

Abstract
C7−H‐functionalized indoles are ubiquitous structural units of biological and pharmaceutical compounds for numerous antiviral agents against SARS‐CoV or HIV‐1. Thus, achieving site‐selective functionalizations of the C7−H position of indoles, while discriminating among other bonds, is in high demand. Herein, we disclose site‐selective C7−H activations of indoles by ruthenium(II) biscarboxylate catalysis under mild conditions. Base‐assisted internal electrophilic‐type substitution C−H ruthenation by weak O‐coordination enabled the C7−H functionalization of indoles and offered a broad scope, including C−N and C−C bond formation. The versatile ruthenium‐catalyzed C7−H activations were characterized by gram‐scale syntheses and the traceless removal of the directing group, thus providing easy access to pharmaceutically relevant scaffolds. Detailed mechanistic studies through spectroscopic and spectrometric analyses shed light on the unique nature of the robust ruthenium catalysis for the functionalization of the C7−H position of indoles.
Keywords: alkenylation, amidation, C−H activation, indoles, ruthenium
Lucky seven: The challenging C7−H activation of indoles has been accomplished by ruthenium(II) catalysis. The versatile ruthenium catalysis allowed C−N andC−C bond formation, scalable reactions, and the traceless removal of the directing group. Detailed mechanistic investigations based on diverse analysis tools shed light on a novel mode of action.

Introduction
Indoles are key structural motifs in a plethora of biorelevant compounds, drugs, and pharmaceuticals.1 In particular, C7‐functionalized indole scaffolds exhibit a number of antiviral activities such as inhibitors of SARS‐CoV, HIV‐1, influenza virus A/Hanfang/359/95 (H3N2), and HSV II.2 As a consequence, there is a continued high demand for general strategies that provide easy access to C7‐substituted indoles in a sustainable fashion.3 Major advances in C−H activation have been witnessed in recent years.4 In particular, carboxylate‐assisted5 ruthenium(II) catalysis6 has emerged as a robust tool for a broad variety of C−H functionalizations, including alkylation,7 alkenylation,8 arylations,9 alkyne annulations,10 amidation,11 hydrogen‐isotope exchange,12 and meta‐C−H functionalizations.13 Despite the recent advances in versatile and cost‐effective ruthenium catalysis, the challenging C7−H functionalization14 of indoles has thus far remained elusive, which mainly stems from the formation of unfavorable six‐membered metallacycles.15 Pioneering contributions in iridium, rhodium, and palladium catalysis have been made by the groups of Chang,16 Ma,17 Shi,18 and others (Figure 1).19
Figure 1.

Selected antiviral compounds bearing C7−functionalized indoles.
Within our program on sustainable C−H activation, we have now developed the first ruthenium(II) biscarboxylate catalyzed C7−H bond activation of indoles by a weakly coordinating20 pivaloyl directing group via the formation of unfavorable six‐membered ruthenacycles. Notable features of our strategy include a) unprecedented carboxylate‐assisted ruthenium‐catalyzed C7−H activation of indoles, b) expedient C7−H activations enabling amidations and alkenylations under exceedingly mild conditions, c) deep mechanistic insight, which provides solid evidence for the site‐selective21 formation of six‐membered ruthenacycles over common five‐membered ruthenacycles, d) detailed kinetic studies by spectroscopic and spectrometric techniques that elucidate the formation of a ruthenium amide in a key step prior to C−H scission (Figure 2).
Figure 2.

Ruthenium(II) biscarboxylate catalyzed C7−H activations of indoles.
Results and Discussion
We initiated our studies by probing various reaction conditions for the envisioned ruthenium(II)‐catalyzed C7−H activation of N‐pivaloylindole 1 a with tosylazide 2 a (Table 1 and Table S1 in the Supporting Information). We were delighted to observe that the desired C7−H‐amidated product 3 aa was exclusively obtained in TFE as the solvent by using the cationic ruthenium(II) complex generated in situ from Ru(OAc)2(p‐cymene) and a silver salt (entry 1). Interestingly, the C7−H‐amidated product 3 aa was selectively formed as the sole product, while the C2−H bond remained entirely unmodified. The commercially available ruthenium(II) catalyst [RuCl2(p‐cymene)]2 fell short in providing the desired product (entry 2), clearly highlighting the importance of the ruthenium(II) biscarboxylate catalysis regime. We additionally employed other well‐defined ruthenium complexes, and sterically demanding ligands proved to be less effective (entries 3–5). Reactions conducted in HFIP or DCE as the solvent gave unsatisfactory results (entries 6 and 7). Control experiments revealed the essential role of the silver salt for solely forming a cationic ruthenium(II) carboxylate (entries 8–10).
Table 1.
Optimization studies for the C7−H activation of indoles.[a]

|
Entry |
Deviation from standard conditions |
Yield [%] |
|---|---|---|
|
1 |
none |
78 |
|
2 |
[RuCl2(p‐cymene)]2 instead of Ru(OAc)2(p‐cymene) |
0 |
|
3 |
Ru(OPiv)2(p‐cymene) instead of Ru(OAc)2(p‐cymene) |
60 |
|
4 |
Ru(O2CMes)2(p‐cymene) instead of Ru(OAc)2(p‐cymene) |
52 |
|
5 |
Ru(O2CAd)2(p‐cymene) instead of Ru(OAc)2(p‐cymene) |
48 |
|
6 |
HFIP instead of TFE |
63 |
|
7 |
DCE instead of TFE |
25 |
|
8 |
8 mol % instead of 20 mol % of AgSbF6 |
56 |
|
9 |
NaSbF6 instead of AgSbF6 |
7 |
|
10 |
AgCl instead of AgSbF6 |
0 |
[a] Reaction conditions: 1 a (0.25 mmol), 2 a (0.75 mmol), catalyst (10 mol %), AgSbF6 (20 mol %), TFE (1.0 mL), 40 °C, 24 h; yield of isolated product is given.
Thereafter, we probed the effect of changing the indole N‐substituent in substrates 1 (Scheme 1). Hence, a variety of ketones, amides, esters, phosphine oxides, sulfones, and pyridines were subjected to the established reaction conditions for the C7−H activation of indoles. Interestingly, only the N‐pivaloylindole 1 a underwent the C7−H amidation process. In contrast, other groups fell short in delivering the corresponding amidated products 3, thus illustrating the importance of steric and electronic effects of the N‐substituent for inducing the C7−H activation of indoles.
Scheme 1.
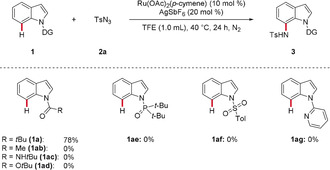
Examination of the N‐substitution pattern.
With the optimal reaction conditions in hand, we next explored the versatility of the C7−H amidation with a set of representative indoles 1 and azides 2 (Scheme 2). Differently substituted indoles 1 bearing alkyl, alkoxy, and halogen groups at the C3‐, C4‐, C5‐, and C6‐positions were site‐selectively transformed into the desired C7−H products. The ruthenium(II)‐catalyzed C7−H activation also tolerated various azides 2 bearing arenesulfonyl and alkanesulfonyl groups.
Scheme 2.

Scope of the C7−H amidation of indoles 1.
The robustness of the ruthenium(II) biscarboxylate catalyzed C7−H activation was further reflected by the C7−H alkenylation of N‐pivaloylindoles 1 with acrylates 4 through a redox‐active process (Scheme 3). Thus, the site‐selective alkenylated products 5 proved to be viable. In contrast, attempted fluorinations and trifluoromethylations have led thus far to less satisfactory results. However, a variety of substituted indoles 1 and acrylates 4 were tolerated, thus efficiently transforming into the desired indole‐7‐alkenyl derivatives 5 with excellent site selectivity.
Scheme 3.

Scope of the C7−H olefination of indoles 1.
The practical utility of the established carboxylate‐assisted ruthenium(II)‐catalyzed C7−H activation was next substantiated by the traceless removal of the N‐pivaloyl motif at room temperature (Scheme 4). It is noteworthy that the traceless removal of the N‐pivaloyl directing group could also proceed sequentially in a one‐pot strategy.
Scheme 4.

Traceless removal of the N‐pivaloyl group in a one‐pot fashion.
Indeed, the ruthenium(II)‐catalyzed C7−H amidation and alkenylation could be easily conducted on a gram scale without significant loss of the catalytic efficacy, thereby highlighting the robustness of the ruthenium(II) biscarboxylate catalysis (Scheme 5).
Scheme 5.

Gram‐scale reaction for the C7−H activation of indoles.
Flow technology provides an avenue for increasing the capability of chemical transformations in terms of improved heat and mass transfer.22 Thus, the scalability of the C7−H activation of indoles 1 was substantiated in a flow set‐up, whereby the desired amidated product 3 aa was obtained without loss of efficiency or selectivity (Scheme 6).23
Scheme 6.

Scalable flow reaction for the C7−H amidation of indoles.
Given the unique features of the unprecedented carboxylate‐assisted ruthenium(II)‐catalyzed C7−H activation, we became intrigued to delineating its mode of action (Scheme 7). To this end, intermolecular competition experiments with differently substituted N‐pivaloylindoles 1 were conducted (Scheme 7 a‐I).23 The competition experiment showed that the electron‐rich substrate possessed an inherent higher reactivity, thus suggesting that a concerted metalation/deprotonation (CMD) was less likely,24 while providing support for a base‐assisted internal electrophile‐type substitution (BIES) manifold.25 Additionally, a Hammett correlation was found in electronically differentiated substrates with a substituent in the meta‐position to the reaction center, thereby indicating that an electrophilic mechanism might occur in the C−H bond scission step (Scheme 7 a‐II).23 Furthermore, we performed the C7−H activation in the presence of isotopically labeled TFE (Scheme 7 a‐III).23 H/D scrambling in the C7‐ and C2‐positions was observed in the absence of substrate 2 a, which indicates the reversible nature of the C−H bond cleavage event. In sharp contrast, a H/D exchange experiment in the presence of 2 a provided evidence of the facile and irreversible formation of a new C−N bond, in agreement with the excellent site selectivity of the ruthenium‐catalyzed C7−H activation of indoles.
Scheme 7.

Detailed experimental and analytical mechanistic investigations.
Subsequently, a reaction performed with substrate 1 a and isotopically labeled compound [D]4‐1 a showed a primary kinetic isotope effect (KIE) of k H/k D≈1.1, thus providing support for a fast C7−H scission (Scheme 7 a‐IV).23 In good agreement with this finding, detailed kinetic experiments unraveled a zero‐order dependence of the reaction rate on the N‐pivaloylindole 1 a, but a first‐order dependence was observed for the concentration of the tosylazide 2 a and Ru(OAc)2(p‐cymene) (Scheme 7 a‐V).23
Furthermore, in operando NMR studies revealed the consumption of substrate 1 a and 2 a with concomitant formation of the desired product 3 aa, accompanied by a small induction period (Scheme 7 b‐I).23 The successive formation and consumption of three ruthenium species were also observed in the 1H NMR spectra during catalysis, one of which potentially corresponds to the catalyst resting state. Therefore, the catalytic experiment was also monitored by high‐resolution electrospray ionization‐mass spectrometry as an attempt to obtain further insight into the ruthenium speciation during the catalysis (Scheme 7 b‐II).23 Furthermore, X‐ray diffraction analysis showed the ruthenium species to be a dimeric ruthenium complex (Ru2‐I), with an exact mass of 728.0533, which is likely part of a deactivation pathway rather than a catalytic intermediate (Scheme 7 b‐III).23
On the basis of our experimental mechanistic studies, a plausible catalytic cycle for the carboxylate‐assisted ruthenium(II)‐catalyzed C7−H indole activation was proposed (Scheme 8). The mechanistic rationale commences with the coordination of substrate 2 a to active ruthenium catalyst A to form ruthenium amide intermediate B,26 which has often been described as the subsequent step to C−H activation. Presumably, C−H activation occurs through a base‐assisted internal electrophilic‐type substitution (BIES) mechanism, which preferentially leads to ruthenium(II) species D rather than D′. The intermediate D is transformed into ruthenium(II) amido intermediate E by amido insertion.11c, 27 Finally, a proto‐demetalation affords product 3 aa and regenerates the active catalyst A.
Scheme 8.

Proposed catalytic cycle.
Conclusion
In summary, we have reported the first well‐defined carboxylate‐assisted ruthenium‐catalyzed C7−H activation of indoles via the formation of challenging six‐membered ruthenacycles. Thus, the ruthenium(II) biscarboxylate catalyst enabled C−N and C−C bond formation with excellent levels of site selectivity under exceedingly mild conditions. The robustness and selectivity of the ruthenium catalysis were reflected by the traceless removal of the directing group and gram‐scale reactions. Detailed kinetic studies, including by NMR spectroscopy and mass spectrometry, highlighted the importance of ruthenium nitrenoid intermediates for the unique selectivity for the C7 indole position.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Generous support from the Kwanjeong Educational Foundation (fellowship to I.C.) and the DFG (Gottfried‐Wilhelm‐Leibniz‐Preis to L.A. and SPP1807) is gratefully acknowledged.
I. Choi, A. M. Messinis, L. Ackermann, Angew. Chem. Int. Ed. 2020, 59, 12534.
In memory of Rolf Huisgen
Contributor Information
Isaac Choi, http://www.ackermann.chemie.uni‐goettingen.de/index.html.
Prof. Dr. Lutz Ackermann, Email: Lutz.Ackermann@chemie.uni-goettingen.de.
References
- 1.
- 1a. Srivastava A., Pandeya S., Int. J. Curr. Pharm. Rev. Res. 2011, 4, 5–8; [Google Scholar]
- 1b. Kochanowska-Karamyan A. J., Hamann M. T., Chem. Rev. 2010, 110, 4489–4497; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. de Sa Alves F. R., Barreiro E. J., Manssour Fraga C. A., Mini-Rev. Med. Chem. 2009, 9, 782–793; [DOI] [PubMed] [Google Scholar]
- 1d. Gribble G. W., J. Chem. Soc. Perkin Trans. 1 2000, 1045–1075. [Google Scholar]
- 2.
- 2a. Zhang M.-Z., Chen Q., Yang G.-F., Eur. J. Med. Chem. 2015, 89, 421–441; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Yeung K.-S., Qiu Z., Xue Q., Fang H., Yang Z., Zadjura L., D′Arienzo C. J., Eggers B. J., Riccardi K., Shi P.-Y., Gong Y.-F., Browning M. R., Gao Q., Hansel S., Santone K., Lin P.-F., Meanwell N. A., Kadow J. F., Bioorg. Med. Chem. Lett. 2013, 23, 198–202; [DOI] [PubMed] [Google Scholar]
- 2c. Chen M., Gan L., Lin S., Wang X., Li L., Li Y., Zhu C., Wang Y., Jiang B., Jiang J., Yang Y., Shi J., J. Nat. Prod. 2012, 75, 1167–1176; [DOI] [PubMed] [Google Scholar]
- 2d. Ghosh A. K., Gong G., Grum-Tokars V., Mulhearn D. C., Baker S. C., Coughlin M., Prabhakar B. S., Sleeman K., Johnson M. E., Mesecar A. D., Bioorg. Med. Chem. Lett. 2008, 18, 5684–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shah T. A., De P. B., Pradhan S., Punniyamurthy T., Chem. Commun. 2019, 55, 572–587. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Gandeepan P., Muller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452; [DOI] [PubMed] [Google Scholar]
- 4b. Kalepu J., Gandeepan P., Ackermann L., Pilarski L. T., Chem. Sci. 2018, 9, 4203–4216; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Park Y., Kim Y., Chang S., Chem. Rev. 2017, 117, 9247–9301; [DOI] [PubMed] [Google Scholar]
- 4d. Kim D.-S., Park W.-J., Jun C.-H., Chem. Rev. 2017, 117, 8977–9015; [DOI] [PubMed] [Google Scholar]
- 4e. Dong Z., Ren Z., Thompson S. J., Xu Y., Dong G., Chem. Rev. 2017, 117, 9333–9403; [DOI] [PubMed] [Google Scholar]
- 4f. Zheng Q.-Z., Jiao N., Chem. Soc. Rev. 2016, 45, 4590–4627; [DOI] [PubMed] [Google Scholar]
- 4g. Gensch T., Hopkinson M. N., Glorius F., Wencel-Delord J., Chem. Soc. Rev. 2016, 45, 2900–2936; [DOI] [PubMed] [Google Scholar]
- 4h. Daugulis O., Roane J., Tran L. D., Acc. Chem. Res. 2015, 48, 1053–1064; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4i. Kakiuchi F., Kochi T., Murai S., Synlett 2014, 25, 2390–2414; [Google Scholar]
- 4j. Rouquet G., Chatani N., Angew. Chem. Int. Ed. 2013, 52, 11726–11743; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11942–11959; [Google Scholar]
- 4k. Li B., Dixneuf P. H., Chem. Soc. Rev. 2013, 42, 5744–5767; [DOI] [PubMed] [Google Scholar]
- 4l. Chen D. Y.-K., Youn S. W., Chem. Eur. J. 2012, 18, 9452–9474; [DOI] [PubMed] [Google Scholar]
- 4m. Giri R., Shi B.-F., Engle K. M., Maugel N., Yu J.-Q., Chem. Soc. Rev. 2009, 38, 3242–3272; [DOI] [PubMed] [Google Scholar]
- 4n. Ackermann L., Vicente R., Kapdi A. R., Angew. Chem. Int. Ed. 2009, 48, 9792–9826; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9976–10011. [Google Scholar]
- 5. Ackermann L., Chem. Rev. 2011, 111, 1315–1345. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Nareddy P., Jordan F., Szostak M., ACS Catal. 2017, 7, 5721–5745; [Google Scholar]
- 6b. Leitch J. A., Frost C. G., Chem. Soc. Rev. 2017, 46, 7145–7153; [DOI] [PubMed] [Google Scholar]
- 6c. Bruneau C., Dixneuf P. H., Top. Organomet. Chem. 2015, 55, 137–188; [Google Scholar]
- 6d. Louillat M.-L., Patureau F. W., Chem. Soc. Rev. 2014, 43, 901–910; [DOI] [PubMed] [Google Scholar]
- 6e. Li B., Dixneuf P. H., Top. Organomet. Chem. 2014, 48, 119–193; [Google Scholar]
- 6f. Arockiam P. B., Bruneau C., Dixneuf P. H., Chem. Rev. 2012, 112, 5879–5918; [DOI] [PubMed] [Google Scholar]
- 6g. Ackermann L., Vicente R., Top. Curr. Chem. 2009, 292, 211–229. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Schinkel M., Wallbaum J., Kozhushkov S. I., Marek I., Ackermann L., Org. Lett. 2013, 15, 4482–4484; [DOI] [PubMed] [Google Scholar]
- 7b. Schinkel M., Marek I., Ackermann L., Angew. Chem. Int. Ed. 2013, 52, 3977–3980; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4069–4072; [Google Scholar]
- 7c. Rouquet G., Chatani N., Chem. Sci. 2013, 4, 2201–2208; [Google Scholar]
- 7d. Ackermann L., Novák P., Vicente R., Hofmann N., Angew. Chem. Int. Ed. 2009, 48, 6045–6048; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6161–6164; [Google Scholar]
- 7e. Ackermann L., Novák P., Org. Lett. 2009, 11, 4966–4969; [DOI] [PubMed] [Google Scholar]
- 7f. Murai S., Kakiuchi F., Sekine S., Tanaka Y., Kamatani A., Sonoda M., Chatani N., Nature 1993, 366, 529–531; [Google Scholar]
- 7g. Lewis L. N., Smith J. F., J. Am. Chem. Soc. 1986, 108, 2728–2735. [Google Scholar]
- 8.
- 8a. Manikandan R., Madasamy P., Jeganmohan M., ACS Catal. 2016, 6, 230–234; [Google Scholar]
- 8b. Bechtoldt A., Tirler C., Raghuvanshi K., Warratz S., Kornhaaß C., Ackermann L., Angew. Chem. Int. Ed. 2016, 55, 264–267; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 272–275; [Google Scholar]
- 8c. Suzuki C., Hirano K., Satoh T., Miura M., Org. Lett. 2013, 15, 3990–3993; [DOI] [PubMed] [Google Scholar]
- 8d. Kozhushkov S. I., Ackermann L., Chem. Sci. 2013, 4, 886–896; [Google Scholar]
- 8e. Hashimoto Y., Ortloff T., Hirano K., Satoh T., Bolm C., Miura M., Chem. Lett. 2012, 41, 151–153; [Google Scholar]
- 8f. Hashimoto Y., Hirano K., Satoh T., Kakiuchi F., Miura M., Org. Lett. 2012, 14, 2058–2061; [DOI] [PubMed] [Google Scholar]
- 8g. Ackermann L., Wang L., Wolfram R., Lygin A. V., Org. Lett. 2012, 14, 728–731; [DOI] [PubMed] [Google Scholar]
- 8h. Cheng K., Yao B., Zhao J., Zhang Y., Org. Lett. 2008, 10, 5309–5312. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Simonetti M., Cannas D. M., Just-Baringo X., Vitorica-Yrezabal I. J., Larrosa I., Nat. Chem. 2018, 10, 724–731; [DOI] [PubMed] [Google Scholar]
- 9b. Nareddy P., Jordan F., Szostak M., Chem. Sci. 2017, 8, 3204–3210; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Nareddy P., Jordan F., Brenner-Moyer S. E., Szostak M., ACS Catal. 2016, 6, 4755–4759; [Google Scholar]
- 9d. Sollert C., Devaraj K., Orthaber A., Gates P. J., Pilarski L. T., Chem. Eur. J. 2015, 21, 5380–5386; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Aihara Y., Chatani N., Chem. Sci. 2013, 4, 664–670; [Google Scholar]
- 9f. Ferrer Flegeau E., Bruneau C., Dixneuf P. H., Jutand A., J. Am. Chem. Soc. 2011, 133, 10161–10170; [DOI] [PubMed] [Google Scholar]
- 9g. Ackermann L., Vicente R., Potukuchi H. K., Pirovano V., Org. Lett. 2010, 12, 5032–5035; [DOI] [PubMed] [Google Scholar]
- 9h. Özdemir I., Demir S., Çetinkaya B., Gourlaouen C., Maseras F., Bruneau C., Dixneuf P. H., J. Am. Chem. Soc. 2008, 130, 1156–1157; [DOI] [PubMed] [Google Scholar]
- 9i. Ackermann L., Vicente R., Althammer A., Org. Lett. 2008, 10, 2299–2302; [DOI] [PubMed] [Google Scholar]
- 9j. Ackermann L., Org. Lett. 2005, 7, 3123–3125; [DOI] [PubMed] [Google Scholar]
- 9k. Kakiuchi F., Kan S., Igi K., Chatani N., Murai S., J. Am. Chem. Soc. 2003, 125, 1698–1699; [DOI] [PubMed] [Google Scholar]
- 9l. Oi S., Fukita S., Hirata N., Watanuki N., Miyano S., Inoue Y., Org. Lett. 2001, 3, 2579–2581. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Ackermann L., Acc. Chem. Res. 2014, 47, 281–295; [DOI] [PubMed] [Google Scholar]
- 10b. Ackermann L., Lygin A. V., Hofmann N., Angew. Chem. Int. Ed. 2011, 50, 6379–6382; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6503–6506; [Google Scholar]
- 10c. Chinnagolla R. K., Jeganmohan M., Chem. Commun. 2012, 48, 2030–2032; [DOI] [PubMed] [Google Scholar]
- 10d. Chinnagolla R. K., Pimparkar S., Jeganmohan M., Org. Lett. 2012, 14, 3032–3035. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Chen C., Kim M. H., Hong S. H., Org. Chem. Front. 2015, 2, 241–247; [Google Scholar]
- 11b. Thirunavukkarasu V. S., Raghuvanshi K., Ackermann L., Org. Lett. 2013, 15, 3286–3289; [DOI] [PubMed] [Google Scholar]
- 11c. Kim J., Kim J., Chang S., Chem. Eur. J. 2013, 19, 7328–7333; [DOI] [PubMed] [Google Scholar]
- 11d. Murahashi S., Naota T., Saito E., J. Am. Chem. Soc. 1986, 108, 7846–7847. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Müller V., Weck R., Derdau V., Ackermann L., ChemCatChem 2020, 12, 100–104; [Google Scholar]
- 12b. Bechtoldt A., Ackermann L., ChemCatChem 2019, 11, 435–438. [Google Scholar]
- 13.
- 13a. Li G., Jia C., Cai X., Zhong L., Zou L., Cui X., Chem. Commun. 2020, 56, 293–296; [DOI] [PubMed] [Google Scholar]
- 13b. Korvorapun K., Kaplaneris N., Rogge T., Warratz S., Stückl A. C., Ackermann L., ACS Catal. 2018, 8, 886–892; [Google Scholar]
- 13c. Li B., Fang S.-L., Huang D.-Y., Shi B.-F., Org. Lett. 2017, 19, 3950–3953; [DOI] [PubMed] [Google Scholar]
- 13d. Teskey C. J., Lui A. Y. W., Greaney M. F., Angew. Chem. Int. Ed. 2015, 54, 11677–11680; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11843–11846; [Google Scholar]
- 13e. Hofmann N., Ackermann L., J. Am. Chem. Soc. 2013, 135, 5877–5884; [DOI] [PubMed] [Google Scholar]
- 13f. Saidi O., Marafie J., Ledger A. E. W., Liu P. M., Mahon M. F., Kociok-Köhn G., Whittlesey M. K., Frost C. G., J. Am. Chem. Soc. 2011, 133, 19298–19301. [DOI] [PubMed] [Google Scholar]
- 14. Fukuda T., Maeda R., Iwao M., Tetrahedron 1999, 55, 9151–9162. [Google Scholar]
- 15.
- 15a. Bu Q., Rogge T., Kotek V., Ackermann L., Angew. Chem. Int. Ed. 2018, 57, 765–768; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 773–776; [Google Scholar]
- 15b. Gandeepan P., Koeller J., Ackermann L., ACS Catal. 2017, 7, 1030–1034; [Google Scholar]
- 15c. Zhu R.-Y., Farmer M. E., Chen Y.-Q., Yu J.-Q., Angew. Chem. Int. Ed. 2016, 55, 10578–10599; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10734–10756; [Google Scholar]
- 15d. Jaiswal Y., Kumar Y., Thakur R., Pal J., Subramanian R., Kumar A., J. Org. Chem. 2016, 81, 12499–12505; [DOI] [PubMed] [Google Scholar]
- 15e. Shen P.-X., Wang X.-C., Wang P., Zhu R.-Y., Yu J.-Q., J. Am. Chem. Soc. 2015, 137, 11574–11577; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15f. Deb A., Bag S., Kancherla R., Maiti D., J. Am. Chem. Soc. 2014, 136, 13602–13605; [DOI] [PubMed] [Google Scholar]
- 15g. Park J., Kim M., Sharma S., Park E., Kim A., Lee S. H., Kwak J. H., Jung Y. H., Kim I. S., Chem. Commun. 2013, 49, 1654–1656; [DOI] [PubMed] [Google Scholar]
- 15h. Ackermann L., Diers E., Manvar A., Org. Lett. 2012, 14, 1154–1157; [DOI] [PubMed] [Google Scholar]
- 15i. Yeung C. S., Zhao X., Borduas N., Dong V. M., Chem. Sci. 2010, 1, 331–336. [Google Scholar]
- 16.
- 16a. Kim Y., Park Y., Chang S., ACS Cent. Sci. 2018, 4, 768–775; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Kim Y., Park J., Chang S., Org. Lett. 2016, 18, 1892–1895. [DOI] [PubMed] [Google Scholar]
- 17. Xu L., Zhang C., He Y., Tan L., Ma D., Angew. Chem. Int. Ed. 2016, 55, 321–325; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 329–333. [Google Scholar]
- 18.
- 18a. Borah A. J., Shi Z., J. Am. Chem. Soc. 2018, 140, 6062–6066; [DOI] [PubMed] [Google Scholar]
- 18b. Yang Y., Qiu X., Zhao Y., Mu Y., Shi Z., J. Am. Chem. Soc. 2016, 138, 495–498. [DOI] [PubMed] [Google Scholar]
- 19. Song Z., Antonchick A. P., Org. Biomol. Chem. 2016, 14, 4804–4808. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Kalepu J., Pilarski L. T., Molecules 2019, 24, 830; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. De Sarkar S., Liu W., Kozhushkov S. I., Ackermann L., Adv. Synth. Catal. 2014, 356, 1461–1479; [Google Scholar]
- 20c. Engle K. M., Mei T.-S., Wasa M., Yu J.-Q., Acc. Chem. Res. 2012, 45, 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Fosu S. C., Hambira C. M., Chen A. D., Fuchs J. R., Nagib D. A., Chem 2019, 5, 417–428; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Liao K., Yang Y.-F., Li Y., Sanders J. N., Houk K. N., Musaev D. G., Davies H. M. L., Nat. Chem. 2018, 10, 1048–1055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Hirano K., Miura M., Chem. Sci. 2018, 9, 22–32; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21d. Brandhofer T., García Mancheño O., Eur. J. Org. Chem. 2018, 6050–6067; [Google Scholar]
- 21e. Ma W., Gandeepan P., Li J., Ackermann L., Org. Chem. Front. 2017, 4, 1435–1467; [Google Scholar]
- 21f. Song W., Kozhushkov S. I., Ackermann L., Angew. Chem. Int. Ed. 2013, 52, 6576–6578; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6706–6708; [Google Scholar]
- 21g. Neufeldt S. R., Sanford M. S., Acc. Chem. Res. 2012, 45, 936–946; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21h. Davies H. M. L., Morton D., Chem. Soc. Rev. 2011, 40, 1857–1869; [DOI] [PubMed] [Google Scholar]
- 21i. Phipps R. J., Grimster N. P., Gaunt M. J., J. Am. Chem. Soc. 2008, 130, 8172–8174. [DOI] [PubMed] [Google Scholar]
- 22. Santoro S., Ferlin F., Ackermann L., Vaccaro L., Chem. Soc. Rev. 2019, 48, 2767–2782. [DOI] [PubMed] [Google Scholar]
- 23.For detailed information, see the Supporting Information.
- 24.
- 24a. Wang L., Carrow B. P., ACS Catal. 2019, 9, 6821–6836; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Lapointe D., Fagnou K., Chem. Lett. 2010, 39, 1118–1126; [Google Scholar]
- 24c. García-Cuadrado D., Braga A. A. C., Maseras F., Echavarren A. M., J. Am. Chem. Soc. 2006, 128, 1066–1067. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Naksomboon K., Poater J., Bickelhaupt F. M., Fernández-Ibáñez M. Á., J. Am. Chem. Soc. 2019, 141, 6719–6725; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Tan E., Quinonero O., Elena de Orbe M., Echavarren A. M., ACS Catal. 2018, 8, 2166–2172; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Zell D., Bursch M., Müller V., Grimme S., Ackermann L., Angew. Chem. Int. Ed. 2017, 56, 10378–10382; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10514–10518; [Google Scholar]
- 25d. Ma W., Mei R., Tenti G., Ackermann L., Chem. Eur. J. 2014, 20, 15248–15251. [DOI] [PubMed] [Google Scholar]
- 26. Hong S. Y., Park Y., Hwang Y., Kim Y. B., Baik M.-H., Chang S., Science 2018, 359, 1016–1021. [DOI] [PubMed] [Google Scholar]
- 27. Park S. H., Kwak J., Shin K., Ryu J., Park Y., Chang S., J. Am. Chem. Soc. 2014, 136, 2492–2502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary