

Abstract
Psilocybin, the principal indole alkaloid of Psilocybe mushrooms, is currently undergoing clinical trials as a medication against treatment‐resistant depression and major depressive disorder. The psilocybin supply for pharmaceutical purposes is met by synthetic chemistry. We replaced the problematic phosphorylation step during synthesis with the mushroom kinase PsiK. This enzyme was biochemically characterized and used to produce one gram of psilocybin from psilocin within 20 minutes. We also describe a pilot‐scale protocol for recombinant PsiK that yielded 150 mg enzyme in active and soluble form. Our work consolidates the simplicity of tryptamine chemistry with the specificity and selectivity of enzymatic catalysis and helps provide access to an important drug at potentially reasonable cost.
Keywords: bioorganic chemistry, biosynthesis, enzymes, fermentation, kinase, phosphorylation
The benefits of bulk: Future use of psilocybin as a medication will entail increased demand. We present a gram‐scale synthetic/biocatalytic approach in which the mushroom kinase PsiK replaces tedious chemical phosphorylation. A pilot‐scale protocol to make three‐digit milligram amounts of pure and active recombinant PsiK available is also described.
Introduction
The indole alkaloid psilocybin (1, Scheme 1) is the principal natural product of hallucinogenic Psilocybe mushrooms.1 Pharmacologically, it represents the stable prodrug of the actual active, yet unstable 4‐dephosphorylated analogue psilocin (2), which targets mainly the human 5‐HT2A‐receptor as a partial agonist.2 Modern scientific therapeutics rediscovered 1 as a valuable drug candidate.3 Phase 2 clinical trials have shown favorable results in the treatment of several indications, and 1 has received Breakthrough Therapy designation by the United States Food and Drug Administration (US FDA) in support of both treatment‐resistant depression and major depressive disorder. Other indications include cancer‐related existential distress and substance dependencies.4
Scheme 1.
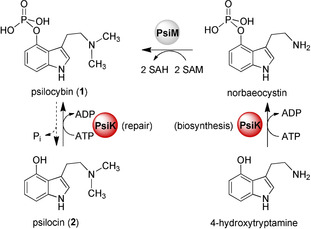
Dual repair and biosynthetic function of P. cubensis PsiK (red). The enzyme catalyzes 4‐hydroxtryptamine phosphorylation to norbaeocystin during 1 biosynthesis or repairs 1 by re‐phosphorylation of 2. In the mushrooms, the methyltransferase PsiM (grey) completes 1 biosynthesis after the phosphorylation step.
Given its status as a candidate drug, the demand for medical‐grade 1 is increasing. Traditionally, this compound is accessible through chemical synthesis.1, 5, 6 Identification of the 1 biosynthetic genes and enzymes in Psilocybe mushrooms, including the kinase gene psiK, opened up new avenues for the production of 1. Heterologously produced enzymes were used to biocatalytically synthesize 1 in vitro.7 In vivo production in microbial hosts has also proven successful.8 In addition, the in vivo approach is attractive because of its scalability to pilot‐scale bioreactors and the availability of cofactors. However, in view of registration as a pharmaceutical, the use of a cell‐based production system entails intensive procedures to remove cellular components and debris that are potentially allergenic or toxic. Furthermore, unlike the in vitro or synthetic approaches, the targeted compound needs to be purified from complex matrices, which may involve a cell lysate or broth.
Herein, we describe the first combined chemical/biocatalytic synthesis of 1. This cell‐free in vitro procedure combines the facile synthetic access to 2 with side‐product‐free quantitative conversion through PsiK‐mediated biocatalysis in a subsequent step, thus eliminating heavy‐metal‐dependent hydrogenolysis and low yielding phosphorylation chemistry, to produce gram‐scale amounts of 1. To facilitate future development of the biotechnological processes, we present a thorough biochemical characterization of P. cubensis PsiK and a protocol to heterologously produce and purify three‐digit milligram amounts of PsiK in soluble and active form.
Results and Discussion
First, we produced PsiK in a small scale as a hexahistidine‐tagged fusion using E. coli KRX×pJF236 and investigated its steady‐state kinetic properties. Phosphate‐buffered in vitro assays were analyzed for formation of 1 by UHPLC‐MS. Previous work confirmed a dual biosynthetic and protective role for PsiK as it accepts both 4‐hydroxytryptamine and 2 as substrates (Scheme 1). The former represents the phosphate acceptor during biosynthesis. The repair reaction, which is the relevant reaction for our study, includes phosphorylation of 2 back to 1, thus removing instable and oligomerization‐prone 2 from the cell. Oligomeric 2 causes the blueing reaction of injured “magic” mushrooms and binds to proteins, which would be fatal to the intact fungal mycelium.9 To protect cell integrity, we expected that the velocity of the repair direction should at least equal that of the biosynthetic direction.
In its native form, P. cubensis PsiK is a 40.4 kDa enzyme of 362 aa. Sequence alignment with other characterized or putative small‐molecule kinases (Figure S1) identified a canonical ATP binding domain that comprises a conserved motif 57KHA59. In the structure of the Arabidopsis thaliana (mouse‐ear cress) 5‐methylthioribose kinase (Protein Data Bank, accession # 2PYW), the equivalent lysine residue in the motif 63KQA66 ionically interacted with the α‐phosphoryl group of ADP. In the Arabidopsis enzyme, the conserved aspartate residue D255 was shown to coordinate bivalent Mg2+ cations in the active site.10 The equivalent residue in PsiK is D224.
To verify its function, we engineered the cloned psiK cDNA to replace D224 with alanine. PsiKD224A was catalytically inactive when 2 was offered as substrate and not even traces of 1 were detected (Figure 1). This result suggests a similar cation‐coordinating role for D224 in the active site.
Figure 1.
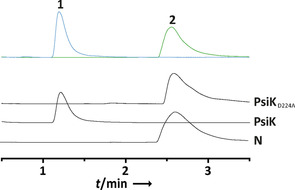
UHPLC‐MS analysis of in vitro activity assays with recombinantly produced PsiK and its variant PsiKD224A. Top trace: overlaid chromatograms of authentic 1 and 2 standards. N: negative control with heat‐treated enzyme.
Optimum turnover was found at 40 °C and at pH 7.0. Furthermore, PsiK activity depended on Mg2+. In contrast, with Co2+ as divalent cation, the turnover dropped to 11.5 % and the enzyme remained nearly inactive (<5 %) with Ca2+, Cu2+, Ni2+, Mn2+, or Zn2+. PsiK followed a sigmoid kinetic response to increasing 2 and 4‐hydroxytryptamine concentrations, with K 0.5 values of 72 μm and 67 μm, and a k cat of 16.1 s−1 and 4.5 s−1 for these substrates (Figure S2). This translates into catalytic efficiencies (k cat/K 0.5) of 2.24×105 and 6.67×104 m −1 s−1, respectively, with 2 and 4‐hydroxytryptamine as acceptor substrates. The K 0.5 value for ATP was 89 μm. The above values identified the repair function of PsiK as more than three times as efficient as the biosynthetic one. Size‐exclusion chromatography showed that active PsiK is monomeric (Figure 2). This situation is reminiscent of glucokinase, which has a single binding site for its phosphate acceptor substrate (d‐glucose), yet shows a sigmoid kinetic, which is a characteristic of positive cooperativity.11
Figure 2.
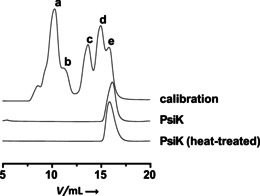
Size‐exclusion chromatography with native and heat‐treated 4‐hydroxytryptamine/psilocin kinase PsiK. Both heat‐inactivated and active PsiK eluted at 15.9 mL (corresponding to ca. 43 kDa). The calculated mass of monomeric N‐terminally His‐tagged PsiK is 44.0 kDa. Calibration trace: a) 669 kDa (thyroglobulin), b) 440 kDa (ferritin), c) 158 kDa (aldolase), d) 75 kDa (conalbumin), e) 43 kDa (ovalbumin).
With a future large‐scale process development in mind, a stirred‐tank fermentation and purification regime was established. E. coli KRX × pJF23 was performed batch‐wise in a 75 L reactor, filled with 30 L yeast‐peptone‐glucose medium. After harvest, cell disruption and FPLC‐based Ni2+ affinity chromatography, 150.2 mg pure PsiK was obtained (Figure S3).
Subsequently, we tested PsiK's storability and stability at low temperatures in phosphate buffer, which was amended with 10 % glycerol, for 30 d and we found the PsiK activity was fully retained (Figure S4). For the use as substrate in a PsiK‐based production assay, 2 was synthesized via a recently published protocol,6 essentially following Speeter–Anthony chemistry.12 Commercial 4‐acetoxyindole underwent acylation and amidation to yield 3‐(2‐(dimethylamino)‐2‐oxoacetyl)‐1H‐indol‐4‐yl acetate, which was reduced with lithium aluminum hydride to afford 2 in 73 % overall yield. When 2 mm (725 mg) of 2, 4 mm ATP, and 1.89 μm PsiK were used, we observed a near‐complete phosphorylation reaction after 10 min, and quantitative conversion into 1 within 20 min (Figure 3 A). Workup included preparative reversed‐phase chromatography and a subsequent solvent/antisolvent precipitation in acidic (pH 4) H2O and acetone. After washing the precipitate, 893 mg (i.e., an isolated yield of 88.5 %) of pure 1 was obtained (Figure 3 B). The identity of the biocatalytically produced 1 was confirmed by 1H and 13C NMR spectroscopy (Figure S5, Table S1), which yielded data that was consistent with published values.13
Figure 3.
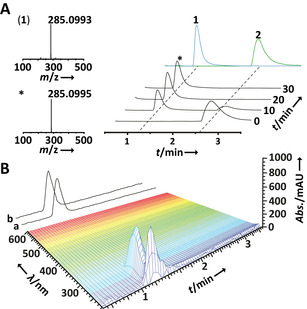
A) Progress of the gram‐scale synthetic/biocatalytic production of 1 from 2, as verified by UHPLC and HR‐ESIMS analyses of an aliquot. Top trace: overlaid individual chromatograms of authentic 1 and 2. B) 3D plot of UHPLC diode array and total ion chromatogram (TIC) data of the singly chromatographed 1 production assay. TIC trace a) ESI (+); trace b) ESI (−) mode.
The structural elucidation and synthesis of 1 and 2 were first described by Albert Hofmann.1 Various improved protocols have been developed since,5 however, the synthesis has still remained problematic due in part to scalability in large‐scale manufacturing, instability during the isolation of 2, in‐process impurities during their synthesis, hydrolysis of 1 during recrystallization, and the lability of the O,O‐dibenzyl ester intermediate. Regardless of the protocol, the final deprotection involves a heavy metal (Pd/C), which is not preferred from a regulatory affairs perspective for medical grade compounds. Furthermore, our approach avoids corrosive and pyrophoric n‐butyllithium. Our route to provide upper milligram to gram amounts of 1 combines the simplicity of traditional tryptamine chemistry with the power of biocatalysis to eliminate heavy metals and the notoriously problematic phosphorylation step, while also circumventing cell‐based production. Demand for 1 is expected to increase. To translate from gram‐ to kilogram‐scale, further optimization, such as reduced solvent use and an alternative to the preparative reversed‐phase HPLC‐based purification procedure, may be required. Also, selective filtration‐precipitation‐recrystallization may help make this process more amenable to scale‐up. With these changes, and given the outstanding selectivity of the enzymatic phosphorylation step, we expect drastically reduced pricing for kilogram‐scale 1 production.
Conclusion
We expect our results will motivate bioprocess research to further develop and scale up the presented protocol. Therefore, the current work is directed towards further upscaling, and optimizing the post‐biocatalysis work‐up. We conclude that our research helps provide 1 at a reasonable cost, particularly to support non‐profit endeavors that are geared towards re‐introducing this invaluable natural product to pharmaceutical and clinical use.
Experimental Section
General procedures: Molecular biology methods followed the instructions of the manufacturers of kits and enzymes (NEB, Promega). Chemicals, media ingredients and solvents were purchased from Carbolution Chemicals, Roth, Sigma–Aldrich, VWR, and Deutero, except 2, which was synthesized by following a published procedure.6 Authentic 1 was isolated from P. cubensis carpophores.14
Site‐directed mutagenesis: The gene encoding PsiKD224A was created by PCR‐amplifying psiK from plasmid pJF23.7a The psiK gene was amplified in two fragments that overlapped in the portion to be mutated. The 5′‐end of fragment 1 and the 3′‐end of fragment 2 overlapped with the polylinker of plasmid pET28a, linearized by BamHI and HindIII restriction. The fragments were assembled using the NEBuilder HiFi DNA Assembly Kit to yield plasmid pJF70. Oligonucleotides to amplify fragment 1 (448 bp) were oJF159 (5′‐CATGGCGGCCCTGTGGAGTG‐3′) and oJF161 (5′‐CTCGAGTGCGGCCGCAAG‐3′). Fragment 2 (699 bp) was amplified using primers oJF160 (5′‐CCACAGGGCCGCCATGACAAG‐3′) and oJF162 (5′‐CAGCAAATGGGTCGCGGATC‐3′). For PCR, 200 μm each dNTP, 1 ng template DNA, and 1 U Phusion DNA polymerase (NEB) in HF buffer and MgCl2, supplied with the enzyme, was used. Thermal cycling was for both fragments: initial hold at 98 °C for 1 min, 32 cycles of 98 °C for 60 s, 67 °C for 30 s, and 72 °C for 60 s, and a terminal hold at 72 °C for 5 min. The PCR products were purified by agarose gel electrophoresis and subsequent elution with Promega's Wizard SV Gel and PCR Clean‐Up System.
Heterologous production of PsiK: PsiK and PsiKD224A were produced in E. coli KRX (Promega), transformed with expression plasmids pJF23 or pJF70. Small‐scale production and purification was carried out as described.15 To produce seed cultures for pilot‐scale fermentations (below), E. coli KRX×pJF23 was grown overnight in 20 mL LB medium, amended with kanamycin (50 μg mL−1), and shaken at 37 °C and 180 rpm. Cells were centrifuged (11 000×g, 10 min, 4 °C), washed with phosphate buffered saline (PBS), and stored in 10 % (v/v) glycerol at −80 °C until use as inoculum.
Pilot‐scale PsiK production: The inoculum was transferred to a 2 L shake flask, filled with 200 mL LB medium, for 12 h, at 37 °C and 180 rpm. This seed culture was inoculated with an OD600 of 0.05 and transferred to the main culture with a final OD600 of 3.5. The main batch culture (30 L, 37 °C) was carried out in a 75 L stirred tank reactor (C75, bbi, Berlin, Germany), using YPD medium (20 g L−1 glycerol, 20 g L−1 tryptone, 10 g L−1 yeast extract). The dissolved oxygen tension was controlled by the stirring rate at a minimum value of 20 %. The initial OD600 was 0.023. PsiK production was induced after 10 h by adding 1 g L−1 l‐rhamnose. Upon induction, the temperature was shifted to 16 °C. For fermentation details, see Figure S6. The fermentation continued for another 14 h before the broth was centrifuged at 14 000×g for 30 min at 4 °C. Subsequently, the biomass was suspended in 5 L lysis buffer (50 mm sodium dihydrogen phosphate, 300 mm NaCl), amended with 20 mm imidazole, homogenized at 850 bar in a high‐pressure Gaulin flow homogenizer which was rinsed with another 1 L of lysis buffer, centrifuged at 14 000×g for 45 min at 4 °C and finally filtered (0.45 μm). PsiK was further purified by immobilized metal affinity chromatography on a FPLC system (ÄktaPilot, GE Healthcare), equipped with a FineLINE Pilot 35 column filled with 200 mL Ni‐Sepharose FF. The flow rate was 20 mL min−1. The system was equilibrated in lysis buffer before 6 L of supernatant were loaded. The column was washed step‐wise with a gradient of 30 mm (2 column volumes), 40 mm (1 cv), and 50 mm imidazole (2 cv), using the same buffer. The final elution was conducted with 500 mm imidazole, resulting in a 200 mL fraction. The eluted enzyme was rebuffered on a Amicon Ultra 15 centrifugal filter unit (Merck), equilibrated with phosphate buffer (50 mm, pH 7.0), and stored at −80 °C with 10 % glycerol until used for production of 1. The protein concentration was determined using Bradford's method.16 The total yield was 150.2 mg pure PsiK. Prior to use for assays, PsiK was rebuffered by FPLC (below).
Size‐exclusion chromatography: FPLC (Äkta Pure 25, GE Healthcare) fitted to a Superdex 200 increase 10/300 GL column with 24 mL bed volume was used 1) to determine whether PsiK was active as a mono‐ or multimer, and 2) to rebuffer PsiK. Prior to sample loading, PsiK was concentrated on a centrifugal filter unit (10 kDa cut off, Amicon Ultra 15, Merck). Elution was with phosphate buffer (10 mm sodium dihydrogen phosphate, 140 mm NaCl, pH 7.5) at a flow of 0.5 mL min−1. For rebuffering and subsequent use in assays, PsiK was eluted with assay buffer (below).
In vitro PsiK assays: Assays to detect activity or to record kinetics were set up in sodium dihydrogen phosphate buffer (50 mm, pH 7.0) and 3 mm β‐mercaptoethanol in a final volume of 50 μL. ATP and MgCl2 or chlorides of other bivalent cations (2 mm each) and either 4‐hydroxytryptamine or 2 (1 mm) were added as substrate, PsiK was added at 1 μm. The reactions were incubated for 10 min at 40 °C. Subsequently, the assays were lyophilized, the residue was dissolved in 50 μL MeOH, centrifuged (11 000 × g, 10 min, 4 °C), filtered, and chromatographically analyzed (below). To determine optima, pH and temperature were varied between pH 6–8 and 20–50 °C. To investigate the storability of PsiK, aliquots were stored with or without additional glycerol (10 %, v/v) at 4, −20 and −80 °C and analyzed by the above assay after 14 and 30 days.
Analytical liquid chromatography: UHPLC‐ESIMS‐analyses of in vitro assays were performed with an Agilent Infinity 1290 chromatograph with a Luna Omega Polar C18 column (100×2.1 mm, 1.6 μm particle size, Phenomenex), and coupled to an Agilent 6130 Single Quadrupole mass detector. The flow was 0.5 mL min−1. Diode array detection was between λ=200–600 nm. Chromatograms were extracted at λ=280 nm. Solvent A was 0.1 % (v/v) formic acid in H2O, solvent B was acetonitrile (ACN). The linear gradient was initially 1 % B, increased to 10 % within 3 min, and to 100 % B in 1 min. LC‐HRESIMS analysis in positive and negative mode with purified substances were performed with a Thermo Accela chromatograph coupled to an Exactive Orbitrap mass spectrometer, as described.14
Gram‐scale production of 1: Scaled‐up production was carried out in a 1.8 L reaction in sodium dihydrogen phosphate buffer (50 mm, pH 7.0). Before adding enzyme and substrates, the reaction buffer was pre‐warmed to 40 °C. Subsequently, 725 mg 2 (2 mm), 3.97 g ATP disodium salt (4 mm), 1.46 g MgCl2 ⋅6H2O (4 mm) and 422 mg β‐mercaptoethanol (3 mm) were added. The reaction was initiated by adding 149.7 mg of purified PsiK (1.89 μm). Samples (50 μL) were taken in triplicates in 10 min intervals to monitor product formation by UHPLC‐MS. After 90 min, the solvent was evaporated under reduced pressure, and the residue was dissolved in 500 mL MeOH, centrifuged (3 200×g, 15 min, 4 °C) also to remove precipitated protein and ADP, and filtered. The solvent was evaporated under reduced pressure. For chromatographic purification of 1, the residue was dissolved in H2O/acetonitrile (ACN, 9:1, v/v) with 0.1 % trifluoroacetic acid (TFA), which yielded 893 mg (88.5 %) of pure 1.
Preparative liquid chromatography: Compound 1 was purified with an Agilent 1260 series instrument, equipped with a Zorbax Eclipse XDB C18 column (250×21.2 mm, 10 μm). Solvent A was 0.1 % TFA in H2O, solvent B was ACN. A linear gradient 5–100 % B within 20 min at a flow of 20 mL min−1 was applied. Further workup included solvent/antisolvent precipitation in 20 mL acidic (pH 4) H2O and 30 mL acetone. The precipitate was washed with cold acetone. To verify purity, 1H and 13C NMR spectra of purified 1, dissolved in D2O, were recorded at 500 and 125 MHz, respectively, and 300 K with a Bruker Avance III spectrometer. Chemical shifts were referenced to residual non‐deuterated solvent (δ H=4.80 ppm).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Karin Burmeister, Karsten Willing, Andrea Perner and Heike Heinecke (Hans‐Knöll‐Institute Jena) for technical assistance, and for recording high‐resolution mass and NMR spectra, respectively. C.L. acknowledges a doctoral fellowship by the International Leibniz Research School (ILRS) for Microbial Interactions. This work was supported by the Deutsche Forschungsgemeinschaft (DFG grant HO2515/7‐1 to D.H.) and by the Usona Institute (Madison, WI). D.H. is also supported by the DFG Collaborative Research Center ChemBioSys 1127.
J. Fricke, R. Kargbo, L. Regestein, C. Lenz, G. Peschel, M. A. Rosenbaum, A. Sherwood, D. Hoffmeister, Chem. Eur. J. 2020, 26, 8281.
References
- 1.
- 1a. Hofmann A., Heim R., Brack A., Kobel H., Experientia 1958, 14, 107–109; [DOI] [PubMed] [Google Scholar]
- 1b. Hofmann A., Heim R., Brack A., Kobel H., Frey A., Ott H., Petrzilka T., Troxler F., Helv. Chim. Acta 1959, 42, 1557–1572. [Google Scholar]
- 2. Geiger H. A., Wurst M. G., Daniels R. N., ACS Chem. Neurosci. 2018, 9, 2438–2447. [DOI] [PubMed] [Google Scholar]
- 3. Vollenweider F. X., Kometer M., Nat. Rev. Neurosci. 2010, 11, 642–651. [DOI] [PubMed] [Google Scholar]
- 4. Johnson M. W., Griffiths R. R., Neurotherapeutics 2017, 14, 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Nichols D. E., Frescas S., Synthesis 1999, 6, 935–938; [Google Scholar]
- 5b. Shirota O., Hakamata W., Goda Y., J. Nat. Prod. 2003, 66, 885–887; [DOI] [PubMed] [Google Scholar]
- 5c. Bartolucci S., Mari M., Di Gregorio G., Piersanti G., Tetrahedron 2016, 72, 2233–2238; [Google Scholar]
- 5d. Sakagami H., Ogasawara K., Heterocycles 1999, 51, 1131–1135; [Google Scholar]
- 5e. Hu C., Qin H., Cui Y., Jia Y., Tetrahedron 2009, 65, 9075–9080; [Google Scholar]
- 5f. Gathergood N., Scammells P. J., Org. Lett. 2003, 5, 921–923. [DOI] [PubMed] [Google Scholar]
- 6. Sherwood A. M., Meisenheimer P., Tarpley G., Kargbo R. B., Synthesis 2020, 52, 688–694. [Google Scholar]
- 7.
- 7a. Fricke J., Blei F., Hoffmeister D., Angew. Chem. Int. Ed. 2017, 56, 12352–12355; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12524–12527; [Google Scholar]
- 7b. Reynolds H. T., Vijayakumar V., Gluck-Thaler E., Korotkin H. B., Matheny P. B., Slot J. C., Evol. Lett. 2018, 2, 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Hoefgen S., Lin J., Fricke J., Stroe M., Mattern D. J., Kufs J. E., Hortschansky P., Brakhage A. A., Hoffmeister D., Valiante V., Metab. Eng. 2018, 48, 44–51; [DOI] [PubMed] [Google Scholar]
- 8b. Adams A. M., Kaplan N. A., Wei Z., Brinton J. D., Monnier C. S., Enacopol A. L., Ramelot T. A., Jones J. A., Metab. Eng. 2019, 56, 111–119. [DOI] [PubMed] [Google Scholar]
- 9. Lenz C., Wick J., Braga D., García-Altares M., Lackner G., Hertweck C., Gressler M., Hoffmeister D., Angew. Chem. Int. Ed. 2020, 59, 1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ku S.-Y., Cornell K. A., Howell P. L., BMC Struct. Biol. 2007, 7, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Porter C. M., Miller B. G., Bioorg. Chem. 2012, 43, 44–50; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Pollard-Knight D., Cornish-Bowden A., Mol. Cell. Biochem. 1982, 44, 71–80. [DOI] [PubMed] [Google Scholar]
- 12. Speeter M. E., Anthony W. C., J. Am. Chem. Soc. 1954, 76, 6208–6210. [Google Scholar]
- 13. Blei F., Baldeweg F., Fricke J., Hoffmeister D., Chem. Eur. J. 2018, 24, 10028–10031. [DOI] [PubMed] [Google Scholar]
- 14. Lenz C., Wick J., Hoffmeister D., J. Nat. Prod. 2017, 80, 2835–2838. [DOI] [PubMed] [Google Scholar]
- 15. Fricke J., Sherwood A., Kargbo R., Orry A., Blei F., Naschberger A., Rupp B., Hoffmeister D., ChemBioChem 2019, 20, 2824–2829. [DOI] [PubMed] [Google Scholar]
- 16. Bradford M., Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary