

Abstract
Miniaturization and acceleration of synthetic chemistry is an emerging area in pharmaceutical, agrochemical, and materials research and development. Herein, we describe the synthesis of iminopyrrolidine‐2‐carboxylic acid derivatives using chiral glutamine, oxo components, and isocyanide building blocks in an unprecedented Ugi‐3‐component reaction. We used I‐DOT, a positive‐pressure‐based low‐volume and non‐contact dispensing technology to prepare more than 1000 different derivatives in a fully automated fashion. In general, the reaction is stereoselective, proceeds in good yields, and tolerates a wide variety of functional groups. We exemplify a pipeline of fast and efficient nanomole‐scale scouting to millimole‐scale synthesis for the discovery of a useful novel reaction with great scope.
Keywords: automation, miniaturization, multicomponent reactions, nanoscale synthesis, sustainable chemistry
The synthesis of iminopyrrolidine‐2‐carboxylic acid derivatives using chiral glutamine, oxo components, and isocyanide building blocks in an unprecedented Ugi 3‐component reaction is described. I‐DOT was used to prepare more than 1000 different derivatives in a fully automated fashion. The reaction is stereoselective, proceeds in good yields, and tolerates a wide variety of functional groups.

Synthesis and exploration of chemical space is usually a sequential, expensive, and slow process. However, acceleration and miniaturization of synthetic chemistry is becoming more and more important for the timely discovery of novel drugs and materials in an age of big data and artificial intelligence.1 For example, nanoscale optimization procedures to explore the scope and limitations of Pd‐catalyzed cross‐coupling reactions were recently described.2 Miniaturization of organic chemistry to a nanoscale is not only time‐ and cost‐effective but also dramatically reduces reagent and solvent consumption and, thus, the ecological footprint of chemistry.3
As part of our ongoing interest in proteolysis targeting chimeras (PROTACs), we aimed to produce libraries centered around the glutarimide pharmacophore addressing the E3 ligase Cereblon using multicomponent reactions (MCR).4 For this, we went back to a report of the use of glutamine (Gln) in the Ugi reaction (U‐5C‐4CR), yielding highly substituted glutarimide derivatives (Figure 1 A).5 Surprisingly, the repetition of the reaction in our hands resulted in the formation of hitherto unprecedented iminopyrrolidine‐2‐carboxylic acid derivatives (P), as proven by an X‐ray structure analysis (Figure 1 A,C).
Figure 1.
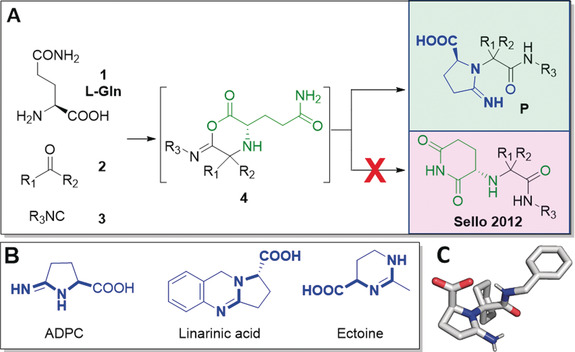
Evolution of iminopyrrolidines. A) The Ugi‐type reaction of l‐Gln with oxo components and isocyanides yields iminopyrrolidines (P) and not glutarimides. B) Biologically active compounds based on the iminopyrrolidine pharmacophore. C) Crystal structure of compound I‐D18, proving the structural outcome of the U‐3CR.20
Opportunistically, we figured that the iminopyrrolidine moiety shares key pharmacophoric and structural features of a number of biologically relevant compounds (Figure 1 B).6 For example (−)‐linarinic acid has anti‐ischemia‐properties, ectoine is an osmoprotectant that helps organisms survive extreme osmotic stress,7 and 5‐amino‐3,4‐dihydro‐2H‐pyrrole‐2‐carboxylate (ADPC) is an artificially engineered osmoprotectant.8 Traditional methods for the synthesis of ADPC start from the commercial pyroglutamatic or glutamic acid exhibiting a long target synthesis time, harsh reaction conditions, difficult‐to‐obtain and expensive starting materials and reagents, and limited diversity of substituents.9 Thus, novel synthetic routes towards the iminopyrrolidine moiety are in high demand. We felt that our newly discovered Ugi‐pathway towards the iminopyrrolidine scaffold shows unique advantages and is, therefore, worthwhile to further exploit.
Initially, we applied various reaction conditions in order to find the most suitable protocol to obtain the target molecules (Supporting Information, Table S1). For this purpose, we used l‐glutamine, cyclohexanone A12, and benzyl isocyanide B23 in the Ugi reaction to yield I‐D18. An extensive survey of solvent, concentration, and time revealed the optimized reaction conditions of the “green” solvent mixture of ethanol/water 1:1 at room temperature for 16 h at a reactant concentration of 0.5 m (Scheme 1). The desired product I‐D18 was formed in 83 % isolated yield under mild room temperature conditions.
Scheme 1.
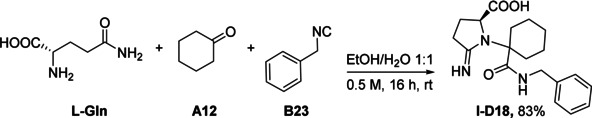
Optimized condition for the U‐5C‐3CR.
Missing structural complexity and broad functional‐group compatibility are well‐described issues of many synthetic methods, which has also been named the “dark space of chemical reactions”.2b Therefore a platform that can enable systematic reaction evaluation and data capture to survey the dark space of chemical reactions and help to define practical limitations of chemical reactions is of great use. Following our encouraging initial results, we tested the generality of the one‐pot cyclization protocol on a diverse set of 44 oxo components and 44 different isocyanides using automated nanoscale synthesis (Figure 2), not taking into account stereochemistry, theoretically 1936 combinations. The building block complexity ranged from simple aliphatic (A1, A5) to polycyclic (B10), 3‐ to 6‐membered ring size (A13, A3, A42, B24), from aromatic to heterocyclic, including indole (A41, B26), tetrahydropyran (A9), tetrahydrofuran (B14), furan (A30), morpholine (B5, B22), piperazine (A17), thiophene (A24). Multiple functional groups were tested for reaction compatibility, including hydroxyl (A2), ether (B42), thioether (A7), amine (B5, B22), ester (B1, B31, B37), carbamate (B9, B11), acetal (B12), nitrile (A14, B43), ene (A15, A19, A29), azide (B32), and halogens (A34, A36, A37, A44, B15).
Figure 2.
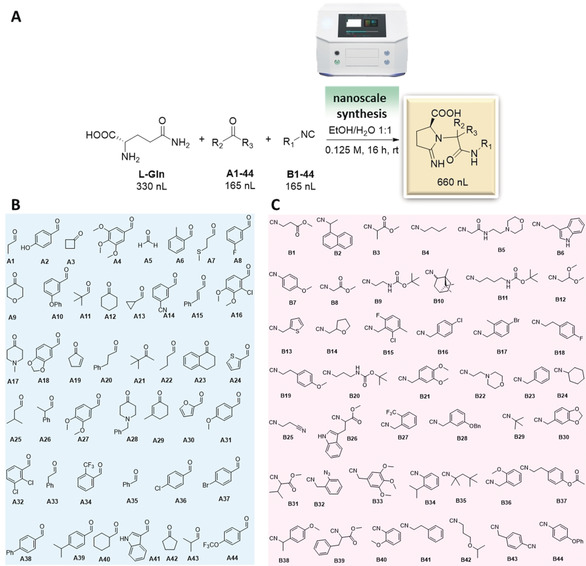
Diversity‐oriented nanoscale synthesis of iminopyrrolidines. A) Reaction condition for nanoscale synthesis. B) Oxo component building blocks. C) Isocyanide building blocks.
The synthesis was performed with a nano dispensing instrument based on the Immediate drop‐on‐demand technology (I‐DOT), a non‐contact, pressure‐based dispensing technology (Figure 2 A).10 Applying a well‐defined pressure pulse on top of a microliter plate with holes in the bottom of each well forms a highly precise nanoliter droplet that is released into any target plate. We used 384‐well polypropylene destination plates for our syntheses. Each destination plate was charged with a total of 660 nL of reagents with a final concentration of 0.125 M. Three 384‐well plates were filled using an algorithm developed in‐house that allows for random combinations of building block dispersions thus removing any synthesis bias (Supporting Information, Page S5). The total length of the dispensing procedure was 15 min per 384 well‐plate.
Next, the analysis of the plates was performed using direct mass spectrometry (MS) (Figure 3 and Supporting Information, Pages S91 and S92). As described recently, the reaction mixtures were injected directly into the mass spectrometer after 16 h reaction time.11 The reaction success was automatically categorized using our software developed in‐house according to three different classes (Supporting Information, Page S43). Reactions showing a major MS peak for the M+H, M+Na, M+K, showing the corresponding peaks but not as highest peaks or not showing the compounds peaks at all were classified as green, yellow, and blue, respectively (Figure 3 and Supporting Information, Figure S3).
Figure 3.
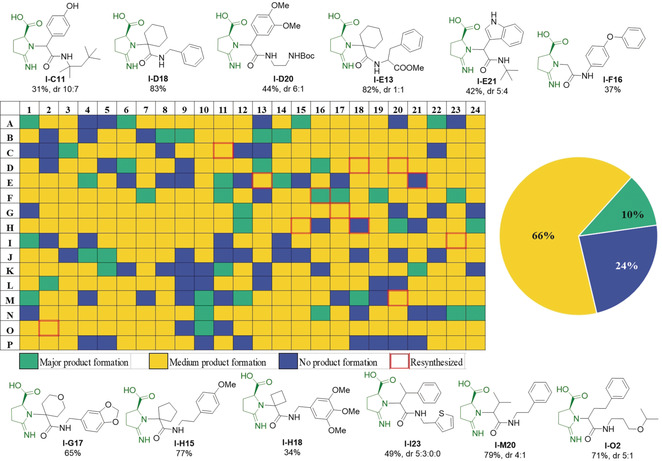
A 384‐well synthesis plate, direct MS‐based quality control, and resynthesized compounds on a mmol scale with isolated yields. I‐E21 and I‐H18 are shown blue because of I‐DOT reagent transfer failure, however their synthesis showed medium product formation.
12 compounds from a 384‐well plate were randomly selected for resynthesis on a mmol scale to determine yields, for thorough analytical characterization and further investigation of the stereochemistry of the reaction (Figure 3 and Supporting Information, Figure S6). Cyclic ketones gave good yields (I‐D18, 83 %, and I‐E13, 82 %), less so cyclobutanone (I‐H18, 34 %). Furthermore, aldehydes gave poor yields at room temperature, while a slight increase of temperature to 40 °C enhanced the yields (I‐C11, I‐D20, I‐E21, I‐F16, I‐I23, I‐M20, I‐O2). In comparison with aromatic aldehydes (I‐C11, I‐D20), aliphatic aldehydes gave higher yields (I‐I23, I‐M20, I‐O2). Paraformaldehyde, in combination with aromatic isocyanide, resulted in I‐F16 in only 37 % yield. Benzyl isocyanides with electron‐donating groups, as well as all aliphatic isocyanides, were applied in this reaction to give the corresponding products with good to moderate yields. Amino acid isocyanide furnished product in good yields (I‐E13, 82 %).
When chiral substrates are used in the Ugi reaction, such as α‐amino acids, there is uncertainty about stereo retention and racemization during the reaction.7a, 12 Thus, a critical question is whether the stereochemical integrity of the chiral glutamine center is maintained during the reaction and whether the formation of the 5‐membered ring takes place without racemization. Therefore, the synthesis of I‐D18 using enantiopure d‐Gln and l‐Gln, and racemic d/l‐Gln was carried out under the same reaction conditions. The corresponding products obtained were subjected to chiral SFC‐MS analysis (SFC= supercritical fluid chromatography, Supporting Information, Figure S7). The results indicated that the reaction is working with stereo retention in the Gln stereocenter. In the case of prochiral oxo‐component usage, diastereomers are formed (Figure 3).
A plausible reaction mechanism is shown in Scheme 2. Firstly, glutamine reacts with the oxo‐component to form the imine. Protonation by the carboxylic‐acid group activates the imine, forming the iminium ion (A) and prepares for the nucleophilic addition of the isocyanide to give the nitrilium ion (B). The nucleophilic trapping of this intermediate (B) by the carboxylic acid counter anion affords the cyclic imidoyl species (C). This intermediate undergoes a “vicarious Mumm‐rearrangement” due to the impossibility of the Mumm‐rearrangement to occur (which would lead to highly strained α‐lactams). Then, the intermediate (D) forms, which undergoes the intramolecular nucleophilic attack of the amide carbon by the secondary amine to give the final product (P). The products are charged zwitterions and in the solid‐state form an intramolecular hydrogen bond between the carboxyl group and the exocyclic imine featuring an 8‐membered ring (Supporting Information, Figure S8).
Scheme 2.
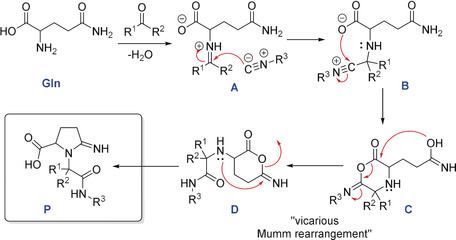
Proposed reaction mechanism.
The toolbox of transformations used in modern drug discovery is heavily biased toward fewer than ten transformations.13 The overuse of these most popular reactions in medicinal chemistry leads to a limited, crowded, and narrow chemistry space.14 Commercial availability of reagents, high robustness of the reactions, and pressure on delivery were proposed as reasons for the overuse of a handful of reactions, while many new synthetic methodologies are neglected.13a However, new synthetic methodologies result in making and exploring structures that were previously inaccessible.15 Amongst those are multicomponent reactions that have the benefit of simple one‐pot procedures, almost unlimited scaffold diversity, and great functional group compatibility.16 Herein, we describe such a novel transformation leading to unprecedented iminopyrrolidines in a one‐pot MCR procedure. The products are non‐planar zwitterionic amino acids formed by the stereo‐selective and ‐preserving reaction of chiral glutamine with oxo components and isocyanides according to a complex Ugi reaction mechanism. We used automated I‐DOT nano dispensing to perform a “real world” evaluation of the new reaction in more than 1000 examples. The benefit of the nanoscale synthesis is a rapid automated evaluation of the reaction, synthesis of a large chemical space while saving lots of chemicals and potentially environmentally hazardous solvent, thus considerably reducing the ecological footprint of the chemistry. The total chemical consumption in precious reagents and solvent was less than 0.6 mg for the 1152 reactions performed on a nanoscale, which compares very favorable to approximately 400 g of reagents and 12 L of solvents when the same number of reactions is done on a mmol scale. The automation aspect of our work synergizes and is of equal importance to synthesize a large chemical space that is clearly beyond human capabilities.17 Moreover, it reduces errors, increases speed and safety, leads to better reproducibility, and more efficient and cheaper workflow.18 Finally, the big data generated can be potentially leveraged by machine‐learning software.19 Altogether the results of our miniaturized, automated, and accelerated platform established a useful new reaction towards drug‐like compounds with great functional group compatibility.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
S.S. was supported by a postdoctoral research fellowship from Kankerbestrijding (KWF grant agreement No 10504). This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement No. 754425. Moreover, funding was coming from the National Institute of Health (2R01GM097082‐05), the European Lead Factory (IMI) under grant agreement number 115489, the Qatar National Research Foundation (NPRP6‐065‐3‐012), the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska‐Curie (ITN “Accelerated Early stage drug dIScovery”, grant agreement No 675555; Cofund ALERT, (665250)). We thank Robin van der Straat, Hylke Middel, and Alexander Jurkowitsch for assistance in the chiral separation, helping with software development, and for technical assistance with the I‐DOT, respectively.
A. Osipyan, S. Shaabani, R. Warmerdam, S. V. Shishkina, H. Boltz, A. Dömling, Angew. Chem. Int. Ed. 2020, 59, 12423.
In memory of Rolf Huisgen
References
- 1. Mattes D. S., Jung N., Weber L. K., Bräse S., Breitling F., Adv. Mater. 2019, 31, 1806656. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Uehling M. R., King R. P., Krska S. W., Cernak T., Buchwald S. L., Science 2019, 363, 405; [DOI] [PubMed] [Google Scholar]
- 2b. Lin S., Dikler S., Blincoe W. D., Ferguson R. D., Sheridan R. P., Peng Z., Conway D. V., Zawatzky K., Wang H., Cernak T., Davies I. W., DiRocco D. A., Sheng H., Welch C. J., Dreher S. D., Science 2018, 361, eaar6236; [DOI] [PubMed] [Google Scholar]
- 2c. Gesmundo N. J., Sauvagnat B., Curran P. J., Richards M. P., Andrews C. L., Dandliker P. J., Cernak T., Nature 2018, 557, 228–232; [DOI] [PubMed] [Google Scholar]
- 2d. Cernak T., Gesmundo N. J., Dykstra K., Yu Y., Wu Z., Shi Z.-C., Vachal P., Sperbeck D., He S., Murphy B. A., Sonatore L., Williams S., Madeira M., Verras A., Reiter M., Lee C. H., Cuff J., Sherer E. C., Kuethe J., Goble S., Perrotto N., Pinto S., Shen D.-M., Nargund R., Balkovec J., DeVita R. J., Dreher S. D., J. Med. Chem. 2017, 60, 3594–3605; [DOI] [PubMed] [Google Scholar]
- 2e. Buitrago Santanilla A., Regalado E. L., Pereira T., Shevlin M., Bateman K., Campeau L.-C., Schneeweis J., Berritt S., Shi Z.-C., Nantermet P., Liu Y., Helmy R., Welch C. J., Vachal P., Davies I. W., Cernak T., Dreher S. D., Science 2015, 347, 49; [DOI] [PubMed] [Google Scholar]
- 2f. Perera D., Tucker J. W., Brahmbhatt S., Helal C. J., Chong A., Farrell W., Richardson P., Sach N. W., Science 2018, 359, 429; [DOI] [PubMed] [Google Scholar]
- 2g. Collins K. D., Gensch T., Glorius F., Nat. Chem. 2014, 6, 859–871. [DOI] [PubMed] [Google Scholar]
- 3. Wong H., Cernak T., Curr. Opin. Green Sustain. Chem. 2018, 11, 91–98. [Google Scholar]
- 4. Konstantinidou M., Li J., Zhang B., Wang Z., Shaabani S., Ter Brake F., Essa K., Dömling A., Expert Opin. Drug Discovery 2019, 14, 1255–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Totaro K. A., Okandeji B. O., Sello J. K., ChemBioChem 2012, 13, 987–991. [DOI] [PubMed] [Google Scholar]
- 6. Mukherjee P., Cinelli M. A., Kang S., Silverman R. B., Chem. Soc. Rev. 2014, 43, 6814–6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Appel D., Lentzen G., J. Enzyme Inhib. Med. Chem. 2009, 24, 1106–1108; [DOI] [PubMed] [Google Scholar]
- 7b. He Y.-Z., Gong J., Yu H.-Y., Tao Y., Zhang S., Dong Z.-Y., Microb. Cell Fact. 2015, 14, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Witt E. M. H. J., Davies N. W., Galinski E. A., Appl. Microbiol. Biotechnol. 2011, 91, 113–122. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Castellanos L., Duque C., Zea S., Espada A., Rodríguez J., Jiménez C., Org. Lett. 2006, 8, 4967–4970; [DOI] [PubMed] [Google Scholar]
- 9b. Lee M., Lown J. W., J. Org. Chem. 1987, 52, 5717–5721; [Google Scholar]
- 9c. Tian Y., Ma C., Feng L., Zhang L., Hao F., Pan L., Cheng M., Arch. Pharm. 2012, 345, 423–430. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Benz M., Molla M. R., Böser A., Rosenfeld A., Levkin P. A., Nat. Commun. 2019, 10, 2879; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Schober L., Büttner E., Laske C., Traube A., Brode T., Traube A. F., Bauernhansl T., J. Lab. Autom. 2015, 20, 154–163. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Wleklinski M., Falcone C. E., Loren B. P., Jaman Z., Iyer K., Ewan H. S., Hyun S.-H., Thompson D. H., Cooks R. G., Eur. J. Org. Chem. 2016, 5480–5484; [Google Scholar]
- 11b. Shaabani S., Xu R., Ahmadianmoghaddam M., Gao L., Stahorsky M., Olechno J., Ellson R., Kossenjans M., Helan V., Dömling A., Green Chem. 2019, 21, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Khoury K., Sinha M. K., Nagashima T., Herdtweck E., Dömling A., Angew. Chem. Int. Ed. 2012, 51, 10280–10283; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10426–10429; [Google Scholar]
- 12b. Berłożecki S., Szymański W., Ostaszewski R., Synth. Commun. 2008, 38, 2714–2721; [Google Scholar]
- 12c. Ugi I., Demharter A., Hörl W., Schmid T., Tetrahedron 1996, 52, 11657–11664. [Google Scholar]
- 13.
- 13a. Brown D. G., Boström J., J. Med. Chem. 2016, 59, 4443–4458; [DOI] [PubMed] [Google Scholar]
- 13b. Schneider N., Lowe D. M., Sayle R. A., Tarselli M. A., Landrum G. A., J. Med. Chem. 2016, 59, 4385–4402. [DOI] [PubMed] [Google Scholar]
- 14. Barker A., Kettle J. G., Nowak T., Pease J. E., Drug Discovery Today 2013, 18, 298–304. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Blakemore D. C., Castro L., Churcher I., Rees D. C., Thomas A. W., Wilson D. M., Wood A., Nat. Chem. 2018, 10, 383–394; [DOI] [PubMed] [Google Scholar]
- 15b. Boström J., Brown D. G., Young R. J., Keserü G. M., Nat. Rev. Drug Discovery 2018, 17, 709–727. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Dömling A., Chem. Rev. 2006, 106, 17–89; [DOI] [PubMed] [Google Scholar]
- 16b. Dömling A., Wang W., Wang K., Chem. Rev. 2012, 112, 3083–3135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c. Bienaymé H., Hulme C., Oddon G., Schmitt P., Chem. Eur. J. 2000, 6, 3321–3329; [DOI] [PubMed] [Google Scholar]
- 16d. Cioc R. C., Ruijter E., Orru R. V. A., Green Chem. 2014, 16, 2958–2975. [Google Scholar]
- 17. Jensen K. F., Coley C. W., Eyke N. S., Angew. Chem. Int. Ed. 2019, 10.1002/anie.201909987; [DOI] [Google Scholar]; Angew. Chem. 2019, 10.1002/ange.201909987. [DOI] [Google Scholar]
- 18. Coley C. W., Eyke N. S., Jensen K. F., Angew. Chem. Int. Ed. 2019, 10.1002/anie.201909989; [DOI] [Google Scholar]; Angew. Chem. 2019, 10.1002/ange.201909989. [DOI] [Google Scholar]
- 19. Chen H., Engkvist O., Wang Y., Olivecrona M., Blaschke T., Drug Discovery Today 2018, 23, 1241–1250. [DOI] [PubMed] [Google Scholar]
- 20.CCDC 1940862 contains the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary