates from cells expressing FLAG–β2V2R mixed with purified βarr1 and FABs, followed by co-immunoprecipitation (co-IP) and detection of the receptor as a readout of complex formation. We observed that Fab30 and the additional FABs selectively pulldown β2V2R upon agonist stimulation through the formation of receptor–βarr1 complex (Fig. 1, A–D). A control FAB that does not interact with βarr1 failed to yield any detectable signal in the co-IP experiment (Fig. 1, A and B).
Figure 1.

Synthetic FABs that recognize β2V2R–βarr1 complex. A, Fab30 selectively recognizes agonist-induced β2V2R–βarr1 complex as assessed by co-immunoprecipitation. Sf9 cells expressing FLAG-tagged β2V2R and GRK2CAAX were stimulated with either carazolol (1 μm) or BI-167107 (100 nm), lysed, and mixed with purified βarr1 and Fab30 (or a control Fab). Subsequently, Fab was immunoprecipitated using protein L–agarose beads, and co-purification of the receptor was visualized by Western blotting (WB) using HRP-coupled anti-FLAG M2 antibody. Fabs were detected by Coomassie staining. B, densitometry-based quantification of Western blotting signal in A presented as means ± S.E. of four independent experiments normalized with respect to maximal response (treated as 100%). C, the ability of additional Fabs to recognize agonist-induced β2V2R–βarr1 complex assessed by co-immunoprecipitation following the protocol mentioned above. D, densitometry-based quantification of Western blotting signal in C presented as means ± S.E. of four independent experiments normalized with respect to carazolol condition (treated as 1). The data in B and D were analyzed using one-way ANOVA. ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05.
Fab30 reports the formation of βarr1 complex for multiple chimeric GPCRs
Before proceeding to generate potentially generic intrabody sensors from these FABs, we evaluated their ability to recognize βarr1 complex with other GPCRs. Considering that these FABs were selected against V2Rpp-bound βarr1, we reasoned that they should detect βarr1 complex for other chimeric GPCRs harboring the V2R C terminus, similar to that in β2V2R. We generated six different chimeric GPCRs including the members from different subclasses such as chemokine (CCR2-V2R), adrenergic (α2B-V2R), complement (C5aR1-V2R), muscarinic (M5-V2R), and dopamine (D2-V2R and D5-V2R) receptors. Some of these receptors, such as M5R, α2BR, and D2R, contain large third intracellular loops, whereas others have relatively shorter third intracellular loops. We tested the ability of Fab30, which was most effective among all the FABs, to report the formation of receptor–βarr1 complex in co-IP assay for these receptors. As presented in Fig. 2 (A–F), we observed that Fab30 efficiently recognized βarr1 for every chimeric GPCR tested here, similar to that of β2V2R. This finding allowed us to conceive that these FABs should work as generic intrabody sensors of βarr1 interaction and trafficking in cellular context for a broad set of chimeric GPCRs.
Figure 2.
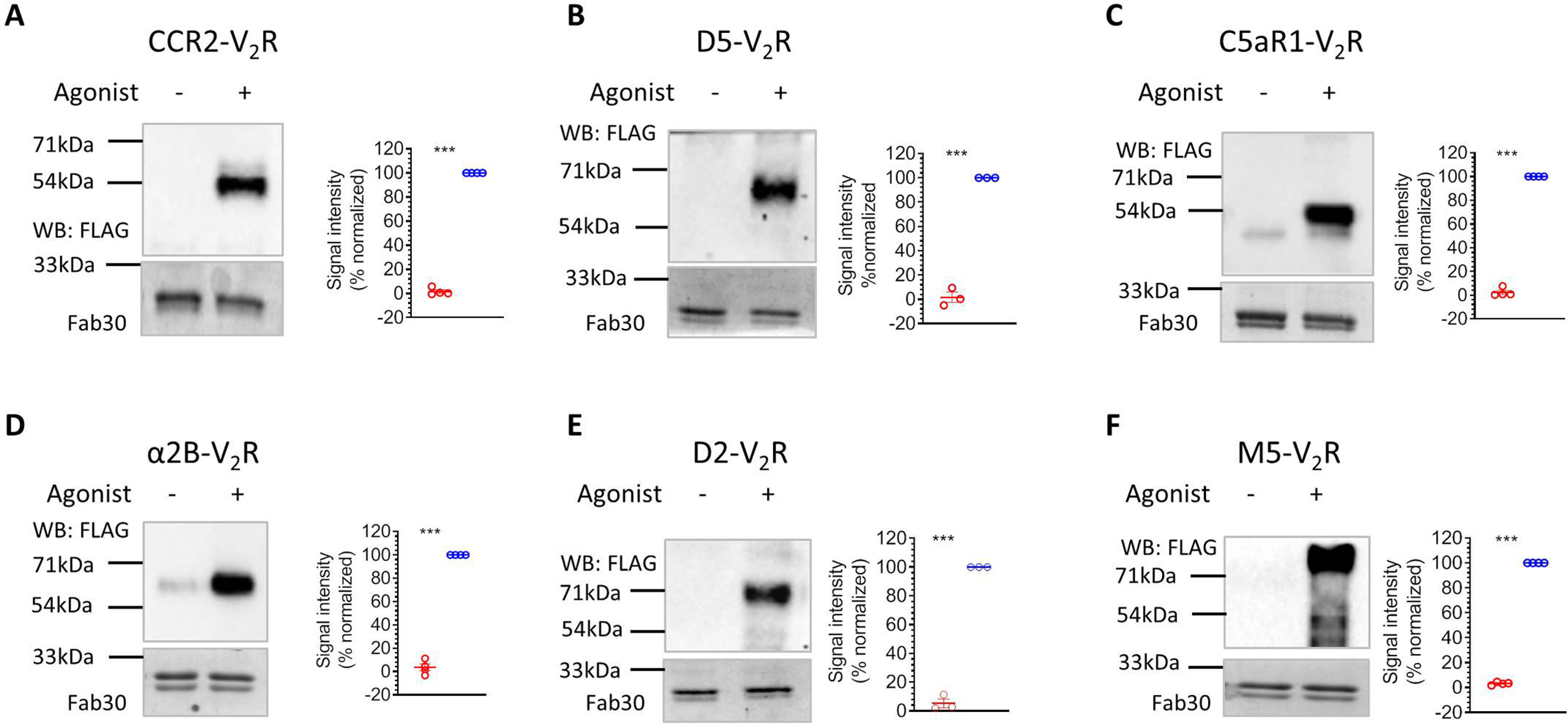
Fab30 reports agonist-induced interaction of βarr1 with chimeric GPCRs. A–F, HEK-293 cells expressing N-terminally FLAG-tagged chimeric GPCRs harboring the V2R C terminus and βarr1 were stimulated with corresponding agonists (100 nm CCL7, 20 μm dopamine, 100 nm C5a, 20 μm epinephrine, 20 μm dopamine, and 20 μm carbachol, respectively), lysed, and mixed with purified Fab30. Subsequently, Fab30 was immunoprecipitated using protein L–agarose beads, and co-purification of the receptor was visualized by Western blotting (WB) using HRP-coupled anti-FLAG M2 antibody. Fabs were detected by Coomassie staining. The graphs in every panel show densitometry-based quantification of Western blotting signal presented as means ± S.E. of four independent experiments (three for D5V2R and D2V2R) normalized with respect to maximal response (treated as 100%) and analyzed using one-way ANOVA. ***, p < 0.001.
Conversion of FABs into intrabodies and their expression analysis
To develop these FABs into cellular sensors of βarr1 activation and trafficking, it is required to express them in functional form in the cytoplasm as intrabodies. We therefore converted the selected FABs into single-chain variable fragments (ScFvs) by connecting the variable domains of their heavy and light chains through a previously optimized flexible linker (12) and then expressed them in HEK-293 cells as intrabodies, either with a C-terminal HA tag or as YFP fusion (Fig. 3, A–D). We observed robust expression of two of these intrabodies namely intrabody30 (Ib30) and intrabody4 (Ib4) in HEK-293 cells, whereas others displayed relatively weaker expression (Fig. 3B). For YFP-tagged intrabodies, we observed cytoplasmic as well as nuclear localization (Fig. 3C–D). The underlying reason for nuclear localization of the intrabodies is not apparent to us, although a previous study has also reported nuclear localization of an intrabody targeting β2-adrenergic receptor (27).
Figure 3.
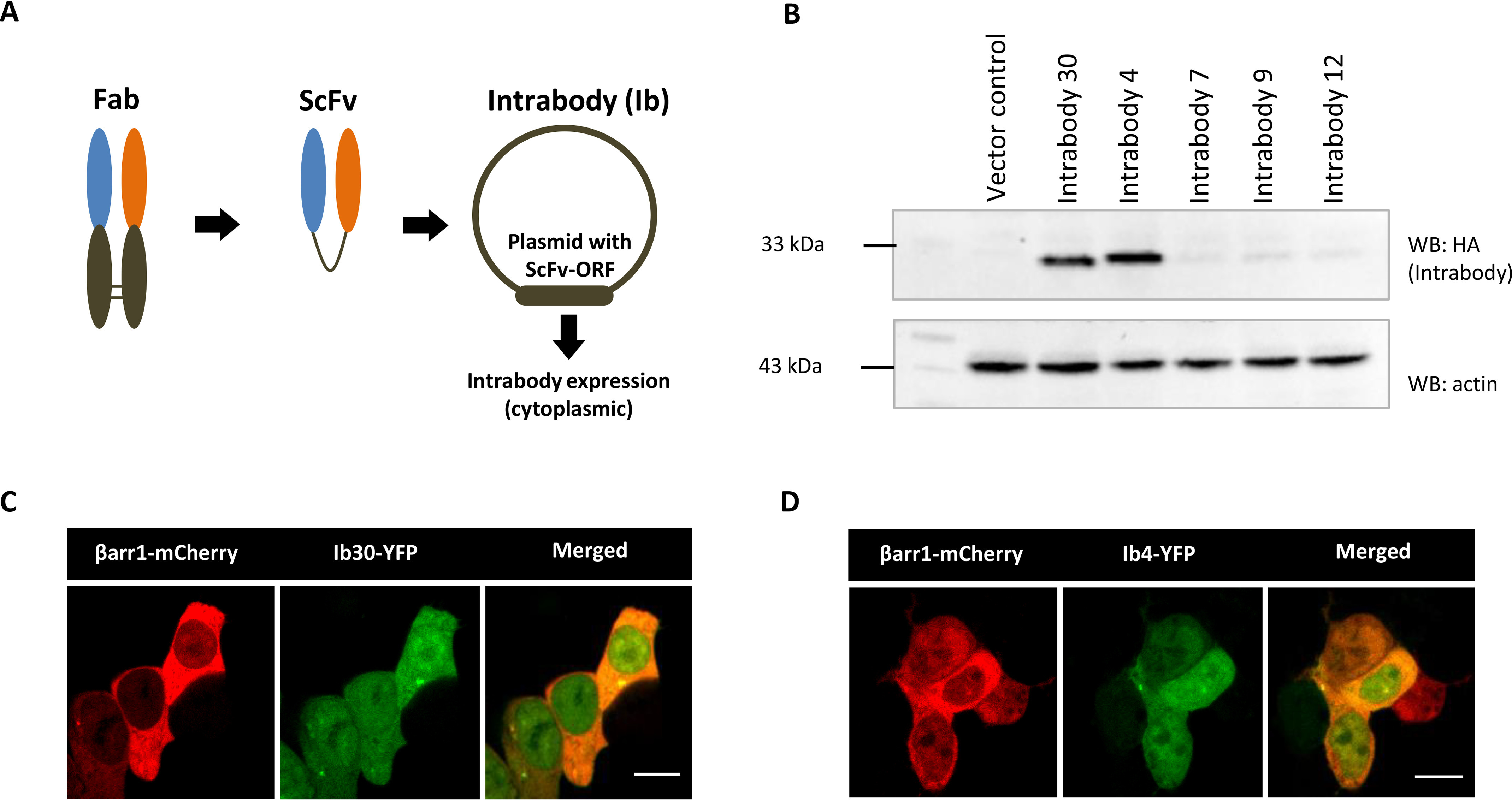
Conversion of FABs into intrabodies and their expression analysis. A, schematic representation of conversion of FABs into ScFv format for intracellular expression as intrabodies. B, expression profile of intrabodies in HEK-293 cells visualized by Western blotting (WB). Lysate prepared from HEK-293 cells expressing the indicated intrabodies with C terminus HA tag were separated on SDS-PAGE followed by visualization using anti-HA antibody. C and D, intracellular expression of Ib30–YFP/Ib4–YFP and βarr1–mCherry as visualized by confocal microscopy. HEK-293 cells expressing the corresponding plasmids were subjected to live cell imaging, and it revealed localization of Ib30–YFP and Ib4–YFP in both cytoplasm and nucleus. Scale bar, 10 μm.
Ib30 and Ib4 report the interaction of βarr1 with β2V2R and trafficking
We next tested whether intrabodies can report the formation of receptor–βarr1 complex in a cellular context. We first co-expressed β2V2R, βarr1, and HA-tagged intrabodies in HEK-293 cells, stimulated the cells with either an agonist (isoproterenol) or inverse agonist (carazolol), and immunoprecipitated the intrabodies using the HA tag. We observed that both intrabodies, i.e. Ib30 and Ib4, recognized the β2V2R–βarr1 complex upon agonist stimulation, although Ib30 was relatively more efficient (Fig. 4, A and B). We also tested the ability of Ib30 to recognize the β2V2R–βarr1 complex formed upon stimulation of the receptor with a set of ligands with varying efficacies. Importantly, we observed that the level of recognition of the β2V2R–βarr1 complex by Ib30 mirrors the efficacy of the ligands (Fig. 4, C and D). This observation underscores the ability of Ib30 to report the formation of pharmacologically relevant receptor–βarr1 complex and corroborates its suitability as a reliable sensor of receptor–βarr1 interaction.
Figure 4.
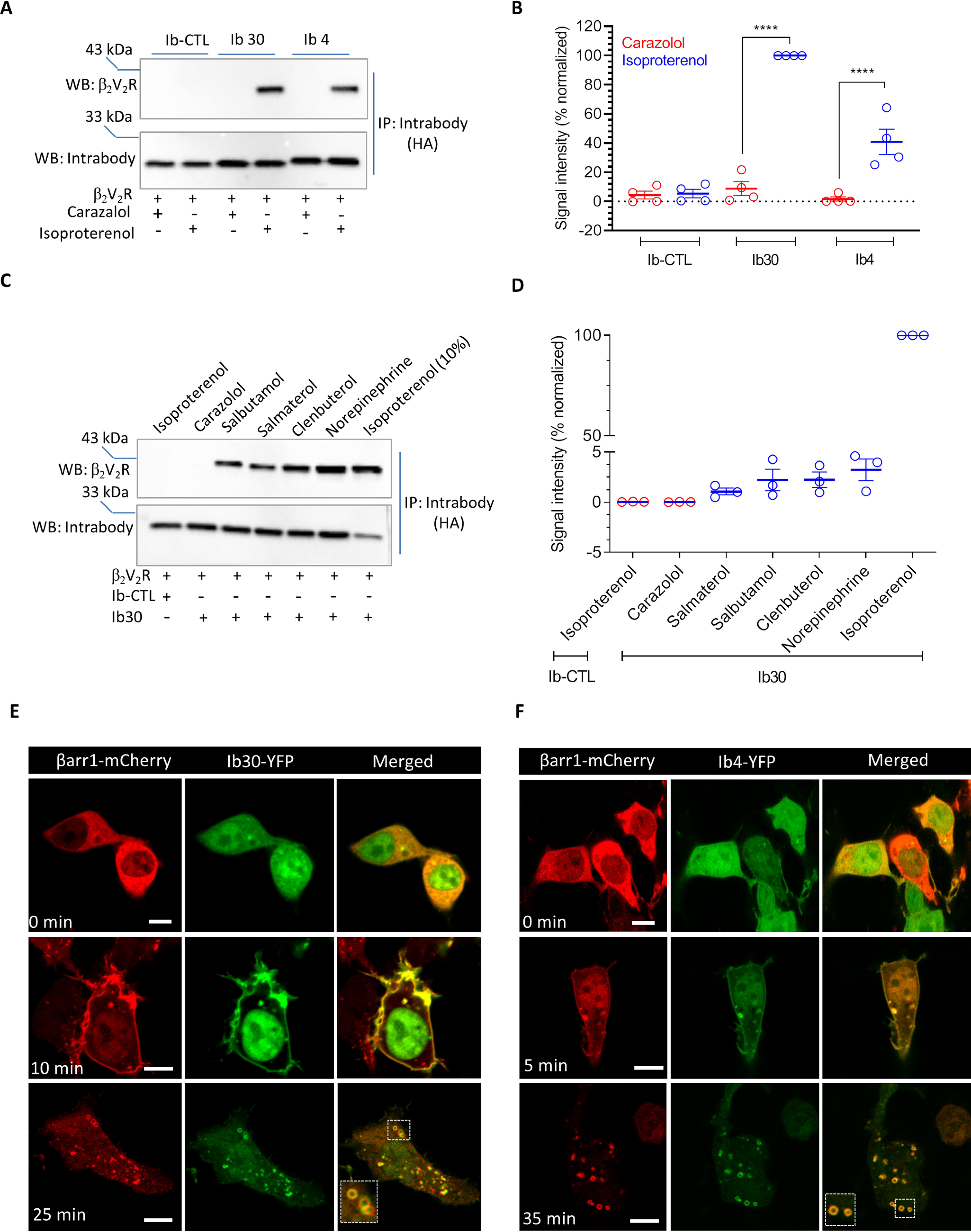
Intrabodies report agonist-induced formation of β2V2R–βarr1 complex and trafficking of βarr1 upon β2V2R stimulation. A, the ability of intrabodies (Ib30 and Ib4) to recognize receptor-bound βarr1 upon agonist stimulation. HEK-293 cells expressing β2V2R, βarr1, and Ib30/Ib4/Ib–CTL were stimulated with either inverse agonist (carazolol; 1 μm) or agonist (isoproterenol; 10 μm) followed by co-IP using anti-HA antibody agarose. Subsequently, the proteins were visualized by Western blotting (WB) using anti-FLAG M2 antibody and anti-HA antibody. B, densitometry-based quantification of the data in A presented as means ± S.E. from four independent experiments normalized with maximal response (treated as 100%) and analyzed using one-way ANOVA. ****, p < 0.0001. C, the ability of Ib30 to report the formation of receptor–βarr1 complex mirrors ligand efficacy. HEK-293 cells expressing β2V2R, βarr1, and Ib30 (or Ib–CTL) were stimulated with saturating concentrations of the indicated ligands followed by co-immunoprecipitation and Western blotting as mentioned above. For isoproterenol condition, which yielded maximal signal, only 10% of the total elution from the co-IP is loaded on the gel to avoid signal saturation. D, densitometry-based quantification of the data in C presented as means ± S.E. from three independent experiments normalized with respect to maximal response (treated as 100%). E and F, HEK-293 cells expressing β2V2R, βarr1–mCherry, and YFP-tagged Ib30/Ib4 were stimulated with isoproterenol (10 μm), and the localization of βarr1 and intrabodies was visualized using confocal microscopy at the indicated time points. PCCs were measured to assess the co-localization of βarr1 and Ib30 using JACoP plugin in ImageJ. The following values were obtained: for Ib30, 0.28 ± 0.03 from 13 cells, 0.74 ± 0.05 from 9 cells, and 0.76 ± 0.02 from 29 cells for the upper, middle, and lower panels, respectively, with four independent experiments; and for Ib4, 0.24 ± 0.03 from 10 cells, 0.84 ± 0.03 from 9 cells, and 0.94 ± 0.01 from 20 cells for the upper, middle, and lower panels, respectively, with three independent experiments. Scale bar, is 10μm.
To probe the utility of intrabodies to monitor βarr1 trafficking upon receptor stimulation, we co-expressed β2V2R, βarr1–mCherry, and YFP-tagged intrabodies in HEK-293 cells and followed the localization of βarr1 and intrabodies using confocal microscopy after agonist treatment (Fig. 4, E and F). As expected, activation of β2V2R resulted in a typical class B pattern of βarr1 translocation, and interestingly, the intrabodies followed the localization of βarr1 and displayed robust co-localization (Fig. 4, E and F). We observed that Ib30 and Ib4 were first translocated to the cell surface from the cytoplasm, and upon sustained agonist stimulation, they were localized in the intracellular vesicles. Taken together, these findings demonstrate the usefulness of intrabodies as yet another tool to monitor the formation of the receptor–βarr1 complex in vitro and βarr1 trafficking in the cellular context.
Intrabodies also report the interaction and trafficking of βarr1 upon V2R stimulation
Because the intrabodies are derived from FABs selected against V2Rpp-bound βarr1, we anticipated that they should be able to report agonist-induced βarr1 interaction and trafficking for V2R as well. Accordingly, we tested the ability of Ib30 and Ib4 to detect the formation of the V2R–βarr1 complex in vitro and report agonist-induced translocation of βarr1 in a cellular context (Fig. 5, A–E). We observed a pattern very similar to that of β2V2R described above in both the co-immunoprecipitation experiment and confocal microscopy (Fig. 5, A–E). That is, Ib30 and Ib4 selectively recognized V2R–βarr1 complex upon agonist stimulation and followed the localization pattern of βarr1 upon agonist stimulation as reflected by translocation to the cell surface first followed by localization in intracellular vesicles. An additional band was observed on the Western blot in the co-IP experiment, which migrates below the V2R band, but its origin is currently not clear to us.
Figure 5.

Intrabodies report the formation of V2R–βarr1 complex and trafficking of βarr1 upon V2R stimulation. A, the ability of intrabodies (Ib30 and Ib4) to recognize V2R-bound βarr1 upon agonist stimulation. HEK-293 cells expressing V2R, βarr1, and Ib30/Ib4/Ib–CTL were stimulated with either inverse agonist (tolvaptan; 100 nm) or agonist (AVP; 100 nm) followed by co-immunoprecipitation (co-IP) using anti-HA antibody agarose. Subsequently, the proteins were visualized by Western blotting (WB) using anti-FLAG M2 antibody and anti-HA antibody. B, densitometry-based quantification of the data in A presented as means ± S.E. from four independent experiments normalized with maximal response (treated as 100%) and analyzed using one-way ANOVA with Bonferroni post test. ****, p < 0.0001. C and D, HEK-293 cells expressing V2R, βarr1–mCherry, and YFP-tagged Ib30/Ib4 were stimulated with AVP (100 nm), and the localization of βarr1 and intrabodies was visualized using confocal microscopy at the indicated time points. PCCs were measured to assess the co-localization of βarr1 and Ib30 using JACoP plugin in ImageJ. The following values were obtained: for Ib30, 0.31 ± 0.02 from 16 cells, 0.81 ± 0.03 from 16 cells, and 0.80 ± 0.02 from 20 cells for the upper, middle, and lower panels, respectively, with six independent experiments; and for Ib4, 0.27 ± 0.02 from 20 cells, 0.74 ± 0.02 from 21 cells, and 0.75 ± 0.01 from 47 cells for the upper, middle, and lower panels, respectively, with three independent experiments. E, time-lapse confocal imaging of HEK-293 cells expressing V2R, βarr1–mCherry, and Ib30–YFP to demonstrate agonist-induced translocation of βarr1 and Ib30 in the same cells over time. A representative image panel from three independent experiments is shown here. Scale bar, 10 μm.
We also measured the ability of Ib30 to recognize endogenous βarr1 upon agonist stimulation of V2R and observed a robust interaction in co-immunoprecipitation assay (Fig. 6, A and B). Furthermore, we evaluated the translocation pattern of Ib30–YFP upon agonist stimulation for β2V2R and V2R in HEK-293 cells where βarr1 is overexpressed without any modification. As presented in Fig. 6C, Ib30–YFP was robustly localized to intracellular vesicles after agonist stimulation, which is reminiscent of the typical translocation pattern of βarr1 for these receptors. These data further strengthen the utility of intrabody sensors described here in monitoring βarr1 recruitment and trafficking.
Figure 6.

Intrabody30 recognizes receptor-bound endogenous βarr1 and reports the trafficking of native βarr1. A, the ability of intrabody Ib30 to recognize V2R-bound endogenous βarr1 upon agonist stimulation. HEK-293 cells expressing V2R and HA-tagged Ib30/Ib–CTL were stimulated with either inverse agonist (tolvaptan; 100 nm) or agonist (AVP; 100 nm) followed by co-IP using anti-HA antibody agarose. Subsequently, the proteins were visualized by Western blotting using anti-βarr and anti-HA antibodies. B, densitometry-based quantification of the data in A presented as means ± S.E. from three independent experiments normalized with maximal response (treated as 100%) and analyzed using one-way ANOVA. ****, p < 0.0001. C, HEK-293 cells expressing β2V2R/V2R and Ib30–YFP were stimulated with isoproterenol (10 μm) and AVP (100 nm), respectively, and the localization of Ib30–YFP was visualized using confocal microscopy. Representative images from three independent experiments are shown here. Scale bar, 10 μm.
Intrabodies do not alter βarr recruitment, receptor endocytosis, G-protein coupling, and ERK1/2 phosphorylation
For the intrabodies to be reliable sensors of βarr recruitment and trafficking, it is important that they do not significantly alter βarr recruitment, receptor endocytosis, and G-protein coupling. Therefore, we first measured agonist-induced recruitment of βarr1 to V2R in presence of either a control intrabody (Ib–CTL) or Ib30/Ib4 using an intermolecular BRET assay. As presented in Fig. 7A, we did not observe any significant difference in βarr1 recruitment. Next, to probe whether V2R is co-localized with Ib30 and βarr1 on intracellular vesicles, we performed three-color confocal imaging on HEK-293 cells expressing FLAG–V2R, βarr1–YFP, and Ib30–HA after agonist stimulation (Fig. 7B). Expectedly, we observed a robust co-localization of V2R, βarr1, and Ib30 on intracellular vesicles, suggesting that Ib30 does not alter the normal trafficking pattern of receptor–βarr1 complex in a cellular context. This is further corroborated by the pattern of V2R co-localization with the early endosomal markers EEA1 and APPL1, which remains unaltered in presence of Ib–CTL versus Ib30 (Fig. 7, C and D). Furthermore, we also measured βarr1 trafficking to endosomes upon V2R activation using an enhanced bystander BRET set-up (15) in presence of either Ib–CTL or Ib4/Ib30. Although we did not observe a significant difference in EC50 values (Fig. 7E), Ib4/Ib30 appear to stabilize endosomal localization of βarr1 as reflected by ΔBRET signal (Fig. 7F). This observation is particularly relevant if the intrabody sensors are used in the context of receptor recycling where they might slow down receptor recycling to the plasma membrane, and it would be interesting to probe this aspect further in future studies.
Figure 7.

Effect of intrabodies on βarr1 recruitment, V2R endocytosis, and endosomal localization of βarr1. A, intrabodies do not significantly alter agonist-induced βarr1 recruitment to V2R as assessed in intermolecular BRET assay. HEK-293 cells expressing V2R–venus, βarr1–RlucII, and the indicated intrabodies were stimulated with varying doses of AVP, and the levels of BRET signal were recorded using a plate reader. The data represent means ± S.E.M. from three independent experiments, each performed in triplicate. B, Ib30 co-localizes with internalized V2R and βarr1 upon agonist stimulation as visualized using confocal microscopy of HEK-293 cells expressing FLAG–V2R, βarr1-YFP, and Ib30–HA. The merged image shows co-localization of all three protein upon receptor internalization. The cells were “fed” anti-FLAG M2 antibody prior to agonist stimulation (AVP 100 nm, 12 min) and were subsequently fixed, permeabilized, treated with HA antibody, and imaged (PCC of V2R and βarr1 in unstimulated cells = 0.38 ± 0.03 and in stimulated cells = 0.88 ± 0.03, Ib30 and V2R in unstimulated cells = 0.29 ± 0.04 and in stimulated cells = 0.83 ± 0.01, and βarr1 with Ib30 in unstimulated cells = 0.43 ± 0.08 and in stimulated cells = 0.63 ± 0.04, no. of cells = 3). A representative image of n = 3 cells/condition is shown here. Scale bar, 5 μm. C, Ib30 does not significantly alter agonist-induced internalization of V2R as assessed by confocal microscopy. Comparative analysis of V2R co-localization with two early endosomal markers, EEA1 and APPL1, upon agonist stimulation was performed in the presence of either Ib–CTL or Ib30. Cells expressing FLAG–V2R and Ib30–HA were treated with anti-FLAG antibody prior to agonist stimulation (AVP, 100 nm, 3–12 min) followed by fixation, permeabilization, and staining for endosomal markers APPL1 or EEA1 (Pearson's coefficient of V2R and EEA1 in Ib–CTL cells = 0.70 ± 0.01 and in Ib30 cells = 0.42 ± 0.01, no. of cells = 4, and Pearson's coefficient of V2R and APPL1 in Ib–CTL cells = 0.69 ± 0.07 and in Ib30 cells = 0.30 ± 0.07, no. of cells = 4). D, co-localization was also measured by manual counting of punctae in confocal images, and quantified data representing means ± S.E. from four different cells per condition are presented. E, an intermolecular BRET assay to measure the effect of intrabodies on the endosomal localization of βarr1 upon agonist stimulation. HEK-293 cells expressing V2R, βarr1–RlucII, rGFP-FYVE, and the indicated intrabodies were stimulated with varying doses of AVP, and the levels of BRET signal were recorded using a plate reader. The data represent means ± S.E.M. from four independent experiments, each performed in triplicate. F, agonist-induced change in BRET signal (i.e. the difference in BRET signal between the highest and the lowest AVP doses) as measured in panel E is presented as ΔBRET and analyzed using one-way ANOVA. **, p < 0.01; ***, p < 0.001.
We next measured the effect of intrabodies on Gαs coupling to the V2R using cAMP response as a readout. Once again, we did not observe any significant difference in cAMP dose response or time kinetics for Ib–CTL versus Ib30/Ib4 conditions (Fig. 8, A and B). Finally, we also evaluated the effect of intrabodies on agonist-induced ERK1/2 MAP kinase activation, a prototypical readout of V2R signaling, and did not detect a significant alteration by the intrabodies (Fig. 8, C and D). Taken together, these data establish that intrabodies do not have a major effect on transducer coupling and receptor endocytosis, making them suitable sensors to record βarr1 interaction and trafficking for GPCRs.
Figure 8.
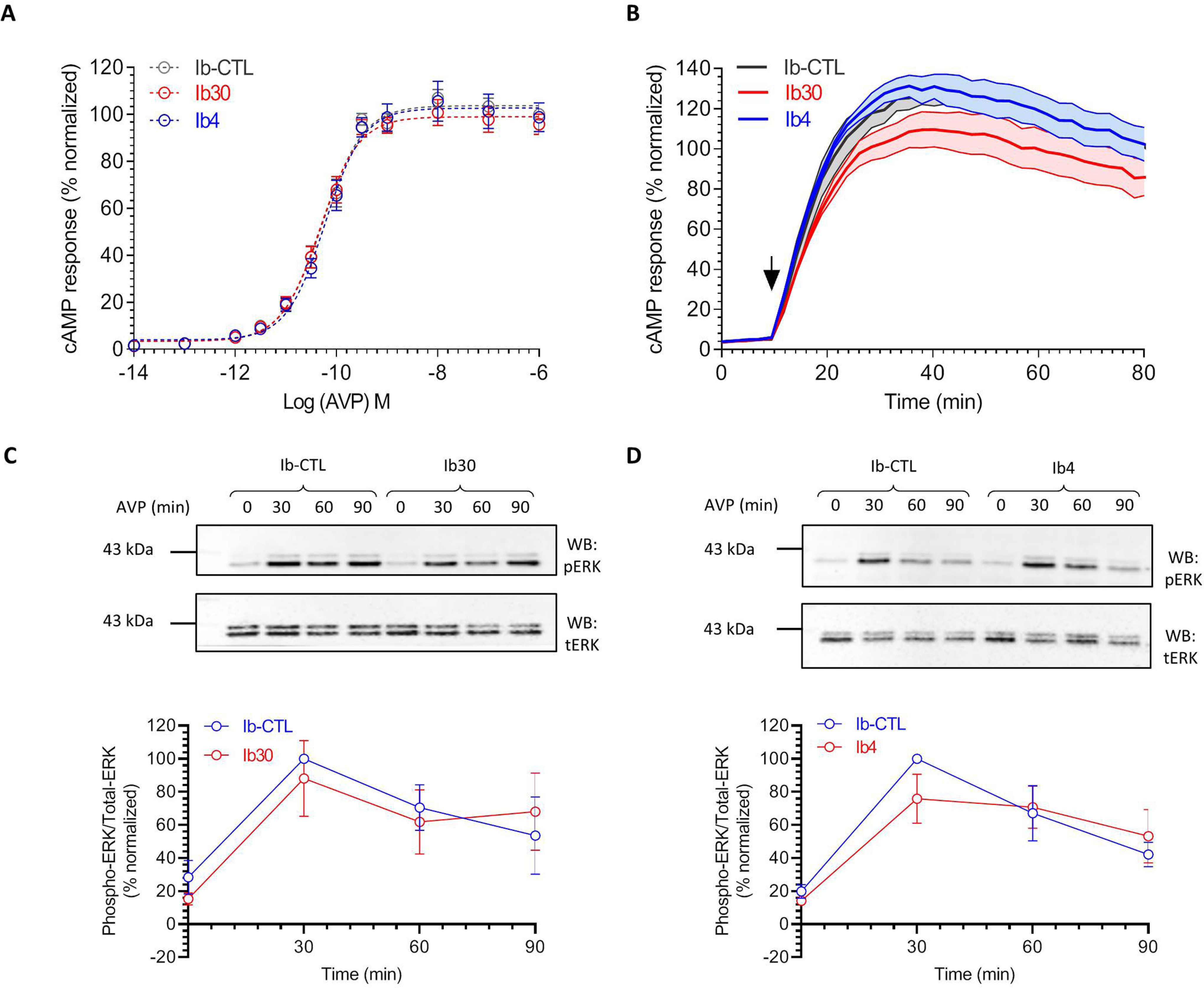
Effect of intrabodies on G-protein coupling and ERK1/2 phosphorylation. A, Ib30 does not significantly alter Gαs coupling of V2R as reflected by cAMP response. HEK-293 cells expressing V2R, the indicated intrabodies, and a luciferase-based cAMP biosensor (F22) were stimulated with varying doses of AVP, and the levels of cAMP were measured in terms of bioluminescence using a microplate reader. The data are normalized with respect to the maximal response obtained in presence of Ib–CTL (treated as 100%), and the graph represents means ± S.E. of three independent experiments, each performed in duplicate. B, time course of agonist-induced cAMP response in HEK-293 cells expressing V2R and the indicated intrabodies. The data are derived from the experiments described in A at an AVP concentration of 100 nm. C and D, intrabodies do not significantly alter agonist-induced ERK1/2 MAP kinase phosphorylation. HEK-293 cells expressing V2R and the indicated intrabodies were stimulated with AVP (100 nm) for the indicated time points followed by detection of ERK1/2 phosphorylation using Western blotting (WB). Representative images from four independent experiments are shown here, and densitometry-based quantification of data, normalized with Ib–CTL, with the 30-min condition treated as 100%, is presented in the lower panels.
Ib30 as a generic sensor of agonist-induced βarr1 trafficking for multiple chimeric GPCRs
Taking lead from the ability of Fab30 to recognize βarr1 complex with several chimeric GPCRs as presented in Fig. 2, we next evaluated Ib30 as a sensor to report βarr1 trafficking for these chimeric GPCRs in cellular context. Similar to previous experiments, we co-expressed the chimeric receptors with βarr1–mCherry and Ib30–YFP in HEK-293 cells and followed the localization of βarr1 and intrabodies using confocal microscopy after agonist treatment (Fig. 9, A–F). We observed that similar to β2V2R, Ib30 followed βarr1 translocation pattern by first localizing to the cell surface followed by trafficking into intracellular vesicles for all of these chimeric receptors (Fig. 9, A–F). It is worth noting here that the receptors used in Fig. 9 (A–C) contain most of the phosphorylation sites in their C terminus, whereas their third intracellular loops are relatively small. On the other hand, receptors included in Fig. 9 (D–F), harbor a larger third intracellular loop, which also contains most of the potential phosphorylation sites, and their C terminus is relatively smaller. Therefore, the data presented in Fig. 9 not only demonstrate the generality of Ib30 as a sensor to monitor agonist-induced βarr1 recruitment and trafficking for chimeric GPCRs but also its versatility for receptors differing in terms of their C terminus and intracellular loops.
Figure 9.
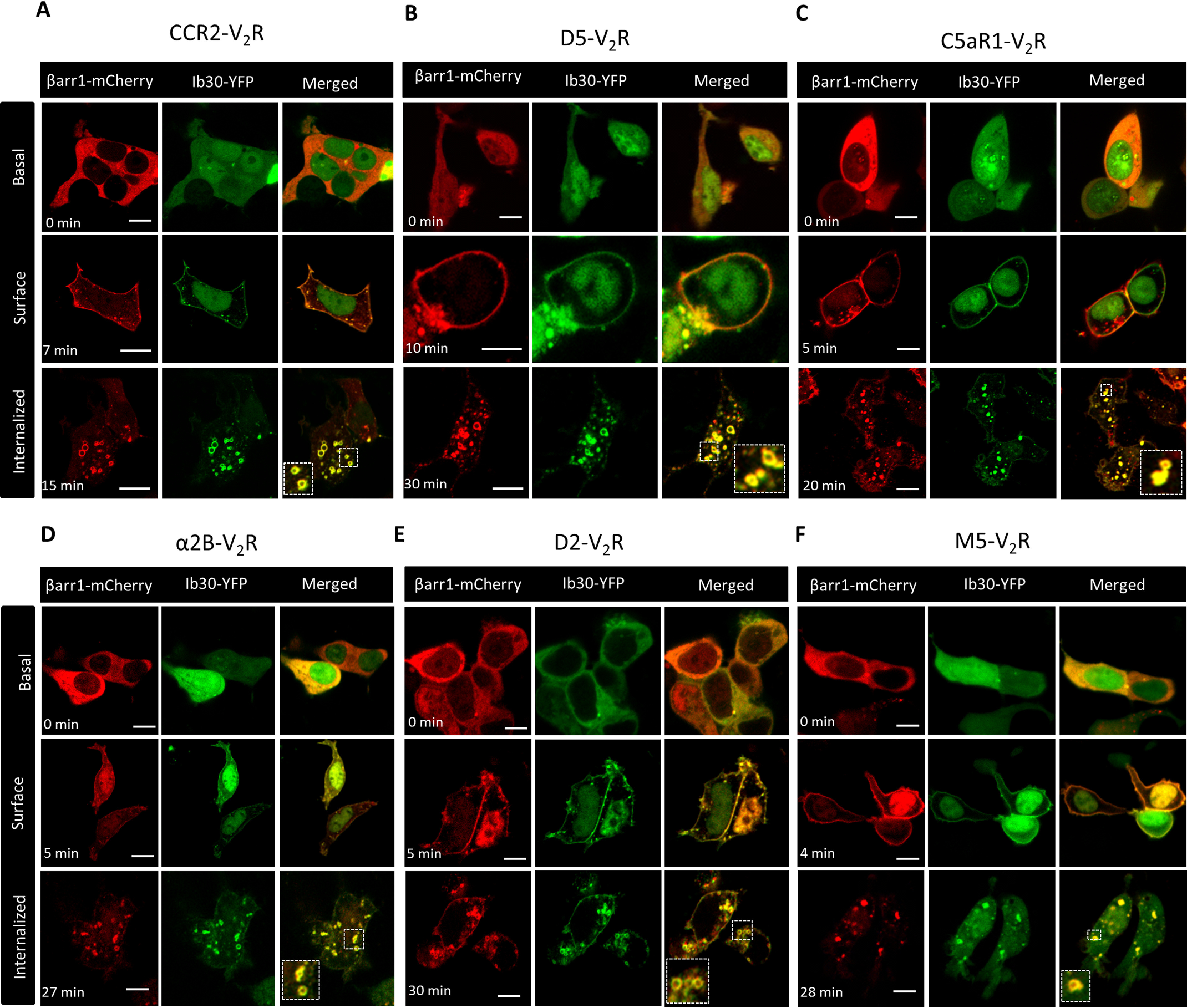
Ib30 reports agonist-induced trafficking of βarr1 for chimeric GPCRs. A–F, HEK-293 cells expressing the indicated chimeric GPCRs with V2R C terminus, βarr1–mCherry, and Ib30–YFP were stimulated with saturating concentration of respective agonists (100 nm CCL7, 20 μm dopamine, and 100 nm C5a, 20 μm epinephrine, 20 μm dopamine, and 20 μm carbachol, respectively), and the localization of βarr1 and Ib30 was visualized using confocal microscopy at the indicated time points. Scale bar, 10 μm. PCCs were measured to assess the co-localization of βarr1 and Ib30 using JACoP plugin in ImageJ, and the values for the upper, middle, and lower panels, respectively, are presented here. The following values were obtained: for CCR2V2R, 0.21 ± 0.02 from 17 cells, 0.84 ± 0.06 from 5 cells, and 0.83 ± 0.02 from 26 cells, with four independent experiments; for D5V2R, 0.36 ± 0.04 from 9 cells, 0.87 ± 0.04 from 6 cells, and 0.82 ± 0.03 from 30 cells, with three independent experiments; for C5aR1V2R, 0.31 ± 0.03 from 34 cells, 0.87 ± 0.01 from 40 cells, and 0.85 ± 0.01 from 53 cells with four independent experiments; for α2BV2R, 0.30 ± 0.04 from 7 cells, 0.90 ± 0.02 from 8 cells, and 0.91 ± 0.02 from 11 cells with four independent experiments; for D2V2R, 0.27 ± 0.04 from 18 cells, 0.88 ± 0.02 from 11 cells, and 0.83 ± 0.03 from 13 cells with three independent experiments; and for M5V2R, 0.27 ± 0.02 from 15 cells, 0.79 ± 0.04 from 22 cells, and 0.82 ± 0.05 from 9 cells with four independent experiments. Scale bar, 10 μm.
Ib30 sensor suggests conformational diversity in GPCR–βarr1 complexes
Finally, we evaluated the ability of the Ib30 sensor to report the trafficking of βarr1 for a set of GPCRs without the fusion of V2R-tail. We observed that Ib30–YFP followed agonist-induced translocation pattern of βarr1 for several different receptors including the complement C5a receptor 1 (C5aR1), the neurotensin receptor 1 (NTSR1), the muscarinic acetylcholine receptor subtype 2 (M2R), and the atypical chemokine receptor subtype 2 (ACKR2) (Fig. 10, A–D). We also validated the ability of Ib30 to recognize receptor-bound βarr1 for C5aR1 and ACKR2 by co-immunoprecipitation experiment (Fig. 10, E and F). These findings suggest that Ib30 can act as a sensor for monitoring agonist-induced βarr1 translocation for at least some GPCRs with their native C terminus as well. Interestingly, however, we observed that Ib30 did not robustly follow βarr1 translocation for the bradykinin subtype 2 receptor (B2R) upon agonist stimulation (Fig. 10G), although there was clear translocation of βarr1, first to the plasma membrane and then in intracellular vesicles. Taken together, these data potentially hint at conformational differences in GPCR–βarr1 complexes, even if the overall recruitment patterns are apparently similar. Future studies focused on measuring conformational differences in different GPCR–βarr complexes may provide additional insights and possibly link the conformational diversity to functional outcomes.
Figure 10.
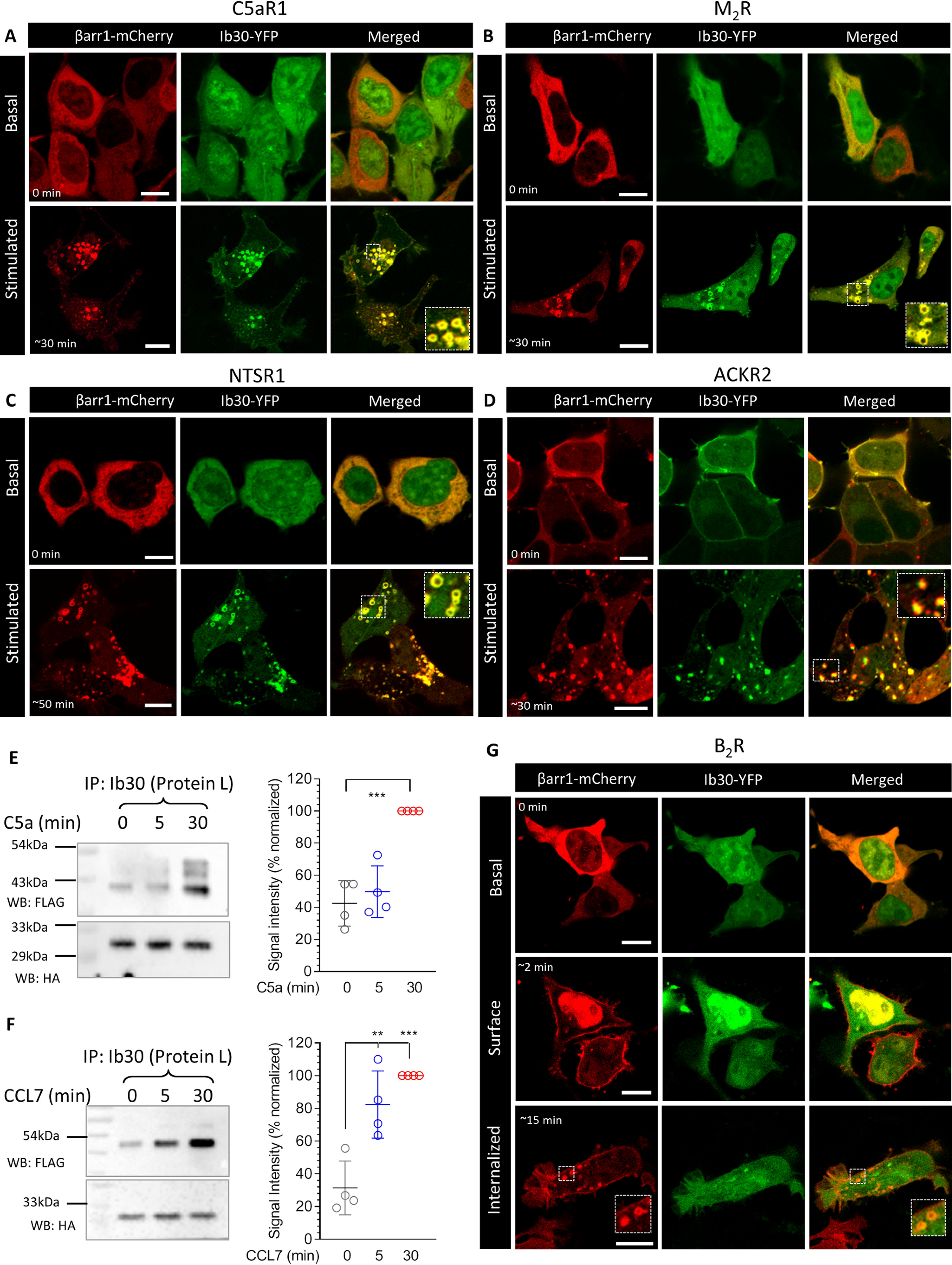
Ib30 reports agonist-induced trafficking of βarr1 for several unmodified GPCRs. A–D, HEK-293 cells expressing the indicated receptor, βarr1–mCherry and Ib30–YFP were stimulated with saturating concentration of respective agonists (100 nm C5a, 20 μm carbachol, 100 nm NTS1, and 100 nm CCL7, respectively), and the localization of βarr1 and Ib30 was visualized using confocal microscopy at the indicated time points. PCCs were measured to assess the co-localization of βarr1 and Ib30 using JACoP plugin in ImageJ, and the values for the unstimulated and stimulated conditions, respectively, are presented here. The following values were obtained: for C5aR1, 0.27 ± 0.03 from 20 cells and 0.75 ± 0.03 from 25 cells with five independent experiments; for M2R, 0.30 ± 0.04 from 8 cells and 0.85 ± 0.02 from 25 cells with four independent experiments; for NTSR1, 0.24 ± 0.04 from 15 cells and 0.87 ± 0.01 from 16 cells with three independent experiments; and for ACKR2, 0.88 ± 0.02 from 9 cells and 0.81 ± 0.01 from 29 cells with three independent experiments. Scale bar, 10 μm. For ACKR2, we observed significant membrane localization of βarr1 and Ib30, even before agonist treatment, which results into higher PCC values for unstimulated condition. E and F, HEK-293 cells expressing the C5aR1 and ACKR2, respectively, together with βarr1 and Ib30 were stimulated with either respective agonists (100 nm) for the indicated time points followed by co-IP using protein L–agarose beads. Subsequently, the proteins were visualized by Western blotting (WB) using anti-FLAG M2 antibody and anti-HA antibody. The right panels show densitometry-based quantification of four independent experiments normalized with signal at 30 min (treated as 100%) and analyzed using one-way ANOVA. **, p < 0.01; ***, p < 0.001. G, Ib30 does not follow agonist-induced translocation of βarr1 for the B2R as assessed by confocal microscopy on HEK-293 cells expressing B2R, βarr1–mCherry, and Ib30–YFP and stimulated with 100 nm bradykinin. The PCCs in the upper, middle, and lower panels were 0.33 ± 0.03 from 15 cells, 0.34 ± 0.03 from 20 cells, and 0.34 ± 0.04 from 16 cells, respectively, based on five independent experiments. Scale bar, 10 μm.
Discussion
Monitoring βarr interaction and subsequent trafficking has been used extensively to study the activation and regulatory framework of GPCRs. A number of approaches are commonly utilized for this including direct fusion of fluorescent proteins to βarrs (4), resonance energy transfer (FRET/BRET)–based assays (14, 28), enzyme complementation methods (16), and reporter assays (17, 18). Each of these methods necessitates a significant engineering and modification of the receptor, the βarr, or both. Intrabody sensors described here recognize receptor-bound βarr1 and report its trafficking in cellular context without the need for any modification of βarr1.
Although we observe that the intrabody sensors are capable of recognizing βarr1 for several GPCRs without the modification of their C termini, a potential drawback is that they are not likely to be universal for every GPCR as reflected for B2R in Fig. 10G. On the other hand, these intrabody sensors are able to recognize βarr1 more generally in the context of chimeric GPCRs harboring the V2R C terminus. It is conceivable that a similar strategy can be employed for other GPCRs as well by using, for example, phosphopeptides derived from the corresponding receptors. It is also worth noting here that many of the βarr assays such as PRESTO-TANGO also utilize chimeric GPCRs with V2R C terminus (V2R tail) (18). Engineering V2R tail typically imparts a class B pattern on GPCRs and thereby makes the detection of βarr1 interaction more robust compared with the unmodified receptors (29). It is also important to note that of five different FABs tested here, only two expressed efficiently as intrabodies in the cytoplasm. Therefore, starting with a larger number of FABs may be desirable to obtain more functional intrabodies in future endeavors.
Considering that YFP fusion does not alter the ability of intrabodies to interact with βarr1 and follow their translocation, it is also conceivable that they can be adapted in resonance energy transfer assays, or even in NanoBit format, for quantitative measurements of receptor–βarr1 interaction. Such strategies may yield even more sensitive versions of these intrabody sensors compared with approaches utilized here. In addition, although the intrabody sensors developed here are specific to βarr1 (25), it is plausible to design and develop similar intrabodies for βarr2 as well. Such an effort may help uncover novel insights into the functional divergence of the two βarr isoforms (30). Another interesting aspect of GPCR–βarr1 interaction is the ability of differential receptor phosphorylation patterns to induce distinct functional conformations in βarrs (31, 32). For several GPCRs, different phosphorylation patterns arising in ligand-specific, cell type–specific, and kinase-specific manners have been mapped and correlated with βarr mediated functional outcomes (33–35). Thus, it is tantalizing to hypothesize that intrabodies designed against different phosphopeptides derived from a given receptor may illuminate interesting attributes of receptor signaling and regulation in future. In conclusion, our study expands the currently available toolbox to monitor GPCR–βarr interaction and trafficking, and the intrabody sensors described here should facilitate drawing novel insights into GPCR signaling and regulatory paradigms.
Experimental procedures
General reagents, plasmids, and cell culture
HEK-293 cells (ATCC) were maintained in Dulbecco's modified Eagle's medium containing 10% FBS and penicillin/streptomycin (100 units/ml) at 37 °C in 5% CO2. Transient transfection of plasmids was performed using PEI, and the cells were typically assayed 48 h post-transfection. The plasmids encoding FLAG–β2V2R, FLAG–V2R, Ib–CTL–HA, Ib4–HA, Ib30–HA, βarr1–mCherry have been described previously (25). YFP-tagged intrabodies were generated by subcloning their coding region in pCMV6–AC–YFP vector. The chimeric GPCRs were generated by grafting the V2R-tail sequence at residues 324 in CCR2, 443 in α2BR, 443 in D2R, 379 in D5R, 514 in M5R, and 326 in C5aR1. All constructs were verified by DNA sequencing. The antibodies were purchased from Sigma (HRP-coupled mouse anti-FLAG M2), Cell Signaling Technology (βarrs), Santa Cruz Biotechnology (rabbit anti-HA), and Thermo Fisher (goat anti-rabbit Alexa Fluor 647 and goat anti-mouse Alexa Fluor 555). Other general chemicals were purchased from Sigma, APExBIO, and local suppliers. Recombinant human CCL7 was purified following a previously published protocol (36).
Co-immunoprecipitation assay
To probe the reactivity of FABs toward β2V2R (Fig. 1), Sf9 cells expressing FLAG-tagged receptor were lysed and incubated with purified βarr1 and FABs. For the co-IP data presented in Fig. 2, the plasmids encoding FLAG-tagged receptor and βarr1 were transfected in HEK-293 cells. 48 h post-transfection, the cells were serum-starved for 4–6 h, stimulated with agonist, lysed by Dounce homogenizer, and incubated with FAB30 for 1 h at room temperature. Subsequently, the receptor–βarr1–FAB complex were solubilized with 1% MNG for 1 h and centrifuged to collect the clarified solubilized complex, and 20 μl of pre-equilibrated (in 20 mm HEPES, 150 mm NaCl, pH 7.4 buffer) Protein L beads (GE Healthcare) were added. After additional 1 h of incubation, the beads were washed three times with wash buffer (20 mm HEPES, 150 mm NaCl, pH 7.4, 0.01% MNG) and eluted with 2× SDS loading buffer. Eluted samples were run on 12% SDS-PAGE, and the receptors were detected using HRP-coupled anti-FLAG M2 antibody, whereas the FABs were visualized using Coomassie staining.
To assess the ability of intrabodies to report the formation of receptor–βarr1 complex (Figs. 4 and 6, A and B), HEK-293 cells expressing the FLAG-tagged receptor, βarr1, and HA-tagged intrabodies were stimulated with saturating concentration of indicated ligands for 30 min at 37 °C. Afterward, the cells were lysed in Nonidet P-40 lysis buffer (50 mm Tris, 150 mm NaCl, 1× PhosStop, 1× Protease inhibitor, 1% Nonidet P-40) followed by incubation with 20 μl of pre-equilibrated HA beads (Sigma, A-2095) for 2 h at 4 °C. The beads were washed three times with wash buffer (20 mm HEPES, 150 mm NaCl, pH 7.4), eluted with 2× SDS loading buffer, and proteins were visualized by Western blotting (HRP-coupled anti-FLAG M2 antibody at 1:2000 dilution and anti-HA antibody, sc-805 from Santa Cruz Biotechnology at 1:5000 dilution).
Confocal microscopy
To monitor the translocation of βarr1 and intrabodies by confocal microscopy (Figs. 3, C and D; 4, E and F; 5, C–E; 6C; 9, A–F; and 10, A–D and G), HEK-293 cells were transfected with plasmids encoding the indicated receptor, βarr1–mCherry, and YFP-tagged intrabodies. 24 h postinfection, the cells were seeded onto confocal dishes (GenetiX; catalog no. 100350) pretreated with 0.01% poly-d-lysine (Sigma). After another 24 h, the cells were serum-starved for 4–6 h prior to stimulation with saturating concentration of indicated agonists. For live cell confocal imaging, we used Zeiss LSM 710 NLO confocal microscope, and samples were housed on a motorized XY stage with a CO2 enclosure and a temperature-controlled platform equipped with 32× array GaAsP descanned detector (Zeiss). YFP was excited with a diode laser at 488-nm laser line, whereas mCherry was excited at 561 nm. Laser intensity and pinhole settings were kept in the same range for parallel set of experiments, and spectral overlap for any two channels was avoided by adjusting proper filter excitation regions and bandwidths. Images were scanned using the line scan mode, and the images were finally processed in ZEN lite (ZEN-blue/ZEN-black) software suite from ZEISS. Co-localization was analyzed by calculating Pearson's correlation coefficient (PCC) between the indicated channels using JACoP plugin in ImageJ software (37). At least three regions of interest per cell were analyzed, and the means ± S.E. of PCCs are presented in the respective figure legends together with the number of cells and independent experiments.
For three-color imaging (Fig. 7B) and co-localization with early endosomal markers (Fig. 7C), receptor imaging of live or fixed cells was monitored by “feeding” cells with anti-FLAG antibody (15 min, 37 °C) in phenol red–free Dulbecco's modified Eagle's medium prior to agonist treatment. Fixed cells were washed three times in PBS, 0.04% EDTA to remove FLAG antibody bound to the remaining surface receptors, fixed using 4% PFA (20 min at room temperature), permeabilized, and stained using HA primary antibody followed by Alexa Fluor 555 or 647 secondary antibodies. For co-localization of FLAG–V2R with endosomal markers, the cells were treated as above except incubated with either of the following primary antibodies postpermeabilization: EEA1 (rabbit anti-EEA1 antibody from Cell Signaling Technology) or APPL1 (rabbit anti-APPL1 antibody from Cell Signaling Technology). The cells were imaged using a TCS-SP5 confocal microscope (Leica) with a 63× 1.4 numerical aperture objective and solid-state lasers of 488, 561, and/or 642 nm as light sources. Leica LAS AF image acquisition software was utilized. All subsequent raw-image tiff files were analyzed using ImageJ or LAS AF Lite (Leica), and co-localization was measured by calculating the PCC using JACoP plugin in ImageJ software as mentioned above.
GloSensor assay and ERK1/2 phosphorylation
To measure the effect of intrabodies on Gαs coupling, if any, we measured agonist-induced cAMP response in GloSensor assay following a previously described protocol (25). Briefly, HEK-293 cells were transfected with plasmids encoding the V2R, the luciferase-based cAMP biosensor (pGloSensorTM-22F plasmid), and the intrabodies. 16 h post-transfection, the medium was aspirated, and the cells were flushed and pooled together in assay buffer containing 1× Hanks balanced salt solution, pH 7.4, and 20 mm of HEPES. Cell density was measured and adjusted such as to yield ∼125,000 cells in 100 μl. The cells were pelleted at 2000 rpm for 3 min to remove the assay buffer, and then the pellet was resuspended in the desired volume of sodium luciferin solution prepared in the same assay buffer. After seeding the cells in a 96-well plate, the plate was incubated at 37 °C for 90 min followed by an additional incubation of 30 min at room temperature. Subsequently, various doses of the indicated ligand were added to the cells, and the luminescence reading was recorded using a microplate reader (Victor × 4; Perkin Elmer). Agonist-induced phosphorylation of ERK1/2 MAP kinase was measured by Western blotting following a previously described protocol (38).
BRET assay
For measuring βarr1 recruitment and endosomal localization by BRET (Fig. 7A, E and F), transfections were performed on HEK-293 cells seeded (40,000 cells/100 µl/well) in 96-well white microplates (Greiner) using PEI at a ratio of 4:1 (PEI: DNA). To monitor V2R–βarr1 interaction, we used βarr1–RlucII and V2R–YFP plasmids described previously (39). To monitor endosomal translocation of βarr1, we used enhanced bystander BRET, in which the BRET acceptor (Renilla GFP; rGFP) is fused to the FYVE domain from endofin protein targeted to early endosomes (rGFP-FYVE) and βarr1 fusion with the BRET donor RlucII (15). 48 h post-transfection, the culture medium was removed, and the cells were washed with Dulbecco's PBS and replaced by Hanks' balanced salt solution. Afterward, the cells were stimulated with increasing concentrations of arginine vasopressin (AVP) for 10 min, and 2.5 μm coelenterazine H (BRET1) or coelenterazine 400a (BRET2) was added 5 min before BRET measurement. BRET signals were recorded on a Mithras (Berthold Scientific) microplate reader equipped with the following filters: 480/20 nm (donor) and 530/20 nm (acceptor) for BRET1 and 400/70 nm (donor) and 515/20 nm (acceptor) for BRET2. The BRET signal was determined as the ratio of the light emitted by the energy acceptor over the light emitted by energy donor. Raw BRET values are presented in Fig. 7 (A and E), whereas agonist-induced change in BRET signal (ΔBRET) obtained by calculating the difference in BRET values for the highest and lowest concentrations of AVP is presented in Fig. 7F.
Statistical analysis and data presentation
The quantified data were plotted and analyzed using GraphPad Prism software, and the details of experimental replicates and statistical analysis are mentioned in the corresponding figure legends.
Data availability
All data are available in the article.
Acknowledgments
We thank the members of our laboratories for critical reading of the manuscript.
Author contributions—M. Baidya, P. K., H. D.-A., S. P., and A. K. S. conceptualization; M. Baidya, P. K., B. S., S. S., A. C. H., M. Bouvier, and A. K. S. data curation; M. Baidya, P. K., H. D.-A., M. C., A. S., D. R., and A. K. S. validation; M. Baidya, H. D.-A., B. S., S. S., M. C., A. S., D. R., and A. K. S. investigation; M. Baidya and P. K. visualization; M. Baidya, P. K., H. D.-A., S. P., B. S., S. S., M. C., A. S., D. R., A. C. H., M. Bouvier, and A. K. S. methodology; M. Baidya, P. K., H. D.-A., S. P., B. S., S. S., M. C., A. S., D. R., A. C. H., M. Bouvier, and A. K. S. writing-review and editing; A. C. H., M. Bouvier, and A. K. S. supervision; A. K. S. resources; A. K. S. formal analysis; A. K. S. funding acquisition; A. K. S. writing-original draft; A. K. S. project administration.
Funding and additional information—This work was supported by DBT Wellcome Trust India Alliance Intermediate Fellowship IA/I/14/1/501285 (to A. K. S.), Innovative Young Biotechnologist Award BT/08/IYBA/2014-3 from the Department of Biotechnology, Government of India (to A. K. S.), a Lady Tata Memorial Trust Young Researcher Award (to A. K. S.), Science and Engineering Research Board Grant SB/SO/BB-121/2013 (to A. K. S.), and Council of Scientific and Industrial Research Grant 37[1637]14/EMR-II. Dr. Shukla is an EMBO Young Investigator. Drs. Hemlata Dwivedi and Mithu Baidya were supported by National Post-Doctoral Fellowship of Science and Engineering Research Board Grants PDF/2016/002930 and PDF/2016/2893. Dr. Ashish Srivastava is supported by Wellcome Trust/DBT India Alliance Early Career Fellowship Grant IA/E/17/1/503687. Dr. Hanyaloglu is supported by Genesis Research Trust Grant P73441 and Biotechnology and Biological Sciences Research Council Grants BB/N016947/1 and BB/S001565/1.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G-protein–coupled receptor
- βarr
- β-arrestin
- FAB
- antibody fragment
- BRET
- bioluminescence resonance energy transfer
- ERK
- extracellular signal-regulated kinase
- MAP
- mitogen-activated protein
- ANOVA
- analysis of variance
- co-IP
- co-immunoprecipitation
- HA
- hemagglutinin
- YFP
- yellow fluorescent protein
- Ib
- intrabody
- CTL
- control
- C5aR1
- C5a receptor 1
- NTSR1
- neurotensin receptor 1
- M2R
- muscarinic acetylcholine receptor subtype 2
- ACKR2
- atypical chemokine receptor subtype 2
- B2R
- bradykinin subtype 2 receptor
- PEI
- polyethyleneimine
- HRP
- horseradish peroxidase
- PCC
- Pearson's correlation coefficient
- RlucII
- Renilla luciferase II
- ScFv
- single-chain variable fragment
- AVP
- arginine vasopressin.
References
- 1. Bockaert J., and Pin J. P. (1999) Molecular tinkering of G protein–coupled receptors: an evolutionary success. EMBO J. 18, 1723–1729 10.1093/emboj/18.7.1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kang D. S., Tian X., and Benovic J. L. (2014) Role of β-arrestins and arrestin domain–containing proteins in G protein–coupled receptor trafficking. Curr. Opin. Cell Biol. 27, 63–71 10.1016/j.ceb.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lefkowitz R. J., and Shenoy S. K. (2005) Transduction of receptor signals by β-arrestins. Science 308, 512–517 10.1126/science.1109237 [DOI] [PubMed] [Google Scholar]
- 4. Oakley R. H., Laporte S. A., Holt J. A., Caron M. G., and Barak L. S. (2000) Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein–coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 10.1074/jbc.M910348199 [DOI] [PubMed] [Google Scholar]
- 5. Freedman N. J., and Lefkowitz R. J. (1996) Desensitization of G protein–coupled receptors. Recent Prog. Horm. Res. 51, 319–351 [PubMed] [Google Scholar]
- 6. Grundmann M., Merten N., Malfacini D., Inoue A., Preis P., Simon K., Rüttiger N., Ziegler N., Benkel T., Schmitt N. K., Ishida S., Müller I., Reher R., Kawakami K., Inoue A., et al. (2018) Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 9, 341 10.1038/s41467-017-02661-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gurevich V. V., and Gurevich E. V. (2018) Arrestin-mediated signaling: Is there a controversy? World J. Biol. Chem. 9, 25–35 10.4331/wjbc.v9.i3.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gutkind J. S., and Kostenis E. (2018) Arrestins as rheostats of GPCR signalling. Nat. Rev. Mol. Cell Biol. 19, 615–616 10.1038/s41580-018-0041-y [DOI] [PubMed] [Google Scholar]
- 9. Luttrell L. M., Wang J., Plouffe B., Smith J. S., Yamani L., Kaur S., Jean-Charles P. Y., Gauthier C., Lee M. H., Pani B., Kim J., Ahn S., Rajagopal S., Reiter E., Bouvier M., et al. (2018) Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 11, eaat7650 10.1126/scisignal.aat7650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ranjan R., Dwivedi H., Baidya M., Kumar M., and Shukla A. K. (2017) Novel structural insights into GPCR–β-arrestin interaction and signaling. Trends Cell Biol. 27, 851–862 10.1016/j.tcb.2017.05.008 [DOI] [PubMed] [Google Scholar]
- 11. Gurevich V. V., and Gurevich E. V. (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 25, 105–111 10.1016/j.tips.2003.12.008 [DOI] [PubMed] [Google Scholar]
- 12. Shukla A. K., Westfield G. H., Xiao K., Reis R. I., Huang L. Y., Tripathi-Shukla P., Qian J., Li S., Blanc A., Oleskie A. N., Dosey A. M., Su M., Liang C. R., Gu L. L., Shan J. M., et al. (2014) Visualization of arrestin recruitment by a G-protein–coupled receptor. Nature 512, 218–222 10.1038/nature13430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Angers S., Salahpour A., Joly E., Hilairet S., Chelsky D., Dennis M., and Bouvier M. (2000) Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. U.S.A. 97, 3684–3689 10.1073/pnas.97.7.3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Charest P. G., Terrillon S., and Bouvier M. (2005) Monitoring agonist-promoted conformational changes of β-arrestin in living cells by intramolecular BRET. EMBO Rep 6, 334–340 10.1038/sj.embor.7400373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Namkung Y., Le Gouill C., Lukashova V., Kobayashi H., Hogue M., Khoury E., Song M., Bouvier M., and Laporte S. A. (2016) Monitoring G protein–coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 7, 12178 10.1038/ncomms12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bassoni D. L., Raab W. J., Achacoso P. L., Loh C. Y., and Wehrman T. S. (2012) Measurements of β-arrestin recruitment to activated seven transmembrane receptors using enzyme complementation. Methods Mol. Biol. 897, 181–203 10.1007/978-1-61779-909-9_9 [DOI] [PubMed] [Google Scholar]
- 17. Barnea G., Strapps W., Herrada G., Berman Y., Ong J., Kloss B., Axel R., and Lee K. J. (2008) The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U.S.A. 105, 64–69 10.1073/pnas.0710487105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kroeze W. K., Sassano M. F., Huang X. P., Lansu K., McCorvy J. D., Giguère P. M., Sciaky N., and Roth B. L. (2015) PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 10.1038/nsmb.3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumari P., Srivastava A., Banerjee R., Ghosh E., Gupta P., Ranjan R., Chen X., Gupta B., Gupta C., Jaiman D., and Shukla A. K. (2016) Functional competence of a partially engaged GPCR-β-arrestin complex. Nat. Commun. 7, 13416 10.1038/ncomms13416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumari P., Srivastava A., Ghosh E., Ranjan R., Dogra S., Yadav P. N., and Shukla A. K. (2017) Core engagement with β-arrestin is dispensable for agonist-induced vasopressin receptor endocytosis and ERK activation. Mol. Biol. Cell 28, 1003–1010 10.1091/mbc.E16-12-0818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cahill T. J. 3rd, Thomsen A. R., Tarrasch J. T., Plouffe B., Nguyen A. H., Yang F., Huang L. Y., Kahsai A. W., Bassoni D. L., Gavino B. J., Lamerdin J. E., Triest S., Shukla A. K., Berger B., Little J. T. 4th, et al. (2017) Distinct conformations of GPCR–β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. U.S.A. 114, 2562–2567 10.1073/pnas.1701529114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiao K., Shenoy S. K., Nobles K., and Lefkowitz R. J. (2004) Activation-dependent conformational changes in β-arrestin 2. J. Biol. Chem. 279, 55744–55753 10.1074/jbc.M409785200 [DOI] [PubMed] [Google Scholar]
- 23. Nobles K. N., Guan Z., Xiao K., Oas T. G., and Lefkowitz R. J. (2007) The active conformation of β-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of β-arrestins1 and -2. J. Biol. Chem. 282, 21370–21381 10.1074/jbc.M611483200 [DOI] [PubMed] [Google Scholar]
- 24. Shukla A. K., Manglik A., Kruse A. C., Xiao K., Reis R. I., Tseng W. C., Staus D. P., Hilger D., Uysal S., Huang L. Y., Paduch M., Tripathi-Shukla P., Koide A., Koide S., Weis W. I., et al. (2013) Structure of active β-arrestin-1 bound to a G-protein–coupled receptor phosphopeptide. Nature 497, 137–141 10.1038/nature12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghosh E., Dwivedi H., Baidya M., Srivastava A., Kumari P., Stepniewski T., Kim H. R., Lee M. H., van Gastel J., Chaturvedi M., Roy D., Pandey S., Maharana J., Guixa-Gonzalez R., Luttrell L. M., et al. (2019) Conformational sensors and domain swapping reveal structural and functional differences between β-arrestin isoforms. Cell Rep. 28, 3287–3299 10.1016/j.celrep.2019.08.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghosh E., Srivastava A., Baidya M., Kumari P., Dwivedi H., Nidhi K., Ranjan R., Dogra S., Koide A., Yadav P. N., Sidhu S. S., Koide S., and Shukla A. K. (2017) A synthetic intrabody-based selective and generic inhibitor of GPCR endocytosis. Nat. Nanotechnol. 12, 1190–1198 10.1038/nnano.2017.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Irannejad R., Tomshine J. C., Tomshine J. R., Chevalier M., Mahoney J. P., Steyaert J., Rasmussen S. G., Sunahara R. K., El-Samad H., Huang B., and von Zastrow M. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 10.1038/nature12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haider R. S., Godbole A., and Hoffmann C. (2019) To sense or not to sense-new insights from GPCR-based and arrestin-based biosensors. Curr. Opin. Cell Biol. 57, 16–24 10.1016/j.ceb.2018.10.005 [DOI] [PubMed] [Google Scholar]
- 29. Oakley R. H., Laporte S. A., Holt J. A., Barak L. S., and Caron M. G. (2001) Molecular determinants underlying the formation of stable intracellular G protein–coupled receptor–β-arrestin complexes after receptor endocytosis. J. Biol. Chem. 276, 19452–19460 10.1074/jbc.M101450200 [DOI] [PubMed] [Google Scholar]
- 30. Srivastava A., Gupta B., Gupta C., and Shukla A. K. (2015) Emerging functional divergence of β-arrestin isoforms in GPCR function. Trends Endocrinol. Metab. 26, 628–642 10.1016/j.tem.2015.09.001 [DOI] [PubMed] [Google Scholar]
- 31. Shukla A. K., Violin J. D., Whalen E. J., Gesty-Palmer D., Shenoy S. K., and Lefkowitz R. J. (2008) Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 9988–9993 10.1073/pnas.0804246105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., and Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51 10.1126/scisignal.2001707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Butcher A. J., Prihandoko R., Kong K. C., McWilliams P., Edwards J. M., Bottrill A., Mistry S., and Tobin A. B. (2011) Differential G-protein–coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 286, 11506–11518 10.1074/jbc.M110.154526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reiter E., Ahn S., Shukla A. K., and Lefkowitz R. J. (2012) Molecular mechanism of β-arrestin–biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197 10.1146/annurev.pharmtox.010909.105800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tobin A. B., Butcher A. J., Kong K. C. (2008) Location, location, location…: site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 29, 413–420 10.1016/j.tips.2008.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goncharuk M. V., R D., Dubinnyi M. A., Nadezhdin K. D., Srivastava A., Baidya M., Agnihotri-Dwivedi H., As A., and Shukla A. K. (2020) Purification of native CCL7 and its functional interaction with selected chemokine receptors. Protein Expr. Purif. 171, 105617 10.1016/j.pep.2020.105617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bolte S., and Cordelieres F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. (Oxford) 224, 213–232 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 38. Kumari P., Dwivedi H., Baidya M., and Shukla A. K. (2019) Measuring agonist-induced ERK MAP kinase phosphorylation for G-protein–coupled receptors. Methods Cell. Biol. 149, 141–153 10.1016/bs.mcb.2018.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rochdi M. D., Vargas G. A., Carpentier E., Oligny-Longpré G., Chen S., Kovoor A., Gitelman S. E., Rosenthal S. M., von Zastrow M., and Bouvier M. (2010) Functional characterization of vasopressin type 2 receptor substitutions (R137H/C/L) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: implications for treatments. Mol. Pharmacol. 77, 836–845 10.1124/mol.109.061804 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available in the article.