

Abstract
Transmembrane proteins are membrane-anchored proteins whose topologies are important for their functions. These properties enable regulation of certain transmembrane proteins by regulated intramembrane proteolysis (RIP) and regulated alternative translocation (RAT). RIP enables a protein fragment of a transmembrane precursor to function at a new location, and RAT leads to an inverted topology of a transmembrane protein by altering the direction of its translocation across membranes during translation. RIP mediated by site-1 protease (S1P) and site-2 protease (S2P) is involved in proteolytic activation of membrane-bound transcription factors. In resting cells, these transcription factors remain in the endoplasmic reticulum (ER) as inactive transmembrane precursors. Upon stimulation by signals within the ER, they are translocated from the ER to the Golgi. There, they are cleaved first by S1P and then by S2P, liberating their N-terminal domains from membranes and enabling them to activate genes in the nucleus. This signaling pathway regulates lipid metabolism, unfolded protein responses, secretion of extracellular matrix proteins, and cell proliferation. Remarkably, ceramide-induced RIP of cAMP response element–binding protein 3–like 1 (CREB3L1) also involves RAT. In resting cells, RIP of CREB3L1 is blocked by transmembrane 4 L6 family member 20 (TM4SF20). Ceramide inverts the orientation of newly synthesized TM4SF20 in membranes through RAT, converting TM4SF20 from an inhibitor to an activator of RIP of CREB3L1. Here, I review recent insights into RIP of membrane-bound transcription factors, focusing on CREB3L1 activation through both RIP and RAT, and discuss current open questions about these two signaling pathways.
Keywords: ceramide, endoplasmic reticulum, Golgi, protein translocation, proteolysis, transcription factor, transmembrane domain, transport, RIP, RAT, endoplasmic reticulum (ER), RAT, topology
Regulated intramembrane proteolysis (RIP) is a signal transduction mechanism that generates regulatory molecules from transmembrane proteins through proteolysis. Such cleavage liberates cytoplasmic domains from transmembrane precursors, allowing the cleaved fragments to function at a new location (1, 2). RIP is catalyzed by four different families of proteases that cleave within a transmembrane domain: site-2 protease (S2P), γ-secretase, signal peptide peptidase, and rhomboid (2). Except for rhomboid, the intramembrane proteolysis catalyzed by these proteases does not occur until the bulk of the protein on the extracytoplasmic (lumenal or extracellular) side is removed by a primary cleavage catalyzed by a different protease (2–6). RIP is known to activate hundreds of transmembrane proteins, influencing processes as diverse as cellular differentiation, lipid metabolism, and immune defense (2). Among these proteins, a family of membrane–bound transcription factors are activated by S2P in mammalian cells to transmit signals from the endoplasmic reticulum (ER) to the nucleus to regulate gene expression
ER is the subcellular organelle where transmembrane and secretory proteins are produced (7) and the majority of lipids are synthesized (8). To carry out these functions properly, ER must communicate with the nucleus to ensure that genes required for these functions are expressed at appropriate levels. One of the mechanisms to transmit signals from the ER to nucleus is through proteolytic activation of membrane-bound transcription factors. These transcription factors are produced as inactive membrane-bound precursors in the ER. After stimulation by specific signals generated from the ER, they are transported from the ER to Golgi, where their luminal domains are cleaved by site-1 protease (S1P). This cleavage enables a second cleavage catalyzed by S2P, which liberates their N-terminal transcription activation domain from membranes, allowing it to enter the nucleus, where it activates target genes (9–13) (Fig. 1). All of these membrane-bound transcription factors share the following features: 1) they contain an N-terminal domain capable of functioning as a transcription factor; 2) they contain an S1P recognition motif RXX(R/L) in the lumen in which the last amino acid of the motif is the cleavage site for S1P (14, 15); and 3) they contain a helical destabilization motif required for S2P-catalyzed cleavage, such as NP, NXXP, or PXXP sequence in the transmembrane helix C-terminal to the cleavage site (13, 16, 17). However, RIP of these transcription factors is stimulated by different signals generated in the ER to activate their unique set of target genes.
Figure 1.
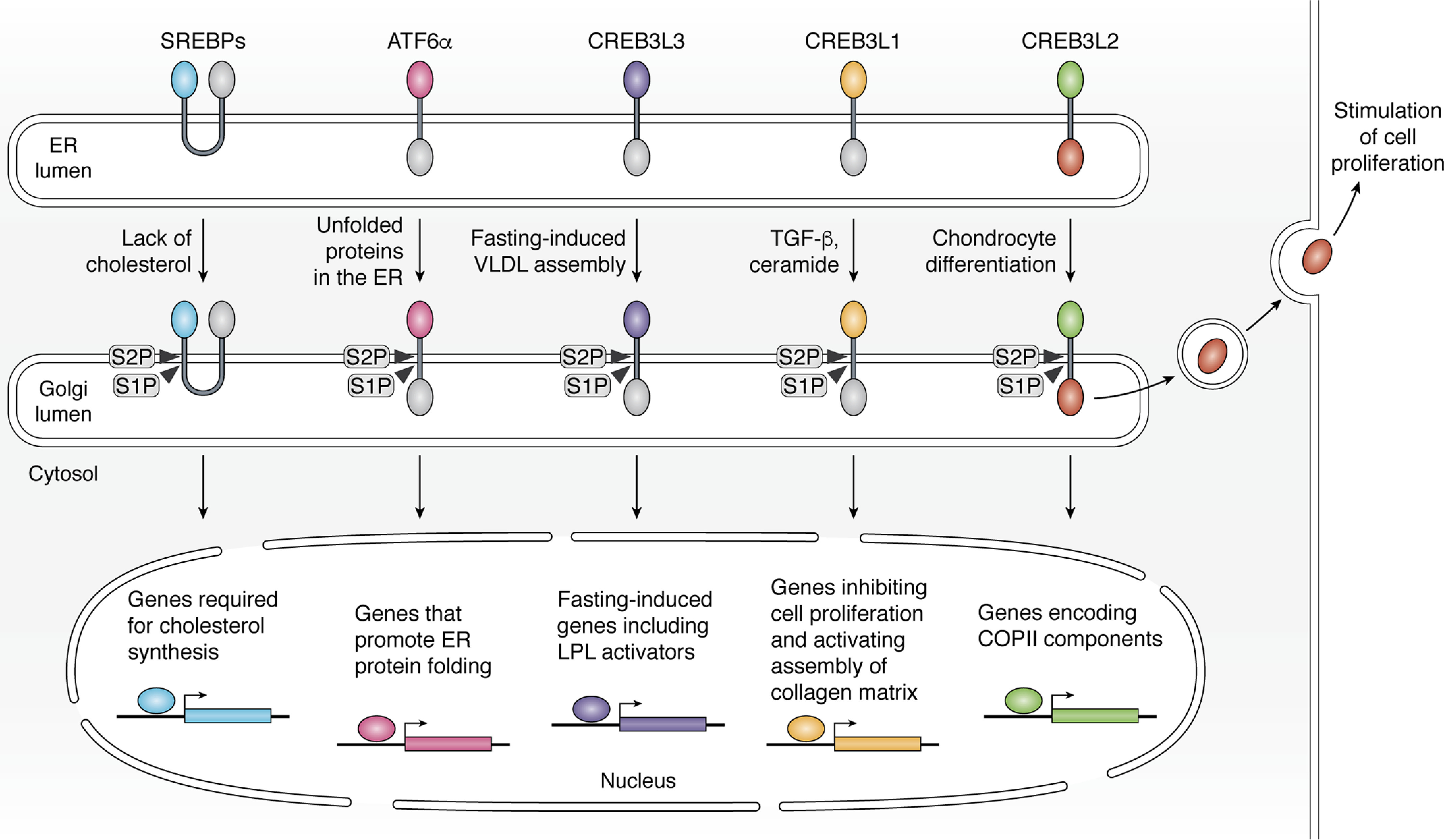
Graphic illustration of RIP mediated by S1P and S2P. Cholesterol deprivation triggers RIP of SREBP to activate genes required for cholesterol synthesis and uptake. Accumulation of unfolded proteins in the ER stimulates RIP of ATF6α to activate genes facilitating protein folding in the ER. Fasting-induced VLDL assembly triggers RIP of CREB3L3 to activate fasting-induced genes, including activators for LPL that hydrolyze VLDL particles. Cytokines of the TGF-β family and ceramide induce RIP of CREB3L1 to stimulate expression of genes that inhibit cell proliferation and that activate assembly of collagen-containing matrix. Chondrocyte differentiation stimulates RIP of CREB3L2 to activate genes encoding COPII components. The luminal C-terminal fragment of CREB3L2 released from S1P-catalyzed cleavage is secreted out of cells to stimulate proliferation of neighboring cells.
Remarkably, recent studies on RIP of cAMP response element–binding protein 3–like 1 (CREB3L1), one of the membrane-bound transcription factors activated by S1P/S2P, revealed another mechanism that regulates transmembrane proteins. This mechanism, which is designated as regulated alternative translocation (RAT), leads to inverted topology of newly synthesized transmembrane 4 L6 family member 20 (TM4SF20), a polytopic transmembrane protein. This topological inversion turns TM4SF20 from an inhibitor to an activator for RIP of CREB3L1 (18).
In this review, I will first provide an overview on several membrane-bound transcription factors activated by S1P/S2P to illustrate how RIP transmits signals from the ER to the nucleus to regulate gene expression. I will then focus on CREB3L1 as an example to illustrate how the membrane-bound transcription factor is activated through RIP and RAT. Finally, I will discuss questions that need to be resolved to understand these signaling pathways more thoroughly.
Transcription factors activated through RIP
SREBPs
Sterol regulatory element–binding proteins (SREBPs) are the master regulators that control cholesterol and fatty acid homeostasis in mammalian cells (19–21). Whereas SREBP-1a and SREBP-1c are the products of one gene generated from different promoters, SREBP-2 is encoded by a different gene. Unlike any other transcription factors activated by S1P/S2P that contain a single transmembrane helix, SREBPs are inserted into membranes with two transmembrane helices in a hairpin fashion, with both the N- and C-terminal ends facing cytosol (Fig. 1). The N-terminal domains of SREBPs are basic helix-loop-helix-leucine zipper transcription factors, whereas the C-terminal domains mediate association with a membrane protein called SREBP cleavage–activating protein (SCAP) (22) (Fig. 2A). In cells depleted of cholesterol, SCAP is incorporated into COPII-coated vesicles, the vehicle that transports proteins from ER to Golgi (23) (Fig. 2A). The translocation of the SCAP/SREBPs complex from ER to Golgi allows SREBPs to be cleaved by S1P followed by S2P (11, 24, 25) (Fig. 1). These cleavages liberate the N-terminal domains of SREBPs from membranes, allowing them to enter the nucleus, where they activate transcription of all genes required for cholesterol synthesis and uptake (26) (Fig. 1). In cells loaded with cholesterol, the sterol is accumulated in ER membranes. Upon exceeding 5% of total ER lipids (molar basis), cholesterol in the ER binds to SCAP (27–29), a reaction causing association of SCAP with insulin-induced gene (INSIG) proteins, a family of transmembrane proteins localized in the ER (30, 31) (Fig. 2A). This binding blocks the interaction between SCAP and components of the COPII coat. Consequently, the SCAP/SREBPs complex is retained in the ER (Fig. 2A), making SREBPs inaccessible to cleavage catalyzed by Golgi-localized S1P and S2P (23, 30, 32). As a result, transcription of the genes required for cholesterol synthesis and uptake declines. Thus, the cholesterol-mediated inhibition of RIP of SREBPs plays a critical role in feedback inhibition of cholesterol synthesis and uptake (33).
Figure 2.
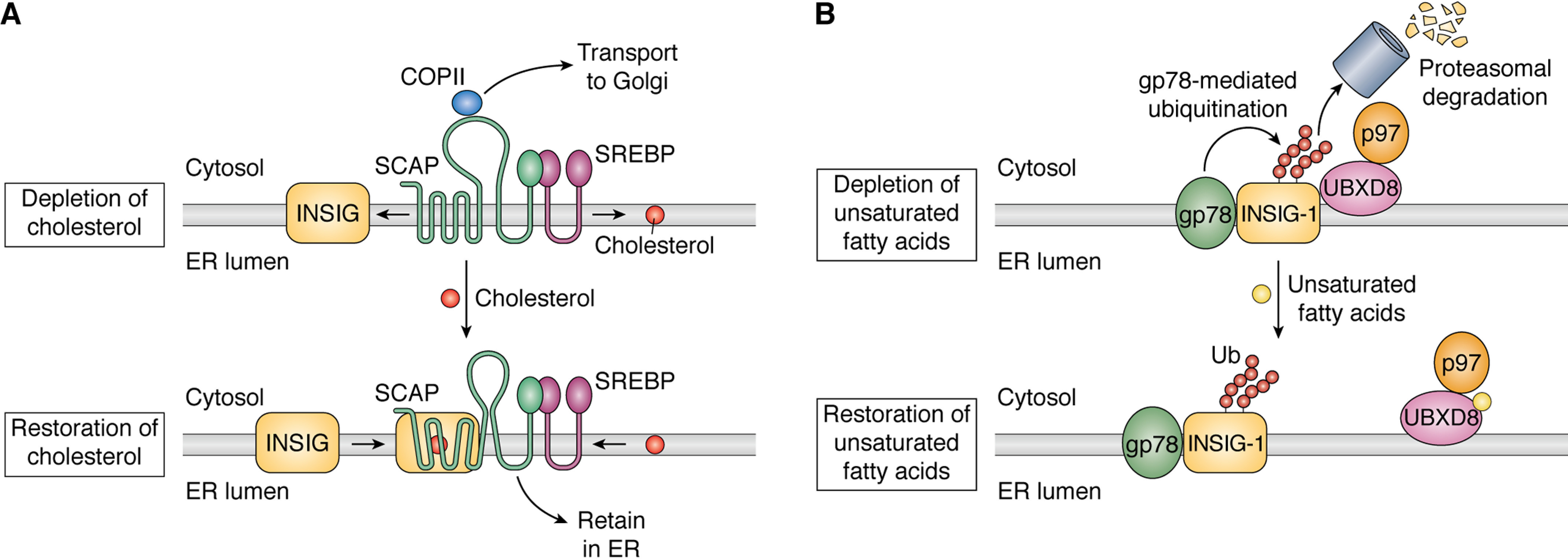
Cholesterol and unsaturated fatty acid–regulated RIP of SREBP. A, in cholesterol-depleted cells, SCAP is dissociated from INSIG proteins and cholesterol. SCAP under this condition interacts with components of COPII, allowing the SCAP/SREBP complex to be incorporated into COPII-coated vesicles so that SREBP is delivered to Golgi for proteolytic activation by S1P and S2P. In cells replete with cholesterol, interaction with cholesterol causes SCAP to bind INSIG proteins. This interaction leads to a conformational change in SCAP that inhibits its incorporation into the COPII-coated vesicles. The SCAP/SREBP complex is thus retained in the ER, preventing proteolytic activation of SREBP. B, in cells depleted of unsaturated fatty acids, INSIG-1 is ubiquitinated by gp78 and interacts with UBXD8, a p97-associated protein. The ubiquitination and p97 recruitment cause INSIG-1 to be rapidly degraded by proteasomes. In cells replete with unsaturated fatty acids, the fatty acids bind to UBXD8, leading to dissociation of UBXD8 from INSIG-1. In the absence of UBXD8-mediated recruitment of p97, INSIG-1 is stabilized, making cholesterol more effective in inhibiting proteolytic activation of SREBP.
In addition to genes involved in cholesterol synthesis and uptake, SREBPs, particularly SREBP-1a and SREBP-1c, also activate genes involved in synthesis of unsaturated fatty acids (26). Thus, unsaturated fatty acids also exert feedback inhibition on SREBP-1a and SREBP-1c. In mice, expression of SREBP-1c, the predominant isoform of SREBP-1 expressed in liver, is driven by liver X receptor (LXR), a ligand-activated nuclear receptor (34). Polyunsaturated fatty acids inhibit expression of SREBP-1c by acting as an antagonist to LXR (35, 36). Unsaturated fatty acids also inhibit cleavage of SREBP-1a in cultured cells (37). In cells depleted of fatty acids, INSIG-1, the predominant isoform of INSIG proteins expressed in cultured cells, is ubiquitinated by gp78 (38) and binds to ubiquitin regulatory X domain–containing protein 8 (UBXD8), a membrane protein that associates with the ER-associated degradation (ERAD) co-factor p97 (39) (Fig. 2B). Ubiquitination and p97 recruitment causes rapid degradation of INSIG-1 by proteasomes through ERAD in these cells (39) (Fig. 2B). Unsaturated fatty acids directly bind to UBXD8, causing dissociation of UBXD8 from INSIG-1 (39, 40) (Fig. 2B). The resultant stabilization of INSIG-1 leads to increased retention of the SCAP/SREBPs complex in the ER, making cholesterol more effective in inhibiting cleavage of SREBP-1a in cells loaded with unsaturated fatty acids (39).
ATF6
Between the two isoforms of ATF6, ATF6α is much better characterized. In resting cells, ATF6α remains as an inactive transmembrane precursor in the ER. Upon accumulation of unfolded proteins in the ER lumen, ATF6α is incorporated into COPII-coated vesicles and translocated from the ER to the Golgi (12, 41), where it is cleaved by S1P followed by S2P (13) (Fig. 1). These cleavages release the N-terminal domain of ATF6α from membranes, allowing it to enter the nucleus, where it activates transcription of BiP and other genes whose products assist folding of ER proteins (42, 43). Thus, RIP of ATF6α has been established as one of the three signaling pathways responsive to ER stress (44).
In addition to unfolded proteins accumulated in the ER lumen, RIP of ATF6α can also be triggered by signals generated in ER membranes. RIP of ATF6α was stimulated by overexpression of a tail-anchored transmembrane protein without any luminal domain or accumulation of dihydroceramide in ER membranes. Interestingly, these treatments only induced proteolytic activation of ATF6α but not the other two branches of the ER stress response, namely splicing of X-box–binding protein 1 and activation of dsRNA-dependent protein kinase-like ER kinase (45, 46). A mutation in the transmembrane helix of ATF6α impaired dihydroceramide-induced RIP of the protein but not that caused by accumulation of unfolded proteins in the ER lumen. In contrast, a mutation in the luminal domain of ATF6α abolished RIP of the protein induced by unfolded proteins in the ER lumen but not that triggered by dihydroceramide (46). These observations suggest that signals generated in the ER lumen and membrane are independently sensed by the luminal and transmembrane domain of ATF6α, respectively.
In contrast to ATF6α, ATF6β is much less well-characterized. Whereas ER stress also induces RIP of ATF6β in cultured cells, the cleaved nuclear form of the protein does not activate transcription of ER chaperons. Instead, it inhibits transcription of these genes by antagonizing the activity of the cleaved nuclear form of ATF6α (47). In a mouse model of metaphyseal chondrodysplasia type Schmid caused by expression of a misfolded mutant type X collagen in chondrocytes, the severity of the disease was increased by knockout of ATF6α but decreased by ablation of ATF6β (48). These results suggest that ATF6β may serve as a transcriptional repressor to antagonize the strength and duration of ATF6α activity that protects cells from ER stress. However, RIP of ATF6β in mouse cardiomyocytes triggered by hemodynamic stress induced expression of genes overlapping with that induced by ATF6α including ER chaperones (49). Thus, ATF6β may have different functions in different tissues.
CREB3L3
CREB3L3, also known as CREB-H, is primarily expressed in livers and intestines. Earlier studies demonstrated that proinflammatory cytokines and lipopolysaccharide (LPS) triggered RIP of CREB3L3 in hepatocytes to induce expression of acute phase response genes such as C-reactive protein and serum amyloid P-component (50). Recent studies have focused on the metabolic consequence of CREB3L3 activation. Expression and RIP of CREB3L3 in mice is stimulated by fasting (51, 52). During fasting, fatty acids released from adipocytes enter livers, causing hepatic accumulation of triglycerides (TGs). These TGs then enter the ER lumen, where they are packaged into very low-density lipoprotein (VLDL) particles. The nascent VLDL particles produced in the ER lumen are transported to Golgi and secreted out of hepatocytes through exocytosis as a vehicle that delivers lipids to peripheral tissues (53). It appears that VLDL assembly in the ER lumen is the signal to trigger RIP of CREB3L3 (Fig. 1), as treatments inhibiting this reaction blocked fasting-induced proteolytic activation of the protein (54). Because nascent VLDL particles are transported from the ER to Golgi by specialized vesicles (55), it will be interesting to determine whether CREB3L3 is co-delivered with VLDL by these vesicles to Golgi for proteolytic activation by S1P/S2P.
Once cleaved by S1P and S2P, the N-terminal domain of CREB3L3 activates genes known to be activated during fasting (Fig. 1). These genes include fibroblast growth factor 21, a fasting-induced hormone that stimulates hepatic fatty acid oxidation and ketogenesis (56); Insig-2, the Insig isoform expressed in fasting livers (51); and genes required for gluconeogenesis (57). More importantly, VLDL assembly–triggered RIP of CREB3L3 coordinates secretion of VLDL with hydrolysis of the lipoprotein particles in circulation. To use fatty acids stored in VLDL by peripheral tissues, TGs stored in VLDL must be hydrolyzed by lipoprotein lipase (LPL) so that fatty acids can be released from the lipoprotein particle (58). Following activation through RIP, the N-terminal domain of CREB3L3 stimulates expression of apolipoproteins such as apoA2, apoA4, and apoA5 that activate LPL activity (59, 60). Consequently, CREB3L3 deficiency in both mice and humans leads to development of hypertriglyceridemia caused by insufficient LPL activity (59).
CREB3L2
CREB3L2, also known as BBF2H7, is best characterized in chondrocytes. During chondrocyte differentiation, CREB3L2 is cleaved by S1P. This cleavage has two consequences: 1) like other transcription factors activated through RIP, it produces the membrane-bound N-terminal fragment of CREB3L2 as a substrate for S2P. The S2P-catalyzed cleavage releases the N-terminal domain of CREB3L2 from membranes so that it can enter the nucleus to stimulate transcription of genes encoding components of the COPII coat, such as various isoforms of Sec23 and Sec24 (61) (Fig. 1). Expression of these genes enlarges COPII-coated vesicles to accommodate the bulky type II collagen, the secretion of which is crucial for differentiation of chondrocytes (61, 62); 2) the S1P-catalyzed cleavage releases the C-terminal domain of the protein from membranes, allowing it to be secreted out of the cells to stimulate proliferation of neighboring chondrocytes by activating the hedgehog signaling pathway (63) (Fig. 1). Thus, the N- and C-terminal fragments of CREB3L2 generated through RIP simultaneously stimulate chondrocyte differentiation and proliferation in developing cartilage, respectively. As a result, mice deficient in CREB3L2 developed severe chondrodysplasia (61).
CREB3L2 is frequently fused with fused in sarcoma (FUS) in low-grade fibromyxoid sarcoma (LGFMS), a malignant soft tissue tumor through chromosome rearrangement (64). In these tumors, the N-terminal domain of FUS is fused in frame with CREB3L2 at a position N-terminal to its DNA-binding domain (65). Because overexpression of transcription factors activated by RIP leads to unregulated proteolytic activation presumably by overpowering the regulatory machinery that retains them in the ER (13, 66), the FUS-CREB3L2 fusion protein driven by the strong FUS promoter is likely to be constitutively cleaved. The cleaved N-terminal domain of CREB3L2 fused with FUS does not activate target genes of CREB3L2. Instead, the fusion protein activates CD24, a marker for LGFMS, the function of which in tumor development remains obscure (67). Inasmuch as expression of genes downstream of hedgehog signaling is elevated in LGFMS (67), the increased secretion of the C-terminal fragment of CREB3L2 (an activator for hedgehog signaling) produced by unregulated cleavage of the fusion protein may also contribute to development of the tumor.
RIP of CREB3L1 activated by RAT of TM4SF20
The physiological function of CREB3L1, also known as OASIS, was first determined from the study of mice deficient in the gene. Creb3l1−/− mice exhibited severe osteopenia owing to insufficiency of type 1 collagen in bone matrix (68). RIP of CREB3L1 in osteoblasts was shown to be stimulated by bone morphogenetic protein 2 (BMP-2), a member of the transforming growth factor β (TGF-β) family of cytokines that is critical for bone development (68). The cleaved nuclear form of CREB3L1 in turn activates transcription of genes involved in assembly of the collagen matrix, including collagen 1α1 (Col1a1) that underlies bone formation (17, 68, 69) (Fig. 1). The crucial role of CREB3L1 for bone development in humans is supported by the observation that people with homozygous mutations inactivating CREB3L1 exhibit osteogenesis imperfecta, a disease caused by insufficient deposition of collagen in bone matrix (70–73).
TGF-β, a cytokine homologous to BMP-2, induces collagen synthesis during tissue repair and fibrosis (74). Like BMP-2, TGF-β triggers RIP of CREB3L1 in cultured cells (75). TGF-β induces RIP of CREB3L1 by inhibiting expression of TM4SF20, an inhibitor for proteolytic activation of CREB3L1 (75).
RIP of CREB3L1 is also essential for doxorubicin, a drug extensively used for cancer chemotherapy (76), to inhibit proliferation of cancer cells. Doxorubicin stimulates production of ceramide, which in turn triggers RIP of CREB3L1, allowing the cleaved nuclear form of the protein to activate p21 and other genes that inhibit cell proliferation (17, 77) (Fig. 1). The importance of this CREB3L1-mediated signaling pathway was demonstrated by the finding that at clinically relevant doses, doxorubicin was much more effective in cancer cells that expressed high levels of CREB3L1 than in those expressing low levels of the gene. This correlation was observed in cancer cells cultured in vitro, in xenograft tumors established in mice, and in human tumor samples (77–80). These results suggest that CREB3L1 may serve as a biomarker to predict sensitivity to doxorubicin.
RIP of CREB3L1 is critical for ceramide to inhibit cell proliferation (77). In contrast to TGF-β, ceramide does not affect expression of TM4SF20. Instead, ceramide leads to inverted topology of newly synthesized TM4SF20 (18). This topological inversion converts TM4SF20 from an inhibitor to an activator for RIP of CREB3L1 (18).
The two orientations of TM4SF20 are illustrated in Fig. 3 (A and B). In the absence of ceramide, the N terminus of TM4SF20 is located in the ER lumen, and the sequence N-terminal to the first transmembrane helix is cleaved off co-translationally by the signal peptidase. Under this configuration, the loop that contains three potential sites for N-linked glycosylation is located in the cytosol, where glycosylation cannot occur. This form of the protein is designated as TM4SF20(A) (18). When cells are treated with ceramide, the N terminus of newly synthesized TM4SF20 is located in the cytosol and the Asn-containing loop is located in the ER lumen, where it is glycosylated. This form of the protein is designated as TM4SF20(B) (18). It should be emphasized that ceramide leads to inverted topology of newly synthesized TM4SF20 but does not flip the topology of pre-existing TM4SF20. Because ceramide alters the direction through which TM4SF20 is translocated across membranes during its synthesis, this regulatory process is designated as regulated alternative translocation (RAT) (18).
Figure 3.

Ceramide-induced RAT of TM4SF20. A, in the absence of ceramide, the first transmembrane helix of TM4SF20 is translocated by the type III insertion mechanism. This type of insertion causes the sequence N-terminal to the transmembrane helix to be translocated into the ER lumen through an unknown mechanism, allowing it to be cleaved by signal peptidase. The nascent peptide C-terminal to the first transmembrane helix is synthesized in the cytosol. This type of translocation produces TM4SF20(A). This form of the protein is not glycosylated because the loop containing the three potential N-linked glycosylation sites is located in cytosol. TM4SF20(A) inhibits RIP of CREB3L1. B, in the presence of ceramide, the first transmembrane helix of TM4SF20 is translocated by the type II insertion mechanism, during which the nascent peptide C-terminal to the transmembrane helix is pushed through the Sec61 translocon by the ribosome and embedded into the ER lumen. This type of translocation produces TM4SF20(B), in which the sequence N-terminal to the transmembrane helix remains intact in the cytosol. TM4SF20(B) is glycosylated and activates RIP of CREB3L1. A and B, the N-terminal loop, the first transmembrane helix, the loop between the first and second transmembrane helix, and the rest of TM4SF20 are highlighted in orange, yellow, red, and black, respectively. The arrow and dashed lines indicate the direction where nascent polypeptide is extended.
In eukaryotic cells, the topology of polytopic membrane proteins is primarily determined by the direction through which the first transmembrane helix is translocated across ER membranes (81). The translocation process has been categorized into three classes. Type I insertion refers to proteins that contain a cleavable ER-targeting signal peptide N-terminal to the first transmembrane helix. The nascent signal peptide binds to the signal recognition particle that directs the ribosome/nascent polypeptide complex to the ER membranes, enabling sequence C-terminal to the signal peptide to be transported into the ER lumen through the Sec61 ER translocon. Following the translocation, the signal peptide is cleaved from the mature protein by the signal peptidase (7, 82). The other two insertions do not use a signal peptide. Instead, their insertions are initiated by recognition of the hydrophobic sequence present in the first transmembrane helix of nascent peptides by the signal recognition particle, which directs the nascent peptide/ribosome complex to the Sec61 ER translocon. The crystal structure of the translocation complex suggests that insertion of the hydrophobic transmembrane helix into ER membranes adjacent to the Sec61 translocon is the leading event that initiates the translocation process (81) (Fig. 3). In type II insertion, the N terminus of transmembrane proteins is in the cytosol (82). In type III insertion, the orientation of the transmembrane helix is reverted so that the N terminus of the proteins is in the ER lumen (82).
TM4SF20(A) cannot be inserted into membranes via the type I insertion because the N-terminal peptide does not serve as a signal peptide. This conclusion is supported by the following lines of evidence: First, the N-terminal sequence of TM4SF20 does not contain any of the hallmarks characteristic for a signal peptide (83); second, the N-terminal sequence of TM4SF20 did not function as a signal peptide when it was substituted for the endogenous signal sequence of alkaline phosphatase (18); third, the N-terminal sequence of TM4SF20 could be replaced by other peptides without altering the topology of TM4SF20(A) and ceramide-induced RAT of TM4SF20 (18). Even though it is not a signal sequence, the N-terminal peptide of TM4SF20 is cleaved by signal peptidase (18), apparently because the peptide is accessible to the protease in the ER lumen. This type of cleavage catalyzed by the signal peptidase has been reported in proteolytic processing of hepatitis C virus protein in which the protease cleaves the viral polyprotein precursor at multiple sites in the ER lumen distal to the N-terminal sequence (84).
Because the N terminus of TM4SF20(A) is in the ER lumen yet the protein is not inserted through the type I insertion, the first transmembrane helix of TM4SF20(A) is translocated through membranes via the type III insertion (Fig. 3A). In the presence of ceramide, the first transmembrane helix of TM4SF20(B) is inserted into membranes via the type II insertion (Fig. 3B). Thus, RAT changes the translocation of the first transmembrane helix of TM4SF20 from the type III to type II insertion. Whereas both of these insertions have been observed in other membrane proteins, TM4SF20 is the first recognized example in which the two insertion types are interconvertible in a regulated manner.
Unlike the well-characterized type II insertion through which the hydrophilic sequence C-terminal to the transmembrane helix is pushed through the translocon by the ribosome, the mechanism by which the hydrophilic sequence N-terminal to the transmembrane helix reaches ER lumen during the type III insertion remains obscure. If this process is mediated by Sec61, then the N-terminal hydrophilic sequence must be unfolded and pulled through the translocation channel by forces other than peptide elongation (81). Other studies suggest that this process could be Sec61-independent (85–87). One clue is that this process may require translocating chain-associated membrane protein 2 (TRAM2), which is highly homologous to TRAM1, an accessory protein that functions together with the Sec61 translocon (81, 88–90). RNAi-mediated knockdown of TRAM2 but not TRAM1 enabled production of TM4SF20(B) even in the absence of ceramide (18). This observation suggests that TRAM2 may play a specific role in type III insertion of the first transmembrane helix of TM4SF20(A). It is interesting that TRAM2, and all TRAM protein, contains a TLC domain that is postulated to bind ceramide or related sphingolipids (91). These observations raise the possibility that TRAM2 is the sensor that allows ceramide to block synthesis of TM4SF20(A).
Because both the type II and III insertions are initiated by contact of the first transmembrane helix with the ER translocon, the first transmembrane helix of TM4SF20 should be critical for RAT of the protein. Indeed, replacing the signal peptide of alkaline phosphatase with the N-terminal sequence of TM4SF20 that contains the first transmembrane helix led to ceramide-induced topological inversion of the fusion protein (18). Mutagenesis analysis revealed that RAT of TM4SF20 required a Gly and an Asn separated by 3 residues (designated as the GXXXN motif) present in the first transmembrane helix. When the Gly or Asn within the motif was mutated to Leu, TM4SF20 failed to adopt the A configuration, and it was constitutively in the B topology, even in the absence of ceramide (18, 92).
Similar to TM4SF20(A), the first transmembrane helix of the majority of GPCRs is inserted into membranes through the type III orientation (93, 94). The first transmembrane helix of several of these receptors contains the GXXXN motif (95). One of these GPCRs is C-C chemokine receptor 5 (CCR5). In unstimulated macrophages, this receptor adopts a topology consistent with that of GPCRs so it can function as a chemokine receptor (96). When the macrophages were stimulated with LPS, the increased production of dihydroceramide triggered RAT of CCR5, and the protein with the inverted topology no longer functioned as a chemokine receptor (95). This finding may explain the well-known observation that LPS-activated macrophages are insensitive to chemotaxis (97). The findings also suggest that RAT may be a widespread mechanism to regulate transmembrane proteins.
Perspective
RIP of membrane-bound transcription factors is ultimately achieved by regulated transport of these proteins from the ER to Golgi upon stimulation by signals generated from the ER. Except for SREBPs, the exact signaling mechanisms that trigger RIP of the other membrane-bound transcription factors are not well-defined. RIP of most if not all of the membrane-bound transcription factors can be stimulated by treatments with pharmacological compounds, such as tunicamycin or thapsigargin, that induce ER stress. Thus, it is tempting to conclude that all of these transcription factors are ER stress transducers. However, the term ER stress is too broad to categorize the signals that activate RIP of the membrane-bound transcription factors, as distinct signals that activate RIP of different membrane-bound transcription factors (e.g. alteration in lipid composition of ER membranes that activates SREBPs, accumulation of unfolded proteins in the ER lumen that activates ATF6, and increased synthesis of extracellular matrix proteins that activates RIP of CREB3L1 and CREB3L2) may all be considered as ER stress. Thus, identification of the exact ER-generated signals that activate RIP of the individual membrane-bound transcription factor will be the primary challenge in the future to understand the signal transduction pathways mediated by these proteins.
Another important question regarding RIP is whether S2P is equally active in cleaving different membrane-bound transcription factors. Previous reports showed that the membrane-bound intermediate form, which represents a fragment of these transcription factors that has been cleaved by S1P but not S2P (Fig. 1), was present for CREB3L1 but not for SREBPs or ATF6α in cells expressing WT S2P (13, 25, 77, 98). This observation suggests that CREB3L1 may be a less efficient substrate for S2P than other membrane-bound transcription factors. Consistent with this hypothesis, S2P(L505F), a point mutation that partially inactivates S2P, diminished cleavage of CREB3L1 but not ATF6 in cultured cells (95). Patients expressing this mutant of S2P developed osteogenesis imperfecta, the same phenotype exhibited by individuals deficient in CREB3L1 (98). In contrast, subjects with mutations that inactivate S2P completely develop IFAP (ichthyosis follicularis atrichia photophobia), a syndrome that has other abnormalities in addition to osteogenesis imperfecta, presumably caused by deficiency in activating membrane-bound transcription factors besides CREB3L1 (99). Understanding why S2P is less efficient in cleaving CREB3L1 may provide strategies to treat these genetic diseases.
Accumulation of ceramide and/or dihydroceramide in ER membranes triggers RIP of ATF6α and CREB3L1 (46, 77). Whereas the mechanism for dihydroceramide-induced RIP of ATF6α remains obscure, ceramide activates RIP of CREB3L1 by inverting the topology of TM4SF20 through altering the direction by which TM4SF20 is translocated into ER membranes (18). Whereas regulation of protein translocation was proposed more than a decade ago (100), ceramide-induced RAT of TM4SF20 discovered recently is the first example that this regulation indeed takes place in mammalian cells. This discovery raises more questions on regulation of protein translocation: if the type III insertion responsible for production of TM4SF20(A) is Sec61-dependent, what is the driving force and unfolding mechanism that allows the N-terminal sequence to be pulled through the translocon? If not, what is the translocation machinery responsible for the type III insertion? Are TRAM proteins the ceramide sensors that control RAT? Perhaps the most important question is how many transmembrane proteins are subjected to topological regulation. The reports published so far suggest that transmembrane proteins regulated through RAT contain a GXXXN motif in the first transmembrane helix (18, 95). However, TM4SF4, another transmembrane protein that contains the same motif in the first transmembrane helix, does not undergo RAT (92). These observations suggest that the GXXXN motif present in the first transmembrane helix may be required but not sufficient to induce RAT. It appears that a proteome-wide approach capable of measuring topology of transmembrane proteins globally is needed to systematically identify transmembrane proteins subjected to topological regulation.
Acknowledgments
I thank Drs. Michael Brown and Joseph Goldstein for constant support and helpful discussion and Nancy Heard for graphic illustration.
Funding and additional information—This work was supported by National Institutes of Health Grants GM-116106 and HL-20948 and Welch Foundation Grant I-1832 (to J. Y.). The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The author declares that he has no conflicts of interest with the contents of this article.
- RIP
- regulated intramembrane proteolysis
- ATF6
- activating transcription factor 6
- BMP-2
- bone morphogenetic protein 2
- CREB3L
- cAMP response element–binding protein 3–like
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- FUS
- fused in sarcoma
- INSIG
- insulin-induced gene
- LGFMS
- low-grade fibromyxoid sarcoma
- LPL
- lipoprotein lipase
- LPS
- lipopolysaccharide
- LXR
- liver X receptor
- RAT
- regulated alternative translocation
- S1P
- site-1 protease
- S2P
- site-2 protease
- SCAP
- SREBP cleavage–activating protein
- SREBP
- sterol regulatory element–binding protein
- TG
- triglyceride
- TGF-β
- transforming growth factor-β
- TM4SF20
- transmembrane 4 L six family member 20
- TRAM
- translocating chain-associated membrane protein
- UBXD8
- ubiquitin regulatory X domain–containing protein 8
- VLDL
- very low-density lipoprotein
- GPCR
- G protein–coupled receptor
- CCR5
- C-C chemokine receptor 5.
References
- 1. Brown M., Ye J., Rawson R., and Goldstein J. (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100, 391–398 10.1016/S0092-8674(00)80675-3 [DOI] [PubMed] [Google Scholar]
- 2. Ye J. (2013) Regulated intramembrane proteolysis. in Encyclopedia of Biological Chemistry (Lennarz W., and Lane D., eds) 2nd Ed., pp. 50–55, Academic Press, Waltham, MA [Google Scholar]
- 3. Wolfe M. S. (2020) Substrate recognition and processing by γ-secretase. Biochim. Biophys. Acta 1862, 183016 10.1016/j.bbamem.2019.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Spinazzi M., and De Strooper B. (2016) PARL: the mitochondrial rhomboid protease. Semin. Cell Dev. Biol. 60, 19–28 10.1016/j.semcdb.2016.07.034 [DOI] [PubMed] [Google Scholar]
- 5. Rawson R. B. (2013) The site-2 protease. Biochim. Biophys. Acta 1828, 2801–2807 10.1016/j.bbamem.2013.03.031 [DOI] [PubMed] [Google Scholar]
- 6. Mentrup T., Loock A.-C., Fluhrer R., and Schröder B. (2017) Signal peptide peptidase and SPP-like proteases—possible therapeutic targets? Biochim. Biophys. Acta 1864, 2169–2182 10.1016/j.bbamcr.2017.06.007 [DOI] [PubMed] [Google Scholar]
- 7. Zimmermann R., Eyrisch S., Ahmad M., and Helms V. (2011) Protein translocation across the ER membrane. Biochim. Biophys. Acta 1808, 912–924 10.1016/j.bbamem.2010.06.015 [DOI] [PubMed] [Google Scholar]
- 8. Balla T., Sengupta N., and Kim Y. J. (2020) Lipid synthesis and transport are coupled to regulate membrane lipid dynamics in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1865, 158461 10.1016/j.bbalip.2019.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sakai J., Rawson R. B., Espenshade P. J., Cheng D., Seegmiller A. C., Goldstein J. L., and Brown M. S. (1998) Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol. Cell 2, 505–514 10.1016/S1097-2765(00)80150-1 [DOI] [PubMed] [Google Scholar]
- 10. Rawson R. B., Zelenski N. G., Nijhawan D., Ye J., Sakai J., Hasan M. T., Chang T. Y., Brown M. S., and Goldstein J. L. (1997) Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell 1, 47–57 10.1016/S1097-2765(00)80006-4 [DOI] [PubMed] [Google Scholar]
- 11. Nohturfft A., Yabe D., Goldstein J. L., Brown M. S., and Espenshade P. J. (2000) Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell 102, 315–323 10.1016/S0092-8674(00)00037-4 [DOI] [PubMed] [Google Scholar]
- 12. Shen J., Chen X., Hendershot L., and Prywes R. (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111 10.1016/S1534-5807(02)00203-4 [DOI] [PubMed] [Google Scholar]
- 13. Ye J., Rawson R. B., Komuro R., Chen X., Davé U. P., Prywes R., Brown M. S., and Goldstein J. L. (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 10.1016/S1097-2765(00)00133-7 [DOI] [PubMed] [Google Scholar]
- 14. Espenshade P. J., Cheng D., Goldstein J. L., and Brown M. S. (1999) Autocatalytic processing of Site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 274, 22795–22804 10.1074/jbc.274.32.22795 [DOI] [PubMed] [Google Scholar]
- 15. Duncan E. A., Brown M. S., Goldstein J. L., and Sakai J. (1997) Cleavage site for sterol-regulated protease localized to a Leu-Ser bond in the lumenal loop of sterol regulatory element-binding protein-2. J. Biol. Chem. 272, 12778–12785 10.1074/jbc.272.19.12778 [DOI] [PubMed] [Google Scholar]
- 16. Ye J., Davé U. P., Grishin N. V., Goldstein J. L., and Brown M. S. (2000) Asparagine-proline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by Site-2 protease. Proc. Natl. Acad. Sci. U.S.A. 97, 5123–5128 10.1073/pnas.97.10.5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Denard B., Seemann J., Chen Q., Gay A., Huang H., Chen Y., and Ye J. (2011) The membrane-bound transcription factor CREB3L1 is activated in response to virus infection to inhibit proliferation of virus-infected cells. Cell Host Microbe 10, 65–74 10.1016/j.chom.2011.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen Q., Denard B., Lee C.-E., Han S., Ye J. S., and Ye J. (2016) Inverting the topology of a transmembrane protein by regulating the translocation of the first transmembrane helix. Mol. Cell 63, 567–578 10.1016/j.molcel.2016.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goldstein J. L., and Brown M. S. (2015) A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161, 161–172 10.1016/j.cell.2015.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ye J., and DeBose-Boyd R. A. (2011) Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 3, a004754 10.1101/cshperspect.a004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DeBose-Boyd R. A., and Ye J. (2018) SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem. Sci. 43, 358–368 10.1016/j.tibs.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sakai J., Nohturfft A., Cheng D., Ho Y. K., Brown M. S., and Goldstein J. L. (1997) Identification of complexes between the COOH-terminal domains of sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage-activating protein. J. Biol. Chem. 272, 20213–20221 10.1074/jbc.272.32.20213 [DOI] [PubMed] [Google Scholar]
- 23. Sun L.-P., Seemann J., Goldstein J. L., and Brown M. S. (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 6519–6526 10.1073/pnas.0700907104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeBose-Boyd R. A., Brown M. S., Li W.-P., Nohturfft A., Goldstein J. L., and Espenshade P. J. (1999) Transport-dependent proteolysis of SREBP: relocation of Site-1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell 99, 703–712 10.1016/S0092-8674(00)81668-2 [DOI] [PubMed] [Google Scholar]
- 25. Sakai J., Duncan E. A., Rawson R. B., Hua X., Brown M. S., and Goldstein J. L. (1996) Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046 10.1016/S0092-8674(00)81304-5 [DOI] [PubMed] [Google Scholar]
- 26. Horton J. D., Shah N. A., Warrington J. A., Anderson N. N., Park S. W., Brown M. S., and Goldstein J. L. (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U.S.A. 100, 12027–12032 10.1073/pnas.1534923100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Radhakrishnan A., Sun L.-P., Kwon H. J., Brown M. S., and Goldstein J. L. (2004) Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol. Cell 15, 259–268 10.1016/j.molcel.2004.06.019 [DOI] [PubMed] [Google Scholar]
- 28. Motamed M., Zhang Y., Wang M. L., Seemann J., Kwon H. J., Goldstein J. L., and Brown M. S. (2011) Identification of luminal loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. J. Biol. Chem. 286, 18002–18012 10.1074/jbc.M111.238311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Radhakrishnan A., Goldstein J. L., McDonald J. G., and Brown M. S. (2008) Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 8, 512–521 10.1016/j.cmet.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang T., Espenshade P. J., Wright M. E., Yabe D., Gong Y., Aebersold R., Goldstein J. L., and Brown M. S. (2002) Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500 10.1016/S0092-8674(02)00872-3 [DOI] [PubMed] [Google Scholar]
- 31. Yabe D., Brown M. S., and Goldstein J. L. (2002) Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 99, 12753–12758 10.1073/pnas.162488899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun L.-P., Li L., Goldstein J. L., and Brown M. S. (2005) Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 280, 26483–26490 10.1074/jbc.M504041200 [DOI] [PubMed] [Google Scholar]
- 33. Brown M. S., and Goldstein J. L. (2009) Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J. Lipid Res. 50, S15–S27 10.1194/jlr.R800054-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liang G., Yang J., Horton J. D., Hammer R. E., Goldstein J. L., and Brown M. S. (2002) Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 277, 9520–9528 10.1074/jbc.M111421200 [DOI] [PubMed] [Google Scholar]
- 35. Ou J., Tu H., Shan B., Luk A., DeBose-Boyd R. A., Bashmakov Y., Goldstein J. L., and Brown M. S. (2001) Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc. Natl. Acad. Sci. U.S.A. 98, 6027–6032 10.1073/pnas.111138698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim C.-W., Addy C., Kusunoki J., Anderson N. N., Deja S., Fu X., Burgess S. C., Li C., Ruddy M., Chakravarthy M., Previs S., Milstein S., Fitzgerald K., Kelley D. E., and Horton J. D. (2017) Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26, 394–406.e6 10.1016/j.cmet.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hannah V. C., Ou J., Luong A., Goldstein J. L., and Brown M. S. (2001) Unsaturated fatty acids down-regulate SREBP isoforms 1a and 1c by two mechanisms in HEK-293 Cells. J. Biol. Chem. 276, 4365–4372 10.1074/jbc.M007273200 [DOI] [PubMed] [Google Scholar]
- 38. Lee J. N., Song B., DeBose-Boyd R. A., and Ye J. (2006) Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J. Biol. Chem. 281, 39308–39315 10.1074/jbc.M608999200 [DOI] [PubMed] [Google Scholar]
- 39. Lee J. N., Zhang X., Feramisco J. D., Gong Y., and Ye J. (2008) Unsaturated fatty acids inhibit proteasomal degradation of Insig-1 at a postubiquitination step. J. Biol. Chem. 283, 33772–33783 10.1074/jbc.M806108200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee J. N., Kim H., Yao H., Chen Y., Weng K., and Ye J. (2010) Identification of Ubxd8 protein as a sensor for unsaturated fatty acids and regulator of triglyceride synthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 21424–21429 10.1073/pnas.1011859107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schindler A. J., and Schekman R. (2009) In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. U.S.A. 106, 17775–17780 10.1073/pnas.0910342106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Haze K., Yoshida H., Yanagi H., Yura T., and Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoshida H., Matsui T., Yamamoto A., Okada T., and Mori K. (2001) XBP1 mRNA Is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 10.1016/S0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]
- 44. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 45. Maiuolo J., Bulotta S., Verderio C., Benfante R., and Borgese N. (2011) Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc. Natl. Acad. Sci. U.S.A. 108, 7832–7837 10.1073/pnas.1101379108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tam A. B., Roberts L. S., Chandra V., Rivera I. G., Nomura D. K., Forbes D. J., and Niwa M. (2018) The UPR activator ATF6 responds to proteotoxic and lipotoxic stress by distinct mechanisms. Dev. Cell 46, 327–343.e7 10.1016/j.devcel.2018.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thuerauf D. J., Morrison L., and Glembotski C. C. (2004) Opposing roles for ATF6α and ATF6β in endoplasmic reticulum stress response gene induction. J. Biol. Chem. 279, 21078–21084 10.1074/jbc.M400713200 [DOI] [PubMed] [Google Scholar]
- 48. Forouhan M., Mori K., and Boot-Handford R. P. (2018) Paradoxical roles of ATF6α and ATF6β in modulating disease severity caused by mutations in collagen X. Matrix Biol. 70, 50–71 10.1016/j.matbio.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Correll R. N., Grimes K. M., Prasad V., Lynch J. M., Khalil H., and Molkentin J. D. (2019) Overlapping and differential functions of ATF6α versus ATF6β in the mouse heart. Sci. Rep. 9, 2059 10.1038/s41598-019-39515-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D. T., Back S. H., and Kaufman R. J. (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124, 587–599 10.1016/j.cell.2005.11.040 [DOI] [PubMed] [Google Scholar]
- 51. Wang H., Zhao M., Sud N., Christian P., Shen J., Song Y., Pashaj A., Zhang K., Carr T., and Su Q. (2016) Glucagon regulates hepatic lipid metabolism via cAMP and Insig-2 signaling: implication for the pathogenesis of hypertriglyceridemia and hepatic steatosis. Sci. Rep. 6, 32246 10.1038/srep32246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nakagawa Y., Satoh A., Tezuka H., Han S-I., Takei K., Iwasaki H., Yatoh S., Yahagi N., Suzuki H., Iwasaki Y., Sone H., Matsuzaka T., Yamada N., and Shimano H. (2016) CREB3L3 controls fatty acid oxidation and ketogenesis in synergy with PPARα. Sci. Rep. 6, 39182 10.1038/srep39182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gibbons G. F., Wiggins D., Brown A. M., and Hebbachi A. M. (2004) Synthesis and function of hepatic very-low-density lipoprotein. Biochem. Soc. Trans. 32, 59–64 10.1042/bst0320059 [DOI] [PubMed] [Google Scholar]
- 54. Cheng D., Xu X., Simon T., Boudyguina E., Deng Z., VerHague M., Lee A.-H., Shelness G. S., Weinberg R. B., and Parks J. S. (2016) Very low density lipoprotein assembly is required for cAMP-responsive element-binding protein H processing and hepatic apolipoprotein A-IV expression. J. Biol. Chem. 291, 23793–23803 10.1074/jbc.M116.749283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Siddiqi S. A. (2008) VLDL exits from the endoplasmic reticulum in a specialized vesicle, the VLDL transport vesicle, in rat primary hepatocytes. Biochem. J. 413, 333–342 10.1042/BJ20071469 [DOI] [PubMed] [Google Scholar]
- 56. Kim H., Mendez R., Zheng Z., Chang L., Cai J., Zhang R., and Zhang K. (2014) Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor α to regulate metabolic hormone FGF21. Endocrinology 155, 769–782 10.1210/en.2013-1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee M.-W., Chanda D., Yang J., Oh H., Kim S. S., Yoon Y.-S., Hong S., Park K.-G., Lee I.-K., Choi C. S., Hanson R. W., Choi H.-S., and Koo S.-H. (2010) Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 11, 331–339 10.1016/j.cmet.2010.02.016 [DOI] [PubMed] [Google Scholar]
- 58. Olivecrona G. (2016) Role of lipoprotein lipase in lipid metabolism. Curr. Opin. Lipidol. 27, 233–241 10.1097/MOL.0000000000000297 [DOI] [PubMed] [Google Scholar]
- 59. Lee J. H., Giannikopoulos P., Duncan S. A., Wang J., Johansen C. T., Brown J. D., Plutzky J., Hegele R. A., Glimcher L. H., and Lee A.-H. (2011) The transcription factor cyclic AMP–responsive element–binding protein H regulates triglyceride metabolism. Nat. Med. 17, 812–815 10.1038/nm.2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu X., Park J.-G., So J.-S., Hur K. Y., and Lee A.-H. (2014) Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J. Lipid Res. 55, 850–859 10.1194/jlr.M045104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Saito A., Hino S-I., Murakami T., Kanemoto S., Kondo S., Saitoh M., Nishimura R., Yoneda T., Furuichi T., Ikegawa S., Ikawa M., Okabe M., and Imaizumi K. (2009) Regulation of endoplasmic reticulum stress response by a BBF2H7-mediated Sec23a pathway is essential for chondrogenesis. Nat. Cell Biol. 11, 1197–1204 10.1038/ncb1962 [DOI] [PubMed] [Google Scholar]
- 62. Ishikawa T., Toyama T., Nakamura Y., Tamada K., Shimizu H., Ninagawa S., Okada T., Kamei Y., Ishikawa-Fujiwara T., Todo T., Aoyama E., Takigawa M., Harada A., and Mori K. (2017) UPR transducer BBF2H7 allows export of type II collagen in a cargo- and developmental stage–specific manner. J. Cell Biol. 216, 1761–1774 10.1083/jcb.201609100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saito A., Kanemoto S., Zhang Y., Asada R., Hino K., and Imaizumi K. (2014) Chondrocyte proliferation regulated by secreted luminal domain of ER stress transducer BBF2H7/CREB3L2. Mol. Cell 53, 127–139 10.1016/j.molcel.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 64. Arbajian E., Puls F., Antonescu C. R., Amary F., Sciot R., Debiec-Rychter M., Sumathi V. P., Järås M., Magnusson L., Nilsson J., Hofvander J., and Mertens F. (2017) In-depth genetic analysis of sclerosing epithelioid fibrosarcoma reveals recurrent genomic alterations and potential treatment targets. Clin. Cancer Res. 23, 7426–7434 10.1158/1078-0432.CCR-17-1856 [DOI] [PubMed] [Google Scholar]
- 65. Mohamed M., Fisher C., and Thway K. (2017) Low-grade fibromyxoid sarcoma: clinical, morphologic and genetic features. Ann. Diagn. Pathol. 28, 60–67 10.1016/j.anndiagpath.2017.04.001 [DOI] [PubMed] [Google Scholar]
- 66. Hua X., Sakai J., Brown M. S., and Goldstein J. L. (1996) Regulated cleavage of sterol regulatory element binding proteins requires sequences on both sides of the endoplasmic reticulum membrane. J. Biol. Chem. 271, 10379–10384 10.1074/jbc.271.17.10379 [DOI] [PubMed] [Google Scholar]
- 67. Möller E., Hornick J. L., Magnusson L., Veerla S., Domanski H. A., and Mertens F. (2011) FUS-CREB3L2–positive sarcomas show a specific gene expression profile with upregulation of CD24 and FOXL1. Clin. Cancer Res. 17, 2646–2656 10.1158/1078-0432.CCR-11-0145 [DOI] [PubMed] [Google Scholar]
- 68. Murakami T., Saito A., Hino S-I., Kondo S., Kanemoto S., Chihara K., Sekiya H., Tsumagari K., Ochiai K., Yoshinaga K., Saitoh M., Nishimura R., Yoneda T., Kou I., Furuichi T., et al. (2009) Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol. 11, 1205–1211 10.1038/ncb1963 [DOI] [PubMed] [Google Scholar]
- 69. Vellanki R. N., Zhang L., Guney M. A., Rocheleau J. V., Gannon M., and Volchuk A. (2010) OASIS/CREB3L1 induces expression of genes involved in extracellular matrix production but not classical endoplasmic reticulum stress response genes in pancreatic β-cells. Endocrinology 151, 4146–4157 10.1210/en.2010-0137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lindahl K., Åström E., Dragomir A., Symoens S., Coucke P., Larsson S., Paschalis E., Roschger P., Gamsjaeger S., Klaushofer K., Fratzl-Zelman N., and Kindmark A. (2018) Homozygosity for CREB3L1 premature stop codon in first case of recessive osteogenesis imperfecta associated with OASIS-deficiency to survive infancy. Bone 114, 268–277 10.1016/j.bone.2018.06.019 [DOI] [PubMed] [Google Scholar]
- 71. Guillemyn B., Kayserili H., Demuynck L., Sips P., De Paepe A., Syx D., Coucke P. J., Malfait F., and Symoens S. (2019) A homozygous pathogenic missense variant broadens the phenotypic and mutational spectrum of CREB3L1-related osteogenesis imperfecta. Hum. Mol. Genet. 28, 1801–1809 10.1093/hmg/ddz017 [DOI] [PubMed] [Google Scholar]
- 72. Cayami F. K., Maugeri A., Treurniet S., Setijowati E. D., Teunissen B. P., Eekhoff E. M. W., Pals G., Faradz S. M., and Micha D. (2019) The first family with adult osteogenesis imperfecta caused by a novel homozygous mutation in CREB3L1. Mol. Genet. Genomic Med. 7, e823 10.1002/mgg3.823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Symoens S., Malfait F., D'hondt S., Callewaert B., Dheedene A., Steyaert W., Bächinger H. P., De Paepe A., Kayserili H., and Coucke P. J. (2013) Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J. Rare Dis. 8, 154–154 10.1186/1750-1172-8-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen Q., Lee C.-E., Denard B., and Ye J. (2014) Sustained induction of collagen synthesis by TGF-β requires regulated intramembrane proteolysis of CREB3L1. PLoS ONE 9, e108528 10.1371/journal.pone.0108528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang F., Teves S. S., Kemp C. J., and Henikoff S. (2014) Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta 1845, 84–89 10.1016/j.bbcan.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Denard B., Lee C., and Ye J. (2012) Doxorubicin blocks proliferation of cancer cells through proteolytic activation of CREB3L1. eLife Sci. 1, e00090 10.7554/elife.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Denard B., Pavia-Jimenez A., Chen W., Williams N. S., Naina H., Collins R., Brugarolas J., and Ye J. (2015) Identification of CREB3L1 as a biomarker predicting doxorubicin treatment outcome. PLoS ONE 10, e0129233 10.1371/journal.pone.0129233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Denard B., Jiang S., Peng Y., and Ye J. (2018) CREB3L1 as a potential biomarker predicting response of triple negative breast cancer to doxorubicin-based chemotherapy. BMC Cancer 18, 813 10.1186/s12885-018-4724-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xiao W., Liang Y., Que Y., Li J., Peng R., Xu B., Wen X., Zhao J., Guan Y., and Zhang X. (2019) Comparison of the MAID (AI) and CAV/IE regimens with the predictive value of cyclic AMP-responsive element-binding protein 3 like protein 1 (CREB3L1) in palliative chemotherapy for advanced soft-tissue sarcoma patients. J. Cancer 10, 3517–3525 10.7150/jca.28734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rapoport T. A., Li L., and Park E. (2017) Structural and mechanistic insights into protein translocation. Annu. Rev. Cell Dev. Biol. 33, 369–390 10.1146/annurev-cellbio-100616-060439 [DOI] [PubMed] [Google Scholar]
- 82. Lodish H., Berk A., Kaiser C., Krieger M., Scott M., Bretscher A., Ploegh H., and Matsudaira P. (2007) Molecular Cell Biology, 6th Ed., pp. 700–702, W. H. Freeman, New York [Google Scholar]
- 83. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 84. Hijikata M., Kato N., Ootsuyama Y., Nakagawa M., and Shimotohno K. (1991) Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. U.S.A. 88, 5547–5551 10.1073/pnas.88.13.5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chitwood P. J., Juszkiewicz S., Guna A., Shao S., and Hegde R. S. (2018) EMC is required to initiate accurate membrane protein topogenesis. Cell 175, 1507–1519e1516 10.1016/j.cell.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. McKenna M., Simmonds R. E., and High S. (2017) Mycolactone reveals the substrate-driven complexity of Sec61-dependent transmembrane protein biogenesis. J. Cell Sci. 130, 1307–1320 10.1242/jcs.198655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Morel J.-D., Paatero A. O., Wei J., Yewdell J. W., Guenin-Macé L., Van Haver D., Impens F., Pietrosemoli N., Paavilainen V. O., and Demangel C. (2018) Proteomics reveals scope of mycolactone-mediated Sec61 blockade and distinctive stress signature. Mol. Cell. Proteomics 17, 1750–1765 10.1074/mcp.RA118.000824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Görlich D., Hartmann E., Prehn S., and Rapoport T. A. (1992) A protein of the endoplasmic reticulum involved early in polypeptide translocation. Nature 357, 47–52 10.1038/357047a0 [DOI] [PubMed] [Google Scholar]
- 89. Voigt S., Jungnickel B., Hartmann E., and Rapoport T. A. (1996) Signal sequence-dependent function of the TRAM protein during early phases of protein transport across the endoplasmic reticulum membrane. J. Cell Biol. 134, 25–35 10.1083/jcb.134.1.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Do H., Falcone D., Lin J., Andrews D. W., and Johnson A. E. (1996) The cotranslational integration of membrane proteins into the phospholipid bilayer is a multistep process. Cell 85, 369–378 10.1016/S0092-8674(00)81115-0 [DOI] [PubMed] [Google Scholar]
- 91. Winter E., and Ponting C. P. (2002) TRAM, LAG1 and CLN8: members of a novel family of lipid-sensing domains? Trends Biochem. Sci. 27, 381–383 10.1016/S0968-0004(02)02154-0 [DOI] [PubMed] [Google Scholar]
- 92. Wang J., Kinch L. N., Denard B., Lee C.-E., Esmaeilzadeh Gharehdaghi E., Grishin N., and Ye J. (2019) Identification of residues critical for topology inversion of the transmembrane protein TM4SF20 through regulated alternative translocation. J. Biol. Chem. 294, 6054–6061 10.1074/jbc.RA119.007681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Guan X. M., Kobilka T. S., and Kobilka B. K. (1992) Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J. Biol. Chem. 267, 21995–21998 [PubMed] [Google Scholar]
- 94. Von Heijne G. (2006) Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 7, 909–918 10.1038/nrm2063 [DOI] [PubMed] [Google Scholar]
- 95. Denard B., Han S., Kim J., Ross E. M., and Ye J. (2019) Regulating G protein-coupled receptors by topological inversion. eLife 8, e40234 10.7554/eLife.40234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Oppermann M. (2004) Chemokine receptor CCR5: insights into structure, function, and regulation. Cell. Signal. 16, 1201–1210 10.1016/j.cellsig.2004.04.007 [DOI] [PubMed] [Google Scholar]
- 97. Biswas S. K., and Lopez-Collazo E. (2009) Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30, 475–487 10.1016/j.it.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 98. Lindert U., Cabral W. A., Ausavarat S., Tongkobpetch S., Ludin K., Barnes A. M., Yeetong P., Weis M., Krabichler B., Srichomthong C., Makareeva E. N., Janecke A. R., Leikin S., Röthlisberger B., Rohrbach M., et al. (2016) MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat. Commun. 7, 11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Oeffner F., Fischer G., Happle R., König A., Betz R. C., Bornholdt D., Neidel U., Boente M. D C., Redler S., Romero-Gomez J., Salhi A., Vera-Casaño A., Weirich C., and Grzeschik K.-H. (2009) IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am. J. Hum. Genet. 84, 459–467 10.1016/j.ajhg.2009.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hegde R. S., and Kang S.-W. (2008) The concept of translocational regulation. J. Cell Biol. 182, 225–232 10.1083/jcb.200804157 [DOI] [PMC free article] [PubMed] [Google Scholar]