

Abstract
Oliceridine is a G protein–biased ligand at the μ‐opioid receptor in development for treatment of moderate to severe acute pain. A phase 1, open‐label, single‐dose study investigated the pharmacokinetics and safety of oliceridine 0.5 mg intravenous (IV) in subjects with end‐stage renal disease (ESRD, n = 9) versus 1 mg in healthy controls (n = 8). A second phase 1, open‐label, single‐dose study investigated the pharmacokinetics and safety of a 0.5‐mg IV dose in hepatic impairment (mild, n = 10; moderate, n = 10; severe, n = 6) versus 1 mg in healthy controls (n = 8). The controls were sex and age (±10 years) matched. In ESRD versus healthy subjects, no difference in clearance was observed between ESRD patients and subjects with normal renal function. Oliceridine clearance and AUC were not affected by hepatic impairment. Half‐life (hours; GM [%CV]) increased in subjects with moderate (4.3 [44.1]) and severe (5.8 [41.2]) impairment versus mild impairment (2.6 [20.0]) and healthy subjects (2.1 [11.3]). Volume of distribution was increased with the degree of hepatic impairment. All adverse events were mild and generally consistent with the known safety profile of oliceridine. No dose adjustment is needed in patients with renal impairment or in patients with mild or moderate hepatic impairment. Initial dose reduction should be considered in severe hepatic impairment, and patients may require fewer doses of oliceridine due to the longer half‐life observed in these patients.
Keywords: G protein–biased ligand, hepatic impairment, oliceridine, pharmacokinetics, renal impairment
Conventional opioids are widely employed for the management of moderate to severe acute pain, particularly postsurgical pain. In 2017, approximately 45 million patients in the United States received intravenous (IV) opioids in hospital settings. Studies indicate that patient satisfaction as well as the duration of hospital stays are associated with the extent and duration of pain control.1, 2 Opioid agonists such as morphine, hydromorphone, and fentanyl produce analgesia by binding to the μ‐opioid receptor. The μ‐opioid receptor is a G protein–coupled receptor that is predominantly expressed in the central nervous system and bowel. Dose‐limiting effects of opioids frequently occur1 and include sedation, respiratory depression, and gastrointestinal effects such as nausea, vomiting, and constipation. In addition, dosing of opioid agonists for more than 7 days can result in tolerance and dependence.3 Current concerns about side‐effect limitations in opioid dosing with inadequate analgesia, development of tolerance, opioid dependence, and opioid use disorder have led to a search for compounds that are more selective for analgesia and have fewer or less severe adverse effects.
There has been interest in developing drugs that selectively signal receptors or subsets of receptors, and this is sometimes referred to as “biased agonism.”4 Opioid ligands bind to μ receptors and nonselectively activate 2 separate intracellular signaling pathways: the G protein pathway that is responsible for analgesia and the β‐arrestin pathway that is responsible for opioid‐related adverse events (AEs)5 and inhibition of G protein‐mediated analgesia.5, 6, 7 β‐Arrestin–mediated opioid‐related AEs narrow the therapeutic window of conventional opioids and may limit the dosing required to achieve analgesic efficacy. G protein–biased ligands stimulate the μ receptor in a differential manner, with a preference toward the G protein pathway over the β‐arrestin pathway. Data from animal studies suggest that this differential signaling decreases the incidence and severity of respiratory depression and gastrointestinal side effects while it maintains the analgesia observed with traditional opioids.8
Oliceridine (TRV130) is the first of a new class of small‐molecule G protein–biased ligands at the μ opioid receptor that are centrally acting synthetic analgesics in development for the treatment of moderate to severe acute pain.3, 9 The compound is a μ opioid receptor ligand biased toward G protein and away from β‐arrestin postreceptor signaling and activates G protein while causing low β‐arrestin recruitment to the μ receptor. Preclinical and early clinical studies show that this novel mechanism of action can optimize μ opioid receptor pharmacology with subsequent equivalent or greater analgesia than conventional intravenous opioids and fewer respiratory and gastrointestinal adverse events.2, 8, 10, 11 At the time of this publication, oliceridine is an investigational product for the management of moderate to severe acute pain and not approved by The US Food and Drug Administration.
Metabolic clearance, primarily by oxidation with subsequent glucuronidation, is the major route of elimination of oliceridine. Oliceridine is hepatically metabolized by cytochrome P450 (CYP) 3A4 and CYP2D6, with a 50:50 contribution by each enzyme.2 Its major metabolites, TRV0109662 (the primary amine, 17% of total drug‐related material) and M22 (a stable ether glucuronide, 62% of total drug‐related material), are inactive at the μ receptor (data on file). Renal clearance is 2% to 5% of total oliceridine clearance (CL). Approximately 70% of the metabolites are eliminated in the urine, and the remainder is eliminated in the feces. In human plasma the free fraction of oliceridine is 23%.
As part of the development process, oliceridine pharmacokinetics (PK) in adults with chronic kidney disease stage 5 who were on chronic dialysis (end‐stage renal disease, ESRD) and adults with varying degrees of hepatic dysfunction were evaluated and compared with oliceridine disposition in healthy adults. Safety and tolerability in these populations were also assessed.
The aim of this article is to report the PK characteristics, safety, and tolerability of oliceridine in renal and hepatic impairment as determined in 2 phase 1 studies. The primary objective of the renal study was to evaluate the single‐dose PK of oliceridine and its primary inactive metabolite, TRV0109662, in adults with ESRD compared with subjects with normal renal function. The primary objective of the hepatic study was to evaluate the single‐dose PK of oliceridine and its metabolite TRV0109662 in adults with mild, moderate, or severe hepatic impairment compared with adults with normal hepatic function. A secondary objective in both studies was to evaluate the single‐dose safety and tolerability of oliceridine in subjects with renal or hepatic impairment in comparison with healthy subjects.
Subjects and Methods
The studies were designed in accordance with US Food and Drug Administration Guidances for Industry: Pharmacokinetics in Patients With Impaired Renal Function 12 and Pharmacokinetics in Patients With Impaired Hepatic Function.13 The impaired renal function study protocol received institutional review board approval (IntegReview, Austin, TX), and the hepatic study received independent ethics committee approvals (Czech site: Etická komise IKEM a TN Thomayerova nemocnice [Ethics Committee of the Institute for Clinical and Experimental Medicine and Thomayer Hospital]; Slovak site: Etická komisia Bratislavského samosprávneho kraja [Ethics Committee of Bratislava Self‐Governing Region]). The studies were conducted in accordance with all appropriate regulatory requirements and conducted in accordance with current Good Clinical Practice and Committee for Medicinal Products for Human Use (CHMP)/ International Conference on Harmonisation 135/95, all appropriate subject privacy requirements, and the ethical principles outlined in the Declaration of Helsinki. Informed consent for the hepatic study also included any additional elements required by local regulations. Each subject provided verbal and written informed consent before study participation.
Sample sizes were based on an estimation approach rather than a formal hypothesis‐testing approach. Eight subjects per group were targeted for enrollment to ensure evaluable PK data on at least 6 subjects per group. A matched‐subject design was used to ensure demographic balance.
Inclusion and Exclusion Criteria
For both studies, inclusion criteria were men or women aged 18 to 80 years (controls were required to be within ±10 years of the matching subjects) with body mass index of 18.0 to 35.0 kg/m2, a minimum weight of 50 kg, and to have been determined by the investigator to be appropriate to participate in the studies. Control subjects were healthy as determined by medical history, physical examination, laboratory testing, and 12‐lead ECG. For the renal study, healthy subjects were required to have a creatinine clearance ≥90 mL/min as calculated using the Cockcroft‐Gault equation.14 For the hepatic study, healthy subjects were required to have a calculated creatinine clearance ≥60 mL/min.14
Exclusion criteria for both studies included clinically significant ECG abnormalities or cardiovascular disease, history of seizures, clinically significant immune‐mediated hypersensitivity reaction to opioids, drug, or alcohol abuse within the past 6 months, positive urine drug screen or alcohol breathalyzer, use of a prohibited medication, pregnant or breastfeeding women, smoking >10 cigarettes (or equivalent) per day and inability to refrain from smoking during the first 4 hours after dosing, serology indicative of human immunodeficiency virus, hepatitis B surface antigen, or hepatitis C virus. CYP2D6 poor metabolizers (determined by blood genotyping) were excluded because oliceridine exposure in these populations might increase nonproportionally.
For the renal impairment study, renal function was determined by medical history, physical examination, and laboratory testing at screening, and met the following criteria: having chronic kidney disease stage 5 on a stable hemodialysis program (ESRD) defined by single pool measure of clearance per dialysis (kt/V, factored for subject size) with an average of ≥1.2. Subjects on hemodialysis who were hepatitis C positive could enroll if they had normal liver function tests and no evidence of clinically significant hepatic disease. In the hepatic impairment study, subjects with hepatic impairment were limited in age to 18 to 65 years and had a confirmed and documented diagnosis of cirrhosis due to parenchymal liver disease. The subjects had to have stable hepatic impairment as judged by the investigator.
Additional exclusion criteria for subjects in the hepatic impairment study included primary biliary cirrhosis, men or women of reproductive age who were not surgically sterile, using 2 forms of contraception or practicing abstinence for 90 days before and after oliceridine administration, and creatinine clearance <60 mL/min. Additional exclusion criteria for hepatically impaired subjects included a history of clinically significant esophageal bleeding, severe hepatic encephalopathy, history of liver transplantation, advanced ascites or ascites requiring paracentesis or albumin supplementation, hemoglobin <105 g/L, uncontrolled hypertension, and clinically significant hypotension. Additional exclusion criteria for healthy subjects included positive serology for hepatitis B surface antigen or hepatitis C virus antibody.
Prohibited medications in both studies included medications or supplements known to be moderate or strong inducers or inhibitors of CYP3A4 or CYP2D6 taken within 30 days before dosing or 5 half‐lives (whichever was longer), over‐the‐counter medications and herbal remedies, opioids taken within 72 hours (or 5 half‐lives, whichever was longer) before dosing and through the last PK collection, chronic opioid therapy, and strenuous activity, sunbathing, or contact sports from 96 hours before admission through the final follow‐up visit.
The following prior or concomitant medications were allowed in both studies and included medications or supplements known to be substrates of CYP3A4 or CYP2D6, or with a mild or weak degree of induction or inhibition of CYP3A4 or CYP2D6, blood lipid–regulating and blood pressure–regulating agents if treatment was stable for 30 days before screening, oral contraceptives, hormone replacement therapy (for menopausal women), diuretics, β‐blockers, topically applied medication, and occasional use of metoclopramide, ibuprofen, or acetaminophen. Additional permitted medications in the renal impairment study included those that were part of the routine care for subjects with ESRD, with allowance for changes in hemodialysis medications in accordance with existing clinical unit protocols. Prior tetrahydrocannabinol use was also allowed.
Study Design
Renal Impairment
The renal impairment study was a phase 1, open‐label, unmasked, parallel‐group, multicenter study to evaluate oliceridine PK, safety and tolerability in subjects with chronic kidney disease stage 5 on chronic hemodialysis. The study compared subjects who regularly underwent hemodialysis as part of their treatment for chronic kidney disease stage 5 (ESRD) with healthy controls, age‐ and sex‐matched at a ratio of 1:1. This was designed as a 2‐part study, but only the first part was conducted. A second part to the study was to be conducted if the ESRD group had a >50% increase in mean peak or total exposure to oliceridine compared with the healthy control group. Because the difference in mean peak and total exposure between ESRD patients and healthy subjects was <50%, further study in less severe renal impairment was not undertaken.
The study was conducted at 2 DaVita Clinical Research study centers in the United States, 1 site in Minneapolis, Minnesota and 1 site in Lakewood, Colorado.
Subjects were admitted to the research unit within 30 days of screening. All medications were withheld for 2 hours before and 2 hours after dose. All subjects were given a light breakfast containing less than 30% of calories from fats before dosing. Water was permitted ad libitum. Subjects were required to remain seated or semirecumbent for at least 4 hours after study medication administration. Dosing and study participation were coordinated with the subject's regularly scheduled dialysis treatments such that dialysis did not occur during the study.
Subjects with ESRD received a single oliceridine 0.5‐mg dose infused over 2 minutes. Healthy controls received a single oliceridine 1‐mg dose infused over 2 minutes. The 0.5‐mg dose was chosen to provide an appropriate safety margin to cover a possible increase in exposure in subjects with renal impairment. Blood was collected for PK analyses at times 0 (before dosing), 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 10, 12, 18, 24, and 36 hours after drug administration. Safety data were collected at intervals until the end of the study.
A total of 17 subjects were enrolled and completed the protocol. There were 9 subjects in the ESRD group. Nine were included in the safety analysis (anyone who received a dose of oliceridine). One subject in the ESRD group was replaced due to problems with blood sampling, so 9 were included in the PK analysis (all subjects who had sufficient data to calculate at least 1 PK parameter).
Hepatic Impairment
The hepatic impairment study was a phase 1, open‐label, parallel‐group, multicenter study to evaluate oliceridine PK, safety, and tolerability in adults with mild, moderate, and severe hepatic impairment compared with healthy subjects. Healthy controls were matched by sex, age, and body mass index.
The study was conducted at 2 clinical sites: Pharmaceutical Research Associates CZ, sro, Prague, Czech Republic and Summit Clinical Research, sro, Bratislava, Slovak Republic.
Assignment of the subjects to a hepatic impairment group was based on Child‐Pugh classification at screening.15 The hepatic impairment groups were mild (Child‐Pugh score 5‐6 points), moderate (Child‐Pugh score 7‐9 points), and severe hepatic impairment (Child‐Pugh score 10‐15 points).
Subjects were admitted to the research unit within 28 days of screening. Subjects with mild hepatic, moderate, or severe hepatic impairment received a dose of oliceridine 0.5mg infused over 2 minutes. Healthy subjects received a dose of oliceridine 1mg infused over 2 minutes. Blood samples of 3mL each were collected for PK assessments predose, at the end of infusion, and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 7, 8, 9, 10, 12, 24, 36, and 48 hours after dose administration. Blood samples were taken via an indwelling IV catheter or by direct venipuncture into K2‐EDTA (plasma). The blood for PK assessments was drawn from the arm contralateral to that used for IV infusion of the study drug.
A total of 34 subjects were enrolled and completed the protocol. Thirty‐four subjects were included in the safety data analysis: 10 with mild hepatic impairment, 10 with moderate hepatic impairment, 6 with severe hepatic impairment, and 8 with normal hepatic function. In the PK analysis set, 8 subjects (80%) were included in each of the mild and moderate hepatic impairment groups. The plasma concentrations for oliceridine and TRV0109662 of 2 subjects in each of the mild and moderate impairment groups were below the limit of quantification. No PK parameters could be calculated for these subjects, and additional subjects were enrolled as replacements. Subsequent investigation into these patients determined that they had not been dosed with oliceridine. In all subjects, TRV0109662 was quantifiable in only a few samples, and so the results are not reported here.
Sample, Pharmacokinetic, and Statistical Analyses
The concentrations of oliceridine and TRV0109662 in human plasma containing K2‐EDTA as an anticoagulant were determined using supported‐liquid extraction followed by analysis using high‐performance liquid chromatography, followed by tandem mass spectrometric detection (LC‐MS/MS). Analysis was done at Covance Indianapolis Bioanalytical (Indianapolis, Indiana) from June 2016 through January 2017. All samples were analyzed within 216 days of collection following storage at –60°C to –80°C. Long‐term frozen matrix stability has been determined for 366 days when stored at –60°C to –80°C.
Oliceridine and the internal standard TRV0110813A:2 were extracted from human plasma by supported liquid extraction. After evaporation under nitrogen, the residue was reconstituted and analyzed using LC‐MS/MS. Detection was done using MS/MS with a Sciex API 5500 using positive‐ion electrospray. The transitions monitored were 387.3→127.1 for oliceridine, 261.4→132.1 for TRV0109662, and 391.3→131.1 for the internal standard. The column used was a Waters Acquity BEH CIS, 100 × 2.1 mm, 1.7 µm particle size (Waters, Milford, Massachusetts). The mobile phase used was 0.1% formic acid in methanol. The method was fully validated (selectivity, carryover, linearity of calibration curve, precision, accuracy, recovery, matrix stability, reinjection reproducibility) for concentrations ranging from 0.0500 ng/mL (lower limit of quantification [LOQ]) to 50.0 ng/mL (upper LOQ) according to FDA guidelines. All data were acquired using Applied Biosystems/MDS‐Sciex Analyst Version 1.5, processed, and reported using Watson Version 7.3.0.01 TM (Thermo Fisher Scientific, Inc, Waltham, Massachusetts). Neither plasma protein binding nor unbound plasma oliceridine was measured.
For oliceridine, both intra‐ and interday precision were <10%. For the LOQ, the precision was <6%. Intra‐ and interday accuracy was <6%. For TRV0109662, both intra‐ and interday precision were ≤15%. For the LOQ, the precision was <10%. For the metabolite, intra‐ and interday accuracy was <6%; <15% at the LOQ. Analytical runs were considered acceptable if at least one half of the undiluted quality control samples at each concentration and two thirds of all undiluted quality control samples in the curve range were within the range of ±15.0% of the nominal concentration.
In the hepatic impairment study 73 were samples reanalyzed to test the reproducibility of the method. Ninety‐six percent (n = 70 of 73) of the repeat results and original results were within 20.0% of the mean of the 2 values and within the acceptance criteria. In the renal study 24 samples were reanalyzed to test the reproducibility with 100% of the repeat results and original results within 20.0% of the mean of the 2 values and within the acceptance criteria.
PK Analyses
PK parameters were calculated by standard noncompartmental methods with an IV bolus input using validated Phoenix WinNonlin 6.3 or later (Certara USA, Inc, Princeton, New Jersey) and using actual dosing and sampling times. The primary PK parameters calculated for oliceridine were total CL, maximum plasma concentration (Cmax), area under the concentration‐time curve from time 0 up to infinity with extrapolation of the terminal phase (AUC0‐∞), and terminal elimination half‐life (t½). Secondary PK parameters calculated were AUC up to time t (AUC0‐t), where t is the last time point with concentrations above the lower LOQ and volume of distribution. Both Cmax and AUC were normalized by dose because the pharmacokinetics of oliceridine has been shown to be linear in this range.2
Statistical Analyses
Safety analyses and statistical programming were performed using SAS (SAS Institute, Cary, North Carolina) version 9.1.3 or higher. All plasma concentrations reported as “no result” were treated as missing. For summary of plasma concentrations, plasma concentrations below the limit of quantification were treated as 0 for the calculation of all summary statistics except for the calculation of geometric mean and percentage coefficient of variation of the geometric mean, for which they were treated as missing. Missing data were not imputed. Demographic and subject characteristics and PK parameters were analyzed using descriptive statistics.
For the renal impairment study, ANCOVA was used to compare the primary PK parameters (CL, Cmax, AUC0‐t, and AUC0‐∞) with pairwise comparisons between the ESRD and healthy‐renal‐function groups. The values of CL were log transformed, and the values of Cmax, AUC0‐t, and AUC0‐∞ were dose‐adjusted and log transformed. For the hepatic impairment study, ANOVA was used to compare the primary PK parameters of oliceridine (CL, Cmax, AUC0‐∞) between the healthy controls and each of the hepatic impairment groups. Before analysis, the values of CL were log transformed, and the values of Cmax and AUC0‐∞ were dose‐adjusted and log transformed. Geometric least‐squares means were used to calculate the ratios of primary PK parameters in each hepatic impairment group to those in the control group, along with 90%CIs. The relationship between log transformed PK parameters of oliceridine and parameters of the Child‐Pugh score was explored by a linear regression approach.
Safety Analyses
The safety analyses included all subjects who had received at least 1 dose of oliceridine. Safety and tolerability were assessed by monitoring AEs, clinical laboratory data, vital signs, 12‐lead ECGs, oxygen saturation (in the hepatic impairment study), and physical examinations. All AEs were listed for each subject with the system organ class and preferred term assigned to the AEs and coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 19.0.16 MedDRA terminology is the international medical terminology developed under the auspices of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Adverse event summaries summarized only treatment‐emergent adverse events, defined as AEs, not present before the start of study medication, or AEs present before study medication that worsened after starting study medication.
Results
Demographic data for subjects enrolled in the studies are shown in Table 1.
Table 1.
Summary of Baseline Demographics and Subject Characteristics in the Renal Impairment Study and Hepatic Impairment Study
| Renal Impairment Study | |||
|---|---|---|---|
| Parameter | ESRD Subjects | Healthy Subjects | Total |
| Number | 9 | 8 | 17 |
| Oliceridine dose (IV) | 0.5 mg | 1 mg | |
| Sex, n (%) | |||
| Male | 8 (88.9) | 7 (87.5) | 15 (88.2) |
| Female | 1 (11.1) | 1 (12.5) | 2 (11.8) |
| Race, n (%) | |||
| Black | 4 (44.4) | 5 (62.5) | 9 (52.9) |
| White | 3 (33.3) | 2 (25.0) | 5 (29.4) |
| Other | 2 (22.2) | 1 (12.5) | 3 (17.6) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 2 (22.2) | 2 (25.0) | 4 (23.5) |
| Not Hispanic or Latino | 7 (77.8) | 6 (75.0) | 13 (76.5) |
| Age, y | |||
| Mean (SD) | 49.6 (5.70) | 45.9 (6.96) | 47.8 (6.41) |
| Median | 50.0 | 45.5 | 47.0 |
| Min‐max | 42‐60 | 34‐54 | 34‐60 |
| Weight, kg | |||
| Mean (SD) | 84.7 (13.2) | 84.4 (9.8) | 84.5 (11.3) |
| Median | 86.7 | 83.8 | 85.4 |
| Min‐max | 65.6‐101.3 | 70.6‐96.8 | 65.6‐101.3 |
| Body mass index (kg/m2) | |||
| Mean (SD) | 27.7 (5.2) | 27.2 (4.7) | 27.4 (4.8) |
| Median | 27.0 | 27.5 | 27.4 |
| Min‐max | 20.7‐34.5 | 20.4‐33.7 | 20.4‐34.5 |
| eGFR (mL/[min/1.73 m2]) | |||
| Mean (SD) | 7.8 (3.4) | 100.3 (20.8) | 51.3 (49.6) |
| Median | 8.0 | 101.5 | 14.0 |
| Min‐max | 4‐14 | 69‐138 | 4‐138 |
| CLcr (mL/min) | |||
| Mean (SD) | 13.0 (3.94) | 120.9 (21.01) | 63.8 (57.28) |
| Median | 13.0 | 118.0 | 19.0 |
| Min‐max | 7‐19 | 95‐157 | 7‐157 |
| Hepatic Impairment Group | Healthy Subjects | |||
|---|---|---|---|---|
| Hepatic impairment | Mild | Moderate | Severe | Normal |
| Child‐Pugh class | 5‐6 | 7‐9 | 10‐15 | Not applicable |
| Number | 10 | 10 | 6 | 8 |
| Oliceridine dose (IV) | 0.5 mg | 0.5 mg | 0.5 mg | 1 mg |
| Sex | ||||
| Male, n (%) | 6 (60) | 7 (70) | 4 (66.7) | 4 (50) |
| Female, n (%) | 4 (40) | 3 (30) | 2 (33.3) | 4 (50) |
| Race | ||||
| White, n (%) | 10 (100) | 10 (100) | 6 (100) | 8 (100) |
| Ethnicity | ||||
| Not Hispanic or Latino | 10 (100) | 10 (100) | 6 (100) | 8 (100) |
| Age, y | ||||
| Mean (SD) | 55.7 (9.8) | 58.3 (10.2) | 54.8 (4.2) | 55.5 (9.7) |
| Median | 56 | 62 | 54 | 58 |
| Min‐max | 36‐71 | 33‐67 | 49‐61 | 42‐66 |
| Weight (kg) | ||||
| Mean (SD) | 77.8 (19.5) | 80.5 (18.5) | 78.3 (23.0) | 78.3 (15.5) |
| Median | 73.4 | 75.0 | 74.8 | 82.0 |
| Min‐max | 55.0‐114.0 | 53.0‐118.0 | 57.0‐119.5 | 58.0‐101.0 |
| Body mass index (m/kg2) | ||||
| Mean (SD) | 26.9 (4.8) | 27.2 (5.2) | 26.5 (5.9) | 27.5 (3.8) |
| Median | 26.8 | 26.4 | 29.0 | 28.8 |
| Min‐max | 20.8‐34.4 | 21.0‐34.1 | 19.0‐33.3 | 21.2‐31.5 |
CLcr indicates creatinine clearance as calculated by Cockcroft‐Gault equation; eGFR, estimated glomerular filtration rate; ESRD, end‐stage renal disease (chronic kidney disease stage 5 on chronic hemodialysis); IV, intravenous.
The PK results for oliceridine in healthy volunteers and ESRD patients are shown in Table 2 and Figure 1. There was no clinically meaningful difference in any PK parameter between ESRD and healthy subjects. Mean oliceridine clearance in ESRD subjects (49.2 L/h) was 81.2% of that observed in healthy subjects (55.3 L/h). Mean oliceridine exposure in ESRD subjects was approximately 20% higher than that observed in subjects with normal renal function; however, the mean exposures observed in ESRD subjects were well within that previously reported for oliceridine in healthy subjects. The inactive metabolite TRV0109662 was measured because it is a major metabolite of oliceridine (comprising ≥10% of total by AUC). As expected, TRV0109662 was quantifiable in only a few samples, and so the results are not shown.
Table 2.
Plasma Oliceridine Noncompartmental Pharmacokinetic Parameters and Summary Statistics for the End‐Stage Renal Disease Group Versus Healthy Subjects
| ESRD (n = 8) | Healthy Subjects (n = 8) | |||||
|---|---|---|---|---|---|---|
| Pharmacokinetic Parameter | Arithmetic Mean (SD) | Geometric Mean (%CVb) | Arithmetic Mean (SD) | Geometric Mean (%CVb) | Ratio of Geometric Meansa | 90%CI for Ratio of Geometric Means |
| Cmax (ng/mL) | 9.96 (1.48)b | 9.87 (14.8)b | 8.97 (1.95) | 8.79 (22.4) | 113 | 95.4‐133.2 |
| Tmax (ng/mL)c | 0.25 (0.25‐0.25)c | 0.25 (0.25‐0.25)c | 0.25 (0.25‐0.25)c | 0.25 (0.25‐0.25)c | … | … |
| AUC0‐t (ng∙h/mL) | 19.7 (5.56)b | 19.1 (27.3)b | 18.0 (3.14) | 17.8 (17.8) | 108 | 90.1‐130.2 |
| AUC0‐∞ (ng∙h/mL) | 21.0 (5.97)b | 20.3 (28.3)b | 18.3 (3.21) | 18.1 (17.8) | 123 | 102.1‐148.4 |
| CL (L/h) | 50.8 (13.4) | 49.2 (28.3) | 56.1 (10.0) | 55.3 (17.8) | 81.2 | 67.4‐98.0 |
| t½ (h) | 3.11 (0.897) | 2.99 (31.9) | 2.44 (0.741) | 2.34 (31.9) | 128 | 95.9‐169.8 |
| Vz (L) | 219 (57.9) | 212 (27.8) | 196 (59.4) | 187 (34.5) | … | … |
AUC0‐t indicates area under the plasma concentration‐vs‐time curve from time 0 to time of last quantifiable concentration after dosing; AUC0‐∞, area under the plasma concentration‐vs‐time curve extrapolated from time 0 to infinity; BMI, body mass index; CL, total clearance; Cmax, maximum observed plasma drug concentration; %CVb, coefficient of variation between subjects; ESRD, end‐stage renal disease (chronic kidney disease stage 5 on chronic hemodialysis); t½, terminal elimination half‐life; Tmax, time to reach maximum plasma concentration; Vz, volume of distribution.
Ratio of geometric means for ESRD subjects/healthy subjects, determined by ANCOVA. The ANCOVA model includes renal function as a factor and the following covariates. AUC0‐∞ and CL: sex, age, and BMI; AUC(0‐t): sex and BMI; Cmax: BMI; t½: no covariates.
Dose‐normalized to 1 mg.
Expressed as median (min‐max).
Figure 1.
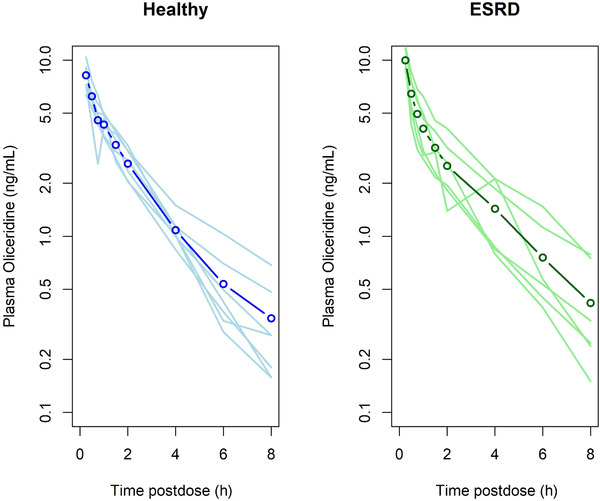
Dose‐normalized mean plasma oliceridine concentration‐vs‐time profiles in healthy subjects with normal kidney function (left panel) and in patients with end‐stage renal disease (right panel). The heavy line with open circles represents the mean concentration for each group. The lighter solid lines are the individual plasma concentration‐vs‐time curves for each individual in the group.
The pharmacokinetic results for the hepatic impairment study are shown in Table 3 and Figure 2. Clearance and dose‐normalized AUC showed no change with the degree of hepatic impairment. Dose‐normalized Cmax was significantly lower in the severe‐hepatic‐impairment group compared with the other groups. The mean Cmax in severe impairment was 24% of the Cmax seen in subjects with normal hepatic function. Half‐life increased with the degree of hepatic impairment, with a 2.79‐fold increase in half‐life observed in the severe hepatic impairment group relative to healthy subjects. Similarly, volume of dostribution increased with the degree of hepatic impairment, increasing from a mean of 129.3 L in healthy volunteers to 370.4 L in the severe group.
Table 3.
Plasma Oliceridine Noncompartmental Pharmacokinetic Parameters and Summary Statistics for the Hepatic Impairment and Healthy Subjects
| Mild | Moderate | Severe | Healthy | |
|---|---|---|---|---|
| Pharmacokinetic Parameter | (N = 10) | (N = 10) | (N = 6) | (N = 8) |
| Arithmetic mean ± SD; geometric mean (%CVb) | ||||
| Cmax (ng/mL)a | 62.3 ± 48.8 | 46.6 ± 19.4 | 11.9 ± 10.6 | 54.1 ± 59.5 |
| 41.4 (78.4) | 41.9 (41.6) | 8.4 (89.5) | 34.8 (109) | |
| AUC0‐t (ng∙h/mL)a | 23.3 ± 7.9 | 29.9 ± 11.3 | 23.4 ± 10.3 | 24.1 ± 7.5 |
| 21.8 (33.8) | 28.2 (37.9) | 21.8 (44.0) | 23.3 (30.8) | |
| AUC0‐∞ (ng∙h/mL)a | 24.1 ± 8.2 | 31.2 ± 11.5 | 25.5 ± 10.6 | 24.5 ± 7.5 |
| 22.50(33.9) | 29.5 (36.9) | 23.9 (41.6) | 23.7 (30.5) | |
| CL (L/h) | 48.4 ± 23.7 | 35.7 ± 11.5 | 44.3 ± 16.2 | 43.7 ± 11.9 |
| 44.5 (48.9) | 33.9 (32.1) | 41.8 (36.5) | 42.3 (27.2) | |
| t½ (h) | 2.7 ± 0.53 | 4.7 ± 2.1 | 6.3 ± 2.6 | 2.1 ± 0.23 |
| 2.6 (20.0) | 4.3 (44.1) | 5.8 (41.2) | 2.1 (11.3) | |
| Vz (L) | 181.3 ± 81.2 | 214.6 ± 39.1 | 370.4 ± 130.3 | 129.3 ± 27.9 |
| 167.3 (44.8) | 211.5 (18.2) | 347.9 (35.2) | 126.1 (21.6) | |
| GM Ratio Mild:Healthy (90%CI) | GM Ratio Moderate:Healthy (90%CI) | GM Ratio Severe:Healthy (90%CI) | ||
| Cmax (ng/mL)a | 1.19 (0.54, 2.60) | 1.20 (0.55, 2.62) | 0.24 (0.10, 0.56) | |
| AUC0‐t (ng∙h/mL)a | 0.93 (0.68, 1.28) | 0.99 (0.71, 1.38) | 0.93 (0.66, 1.310 | |
| AUC0‐∞ (ng∙h/mL)a | 0.95 (0.70, 1.29) | 1.25 (0.92, 1.70) | 1.01 (0.72, 1.41) | |
| CL (L/h) | 1.05 (0.77, 1.43) | 0.80 (0.59, 1.09) | 0.99 (0.71, 1.38) | |
| t½ (h) | 1.26 (0.96, 1.66) | 2.09 (1.59, 2.75) | 2.79 (2.08, 3.74) | |
AUC0‐t indicates area under the plasma concentration‐vs‐time curve from time 0 to time of last quantifiable concentration after dosing; AUC0‐∞, area under the plasma concentration‐vs‐time curve extrapolated from time 0 to infinity; CL, total clearance; Cmax, maximum observed plasma drug concentration; %CVb, coefficient of variation between subjects; GM, geometric mean; t½, terminal elimination half‐life; Vz, volume of distribution.
Cmax and AUCs were dose‐normalized to a 1‐mg dose; ratio of GM determined by ANOVA.
Figure 2.
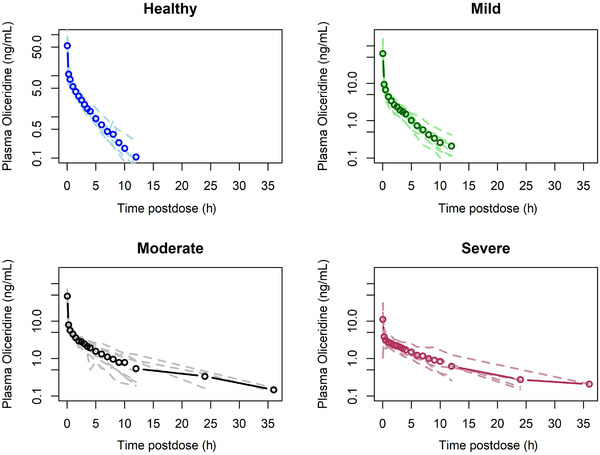
Dose‐normalized mean plasma oliceridine concentration‐vs‐time profiles in mild (n = 8), moderate (n = 8), and severe (n = 6) hepatic impairment, compared with healthy subjects (n = 8). The heavy line with open circles represents the mean concentration for each group. The lighter dashed lines are the individual plasma concentration‐vs‐time curves for each individual in the group.
Safety
Treatment emergent AEs (TEAEs) are shown in Table 4. Overall, 6 of 17 subjects (35.3%) in the renal study experienced a total of 11 mild TEAEs during the study. The TEAEs occurred more frequently in the 1‐mg dose group compared with the 0.5‐mg dose group. The most common adverse events were nausea, fatigue, and euphoria. All adverse events were judged by the investigator as being mild in severity, and no subject withdrew due to a TEAE. The majority of TEAEs were considered by the Investigator to be possibly or probably related to oliceridine, and all TEAEs resolved by the end of the study.
Table 4.
Summary of Treatment‐Emergent Adverse Events With Single‐Dose Intravenous Oliceridine in Healthy Subjects and Subjects With End‐Stage Renal Disease or Hepatic Impairment
| Renal Impairment Study | ||
|---|---|---|
| ESRD | Healthy | |
| Number of subjects in group | 9 | 8 |
| Oliceridine dose | 0.5 mg | 1 mg |
| Subjects with at least 1 TEAE, n (%), no. of events | 2 (22%), 2 | 4 (50%), 9 |
| Subjects with TEAEs by maximum severity | ||
| Mild | 2 (22%), 2 | 4 (50%), 9 |
| Moderate | 0 | 0 |
| Severe | 0 | 0 |
| Adverse event | ||
| Nausea | 1 (11%), 1 | 2 (25%), 3 |
| Fatigue | 1 (11%), 1 | 2 (25%), 2 |
| Feeling of relaxation | 0 | 1 (12.5%), 1 |
| Pain in extremity | 0 | 1 (12.5%), 1 |
| Dizziness | 0 | 1 (12.5%), 1 |
| Headache | 0 | 1 (12.5%), 1 |
| Euphoria | 0 | 2 (25%), 2 |
| Hepatic Impairment Study | ||||
|---|---|---|---|---|
| Mild | Moderate | Severe | Healthy | |
| Number of subjects in group | 10 | 10 | 6 | 8 |
| Oliceridine dose | 0.5 mg | 0.5 mg | 0.5 mg | 1 mg |
| Subjects with at least 1 TEAE, n (%), no. of events | 2 (20%), 2 | 0 (0%), 0 | 0 (0%), 0 | 0 (0%), 0 |
| Subjects with TEAEs by maximum severity | ||||
| Mild | 2 (20%), 2 | 0 (0%), 0 | 0 (0%), 0 | 0 (0%), 0 |
| Moderate | 0 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 |
| Adverse event | ||||
| Somnolence | 2 (20%), 2 | 0 (0%), 0 | 0 (0%), 0 | 0 (0%), 0 |
ESRD indicates end‐stage renal disease; TEAE, treatment‐emergent adverse event.
In the hepatic impairment study only 2 TEAEs of somnolence were reported in 2 subjects in the mild hepatic impairment group. These were judged by the investigator to be mild in intensity and lasted for approximately 1 hour. Both resolved completely.
Oliceridine had no measurable impact on laboratory parameters, vital signs, ECG parameters, or oxygen saturation that could be attributed to hepatic impairment.
Discussion
The results of the renal study suggest that in patients with severe renal impairment categorized by ESRD, there was no clinically relevant change in oliceridine clearance (>50% difference) or other PK parameters compared with healthy age‐ and sex‐matched controls. Because renal clearance of oliceridine is only 2% to 4% of total clearance, these results were expected. Although it has been reported that patients with chronic kidney disease have reduced CYP2D6 activity,17 this was not observed in our study. Both clearance and plasma exposure were similar for ESRD subjects compared with healthy subjects, and by 24 hours, no subjects in either group had quantifiable oliceridine plasma concentrations. Therefore, dosage adjustment is not required in patients with any stage of kidney disease. Because conventional opioids such as morphine and hydromorphone are metabolized to form active metabolites that are renally excreted,18 and these metabolites can accumulate over time in patients with kidney disease, a medication such as oliceridine that has no active metabolites and is not renally cleared, may be particularly advantageous in this population as well as in the elderly, most of whom have some degree of renal impairment.19
Oliceridine is hepatically metabolized by CYP3A4 and CYP2D6, with a 50:50 contribution by each enzyme.2 In an earlier study healthy subjects given single IV oliceridine doses of 0.25 mg to 6 mg had mean CL values that ranged from 36 to 47.2 L/h in CYP2D6 extensive metabolizers and 19.6 to 24.7 L/h in CYP2D6 poor metabolizers. Thus, before conduct of this study, it was expected that oliceridine clearance might be reduced in hepatic impairment. In fact, across the spectrum of hepatic impairment, there were no clinically meaningful differences in CL or dose‐normalized AUC as compared with healthy subjects.
Hepatic impairment results in a reduction in both the intrinsic clearance of oliceridine (as a result of the reduction of functional hepatic cell mass) and decreased hepatic blood flow due to fibrosis and scarring of the liver. Free drug concentrations were not measured in the current study. In the absence of free drug concentrations, one may look at measured t½. Oliceridine concentrations clearly increase as hepatic function decreases, and this is the expected finding for a drug that is hepatically metabolized. These changes reflect the reduced metabolic capacity of the liver and are not confounded by measuring only total oliceridine concentrations. From a clinical standpoint, the observed effect of hepatic impairment on t½ can be used when determining dosing regimens for patients.
Similarly, the volume of distribution (Vd,plasma) is affected by an increased free fraction of drug and the presence of ascites in hepatically impaired patients. This is furthered by the fact that hypoalbuminemia (responsible for the increased free fraction of protein‐bound drugs such as oliceridine) also directly causes ascites.
A consideration of the well‐stirred model (1),
| (1) |
where QH is hepatic blood flow, CLint is intrinsic clearance, and fuP is the fraction of drug unbound in the plasma, shows that for drugs where QH>> fuP*CLint (which is the case for the majority of drugs), the above equation20 reduces to
| (2) |
Because it is unbound drug that is subject to hepatic elimination, the well‐stirred model suggests that an increased free fraction alone will result in an increase in unbound clearance under conditions of hypoalbuminemia, thus leading to no change in unbound drug concentrations.21 Total body clearance will decrease in hepatic impairment as a result of a decrease in CLint (due to the reduction of functional hepatic cell mass). This decrease in total body clearance should in turn result in an increase in AUC. However, because of the increase in free fraction (in the presence of decreased capacity to metabolize oliceridine) and the presence of ascites, the volume of distribution will also increase. Equation 3 shows the relationship between plasma and tissue binding and tissue volume:22
| (3) |
Vp is plasma volume, fuP is the free fraction in plasma, fuT is the free fraction in tissue, and V’T is the tissue volume. An increase in either fuP (due to hypoalbuminemia) or in V’T (due to the presence of ascites) will increase the overall volume of distribution, Vd,plasma. If decreases in CLint and increases in Vd,plasma change by roughly the same magnitude (eg, fuP increases by approximately 30% and CLint decreases by approximately 30%), then the AUC may not reflect the decrease in CLint due to liver disease. In that case the observed oliceridine t½ would still be expected to show a clear increase with the degree of hepatic impairment. This was observed in the present study.
Medications used for treatment of acute pain are dosed on an as‐needed basis, with no fixed dose or dosing interval, and therefore adjustment of the initial dose is not required in mild or moderate hepatic impairment. Because the observed t½ of oliceridine is extended in subjects with hepatic impairment, these patients may require fewer doses than those with normal hepatic function. In severe hepatic impairment, consideration should be made to reducing the initial dose, followed by careful monitoring, as these patients will require fewer doses of oliceridine.
Among the 65 subjects enrolled in these 2 studies, a single IV infusion of oliceridine was safe, and the observed TEAEs were of mild intensity. These safety results are consistent with reports from phase 1, 2, and 3 studies of oliceridine, in which decreased opioid‐related AEs, including respiratory depression and postoperative nausea and vomiting, were observed,23, 24, 25 and consistent with AEs commonly observed following the administration of opioids.
Conclusions
The results of these studies indicate that no dose adjustment of oliceridine is needed in patients with renal dysfunction or in patients with mild to moderate hepatic dysfunction. In severe hepatic impairment, consideration of initial dose reduction followed by careful monitoring should be done, as these patients will require fewer doses of oliceridine. Among the healthy adults and those with ESRD or hepatic impairment in these studies, oliceridine was safe and caused only mild‐intensity adverse effects.
Conflicts of Interest
At the time this work was performed, Kelly A. Arscott, Kristina Cochrane, Franck Skobieranda, David Burt, and Michael Fossler were employees of Trevena, Inc. Anne N. Nafziger has no conflicts of interest or financial disclosures.
Disclosure
Both studies were sponsored by Trevena, Inc, Chesterbrook, PA, which is developing oliceridine for the treatment of moderate to severe acute pain.
Acknowledgments
The MedDRA trademark is registered by the International Federation of Pharmaceutical Manufacturers & Associations on behalf of the International Conference on Harmonisation.
References
- 1. Gan TJ. Poorly controlled postoperative pain: prevalence, consequences, and prevention. J Pain Res. 2017;10:2287‐2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soergel DG, Subach RA, Sadler B, et al. First clinical experience with TRV130: pharmacokinetics and pharmacodynamics in healthy volunteers. J Clin Pharmacol. 2014;54(3):351‐357. [DOI] [PubMed] [Google Scholar]
- 3. US Food and Drug Administration . Extended‐release (ER) and long‐acting (LA) opioid analgesics risk evaluation and mitigation strategy (REMS). Silver spring, MD: US Food and Drug Administration; June 2015. [Google Scholar]
- 4. Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72(6):1393‐1401. [DOI] [PubMed] [Google Scholar]
- 5. Raehal KM, Walker JK, Bohn LM. Morphine side effects in β‐arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314(3):1195‐1201. [DOI] [PubMed] [Google Scholar]
- 6. Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking β‐arrestin 2. Science. 1999;286(5449):2495‐2498. [DOI] [PubMed] [Google Scholar]
- 7. DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. β‐Arrestins and cell signaling. Annu Rev Physiol. 2007;69:483‐510. [DOI] [PubMed] [Google Scholar]
- 8. DeWire SM, Yamashita DS, Rominger DH, et al. A G protein‐biased ligand at the μ‐opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344(3):708‐717. [DOI] [PubMed] [Google Scholar]
- 9. Fossler MJ, Sadler BM, Farrell C, Burt DA. Oliceridine (TRV130), a novel G protein–biased ligand at the μ‐opioid receptor, demonstrates a predictable relationship between plasma concentrations and pain relief. I: Development of a pharmacokinetic/pharmacodynamic model. J Clin Pharmacol. 2018;58(6):750‐761. [DOI] [PubMed] [Google Scholar]
- 10. Soergel DG, Subach RA, Burnham N, et al. Biased agonism of the μ‐opioid receptor by TRV130 increases analgesia and reduces on‐target adverse effects versus morphine: a randomized, double‐blind, placebo‐controlled, crossover study in healthy volunteers. Pain. 2014;155(9):1829‐1835. [DOI] [PubMed] [Google Scholar]
- 11. Viscusi ER, Webster L, Kuss M, et al. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ‐opioid receptor, for the intravenous treatment of acute pain. Pain. 2016;157(1):264‐272. [DOI] [PubMed] [Google Scholar]
- 12. CDER. Pharmacokinetics in Patients With Impaired Renal Function—Study Design, Data Analysis, and Impact on Dosing and Labeling. Rockville, MD: US Food and Drug Administration; 2010:1‐17. [Google Scholar]
- 13. DBER. Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing and Labeling. Rockville MD: US Food and Drug Administration; 2003:1‐19. [Google Scholar]
- 14. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31‐41. [DOI] [PubMed] [Google Scholar]
- 15. Pugh RN, Murray‐Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646‐649. [DOI] [PubMed] [Google Scholar]
- 16.McLean, VA:: International Federation of Pharmaceutical Manufacturers & Associations; 2016.. MedDRA: Medical Dictionary for Regulatory Activities, version 19.0. [Google Scholar]
- 17. Yoshida K, Sun B, Zhang L, et al. Systematic and quantitative assessment of the effect of chronic kidney disease on CYP2D6 and CYP3A4/5. Clin Pharmacol Ther. 2016;. 100(1):75‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nafziger AN, Barkin RL. Opioid therapy in acute and chronic pain. J Clin Pharmacol. 2018;58(9):1111‐1122. [DOI] [PubMed] [Google Scholar]
- 19. Lötsch J. Opioid metabolites. J Pain Symptom Manage. 2005;29(5 suppl):S10‐S24. [DOI] [PubMed] [Google Scholar]
- 20. Benet LZ, Hoener BA. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115‐121. [DOI] [PubMed] [Google Scholar]
- 21. Greenblatt DJ, Sellers EM, Koch‐Weber J. Importance of protein binding for the interpretation of serum or plasma drug concentrations. J Clin Pharmacol. 1982;22:259‐263. [DOI] [PubMed] [Google Scholar]
- 22. Schmidt S, Gonzalez D, Derendorf H. Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci. 2010;99(3):1107‐1122. [DOI] [PubMed] [Google Scholar]
- 23. Singla N, Minkowitz HS, Soergel DG, et al. A randomized, phase IIb study investigating oliceridine (TRV130), a novel μ‐receptor G‐protein pathway selective (μ‐GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J Pain Res. 2017;10:2413‐2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Viscusi ER, Skobieranda F, Soergel DG, Cook E, Burt DA, Singla N. APOLLO‐1: a randomized placebo and active‐controlled phase III study investigating oliceridine (TRV130), a G protein‐biased ligand at the μ‐opioid receptor, for management of moderate‐to‐severe acute pain following bunionectomy. J Pain Res. 2019;12:927‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Singla NK, Skobieranda F, Soergel DG, et al. APOLLO‐2: a randomized, placebo and active‐controlled phase III study investigating oliceridine (TRV130) a G protein‐biased ligand at the μ‐opioid receptor, for management of moderate to severe acute pain following abdominoplasty. Pain Practice. 2019;19(7):715‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]