

Summary
Background
Agonists of 5‐hydroxytryptamine 4 receptor are potential agents for irritable bowel syndrome with predominant constipation (IBS‐C). However, only tegaserod has been approved for a very limited population in the US.
Aim
To evaluate the efficacy and safety of minesapride in patients with Rome IV defined IBS‐C.
Methods
A double‐blind, placebo‐controlled, dose‐finding study was performed. Overall, 411 patients were randomised to receive minesapride at 10, 20 or 40 mg/d, or placebo for 12 weeks. The primary endpoint was Food and Drug Administration (FDA) composite endpoint (responder: a patient who reported an increase in one or more complete spontaneous bowel movements from baseline and improvement of ≥30% from baseline in weekly average of worst abdominal pain score, both in the same week for ≥6/12 weeks).
Results
The FDA composite responder rate was 13.6% (14/103) in the placebo group, 13.6% (14/103) in the 10 mg group, 19.2% (20/104) in the 20 mg group and 14.9% (15/101) in the 40 mg group, and no dose‐response relationship was found. A greater percentage of minesapride 40 mg‐treated patients than placebo‐treated patients met both responder requirements for ≥9/12 weeks as the stricter composite evaluation (P < 0.05). Furthermore, minesapride 40 mg significantly increased SBM frequency compared with placebo (adjusted P < 0.001 at Week 12). The most common adverse event was mild diarrhoea.
Conclusions
Minesapride was safe and well‐tolerated. Although the primary endpoint was negative, minesapride 40 mg is likely to improve the stricter composite endpoint and SBM frequency.
Japan Pharmaceutical Information Center Number: Japic CTI‐163459.
1. INTRODUCTION
Irritable bowel syndrome (IBS) is a functional gastrointestinal disorder characterised by recurrent abdominal pain associated with changes in bowel habits. 1 IBS is divided into four subtypes in the Rome IV criteria. 2 Although the Rome criteria specify strict criteria, in practice most patients are diagnosed by report rather than daily diary which actually shows more normal days than most patients recall. Given that, Rome IV recommended basing subtyping on days with abnormal stool. Of the four subtypes, IBS with predominant constipation (IBS‐C) is defined by hard or lumpy stools accounting for >25% of defaecations, and loose or watery stools accounting for <25% of defaecations, in addition to abdominal pain. 1
IBS patients have significantly worse health‐related quality of life (QOL). 3 , 4 Epidemiological studies have shown that IBS affects 8.8% of the adult population globally. 5 The economic burden of moderate to severe IBS‐C patients is high. A study conducted in European countries showed that the annual direct cost (combined cost to healthcare system and patient) is €1421.7‐€2487.1. 6
Serotonin 4 (5‐hydroxytryptamine 4:5‐HT4) receptor agonists are believed to exert their therapeutic effect by acting on the 5‐HT4 receptors in the gastrointestinal tract and lead to the release of acetylcholine, thereby promoting gastrointestinal motility. 7 In fact, tegaserod can be used for a subpopulation of IBS‐C patients (female patients less than 65 years of age with contraindications to account for cardiovascular risk) in the US. 8 , 9 A 5‐HT4 receptor agonist prucalopride is known to increase the stool frequency and accelerate colonic transit. 10 For chronic idiopathic constipation, prucalopride has proven efficacy 11 , 12 and has been approved globally. However, the efficacy for IBS‐C patients has not been evaluated in any large‐scaled randomised controlled trial (RCT). Mosapride has been reported to increase bowel frequency in patients with chronic constipation due to Parkinson's disease 13 or diabetes mellitus. 14 Actually, mosapride activates colorectal motility measured with a barostat in IBS‐C patients. 15 However, RCTs with larger sample size are lacking. There is no approved 5‐HT4 agonist for IBS‐C, other than tegaserod for a limited population in the US. Therefore, new 5‐HT4 agonists are required for IBS‐C patients.
Minesapride (DSP‐6952) is a novel 5‐HT4 receptor partial agonist with high selectivity. The affinity for the 5‐HT4 receptors is almost comparable to that in prucalopride. 16 Minesapride has potent enterokinetic effects and shows minimal effects on human Ether‐a‐go‐go related gene potassium channels in pre‐clinical studies. 16 , 17 In clinical studies, a phase 1 (first in human) study showed minesapride exposure in plasma increased in a dose‐proportional manner with acceptable safety profile up to 120 mg/d. 18 A QT/QTc study conducted according to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline showed that minesapride has no effect on QT prolongation. 19 In an early phase 2 study, Rome III‐defined patients with IBS‐C received minesapride at 1, 4, 12 or 40 mg/d once daily for 4 weeks. 20 Minesapride 40 mg/d led to increases in the number of complete spontaneous bowel movements (CSBMs), improvement in abdominal symptoms, and overall improvement in IBS symptoms, with favourable tolerability. 20 On the basis of these findings, minesapride 40 mg/d is expected to be efficacious in the treatment of IBS‐C. The aim of the present study was to test the hypothesis that minesapride would show efficacy and safety in a dose‐dependent manner during the treatment period for 12 weeks in patients with IBS‐C diagnosed according to Rome IV criteria.
2. METHODS
2.1. Patients
Female and male outpatients aged 20‐64 years were eligible to participate if they met Rome IV criteria 2 for IBS‐C. There was no organic disorder on examinations (eg colonoscopy) performed within 5 years before screening. During the week immediately before randomisation, patients had to report an average score ≥3.0 for daily abdominal pain at its worst evaluation on an 11‐point (0: none, 10: very severe) numerical rating scale, an average of <3 CSBMs per week, 21 and an average of ≤5 spontaneous bowel movements (SBMs) per week. Key exclusion criteria included a past or current history of serious diseases that is inappropriate for study participation; a past or current history of diseases that might affect the gastrointestinal transit or function of the large intestine; severe depression or anxiety symptoms judged to affect the assessments of the drug effects; and a past or current history of hyperthyroidism, hypothyroidism or malignancy. Patients who underwent gastrointestinal surgery were also excluded. All criteria including other inclusion and exclusion criteria are provided in Tables S1 and S2.
2.2. Study design
This late phase 2 study was a multicentre, placebo‐controlled, randomised, double‐blind, parallel‐group, comparative study in patients with IBS‐C. This study consisted of the following three periods: placebo run‐in period (2 weeks), treatment period (12 weeks), and follow‐up period (4 weeks). In the run‐in period, a single‐blind placebo was orally administered once daily after breakfast for 2 weeks. In the treatment period, the subjects who had met the eligibility criteria at the end of the run‐in period and had entered the treatment period were randomised in a 1:1:1:1 ratio to receive double‐blind treatment with minesapride at 10, 20, 40 mg/d or placebo once daily after breakfast for 12 weeks. The follow‐up period was defined as 4 weeks from the day following either the end of the treatment period or examination at discontinuation (Figure 1A). Randomisation was done using an interactive web response system, was not stratified, and the block size was 12. The randomisation schedule was generated by the vendor responsible for the system and approved by sponsor. For each dose, tablets and packaging were identical in size, shape, colour, and appearance. All patients, investigators, site stuffs and the study sponsor were masked to treatment assignments. According to the protocol, sodium picosulfate, bisacodyl or enema could be used as a rescue medication according to the protocol (Supplementary Text S1).
FIGURE 1.

(A) Design of the study. (B) Patients disposition
When any subject had a stool consistency of Bristol Stool Form Scale (BSFS) 22 type 6 or 7 on at least two consecutive days after administration of the study drug, the study treatment could be temporarily suspended as necessary at the discretion of the Investigator. Administration of the study drug was to be discontinued if the subject required a third occasion of rescue medication between the start of the run‐in period and the end of the treatment period, or the subject continued to have a stool consistency of BSFS type 6 or 7 even after study drug interruption for a reasonable time period.
Drugs used to treat gastrointestinal symptoms or affect IBS symptoms were prohibited from two days before screening until the end of the follow‐up period. Colonoscopy, sigmoidoscopy, lower gastrointestinal series, upper gastrointestinal series, stool disimpaction and non‐drug therapies to improve IBS symptoms were also prohibited from three days before screening until the end of the follow‐up period. Other restrictions are described in Supplementary Text S2.
The study was conducted between January 2017 and October 2017 (from the first informed consent to the last observation) at 43 medical institutions in Japan and was reviewed and approved by the central institutional review board (Supplementary Listing).
This study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and in compliance with the protocol, ICH‐good clinical practice, and related notifications. The investigator ensured that potential subjects were given full and adequate verbal and written information regarding the nature, purpose, and possible risks and benefits of the study. The investigator obtained a signed informed consent form from all subjects before any study procedures were performed. This study was registered with Japan Pharmaceutical Information Center, number Japic CTI‐163459. All authors had access to the study data and reviewed and approved the final manuscript.
2.3. Study assessments
Daily electronic patient‐reported outcomes assessments included symptom ratings of worst abdominal pain, abdominal discomfort, and abdominal bloating (all abdominal symptoms were measured using an 11‐point numerical rating scale), as well as the number of bowel movements (BMs) and whether rescue medication was used. Each BM was assessed for sensation of incomplete evacuation (yes/no), stool consistency (BSFS) and severity of straining rated on a 11‐point numerical rating scale from 0 (none) to 10 (very severe). Weekly assessments by electronic patient‐reported outcomes included adequate relief of IBS‐C symptoms in the last 1 week (yes/no) and global relief of IBS‐C symptoms in the last 1 week (7‐point ordinal scale: 1 = completely relieved, 2 = considerably relieved, 3 = slightly relieved, 4 = unchanged, 5 = slightly worsened, 6 = considerably worsened, and 7 = worsened most seriously). Each subject assessed the individual items of the IBS Severity Index Japanese version (IBS‐SI, IBS‐Severity Scoring System; IBS‐SSS), 23 , 24 IBS Quality of Life measure Japanese version (IBS‐QOL), 25 , 26 and EuroQoL 5 Dimensions‐5 Levels Japanese version (EQ‐5D‐5L) 27 at the end of the run‐in period, at Weeks 4 and 8 of the treatment period, and the end of the treatment period (or at discontinuation).
Clinical laboratory tests, vital sign assessments, body weight assessments and 12‐lead electrocardiogram (ECG) were conducted at screening, the end of the run‐in period, Week 2, Week 4 and Week 8 of the treatment period, the end of the treatment period (or at discontinuation), and the end of the follow‐up period. Adverse events (AEs) were recorded from the first day of administration of the study drug for the run‐in period to the end of the follow‐up period. Among them, treatment‐emergent AEs were defined as adverse events with a start date on or after Day 1 (ie the day following the end of the run‐in period).
The definition of diarrhoea is given in Supplementary Text S3. The number and percentage of subjects who had World Health Organization (WHO)‐defined diarrhoea (BSFS type 6 or 7 stools 3 or more times a day) were calculated as well.
Blood samples were collected for plasma drug concentration measurements in individual subjects at Week 2 and at the end of the treatment period.
2.4. Study endpoints
The primary endpoint was set in accordance with recommendations in the Food and Drug Administration (FDA) Guidance for Industry: Irritable Bowel Syndrome. 21 This endpoint was a responder rate defined as the percentage of responders. Responders were patients who reported (a) an increase of at least one CSBM per week from baseline (the CSBM response criterion) and (b) a decrease in the weekly average score of worst abdominal pain in the past 24 hours (measured daily) of at least 30 percent compared with the baseline weekly average (the abdominal pain response criterion), both in the same week for ≥6/12 weeks. The secondary endpoints were composite response for at least 9/12 weeks, CSBM, SBMs and others. The composite responder rate for 9/12 weeks or more is the stricter evaluation than the primary endpoint because the patients needed to have both the CSBM response and abdominal pain response in the same week for at least 75% of the treatment period. CSBMs and SBMs per week were calculated on the basis of the bowel movements recorded in the symptom diary. An SBM was defined as a bowel movement that occurred without use of rescue medication. A CSBM was defined as an SBM without a sensation of incomplete evacuation. On the day of and the day following the use of rescue medication, the numbers of bowel movements, CSBMs and SBMs were regarded as zero for the calculation. For any week in which the bowel movements were appropriately recorded on ≤4 days, CSBMs per week, SBMs per week, the weekly average score of straining during bowel movement, and the weekly average score of stool consistency were regarded as missing. For any week in which the abdominal symptoms (ie abdominal pain, abdominal discomfort and abdominal bloating) were appropriately recorded on ≤4 days, the weekly average score of abdominal symptoms was regarded as missing. Further information is described in Supplementary Text S4.
2.5. Statistical analysis
This study was intended to collect information required to determine the phase 3 study dose and design. Thus, on the basis of the results from a previous early phase 2 study, 20 and also in light of the feasibility of this study, the sample size per group was set to 100. The primary analysis population for the efficacy analysis was the modified intention‐to‐treat (mITT) population. The mITT population consisted of all patients who were randomised and received the study drug for the treatment period, and had a baseline and at least one post baseline measurement of the number of bowel movements and abdominal pain score, regardless of any protocol deviation.
As for the primary endpoint, paired comparison was made between each minesapride group and the placebo group, in terms of the percentage of the responders meeting both the CSBM response criterion and the abdominal pain response criterion for at least 6 weeks in the treatment period. For the comparison, the difference and its 95% confidence interval (CI) were calculated, and the P value was calculated using the Fisher's exact test.
Regarding CSBM, SBM, abdominal pain, abdominal discomfort, abdominal bloating, straining during bowel movement, stool consistency, global relief of IBS‐C symptoms, IBS‐SI overall score, IBS‐QOL overall score, and EQ‐5D‐5L index change from baseline, the mixed model for repeated measures analysis was performed, and the model included treatment group and visit as fixed effects, baseline as a covariate, and treatment‐visit interaction for paired comparison between each minesapride group and the placebo group. Within‐subject correlation was calculated using the unstructured covariance matrix. The degree of freedom was calculated using the Kenward‐Roger's approximation. When iterative calculations did not converge with this analysis model, then a robust sandwich estimator was to be used for estimation of the standard deviation in the fixed effect, and then the covariance matrixes were to be used in the following order, and the first model with convergence was to be employed: heterogeneous Toeplitz covariance matrix, heterogeneous first‐order autoregressive covariance matrix, Toeplitz covariance matrix.
In the primary endpoint, the Hochberg's method was used for multiplicity adjustment for comparison of multiple treatment groups to maintain the overall type 1 error at 5%. A similar method was used also on the other endpoints for adjustment of multiplicity in the statistical testing. However, no multiplicity adjustment was performed for statistical testing of multiple tests or multiple time points of analysis. To evaluate dose response, the Cochran‐Armitage trend test with six weights as follows was tested; (placebo, minesapride 10, 20, 40) = (−3, −1, 1, 3), (−3, 1, 1, 1), (−5, −1, 3, 3), (−3, −3, 1, 5), (−1, −1, 1, 1) and (−1, −1, −1, 3). Subgroup analysis methods are described in Supplementary Text S5.
3. RESULTS
3.1. Patients disposition and characteristics
The disposition of the study subjects is presented in Figure 1B. All randomised patients (411 patients) who received the study drug during the treatment period were included in the mITT population. There were 386 (93.9%) patients who completed the treatment period. The most common reason for discontinuation was withdrawal of consent, reported in 13 patients.
The summary of the main demographic data in the mITT population is provided in Table 1. Female subjects accounted for 86.4%, and patient age ranged from 20 to 64 years, with a mean age of 42.5 years. The mean duration (±SD) of IBS‐C was 14.5 (±11.2) years. The mean weekly number of CSBMs at baseline was 0.34 (±0.63), and the mean weekly number of SBMs was 2.92 (±1.17). The mean abdominal pain score at baseline was 5.59 (±1.53). The mean baseline values showed no substantial differences across the four treatment groups.
TABLE 1.
Baseline demographic and clinical characteristics—mITT population
| Placebo | Minesapride 10mg | Minesapride 20mg | Minesapride 40mg | Total | |
|---|---|---|---|---|---|
| N = 103 | N = 103 | N = 104 | N = 101 | N = 411 | |
| Sex | |||||
| Female | 85 (82.5%) | 92 (89.3%) | 87 (83.7%) | 91 (90.1%) | 355 (86.4%) |
| Male | 18 (17.5%) | 11 (10.7%) | 17 (16.3%) | 10 (9.9%) | 56 (13.6%) |
| Age, y | 43.6 ± 11.0 | 43.2 ± 9.4 | 42.5 ± 10.6 | 40.7 ± 10.1 | 42.5 ± 10.3 |
| Weight, kg | 56.6 ± 10.5 | 55.9 ± 10.2 | 55.0 ± 9.5 | 56.5 ± 10.7 | 56.0 ± 10.2 |
| BMI, kg/m2 | 21.9 ± 3.6 | 21.8 ± 3.4 | 21.3 ± 2.7 | 22.1 ± 3.6 | 21.7 ± 3.4 |
| Duration of IBS‐C, y | 14.9 ± 12.3 | 14.6 ± 10.9 | 16.6 ± 11.5 | 11.7 ± 9.6 | 14.5 ± 11.2 |
| Frequency of CSBM, counts/week | 0.37 ± 0.66 | 0.34 ± 0.63 | 0.42 ± 0.66 | 0.25 ± 0.54 | 0.34 ± 0.63 |
| Frequency of SBM, counts/week | 2.89 ± 1.12 | 2.98 ± 1.13 | 2.82 ± 1.25 | 2.98 ± 1.20 | 2.92 ± 1.17 |
| Stool consistency (BSFS) | 1.91 ± 0.61 | 1.87 ± 0.63 | 1.90 ± 0.64 | 1.81 ± 0.65 | 1.87 ± 0.63 |
| Abdominal symptoms | |||||
| Pain | 5.67 ± 1.58 | 5.79 ± 1.49 | 5.54 ± 1.63 | 5.37 ± 1.41 | 5.59 ± 1.53 |
| Discomfort | 6.12 ± 1.66 | 6.28 ± 1.56 | 5.94 ± 1.62 | 5.94 ± 1.60 | 6.07 ± 1.61 |
| Bloating | 6.31 ± 1.61 | 6.45 ± 1.57 | 6.05 ± 1.63 | 6.18 ± 1.69 | 6.25 ± 1.63 |
| IBS‐SI (IBS‐SSS) overall score | 316.7 ± 83.4 | 320.0 ± 71.7 | 303.2 ± 74.9 | 302.4 ± 75.8 | 310.6 ± 76.7 |
| IBS‐QOL overall score | 77.5 ± 18.6 | 74.9 ± 18.0 | 77.9 ± 16.2 | 77.6 ± 18.1 | 77.0 ± 17.7 |
| EQ‐5D‐5L index value | 0.90 ± 0.10 | 0.91 ± 0.09 | 0.91 ± 0.09 | 0.90 ± 0.10 | 0.90 ± 0.09 |
Data are presented as n (%) or mean ± SD.
Abbreviations: BMI, body mass index; BSFS, Bristol Stool Form Scale; CSBM, complete spontaneous bowel movement; EQ‐5D‐5L, Euro‐QOL 5 Dimensions‐5 Levels Japanese version; IBS‐C, irritable bowel syndrome with constipation; IBS‐QOL, IBS quality of life measure; IBS‐SI, IBS Severity Index; IBS‐SSS, IBS Severity Scoring System; SBM, spontaneous bowel movement.
3.2. Efficacy
The primary endpoint was the responder rate defined as the percentage of responders (ie patients who met the CSBM response criterion and the abdominal pain response criterion, both in the same week for ≥6/12 weeks [FDA composite endpoint]). The responder rate was 13.6% (14/103) in the placebo group, 13.6% (14/103) in the 10 mg group, 19.2% (20/104) in the 20 mg group, and 14.9% (15/101) in the 40 mg group (Table 2), with the effect showing no dose‐response relationship (Table S3A).
TABLE 2.
Responder rate analyses (at least 50% [at least 6/12 weeks] responder)
|
Placebo N = 103 |
Minesapride | |||
|---|---|---|---|---|
|
10 mg N = 103 |
20 mg N = 104 |
40 mg N = 101 |
||
| Composite endpoint (responders who met ≥30% abdominal pain reduction and increase ≥1 CSBM from baseline in the same week for ≥6/12 weeks), n/N (%) | 14/103 (13.6) | 14/103 (13.6) | 20/104 (19.2) | 15/101 (14.9) |
| 95% CI | 7.6, 21.8 | 7.6, 21.8 | 12.2, 28.1 | 8.6, 23.3 |
| Difference vs placebo, % | 0.0 | 5.6 | 1.3 | |
| P value vs placebo | 1.000 | 0.349 | 0.843 | |
| Adjusted P value vs placebo | 1.000 | 1.000 | 1.000 | |
| Responders who met ≥30% abdominal pain reduction from baseline for ≥6/12 weeks, n/N (%) | 30/103 (29.1) | 26/103 (25.2) | 29/104 (27.9) | 30/101 (29.7) |
| 95% CI | 20.6, 38.9 | 17.2, 34.8 | 19.5, 37.5 | 21.0, 39.6 |
| Difference vs placebo, % | −3.9 | −1.2 | 0.6 | |
| P value vs placebo | 0.639 | 0.878 | 1.000 | |
| Adjusted P value vs placebo | 1.000 | 1.000 | 1.000 | |
| Responders who met increase ≥1 CSBM from baseline for ≥6/12 weeks, n/N (%) | 39/103 (37.9) | 43/103 (41.7) | 47/104 (45.2) | 38/101 (37.6) |
| 95% CI | 28.5, 48.0 | 32.1, 51.9 | 35.4, 55.3 | 28.2, 47.8 |
| Difference vs placebo, % | 3.9 | 7.3 | −0.2 | |
| P value vs placebo | 0.669 | 0.324 | 1.000 | |
| Adjusted P value vs placebo | 1.000 | 0.973 | 1.000 | |
The 95% confidence interval of the responder rate was calculated using the Clopper‐Pearson method. For the comparison, the treatment difference and its exact 95% confidence interval were calculated, and the P value was calculated using the Fisher's exact test. The Hochberg's method was used for adjustment of multiplicity in the statistical testing for comparison of multiple minesapride groups and the placebo group.
Abbreviations: CI, confidence interval; CSBM, complete spontaneous bowel movement.
To clarify the change in placebo response change over time, a post hoc analysis was conducted. In this analysis, the percentage of responders (ie patients who met the CSBM response criterion and the abdominal pain response criterion in the same week for ≥2 weeks in the first 4 weeks [Week 1‐4], ≥3 weeks in the first 6 weeks [Week 1‐6], ≥4 weeks in the first 8 weeks [Week 1‐8], and ≥5 weeks in the first 10 weeks [Week 1‐10]) were calculated in addition to ≥6/12 weeks. The results showed that the greater difference from the placebo group was detected in the 20 and 40 mg groups up to 8 weeks (Figure S1).
Regarding secondary endpoints, the percentage of the responders meeting the CSBM response criterion and the abdominal pain response criterion in the same week for ≥9/12 weeks was 3.9% in the placebo group, 3.9% in the 10 mg group, 8.7% in the 20 mg group and 12.9% in the 40 mg group. The responder rate in the 40 mg group was greater than that in the placebo group (P < 0.05, adjusted P = 0.070) (Figure 2). The contrast with the lowest P value was [−3, −3, 1, 5]. (P = 0.004; Table S3B). In all minesapride groups, the frequency of SBMs tended to increase (improve) from Week 1. The change from baseline in the number of SBMs at Week 12 (LS mean ± SE) was 0.84 ± 0.20 in the placebo group, 1.33 ± 0.19 in the 10 mg group, 1.45 ± 0.19 in the 20 mg group, and 1.89 ± 0.20 in the 40 mg group. A significant difference was shown between the 40 mg group and the placebo group throughout the treatment period (Adjusted P < 0.001 at Week 12) (Figure 3).
FIGURE 2.

Responder rate analyses (at least 75% [at least 9/12 weeks] responder). (A) The percentage of responders: patients who met the CSBM response criterion for ≥9/12 weeks. (B) The percentage of responders: patients who met the abdominal pain response criterion for ≥9/12 weeks. (C) Composite endpoint (≥30% abdominal pain reduction and increase ≥1 CSBM from baseline in the same week for ≥9/12 weeks). *P value = 0.023 vs placebo (Adjusted P = 0.070). The 95% confidence interval of the responder rate was calculated using the Clopper‐Pearson method. For the comparison, the treatment difference and its exact 95% confidence interval were calculated, and the P value was calculated using the Fisher's exact test. The Hochberg's method was used for adjustment of multiplicity in the statistical testing for comparison of multiple minesapride groups and the placebo group
FIGURE 3.
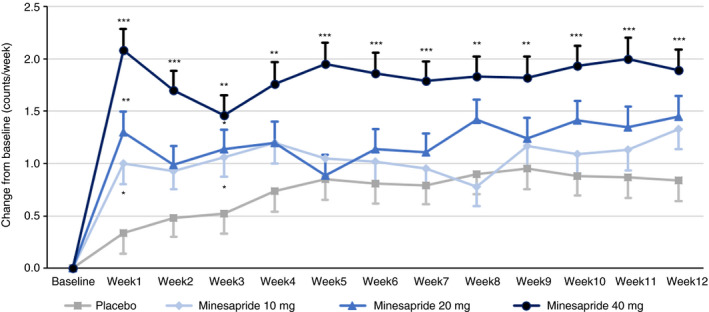
Changes from baseline in the number of SBMs by week. Data are shown as LS mean. Error bars show SE. *Adjusted P < 0.05, **Adjusted P < 0.01, ***Adjusted P < 0.001 vs placebo. The mixed model for repeated measures analysis was performed, and the model included treatment group and visit as fixed effects, baseline as a covariate, and treatment‐visit interaction for paired comparison between each minesapride group and the placebo group
In the subgroup analysis using subjects with fewer than 3 SBMs per week at baseline, the responder rate for ≥6/12 weeks (FDA composite endpoint) and ≥9/12 weeks (stricter composite endpoint) in the 40 mg group were higher than those in the placebo group (stricter composite endpoint: P < 0.05, adjusted P = 0.112) (Figure S2). Furthermore, the percentage of subjects with any SBM within 1 day after the start of administration (ie on the first day of administration) of the study drug for the treatment period was 26.8% in the placebo group, 53.7% in the 10 mg group, 53.8% in the 20 mg group, and 65.7% in the 40 mg group, and all minesapride groups showed significant differences compared with the placebo group (Adjusted P < 0.05 for all minesapride groups) (Table S4).
The changes from baseline in CSBM, abdominal pain, abdominal discomfort, abdominal bloating, straining during bowel movement and stool consistency are shown in Table 3. The frequency of CSBMs tended to increase (improve) after the start of administration of the study drug for the treatment period in the 40 mg group. However, there were no significant differences from the placebo group at Week 12. In terms of other abdominal symptoms (abdominal pain, abdominal discomfort, abdominal bloating, and straining during bowel movement), the change from baseline was greatest in the 40 mg group at Week 12. However, there was no significant difference between the 40 mg group and the placebo group.
TABLE 3.
Changes from baseline in CSBMs, abdominal symptoms and stool consistency
|
Placebo N = 103 |
Minesapride | |||
|---|---|---|---|---|
|
10 mg N = 103 |
20 mg N = 104 |
40 mg N = 101 |
||
| CSBMs (counts/week) | 0.96 (0.20) | 1.22 (0.20) | 1.29 (0.20) | 1.39 (0.21) |
| P value vs placebo | 0.356 | 0.234 | 0.136 | |
| Adjusted P value vs placebo | 0.356 | 0.356 | 0.356 | |
| Abdominal pain | −1.30 (0.20) | −0.93 (0.20) | −1.31 (0.20) | −1.42 (0.21) |
| P value vs placebo | 0.200 | 0.968 | 0.681 | |
| Adjusted P value vs placebo | 0.599 | 0.968 | 0.968 | |
| Abdominal discomfort | −1.36 (0.20) | −0.97 (0.20) | −1.39 (0.20) | −1.46 (0.21) |
| P value vs placebo | 0.171 | 0.909 | 0.740 | |
| Adjusted P value vs placebo | 0.513 | 0.909 | 0.909 | |
| Abdominal bloating | −1.34 (0.21) | −1.06 (0.21) | −1.47 (0.21) | −1.48 (0.21) |
| P value vs placebo | 0.333 | 0.657 | 0.627 | |
| Adjusted P value vs placebo | 0.657 | 0.657 | 0.657 | |
| Straining during BM | −1.45 (0.21) | −1.28 (0.21) | −1.71 (0.21) | −2.06 (0.22) |
| P value vs placebo | 0.562 | 0.384 | 0.044 | |
| Adjusted P value vs placebo | 0.562 | 0.562 | 0.132 | |
| Stool consistency | 1.08 (0.11) | 1.29 (0.10) | 1.32 (0.11) | 1.35 (0.11) |
| P value vs placebo | 0.164 | 0.121 | 0.079 | |
| Adjusted P value vs placebo | 0.164 | 0.164 | 0.164 | |
Data are presented as LS mean (SE).
The mixed model for repeated measures analysis was performed, and the model included treatment group and visit as fixed effects, baseline as a covariate, and treatment‐visit interaction for paired comparison between each minesapride group and the placebo group.
Abbreviations: BM, bowel movement; CSBM, complete spontaneous bowel movement; LS, least squares.
As for changes from baseline in global relief, IBS‐SI, IBS‐QOL, and EQ‐5D‐5L at Week 12, a tendency towards improvement was shown in all minesapride groups. As for IBS‐SI overall score, the change was greater in the 40 mg group, but with no significant difference from the placebo group at Week 12 (P = 0.041, adjusted P = 0.124) (Table S5).
3.3. Safety
The overall incidences of treatment‐emergent AEs are shown in Table 4. The incidence of treatment‐emergent AEs was 46.6% in the placebo group, 48.5% in the 10 mg group, 59.6% in the 20 mg group and 58.4% in the 40 mg group. The incidence of treatment‐related AEs was 23.3% in the placebo group, 35.9% in the 10 mg group, 39.4% in the 20 mg group, 44.6% in the 40 mg group. No deaths occurred in any treatment group. One serious event of iron deficiency anaemia occurred during the follow‐up period in one patient in the 20 mg group, for which a causal relationship to the study drug was ruled out. The incidence of moderate treatment‐emergent AEs was 2.9% in the placebo group, 1.9% in the 10 mg group, 2.9% in the 20 mg group, and 2.0% in the 40 mg group, and was similar across the four treatment groups. Treatment‐emergent AEs leading to discontinuation occurred in one patient in the 20 mg group (dizziness) and two patients in the 40 mg group (tachycardia and diarrhoea). The pulse rate of the patient who experienced tachycardia was 68‐112 beats/min during the study. These events resolved after discontinuation of the study drug. Treatment‐emergent AEs leading to study drug interruption were diarrhoea, nausea, and rash. Of these, diarrhoea occurred in one patient in the placebo group, one patient in the 10 mg group, and four patients in the 40 mg group, while nausea and rash occurred in one patient each in the 40 mg group. All these events resolved. The incidence of common treatment‐emergent AEs is also shown in Table 4. Common adverse events (≥2%) were diarrhoea, nasopharyngitis, abdominal pain, and blood creatine phosphokinase increased. Diarrhoea was the most common treatment‐emergent AE in all treatment groups (29.1% in the placebo group, 37.9% in the 10 mg group, 47.1% in the 20 mg group and 51.5% in the 40 mg group). All reported events of diarrhoea were mild in severity. WHO‐defined diarrhoea occurred only in the minesapride groups (incidence 2.9% in the 10 mg group, 3.8% in the 20 mg/d group, and 6.9% in the 40 mg/d group) (Table S6). Abdominal pain occurred only in the minesapride groups, and was slightly more common in the 20 mg group and the 40 mg group. For all other adverse events, the incidence was similar across the four treatment groups.
TABLE 4.
Summary of treatment‐emergent adverse events
|
Placebo N = 103 |
Minesapride | |||
|---|---|---|---|---|
|
10 mg N = 103 |
20 mg N = 104 |
40 mg N = 101 |
||
| Treatment‐emergent AEs | 48 (46.6) | 50 (48.5) | 62 (59.6) | 59 (58.4) |
| Deaths | 0 | 0 | 0 | 0 |
| Serious AEs | 0 | 0 | 1 (1.0) | 0 |
| AEs leading to discontinuation | 0 | 0 | 1 (1.0) | 2 (2.0) |
| Mild | 45 (43.7) | 48 (46.6) | 59 (56.7) | 57 (56.4) |
| Moderate | 3 (2.9) | 2 (1.9) | 3 (2.9) | 2 (2.0) |
| Severe | 0 | 0 | 0 | 0 |
| Common treatment‐emergent AEs (≥2%) | ||||
| Diarrhoea | 30 (29.1) | 39 (37.9) | 49 (47.1) | 52 (51.5) |
| Abdominal pain | 0 | 1 (1.0) | 4 (3.8) | 3 (3.0) |
| Nasopharyngitis | 5 (4.9) | 5 (4.9) | 9 (8.7) | 6 (5.9) |
| Blood creatine phosphokinase increased | 2 (1.9) | 3 (2.9) | 0 | 2 (2.0) |
Data are presented as n (%).
Abbreviation: AEs, adverse events.
Abnormalities in ECG parameters are summarized in Table S7. The number of subjects assessed as having clinically significant abnormalities after receiving the study drug among the subjects whose baseline ECG was within normal or clinically insignificantly abnormal was 1‐4 in each treatment group, and did not substantially differ across the four treatment groups. No subjects in any treatment group had QTcF >480 ms. The change in QTcF from baseline was >60 ms only in one subject in the 20 mg group. Changes from baseline for ECG parameters are shown in Table S8. In all minesapride groups, heart rate was slightly increased by about 3 beats/min.
In terms of laboratory test values, and vital signs, no clinically problematic changes were observed in any treatment group.
4. DISCUSSION
This is the first study to evaluate the efficacy and safety of novel 5‐HT4 receptor agonist minesapride in patients with Rome IV defined IBS‐C. We previously reported the efficacy and safety of minesapride in patients with IBS‐C but they were diagnosed with Rome III criteria. 20 In the present study, minesapride failed to meet the primary endpoint, and no clear dose‐response relationship was found.
The percentage of responders who met the CSBM response and abdominal pain improvement criteria, both in the same week for ≥9/12 weeks showed a better outcome in the 40‐mg minesapride group than that in the placebo group. Interestingly this is a stricter composite responder evaluation than the primary endpoint (FDA composite endpoint). 21 Minesapride 40 mg group also showed a greater improvement in SBM frequency than the placebo group throughout the treatment period. However, this study could prove a dose‐response relationship neither in the primary endpoint nor in CSBMs as well as abdominal pain. These results suggest that minesapride 40 mg would have the efficacy at least for the stricter composite evaluation and increasing the frequency of SBMs.
It is noteworthy that we could successfully reduce the placebo responder rate down to 3.9% by introducing the stricter composite responder evaluation for 9/12 weeks. According to the meta‐analysis, the pooled placebo response rate is 20%‐40% in IBS. 28 Therefore, the placebo response rate using the composite responder for 9/12 weeks in this study is much lower than that in the earlier reports. Generally, a placebo run‐in period reduces the placebo response. 29 In this study, a two week placebo run‐in period was set before the treatment period. However, the responder rate for the primary endpoint in the placebo group was similar to other studies without a run‐in period. 30 , 31 , 32 The low placebo response rate using the composite response for 9/12 weeks, may have led to detecting the effect of high‐dose minesapride in this study.
As for the effect on SBMs, minesapride showed a robust improvement. The placebo increased SBMs between 0.34 and 0.95 counts/week while 40 mg of minesapride increased SBMs between 1.46 and 2.08 counts/week. These values are almost comparable with those in a recent RCT of approved agents for IBS‐C patients in Japan. 33 Subgroup analysis in patients with fewer than 3 SBMs per week at baseline also suggested that minesapride improved the frequency of SBMs and CSBMs (Figures S3 and S4). Furthermore, a post‐hoc analysis showed that the percentage of patients achieved a mean of 3 or more CSBMs per week over the 12 weeks of treatment in the 40‐mg minesapride was greater than that in the placebo group (22.9% vs 12.2%) (Figure S5). The percentage was almost comparable with that of prucalopride. 34 In addition, minesapride acts as a visceral analgesic based on the non‐clinical study unlike prucalopride. 17 Because the other 5‐HT4 agonists have stimulatory actions on the lower gastrointestinal tract, 8 , 10 , 11 , 12 , 13 , 14 , 15 it is no wonder that minesapride also improves bowel movements in patients with constipation. Minesapride would ameliorate constipation in patients with IBS‐C.
The most common treatment‐emergent AE in this study was diarrhoea, and all reported events of diarrhoea were mild in severity. The percentage of subjects with treatment‐emergent AEs that led to discontinuation or interruption of the study drug interruption was low. The incidence of diarrhoea including diarrhoea in the placebo group was higher compared with that in another study conducted in Japan. 33 In the present study, we regarded all BSFS type 6 or 7 stools as diarrhoea. It would be too sensitive to assess the incidence of diarrhoea for IBS patients. Given the discontinuation rate, the interruption rate, and the severity, diarrhoeas due to minesapride administration would not be problematic. In terms of ECG parameters, there were no clinically problematic changes. Results of a thorough QT study also support that minesapride has no effect on QT prolongation. 19 A patient in the 40 mg group experienced moderate tachycardia and discontinued minesapride. In this study, heart rate was slightly increased from baseline by about 3 beats/min in all minesapride groups. This effect was also reported in the minesapride non‐clinical study and QT/QTc study. 16 , 19 A 5‐HT4 receptor agonist prucalopride also had a tendency to increase heart rate slightly in clinical studies. 11 , 35 A recent observational population‐based cohort study showed no indication of increased risk for major adverse cardiovascular events with new use of prucalopride compared with polyethylene glycol 3350 use. 36 Based on these reports, the slight increase in heart rate would not induce major adverse cardiovascular events. Overall, minesapride was safe and well tolerated at a dose of up to 40 mg for 12 weeks.
There are several strengths in this study. First, this is the first RCT in Japanese with IBS‐C diagnosed with Rome IV criteria. 1 Second, this study depicted some important efficacy including effects on the strict composite responder evaluation and SBMs. An early phase 2a study of minesapride for IBS‐C patients showed positive results for global improvement, adequate relief, CSBMs, and reduction in IBS‐SI. 20 Therefore, these findings suggest it will be necessary to use a larger sample size to ensure detecting a positive phase 3 result. Third, the actual RCT data in IBS patients with placebo run‐in are specific. Design of the study with a placebo run‐in may underestimate the overall effect size. 37 Therefore, RCTs with a placebo run‐in are relatively rare in the area of IBS. Together with the early phase 2a study, 20 this study provides actual data of the placebo run‐in period. These strengths provide unique aspects of this study.
There are some limitations in this study. First, no dose‐response relationship was observed in the primary endpoint. The placebo response was too high to evaluate the efficacy in accordance with the primary endpoint based on FDA guidance. 21 Based on the responder rate analysis by period (Figure S1), minesapride showed dose‐response efficacy for the primary endpoint until 8 weeks. It disappeared at the final analysis because the placebo response increased. Moreover we set the dose range based on the early phase 2a study. 20 However, higher doses (>40 mg/d) might be required to show effects on the primary endpoint. Second, further long‐term safety data are needed to assess the cardiovascular risks.
In conclusion, study results suggested that minesapride was safe and well‐tolerated at doses up to 40 mg/d. Although the primary endpoint was negative, minesapride 40 mg is likely to improve a stricter composite responder evaluation and SBM frequency in IBS‐C patients.
AUTHORSHIP
Guarantor of the article: Tatsuto Hamatani, Shin Fukudo.
Author contributions: Tatsuto Hamatani, Shin Fukudo, Yosuke Nakada, Hiroshi Inada, and Hiroto Miwa designed the study, analysed and assessed the data. Kiyoyasu Kazumori was involved in the analyses, and interpreted the data. All authors critically reviewed the manuscript and approved the final manuscript for submission.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the investigators and staffs in the medical institutions (Supplementary Listing) and patients who attended this study.
Declaration of personal interests: Tatsuto Hamatani, Yosuke Nakada, Hiroshi Inada, and Kiyoyasu Kazumori are employees of Sumitomo Dainippon Pharma Co., Ltd. Shin Fukudo and Hiroto Miwa served as coordinating investigators and received consultation fees from Sumitomo Dainippon Pharma Co., Ltd.
Declaration of funding interests: This study was funded by Sumitomo Dainippon Pharma Co., Ltd.
Hamatani T, Fukudo S, Nakada Y, Inada H, Kazumori K, Miwa H. Randomised clinical trial: minesapride vs placebo for irritable bowel syndrome with predominant constipation. Aliment Pharmacol Ther. 2020;52:430–441. 10.1111/apt.15907
The Handling Editor for this article was Professor Alexander Ford, and it was accepted for publication after full peer‐review.
Tatsuto Hamatani and Shin Fukudo equally contributed to this work.
Funding information
This study was funded by Sumitomo Dainippon Pharma Co., Ltd.
DATA AVAILABILITY STATEMENT
Sumitomo Dainippon Pharma, Co.,Ltd.'s policy on data sharing can be found at https://www.ds‐pharma.com/rd/clinical/clinical_study_data.html
REFERENCES
- 1. Lacy BE, Mearin F, Chang L, et al. Bowel disorders. Gastroenterology. 2016;150:1393‐1407. [DOI] [PubMed] [Google Scholar]
- 2. Mearin F, Lacy BE, Chang L, et al. Bowel disorders In: Drossman DA, Chang L, Chey WD, et al. Rome IV: Functional Gastrointestinal Disorders; Disorders of Gut‐Brain Interaction. 4th ed, Vol II. Raleigh, NC: The Rome Foundation; 2016:967‐1057. [Google Scholar]
- 3. Gralnek IM, Hays RD, Kilbourne A, et al. The impact of irritable bowel syndrome on health‐related quality of life. Gastroenterology. 2000;119:654‐660. [DOI] [PubMed] [Google Scholar]
- 4. Halder SL, Locke GR 3rd, Talley NJ, et al. Impact of functional gastrointestinal disorders on health‐related quality of life: a population‐based case–control study. Aliment Pharmacol Ther. 2004;19:233‐242. [DOI] [PubMed] [Google Scholar]
- 5. Sperber AD, Dumitrascu D, Fukudo S, et al. The global prevalence of IBS in adults remains elusive due to the heterogeneity of studies: a Rome Foundation working team literature review. Gut. 2017;66:1075‐1082. [DOI] [PubMed] [Google Scholar]
- 6. Tack J, Stanghellini V, Mearin F, et al. Economic burden of moderate to severe irritable bowel syndrome with constipation in six European countries. BMC Gastroenterol. 2019;19:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397‐414. [DOI] [PubMed] [Google Scholar]
- 8. Black CJ, Burr NE, Ford AC, et al. Relative efficacy of tegaserod in a systematic review and network meta‐analysis of licensed therapies for irritable bowel syndrome with constipation. Clin Gastroenterol Hepatol. 2020;18:1238‐1239.e1. [DOI] [PubMed] [Google Scholar]
- 9. Highlights of prescribing information (Zelnorm). https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021200Orig1s015lbl.pdf. Accessed March 9, 2020
- 10. De Schryver AMP, Andriesse GI, Samsom M, et al. The effects of the specific 5HT4 receptor agonist, prucalopride, on colonic motility in healthy volunteers. Aliment Pharmacol Ther. 2002;16:603‐612. [DOI] [PubMed] [Google Scholar]
- 11. Camilleri M, Kerstens R, Rykx AN, et al. A placebo‐controlled trial of prucalopride for severe chronic constipation. N Engl J Med. 2008;358:2344‐2354. [DOI] [PubMed] [Google Scholar]
- 12. Ke M, Zou D, Yuan Y, et al. Prucalopride in the treatment of chronic constipation in patients from the Asia‐Pacific region: a randomized, double‐blind, placebo‐controlled study. Neurogastroenterol Motil. 2012;24:999‐e541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Z, Sakakibara R, Odaka T, et al. Mosapride citrate, a novel 5‐HT4 agonist and partial 5‐HT3 antagonist, ameliorates constipation in parkinsonian patients. Mov Disord. 2005;20:680‐686. [DOI] [PubMed] [Google Scholar]
- 14. Ueno N, Inui A, Satoh Y. The effect of mosapride citrate on constipation in patients with diabetes. Diabetes Res Clin Pract. 2010;87:27‐32. [DOI] [PubMed] [Google Scholar]
- 15. Kanazawa M, Watanabe S, Tana C, et al. Effect of 5‐HT(4) receptor agonist mosapride citrate on rectosigmoid sensorimotor function in patients with irritable bowel syndrome. Neurogastroenterol Motil. 2011;23:754‐e332. [DOI] [PubMed] [Google Scholar]
- 16. Tsubouchi T, Kunimatsu T, Tsujimoto S, et al. The in vitro pharmacology and intensive non‐clinical cardiovascular safety studies of a novel 5‐HT4 receptor agonist, DSP‐6952. Eur J Pharmacol. 2018;826:96‐105. [DOI] [PubMed] [Google Scholar]
- 17. Mine Y, Itakura T, Oku S, et al. DSP‐6952, a novel partial 5‐HT4 receptor agonist, inhibits visceral hypersensitivity and ameliorates gastrointestinal dysfunction in experimental animals. Eur J Pharmacol. 2018;826:123‐132. [DOI] [PubMed] [Google Scholar]
- 18. Yumizaki T, Maeda M, Fujita T, et al. A three‐part phase 1 study to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of DSP‐6952 in healthy Japanese subjects and those with ≤3 spontaneous bowel movements per week. Clin Pharmacol Drug Dev. 2020;9:353‐365. [DOI] [PubMed] [Google Scholar]
- 19. Hamatani T, Noda N, Takagaki T, et al. Thorough QT/QTc study shows that a novel 5‐HT4 receptor partial agonist minesapride has no effect on QT prolongation. Clin Pharmacol Drug Dev. 2020. 10.1002/cpdd.778 [DOI] [PubMed] [Google Scholar]
- 20. Fukudo S, Nakamura M, Hamatani T, et al. Efficacy and safety of 5‐HT4 receptor agonist minesapride for irritable bowel syndrome with constipation in a randomized controlled trial. Clin Gastroenterol Hepatol. 2020. 10.1016/j.cgh.2020.03.019. In press [DOI] [PubMed] [Google Scholar]
- 21. Kux L. Department of Health and Human Services, Food and Drug Administration [docket no. FDA– 2012–D–0146]: guidance for industry on irritable bowel syndrome—clinical evaluation of drugs for treatment: availability. Fed Reg. 2012;77:32124‐32125. [Google Scholar]
- 22. O'Donnell LJ, Virjee J, Heaton KW. Detection of pseudodiarrhoea by simple clinical assessment of intestinal transit rate. BMJ. 1990;300:439‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Francis CY, Morris J, Whorwell PJ. The irritable bowel severity scoring system: a simple method of monitoring irritable bowel syndrome and its progress. Aliment Pharmacol Ther. 1997;11:395‐402. [DOI] [PubMed] [Google Scholar]
- 24. Shinozaki M, Kanazawa M, Sagami Y, et al. Validation of the Japanese version of the Rome II modular questionnaire and irritable bowel syndrome severity index. J Gastroenterol. 2006;41:491‐494. [DOI] [PubMed] [Google Scholar]
- 25. Patrick DL, Drossman DA, Frederick IO, et al. Quality of life in persons with irritable bowel syndrome: development and validation of a new measure. Dig Dis Sci. 1998;43:400‐411. [DOI] [PubMed] [Google Scholar]
- 26. Kanazawa M, Drossman DA, Shinozaki M, et al. Translation and validation of a Japanese version of the irritable bowel syndrome‐quality of life measure (IBS‐QOL‐J). Biopsychosoc Med. 2007;1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ikeda S, Shiroiwa T, Igarashi A, et al. Developing a Japanese version of the EQ‐5D‐5L value set. J Natl Inst Public Health. 2015;64:47‐55. [Google Scholar]
- 28. Elsenbruch S, Enck P. Placebo effects and their determinants in gastrointestinal disorders. Nat Rev Gastroenterol Hepatol. 2015;12:472‐485. [DOI] [PubMed] [Google Scholar]
- 29. Pitz M, Cheang M, Bernstein CN, et al. Defining the predictors of the placebo response in irritable bowel syndrome. Clin Gastroenterol Hepatol. 2005;3:237‐247. [DOI] [PubMed] [Google Scholar]
- 30. Chey WD, Lembo AJ, Lavins BJ, et al. Linaclotide for irritable bowel syndrome with constipation: a 26‐week, randomized, double‐blind, placebo‐controlled trial to evaluate efficacy and safety. Am J Gastroenterol. 2012;107:1702‐1712. [DOI] [PubMed] [Google Scholar]
- 31. Rao S, Lembo AJ, Shiff SJ, et al. A 12‐week, randomized, controlled trial with a 4‐week randomized withdrawal period to evaluate the efficacy and safety of linaclotide in irritable bowel syndrome with constipation. Am J Gastroenterol. 2012;107:1714‐1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brenner DM, Fogel R, Dorn SD, et al. Efficacy, safety, and tolerability of plecanatide in patients with irritable bowel syndrome with constipation: results of two phase 3 randomized clinical trials. Am J Gastroenterol. 2018;113:735‐745. [DOI] [PubMed] [Google Scholar]
- 33. Fukudo S, Miwa H, Nakajima A, et al. A randomized‐controlled and long‐term linaclotide study of irritable bowel syndrome with constipation patients in Japan. Neurogastroenterol Motil. 2018;30:e13444. [DOI] [PubMed] [Google Scholar]
- 34. Camilleri M, Piessevaux H, Yiannakou Y, et al. Efficacy and safety of prucalopride in chronic constipation: an integrated analysis of six randomized, controlled clinical trials. Dig Dis Sci. 2016;61:2357‐2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double‐blind, placebo‐ and positive‐controlled thorough QT study. Br J Clin Pharmacol. 2012;73:203‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gilsenan A, Fortuny J, Cainzos‐Achirica M, et al. Cardiovascular safety of prucalopride in patients with chronic constipation: a multinational population‐based cohort study. Drug Saf. 2019;42:1179‐1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Irvine EJ, Tack J, Crowell MD, et al. Design of treatment trials for functional gastrointestinal disorders. Gastroenterology. 2016;150:1469‐1480.e1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Sumitomo Dainippon Pharma, Co.,Ltd.'s policy on data sharing can be found at https://www.ds‐pharma.com/rd/clinical/clinical_study_data.html