

Abstract
Inositol hexakisphosphate (IP6) is an abundant metabolite synthesized from inositol 1,3,4,5,6-pentakisphosphate (IP5) by the single IP5 2-kinase (IP5K). Genetic and biochemical studies have shown that IP6 usually functions as a structural cofactor in protein(s) mediating mRNA export, DNA repair, necroptosis, 3D genome organization, HIV infection, and cullin–RING ligase (CRL) deneddylation. However, it remains unknown whether pharmacological perturbation of cellular IP6 levels affects any of these processes. Here, we performed screening for small molecules that regulate human IP5K activity, revealing that the antiparasitic drug and polysulfonic compound suramin efficiently inhibits IP5K in vitro and in vivo. The results from docking experiments and biochemical validations suggested that the suramin targets IP5K in a distinct bidentate manner by concurrently binding to the ATP- and IP5-binding pockets, thereby inhibiting both IP5 phosphorylation and ATP hydrolysis. NF449, a suramin analog with additional sulfonate moieties, more potently inhibited IP5K. Both suramin and NF449 disrupted IP6-dependent sequestration of CRL by the deneddylase COP9 signalosome, thereby affecting CRL activity cycle and component dynamics in an IP5K-dependent manner. Finally, nontoxic doses of suramin, NF449, or NF110 exacerbate the loss of cell viability elicited by the neddylation inhibitor and clinical trial drug MLN4924/pevonedistat, suggesting synergistic ef-fects. Suramin and its analogs provide structural templates for designing potent and specific IP5K inhibitors, which could be used in combination therapy along with MLN4924/pevonedistat. IP5K is a potential mechanistic target of suramin, accounting for suramin's therapeutic effects.
Keywords: inositol phosphate, inhibitor, E3 ubiquitin ligase, cancer chemoprevention, protein degradation, COP9 signalosome, cullin–RING ligase, IP5K, neddylation, suramin
Inositol hexakisphosphate (IP6), is the most abundant inositol polyphosphate metabolite ubiquitously present from yeast to mammals and plants (1). In plants, IP6 is also known as phytate and can represent up to 1% of the mass of a plant seed. The biosynthesis of IP6 originates from the GPCR second messenger inositol triphosphate, which is stepwise phosphorylated by a series of inositol phosphate kinases (IPKs), including inositol 1,4,5-trisphosphate 3-kinase (IP3K), inositol 1,3,4-triphosphate 5/6-kinase, inositol polyphosphate multikinase (IPMK), and inositol 1,3,4,5,6-pentakisphosphate 2-kinase (IP5K), to generate inositol 1,3,4,5-tetrakisphosphate to IP6 (2). IP6 can be further phosphorylated by IP6 kinases (IP6Ks) and diphosphoinositol–pentakisphosphate kinases, generating inositol pyrophos-phate species (diphosphoinositiol pentakisphosphate and bis-diphosphoinositol tetrakisphosphate) that are less abundant but more dynamic molecules containing energetic pyrophosphate bond(s) (3). The higher inositol polyphosphates are highly conserved from yeast to human but are not as well-studied as inositol triphosphate.
In mammals, IP6 is generated by a single IP5K, which phosphorylates the 2´-OH group of IP5, the only isoform of endogenous IP5 (4, 5). IP6 has been found to play critical structural and regulatory roles in several proteins or protein complexes involved in 3D genome organization (6), DNA repair (7), mRNA editing and export (8, 9), HIV infection (10), and protein ubiquitylation (11–13). Although the biochemical importance of IP6 in the abovementioned processes has been firmly established by structural studies, dissecting the cellular functions of IP6 has been difficult, in part because of the lack of genetic materials to study IP5K, which is essential for embryonic viability in mice (14).
An alternative, nongenetic approach to study the physiology of higher inositol polyphosphates and their biosynthetic kinases is via pharmacologic inhibition. In this regard, small molecule screening has been conducted for mammalian IP3K (15, 16), IPMK (17) and the various IP6K isoforms (18–21). However, a screen for mammalian IP5K inhibitors/activators has not been reported to date, although recent effort has identified active-site probes for plant IP5K (22). The development of a pharmacologic tool to regulate IP5K could be of future therapeutic relevance, because low, physiologic concentrations of IP6 promote tumor cell proliferation (23). Although the underlying mechanism remains unclear, this protumorigenic observation is in line with the earlier-reported anti-apoptotic effect of overexpressed IP5K (24) but is contrary to the known anti-cancer effect of IP6 when applied at high, supraphysiological doses (25). These seemingly contradictory results of historic and recent IP6 research calls for a pharmacologic tool to manipulate cellular IP6 levels and dissect its exact role during carcinogenesis.
Because IP6 is considered a stable metabolite (26), it remains unknown whether continued synthesis by IP5K is required to supply IP6 for its many cellular functions. This question can be answered by acute pharmacologic IP5K blockade. Here, we performed high-throughput screening for small molecules that modulates the catalytic activity of human IP5K (hIP5K). Surprisingly, none of the compounds from the kinase inhibitor library targets IP5K. Rather, we identified suramin, an anti-helminthic drug belonging to the World Health Organization's model list of essential medicine, as an IP5K inhibitor with low-micromolar IC50 values. Mechanistically, suramin inhibits the binding of both ATP and IP5, suggesting an unusual bidentate inhibition mechanism. We further validated that suramin and its more potent analog NF449 inhibit cellular IP5K by assaying for IP6 levels and by examining complex formation between Cullin–RING E3 ligases and the COP9 signalsome (CSN), which employs IP6 as an intermolecular bridge for Cullin deneddylation (11, 27). Moreover, by disrupting IP6-mediated Cullin–RING ligases (CRL) sequestration by CSN, suramin synergizes with the phase III clinical trial drug MLN4924 to prevent cancer cell proliferation.
Results
Identification of suramin as an IP5K inhibitor via unbiased screening
MBP-tagged human IP5K was purified to apparent homogeneity in monomeric form and used for subsequent enzymatic assays (Fig. 1, A and B). The ADP-Glo assay, which measures ADP formation (15), was opted to monitor IP5K catalysis (Fig. 1C). Concentrations of ATP, IP5, and IP5K were optimized such that reactions were terminated at steady state, when the percentage of IP5 converted to IP6, determined by ATP consumption, is ∼50% (Fig. S1A). This turnover rate is further validated by using the PAGE gel method to separate and visualize IP5 and IP6 with 35.5% PAGE (28) (Fig. S1B).
Figure 1.
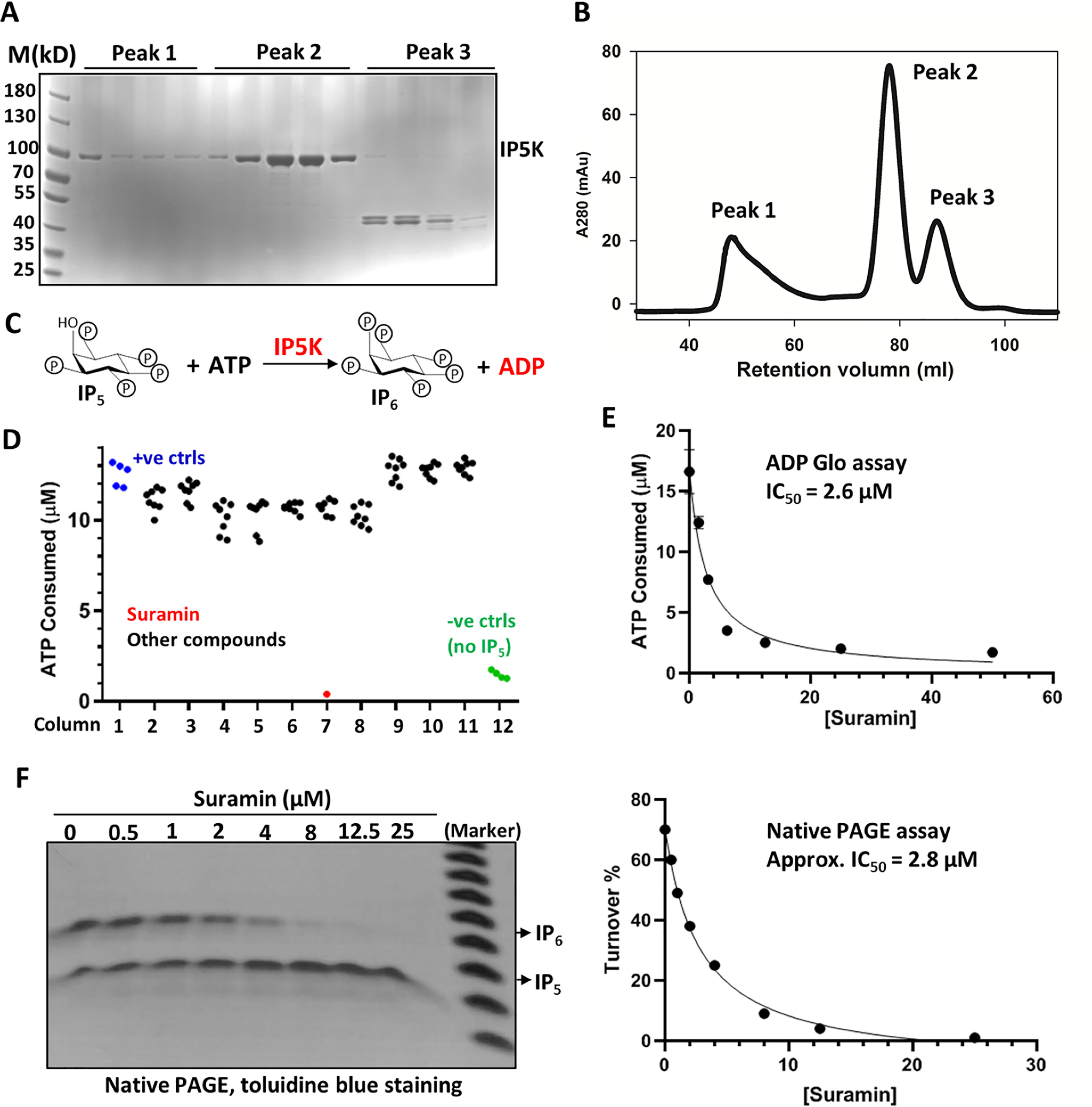
Identification of suramin as an IP5K inhibitor. A and B, SDS-PAGE analysis (A) of purified MBP–His6–tagged human IP5K, after gel filtration (B) on a HiLoad Superdex 200. Fractions corresponding to peak 2 are collected for subsequent assays. C, scheme depicting the enzymatic reaction catalyzed by IP5K, leading to ADP generation. D, primary screening for IP5K kinase regulators. The raw data from the plate containing suramin are shown. E and F, suramin dose-dependently inhibits IP5K activity as measured by the ADP-GloTM kinase assay (E) and the native PAGE assay (F). The data points were fitted in GraphPad with approximate IC50 determined following the equation described under “Experimental procedures.”
The IPKs, including IP5K (29), contain a kinase fold that is distantly related to protein kinases (30–32). We therefore started by screening a library of 243 known protein kinase inhibitors in a 96-well–based assay. To our surprise, none of these kinase inhibitors displayed strong activity against IP5K. Although three compounds (ryuvidine, HP372, and PD169316) do inhibit IP5K to some extent (Table 1), the IC50 values are much higher than that for their cognate kinase targets. Within this screened library, the flavonol quercetin is a pleiotropic kinase inhibitor (33) that has previously been found to inhibit several IPKs, including IPMK, IP6K1, and IP6K2 (17, 18, 20). However, quercetin fails to inhibit IP5K at the various IP5K concentrations tested (Fig. S1C), indicating that the kinase active site of IP5K is quite distinct from the other IPKs and from canonical protein kinases.
Table 1.
The IC50 values of screened compounds that exhibit inhibitory effect on IP5K enzymatic activity
| Compound | KP372 | Ryuvidine | PD169316 | 5-Iodotubericidin | l-Thyroxine | Suramin |
|---|---|---|---|---|---|---|
| IC50 (μm) | 14 ± 3 | 16 ± 4 | 20 ± 4 | 27 ± 8 | 86 ± 23 | 2.6 ± 0.3 |
We then conducted screening against a Food and Drug Administration–approved compound library, a natural product collection, as well as some ChemDiv compounds selected based on virtual screening of IP5K binders using the mouse IP5K crystal structure (34). This primary screen of ∼700 representative compounds revealed suramin, 5-iodotubericin, and l-thyroxine as potential IP5K inhibitors (Fig. 1D and Table 1). Suramin, an anti-parasitic drug approved by the World Health Organization (35), is by far the most potent IP5K inhibitor, with an apparent IC50 of ∼2.6 μm based on the ADP-Glo assay (Fig. 1E) or 2.8 μm based on PAGE gel imaging analysis (Fig. 1F). In comparison, suramin does not inhibit IP6K1 activity until 25 μm (Fig. S1D), suggesting rather specific inhibition of IP5K.
Suramin is a bidentate IP5K inhibitor that competes with both ATP and IP5
To understand how suramin inhibits IP5K, we first examined their binding affinity by isothermal titration calorimetry analysis. Suramin binds IP5K with a dissociation constant (Kd) of ∼1.1 μm (Fig. 2A). This Kd value is well-aligned with suramin's IC50 against IP5K (2.6 μm; Fig. 1E) and is comparable with the Kd(IP5) of IP5K (1.5 μm; Fig. 2G).
Figure 2.
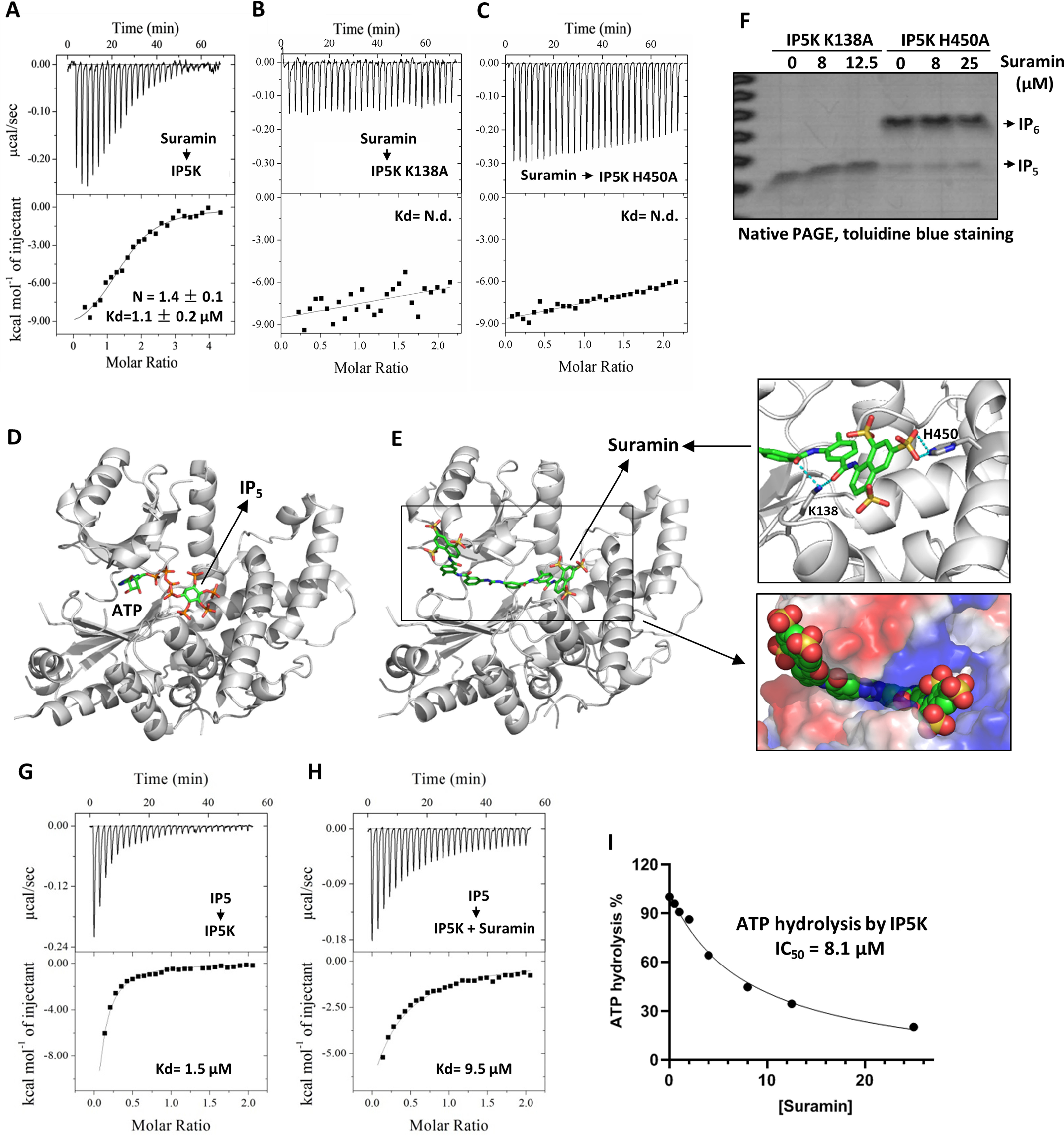
Mechanistic basis of IP5K inhibition by suramin. A–C, quantitative ITC measurement of suramin binding to IP5K WT (A), K138A (B), and H450A (C) mutant proteins. D, the ATP and IP5 binding sites in the crystal structure of IP5K (PDB code 5MW1). E, docking generated structural model of the suramin–IP5K complex with the lowest free energy value. Right top panel, close-up view of the docked model. Potential suramin-interacting residues (Lys138 and His450) are shown as sticks. Right bottom panel, close-up view of the docked model with IP5K shown in surface and suramin shown as spheres. F, catalytic activity of IP5K-K138A and IP5K-H450A in the presence/absence of suramin, measured by the native PAGE assay. G and H, quantitative ITC measurement of IP5–IP5K binding affinity in the absence (G) and presence (H) of suramin (20 μm). I, suramin dose-dependently inhibits the IP5-independent ATP hydrolysis activity of IP5K, measured by the ADP-GloTM kinase assay. N.d., not detected.
To probe the mode of interaction between suramin and IP5K, we performed computational docking. Docked poses with best scores consistently suggest that suramin binds to the catalytic active site of IP5K through extensive polar and nonpolar contacts with residues lining the ATP and IP5-binding pocket (Fig. 2, D and E), resulting in favorable binding free energy (ΔGbind = −14.55 Kcal/mol) for the IP5K–suramin complex. Specifically, one terminal naphthalene trisulfonate moiety of suramin binds to the arginine- and lysine-rich IP5-binding pocket, whereas the benzene ring, amide, and urea functional groups in the middle portion of suramin occupy the ATP-binding pocket (Fig. 2E), rendering suramin a bidentate IP5K inhibitor.
We validated the suramin-binding site by mutagenesis. Residue Lys138, essential for IP5K catalytic activity (Fig. 2F), lies deep in the IP5-binding pocket and interacts with IP5's phosphate groups (29, 34). Lys138 is predicted to also make electrostatic contact with suramin (Fig. 2E). Mutating Lys138 to alanine abolishes IP5K–suramin binding (Fig. 2B), suggesting that suramin indeed occupies the IP5-binding pocket.
Another IP5K residue, His450 (His448 in mice), does not interact with IP5 (29, 34) but makes critical contact with one sulfonate moiety of suramin (Fig. 2E). Similarly to the K138A mutation, the H450A mutation dramatically diminishes IP5K–suramin binding (Fig. 2C). Importantly, suramin no longer inhibits the catalytic activity of the H450A mutant (Fig. 2F and Fig. S2A), indicating that His450 selectively interacts with suramin and not IP5. Together, these data demonstrate that the basic IP5-binding pocket of IP5K also binds suramin, despite the differential requirement of certain residues in this pocket.
We next validated the predicted competition between suramin and IP5. ITC measurement detects avid binding of IP5 to IP5K, with a Kd value (1.5 μm) comparable with the reported Km(IP5) (5) (Fig. 2G). Suramin supplementation decreased the binding affinity of IP5–IP5K by 6.3-fold (Fig. 2H), consistent with steric competition between suramin and IP5 for binding IP5K.
We then set to confirm that suramin also competes with ATP. Our measured IP5K's Km(ATP) is high (41 μm; Fig. S1A), consistent with prior measurement using a different method (5). High Km(ATP) suggests low binding affinity, which precludes reliable examination of suramin–ATP competition by ITC. We therefore took advantage of the observation that IP5K can hydrolyze ATP in the absence of IP5, an activity that still requires the Mg2+-coordinating catalytic residue Asp439 (Fig. S2, B and C), thereby excluding the possibility of nonspecific ATP hydrolysis caused by enzyme impurity. Suramin dose-dependently inhibits this IP5-independent ATP hydrolysis by IP5K, with an IC50 of 8.1 μm (Fig. 2I), supporting competition between suramin and ATP. Together, these data are consistent with the docked model of the suramin–IP5K complex, whereby suramin directly interacts with IP5K in a bidentate manner and sterically blocks the access of both ATP and IP5.
Suramin inhibits hIP5K in vivo
To investigated whether suramin inhibits IP5K in cells, we examined cellular IP6 levels by using the established PAGE gel method (28), which is more accurate than the traditional [3H]inositol-labeling method (36). Although the level of cellular IP6 does not usually fluctuate (1), suramin significantly decreased IP6 levels in two different cell lines (Fig. 3, A and B). These data are in line with the diminishment of cellular IP6 levels upon IP5K knockdown (11) or knockout (12). Together, these data suggest that sustained IP5K catalysis is required to replenish cellular IP6 pool, which is amenable to suramin inhibition.
Figure 3.
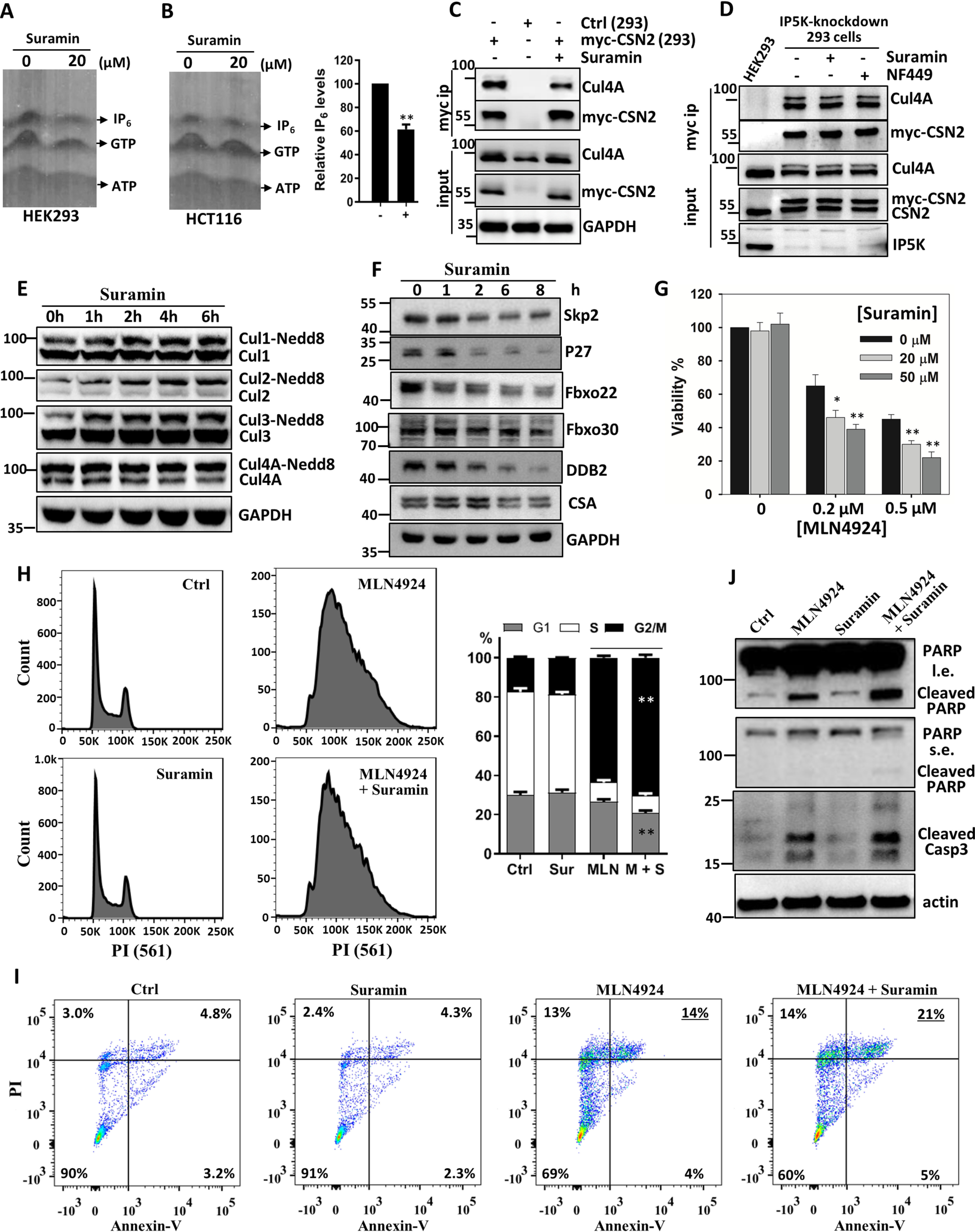
Suramin inhibits IP5K function in vivo. A and B, effect of suramin (20 μm, 12 h) on cellular IP6 levels in HEK293 (A) and HCT116 (B) cells. IP6 was extracted from cells for PAGE separation and toluidine blue staining. B, right panel, bar graph quantification of relative IP6 levels in HCT116 cells after normalization to cell number. C, immunoprecipitation of myc-CSN2 from myc-CSN2-HEK293 stable cell line treated with or without suramin for 8 h. D, immunoprecipitation of myc-CSN2 from myc–CSN2–HEK293 stable cell after IP5K knockdown, with or without suramin or NF449 treatment. E, Western blotting analysis of Cullin neddylation levels in HCT116 cells after treatment with 20 μm suramin for the indicated time periods. F, Western blotting analysis of the CRL substrate receptors and ubiquitylation substrate p27 after treatment with suramin (20 μm) for the indicated time periods. G, effect of MLN4924, alone or in combination with suramin, on cell viability. HCT116 cells were treated with MLN4924, suramin, or both for 48 h to examine cell viability. *, p < 0.05; **, p < 0.01 (Student's t test). H, HCT116 cells were treated with MLN4924 (0.5 μm), suramin (20 μm), or both for 24 h. DNA profiles of treated cells were analyzed by flow cytometry after PI staining. Right panel, bar graph representation of the cell cycle data expressed as means ± standard deviation. **, p < 0.01 (Student's t test). I, HCT116 cells were treated as described for H and then stained with FITC-conjugated annexin V and PI before being analyzed by flow cytometry. J, HeLa cells were treated as described in H. After 40 h, apoptosis marker proteins were blotted. Ctrl, control; Sur, suramin; MLN, MLN4924; M + S, MLN4924 and suramin.
Suramin regulates CRL–CSN complex formation and CRL activation in an IP5K/IP6-dependent manner
We wondered how suramin affects IP5K/IP6 downstream effector(s). IP6 is a multifunctional metabolite that serves as a structural cofactor in several protein and protein complexes (1). We recently identified a “glue”-like role for IP6 in linking CRLs with their cognate deneddylase: the CSN, thereby regulating the neddylation status and catalytic cycle of CRLs (11, 27, 37, 38). Consistent with suramin-mediated IP6 depletion, cellular interactions between CSN and Cullin 4A (Cul4A) are significantly diminished by suramin (Fig. 3C). Importantly, suramin does not decrease CSN binding to Cul4A in IP5K-depleted cells, suggesting that it regulates CRL–CSN complex formation via IP5K inhibition (Fig. 3D).
Disruption of CRL–CSN binding would prevent cullin deneddylation (37). Consistently, suramin time-dependently stimulates the neddylation of Cul1–Cul4 in the HCT116 colorectal cancer cell line (Fig. 3E). This suramin-augmented Cullin neddylation is also observed in WT, but not IP5K−/− HEK293 cells (Fig. S3), suggesting that suramin acts via IP6. Apart from catalyzing deneddylation, CSN binding also protects CRL substrate receptors from self-ubiquitylation because of prolonged activation (37). Skp2, the CRL1 substrate receptor, and p27, a CRL1Skp2 substrate, are also destabilized following suramin treatment (Fig. 3F). To more broadly understand the effect of suramin on CRL dynamics, we examined some other CRL substrate receptors, including the CRL1 substrate receptors Fbxo22 and Fbxo30 (39), and the CRL4 substrate receptors DDB1 and CSA (40). Levels of Fbxo22 and DDB2 decrease markedly after suramin treatment, whereas CSA and Fbxo30 are only weakly affected (Fig. 3F). Overall, these data are consistent with suramin enhancing CRL neddylation and activation but also eliciting activation-induced self-destruction of certain substrate receptors (37, 41, 42). Thus, suramin can perturb IP6-mediated physiological changes, specifically in CRL regulation by CSN.
Suramin sensitizes cancer cells to the neddylation inhibitor pevonedistat/MLN4924
Many CRL ubiquitylation substrates are involved in cell fate determination processes such as apoptosis, cell cycle arrest, senescence, immune evasion, proliferation, and/or migration. Aberrant activation of specific CRLs are common in cancers, which makes them attractive therapeutic targets (43–45). CRL activity requires cycles of neddylation and deneddylation. The CRL neddylation inhibitor MLN4924 (also known as pevonedistat) is under phase III anti-cancer clinical trial (NCT03268954) (46). CSN-mediated sequestration and deneddylation facilitates CRL catalytic cycle by protecting CRL substrate receptor components and by enabling CAND1 binding to nonneddylated CRL for substrate receptor exchange (37, 41, 42, 47, 48). Thus, inhibitors of CSN can also deprotect CRL components, can disrupt CRL activity cycle, and have been explored as anticancer agents (49).
We have previously observed that MLN4924-elicited cytotoxicity is synergized by IP5K knockdown, which disrupts IP6-bridged CRL protection by CSN (11). Given that suramin also induces CSN–CRL dissociation (Fig. 3C) and destabilizes CRL substrate receptors (Fig. 3F), we examined whether suramin will sensitize cancer cells to MLN4924, similarly to IP5K depletion. Although suramin is nontoxic at 10 and 20 μm concentrations, it significantly potentiates the cytostatic effect of MLN4924 on HCT116 cells, at all MLN4924 doses examined (Fig. 3G). Consistently, MLN4924-induced cell cycle arrest is significantly enhanced by suramin (Fig. 3H). Direct measurement of cell death by annexin V/propidium iodide (PI) double staining (Fig. 3I) or by blotting for Caspase-3 and PARP cleavage (Fig. 3J) showed that suramin also augments the apoptotic effects of MLN4924 in HCT116 and HeLa cancer cells. Together, these data suggest that IP5K inhibition and consequent disruption of CRL–CSN complex formation could be employed as combination therapy along with MLN4924.
The suramin analog NF449 is a monodentate but more potent IP5K inhibitor that also synergizes with MLN4924
To further understand the structure–function relationship between IP5K and suramin, we examined the effect of several suramin analogs on IP5K (Fig. 4A). Among them, NF023 weakly inhibits IP5K, whereas NF110 inhibits IP5K to levels comparable with suramin (Fig. 4B). Notably, NF449 is a more potent IP5K inhibitor than suramin (Fig. 4B), with its apparent IC50 (1.1 μm) ∼3-fold lower than that of suramin (Fig. 4C and Fig. 1F). This result seems fitting, with NF449 having more sulfonate moieties that could potentially interact more robustly with the many arginine, lysine, and histidine residues in the IP5-binding pocket of IP5K. In support of more efficient IP5 competition by NF449, IP5–IP5K binding is pronouncedly inhibited by NF449, to levels undetectable by ITC (Fig. S4, A and B), whereas suramin addition reduces IP5–IP5K affinity by ∼6-fold (Fig. 2, D and E).
Figure 4.
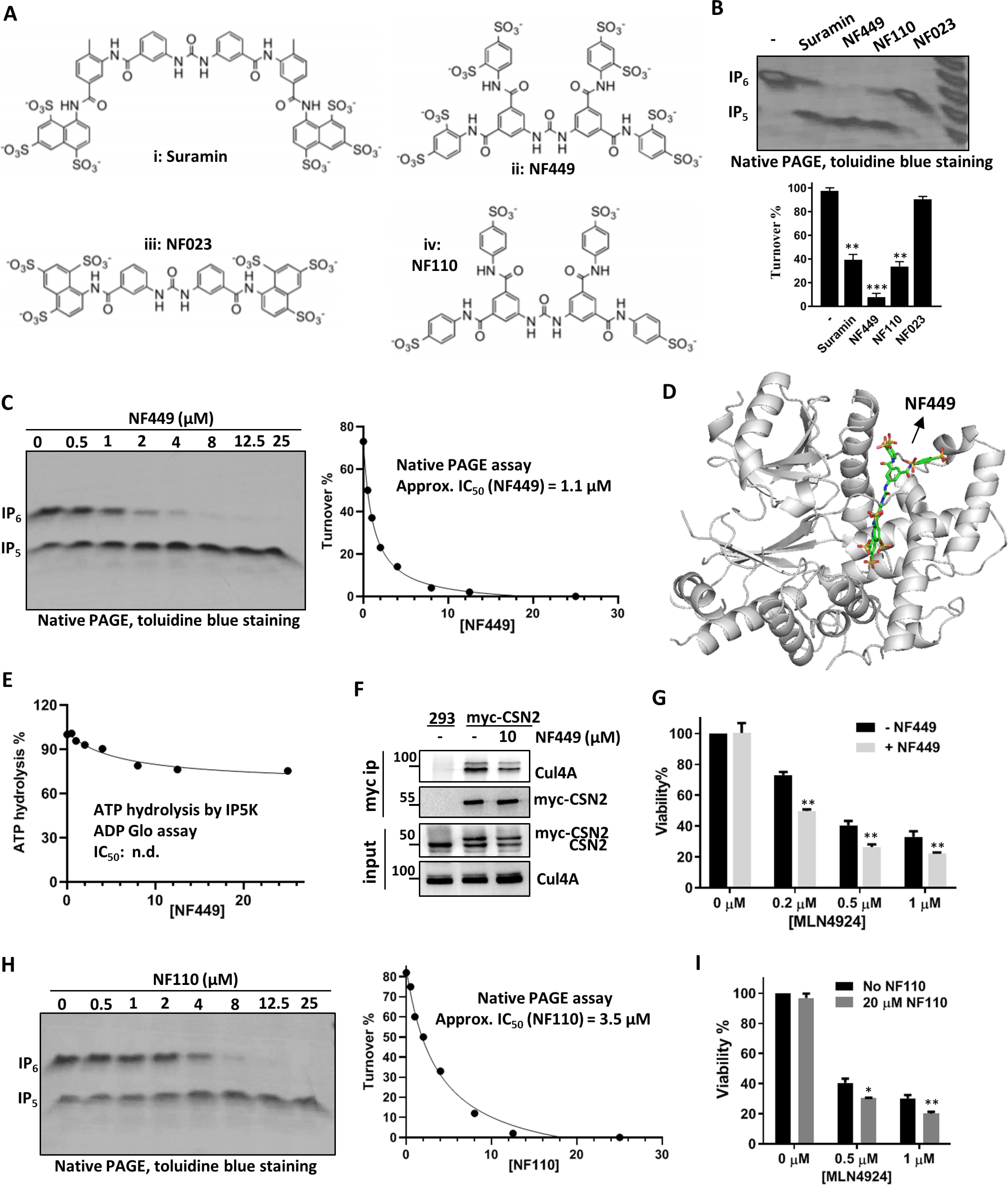
The suramin analogs NF449 and NF110 are also functional IP5K inhibitors. A, structures of suramin (panel i) and its analogs: NF449 (panel ii), NF023 (panel iii), and NF110 (panel iv). B, suramin analogs (10 μm each) modulate IP5K activity. **, p < 0.01; ***, p < 0.001 (Student's t test). C and H, NF449 (C) and NF110 (H) dose-dependently inhibit IP5K activity as measured the native PAGE assay. D, docking model of the NF449–IP5K complex with the lowest free energy value. E, NF449 does not significantly inhibit IP5K hydrolysis of ATP. F, immunoprecipitation of myc-CSN2 from myc-CSN2 stable HEK293 cells treated with or without NF449 (10 μm) for 8 h. G and I, effect of MLN4924, alone or in combination with NF449 (G) or NF110 (I), on HCT116 cell viability. *, p < 0.05; **, p < 0.01 (Student's t test).
The above results led us to hypothesize that NF449 would bind IP5K similarly to suramin, which we probed by docking analysis (Fig. 4D). To our surprise, although optimal docked models of the NF449–IP5K complex have indeed lower free energy values (−15.42 Kcal/mol) than that of the suramin–IP5K complex (−14.55 Kcal/mol), and NF449 occupies only the IP5-binding pocket, without extending to the neighboring ATP-binding site. This model, if correct, predicts that NF449 does not compete with ATP for binding IP5K. Consistent with this notion, in the absence of IP5, NF449, up to 25 μm, barely inhibits the ATP hydrolysis activity of IP5K (Fig. 4E), in sharp contrast to suramin (Fig. 2I).
Despite the distinct mode of IP5K binding, NF449 still disrupts CSN–CRL4 complex formation in WT (Fig. 4F) but not in IP5K-depeleted cells (Fig. 3D), suggesting that it acts via IP5K. Moreover, NF449 exacerbates the cytotoxicity of MLN4924 at all doses tested (Fig. 4G and Fig. S4C), similar to suramin. Finally, NF110, which inhibits IP5K with potency comparable with suramin (Fig. 4H), also synergizes with MLN4924 in reducing cell viability (Fig. 4I). Together, these data suggest that IP5K active-site inhibitors have the general potential to enhancing the cytotoxic effect of MLN4924 against cancer cells.
Discussion
IPKs are small molecule kinases that share the overall kinase fold with conventional protein kinases (18). Despite their ubiquitous presence and versatile functions, IPKs receive relatively less attention as drug targets when compared with protein kinases. The IP5Ks, in particular, are the last few IPK family members without any reported inhibitors. Our study reports the first identification of a class of sulfonic acid–rich compounds, exemplified by the prescribed drug suramin, as human IP5K inhibitors. We further demonstrate that suramin and its analogs inhibit cellular IP6 production and function and are candidate chemotherapy potentiators.
IP5K is notably distinct from the other IPKs. Structurally, IP5K has the most elaborate C-terminal IP-binding lobe, which ensures stringent substrate selectivity (29, 34). Indeed, although the only substrate for IP5K is IP5, other IPKs generally are capable of phosphorylating more than one IP substrate (30). Moreover, an IPK signature PXXXDXKXG motif that binds inositol polyphosphate and is present in other IPKs including IP3K, IPMK, and IP6K, is absent in IP5K, making IP5K the most distant member among the IPK family (30). IP5K is also most divergent from canonical protein kinases in that its N-lobe lacks an acidic residue known to form salt bridge with an ATP-coordinating lysine (34). Consistent with these unique properties of IP5K, we found that quercetin, a common inhibitor of other IPKs and many protein kinases (17, 18, 20, 33), is ineffective against IP5K. On the other hand, suramin, a compound not known to inhibit protein kinases, is now identified as an efficient inhibitor of IP5K but not IP6K1. This effectiveness of suramin likely arises from its rare mechanism of action: as a bidentate inhibitor targeting both the ATP- and IP5-binding sites. Suramin could be an attractive starting point to generate highly selective and potent IP5K inhibitors upon medicinal chemistry optimization.
What kind of biomedical purposes might an IP5K inhibitor serve? First, from a biological perspective, effective reduction of cellular IP6 levels by suramin provides a pharmacologic tool to directly prove that IP6 is being constantly turned over, a conclusion difficult to draw with genetic approaches. Second, from a translational perspective, suramin and its analogs could be explored to target IP6 effector modules, especially those related to diseases. In this regard, the CRLs, one of the major IP6 effectors we recently identified (11, 27, 37), are emerging anticancer targets (43, 44), we found that suramin and NF449 effectively disrupted IP6-dependent CRL regulation by the deneddylase CSN. The lack of effect of suramin and NF449 in IP5K knockdown and knockout cells verified that they specifically target IP5K to regulate CRL neddylation. Interestingly, suramin also interferes with the binding of CRL to the E2 enzyme CDC34 (50), suggesting that it can disrupt CRL catalytic cycle via two different mechanisms. Importantly, we directly demonstrate synergistic cytotoxic effects between nontoxic doses of suramin and MLN4924, a CRL inhibitor under phase III clinical trial (46), suggesting novel means of combination therapy.
Suramin is a century-old drug that has been used mainly to treat parasitic disease such as sleeping sickness caused by trypanosome infection (35). However, attempts has been made to repurpose suramin for treating other diseases, including AIDS (51) and prostate cancer (52), based on favorable pharmacological outcome. The mechanistic target(s) of suramin remain unclear in many of these cases. In view of our findings, it is worth investigating whether parasite or human IP5Ks are targeted by suramin to exert its reported therapeutic effects. Indeed, HIV critically relies on cellular IP6 for its capsid assembly and cell replication (10, 53), calling for reanalysis of suramin's mechanism of action in inhibiting HIV replication.
Experimental procedures
Materials
Suramin was purchased from MCE (HY-B0879A, >99.9% purity), and NF449 was obtained from Millipore (480420, >95% purity). NF110 and NF023 were kind gifts from the Shuwen Liu' laboratory at the School of Pharmaceutical Sciences of Southern Medical University (54). Anti-CSN2 and anti-Fbxo22 antibodies were purchased from Preoteintech (10969-2-AP). Anti-Cul4A, anti-PARP, anti-Skp2, anti-DDB2, and anti-cleaved Caspase3 antibodies were obtained from Cell Signaling Technology. Anti-Cul3 antibody was obtained from Bethyl Laboratories. Anti-IP5K antibody was codeveloped by us and Abcam. Anti-p21 and anti-p27 antibodies were from Santa Cruz. Anti-CSA antibody was from GeneTex. Anti-Fbxo30 antibody was from ABNOVA. IP5 was purchased from Cayman (10007784). MLN4924 was from APEXBIO (B1036). ADP-GLOTM kinase assay was purchased from Promega (V9101).
Recombinant expression and purification of hIP5K and IP6K1
The cDNA encoding hIP5K was obtained as previously described (11). To avoid the formation of inclusion bodies, a truncated version of hIP5K, in which the unstructured C-terminal 21 amino acids are deleted, is amplified using primers hIP5K-FP (atatggatccatggaagaggggaagatg) and hIP5K-RP (cgagcttaaggtctttggcacgtaca) and cloned into pMa-p2X vectors with N-terminal fusion of a maltose-binding protein (MBP)–His6 tandem affinity tag. Recombinant MBP–His6–hIP5K was then expressed in BL21 and purified to apparent homogeneity after nickel–nitrilotriacetic acid affinity chromatography, and size-exclusion chromatography was performed using the AKTA pure apparatus as previously described (55). IP6K1 was purified as previously described (56).
High-throughput screen for IP5K activity modulators by using the ADP glo assay
The screen for hIP5K inhibitors was performed using the Food and Drug Administration–approved drug library and the protein kinase inhibitor library, obtained from the Chinese National Compound Library (RRID:SCR_018723), natural product compound collection from other groups, and virtual screened compounds purchased from ChemDiv.
The kinase ADP-Glo assay kit containing ADP-Glo reagent and kinase detection reagents was purchased from Promega. In this assay, after the hIP5K kinase reaction, unmetabolized ATP was first degraded to AMP, and ADP was then converted back to ATP, which serves a luciferase substrate generating luminescence (15). The assay was then adapted for HTS in 96-well plates to ensure reproducibility. Compounds diluted in DMSO were added at a final concentration of 20 μm into a preoptimized reaction mixture containing 50 μm ATP, 100 nm hIP5K, 50 mm Tris-HCl (pH 7.0), and 5 mm MgCl2, with the addition of 25 μm IP5 initiating the reaction. After 30 min of incubation, an equal volume of ADP-GloTM reagent was added for 40 min to degrade leftover ATP, followed by incubation with kinase detection reagents for another 30 min. For luminescence measurements, a Synergy H1 (Biotek) plate reader was used at 400 nm with an integration time of 40 ms. For validation of the initial hits, compounds were used at serial dilutions in duplicates. To determine IC50, dose-dependent inhibition of ATP consumption data were fitted in GraphPad using the equation: Y = Bottom + (Top − Bottom)/[1 + (X/IC50)], as previously described (57).
Isothermal titration calorimetry
VP-ITC calorimeter (MicroCal) was used to measure the equilibrium dissociation constant (KD) for the interaction between IP5K and IP5, suramin, and NF449. Titration was performed at 25 °C in the buffer (pH 8.0) that contains 20 mm Tris-HCl, 150 mm NaCl, and 10 μm IP5K. The ligands were generally titrated at 100 μm. The data were analyzed after being normalized to the average value of the last three normalized delta hear. Data fitting was based on a OneSites binding model using the embedded software package MicroCal analysis launcher.
PAGE-based assay of IP5K and IP6K kinase activity
ADP Glo assay data were validated by the PAGE method, which allowed direct visualization of IP5 conversion to IP6 on a high-percentage PAGE gel (28). Enzymatic reactions were generally carried out in 1× buffer (20 mm Tris-HCl, pH 7.4, 5 mm MgCl2) with 0.5 μg of IP5K protein, 100 μm IP5, 200 μm ATP, 2 mm DTT at 30 °C for 30 min. Where applied, suramin and NF449 were added to the reaction at concentrations described in the figures. The reactions were stopped by adding 6× loading dye with 20 mm EDTA, and the samples were loaded to 35.5% PAGE gel (33.9 ml of 40% acrylamide, 3.8 ml of 10× Tris-Borate-EDTA buffer, 200 μl of 10% APS, 20 μl of TEMED) for visualization as described in the next section.
IP6 extraction from cells and visualization
The purification, separation, and visualization of IP6 was performed accordingly to established protocol (27). Briefly, the cells were washed twice in PBS and then harvested with 1 m perchloric acid (PA) containing 5 mm EDTA. The samples were vortexed for 10 min and then centrifuged at 18,000 × g for 5 min at 4 °C. At this time, we prepared the TiO2 beads (Titansphere TiO 5 μm; GL Sciences), which were weighed and prepared by washing once in water then once in 1 m PA containing 5 mm EDTA, (4–5 mg for one sample). The supernatants were removed into new Eppendorf tubes, and TiO2 beads were added. The samples were rotated for 15 min at 4 °C. The beads were pelleted by centrifuging at 3500 × g for 1 min and then washed twice in PA with the supernatants discarded. Bound inositol phosphates were then eluted with 200 μl of 5% ammonium hydroxide. The eluents were then vacuum-evaporated to 50 μl and subjected to 35.5% PAGE. Phosphate-rich metabolites were imaged with toluidine blue staining, using commercial IP6 as a control.
Western blotting and coimmunoprecipitation
HEK293 cells stably expressing myc-CSN2 were as described before (12). For CRL–CSN interaction analysis, the cells were treated with 20 μm suramin or 10 μm NF449 for 8 h, with normal HEK 293 cells as control. Myc immunoprecipitation experiments were performed as previously described (57). The samples were loaded to SDS-PAGE gel for Western blotting of the indicated proteins. Where applied, IP5K was knocked down in myc-CSN2 stable HEK293 cells using reagents as previously described (11).
Cell viability assay
HCT116 cells were seeded to 24-well plates with 8 × 104/well and were treated with various drug or drug combinations for 48 h to test the viability of cells by counting. The results were normalized to untreated control wells.
Cell cycle analysis
HCT116 cells were plated in 6-well plate and incubated with 0.5 μm MLN4924, 20 μm suramin, or 0.5 μm MLN4924 plus 20 μm suramin for 24 h. After trypsinization (0.25% trypsin without EDTA), the cells were washed with cold PBS and fixed with 70% ethanol overnight at 4 °C. Fixed cells were centrifuged at 1000 rpm for 5 min to remove ethanol and then resuspended by 50 μg/μl propidium iodide (BD, 550825) and 50 μg/μl RNase in PBS for 30 min on ice protected from light. Cell cycle distributions were determined by flow cytometry (BD FACSCanto) and analyzed using FlowJo 10.
Apoptosis (annexin V–PI) analysis
HCT116 cells were plated in 6-well plate overnight and incubated with 0.5 μm MLN4924, 20 μm suramin, or 0.5 μm MLN4924 with 20 μm suramin for 24 h, respectively. After trypsinization, the cells were washed twice with cold PBS and resuspended in 1× binding buffer at a concentration of 1 × 106 cells/ml. 1 × 105 cells were stained with FITC–annexin V and propidium iodide (BD, 556547) for 15 min at room temperature protected from light. After the addition of 400 μl of binding buffer, the cells were sorted by flow cytometry (BD FACSCanto), and the results were analyzed using FlowJo 10.
Suramin–IP5K docking studies
Many crystal structures of inositol polyphosphate kinase (PDB codes 5MWL, 5MWM, and 5MW8, respectively) are available in Protein Data Bank, and the structure of IP5K with the best resolution of 2.4 Å (PDB code 5MW8) (34) was chosen for molecular docking study. For the docking simulation, the structure of the protein was prepared by adding hydrogen atoms, deleting water molecules, and performing a 100-step energy minimization using CHARMM22/27 force field as described in a previous study (2). Meanwhile, the structure of suramin was downloaded from EMBL-EBI (small molecular code SVR) and optimized using the Optimized Potential for Liquid Simulations force field. Finally, molecular docking was carried out via the SwissDock package (3), in which the default parameters were used. The view of the IP5K–suramin complex was generated by PyMOL (RRID:SCR_000305).
Statistics
Where applied, significances were analyzed by one-way analysis of variance with GraphPad Prism.
Data availability
All data are contained within the article.
Supplementary Material
Acknowledgments
We thank the assistance of Southern University of Science and Technology Core Research Facilities.
This article contains supporting information.
Author contributions—X. Z., Y. S., and X. Y. data curation; X. Z. and Y. S. validation; X. Z., S. S., Y. S., X. Y., and S. H. investigation; X. Z., S. S., and J. W. methodology; X. Z. and F. R. writing-original draft; X. Z., Y. S., and F. R. writing-review and editing; X. Y. formal analysis; J. W. software; J. Z. and F. R. resources; J. Z. and F. R. supervision; J. Z. and F. R. funding acquisition; F. R. conceptualization.
Funding and additional information—This work was supported by Grants KQJSCX20180322152418316, JCYJ20170412153517422, and JCYJ20170817104311912 from the Science and Technology Innovation Commission of Shenzhen Municipal Government (to F. R.), Grants 31872798 and 91853129 (to F. R.) and 81925034 (to J. Z.) from the National Science Foundation of China, and Grant 2018A030313207 from the Department of Science and Technology of Guangdong Province (to F. R.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- IP6
- inositol hexakisphosphate
- IP5
- inositol 1,3,4,5,6-pentakisphosphate
- IP5K
- IP5 2-kinase
- CRL
- cullin–RING ligase
- CSN
- COP9 signalosome
- IPK
- inositol polyphosphate kinase
- IP3K
- inositol 1,4,5-trisphosphate 3-kinase
- IPMK
- inositol polyphosphate multikinase
- IP6K
- IP6 kinase
- hIP5K
- human IP5K
- PI
- propidium iodide
- MBP
- maltose-binding protein
- TEMED
- N,N,N',N'-tetramethylethylenediamine
- PA
- perchloric acid
- PDB
- Protein Bank Data.
References
- 1. Hatch A. J., and York J. D. (2010) SnapShot: inositol phosphates. Cell 143, e1030.e1 10.1016/j.cell.2010.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang Z., Zhao J., Deng W., Chen Y., Shang J., Song K., Zhang L., Wang C., Lu S., Yang X., He B., Min J., Hu H., Tan M., Xu J., et al. (2018) Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 14, 1118–1126 10.1038/s41589-018-0150-0 [DOI] [PubMed] [Google Scholar]
- 3. Grosdidier A., Zoete V., Michielin O. (2011) SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 39, W270–W277 10.1093/nar/gkr366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. York J. D., Odom A. R., Murphy R., Ives E. B., and Wente S. R. (1999) A phospholipase C–dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science 285, 96–100 10.1126/science.285.5424.96 [DOI] [PubMed] [Google Scholar]
- 5. Ives E. B., Nichols J., Wente S. R., and York J. D. (2000) Biochemical and functional characterization of inositol 1,3,4,5, 6-pentakisphosphate 2-kinases. J. Biol. Chem. 275, 36575–36583 10.1074/jbc.M007586200 [DOI] [PubMed] [Google Scholar]
- 6. Ouyang Z., Zheng G., Tomchick D. R., Luo X., and Yu H. (2016) Structural basis and IP6 requirement for Pds5-dependent cohesin dynamics. Mol. Cell 62, 248–259 10.1016/j.molcel.2016.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanakahi L. A., Bartlet-Jones M., Chappell C., Pappin D., and West S. C. (2000) Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell 102, 721–729 10.1016/S0092-8674(00)00061-1 [DOI] [PubMed] [Google Scholar]
- 8. Macbeth M. R., Schubert H. L., Vandemark A. P., Lingam A. T., Hill C. P., and Bass B. L. (2005) Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 309, 1534–1539 10.1126/science.1113150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alcázar-Román A. R., Tran E. J., Guo S., and Wente S. R. (2006) Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat. Cell Biol. 8, 711–716 10.1038/ncb1427 [DOI] [PubMed] [Google Scholar]
- 10. Dick R. A., Zadrozny K. K., Xu C., Schur F. K. M., Lyddon T. D., Ricana C. L., Wagner J. M., Perilla J. R., Ganser-Pornillos B. K., Johnson M. C., Pornillos O., and Vogt V. M. (2018) Inositol phosphates are assembly co-factors for HIV-1. Nature 560, 509–512 10.1038/s41586-018-0396-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scherer P. C., Ding Y., Liu Z., Xu J., Mao H., Barrow J. C., Wei N., Zheng N., Snyder S. H., and Rao F. (2016) Inositol hexakisphosphate (IP6) generated by IP5K mediates cullin-COP9 signalosome interactions and CRL function. Proc. Natl. Acad. Sci. U.S.A. 113, 3503–3508 10.1073/pnas.1525580113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin H., Zhang X., Liu L., Fu Q., Zang C., Ding Y., Su Y., Xu Z., He S., Yang X., Wei X., Mao H., Cui Y., Wei Y., Zhou C., et al. (2020) Basis for metabolite-dependent Cullin-RING ligase deneddylation by the COP9 signalosome. Proc. Natl. Acad. Sci. U.S.A. 117, 4117–4124 10.1073/pnas.1911998117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X., and Rao F. (2019) Are inositol polyphosphates the missing link in dynamic cullin–RING ligase regulation by the COP9 signalosome? Biomolecules 9, 349 10.3390/biom9080349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verbsky J., Lavine K., and Majerus P. W. (2005) Disruption of the mouse inositol 1,3,4,5,6-pentakisphosphate 2-kinase gene, associated lethality, and tissue distribution of 2-kinase expression. Proc. Natl. Acad. Sci. U.S.A. 102, 8448–8453 10.1073/pnas.0503656102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schröder D., Rehbach C., Seyffarth C., Neuenschwander M., Kries J. V., and Windhorst S. (2013) Identification of a new membrane-permeable inhibitor against inositol-1,4,5-trisphosphate-3-kinase A. Biochem. Biophys. Res. Commun. 439, 228–234 10.1016/j.bbrc.2013.08.053 [DOI] [PubMed] [Google Scholar]
- 16. Schröder D., Tödter K., Gonzalez B., Franco-Echevarría E., Rohaly G., Blecher C., Lin H. Y., Mayr G. W., and Windhorst S. (2015) The new InsP3Kinase inhibitor BIP-4 is competitive to InsP3 and blocks proliferation and adhesion of lung cancer cells. Biochem. Pharmacol. 96, 143–150 10.1016/j.bcp.2015.05.004 [DOI] [PubMed] [Google Scholar]
- 17. Mayr G. W., Windhorst S., and Hillemeier K. (2005) Antiproliferative plant and synthetic polyphenolics are specific inhibitors of vertebrate inositol-1,4,5-trisphosphate 3-kinases and inositol polyphosphate multikinase. J. Biol. Chem. 280, 13229–13240 10.1074/jbc.M500545200 [DOI] [PubMed] [Google Scholar]
- 18. Gu C., Stashko M. A., Puhl-Rubio A. C., Chakraborty M., Chakraborty A., Frye S. V., Pearce K. H., Wang X., Shears S. B., and Wang H. (2019) Inhibition of inositol polyphosphate kinases by quercetin and related flavonoids: a structure–Activity analysis. J. Med. Chem. 62, 1443–1454 10.1021/acs.jmedchem.8b01593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Puhl-Rubio A. C., Stashko M. A., Wang H., Hardy P. B., Tyagi V., Li B., Wang X., Kireev D., Jessen H. J., Frye S. V., Shears S. B., and Pearce K. H. (2018) Use of protein kinase–focused compound libraries for the discovery of new inositol phosphate kinase inhibitors. SLAS Discov. 23, 982–988 10.1177/2472555218775323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wormald M., Liao G., Kimos M., Barrow J., and Wei H. (2017) Development of a homogenous high-throughput assay for inositol hexakisphosphate kinase 1 activity. PLoS One 12, e0188852 10.1371/journal.pone.0188852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Padmanabhan U., Dollins D. E., Fridy P. C., York J. D., and Downes C. P. (2009) Characterization of a selective inhibitor of inositol hexakisphosphate kinases: use in defining biological roles and metabolic relationships of inositol pyrophosphates. J. Biol. Chem. 284, 10571–10582 10.1074/jbc.M900752200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Whitfield H., Gilmartin M., Baker K., Riley A. M., Godage H. Y., Potter B. V. L., Hemmings A. M., and Brearley C. A. (2018) A fluorescent probe identifies active site ligands of inositol pentakisphosphate 2-kinase. J. Med. Chem. 61, 8838–8846 10.1021/acs.jmedchem.8b01022 [DOI] [PubMed] [Google Scholar]
- 23. Windhorst S., Lin H., Blechner C., Fanick W., Brandt L., Brehm M. A., and Mayr G. W. (2013) Tumour cells can employ extracellular Ins(1,2,3,4,5,6)P6 and multiple inositol-polyphosphate phosphatase 1 (MINPP1) dephosphorylation to improve their proliferation. Biochem. J. 450, 115–125 10.1042/BJ20121524 [DOI] [PubMed] [Google Scholar]
- 24. Verbsky J., and Majerus P. W. (2005) Increased levels of inositol hexakisphosphate (InsP6) protect HEK293 cells from tumor necrosis factor α- and Fas-induced apoptosis. J. Biol. Chem. 280, 29263–29268 10.1074/jbc.M503366200 [DOI] [PubMed] [Google Scholar]
- 25. Brehm M. A., and Windhorst S. (2019) New options of cancer treatment employing InsP6. Biochem. Pharmacol. 163, 206–214 10.1016/j.bcp.2019.02.024 [DOI] [PubMed] [Google Scholar]
- 26. Shears S. B. (2001) Assessing the omnipotence of inositol hexakisphosphate. Cell. Signal. 13, 151–158 10.1016/S0898-6568(01)00129-2 [DOI] [PubMed] [Google Scholar]
- 27. Wilson M. S., and Saiardi A. (2018) Inositol phosphates purification using titanium dioxide beads. Bio Protoc. 8, e2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Losito O., Szijgyarto Z., Resnick A. C., and Saiardi A. (2009) Inositol pyrophosphates and their unique metabolic complexity: analysis by gel electrophoresis. PLoS One 4, e5580 10.1371/journal.pone.0005580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. González B., Baños-Sanz J. I., Villate M., Brearley C. A., and Sanz-Aparicio J. (2010) Inositol 1,3,4,5,6-pentakisphosphate 2-kinase is a distant IPK member with a singular inositide binding site for axial 2-OH recognition. Proc. Natl. Acad. Sci. U.S.A. 107, 9608–9613 10.1073/pnas.0912979107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shears S. B., and Wang H. (2019) Inositol phosphate kinases: expanding the biological significance of the universal core of the protein kinase fold. Adv. Biol. Regul. 71, 118–127 10.1016/j.jbior.2018.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miller G. J., and Hurley J. H. (2004) Crystal structure of the catalytic core of inositol 1,4,5-trisphosphate 3-kinase. Mol. Cell 15, 703–711 10.1016/j.molcel.2004.08.005 [DOI] [PubMed] [Google Scholar]
- 32. González B., Schell M. J., Letcher A. J., Veprintsev D. B., Irvine R. F., and Williams R. L. (2004) Structure of a human inositol 1,4,5-trisphosphate 3-kinase: substrate binding reveals why it is not a phosphoinositide 3-kinase. Mol. Cell 15, 689–701 10.1016/j.molcel.2004.08.004 [DOI] [PubMed] [Google Scholar]
- 33. Russo G. L., Russo M., and Spagnuolo C. (2014) The pleiotropic flavonoid quercetin: from its metabolism to the inhibition of protein kinases in chronic lymphocytic leukemia. Food Funct. 5, 2393–2401 10.1039/c4fo00413b [DOI] [PubMed] [Google Scholar]
- 34. Franco-Echevarría E., Sanz-Aparicio J., Brearley C. A., González-Rubio J. M., and González B. (2017) The crystal structure of mammalian inositol 1,3,4,5,6-pentakisphosphate 2-kinase reveals a new zinc-binding site and key features for protein function. J. Biol. Chem. 292, 10534–10548 10.1074/jbc.M117.780395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Melhorn H. (2008) Energy-metabolism-disturbing drugs. Encyclopedia of Parasitology, pp. 475–479, Springer, Berlin, Heidelberg [Google Scholar]
- 36. Desfougères Y., Wilson M. S. C., Laha D., Miller G. J., and Saiardi A. (2019) ITPK1 mediates the lipid-independent synthesis of inositol phosphates controlled by metabolism. Proc. Natl. Acad. Sci. U.S.A. 116, 24551–24561 10.1073/pnas.1911431116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rao F., Lin H., and Su Y. (2020) Cullin-RING ligase regulation by the COP9 signalosome: structural mechanisms and new physiologic players. Adv. Exp. Med. Biol. 1217, 47–60 10.1007/978-981-15-1025-0_4 [DOI] [PubMed] [Google Scholar]
- 38. Scott D. C., and Kleiger G. (2020) Regulation of cullin–RING E3 ligase dynamics by inositol hexakisphosphate. Proc. Natl. Acad. Sci. U.S.A. 117, 6292–6294 10.1073/pnas.2001683117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jin J., Cardozo T., Lovering R. C., Elledge S. J., Pagano M., and Harper J. W. (2004) Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 18, 2573–2580 10.1101/gad.1255304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin J., Arias E. E., Chen J., Harper J. W., and Walter J. C. (2006) A family of diverse Cul4-Ddb1–interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 10.1016/j.molcel.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 41. Wolf D. A., Zhou C., and Wee S. (2003) The COP9 signalosome: an assembly and maintenance platform for cullin ubiquitin ligases?. Nat. Cell Biol. 5, 1029–1033 10.1038/ncb1203-1029 [DOI] [PubMed] [Google Scholar]
- 42. Cope G. A., and Deshaies R. J. (2006) Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC Biochem. 7, 1 10.1186/1471-2091-7-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao Y., and Sun Y. (2013) Cullin–RING ligases As attractive anti-cancer targets. Curr. Pharm. Des. 19, 3215–3225 10.2174/13816128113199990300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng J., Guo J., North B. J., Tao K., Zhou P., and Wei W. (2019) The emerging role for Cullin 4 family of E3 ligases in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 1871, 138–159 10.1016/j.bbcan.2018.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu J., Peng Y., Zhang J., Long J., Liu J., and Wei W. (2020) Targeting SCF E3 ligases for cancer therapies. Adv. Exp. Med. Biol. 1217, 123–146 10.1007/978-981-15-1025-0_9 [DOI] [PubMed] [Google Scholar]
- 46. Sekeres M. A., Fram R. J., Hua Z., and Ades L. (2018) Phase 3 study of first line pevonedistat (PEV) + azacitidine (AZA) versus single-agent AZA in patients with higher-risk myelodysplastic syndromes (HR MDS), chronic myelomonocytic leukemia (CMML) or low-blast acute myelogenous leukemia (AML). J. Clin. Oncol. 36, 7077 [Google Scholar]
- 47. Wee S., Geyer R. K., Toda T., and Wolf D. A. (2005) CSN facilitates cullin–RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat. Cell Biol. 7, 387–391 10.1038/ncb1241 [DOI] [PubMed] [Google Scholar]
- 48. Reitsma J. M., Liu X., Reichermeier K. M., Moradian A., Sweredoski M. J., Hess S., and Deshaies R. J. (2017) Composition and regulation of the cellular repertoire of SCF ubiquitin ligases. Cell 171, 1326–1339.e14 10.1016/j.cell.2017.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schlierf A., Altmann E., Quancard J., Jefferson A. B., Assenberg R., Renatus M., Jones M., Hassiepen U., Schaefer M., Kiffe M., Weiss A., Wiesmann C., Sedrani R., Eder J., and Martoglio B. (2016) Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat. Commun. 7, 13166 10.1038/ncomms13166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu K., Chong R. A., Yu Q., Bai J., Spratt D. E., Ching K., Lee C., Miao H., Tappin I., Hurwitz J., Zheng N., Shaw G. S., Sun Y., Felsenfeld D. P., Sanchez R., et al. (2016) Suramin inhibits cullin–RING E3 ubiquitin ligases. Proc. Natl. Acad. Sci. U.S.A. 113, E2011–E2018 10.1073/pnas.1601089113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. De Clercq E. (1987) Suramin in the treatment of AIDS: mechanism of action. Antiviral Res. 7, 1–10 10.1016/0166-3542(87)90034-9 [DOI] [PubMed] [Google Scholar]
- 52. Vogelzang N. J., Karrison T., Stadler W. M., Garcia J., Cohn H., Kugler J., Troeger T., Giannone L., Arrieta R., Ratain M. J., and Vokes E. E. (2004) A phase II trial of suramin monthly x 3 for hormone-refractory prostate carcinoma. Cancer 100, 65–71 10.1002/cncr.11867 [DOI] [PubMed] [Google Scholar]
- 53. Mallery D. L., Faysal K. M. R., Kleinpeter A., Wilson M. S. C., Vaysburd M., Fletcher A. J., Novikova M., Bocking T., Freed E. O., Saiardi A., and James L. C. (2019) Cellular IP6 levels limit HIV production while viruses that cannot efficiently package IP6 are attenuated for infection and replication. Cell Rep. 29, 3983–3996.e4 10.1016/j.celrep.2019.11.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tan S., Li J. Q., Cheng H., Li Z., Lan Y., Zhang T. T., Yang Z. C., Li W., Qi T., Qiu Y. R., Chen Z., Li L., and Liu S. W. (2019) The anti-parasitic drug suramin potently inhibits formation of seminal amyloid fibrils and their interaction with HIV-1. J. Biol. Chem. 294, 13740–13754 10.1074/jbc.RA118.006797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rao F., See R. Y., Zhang D., Toh D. C., Ji Q., and Liang Z. X. (2010) YybT is a signaling protein that contains a cyclic dinucleotide phosphodiesterase domain and a GGDEF domain with ATPase activity. J. Biol. Chem. 285, 473–482 10.1074/jbc.M109.040238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rao F., Xu J., Khan A. B., Gadalla M. M., Cha J. Y., Xu R., Tyagi R., Dang Y., Chakraborty A., and Snyder S. H. (2014) Inositol hexakisphosphate kinase-1 mediates assembly/disassembly of the CRL4-signalosome complex to regulate DNA repair and cell death. Proc. Natl. Acad. Sci. U.S.A. 111, 16005–16010 10.1073/pnas.1417900111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rao F., Cha J., Xu J., Xu R., Vandiver M. S., Tyagi R., Tokhunts R., Koldobskiy M. A., Fu C., Barrow R., Wu M., Fiedler D., Barrow J. C., and Snyder S. H. (2014) Inositol pyrophosphates mediate the DNA-PK/ATM-p53 cell death pathway by regulating CK2 phosphorylation of Tti1/Tel2. Mol. Cell 54, 119–132 10.1016/j.molcel.2014.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article.