

Abstract
NS5B is the RNA-dependent RNA polymerase that catalyzes the replication of the hepatitis C virus genome. It is a major target for antiviral drugs including nucleoside analogs, such as the prodrugs mericitabine and sofosbuvir, which get metabolized to 2′-fluoro-2′C-methylcytidine-5′-triphosphate and 2′-fluoro-2′C-methyluridine-5′-triphosphate, respectively. These analogs act as chain terminators after they are incorporated during RNA synthesis. Recently, it has been shown that NS5B can efficiently remove chain terminators by a nucleotide-mediated excision reaction that rescues RNA synthesis. In this study, we use transient-state kinetics to understand the efficiency of inhibition for five nucleoside analogs. We show that CTP analogs are readily incorporated into a growing primer by NS5B but are also efficiently excised. In contrast, although UMP analogs are more slowly incorporated, the excision of UMP is slow and inefficient, and modifications to the 2′-carbon of the UTP ribose ring further decreased rates of excision to an undetectable level. Taken together, these data suggest that the clinical effectiveness of sofosbuvir is largely a function of being intractable to nucleotide-mediated excision compared with similar nucleoside analogs.
Keywords: hepatitis C virus (HCV), NS5B, inhibition mechanism, enzyme kinetics, RNA polymerase, viral polymerase, hepatitis C virus (HCV), nucleoside/nucleotide analogue, antiviral drug, chain terminator, mericitabine, sofosbuvir
The hepatitis C virus (HCV) infects an estimated 3% of the world's population, with ∼2.4 million people in the United States currently infected (1, 2). Chronic HCV infections lead to hepatic fibrosis, cirrhosis, and hepatocellular carcinoma (3). HCV is a plus-sense single-stranded RNA virus containing a 9.6-kb genome, which encodes a single polyprotein (4). This polyprotein consists of three structural proteins and seven nonstructural proteins, including NS5B, the RNA-dependent RNA polymerase responsible for replicating the viral genome. NS5B catalyzes de novo initiation of RNA synthesis, which is inefficient in vitro but is followed by a transition to an efficient and fast processive elongation phase (5, 6).
NS5B is the target for many antiviral therapies including nucleoside analogs (7–9). These analogs work by mimicking the natural nucleotides and are incorporated during processive replication of the viral genome (10). Once incorporated, the analogs act as chain terminators by hindering polymerization of the next incoming base to prevent further elongation of the nascent RNA strand (11). These analogs include the prodrugs mericitabine and sofosbuvir, which get metabolized to 2′-fluoro-2′C-methylcytidine-5′-triphosphate and 2′-fluoro-2′C-methyluridine-5′-triphosphate, respectively (7, 12, 13).
Recent advancements have afforded formation and isolation of a processive elongation complex of NS5B with RNA in vitro (5). Transient-state kinetic methods have uncovered an efficient ATP-mediated excision reaction, which may play a role in maintaining fidelity during genome replication (14). Interestingly, ATP-mediated excision can also efficiently remove chain terminators, thereby rescuing RNA synthesis. This mechanism was also observed in AZT-resistant variants of HIV RT and is thought to be the main cause of AZT resistance (15–18). However, WT NS5B has been shown to be ∼50-fold more efficient than AZT-resistant forms of HIV RT in removing chain terminators via ATP-mediated excision (14). This raises questions regarding how any chain-terminating nucleoside analog could be effective in treating HCV infections.
In this study, we use transient-state kinetic methods to measure the efficiencies of incorporation and excision of several nucleoside analogs (Fig. 1). We show that CTP and CTP analogs are efficiently incorporated but are also readily excised. Furthermore, although there is a large discrimination against incorporation of the 2′-modified UTP analog compared with unmodified UTP, ATP-dependent excision and pyrophosphorolysis of UTP and its analog are very inefficient.
Figure 1.

Structures of nucleoside analogs. A, structures of the prodrugs mericitabine and sofosbuvir. B, structures of the four nucleoside analogs provided by Janssen Pharmaceutical, Inc. (cytidine analogs) and Gilead Sciences (uridine analog). Analogs were provided with 5′-triphosphate.
Results
Incorporation of NTP and NTP analogs
Analysis of the kinetics of nucleotide incorporation under single-turnover conditions afforded the apparent equilibrium dissociation constants for ground-state binding (Kd,app) and the rate constants for the maximum rate of incorporation (kpol) for CTP and UTP. Although the rates of incorporation of CTP and UTP were sufficiently fast to require the use of rapid-quench-flow methods, the rates of incorporation of the analogs were slow, so the kinetics could be measured using hand-mixing methods. The reactions were measured under single-turnover conditions since the NS5B/9-nt/20-nt elongation complex pauses after the first incorporation because of the absence of the next complementary NTP and the dissociation of RNA from the enzyme is exceedingly slow (5). These measurements afforded the specificity constant (kcat/Km = kpol/Kd,app). The discrimination against incorporation of each analog was defined by the ratio of the specificity constant for the canonical nucleotide divided by that for the corresponding analog. The results for incorporation of CTP and CTP analogs (Fig. 2, A–D) and UTP and the UTP analog (Fig. 3, A and B) were fit based on Scheme 1 using KinTek Explorer software (Austin, TX). The results are summarized in Table 1. The kinetic parameters for incorporation of CTP (kpol = 21 ± 3 s−1, Kd,app = 46 ± 9 µm, kpol/Kd,app = 0.46 ± 0.1 µm−1 s−1) are in agreement with previously reported results under similar conditions (5). The Kd,app for 2′C-Me-2′F-CTP (59 ± 14 µm), 2′C-Me-CTP (61 ± 11 µm), and 4′-azido-CTP (23 ± 7 µm) were similar to that of CTP. However, the rate constant for incorporation for each of the three CTP analogs (1.9 ± 0.4, 1.0 ± 0.2, and 1.5 ± 0.4 s−1) was significantly lower than for unmodified CTP.
Figure 2.
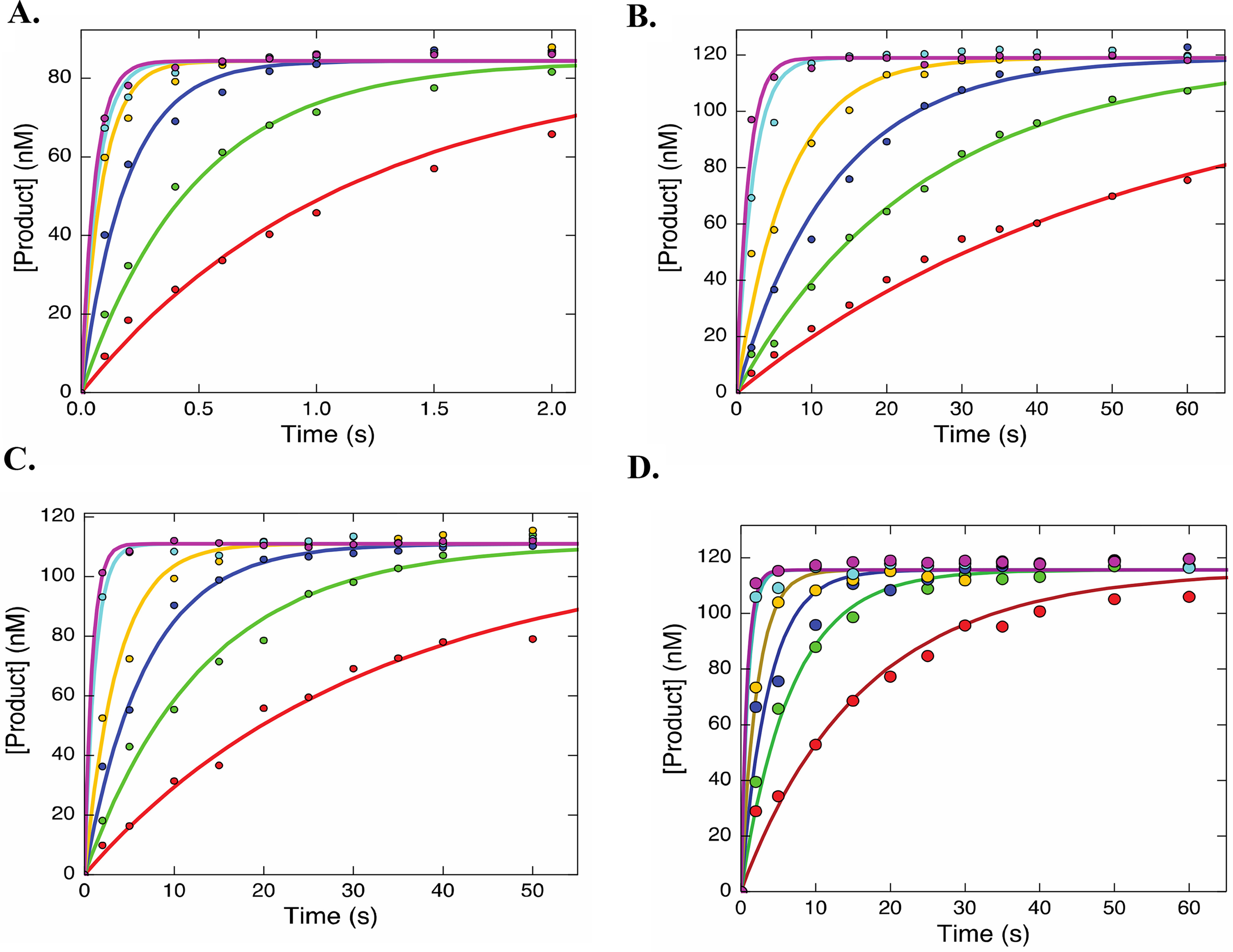
Incorporation of CTP and CTP analogs during elongation. The time courses for incorporation of CTP (2, 5, 15, 50, 100, and 200 μm) (A), 2′C-Me-CTP (1, 2.5, 5, 10, 50, and 100 μm) (B), 2′C-Me-2′F-CTP (1, 2.5, 5, 10, 50, and 100 μm) (C), and 4′-azido-CTP (1, 2.5, 5, 10, 50, and 100 μm) (D) were collected using rapid quenched flow. The data were fit using KinTek Explorer using Scheme 1. The solid line represents the best fit generated during data fitting. The results from the fit are summarized in Table 1.
Figure 3.

Incorporation of UTP and 2′C-Me-2′F-UTP. The plots show the incorporation of UTP (10, 25, 100, 200, and 400 μm) (A) and 2′C-Me-2′F-UTP (12.3, 37, 111, 333, and 1000 μm) (B). UTP incorporation was measured using rapid quenched flow, and incorporation of 2′C-Me-2′F-UTP was measured using hand quench methods. The data were fit using Scheme 1 using KinTek Explorer, and the solid line shows the best fit generated during data fitting. Results are summarized in Table 1.
Scheme 1.

Minimal model for incorporation and pyrophosphorolysis of nucleotide triphosphate. E, enzyme; Rn, an RNA primer n residues in length.
Table 1.
HCV NS5B nucleoside and nucleoside analog incorporation parameters
Shown is a summary of kinetic parameters for incorporation of nucleoside and nucleoside analog triphosphates. The rate constants were derived by fitting the data using Scheme 1. S.E. values were derived by nonlinear regression in globally fitting the data (19). Discrimination is determined using the expression, (kpol/Kd,app)NTP/(kpol/Kd,app)Inhibitor.
| Kd,app | kpol | kpol/Kd,app | Discrimination | |
|---|---|---|---|---|
| μm | s−1 | μm−1 s−1 | ||
| CTP:G | ||||
| CTP | 26 ± 3 | 10 ± 0.8 | 0.38 ± 0.05 | |
| 2′C-Me-CTP | 48 ± 7 | 1.0 ± 0.1 | 0.021 ± 0.004 | 18 ± 4 |
| 2′C-Me-2′F-CTP | 63 ± 11 | 1.9 ± 0.3 | 0.030 ± 0.007 | 13 ± 3 |
| UTP:A | ||||
| UTP | 320 ± 60 | 33 ± 5 | 0.10 ± 0.02 | |
| 2′C-Me-2′F-UTP | 410 ± 50 | 0.3 ± 0.03 | 0.00073 ± 0.0001 | 140 ± 45 |
The kpol and Kd,app for UTP incorporation were 33 ± 5 s−1 and 320 ± 60 µm, respectively, giving a kpol/Kd,app of 0.1 ± 0.02 µm−1 s−1. Similar to the case with the CTP analogs, the Kd,app for incorporation of 2′C-Me-2′F-UTP (410 ± 50 µm) was comparable with that for UTP. The maximum rate for incorporation of the UTP analog was 0.3 ± 0.03 s−1, roughly 100-fold lower than the rate constant for incorporation of UTP so that kpol/Kd,app = 0.0007 ± 0.0001 µm−1 s−1. The enzyme discriminates against incorporation of 2′C-Me-2′F-UTP by a factor 140 ± 45 relative to UTP. These results differ from previously published results for incorporation of the UTP analog (20), where the reported Kd,app = 113 µm, kpol = 0.67 s−1, and kpol/Kd,app = 0.0059 µm−1 s−1 were ∼8-fold more efficient compared with our measurements. This may be due to a difference in the strain used, as well as a difference in the sequence of the RNA template. Nevertheless, both results reflect a large discrimination against incorporation of the analog.
Our results reveal that the modifications of the ribose ring do not influence the apparent binding affinity of the analog; rather, discrimination against these analogs is driven by reduced values of kpol. All nucleoside analogs tested were shown to be effective chain terminators except for 4′-azido-CTP (data not shown). Therefore, the rest of this study focuses on the 2′C-modified nucleoside analogs.
Pyrophosphorolysis of incorporated NMPs and 2′C-modified NMP analogs
To examine the kinetics of excision by pyrophosphorolysis, the NS5B elongation complex was generated in the presence of either the unmodified nucleotide or the 2′C-modified analog to generate an NS5B/10-nt/20-nt enzyme/primer/template complex. We then mixed the complex with 0–2 mm sodium pyrophosphate supplemented with a 1:1 ratio of MgCl2/pyrophosphate. The results were fit using Scheme 1, where K3 is the apparent equilibrium dissociation constant for pyrophosphate, k−2 is the rate constant for pyrophosphorolysis, and k−2/K3 is the specificity constant for pyrophosphorolysis. The results are summarized in Table 2.
Table 2.
HCV NS5B nucleoside and nucleoside analog pyrophosphorolysis parameters
Shown is a summary of kinetic parameters for pyrophosphorolysis of incorporated nucleosides and nucleoside analogs. The rate constants were derived by fitting the data sets using Scheme 1. S.E. values were derived by nonlinear regression in globally fitting the data (19). Confidence contour analysis supported the use of S.E. estimates derived from nonlinear regression.
| Nucleotide | 1/K1 | k2 | K1 k2 | k−2 | K3 | k−2/K3 × 10−6 |
|---|---|---|---|---|---|---|
| μm | s−1 | μm−1 s−1 | s−1 | μm | μm−1s−1 | |
| CTP | 21 ± 1 | 46 ± 4 | 0.46 ± 0.05 | 0.3 ± 0.03 | 410 ± 50 | 730 ± 100 |
| 2′C-Me-CTP | 1.7 ± 0.2 | 54 ± 7 | 0.032 ± 0.006 | 0.4 ± 0.04 | 820 ± 100 | 460 ± 83 |
| 2′C-Me-2′F-CTP | 1.8 ± 0.2 | 56 ± 7 | 0.031 ± 0.006 | 0.3 ± 0.03 | 1600 ± 200 | 180 ± 24 |
| UTP | 28 ± 5 | 310 ± 70 | 0.09 ± 0.02 | 0.007 ± 0.002 | 1700 ± 200 | 4.1 ± 1 |
| 2′C-Me-2′F-UTP | 410a | 0.3a | 0.00073a | ≤0.01 |
a Rate constants for 2′C-Me-2′F-UTP incorporation are from Table 2.
The rate constant for pyrophosphorolysis of CMP-terminated RNA (k−2) was measured to be 0.3 ± 0.03 s−1, and K3 was determined to be 410 ± 50 µm, resulting in a specificity constant of (7.3 ± 1) × 10−4 µm−1 s−1 (Fig. 4A). These results are well within range of previously reported results (14). The rate constants for pyrophosphorolysis for 2′C-Me-CMP (Fig. 4B) and 2′C-Me-2′F-CMP (Fig. 4C) are 0.4 ± 0.04 s−1 and 0.3 ± 0.03 s−1, respectively. These rate constants are comparable to the rate constant for pyrophosphorolysis of CMP. There is a larger difference in the binding of pyrophosphate during excision of the analogs (820 ± 100 µm for 2′C-Me-CMP and 1600 ± 200 µm for 2′C-Me-2′F-CMP) when compared with CMP (410 ± 50 µm). These data indicate that binding of pyrophosphate, instead of the rate constant for excision, accounts for the difference in the specificity constant for pyrophosphorolysis.
Figure 4.
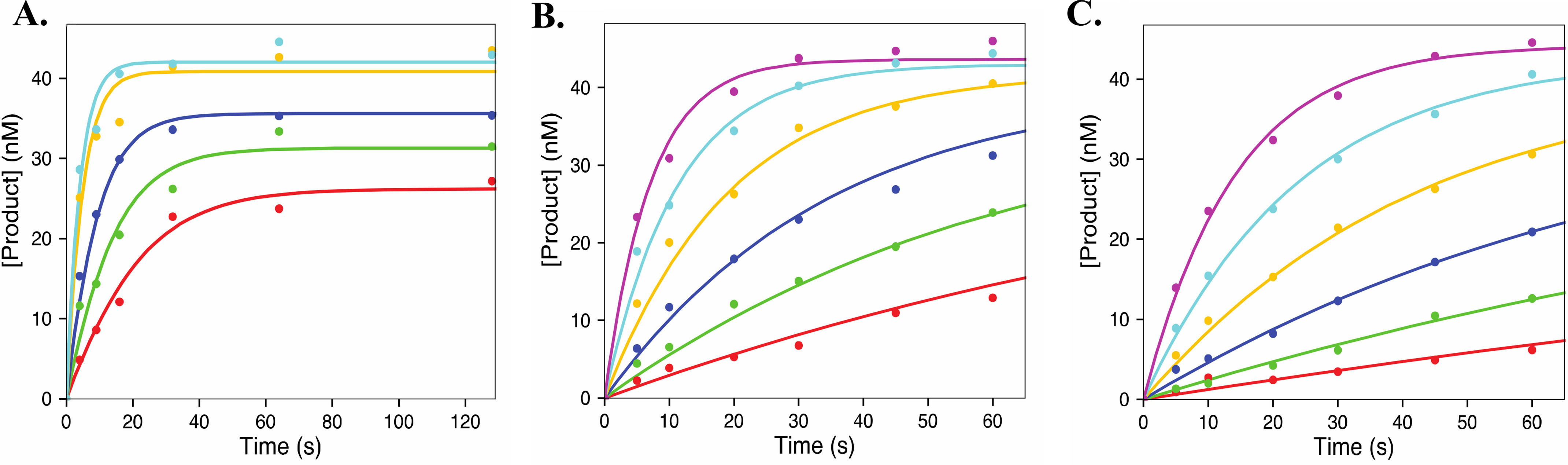
Pyrophosphorolysis of CMP and 2′C Modified CTP analogues. The plots show the pyrophosphorolysis of CMP (31.25, 62.5, 125, 500, and 1000 μm) (A), 2′C-Me-CMP (15.6, 31.3, 62.5, 125, 250, and 500 μm) (B) and 2′C-Me-2′F-CMP (15.6, 31.3, 62.5, 125, 250, and 500 μm) (C). The data were fit using Scheme 2 using KinTek Explorer. Solid lines represent best fit resulting from the fitting process. Results are summarized in Table 2.
According to our results, NS5B catalyzes pyrophosphorolysis of CMP more efficiently than 2′C-Me-CMP (k−2/K3 for CMP is (7.3 ± 1) × 10−4 µm−1 s−1 versus (4.6 ± 0.8) × 10−4 µm−1 s−1). 2′C-Me-2′F-CMP is most resistant to pyrophosphorolysis (k−2/K3 = (1.8 ± 0.2) × 10−4 µm−1 s−1) compared with CMP and 2′C-Me-CMP. It is important to note that the apparent equilibrium constant for pyrophosphate binding will include a term for translocation of the RNA from the nucleotide-binding site to the primer-binding site. A more favorable equilibrium constant for translocation will reduce the apparent affinity for pyrophosphate binding. Thus, 2′C-modified nucleoside analogs may shift the translocation to move the 3′ nucleotide of the RNA away from the reaction center.
Our data demonstrate that the 2′F modification further increases resistance to excision via pyrophosphorolysis. We also noted that the amplitude of the observed reaction depended on pyrophosphate concentration, indicating that the reaction came to equilibrium such that the net reaction was linked to concentration of added pyrophosphate. This could be due to slow release or rebinding of the NTP product of the reaction. We fit the data according to Scheme 1 by allowing the rebinding and reincorporation of the NTP to afford estimates of k2 and the apparent equilibrium constant for NTP rebinding (K1) to yield the specificity constant, K1k2. The measured specificity constant for NTP reincorporation is nearly identical to the results from the nucleotide incorporation data. For this reason, we globally fit data for the kinetics of incorporation and pyrophosphorolysis to obtain the values for rate constants listed in Table 2.
UMP undergoes pyrophosphorolysis significantly less efficiently (see Fig. 6A) compared with CMP and the CMP analogs. The rate constant for pyrophosphorolysis was measured to be 0.007 ± 0.002 s−1, and the apparent affinity for pyrophosphate binding was 1700 ± 200 µm. This slower rate constant and weaker binding results in a specificity constant of (0.041 ± 0.02) × 10−4 µm−1 s−1, ∼178-fold less than the k−2/K3 for pyrophosphorolysis of CMP. As with the pyrophosphorolysis of CMP and CMP analogs, an amplitude dependence was observed with increasing concentration of pyrophosphate due to the reincorporation of UTP. Under these conditions, k2 and K1 are almost identical to the results measured during the UTP incorporation reaction and so we globally fit with data for kinetics of incorporation and pyrophosphorolysis to obtain the values for rate constants listed in Table 2.
Figure 6.

Excision of UTP. The plots show UMP excision by pyrophosphorolysis (250, 500, 1000, and 2000 μm) (A) and ATP-mediated excision (1, 2, 4, and 8 mm) (B). The results were fit to Schemes 2 and 3, respectively, using KinTek Explorer. The solid lines represent the best fit generated during fitting. Results are summarized in Table 2.
We attempted to measure the pyrophosphorolysis of 2′C-Me-2′F-UMP (see Fig. 7A). No pyrophosphorolysis was observed at any pyrophosphate concentration over the time course measured. To set an upper limit on the rate constant for pyrophosphorolysis, we estimate that within the limits of detection, <1% of the input substrate reacted at the highest concentration of pyrophosphate. Accordingly, we set an upper limit of k−2/K3 ≤ 0.0001 × 10−4 µm−1 s−1. Together, these results indicate that efficiency of pyrophosphorolysis depends on the base, and modifications to the 2′-carbon on the ribose ring further increase resistance to pyrophosphorolysis.
Figure 7.

Excision of 2′C-Me-2′F-UTP. A, representative 16% denaturing PAGE separation of UMP pyrophosphorolysis. Bands below the 9 nt band appear due to pyrophosphorolysis occurring at that position. All bands below 10 nucleotides were summed during analysis to obtain the correct concentration of the loss of the 10-nt substrate due to pyrophosphorolysis. B and C, results for pyrophosphorolysis of 2′C-Me-2′F-UMP (B) and ATP-mediated 2′C-Me-2′F-UMP excision (C). Reactions were separated on a 16% polyacrylamide gel containing 8 m urea. The line in B indicates where gels are spliced together. No excision products were detectable during the observed time course. The addition of the next correct nucleotide did not result in extension of the primer strand to an 11-nt product (CTP Ctrl, first lane in B and C), indicating effective chain termination.
ATP-mediated excision of NMP and 2′C-modified NMP analogs
We measured the efficiency of ATP-mediated excision by generating the NS5B/10-nt/20-nt elongation complex as described under “Materials and methods.” We then incubated the complex with 0–8 mm ATP supplemented with a 1:1 molar ratio of MgCl2/ATP. The results were fit using Scheme 2, where k5 is the rate constant for ATP-mediated excision, 1/K4 is the apparent equilibrium dissociation constant for binding ATP during excision, and K4k5 is the specificity constant for ATP-mediated excision. We observed an ATP concentration dependence on the amplitude of the excision reaction, implying a reversible link between ATP binding and the equilibrium end point of the reaction. We account for the amplitude dependence by including the reverse reaction. In other words, the dinucleoside tetraphosphate (Ap4N) product generated during the excision reaction can be reincorporated into the primer strand so the excision reaction comes to equilibrium. The rate constant for Ap4N reincorporation is defined by k−5, the apparent equilibrium dissociation constant is K6, and k−5/K6 defines the specificity constant for reincorporation. Although the apparent Kd and maximum rate of the reaction of Ap4N are not defined, the data provide reasonable limits on the estimate of the specificity constant for reaction of Ap4N, k−5/K6. The results are summarized in Table 3.
Scheme 2.

Minimal model for ATP-mediated excision of incorporated nucleoside monophosphate.
Table 3.
HCV NS5B nucleoside and nucleoside analog ATP-mediated excision
Shown is a summary of kinetic parameters for ATP-meditated excision of incorporated nucleosides and nucleoside analogs. The rate constants were derived by fitting the data sets using Scheme 2. S.E. values were derived by nonlinear regression in globally fitting the data (19). The individual rates for the reverse reactions are not defined by the data, and therefore only the specificity constant (k−5/K6) is able to be determined.
| Nucleotide | 1/K4 | k5 | K4k5 × 10−6 | k−5/K6 |
|---|---|---|---|---|
| μm | s−1 | μm−1 s−1 | μm−1 s−1 | |
| CMP | 2800 ± 400 | 0.043 ± 0.006 | 15 ± 3 | 0.38 ± 0.2 |
| 2′C-Me-CMP | 1600 ± 67 | 0.034 ± 0.002 | 20 ± 1 | 0.47 ± 0.2 |
| 2′C-Me-2′F-CMP | 2100 ± 100 | 0.024 ± 0.002 | 12 ± 1 | 0.92 ± 0.3 |
| UMP | 0.05 ± 0.02 | |||
| 2′C-Me-2′F-UMP | ≤0.004 |
The kinetic parameters for ATP-mediated excision of CMP (Fig. 5A) were comparable to previously published results (k5 = 0.043 ± 0.006 s−1, 1/K4 = 2.8 ± 0.4 mm, K4k5 = (1.5 ± 0.3) × 10−5 µm−1 s−1). The incorporated CTP analogs were excised at slightly slower rates (k5 = 0.034 ± 0.002 s−1 and 0.024 ± 0.002 s−1 for 2′C-Me-CMP and 2′C-Me-2′F-CMP, respectively) when compared with excision of CMP. NS5B has a slightly higher apparent affinity for ATP during excision of 2′C-Me-CMP (1.6 ± 0.67 mm; Fig. 5B), but the apparent affinity remains relatively unchanged during excision of 2′C-Me-2′F-CMP (2.1 ± 0.1 mm; Fig. 5B). After incorporation, CTP and CTP analogs are readily excised via this mechanism. Therefore, the resulting specificity constants indicate that 2′C-Me-CMP is ∼1.3-fold more efficiently excised, K4k5 = (2.0 ± 0.1) × 10−5 µm−1 s−1, whereas 2′C-Me-2′F-CMP is ∼1.3-fold less efficiently excised, K4k5 = (1.2 ± 0.1) × 10−5 µm−1 s−1, compared with CMP.
Figure 5.
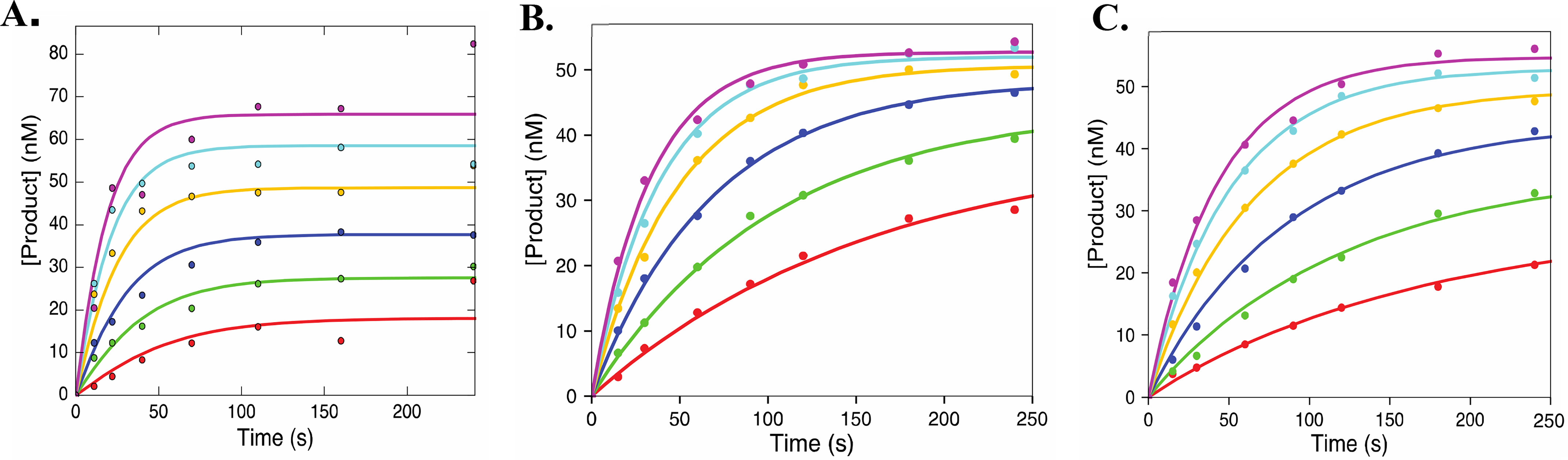
ATP-mediated excision of 2′C-modified CTP analogues. These plots show the ATP-mediated excision of CMP (A), 2′C-Me-CMP (B), and 2′C-Me-2′F-CMP (C). ATP concentrations used were 0.25, 0.5, 1, 2, 4, and 8 mm. The data were fit using Scheme 3 using KinTek Explorer. The solid lines show the best fit generated during the fitting process. Results are summarized in Table 2.
The specificity constant for the reverse of ATP-mediated excision (reaction of Ap4C; k−5/K6 = 0.38 ± 0.2 µm−1 s−1) is similar to values previously reported (5). As with the reincorporation of the Ap4C, estimates for the specificity constant for reincorporation of the Ap4C analogs can be obtained from the data. The specificity constant for the reverse of the ATP-mediated excision for 2′C-Me-CMP (k−5/K6 = 0.47 ± 0.2 µm−1 s−1) is similar to that of Ap4C. For 2′C-Me-2′F-CMP, the efficiency of reincorporation of the dinucleoside is over 2-fold higher compared with Ap4C (k−5/K6 = 0.92 ± 0.3 µm−1 s−1). This indicates that the modifications play a role in increasing the efficiency of reincorporation of the dinucleoside tetraphosphate after ATP-mediated excision.
ATP-mediated excision of UMP is much less efficient than the excision of CMP and its analogs (Fig. 6B). At the highest ATP concentrations, only ∼20% of the input NS5B/10-nt/20-nt complex was excised by the end of the time course, and saturation could not be reached. Therefore, we were only able to estimate the lower limit on the rate constant for excision to calculate the specificity constant from these data. The specificity constant was determined from the ATP concentration dependence of the rate of excision, (0.05 ± 0.02) × 10−6 µm−1 s−1. This result indicates that, as is the case with pyrophosphorolysis, the efficiency of ATP-mediated excision is greatly reduced with UMP compared with CMP. Moreover, upon attempting to measure ATP-mediated excision of 2′C-Me-2′F-UMP, no excision was observed at any concentration of ATP (Fig. 7B). We set upper limits on the rate constant for ATP-mediated excision of 2′C-Me-2′F-UMP by NS5B by estimating a lower limit for detectable product of <1% turnover by the end of the time course. This lower limit gives an estimated K4k5 ≤ 0.004 × 10−6 µm−1 s−1. Therefore, not only does the uracil base lead to resistance to ATP-mediated excision, but also the 2′C modifications further reduce the efficiency of excision. By evading ATP-dependent excision, sofosbuvir provides an effective treatment, whereas mericitabine fails because it is rapidly removed by excision.
Sequence dependence of excision of nucleoside analogs
Previous studies on various polymerases have demonstrated nucleotide incorporation and excision can be influenced by local sequence context (21–24). To survey whether certain sequences are more or less susceptible to incorporation of nucleotide analogues and ATP-mediated excision, a processive elongation assay was performed in the presence of the analogs and a high concentration of ATP. The elongation complex was assembled as indicated under “Materials and methods” using a 45-nt template to generate an NS5B/9-nt/45-nt complex. The 45-nt template was designed to contain multiple opportunities for the analogs to be incorporated. The elongation complex was then mixed with all four NTPs along with a nucleotide analog. The reactions were performed using 50 µm or 3 mm ATP. The results were fit using Scheme 3. According to the template sequence, we expected to see incorporation of the CTP analogs and chain termination at the 11-, 29-, 36-, 40-, and 44-nt position of the primer strand. Both CTP analogs were efficiently incorporated at each position except for the 29-nt position. This may indicate that there is some sequence dependence for incorporation. Nevertheless, the chain terminators were able to inhibit full extension of the primer strand (Figs. 8 and 9). By monitoring the reduction in band intensity over time, excision was observed at the 11-nt position of the primer for both CTP analogs at low ATP (Figs. 8A and 9A) and high ATP (Figs. 8B and 9B). The rate constant for excision at the 11-nt position was 0.088 ± 0.01 and 0.028 ± 0.01 s−1 for 2′C-Me-CTP and 2′C-Me-2′F-CTP, respectively. Little excision was observed at the 36-, 40-, and 44-nt positions. This may indicate that efficiency of the excision of the CTP analogs may depend somewhat on local sequence.
Scheme 3.

Minimal model for processive elongation in the presence of nucleotide analog. The above model is used to test for sequence dependence of ATP-mediated excision after NS5B chain termination. The various species are defined in the inset box.
Figure 8.

Sequence dependence of ATP-mediated 2′C-Me-CTP excision by NS5B. The processive elongation of a primer in the presence of 500 μm 2′C-Me-CTP and 50 μm ATP (A) or 3 mm ATP (B) is shown. Final concentrations for CTP, GTP, and UTP were 50 μm in each experiment. The color of the curves corresponds to nucleotide position in the primer sequence above. The solid lines represent the best fit generated during fitting using Scheme 3.
Figure 9.

Sequence dependence of ATP-mediated of 2′C-Me-2′F-CTP by NS5B. Shown is the processive elongation of a primer in the presence of 500 μm 2′C-Me-2′F-CTP and 50 μm ATP (A) or 3 mm ATP (B). Final concentrations for CTP, GTP, and UTP were 50 μm in each experiment. The color of the curves corresponds to nucleotide position in the primer sequence above. The solid lines represent the best fit generated during fitting using Scheme 3.
For 2′C-Me-2′F-UTP, we expected to observe incorporation at the 10-, 15-, 17-, 18-, 22-, 26-, 28-, 33-, and 43-nt positions of the primer strand. We observed incorporation at each position except for the 43-nt position. Regardless of the ATP concentration, after incorporation of the analog, the curves stay completely flat, indicating that no excision was occurring (Fig. 10). Fitting these results using Scheme 3 yields rate constants for excision that are essentially zero. This demonstrates that the resistance to ATP-mediated excision of the 2′-modified UMP analog appears to be universal and is not dependent on the context of the sequence.
Figure 10.

Sequence dependence of ATP-mediated of 2′C-Me-2′F-UTP by NS5B. The plots show the processive elongation of a primer in the presence of 1 mm 2′C-Me-2′F-UTP with 50 μm ATP (A) or 3 mm ATP (B). CTP, GTP, and UTP were present to a final concentration of 50 μm of each nucleoside triphosphate for both experiments. The color of each curve corresponds to the position in the primer sequence above. The solid lines represent the best fit generated during fitting using Scheme 3.
Discussion
In this study, we set out to understand the kinetic basis for NS5B inhibition by nucleoside analogs and to assess whether they are able to resist ATP-dependent excision. By isolating an active NS5B elongation complex, we were able to determine the kinetics of incorporation for CTP, UTP, and their respective analogs. NS5B discriminates against incorporation of CTP analogs between 6- and 27-fold with respect to incorporation of the correct substrate. However, there is a 140-fold discrimination against incorporation of the 2′C-Me-2′F-UTP analog when compared with incorporation of UTP. This result is ∼3-fold higher than previously reported (140-fold versus 45-fold (20)). This difference could possibly be explained by the difference in the strains used (Con1 strain versus BK strain). When comparing the differences in discrimination, one would conclude that the 2′C-modified CTP analogs would be more effective inhibitors. However, 4′-azido-CTP, which showed the lowest discrimination, allowed for slow elongation on top of the azido-CMP and is therefore not an effective chain terminator. Also, it is known from clinical trials that mericitabine, the prodrug that becomes metabolized into 2′C-Me-2′F-CTP, is less effective at treating HCV infections when compared with sofosbuvir (10, 20). Our data suggest that the effectiveness of the nucleoside analog is not determined by their kinetics of incorporation. Rather, the effectiveness of the nucleoside analog is driven by the ability to resist ATP-dependent excision after incorporation.
Pyrophosphorolysis or ATP-mediated excision reactions could be used to remove chain terminators. In our studies, pyrophosphorolysis occurs more efficiently on the natural CMP base than in the 2′C-Me-CMP analog. The 2′-fluoro modification further decreases the efficiency of pyrophosphorolysis ∼2.5-fold, indicating that the 2′C-modifications play a role in mitigating the reversal of analog incorporation. However, our data demonstrate that the efficiency of pyrophosphorolysis depends on the incorporated base. Pyrophosphorolysis of UMP is ∼245-fold less efficient than that of CMP. Furthermore, our data show that the addition of the 2′C-Me-2′F modification further lowered the efficiency of pyrophosphorolysis to a level undetectable in our assays. Therefore, our data support a model in which the efficiency of pyrophosphorolysis is determined not only by the base, but also by the modifications at the 2′-carbon of the ribose ring.
Pyrophosphorolysis and ATP-mediated excision require that the 3′ end of the RNA primer be in the active site. Following polymerization, the RNA duplex translocates to move the 3′ end out of the active site to allow binding of the next NTP. Thus, translocation will reduce the observed rate of reaction with either ATP or pyrophosphate. It is likely that pyrophosphate (or ATP) will bind only to the untranslocated state. The Kd,app for binding pyrophosphate would be attenuated by the equilibrium constant for translocation. Thus, it is likely that the identity of the base and modifications may alter the rate and the equilibrium constant for translocation. Differences in the translocation equilibrium may explain differences in reactivity in comparing CMP and UMP.
The efficiency of pyrophosphorolysis is 15–50-fold higher than ATP-mediated excision for CMP and CMP analogs and 91-fold higher for UMP. However, the average physiological concentrations of pyrophosphate and ATP are ∼3.5 µm and 3 mm, respectively (25, 26). Therefore, ATP-mediated excision will occur at a faster rate than pyrophosphorolysis under physiological conditions (Fig. 11).
Figure 11.

Comparison of rates of excision reactions under physiological conditions. The chart compares the rates of pyrophosphorolysis and ATP-mediated excision for CMP, 2′-modified CMP analogs, and UMP at physiological concentrations. Rates were calculated using 3.5 μm for pyrophosphate and 3 mm for ATP. The numbers above the bar show the calculated rates × 10−2 s−1. Error bars, S.E.
ATP-mediated excision by HIV RT plays a significant role in the emergence of AZT resistance (16, 27, 28). Our work has demonstrated that WT NS5B catalyzes the ATP-mediated excision of CMP 25-fold more efficiently than AZT-resistant variants of HIV RT (14, 16). The addition of the 2′C-Me modification slightly increased the efficiency of ATP-mediated excision compared with CMP, but the addition of the 2′C-Me-2′F modification mitigated the efficiency of ATP-mediated excision. However, ATP-mediated excision of the analogs is significantly higher when compared with the rates of ATP-mediated excision of AZT by resistant mutants of HIV RT (34-fold higher for 2′C-Me-CMP and 20-fold higher for 2′C-Me-2′F-CMP (16). In contrast, ATP-mediated excision of UMP by NS5B is incredibly inefficient. Our results demonstrated that the efficiency is approximately 10-fold lower than AZT excision by HIV RT. The further addition of the 2′C modifications further mitigated the efficiency of ATP-mediated excision to a level undetectable above background. Estimates of the upper limit of the rate constant for ATP-mediated excision of the UMP analog indicate that the efficiency is at least 130-fold lower than AZT excision by resistant HIV RT variants.
Previous studies have demonstrated that a serine to threonine mutation at the 282-position confers resistance to sofosbuvir (29, 30). However, this mutation has not been observed in any clinical isolates. A low frequency of sofosbuvir-resistant mutations emerged during clinical trials (L159F and V321A), which may have contributed to the treatment failing to mitigate the viral infection (31). These mutants required an intensified treatment to achieve sustained virologic response. Our attempts to study the effect of these mutants have been limited due to an inability to isolate NS5B variants that can make it through the inefficient de novo initiation to generate an active elongation complex. More work is needed to optimize conditions to determine whether the observed resistance is due to an increase in discrimination against incorporation or an increase in the efficiency of excision reactions.
Our work demonstrates that although 2′C-modified CTP analogs are more efficiently incorporated compared with the UTP analog, the UTP analog is a better inhibitor because of its resistance to ATP-dependent excision. Once the UTP analog is incorporated, it exhibits a t½ of greater than 23 h before ATP-mediated excision occurs (Fig. 12). The CTP analogs, however, have a t½ of 30-50 s (Fig. 12) and are therefore less efficient inhibitors under physiological conditions. This resistance is attributed to both the base itself and the additional 2′C-Me-2′F modifications.
Figure 12.

t½ of CMP, UMP, and nucleotide analogs. The chart shows the t½ of each incorporated nucleotide and analog due to ATP-mediated excision. t½ was calculated using the equation, t½ = ln(2)/k, where k is the rate of ATP-mediated excision at physiological concentrations of ATP (Fig. 11).
Materials and methods
Nucleic acids and chemicals
CTP and UTP analogs used in this study were generously provided in triphosphate form by Alios BioPharma (now Janssen Pharmaceutical, Inc.) and Gilead Sciences, respectively. The RNA templates, pGG dimer, and OHGG dimer were purchased from Dharmacon, Inc. (Chicago, IL). The sequences of the RNA templates used are shown in Table 4. The OHGG dimer was labeled with [γ-32P]ATP (PerkinElmer Life Sciences) using T4 polynucleotide kinase from New England Biolabs (Ipswich, MA) according to the protocol recommended by New England Biolabs. The reaction was stopped by incubating at 95 °C for 3 min. The radiolabeled pGG was diluted 10-fold with cold pGG to make a radiolabeled stock solution of a known concentration. Ribonucleoside triphosphates were purchased from Promega (Madison, WI). Tris-HCl buffers, NaCl, MgCl2, and EDTA solutions were purchased from Ambion (Austin, TX). Octyl β-d-glucopyranoside and DTT were purchased from GoldBio (St. Louis, MO).
Table 4.
RNA templates
The underlined base indicates the last template position before the reaction pauses during elongation complex assembly. The base in red indicates the templating base for the next incorporation.
| Template | Sequence |
|---|---|
| 20-nt CG | 3′-CCUAUAUUAGCAAUAUCUAA-5′ |
| 20-nt UA | 3′-CCUCUCUUCAGAAUAUCUAA-5′ |
| 45-nt | 3′-CCUCUCUUCAGUCUACAAUUCAUCUACAGUUCACCGUUCGCCAGU-5′ |
Expression and purification of NS5BΔ21
N-terminal hexa-His-NS5BΔ21 (Con1 strain, GT1b with the C-terminal 21-amino acid membrane anchor domain deleted) was cloned into a pcI(ts, ind+) vector (32) under control of a rightward promoter from bacteriophage λ controlled by a chemically inducible and temperature-inducible λ repressor encoded on the plasmid. Plasmids were transformed into New England Biolabs Turbo Escherichia coli cells and cultured in Terrific Broth at 30 °C overnight. One liter of medium was inoculated with the cultures grown overnight at an A600 of 0.1 and grown at 30 °C until an A600 of 4 was reached. Expression was induced by the addition of nalidixic acid to a final concentration of 50 µg/ml, and the temperature was raised to 37 °C. After 16 h, cells were collected, and the pellets were stored at −80 °C. Cells were resuspended in lysis buffer (50 mm HEPES, pH 7.5, 300 mm NaCl, 2 mm DTT, 20% (v/v) glycerol, 0.1% (w/v) octyl β-d-glucopyranoside, 20 mm imidazole, Pierce EDTA-free protease inhibitor tablets (Thermo Fisher Scientific)) and incubated with 300 µg/ml lysozyme on ice for 20 min. The lysate was sonicated for 20 min on ice using a Branson Sonifier 450 (duty cycle = 20%, output = 5) and then centrifuged at 105,000 × g for 30 min at 4 °C using a Beckman Optima LE-80K Ultracentrifuge. The supernatant was loaded onto a 5-ml HisTrap HP column (GE Healthcare) equilibrated in lysis buffer. NS5BΔ21 was eluted using a gradient of 0–100% elution buffer (50 mm HEPES, pH 7.5, 300 mm NaCl, 2 mm DTT, 20% (v/v) glycerol, 0.1% (w/v) octyl β-d-glucopyranoside, 400 mm imidazole) over 10 column volumes. Peak fractions were pooled, concentrated, and dialyzed into storage buffer (30 mm Tris-HCl, pH 7.5, 400 mm NaCl, 5 mm DTT, 20% (v/v) glycerol, and 0.1% (w/v) octyl β-d-glucopyranoside). The concentration was determined by measuring the absorbance at 280 nm using an extinction coefficient of 170,850 cm−1 m−1 (5). The protein was stored at −80 °C until use.
Assembly and isolation of elongation complex
A reaction containing 12 µm NS5B, radiolabeled 20 µm pGG, 20 µm RNA template was incubated at 30 °C for 1.5 h to form an elongation complex of NS5B with a 9-nt primer and a 20- or 45-nt template (NS5B/9-nt/20-nt or 45-nt). For the 20-nt CG template 50 µm ATP and UTP were added. For the 20-nt UA and 45-nt templates 50 µm ATP and GTP were added. Reactions were carried out in buffer containing 40 mm Tris-HCl, pH 7.0, 40 mm NaCl, 5 mm DTT, and 2 mm MgCl2. The elongation complex was isolated by centrifuging at 16,000 rpm for 5 min using a benchtop centrifuge. The supernatant was discarded, and the pellet was washed twice with wash buffer (40 mm Tris-HCl, pH 7.0, 20 mm NaCl, 5 mm DTT, and 2 mm MgCl2) to remove contaminants. The pellet was resuspended in a buffer containing 40 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm DTT, and 2 mm MgCl2.
Assembly and isolation of NS5B/10-nt/20-nt elongation complex
The reaction described above was performed to generate the NS5B/9-nt/20-nt elongation complex followed by the addition of 20 µm next correct NTP or 100 µm nucleoside analog. The reaction was incubated at 30 °C for 20 s for NTP incorporation or 5 min for nucleoside analog incorporation to generate the NS5B/10-nt/20-nt elongation complex. The elongation complex was pelleted, washed twice, and resuspended as described above.
Measurement of incorporation of nucleoside triphosphate and nucleoside analogs
The elongation complex was assembled, washed, and resuspended in a buffer containing 40 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm DTT, and 2 mm MgCl2. To measure the kinetics of NTP incorporation, resuspended elongation complex was rapidly mixed with an equal volume of solution containing NTP in the same buffer using an RQF-3 rapid-quench-flow instrument (KinTek Corp., Austin, TX). Reactions were quenched in a solution containing 50 mm EDTA and collected and stored in solution containing 50 mm EDTA, 90% formamide, 0.1% bromphenol blue, and 0.1% xylene cyanol FF. To measure the kinetics of nucleoside analog incorporation, resuspended elongation complex was mixed by hand with an equal volume of solution containing nucleoside analog in the same buffer. At given time points, aliquots of the reaction solution were transferred to a quench solution containing 50 mm EDTA, 90% formamide, 0.1% bromphenol blue, and 0.1% xylene cyanol FF. The samples were denatured by incubating at 95 °C for 5 min and loaded onto 16% denaturing polyacrylamide gel containing 7 m urea. Electrophoresis was performed at 100 W and 50 °C using the Bio-Rad Sequi-Gen GT System. Gels were dried at 80 °C for 1 h using a Bio-Rad model 583 Gel Dryer and exposed to a storage phosphor screen. The screen was imaged using a GE Healthcare Typhoon 9400 scanner. Band intensities were quantified using ImageQuant (GE Healthcare). Product formation was calculated as the fractional intensity of each product band relative to the total intensity of all bands in the given lane.
Pyrophosphorolysis and ATP-mediated excision
The NS5B/10-nt/20-nt elongation complex was generated, pelleted, and resuspended in buffer as described above. The elongation complex was mixed with an equal volume of solution containing sodium pyrophosphate or ATP and incubated at 30 °C. For a given time point, an aliquot was removed from the reaction and quenched in solution containing 50 mm EDTA, 90% formamide, 0.1% bromphenol blue, and 0.1% xylene cyanol FF. The samples were denatured by incubating at 95 °C for 5 min and loaded onto a 16% denaturing polyacrylamide gel containing 7 m urea. Electrophoresis, drying, exposure, and quantification were conducted as described in the previous section.
Processive elongation with nucleoside analogs
The NS5B/9-nt/45-nt elongation complex was generated, pelleted, and resuspended in buffer as described above. The elongation complex was mixed with an equal volume of solution containing 50 µm CTP, GTP, UTP, and ATP, and either 500 µm 2′-modified CTP analog or 1 mm 2′F-2′CMe-UTP in the same buffer and allowed to react at 30 °C. To test for ATP-mediated excision, the elongation complex was generated, pelleted, and resuspended in buffer as described previously. It was then mixed with an equal volume of solution containing 50 µm CTP, GTP, and UTP, 3 mm ATP and either 500 µm 2′-modified CTP analog or 1 mm 2′F-2′CMe-UTP in the same buffer and allowed to react at 30 °C. For each given time point, an aliquot was removed from the reaction and quenched in a solution containing 50 mm EDTA, 90% formamide, 0.1% bromphenol blue, and 0.1% xylene cyanol FF. The samples were heat-denatured and loaded onto a 16% denaturing polyacrylamide gel with 7 m urea. Electrophoresis, drying, exposure, and quantification were conducted as described in previous sections. The concentrations of each nt position over time were plotted versus time in Microsoft Excel.
Data analysis
The kinetics of incorporation, pyrophosphorolysis, and ATP-mediated excision were fit using KinTek Explorer (KinTek Corp.) to determine the rate constants of the respective reactions as described previously (19, 33). The kinetics of incorporation were determined by fitting using Scheme 1. To fit the data to determine the Kd,app for NTP binding, the rate constant for NTP or nucleotide analog binding (k1) was assumed to be close to diffusion-limited and locked at 100 µm−1 s−1, and the rate constant for dissociation (k-1) was allowed to vary during fitting. The apparent equilibrium dissociation constant was then calculated by dividing the rate constant for dissociation by the rate constant for binding (Kd,app = 1/K1 = k−1/k1). The maximum rate constant for polymerization (kpol) afforded definition of the rate constant k2. Because the amount of pyrophosphate produced during the reaction is low relative to nucleotide, the reaction is largely irreversible; therefore, k−2 was locked at 0. Pyrophosphate release is fast and not rate-limiting, so the k3 is locked at 100 s−1. The specificity constant (kcat/Km = kpol/Kd,app) was determined by dividing the rate constant for polymerization by the Kd,app for nucleotide binding (k2/Kd,app = K1k2).
The kinetics of pyrophosphorolysis were determined by fitting using the reverse reactions shown in Scheme 1. Pyrophosphorolysis reactions were globally fit, including incorporation reactions, to account for the amplitude dependence observed. The rate constant for pyrophosphate binding (k−3) was assumed to be diffusion-limited and locked at 100 µm−1 s−1. The rate constants for pyrophosphate dissociation (k3) and pyrophosphorolysis (k−2) were allowed to vary during fitting. The Kd,app for pyrophosphate was determined by dividing the rate constant for pyrophosphate release by the rate constant for binding (Kd,app = K3 = k3/k−3). The specificity constant for pyrophosphorolysis (kcat/Km) was determined by dividing the rate constant for pyrophosphorolysis by the Kd,app for pyrophosphate binding (k−2/K3).
The kinetics of ATP-mediated excision were determined by fitting using Scheme 2. To determine the apparent equilibrium dissociation constant for ATP (Kd,app), the rate constant for ATP binding (k4) was assumed to be diffusion-limited and locked at 100 µm−1 s−1. The rate constant for ATP dissociation (k−4) was allowed to vary during the fitting process. The rate constant for the dissociation of ATP was then divided by the rate constant for binding to determine the Kd,app (1/K4 = k−4/k4). The maximum rate constant for ATP-mediated excision was determined by allowing k5 to vary during the fitting process. The specificity constant was determined by multiplying k5 by 1/Kd,app (kcat/Km = K4k5). The rates for the reverse of ATP-mediated excision (k−5) and Ap4N release (k6) were linked at a constant ratio during fitting. Due to the low concentration of Ap4N, the rate constant for Ap4N rebinding (k−6) was assumed to be slow and locked at 1 µm−1 s−1.
For the processive elongation in the presence of nucleotide analog, the concentration of each chain-terminated primer position was plotted versus time in KinTek Explorer. The curves for each primer position were fit by simulation using the processive elongation model in Scheme 3. This model accounts for NTP incorporation (E.Rn → E.Rn+1), chain termination due to nucleotide analog incorporation (E.Rn → E.RX), and possible ATP-mediated excision leading to RNA synthesis rescue (E.RX → E.Rn.Ap4NA → E.Rn).
Acknowledgments
2′C-Me-CTP, 2′C-Me-2′F-CTP, and 4′-azido-CTP were provided by Janssen Pharmaceutical, Inc. 2′C-Me-2′F-UTP was provided by Gilead Sciences.
Author contributions—B. V. and J. L. conceptualization; B. V. and J. L. data curation; B. V. and J. L. formal analysis; B. V. and J. L. investigation; B. V. writing-original draft; K. A. J. resources; K. A. J. supervision; K. A. J. validation; K. A. J. project administration; K. A. J. writing-review and editing.
Funding and additional information—This work was supported by grants from NIAID, National Institutes of Health, Grant 1R01AI110577 (to K. A. J.) and Welch Foundation Grant F-1604 (to K. A. J.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—K. A. J. is president of KinTek Corporation, which provided the RQF-3 rapid quench-flow instrument and KinTek Explorer software used in this study.
- HCV
- hepatitis C virus
- NS5B
- nonstructural protein 5B
- nt
- nucleotide
- Ap4N
- adenine dinucleoside tetraphosphate
- AZT
- azidothymidine.
References
- 1. Hofmeister M. G., Rosenthal E. M., Barker L. K., Rosenber E. S., Barranco M. A., Hall E. W., Edlin B. R., Mermin J., Ward J. W., and Ryerson A. B. (2019) Estimating prevalence of hepatitis C virus infection in the United States, 2013-2016. Hepatology 69, 1020–1031 10.1002/hep.30297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shepard C. W., Finelli L., and Alter M. J. (2005) Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5, 558–567 10.1016/S1473-3099(05)70216-4 [DOI] [PubMed] [Google Scholar]
- 3. Chak E., Talal A. H., Sherman K. E., Schiff E. R., and Saab S. (2011) Hepatitis C virus infection in U.S.A.: an estimate of true prevalence. Liver Int. 31, 1090–1101 10.1111/j.1478-3231.2011.02494.x [DOI] [PubMed] [Google Scholar]
- 4. Ashfaq U. A., Javed T., Rehman S., Nawaz Z., and Riazuddin S. (2011) An overview of HCV molecular biology, replication and immune responses. Virol J. 8, 161 10.1186/1743-422X-8-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jin Z., Leveque V., Ma H., Johnson K. A., and Klumpp K. (2012) Assembly, purification, and pre-steady-state kinetic analysis of active RNA-dependent RNA polymerase elongation complex. J. Biol. Chem. 287, 10674–10683 10.1074/jbc.M111.325530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li J., and Johnson K. A. (2016) Thumb site 2 inhibitors of hepatitis C viral RNA-dependent RNA polymerase allosterically block the transition from initiation to elongation. J. Biol. Chem. 291, 10067–10077 10.1074/jbc.M115.708354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sofia M. J., Bao D., Chang W., Du J., Nagarathnam D., Rachakonda S., Reddy P. G., Ross B. S., Wang P., Zhang H.-R., Bansal S., Espiritu C., Keilman M., Lam A. M., Micolochick Steuer H. M., et al. (2010) Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53, 7202–7218 10.1021/jm100863x [DOI] [PubMed] [Google Scholar]
- 8. Sofia M. J., Chang W., Furman P. A., Mosley R. T., and Ross B. S. (2012) Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J. Med. Chem. 55, 2481–2531 10.1021/jm201384j [DOI] [PubMed] [Google Scholar]
- 9. Götte M., and Feld J. J. (2016) Direct-acting antiviral agents for hepatitis C: structural and mechanistic insights. Nat. Rev. Gastroenterol. Hepatol. 13, 338–351 10.1038/nrgastro.2016.60 [DOI] [PubMed] [Google Scholar]
- 10. Eltahla A. A., Luciani F., White P. A., Lloyd A. R., and Bull R. A. (2015) Inhibitors of the hepatitis C virus polymerase; mode of action and resistance. Viruses 7, 5206–5224 10.3390/v7102868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Appleby T. C., Perry J. K., Murakami E., Barauskas O., Feng J., Aesop C., Fox D. 3rd, Wetmore D. R., McGrath M. E., Ray A. S., Sofia M. J., Swaminathan S., and Edwards T. E. (2015) Structural basis for RNA replication by the hepatitis C virus polymerase. Science 347, 771–775 10.1126/science.1259210 [DOI] [PubMed] [Google Scholar]
- 12. Murakami E., Bao H., Ramesh M., McBrayer T. R., Whitaker T., Micolochick Steuer H. M., Schinazi R. F., Stuyver L. J., Obikhod A., Otto M. J., and Furman P. A. (2007) Mechanism of activation of β-D-2-deoxy-2-fluoro-2-C-methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob. Agents Chemother. 51, 503–509 10.1128/AAC.00400-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. German P., Mathias A., Brainard D., and Kearney B. P. (2016) Clinical pharmacokinetics and pharmacodynamics of ledipasvir/sofosbuvir, a fixed-dose combination tablet for the treatment of hepatitis C. Clin. Pharmacokinet. 55, 1337–1351 10.1007/s40262-016-0397-0 [DOI] [PubMed] [Google Scholar]
- 14. Jin Z., Leveque V., Ma H., Johnson K. A., and Klumpp K. (2013) NTP-mediated nucleotide excision activity of hepatitis c virus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 110, E348–E357 10.1073/pnas.1214924110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boyer P. L., Sarafianos S. G., Arnold E., and Hughes S. H. (2002) Nucleoside analog resistance caused by insertions in the fingers of human immunodeficiency virus type 1 reverse transcriptase involves ATP-mediated excision. J. Virol. 76, 9143–9151 10.1128/jvi.76.18.9143-9151.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer P. R., Matsuura S. E., Mian A. M., So A. G., and Scott W. A. (1999) A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell 4, 35–43 10.1016/S1097-2765(00)80185-9 [DOI] [PubMed] [Google Scholar]
- 17. Sluis-Cremer N., Sheen C.-W., Zelina S., Argoti Torres P. S., Parikh U. M., and Mellors J. W. (2007) Molecular mechanism by which the K70E mutation in human immunodeficiency virus type 1 reverse transcriptase confers resistance to nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 51, 48–53 10.1128/AAC.00683-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tu X., Das K., Han Q., Bauman J. D., Clark A. D. Jr., Hou X., Frenkel Y. V., Gaffney B. L., Jones R. A., Boyer P. L., Hughes S. H., Sarafianos S. G., and Arnold E. (2010) Structural basis of HIV-1 resistance to AZT by excision. Nat. Struct. Mol. Biol. 17, 1202–1209 10.1038/nsmb.1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson K. A., Simpson Z. B., and Blom T. (2009) Global Kinetic Explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 10.1016/j.ab.2008.12.024 [DOI] [PubMed] [Google Scholar]
- 20. Fung A., Jin Z., Dyatkina N., Wang G., Beigelman L., and Deval J. (2014) Efficiency of incorporation and chain termination determines the inhibition potency of 2′-modified nucleotide analogs against hepatitis C virus polymerase. Antimicrob. Agents Chemother. 58, 3636–3645 10.1128/AAC.02666-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bloom L. B., Otto M. R., Eritja R., Reha-Krantz L. J., Goodman M. F., and Beechem J. (1994) Pre-steady-state kinetic analysis of sequence-dependent nucleotide excision by the 3′-exonuclease activity of bacteriophage T4 DNA polymerase. Biochemistry 33, 7576–7586 10.1021/bi00190a010 [DOI] [PubMed] [Google Scholar]
- 22. Mendelman L. V., Boosalis M. S., Petruska J., and Goodman M. F. (1989) Nearest neighbor influences on DNA polymerase insertion fidelity. J. Biol. Chem. 264, 14415–14423 [PubMed] [Google Scholar]
- 23. Pless R. C., and Bessman M. J. (1983) Influence of local nucleotide sequence on substitution of 2-aminopurine for adenine during deoxyribonucleic acid synthesis in vitro. Biochemistry 22, 4905–4915 10.1021/bi00290a006 [DOI] [PubMed] [Google Scholar]
- 24. Petruska J., and Goodman M. F. (1985) Influence of neighboring bases on DNA polymerase insertion and proofreading fidelity. J. Biol. Chem. 260, 7533–7539 [PubMed] [Google Scholar]
- 25. Ryan L. M., Kozin F., and McCarty D. J. (1979) Quantification of human plasma inorganic pyrophosphate. Arthritis Rheum. 22, 886–891 10.1002/art.1780220812 [DOI] [PubMed] [Google Scholar]
- 26. Traut T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 10.1007/BF00928361 [DOI] [PubMed] [Google Scholar]
- 27. Meyer P. R., Matsuura S. E., So A. G., and Scott W. A. (1998) Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 95, 13471–13476 10.1073/pnas.95.23.13471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ray A. S., Murakami E., Basavapathruni A., Vaccaro J. A., Ulrich D., Chu C. K., Schinazi R. F., and Anderson K. S. (2003) Probing the molecular mechanisms of AZT drug resistance mediated by HIV-1 reverse transcriptase using a transient kinetic analysis. Biochemistry 42, 8831–8841 10.1021/bi034435l [DOI] [PubMed] [Google Scholar]
- 29. Migliaccio G., Tomassini J. E., Carroll S. S., Tomei L., Altamura S., Bhat B., Bartholomew L., Bosserman M. R., Ceccacci A., Colwell L. F., Cortese R., de Francesco R., Eldrup A. B., Getty K. L., Hou X. S., et al. (2003) Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 278, 49164–49170 10.1074/jbc.M305041200 [DOI] [PubMed] [Google Scholar]
- 30. Svarovskaia E. S., Dvory-Sobol H., Parkin N., Hebner C., Gontcharova V., Martin R., Ouyang W., Han B., Xu S., Ku K., Chiu S., Gane E., Jacobson I. M., Nelson D. R., Lawitz E., et al. (2014) Infrequent development of resistance in genotype 1–6 hepatitis C virus–infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin. Infect. Dis. 59, 1666–1674 10.1093/cid/ciu697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Svarovskaia E. S., Gane E., Dvory-Sobol H., Martin R., Doehle B., Hedskog C., Jacobson I. M., Nelson D. R., Lawitz E., Brainard D. M., McHutchison J. G., Miller M. D., and Mo H. (2016) L159F and V321A sofosbuvir-associated hepatitis C virus NS5B substitutions. J. Infect. Dis. 213, 1240–1247 10.1093/infdis/jiv564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brandis J. W., and Johnson K. A. (2009) High-cell density shake-flask expression and rapid purification of the large fragment of Thermus aquaticus DNA polymerase I using a new chemically and temperature inducible expression plasmid in Escherichia coli. Protein Expr. Purif. 63, 120–127 10.1016/j.pep.2008.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnson K. A., Simpson Z. B., and Blom T. (2009) FitSpace Explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 10.1016/j.ab.2008.12.025 [DOI] [PubMed] [Google Scholar]