

Abstract
Proteins require an optimal balance of conformational flexibility and stability in their native environment to ensure their biological functions. A striking example is spidroins, spider silk proteins, which are stored at extremely high concentrations in soluble form, yet undergo amyloid‐like aggregation during spinning. Here, we elucidate the stability of the highly soluble N‐terminal domain (NT) of major ampullate spidroin 1 in the Escherichia coli cytosol as well as in inclusion bodies containing fibrillar aggregates. Surprisingly, we find that NT, despite being largely composed of amyloidogenic sequences, showed no signs of concentration‐dependent aggregation. Using a novel intracellular hydrogen/deuterium exchange mass spectrometry (HDX‐MS) approach, we reveal that NT adopts a tight fold in the E. coli cytosol and in this manner conceals its aggregation‐prone regions by maintaining a tight fold under crowded conditions. Fusion of NT to the unstructured amyloid‐forming Aβ40 peptide, on the other hand, results in the formation of fibrillar aggregates. However, HDX‐MS indicates that the NT domain is only partially incorporated into these aggregates in vivo. We conclude that NT is able to control its aggregation to remain functional under the extreme conditions in the spider silk gland.
Keywords: hydrogen/deuterium exchange mass spectrometry, intracellular protein folding, protein aggregation, spider silk
Intracellular protein folding. The N‐terminal domain (NT) from the spider silk protein MaSp1 is highly soluble and facilitates the expression of amyloid‐forming proteins. Here, we use intracellular hydrogen/deuterium exchange mass spectrometry to reveal that NT adopts a tight fold in the cytosol and protects its aggregation‐prone segments. NT is able to remain partially folded inside amyloid‐like aggregates, suggesting that its stability may play a role in spider silk assembly.
Abbreviations
- Aβ40
amyloid β 1–40
- Aβ42
amyloid β 1–42
- ETD
electron‐transfer dissociation
- HDX‐MS
hydrogen/deuterium exchange mass spectrometry
- MaSp1 NT
major ampullate spidroin 1 N‐terminal domain
- nESI
nano‐electrospray ionization
- ThT
thioflavin T
Introduction
Highly ordered protein aggregates termed amyloid‐like fibrils are a hallmark of several human disorders such as Alzheimer’s and Parkinson’s diseases, but also a common feature of some functional protein assemblies, such as spider silk 1, 2. In both cases, aggregation is driven by the ability of individual protein segments to adopt β‐strand conformations and readily assemble into fibrils, for example, via tight, zipper‐like interfaces 3. It has been proposed that proteins have therefore evolved to protect aggregation‐prone segments from spontaneous self‐assembly by trapping them in a specific three‐dimensional fold 4, 5, 6. This ability to ‘self‐chaperone’ is concentration‐dependent, as the number of protein molecules spontaneously exposing aggregation‐prone segments increases with protein concentration 7.
It is therefore remarkable how spider silk, one of the toughest polymers in nature, is able to control its own conversion from a soluble concentrated dope into the β‐strand‐rich fibres that constitute mature silk 8. Spider silk proteins, spidroins, are stored in the sac of the silk gland at concentrations exceeding 40% w/v as micelle‐like oligomers with helical and disordered conformations 9, 10, 11. During spinning, the long repetitive regions of the spidroins are converted into β‐sheet crystals connected by disordered linkers 12. This process is controlled by a small C‐terminal domain, which likely serves to align the spidroins and trigger their α‐helix to β‐strand conversion 13, 14, and a 130‐residue N‐terminal domain (NT), which undergoes pH‐dependent dimerization and connects the spidroin oligomers 15, 16, 17, 18, 19, 20, 21, 22.
Importantly, its role in the spinning process requires that NT does not undergo aggregation, but retains its dimeric α‐helical conformation. This ability is particularly noteworthy as the highly concentrated surrounding proteins are simultaneously converted into β‐strand aggregates. Although it is not clearly established which conformation NT has in the mature silk, the pH shift at the end of the gland’s spinning duct suggests that NT dimerization occurs at the final stages of the spinning process 14.
To understand how the structure of NT is affected by crowding and aggregation, we have developed an intracellular hydrogen/deuterium exchange mass spectrometry (HDX‐MS) approach that enables us to study the global fold of NT in live Escherichia coli in its soluble form, as well as in amyloid‐like inclusion bodies. We find that NT, despite containing multiple predicted steric zipper segments, is refractory to aggregation even at high concentrations. The protein adopts a tight conformation in the cytosol, which serves to protect it from aggregation, and is partially maintained even when the protein is linked to a peptide that forms fibrillar aggregates.
Results and Discussion
NT is resistant to concentration‐dependent aggregation
The assembly of spider silk involves the conversion of soluble spidroins in disordered and α‐helical conformations into β‐sheet‐rich aggregates, which manifests itself in a strong propensity to undergo amyloid‐like aggregation in vitro. As the first step, we sought to understand how this aggregation propensity is encoded in the spidroin sequence. We therefore analysed the distribution of potentially amyloidogenic regions in the three distinct domains (N‐terminal domain, repeat domain and C‐terminal domain) of the full‐length major ampullate spidroin 1 (MaSp1) from the orb‐weaving spider Euprosthenops australis. Amyloidogenic regions were predicted using the ZipperDB server (http://services.mbi.ucla.edu/zipperdb), which identifies sequence motifs able to form self‐complementary ‘steric zippers’ that are the basis for amyloid‐like aggregation (Fig. 1A) 4, 23. The resulting aggregation profiles show that the entire MaSp1 sequence is extraordinarily rich in potentially amyloidogenic sequences. Approximately 65% of the NT sequence is predicted to have a high propensity for β‐strand aggregation. Notably, these segments are almost exclusively located in the helical regions of the protein. The repeat domain shows an even distribution of disordered glycine‐rich and zipper‐forming polyalanine regions that closely match its structure in the mature silk fibre. The MaSp1 C‐terminal domain contains multiple self‐complementary regions, in line with its ability to form fibrils in vitro 14. Therefore, all MaSp1 domains contain significantly more steric zipper sequences than the human and E. coli proteome averages of 18.8% and 22.0%, respectively, as illustrated by the ZipperDB profile of horse heart myoglobin (Fig. 1A) 4.
Figure 1.
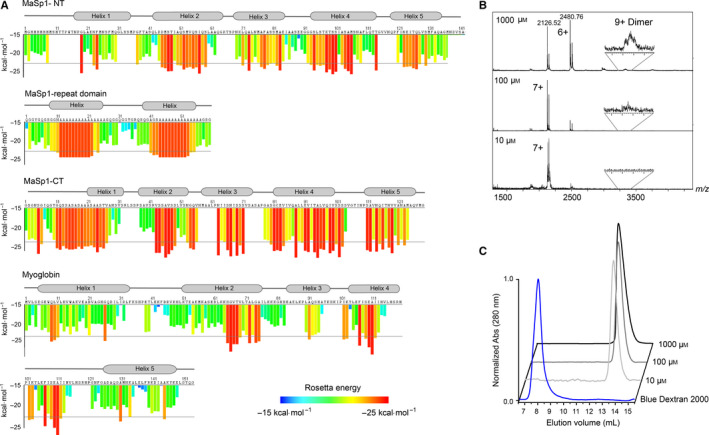
The highly soluble NT domain contains multiple aggregation‐prone segments. (A) Steric zipper profiles of the Eu. australis MaSp1 N‐terminal domain (top), two repeat sequences (second from the top) and the C‐terminal domain (third from the top) show a high density of sequences that are capable of forming steric zippers below the Rosetta energy threshold of −23 kJ·mol−1. For comparison, the steric zipper profile of horse heart myoglobin, an α‐helical protein of similar size, shows less than 30% zipper sequences (bottom). Profiles were generated using the ZipperDB server (http://services.mbi.ucla.edu/zipperdb). The locations of secondary structure elements are indicated above the sequence. For the C‐terminal domain, the secondary structure is based on homology with the Araneus diadematus CT. Notably, the aggregation‐prone sites coincide with the helical segments of the protein. (B) Native MS analysis of NT in 100 mm ammonium acetate, pH 7.5, shows a decrease in the average ion charge but no increase in oligomer formation at high protein concentrations. (C) Size exclusion chromatography of NT was performed at pH 7.5 with protein concentrations of 10, 100 and 1000 μm. The resulting chromatograms show no notable shift in oligomeric state at any concentration.
While aggregation‐prone sequences can be expected in the repeat and CT domains, their presence in the NT domain is surprising. During the preparation of recombinant NT, we previously noted that the protein did not display a tendency to aggregate even at high concentrations. We tested the maximum achievable concentration for NT using spin concentrators and reached 310 mg·mL−1 in phosphate buffer as judged by absorbance at 280 nm. Notably, we found no signs of aggregation according to the absorbance at 320 nm as well as spin separation of soluble and insoluble protein fractions 15, 24.
We used native mass spectrometry to investigate whether NT is susceptible to nonspecific oligomer formation at high concentrations (Fig. 1B). By recording spectra under gentle ionization conditions from native‐like solution conditions, it is possible to monitor concentration‐dependent changes in the oligomeric states of soluble proteins 25. MS spectra of 10, 100 and 1000 μm NT at pH 7.5 show a reduction in average charge from 6.8+ to 6.3+ at the highest concentrations, but only a negligible increase in dimers. No higher oligomers could be observed at the highest protein concentration. Next, we performed size exclusion chromatography of NT at the same concentrations and observed no concentration‐dependent shift in retention time of NT, indicating that the protein remains monomeric also in solution (Fig. 1C). These findings show that despite its potentially amyloidgenic sequence, NT is not susceptible to nonspecific self‐association and remains folded even at high concentrations. The in vitro stability of the NT domain therefore differs significantly from those of the repeat and CT domains, despite their similarly high content in aggregation‐prone sequences.
NT adopts a tight structure inside cells
Since the NT domain has evolved to exert its function in an environment filled with mostly identical proteins, we then asked how NT is able to protect its aggregation‐prone regions from aberrant interactions under crowded physiological conditions. The E. coli cytosol contains up to 40% proteins w/v and has a pH between 7.4 and 7.6 under normal growth conditions 26, 27. When overexpressed, nontoxic small proteins commonly achieve an intracellular concentration above 2 mm and account for about 30% of the total protein content 28. Bacterial overexpression of NT thus captures several features of the native environment of the silk gland.
We used HDX‐MS to compare the global fold of the NT domain in dilute solutions and inside live E. coli. HDX‐MS measures the exchange of backbone amide hydrogens for deuterium, providing information about the backbone structural dynamics and accessibility 29. Previous intracellular HDX‐MS strategies relied on embedding cells in acidic matrix to quench the exchange and ionize the analyte using laser desorption 30. However, this approach has limitations compared to nano‐electrospray ionization (nESI) which allows top‐down sequencing of the labelled proteins. Capitalizing on recent developments in the mass spectrometric analysis of overexpressed proteins directly from bacterial lysates 31, 32, we developed a strategy where bacteria containing the overexpressed protein are diluted in > 100‐fold excess of deuterated buffer to initiate labelling, while maintaining the physiological pH range to avoid distortion of the pH homeostasis of the bacteria. Deuterium penetration of E. coli under standard conditions occurs on the timescale of seconds, as judged by NMR and MS measurements 30, 33. After a fixed time, the labelling reaction is quenched by lysis of the bacteria in cold acidic buffer. The labelled protein is extracted from the lysate by centrifugation and then directly infused into a high‐resolution mass spectrometer to measure deuterium incorporation with isotopic resolution (Fig. 2A). For quenching and lysis, 10% formic acid was used. While the resulting pH of 1.8 of the bacterial lysate is below the optimal quenching pH of 2.2, these conditions were found to enable the detection of the soluble and insoluble proteins used in this study (see Methods).
Figure 2.
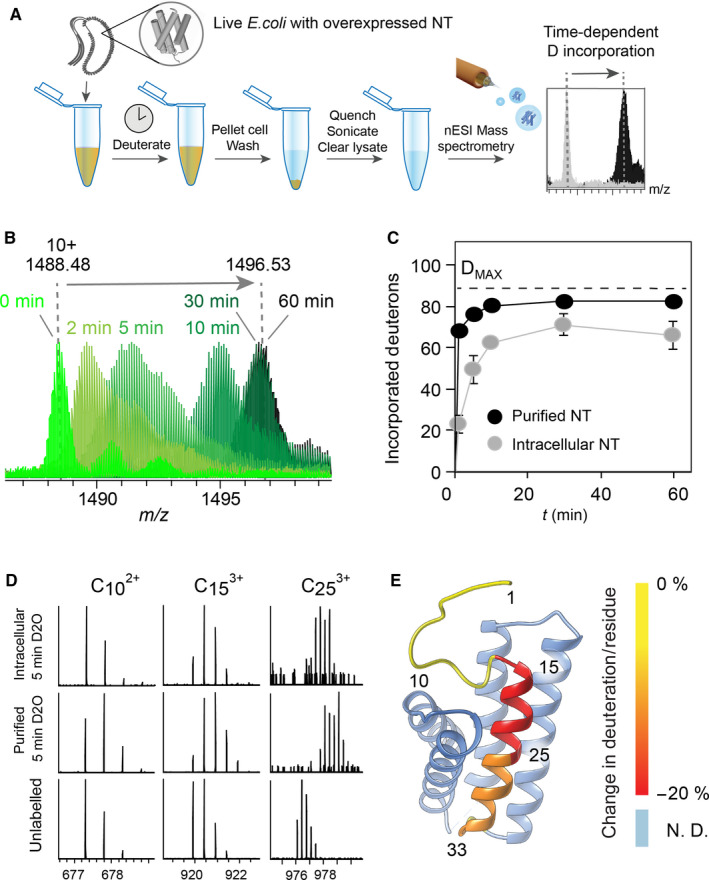
Intracellular NT adopts a tight fold. (A) Schematic representation of the in‐cell HDX‐MS strategy. Live Escherichia coli overexpressing NT are suspended in deuterated PBS. At each time‐point, the exchange is quenched by addition of 10% formic acid, the cells are lysed by sonication, and the soluble fraction is isolated by centrifugation and subjected to direct MS analysis. (B) Overlaying the 10+ charge state in mass spectra of NT deuterated for 0–60 min inside live E. coli shows how the protein shifts to the higher m/z region with increasing incubation time, indicating time‐dependent deuteration. (C) Absolute deuterium uptake of purified NT (black circles) and NT in live E. coli (grey circles) plotted against deuteration time shows slower and lower labelling of the intracellular protein. Error bars indicate standard deviations (n = 6). (D) Deuterated NT was subjected to top‐down sequencing by ETD to reveal the local deuterium uptake in the N‐terminal region of NT. ETD fragments covering residues 1–10, 1–15 and 1–25 show slower deuterium uptake in the intracellular protein than in the purified protein. (E) The relative difference in deuterium uptake between purified and intracellular NT after 5 min of deuteration in solution and inside cells was calculated from the ETD fragments and mapped on the protein structure using ucsf chimera 45. The colour shift from yellow (no difference protection) to red (increased protection inside cells) shows increased protection of helix 1 in the intracellular protein.
nESI‐MS spectra of full‐length NT in quenched bacterial lysates closely match those of the purified protein under denaturing conditions. When the cells were incubated for 1–60 min in deuterated PBS followed by quenching and lysis, we readily observed time‐dependent deuteration. Interestingly, for short labelling times (2 and 5 min), we observe broadening of the isotope distributions, suggesting some conformational heterogeneity (Fig. 2B). To be able to compare the deuterium uptake of intracellular NT to that of the purified protein, we recorded global HDX‐MS of pure NT using similar dilution factors and the same quenching and sonication protocol as for intracellular labelling (see Methods). Importantly, deuteration of the purified protein was carried out at pH 7.5, matching the pH inside the cell. The purified protein reaches near‐complete deuteration within 10 min, in good agreement with previous studies 16. The intracellular protein, on the other hand, exhibited significantly reduced deuterium uptake, reaching maximal labelling after 30 min and incorporating a total of 70 deuterons (Fig. 2C). Next, we performed top‐down sequencing of purified and intracellular NT after 5 min of deuteration by subjecting the protein to electron‐transfer dissociation in the absence of collisional activation to prevent scrambling of the backbone hydrogens 34. In this manner, we could obtain well‐resolved peaks for fragments covering the 33 N‐terminal residues of NT and calculate the average deuterium uptake per residue for each fragment (Fig. 2D). Comparison with unlabelled NT revealed that the N‐terminal helix indeed becomes more protected in the intracellular protein (Fig. 2E).
The increasing protection of the NT backbone from amide hydrogen exchange suggests that the protein adopts a tight fold in the cytosol. Negatively charged proteins can exhibit increased conformational stability due to charge repulsions with their environment 33, 35. We speculate that the negatively charged NT may behave in a similar manner under crowded cytosolic conditions. The ability to undergo structural tightening in the cell is therefore encoded in the primary structure 36. Although the effect likely is nonspecific, it reduces the ability of NT to expose amyloidogenic segments and could in this manner increase its resilience to aggregation.
Fibril formation mediated by an exposed amyloidogenic sequence does not result in global NT aggregation
To test the conformational stability of NT inside cells, we asked whether adding an exposed unstructured amyloidogenic sequence can drive the protein into aggregation. We therefore fused the D40K/K65D NT variant (NT*), which does not dimerize at low pH 24, to the amyloid β 1–40 (Aβ40) peptide separated by an enzymatic cleavage site (Fig. 3A). NT* was chosen since it yields higher expression levels than NT 24, 37 and thus enables detection of the fusion protein in the bacterial lysate. Importantly, NT* has the same structure and charge as the wild‐type protein 24. While both variants were found to have similar solubility in E. coli, we have used the isolated NT* domain for solubility studies 24. Monomeric Aβ40 is disordered in solution and readily forms β‐sheet fibrils under physiological conditions and in the E. coli cytosol 38. We expressed NT* with the C‐terminal Aβ40 extension and an 8‐residue connecting sequence in E. coli and compared its solubility to free NT* expressed under identical conditions. To test whether Aβ40 induces the formation of fibrillar aggregates, we added the β‐sheet‐specific dye thioflavin T (ThT), which increases its fluorescence yield upon binding to amyloid‐like aggregates, to lysates of cells expressing either NT*, NT*‐Aβ40 fusion protein or Met‐Aβ42 as positive control (Fig. 3B). The resulting fluorescence emission spectra reveal the presence of β‐sheet structures in the NT*‐Aβ40‐ and the Met‐Aβ42‐containing lysates. To confirm that NT*‐Aβ40, but not NT*, indeed forms intracellular aggregates, we prepared the insoluble fractions of cells expressing either NT* or the NT*‐Aβ40 fusion protein. SDS/PAGE analysis shows the formation of inclusion bodies composed of NT*‐Aβ40 (Fig. 3C). Since the NT domain and the Aβ40 peptide are separated by an enzymatic cleavage site, we then added the tobacco etching virus (TEV) protease to the inclusion bodies. We observed that the enzyme is readily able to release NT* from the aggregates, suggesting that the cleavage site in the fusion protein remains partially accessible (Fig. 3D). Together, these results indicate that the addition of an unguarded amyloidogenic sequence can drive amyloid‐like aggregation when attached to NT*.
Figure 3.
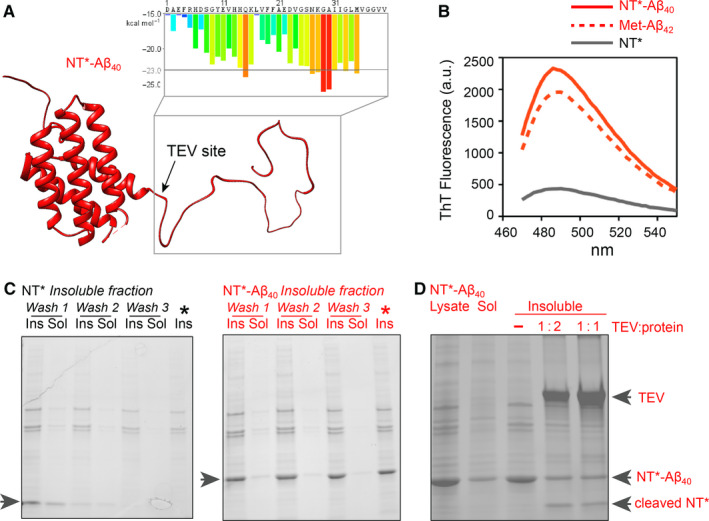
An unguarded amyloidogenic segment triggers fibril formation but not NT aggregation. (A) Predicted structure of NT*‐Aβ40 (red) with the ZipperDB profile of the C‐terminal Aβ40 sequence indicated. (B) Cells expressing NT*‐Aβ40 but not NT* only stain positive for β‐sheet aggregates. Lysates from 1 mL of cells expression NT*‐Aβ40 or NT* were subjected to fluorescence measurements with the amyloid‐specific dye ThT. Fluorescence was detected in the lysate from cells expressing the NT*‐Aβ40 fusion protein (red line), but not NT* alone (grey line). The ThT fluorescence of cell lysate containing Met‐Aβ42 is shown as a positive control (dashed red line). (C) SDS/PAGE of the soluble (sol) and insoluble (insol) fractions of Escherichia coli expressing either NT* or NT*‐Aβ40 shows that the fusion protein is located in inclusion bodies. A representative example from two repeats is shown. (D) Inclusion bodies composed of NT*‐Aβ40 can be cleaved with TEV protease. Inclusion bodies were prepared from the bacterial lysate and mixed with TEV at a 1 : 1 or 1 : 2 ratio (w/w) and incubated for 2 h. TEV was able to cut the aggregated fusion protein at the specific cleavage site between NT* and Aβ40 to release free NT*. A representative example from two repeats is shown.
Next, we used intracellular HDX‐MS to assess whether amyloid‐like aggregation involves the entire fusion protein. To directly compare the deuterium uptake of the aggregated NT*‐Aβ40 to that of soluble NT, deuterium labelling, quenching and MS analysis were performed using a 1 : 1 mixture of cells expressing either NT or the fusion protein (Fig. 4A). At each time‐point, the difference in deuterium incorporation between the two proteins was determined from the shifts of their peaks in the same MS spectrum (Fig. 4B). However, it should be noted that the NT*‐Aβ40 fusion protein gave weaker MS signals than the NT domain, indicating lower solubility. Despite being a total of 48 residues longer, the NT*‐Aβ40 fusion protein exhibited slower deuterium uptake and incorporated up to 20 deuterons less than NT alone (Fig. 4C). The significantly higher protection suggests that the protein is partially inaccessible for deuterium labelling. However, since we are able to cleave the fusion protein inside inclusion bodies (Fig. 3D), we speculated that the protein is only partially incorporated into the aggregates. To gain more insights into the role of the NT domain on aggregation, we first performed intracellular HDX of Met‐Aβ42. Met‐Aβ42 is completely incorporated into fibrillar inclusion bodies in E. coli and can therefore serve as a comparison for the degree of labelling expected for a fully aggregated peptide 39. Indeed, we observed nearly no deuterium incorporation, suggesting near‐total protection from exchange (Fig. 4D). The deuterium uptake of the NT*‐Aβ40 fusion protein is thus intermediate between the soluble NT domain and the fully aggregated Met‐Aβ42 peptide. To test this, we performed top‐down sequencing of NT*‐Aβ40 following 5 and 30 min of deuteration. Due to the lower signal intensity compared to soluble NT (Fig. 4B), we were not able to obtain sufficiently intense MS signals for peptides beyond residue 15 at the beginning of helix 1. However, analysis of the deuterium uptake in this region shows lower deuterium incorporation compared to the nonaggregated protein, as well as a time‐dependent increase in labelling (Fig. 4E). This is particularly noteworthy as the region in question is mostly unstructured in solution, and may indicate a generally low penetration with deuterated solvent.
Figure 4.
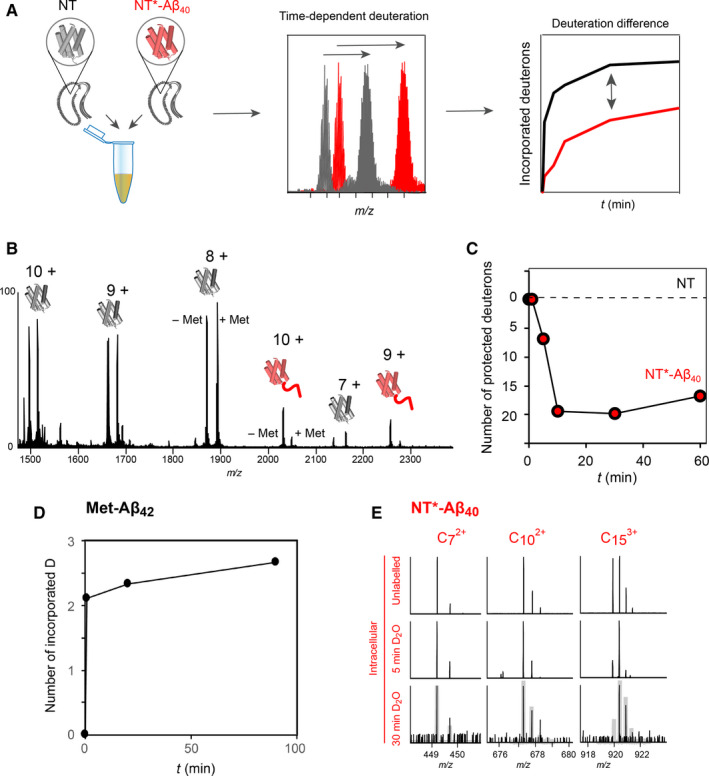
Differential HDX‐MS of intracellular NT*‐Aβ40 aggregates. (A) The strategy for comparative intracellular HDX‐MS. Labelling is performed on a 1 : 1 mixture of bacteria expressing either NT or NT*‐Aβ40, with the subsequent steps as in Fig. 2A. The difference in deuteration between the two proteins is calculated from the same MS spectrum at each time‐point. (B) The mass spectrum of the lysate from a 1 : 1 mixture of bacteria expressing NT or NT*‐Aβ40 shows that both proteins can be detected, but with lower signal intensity for NT*‐Aβ40. (C) Subtracting the absolute deuterium incorporation of NT from that of NT*‐Aβ40 at each time‐point shows that the fusion protein is deuterated slower and incorporates 20 D less than the NT domain alone, suggesting that the aggregated fusion protein is significantly more protected from exchange. (D) Intracellular HDX‐MS of Met‐Aβ42, which is fully incorporated into amyloid‐like fibrils, shows low levels of deuterium incorporation even after 90 min. (E) Top‐down analysis of unlabelled NT*‐Aβ40 (top) and following 5‐min (middle) or 30‐min (bottom) intracellular deuterium labelling. The ETD fragments show that the N‐terminal 15 residues of NT*‐Aβ40 take 30 min to reach the same degree of labelling as intracellular NT after 5 min (shown as grey traces).
Taken together, we conclude that decreased deuterium uptake in the fusion protein relative to NT alone is due to multiple factors. Comparison with Met‐Aβ42 suggests that aggregation of the unguarded Aβ40 moiety drives the fusion protein to form ThT‐positive inclusion bodies. The juxtapositioned NT domain, on the other hand, remains partially accessible to enzymatic cleavage, but displays a lower degree of deuterium labelling than the free protein. Considering the tendency of NT to tighten its fold under crowded conditions, it therefore appears possible that folded NT domains are buried in the large amorphous inclusion bodies, resulting in an intermediate labelling regime compared to soluble NT. It is also possible that the NT domain unfolds and is partially converted in an amyloid‐like state, but this would likely result in inaccessibility for enzymatic release of the entire NT domain. In either case, the presence of the NT domain results in partially accessible and structured intracellular aggregates, providing a possible rationale for its stabilizing effect when used as an expression tag for poorly soluble peptides 24, 37.
Implications for the structure of NT during silk spinning
Although the role of NT as cross‐linking domain during silk spinning is well‐established, its structure in the mature silk has not been elucidated. NMR studies on silk spun from an artificial mini‐spidroin composed of the N‐ and C‐terminal domains as well as two repeat units recently indicated that the NT domain retains its dimeric structure in the fibre 40. Remarkably, the low‐end pH during which NT dimerizes is reached only in the distal region of the spinning duct, shortly before extrusion of the silk 14. At this stage, the spidroin dope is already highly concentrated and exposed to significant shear force, yet NT needs to remain folded and able to undergo specific structural changes in order to exert its function 19, 20. In vitro, NT dimerization is unaffected by concentrations as high as 1 mm 19. In line with these requirements, we have found that NT can still dimerize in a pH‐dependent manner at high concentrations 19, which is particularly remarkable considering its high content in potentially amyloidogenic segments. A possible explanation is provided by our observation that NT adopts a tighter 3‐dimensional fold in response to the crowded intracellular environment. This ability reduces the exposure of aggregation‐prone sites and thus lowers the chance of self‐association as the protein concentration increases 7. Importantly, NT also needs to be able to rapidly undergo pH‐induced conformational changes 41 and likely uses the extremely high methionine content in its sequence to retain the required structural plasticity without comprising stability 42. The ability of NT to safeguard its aggregation‐prone segments under crowded conditions gives rise to its extreme solubility and is therefore a prerequisite for its function in silk assembly.
Methods
Chemicals
All reagents were purchased from Sigma (St. Louis, MO, USA) unless noted otherwise.
Protein expression and purification
BL21(DE3) cells were grown to an OD600 of 0.7 at 30 °C. Expression of NT, NT*, NT*‐Aβ40 or Met‐Aβ42 was induced from a T7 promotor by adding 1 mm IPTG and carried out for 18 h at 20 °C. NT was purified as described previously 19 .
Size exclusion chromatography
Purified NT was diluted to 10, 100 and 1000 µm in running buffer (20 mm NaPi, 154 mm NaCl, 1 mm EDTA, pH 7.5) and applied on a Superdex 75 Increase 10/300 GL column (GE Healthcare, Chicago, IL, USA) using a 250 µL loop. Elution of protein was followed by measuring the absorbance at 280 nm. Blue Dextran 2000 was applied to determine the void volume. SEC curves have been individually corrected for their baseline and normalized for their respective maximum.
Thioflavin T fluorescence
One millilitre of cells expressing either NT*, NT*‐Aβ40 or Met‐Aβ42 was collected at an OD600 of 2.0 by centrifugation at 3000 g for 2 min. Pellets were washed and resuspended in 1 mL cold PBS and cells lysed by sonication (1 min total, 1‐s on, 1‐s off, amplitude 40%). ThT was added from a 1 mm stock solution to a final concentration of 5 μm to 100 μL lysate in black Corning Costar 96‐well plates with clear flat‐bottom wells (Corning, NY, USA). Fluorescence emission spectra were recorded on a Tecan Spark 20M multimode spectrofluorometer (Tecan Instruments, Männedorf, Switzerland). Excitation wavelength was 450 nm with a 1 nm bandwidth, and emission was recorded between 470 and 550 nm in 2 nm steps with 5 nm bandwidth and an integration time of 40 μs.
Preparation of inclusion bodies
One millilitre of E. coli culture expressing either NT* or NT*‐Aβ40 was collected by centrifugation at 3000 g for 2 min, and the supernatant was discarded. The pellet was resuspended in 1 mL 10 mm Tris buffer, pH 7.5. Cells were lysed by sonication on ice (1 min total, 1‐s on, 1‐s off, amplitude 40%), and the insoluble material was separated by centrifugation for 8 min at 21 000 g and 4 °C. Inclusion bodies were further purified through three additional rounds of sonication and centrifugation 43.
TEV cleavage
One millilitre of E. coli culture expressing NT*‐Aβ40 was collected by centrifugation at 3000 g for 2 min, and the supernatant was discarded. The pellet was resuspended in 1 mL 10 mm Tris buffer, pH 7.5. Cells were lysed, and the insoluble material was separated as described previously. Fusion protein cleavage was initiated in 10 mm Tris and 100 mm NaCl buffer, pH 7.5, by adding TEV protease (T4455 Sigma) at 1 : 2 and 1 : 1 ratio (TEV : protein w/w). Samples were incubated for 2.5 h at 30 °C and analysed with SDS/PAGE.
Hydrogen/deuterium exchange
For intracellular HDX, cells from 1 mL of E. coli culture expressing either NT, NT*, NT*‐Aβ40 or Met‐Aβ42 were collected at an OD600 of 2.0 by centrifugation at 3000 g for 2 min. The pellet was resuspended in 1 mL deuterated PBS and incubated at room temperature. Following incubation for 1, 5, 10, 30 or 60 min, the cells were pelleted for 2 min at 3000 g at 4 °C and resuspended in 700 μL quenching solution on ice. In order to most efficiently quench the exchange and be able to directly infuse the quenched lysate into the MS by nano‐ESI, we tested different ratios of H2O/formic acid. Due to the presence of large amounts of cell debris in the lysate, low amounts of formic acid (0.2%) resulted in a pH close to 4.0 leading to protein precipitation and poorly resolved spectra (data not shown). For NT and NT*, 1% formic acid yielded well‐resolved MS spectra; however, we found that when using the same quick sonication protocol, NT*‐Aβ40 or Met‐Aβ42 from inclusion bodies could only be detected at formic acid concentrations of 10% or higher. Since the rate of back exchange depends on the pH of the quenching solution, we therefore performed the quenching reactions for all proteins in this study in 10% formic acid in order to be able to directly compare deuteration. After quenching, cells were immediately lysed by sonication on ice (1 min total, 1‐s on, 1‐s off, amplitude 40%) and the debris was removed by centrifugation for 8 min at 21 000 g and 4 °C. Four microlitre of supernatant was then loaded into a borosilicate capillary (Thermo, Waltham, MA, USA) and directly infused into the mass spectrometer. For HDX of purified NT, 4 μL of 3 mm NT stock solution in 20 mm phosphate buffer, pH 7.5, was diluted in 36 μL deuterated PBS and incubated at room temperature for 1, 5, 10, 30 or 60 min, as well as 24 h as end time‐point. Exchange was quenched by adding 460 μL 10% formic acid to a final protein concentration of 3 μm. The quenched purified protein samples were then subjected to the same centrifugation and sonication regime as the cell lysates. The total handling time from adding quenching solution to MS analysis was kept constant at 14 min for all proteins and conditions.
Mass spectrometry
Deuterium‐exchanged proteins were directly infused into an Orbitrap Fusion Mass Spectrometer (Thermo) using an off‐line nanospray source. The mass spectrometer was operated in intact protein mode. The capillary voltage was 1.7 kV, and the source temperature was 80 °C. Spectra were acquired in the Orbitrap mass analyser operated at a resolution of 120 000 at m/z 200 between m/z 1000 and 6000 and analysed using the excalibur software package (Thermo). For top‐down analysis, the 10+ charge state of NT was selected in the quadrupole region of the mass spectrometer. Fragmentation was performed by electron‐transfer dissociation in the ion trap with a reaction time of 50 ms and no additional collisional activation to prevent backbone hydrogen scrambling. Protein fragments were detected in the Orbitrap between m/z 100 and 2000 and manually assigned using the theoretical fragmentation calculated by the ProteinProspector server (http://prospector.ucsf.edu). The average deuteration for each residue was calculated as described 44.
For native MS of NT, 30 µL of 3 mm NT stock solution in 20 mm phosphate buffer, pH 7.5, was buffer‐exchanged into 1 m ammonium acetate, pH 7.5, using 0.5 mL Zeba Spin columns (Thermo) and diluted to final concentrations of 1000 µm, 100 μm or 10 μm in 1 m ammonium acetate. Protein solutions were directly infused into a Waters LCT ToF Mass Spectrometer equipped with an off‐line nanospray source and modified for intact MS of large protein complexes (MS Vision, Almere, The Netherlands) using coated borosilicate capillaries (Thermo). The capillary voltage was 1.5 kV, the source temperature 20 °C and the cone voltage 100 V. The source pressure was maintained at 1.2 mbar for maximum collisional cooling. Spectra were acquired between m/z 1000 and 20 000 and analysed using masslynx 41 (Waters, Elstree, UK).
Protein structures were visualized using UCSF Chimera 45.
Protein sequences
NT: MGHHHHHHMSHTTPWTNPGLAENFMNSFMQGLSSMPGFTASQLDDMSTIAQSMVQSIQSLAAQGRTSPNKLQALNMAFASSMAEIAASEEGGGSLSTKTSSIASAMSNAFLQTTGVVNQPFINEITQLVSMFAQAGMNDVSA
NT* (mutations in bold): MGHHHHHHMSHTTPWTNPGLAENFMNSFMQGLSSMPGFTASQLDKMSTIAQSMVQSIQSLAAQGRTSPNDLQALNMAFASSMAEIAASEEGGGSLSTKTSSIASAMSNAFLQTTGVVNQPFINEITQLVSMFAQAGMNDVSA
NT*‐Aβ40 (TEV cleavage site underlined, Aβ40 in italics): MGHHHHHHMSHTTPWTNPGL AENFMNSFMQGLSSMPGFTASQLDDMSTIAQSMVQSIQSLAAQGRTSPNKLQALNMAFASSMAEIAASEEGGGSLSTKTSSIASAMSNAFLQTTGVVNQPFINEITQLVSMFAQAGMNDVSAGNSENLYFQ DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV
Met‐Aβ42: MDAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AR, JJ and ML designed the study with input from HJ, SL and DPL. MK, AL, GC and MS expressed and purified protein. MK, MG‐U and ML performed mass spectrometry measurements. CS and NF performed protein characterization. KN, NK, AA and HB provided constructs and reagents. MK, AR, JJ and ML wrote the paper with input from all authors.
Acknowledgements
ML is supported by an Ingvar Carlsson Award from the Swedish Foundation for Strategic Research, a KI Faculty‐funded Career Position, a KI‐StratNeuro Starting Grant and a VR Starting Grant (2019‐01961). CS is supported by a Novo Nordisk Foundation Postdoctoral Fellowship (NNF19OC0055700). SL is supported by Barncancerfonden grant TJ2014‐0013. DPL is supported by VR grant 2013_08807. JJ and AR acknowledge support from the Center for Innovative Medicine at Karolinska Institutet (CIMED), VR and Vinnova. ML gratefully acknowledges technical support from MS Vision, NL.
Contributor Information
Anna Rising, Email: anna.rising@ki.se.
Jan Johansson, Email: janne.johansson@ki.se.
Michael Landreh, Email: michael.landreh@ki.se.
References
- 1. Landreh M, Sawaya MR, Hipp MS, Eisenberg DS, Wüthrich K & Hartl FU (2016) The formation, function and regulation of amyloids: insights from structural biology. J Intern Med 280, 164–176. [DOI] [PubMed] [Google Scholar]
- 2. Greenwald J & Riek R (2010) Biology of amyloid: structure, function, and regulation. Structure 18, 1244–1260. [DOI] [PubMed] [Google Scholar]
- 3. Eisenberg DS & Sawaya MR (2017) Structural studies of amyloid proteins at the molecular level. Annu Rev Biochem 86, 69–95. [DOI] [PubMed] [Google Scholar]
- 4. Goldschmidt L, Teng PK, Riek R & Eisenberg D (2010) Identifying the amylome, proteins capable of forming amyloid‐like fibrils. Proc Natl Acad Sci USA 107, 3487–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teng PK, Anderson NJ, Goldschmidt L, Sawaya MR, Sambashivan S & Eisenberg D (2012) Ribonuclease A suggests how proteins self‐chaperone against amyloid fiber formation. Protein Sci 21, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Landreh M, Johansson J, Rising A, Presto J & Jörnvall H (2012) Control of amyloid assembly by autoregulation. Biochem J 447, 185–192. [DOI] [PubMed] [Google Scholar]
- 7. Chiti F & Dobson CM (2009) Amyloid formation by globular proteins under native conditions. Nat Chem Biol 5, 15–22. [DOI] [PubMed] [Google Scholar]
- 8. Andersson M, Johansson J & Rising A (2016) Molecular sciences silk spinning in silkworms and spiders. Int J Mol Sci 17, 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jin HJ & Kaplan DL (2003) Mechanism of silk processing in insects and spiders. Nature 424, 1057–1061. [DOI] [PubMed] [Google Scholar]
- 10. Parent LR, Onofrei D, Xu D, Stengel D, Roehling JD, Addison JB, Forman C, Amin SA, Cherry BR, Yarger JL et al (2018) Hierarchical spidroin micellar nanoparticles as the fundamental precursors of spider silks. Proc Natl Acad Sci USA 115, 11507–11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andersson M, Jia Q, Abella A, Lee XY, Landreh M, Purhonen P, Hebert H, Tenje M, Robinson CV, Meng Q et al (2017) Biomimetic spinning of artificial spider silk from a chimeric minispidroin. Nat Chem Biol 13, 262–264. [DOI] [PubMed] [Google Scholar]
- 12. Vollrath F & Knight DP (2001) Liquid crystalline spinning of spider silk. Nature 410, 541–548. [DOI] [PubMed] [Google Scholar]
- 13. Hagn F, Eisoldt L, Hardy JG, Vendrely C, Coles M, Scheibel T & Kessler H (2010) A conserved spider silk domain acts as a molecular switch that controls fibre assembly. Nature 465, 239–242. [DOI] [PubMed] [Google Scholar]
- 14. Andersson M, Chen G, Otikovs M, Landreh M, Nordling K, Kronqvist N, Westermark P, Jörnvall H, Knight S, Ridderstråle Y et al (2014) Carbonic anhydrase generates CO2 and H+ that drive spider silk formation via opposite effects on the terminal domains. PLoS Biol 12, e1001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Askarieh G, Hedhammar M, Nordling K, Saenz A, Casals C, Rising A, Johansson J & Knight SD (2010) Self‐assembly of spider silk proteins is controlled by a pH‐sensitive relay. Nature 465, 236–238. [DOI] [PubMed] [Google Scholar]
- 16. Landreh M, Askarieh G, Nordling K, Hedhammar M, Rising A, Casals C, Astorga‐Wells J, Alvelius G, Knight SD, Johansson J et al (2010) A pH‐dependent dimer lock in spider silk protein. J Mol Biol 404, 328–336. [DOI] [PubMed] [Google Scholar]
- 17. Hagn F, Thamm C, Scheibel T & Kessler H (2011) PH‐dependent dimerization and salt‐dependent stabilization of the N‐terminal domain of spider dragline silk – implications for fiber formation. Angew Chemie Int Ed 50, 310–313. [DOI] [PubMed] [Google Scholar]
- 18. Landreh M, Andersson M, Marklund EG, Jia Q, Meng Q, Johansson J, Robinson CV & Rising A (2017) Mass spectrometry captures structural intermediates in protein fiber self‐assembly. Chem Commun 53, 3319–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kronqvist N, Otikovs M, Chmyrov V, Chen G, Andersson M, Nordling K, Landreh M, Sarr M, Jörnvall H, Wennmalm S et al (2014) Sequential pH‐driven dimerization and stabilization of the N‐terminal domain enables rapid spider silk formation. Nat Commun 5, 3254. [DOI] [PubMed] [Google Scholar]
- 20. Schwarze S, Zwettler FU, Johnson CM & Neuweiler H (2013) The N‐terminal domains of spider silk proteins assemble ultrafast and protected from charge screening. Nat Commun 4, 2815. [DOI] [PubMed] [Google Scholar]
- 21. Jaudzems K, Askarieh G, Landreh M, Nordling K, Hedhammar M, Jörnvall H, Rising A, Knight SD & Johansson J (2012) PH‐dependent dimerization of spider silk N‐terminal domain requires relocation of a wedged tryptophan side chain. J Mol Biol 422, 477–487. [DOI] [PubMed] [Google Scholar]
- 22. Gaines WA, Sehorn MG & Marcotte WR (2010) Spidroin N‐terminal domain promotes a pH‐dependent association of silk proteins during self‐assembly. J Biol Chem 285, 40745–40753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D & Eisenberg D (2006) The 3D profile method for identifying fibril‐forming segments of proteins. Proc Natl Acad Sci USA 103, 4074–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kronqvist N, Sarr M, Lindqvist A, Nordling K, Otikovs M, Venturi L, Pioselli B, Purhonen P, Landreh M, Biverstål H et al (2017) Efficient protein production inspired by how spiders make silk. Nat Commun 8, 15504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu Y, Liu H, Saer RG, Zhang H, Meyer CM, Li VL, Shi L, King JD, Gross ML & Blankenship RE (2017) Native mass spectrometry analysis of oligomerization states of fluorescence recovery protein and orange carotenoid protein: two proteins involved in the cyanobacterial photoprotection cycle. Biochemistry 56, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zimmerman SB & Trach SO (1991) Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli . J Mol Biol 222, 599–620. [DOI] [PubMed] [Google Scholar]
- 27. Olsen KN, Budde BB, Siegumfeldt H, Rechinger KB, Jakobsen M & Ingmer H (2002) Noninvasive measurement of bacterial intracellular pH on a single‐cell level with green fluorescent protein and fluorescence ratio imaging microscopy. Appl Environ Microbiol 68, 4145–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Speer SL, Guseman AJ, Patteson JB, Ehrmann BM & Pielak GJ (2019) Controlling and quantifying protein concentration in Escherichia coli . Protein Sci 28, 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marcsisin SR & Engen JR (2010) Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal Bioanal Chem 397, 967–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ghaemmaghami S & Oas TG (2001) Quantitative protein stability measurement in vivo. Nat Struct Biol 8, 879–882. [DOI] [PubMed] [Google Scholar]
- 31. Gan J, Ben‐Nissan G, Arkind G, Tarnavsky M, Trudeau D, Noda Garcia L, Tawfik DS & Sharon M (2017) Native mass spectrometry of recombinant proteins from crude cell lysates. Anal Chem 89, 4398–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cveticanin J, Netzer R, Arkind G, Fleishman SJ, Horovitz A & Sharon M (2018) Estimating interprotein pairwise interaction energies in cell lysates from a single native mass spectrum. Anal Chem 90, 10090–10094. [DOI] [PubMed] [Google Scholar]
- 33. Monteith WB & Pielak GJ (2014) Residue level quantification of protein stability in living cells. Proc Natl Acad Sci USA 111, 11335–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abzalimov RR, Kaplan DA, Easterling ML & Kaltashov IA (2009) Protein conformations can be probed in top‐down HDX MS experiments utilizing electron‐transfer dissociation of protein ions without hydrogen scrambling. J Am Soc Mass Spectrom 20, 1514–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mu X, Choi S, Lang L, Mowray D, Dokholyan NV, Danielsson J & Oliveberg M (2017) Physicochemical code for quinary protein interactions in Escherichia coli . Proc Natl Acad Sci USA 114, 4556–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Danielsson J & Oliveberg M (2017) Comparing protein behaviour in vitro and in vivo, what does the data really tell us? Curr Opin Struct Biol 42, 129–135. [DOI] [PubMed] [Google Scholar]
- 37. Sarr M, Kronqvist N, Chen G, Aleksis R, Purhonen P, Hebert H, Jaudzems K, Rising A & Johansson J (2018) A spidroin‐derived solubility tag enables controlled aggregation of a designed amyloid protein. FEBS J 285, 1873–1885. [DOI] [PubMed] [Google Scholar]
- 38. Sanagavarapu K, Nuske E, Nasir I, Immink JN, Sormani P, Velendruscolo M, Knowles TPJ, Malmendal A, Lago‐Cabaleiro C & Linse S (2019) A method of predicting the in vitro fibril formation propensity of Aβ40. Sci Rep 9, 3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walsh DM, Thulin E, Minogue AM, Gustavsson N, Pang E, Teplow DB & Linse S (2009) A facile method for expression and purification of the Alzheimer’s disease‐associated amyloid β‐peptide. FEBS J 276, 1266–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Otikovs M, Andersson M, Jia Q, Nordling K, Meng Q, Andreas LB, Pintacuda G, Johansson J, Rising A & Jaudzems K (2017) Degree of biomimicry of artificial spider silk spinning assessed by NMR spectroscopy. Angew Chemie Int Ed 56, 12571–12575. [DOI] [PubMed] [Google Scholar]
- 41. Ries J, Schwarze S, Johnson CM & Neuweiler H (2014) Microsecond folding and domain motions of a spider silk protein structural switch. J Am Chem Soc 136, 17136–17144. [DOI] [PubMed] [Google Scholar]
- 42. Heiby JC, Goretzki B, Johnson CM, Hellmich UA & Neuweiler H (2019) Methionine in a protein hydrophobic core drives tight interactions required for assembly of spider silk. Nat Commun 10, 4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang L, Maji SK, Sawaya MR, Eisenberg D & Riek R (2008) Bacterial inclusion bodies contain amyloid‐like structure. PLoS Biol 6, 1791–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaltashov IA, Bobst CE & Abzalimov RR (2009) H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: is there a need for a top‐down approach? Anal Chem 81, 7892–7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC & Ferrin TE (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]