

Abstract
Background
Constitutional or somatic mosaic epimutations are increasingly recognized as a mechanism of gene dysregulation resulting in cancer susceptibility. Beckwith‐Wiedemann syndrome is the cancer predisposition syndrome most commonly associated with epimutation and is extremely variable in its phenotypic presentation, which can include isolated tumors. Because to the authors' knowledge large‐scale germline DNA sequencing studies have not included methylation analysis, the percentage of pediatric cancer predisposition that is due to epimutations is unknown.
Methods
Germline methylation testing at the 11p15.5 locus was performed in blood for 24 consecutive patients presenting with hepatoblastoma (3 patients) or Wilms tumor (21 patients).
Results
Six individuals with Wilms tumor and 1 patient with hepatoblastoma were found to have low‐level gain of methylation at imprinting control 1, and a child with hepatoblastoma was found to have loss of methylation at imprinting control 2. The loss of methylation at imprinting control 2 was found to be maternally inherited, despite not being associated with any detectable genomic alteration.
Conclusions
Overall, 33% of patients (8 of 24 patients) with Wilms tumor or hepatoblastoma were found to have an epigenetic susceptibility that was detectable in the blood. It is interesting to note that low‐level gain of methylation at imprinting control 1 predominantly was detected in females with bilateral Wilms tumors. Further studies in larger cohorts are needed to determine the efficacy of testing all patients with Wilms tumor or hepatoblastoma for 11p15.5 epimutations in the blood as part of DNA analysis because this hallmark of predisposition will not be detected by sequencing‐based approaches and detecting a cancer predisposition may modify treatment.
Keywords: Beckwith‐Wiedemann syndrome, genetic predisposition to disease, hepatoblastoma, methylation, Wilms tumor
Short abstract
In the current study, all patients presenting with Wilms tumor or hepatoblastoma undergo 11p15.5 methylation analysis. Approximately one‐third are found to have an epimutation at this locus that is detectable in peripheral blood.
Introduction
Pediatric cancer is associated with an identifiable heritable predisposition in approximately 10% of patients. 1 , 2 , 3 , 4 This number has been derived from pan‐cancer agnostic sequencing studies, which have focused primarily on relatively highly penetrant mutations detected with sequencing, similar to the large germline sequencing panels that are increasingly being used on a clinical basis in oncology and genetics clinics. However, these sequencing‐based methods do not detect epimutations, and to the best of our knowledge no large‐scale study of pediatric pan‐cancer predisposition to date has included methylation analysis of peripheral blood.
Methylation abnormalities may lead to epigenetic cancer susceptibility (ECS) through different mechanisms. Constitutional epimutations have been reported as a mechanism of gene dysregulation, resulting in cancer predisposition in both developmental disorders and adult‐onset cancer‐specific syndromes. 5 , 6 , 7 , 8 , 9 , 10 , 11 Although aberrant methylation is a common method of gene silencing in many tumor types, 12 , 13 epigenetic abnormalities that are detectable in noncancerous somatic cells consistent with a constitutional or mosaic predisposition appear to be less common but are increasingly recognized. Many cancer predisposition genes that typically are mutated by sequencing errors also can undergo promoter methylation that results in gene silencing, with similar effect. For example, Lynch syndrome typically is caused by point mutations or deletions or duplications in one of the mismatch repair genes; however, both constitutional and mosaic epimutations in the promoters of MLH1 and MSH2 also have been reported. 5 , 6 , 7 Constitutional promoter methylation of the RB1 gene has been documented in a child with unilateral retinoblastoma. 8 Mosaic epimutations of SDHC have been observed in patients with gastrointestinal stromal tumors. 9 In these cases, the epimutation found in the tumor was detectable at a lower level in blood and saliva, suggesting a postzygotic, pretumorigenesis origin for the methylation abnormality. Unlike the aforementioned ECS, Beckwith‐Wiedemann syndrome (BWS) is ECS characterized by overgrowth and predisposition toward embryonal tumors, which most often manifests from 11p15.5 epimutations at imprinting control (IC) regions (IC1 and/or IC2), rather than from methylation alterations at gene promoters or gene level point mutations or copy number alterations. 10 , 11 Unlike methylation alterations at gene promoters, the 11p15.5 epimutations in BWS dysregulate the transcription of a group of genes located adjacent to or within these IC regions, which are known to be dose‐sensitive and typically are tightly regulated based on the parent of origin of each chromosome (Fig. 1).
Figure 1.
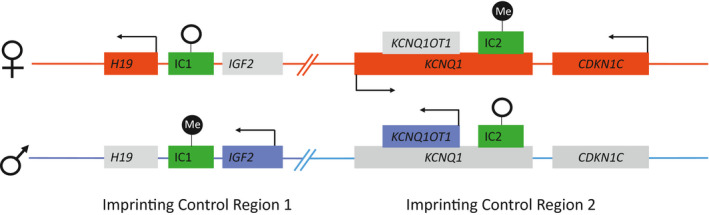
Imprinting control (IC) regions IC1 and IC2 on 11p15.5. Genes that are expressed from the maternal allele (above) are shown in red, genes that are expressed from the paternal allele (below) are shown in blue, and genes that are not expressed are shown in gray. Green boxes indicate the IC regions; black circles, methylation; white circles, the absence of methylation.
Both Wilms tumor and hepatoblastoma, the most common childhood malignant tumors of the kidney and liver, respectively, can occur within the context of well‐described predisposition syndromes, including BWS. 14 In classic BWS, tumor risk varies by molecular subtype. Patients with gain of methylation (GOM) at IC1 (GOM IC1) have the highest risk (28%), whereas patients with loss of methylation (LOM) at IC2 (LOM IC2) have the lowest risk (2.6%). 11 Patients with both GOM IC1 and LOM IC2 due to paternal uniparental isodisomy (pUPD) of chromosome 11p15.5 have an intermediate risk (16%). It is hypothesized that the IC1 region plays a greater role in the development of Wilms tumor and the IC2 region plays a greater role in the development of hepatoblastoma because the majority of tumors that developed in patients with GOM IC1 are Wilms tumors and those in patients with LOM IC2 are hepatoblastoma. 15 Patients with pUPD can develop both Wilms tumor or hepatoblastoma, most likely due to the fact that both the IC1 and IC2 regions are affected.
BWS recently has been redefined as the Beckwith‐Wiedemann spectrum (BWSp), which is especially broad, most likely due to the frequent somatic (rather than predominantly germline) occurrence of underlying epigenetic alterations, often resulting in mosaic results across various tissues. 11 , 16 For example, outside of patients who clearly are syndromic, it has been shown that Wilms tumor can be the presenting sign of BWSp in children who either are without other signs or with subtle other signs that were not previously realized to be related. 17 , 18 Isolated lateralized overgrowth (also known as hemihypertrophy and/or hemihyperplasia) has long been recognized to be associated with a predisposition toward Wilms tumor and hepatoblastoma, regardless of whether a detectable genetic or epigenetic mutation is present. 19 Finally, 11p15.5 alterations are very common somatic occurrences in both patients with Wilms tumor and those with hepatoblastoma. 20 , 21 Therefore, these 2 tumor types represent an especially important pool of patients to screen with 11p15.5 methylation analysis. In the current study, we have presented the results of germline methylation testing of 11p15.5 in individuals with hepatoblastoma or Wilms tumor.
Materials and Methods
All patients presenting to the Clinical Genetics Service and Pediatric Cancer Predisposition Screening Program at Memorial Sloan Kettering Cancer Center with Wilms tumor or hepatoblastoma between January 2016 and May 2019 were selected for analysis. Patients who had a known diagnosis of BWS at the time of their cancer diagnosis were excluded. A patient with hepatoblastoma and a known familial APC mutation was not tested for BWS and therefore also was excluded.
Germline analysis included molecular testing for BWS, which was performed on peripheral blood at the University of Pennsylvania Genetic Diagnostic Laboratory (www.med.upenn.edu/genetics/gdl), a Clinical Laboratory Improvement Amendments (CLIA)–approved laboratory. Testing consisted of methylation‐sensitive, quantitative, real‐time polymerase chain reaction (PCR) of bisulfite‐treated DNA to evaluate methylation levels at IC1 and IC2 22 and a comparative genomic hybridization and single‐nucleotide polymorphism array (SNP) to identify copy number changes and uniparental disomy at 11p15.5, with reflex to Sanger sequencing of the CDKN1C gene if methylation and copy number and/or SNP analysis were normal. 23 , 24 Briefly, 2 independent DNA isolates were collected from each submitted sample for testing and DNA was isolated as previously described. 25 Evaluation of polymorphic short tandem repeats to verify that both independent DNA isolates were collected from the same individual was performed for quality control as described. 26 The EZ DNA Methylation‐Direct kit (Zymo Research, Irvine, California) was used to bisulfite convert 200 ng of genomic DNA. As previously described, allele‐specific, methylated, multiplex, quantitative real‐time PCR was performed using a StepOnePlus Real‐Time PCR System (Thermo Fisher Scientific, Waltham, Massachusetts), and data then were analyzed using the cycle threshold method. 27 Methylation testing was performed on 2 independent DNA isolates from each sample tested.
The amount of methylated and unmethylated DNA was calculated for each sample by interpolation on the standard curve that was run in parallel with the patient samples. The methylation percentage was determined by dividing the amount of methylated DNA by the amount of total DNA. The mean of the assays was used to calculate the methylation index for that patient. A normal range previously was generated by the laboratory by testing a series of unaffected individuals. The mean and standard deviation were calculated for this set of samples. Methylation values were considered pathogenic if they were >2 standard deviations from the normal. Methylation levels that were abnormal but within 10% of the control were considered low‐level mosaic epimutations.
The array was custom designed using Agilent technologies. The array was high‐density and able to detect exonic and intronic copy number changes in CDKN1C, H19, IGF2, KCNQ1, and KCNQ1OT1, as well as intergenic copy number variants in the 11p15.5 region. It also was able to detect copy neutral regions of homozygosity in the targeted region, indicative of pUPD when found in conjunction with GOM IC1 and LOM IC2. 28 Analysis of the targeted region was performed using CytoGenomics software (Agilent Technologies, Santa Clara, California).
When samples were available, patients with positive results in the blood at 11p15.5 also underwent tumor testing including 11p15.5 methylation analysis and whole‐genome sequencing to assess tumor signatures. Familial segregation testing was performed in one case with nonmosaic abnormal results. Germline whole‐genome sequencing was performed on this patient, who was found to have inherited his epimutation from his mother, to look for underlying genomic alterations. The current study was performed under institutional review board–approved studies 12‐245, 06‐107, 17‐575, and 18‐083 at Memorial Sloan Kettering Cancer Center.
Results
A total of 24 patients (21 patients with Wilms tumor and 3 patients with hepatoblastoma) underwent germline assessment including 11p15.5 methylation and copy number and/or SNP analysis (Fig. 2) (Table 1). Overall, 33% of patients (8 of 24 patients) were found to have an epigenetic change at chromosome 11p15.5 that was detectable in the blood. Six individuals with Wilms tumor and 1 patient with hepatoblastoma were found to have low‐level GOM IC1 that was consistent with a molecular diagnosis of mosaic BWSp or ECS (Fig. 3). A second child with hepatoblastoma was found to have LOM IC2. His mother also had LOM IC2; her medical history was notable only for a pancreatic cyst. Both of the proband's maternal grandparents had normal methylation at 11p15.5. No patients were found to have copy number variants at 11p15.5 or pUPD as determined by a comparative genomic hybridization and SNP array, or mutations in CDKN1C as determined by Sanger sequencing in blood.
Figure 2.

Clinical characteristics and peripheral blood methylation results of patients presenting with Wilms tumor (patients 1‐21) or hepatoblastoma (patients 22‐24) who underwent 11p15.5 methylation testing on the blood. Somatic mutations and tumor single‐nucleotide polymorphism (SNP) array results are shown for patients who underwent this testing. Somatic tumor mutations are shown for genes known to be associated with Wilms tumor or genes that were found to be mutated in multiple patients. GOM indicates gain of methylation; IC1, imprinting control 1; IC2, imprinting control 2; LOM; loss of methylation.
Table 1.
Methylation Levels of Patients With GOM at IC1 and LOM at IC2
| Patient No. | IC1 Methylation | Normal Range | IC2 Methylation | Normal Range | |
|---|---|---|---|---|---|
| Patients with Wilms tumor | 1 | 53.81% ± 0.80% | 50.00% ± 1.12% | Normal | |
| 2 | 55.11% ± 2.00% | 50.00% ± 0.98% | Normal | ||
| 3 | 56.28% ± 1.58% | 50.00% ± 1.54% | Normal | ||
| 4 | 54.10% ± 0.88% | 50.00% ±‐ 0.97% | Normal | ||
| 5 | 54.48% ± 0.96% | 50.00% ±‐ 1.08% | Normal | ||
| 6 | 55.24% ± 0.95% | 50.00% ± 1.51% | Normal | ||
| 7 | Normal | Normal | |||
| 8 | Normal | Normal | |||
| 9 | Normal | Normal | |||
| 10 | Normal | Normal | |||
| 11 | Normal | Normal | |||
| 12 | Normal | Normal | |||
| 13 | Normal | Normal | |||
| 14 | Normal | Normal | |||
| 15 | Normal | Normal | |||
| 16 | Normal | Normal | |||
| 17 | Normal | Normal | |||
| 18 | Normal | Normal | |||
| 19 | Normal | Normal | |||
| 20 | Normal | Normal | |||
| 21 | Normal | Normal | |||
| Patients with hepatoblastoma | 22 | 56.23% ± 0.84% | 50.00% ± 1.61% | Normal | |
| 23 | Normal | 40.44% ± 1.68% | 49.99% ± 0.97% | ||
| 24 | Normal | Normal |
Abbreviations: GOM, gain of methylation; IC1, imprinting control 1; IC2, imprinting control 2; LOM, loss of methylation.
Figure 3.
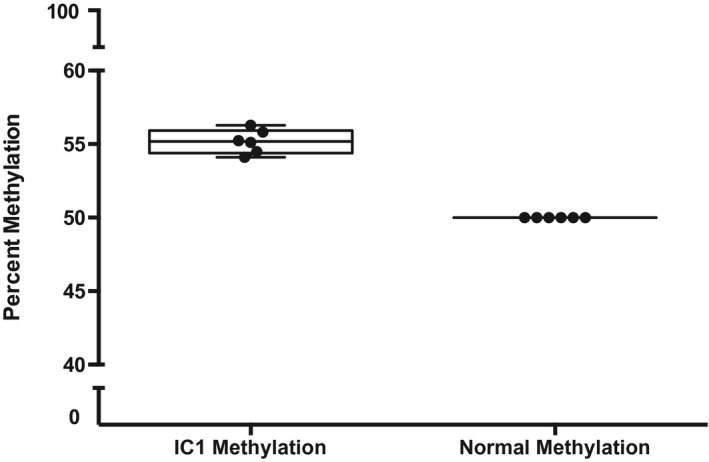
Box and whisker plot showing the percentage methylation at imprinting control (IC) region IC1 on 11p15.5 in patients with hypermethylation at this locus (in red) compared with control samples (in blue).
Patient Characteristics
Five of eight patients (62.5%) with abnormal methylation did not manifest overt clinical findings of BWS apart from their tumor. Two children demonstrated lateralized overgrowth, and a third child had mild hypoglycemia at birth, which spontaneously resolved.
As shown in Figure 2, all 7 patients with GOM IC1 were female, compared with 6 of the 16 patients with methylation in the normal range (38%), whereas the patient with inherited LOM IC2 was male. For patients with epimutations, the average and median age at the time of diagnosis of Wilms tumor both were 4.3 years (range, 25‐78 months). For patients without epimutations, the average age at the time of diagnosis of Wilms tumor was 7.8 years, and the median age was 4.3 years (range, 3 months‐35 years). Patients with hepatoblastoma were diagnosed at an average age of 14 months (range, 4‐31 months). It is interesting to note that the child with hepatoblastoma and LOM IC2 was aged 31 months at the time of diagnosis, which is just above the age range reported in a review of age at the time of hepatoblastoma diagnosis in individuals with BWS. 29
Nephrogenic rests were present in 3 of 15 of the patients with Wilms tumor with normal methylation (20%), whereas all 5 patients with Wilms tumor with GOM IC1 for whom the data were available had nephrogenic rests. One patient with GOM IC1 and Wilms tumor was referred for evaluation as an adult; this individual had been diagnosed with Wilms tumor in the 1980s, prior to the current classification and terminology system, and therefore it was not known whether she had nephrogenic rests. Of the 5 patients with Wilms tumor with epimutations who had nephrogenic rests, 4 of the rests were perilobar and 1 rest was intralobar.
Molecular Analysis of the Tumor Samples
Of the patients with an 11p15.5 epimutation detected in the blood, 5 patients had tumors that were available for further analysis. All 5 tumors had epimutations consistent with what was observed in the blood: 4 patients with Wilms tumor with GOM IC1 and 1 patient with hepatoblastoma with LOM IC2. These 5 patients also had their tumors characterized by whole‐genome sequencing, and analysis of mutational signatures was possible in 3 cases that had >100 somatic mutations detected (the purity of the other 2 samples was too low for analysis). In the 3 cases (2 Wilms tumors and 1 hepatoblastoma), the low mutational burden (0.05‐0.25 mutations per megabase) was mostly attributed to age‐related mutational signature 1, demonstrating the low activity of other mutational processes. 30
Approximately 83.3% of patients (20 of 24 patients) had additional tumor testing ordered by the primary oncologist, as shown in Figure 2. It is interesting to note that 5 patients with Wilms tumor also had loss of heterozygosity of either all of chromosome 11 or of a portion containing 11p15.5 in their tumor sample; all of these patients had normal 11p15.5 methylation in the blood.
Discussion
In the current study, we observed low levels of aberrant 11p15.5 methylation detectable in the blood in a subset of children with Wilms tumor and hepatoblastoma who did not demonstrate phenotypic features or a family history diagnostic of a hereditary cancer syndrome. The presence of an epimutation was most prominent in females with bilateral Wilms tumors. We also identified an inherited 11p15.5 epimutation in a child with hepatoblastoma, with no detectable underlying genomic cause at 11p15.5. It is interesting to note that these low‐level epimutations were detected using methylation‐sensitive, quantitative, real‐time PCR, which is more sensitive than commonly used multiplex ligation–dependent probe amplification–based methods. 11
It is unlikely that the low‐level epimutations found in these patients represented circulating tumor DNA. The timing of blood collection for BWS testing was highly variable in relation to the time of tumor diagnosis and therapy because it was related to the time of genetics referral for each patient. Some patients had blood drawn during treatment, and some had it drawn months or years later. The greatest outlier in this regard was patient 6, whose bilateral Wilms tumors were diagnosed and treated in the 1980s. Her results were very similar to those of the other females with bilateral Wilms tumors despite her blood draw for BWS testing occurring >30 years after her diagnosis. In addition, MacFarland et al did not find any evidence of circulating tumor DNA in the blood in their cohort, despite many positive results in other tissues. 17
To our knowledge, the current study cohort differs from Wilms tumor cohorts described previously 17 , 31 in that all patients in the current study cohort were found to have epimutations on analysis from peripheral blood, whereas patients in the prior cohorts were identified through testing blood, skin, tumor, and healthy kidney, and did not include patients with hepatoblastoma. The most notable difference with regard to the findings of the current study was that none of the patients in our cohort were found to have pUPD, whereas in the study by MacFarland et al 17 approximately one‐half of the cohort was diagnosed with mosaic pUPD. However, the 5 patients in the current study with complete or partial loss of heterozygosity of chromosome 11 detected in tumor may have been somatic mosaic for this abnormality if other samples were analyzed because pUPD is especially likely to be mosaic. The approximately 33% of patients in the current study cohort with positive findings represented what was detectable in the blood, but there is likely to be a higher incidence of somatic mosaicism of BWSp (both epimutations and chromosome abnormalities) when more tissues are included.
Epigenetic defects were identified in 2 of 3 patients with hepatoblastoma. The finding of GOM IC1 in one of the patients with hepatoblastoma was surprising because hepatoblastoma is extremely rare in patients with this molecular subtype. In a large pooled cohort of patients with BWS and tumors, there were no reports of patients with hepatoblastoma with GOM IC1, and to our knowledge, only 1 case has been reported previously. 32 , 33 It is possible that more patients with hepatoblastoma are affected by 11p15.5 epimutations but have not undergone methylation analysis due to a lack of syndromic features; however, given the small number of patients with hepatoblastoma in the current study, we were unable to make more than anecdotal observations. Further studies combining larger numbers of patients with hepatoblastoma are needed to draw conclusions regarding this group.
Although to our knowledge there was no sex discrepancy for unilateral Wilms tumors, it is well established that patients with bilateral disease at the time of presentation are more often female and tend to be younger and to have nephrogenic rests. 34 , 35 It is interesting to note that in the current study cohort, all 4 females with bilateral tumors who underwent testing were found to have low‐level GOM IC1. Although the sample size was small, given that the reason for the increased incidence of bilateral Wilms tumors in females is unknown, the finding of this epimutation warrants further evaluation as being potentially related to a mechanistic molecular explanation. Future studies in larger patient cohorts are needed to fully characterize the degree to which GOM IC1 may play a role in the female predilection toward bilateral Wilms tumors, and the relationship between the development of certain epimutations and female sex.
The majority of epimutations at 11p15.5 that are related to BWSp are considered to be unlikely to be heritable in the absence of a detectable genomic abnormality, although to our knowledge empiric data are modest. 11 The finding reported herein of a family with hereditary LOM IC2 indicates that some cases with an apparently isolated epimutation (via clinically available testing and additional research efforts) still can be passed from one generation to the next. The GC‐rich repetitive nature of the IC2 region may be one limitation to detecting causative genomic alterations for individuals with epimutations at this locus. Further work also is needed to determine the heritability of the low‐level GOM IC1 detected among patients in the current study. In addition to obtaining empiric data, determining whether this epimutation represents a somatic mosaic event, a cryptic germline DNA event for which hypermethylation is a downstream consequence, or a biomarker for a constitutional event will help with elucidating the mechanism of inheritance and allow for more accurate genetic counseling to be provided to these families.
In addition to the diagnostic implications of the genomic findings reported in the current study, therapeutic vulnerabilities also may be revealed through epigenetic analysis. Clinical trials based on molecular profiling currently are available for both pediatric and adult patients with cancer; this subset of patients with epigenetic alterations may be considered for targeted approaches as part of clinical trials when traditional lines of therapy have failed. Because epimutations at 11p15.5 in patients with Wilms tumor and hepatoblastoma are believed to drive IGF2 overexpression, 36 IGF2 inhibitors may be a consideration. 37 IGF2 inhibitors have started to be explored in phase 1 trials, and identifying an underlying epigenetic molecular abnormality in patients with Wilms tumor or hepatoblastoma may provide a therapeutic rationale for this class of targeted therapy. 37 , 38 , 39
Conclusions
Although broad molecular somatic and germline diagnostic evaluations are increasingly available to pediatric patients with cancer, the majority of agnostic germline testing approaches are based on DNA sequencing and will fail to detect the epigenetic alterations underpinning some cancer susceptibility. 4 , 40 , 41 Constitutional or somatic epimutations have been shown to be an alternative mechanism of cancer predisposition in many nonsyndromic cancer susceptibilities as well as in BWSp through different mechanisms of methylation alterations. 6 , 7 , 42 , 43 The current study evaluated methylation abnormalities affecting the imprinted region at 11p15.5 in children with the 2 most commonly associated tumors. The data presented in the current study have expanded on previous work demonstrating that Wilms tumor can be the presenting sign of epimutations associated with BWSp. 17 Further work should be done in larger cohorts of patients with Wilms tumor and hepatoblastoma to determine the efficacy of screening all patients with these tumors for 11p15.5 epimutations. In addition, broader methylation testing of cancer predisposition genes known to be associated with pathogenic epimutations in some cases also may be considered among many cancer types to comprehensively evaluate for predisposition. Larger pan‐cancer studies that include methylation assessment of a broader number of genes are needed to determine the overall percentage of pediatric cancer susceptibilities attributable to methylation abnormalities, both imprinting defects as evaluated herein, and promotor methylation, as has been shown in many other cancer predisposition genes.
Funding Support
No specific funding was disclosed.
Conflict of Interest Disclosures
All authors at Memorial Sloan Kettering Cancer Center were supported by the Memorial Sloan Kettering Cancer Center Support Grant/Core Grant via National Cancer Institute (NCI) grant P30 CA008748. Michael V. Ortiz was supported by Cannonball Kids' cancer, Family and Friends of Caroline Bhatt, and NCI grant K12 CA18474. Karen Cadoo has received travel costs and expenses as well as institutional support for a therapeutic clinical trial from AstraZeneca; institutional support for a therapeutic clinical trial from Syndax Pharmaceuticals; honorarium, travel costs, and expenses from Tessaro; and honorarium from OncLive for work performed outside of the current study. Jennifer M. Kalish was supported by NCI grant K08 CA193915, the St. Baldrick's Foundation, and the Alex's Lemonade Stand Foundation. Michael F. Walsh was supported by the Robert and Kate Niehaus Center for Inherited Cancer Genomics at Memorial Sloan Kettering Cancer Center, the V Foundation for Cancer Research, the Crawford Fund, and the Corning Fund. The other authors made no disclosures.
Author Contributions
Elise M. Fiala and Michael F. Walsh had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Elise M. Fiala, Michael V. Ortiz, and Jennifer A. Kennedy were co‐first authors. Jennifer M. Kalish and Michael F. Walsh were co‐senior authors. Megan Harlan Fleischut and Kaitlyn Tkachuk contributed to the acquisition of the data and review of the article. Concept and design: Elise M. Fiala, Michael V. Ortiz, Jennifer A. Kennedy, Jennifer M. Kalish, and Michael F. Walsh. Acquisition, analysis, or interpretation of the data: Elise M. Fiala, Michael V. Ortiz, Jennifer A. Kennedy, Dominik Glodzik, Megan Harlan Fleischut, Kelly A. Duffy, Evan R. Hathaway, Todd Heaton, Justin T. Gerstle, Peter Steinherz, Neerav Shukla, Nicole McNeer, Nancy Bouvier, Karen Cadoo, Maria I. Carlo, Alicia Latham, Marianne Dubard Gault, Vijai Joseph, Yelena Kemel, Alex Kentsis, Zsofia Stadler, Michael La Quaglia, Elli Papaemmanuil, Danielle Friedman, Arupa Ganguly, Andrew Kung, Kenneth Offit, Jennifer M. Kalish, and Michael F. Walsh. Drafting of the article: Elise M. Fiala and Michael F. Walsh. Critical revision of the article: Elise M. Fiala, Michael V. Ortiz, Jennifer A. Kennedy, Dominik Glodzik, Megan Harlan Fleischut, Kelly A. Duffy, Evan R. Hathaway, Todd Heaton, Justin T. Gerstle, Peter Steinherz, Neerav Shukla, Nicole McNeer, Nancy Bouvier, Karen Cadoo, Maria I. Carlo, Alicia Latham, Marianne Dubard Gault, Vijai Joseph, Yelena Kemel, Alex Kentsis, Zsofia Stadler, Michael La Quaglia, Elli Papaemmanuil, Danielle Friedman, Arupa Ganguly, Andrew Kung, Kenneth Offit, Jennifer M. Kalish, and Michael F. Walsh.
The first 3 authors contributed equally to this article.
We thank the patients and their families and the Robert and Kate Niehaus Center for Inherited Cancer Genomics for funding this work. In addition, we thank Audrey Mauguen, PhD, for input regarding statistical methods.
The copyright line for this article was changed on 11 June 2020 after original online publication.
Contributor Information
Jennifer M. Kalish, KALISHJ@email.chop.edu.
Michael F. Walsh, Email: walshm2@mskcc.org.
References
- 1. Mody RJ, Wu YM, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314:913‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336‐2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walsh M, Wu G, Edmonson M, et al. Incidence of germline mutations in cancer‐predisposition genes in children with hematologic malignancies: a report from the Pediatric Cancer Genome Project. Blood. 2014;124. [Google Scholar]
- 4. Parsons DW, Roy A, Yang Y, et al. Diagnostic yield of clinical tumor and germline whole‐exome sequencing for children with solid tumors. JAMA Oncol. 2016;2:616‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morak M, Schackert HK, Rahner N, et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur J Hum Genet. 2008;16:804‐811. [DOI] [PubMed] [Google Scholar]
- 6. Chan TL, Yuen ST, Kong CK, et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet. 2006;38:1178‐1183. [DOI] [PubMed] [Google Scholar]
- 7. Hitchins MP, Wong JJ, Suthers G, et al. Inheritance of a cancer‐associated MLH1 germ‐line epimutation. N Engl J Med. 2007;356:697‐705. [DOI] [PubMed] [Google Scholar]
- 8. Gelli E, Pinto AM, Somma S, et al. Evidence of predisposing epimutation in retinoblastoma. Hum Mutat. 2019;40:201‐206. [DOI] [PubMed] [Google Scholar]
- 9. Killian JK, Miettinen M, Walker RL, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6:268ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shuman C, Beckwith JB, Weksberg R. Beckwith‐Wiedemann syndrome In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Library of Medicine, National Institutes of Health; 1993. [Google Scholar]
- 11. Brioude F, Kalish JM, Mussa A, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith‐Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14:229‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang L, Cunningham JM, Winters JL, et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63:5209‐5212. [PubMed] [Google Scholar]
- 13. Szyf M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012;4:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalish JM, Doros L, Helman LJ, et al. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res. 2017;23:e115‐e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brioude F, Lacoste A, Netchine I, et al. Beckwith‐Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediat. 2013;80:457‐465. [DOI] [PubMed] [Google Scholar]
- 16. Slatter RE, Elliott M, Welham K, et al. Mosaic uniparental disomy in Beckwith‐Wiedemann syndrome. J Med Genet. 1994;31:749‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. MacFarland SP, Duffy KA, Bhatti TR, et al. Diagnosis of Beckwith‐Wiedemann syndrome in children presenting with Wilms tumor. Pediatr Blood Cancer. 2018;65:e27296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scott RH, Douglas J, Baskcomb L, et al. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet. 2008;40:1329‐1334. [DOI] [PubMed] [Google Scholar]
- 19. Hoyme HE, Seaver LH, Jones KL, Procopio F, Crooks W, Feingold M. Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet. 1998;79:274‐278. [PubMed] [Google Scholar]
- 20. Scott RH, Murray A, Baskcomb L, et al. Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget. 2012;3:327‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suzuki M, Kato M, Yuyan C, et al. Whole‐genome profiling of chromosomal aberrations in hepatoblastoma using high‐density single‐nucleotide polymorphism genotyping microarrays. Cancer Sci. 2008;99:564‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coffee B, Muralidharan K, Highsmith WE Jr, Lapunzina P, Warren ST. Molecular diagnosis of Beckwith‐Wiedemann syndrome using quantitative methylation‐sensitive polymerase chain reaction. Genet Med. 2006;8:628‐634. [DOI] [PubMed] [Google Scholar]
- 23. Lee MP, DeBaun M, Randhawa G, Reichard BA, Elledge SJ, Feinberg AP. Low frequency of p57KIP2 mutation in Beckwith‐Wiedemann syndrome. Am J Hum Genet. 1997;61:304‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li M, Squire J, Shuman C, et al. Imprinting status of 11p15 genes in Beckwith‐Wiedemann syndrome patients with CDKN1C mutations. Genomics. 2001;74:370‐376. [DOI] [PubMed] [Google Scholar]
- 25. Lalonde E, Ebrahimzadeh J, Rafferty K, et al. Molecular diagnosis of somatic overgrowth conditions: a single‐center experience. Mol Genet Genomic Med. 2019;7:e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kessler L, Adams R, Mighion L, Walther S, Ganguly A. Prenatal diagnosis in haemophilia A: experience of the genetic diagnostic laboratory. Haemophilia. 2014;20:e384‐e391. [DOI] [PubMed] [Google Scholar]
- 27. Azzi S, Steunou V, Rousseau A, et al. Allele‐specific methylated multiplex real‐time quantitative PCR (ASMM RTQ‐PCR), a powerful method for diagnosing loss of imprinting of the 11p15 region in Russell Silver and Beckwith Wiedemann syndromes. Hum Mutat. 2011;32:249‐258. [DOI] [PubMed] [Google Scholar]
- 28. Ozyilmaz B, Kirbiyik O, Ozdemir TR, et al. The efficiency of SNP‐based microarrays in the detection of copy‐neutral events at 15q11.2 and 11p15.5 loci. J Pediatr Genet. 2019;9:9‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mussa A, Duffy KA, Carli D, Ferrero GB, Kalish JM. Defining an optimal time window to screen for hepatoblastoma in children with Beckwith‐Wiedemann syndrome. Pediatr Blood Cancer. 2019;66:e27492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chao LY, Huff V, Tomlinson G, Riccardi VM, Strong LC, Saunders GF. Genetic mosaicism in normal tissues of Wilms' tumour patients. Nat Genet. 1993;3:127‐131. [DOI] [PubMed] [Google Scholar]
- 32. Maas SM, Vansenne F, Kadouch DJ, et al. Phenotype, cancer risk, and surveillance in Beckwith‐Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A. 2016;170:2248‐2260. [DOI] [PubMed] [Google Scholar]
- 33. Mussa A, Molinatto C, Baldassarre G, et al. Cancer risk in Beckwith‐Wiedemann syndrome: a systemic review and meta‐analysis outlining a novel (epi)genotype specific histotype targeted screening protocol. J Pediatr. 2016;176:142‐149. [DOI] [PubMed] [Google Scholar]
- 34. Breslow N, Olshan A, Beckwith JB, Green DM. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993;21:172‐181. [DOI] [PubMed] [Google Scholar]
- 35. Charlton J, Irtan S, Bergeron C, Pritchard‐Jones K. Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med. 2017;19:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weksberg R, Shen DR, Fei YL, Song QL, Squire J. Disruption of insulin‐like growth factor 2 imprinting in Beckwith‐Wiedemann syndrome. Nat Genet. 1993;5:143‐150. [DOI] [PubMed] [Google Scholar]
- 37. Maschietto M, Charlton J, Perotti D, et al. The IGF signalling pathway in Wilms tumours–a report from the ENCCA Renal Tumours Biology–driven drug development workshop. Oncotarget. 2014;5:8014‐8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trautmann M, Menzel J, Bertling C, et al. FUS‐DDIT3 fusion protein–driven IGF‐IR signaling is a therapeutic target in myxoid liposarcoma. Clin Cancer Res. 2017;23:6227‐6238. [DOI] [PubMed] [Google Scholar]
- 39. Arnaldez FI, Helman LJ. Targeting the insulin growth factor receptor 1. Hematol Oncol Clin North Am. 2012;26:527‐542, vii‐viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rusch M, Nakitandwe J, Shurtleff S, et al. Clinical cancer genomic profiling by three‐platform sequencing of whole genome, whole exome and transcriptome. Nat Commun. 2018;9:3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Downing JR, Wilson RK, Zhang J, et al. The Pediatric Cancer Genome Project. Nat Genet. 2012;44:619‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hansmann T, Pliushch G, Leubner M, et al. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early‐onset sporadic breast cancer. Hum Mol Genet. 2012;21:4669‐4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tufarelli C, Stanley JA, Garrick D, et al. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet. 2003;34:157‐165. [DOI] [PubMed] [Google Scholar]