

Abstract
Background
Circulating cell‐free tumor DNA (ctDNA)‐based mutation profiling, if sufficiently sensitive and comprehensive, can efficiently identify genomic targets in advanced lung adenocarcinoma. Therefore, the authors investigated the accuracy and clinical utility of a commercially available digital next‐generation sequencing platform in a large series of patients with non–small cell lung cancer (NSCLC).
Methods
Plasma‐based comprehensive genomic profiling results from 8388 consecutively tested patients with advanced NSCLC were analyzed. Driver and resistance mutations were examined with regard to their distribution, frequency, co‐occurrence, and mutual exclusivity.
Results
Somatic alterations were detected in 86% of samples. The median variant allele fraction was 0.43% (range, 0.03%‐97.62%). Activating alterations in actionable oncogenes were identified in 48% of patients, including EGFR (26.4%), MET (6.1%), and BRAF (2.8%) alterations and fusions (ALK, RET, and ROS1) in 2.3%. Treatment‐induced resistance mutations were common in this cohort, including driver‐dependent and driver‐independent alterations. In the subset of patients who had progressive disease during EGFR therapy, 64% had known or putative resistance alterations detected in plasma. Subset analysis revealed that ctDNA increased the identification of driver mutations by 65% over standard‐of‐care, tissue‐based testing at diagnosis. A pooled data analysis on this plasma‐based assay demonstrated that targeted therapy response rates were equivalent to those reported from tissue analysis.
Conclusions
Comprehensive ctDNA analysis detected the presence of therapeutically targetable driver and resistance mutations at the frequencies and distributions predicted for the study population. These findings add support for comprehensive ctDNA testing in patients who are incompletely tested at the time of diagnosis and as a primary option at the time of progression on targeted therapies.
Keywords: anaplastic lymphoma kinase (ALK), circulating cell‐free tumor DNA (ctDNA), epidermal growth factor receptor (EGFR), liquid biopsy, non–small cell lung cancer
Short abstract
Circulating cell‐free tumor DNA‐based liquid biopsy using next‐generation sequencing detects a spectrum of targetable alterations at frequencies expected in patients with advanced non–small cell lung cancer. These findings support the concept of a plasma‐first algorithm at the time of progression on targeted therapy for this population.
Introduction
The practice of precision oncology in metastatic non–small cell lung cancer (NSCLC) requires analyzing a growing set of tumor alterations predictive of drug activity. To identify uncommon but actionable mutations, comprehensive next‐generation sequencing (NGS) approaches are increasingly recommended.1, 2 The National Comprehensive Cancer Network guidelines and the College of American Pathology/International Association for the Study of Lung Cancer/Association for Molecular Pathology guidelines recommend up‐front testing for epidermal growth factor receptor (EGFR) activating mutations, ALK and ROS1 fusions, and BRAF V600E.1, 3 There is also a consensus for testing high‐level MET copy number gain (CNG), MET exon 14 skipping (E14skip) mutations, and RET and NTRK rearrangements, each of which is associated with available therapies, and active clinical trials testing therapies that target ERBB2 (HER2) activating mutations. Although it is not currently linked to an approved targeted agent, the identification of KRAS activating mutations at diagnosis effectively rules out the presence of other actionable driver alterations.4, 5
Although the initial efficacy of tyrosine kinase inhibitors (TKIs) is high in oncogene‐driven NSCLC, eventual acquired resistance is almost universal. The use of liquid biopsy to identify mechanisms of resistance (MORs), such as T790M, is already guideline‐recommended regardless of tissue biopsy feasibility.1, 3 As new generations of targeted agents—characterized by improved kinetics, target specificity, and brain metastasis control—receive US Food and Drug Administration (FDA) approval and transition into the front line, it has become evident that each agent generates a distinct resistance profile that differs from the profiles associated with first‐generation inhibitors.1, 6, 7, 8 For instance, patients with cancers harboring ROS1 fusions often acquire intragene resistance mutations, analogous to ALK resistance mutations, which may be treatable with alternative inhibitors.9 NSCLCs with RET fusions and MET E14skip mutations may acquire gatekeeper mutations, necessitating a change in TKI.10, 11 Clearly, identifying the specific MOR at the time of progression is essential for continued personalized therapy. Furthermore, identifying nontargetable MORs (ie, KRAS or RB1 mutations) may predict lack of response to a next‐generation TKI and require pursuit of alternative strategies. Tools that increase the availability of informative biomarkers, both at baseline and at progression, will be instrumental to improved outcomes in NSCLC.
The sequencing of circulating cell‐free tumor DNA (ctDNA), if sufficiently sensitive and comprehensive, can efficiently identify genomic targets in advanced NSCLC. Although the spectrum and frequency of NSCLC oncogenic driver mutations have been described in tissue4, 12 and their concordance with plasma ctDNA has been well published,13, 14 questions remain regarding how well they can consistently be recapitulated in ctDNA and whether additional information stemming from metastatic tumor heterogeneity may improve diagnostic utility. Here, we describe the spectrum of mutations found in a cohort of more than 8000 patients with NSCLC who were analyzed using a commercially available, comprehensive ctDNA NGS panel (Guardant360; Guardant Health, Inc). We also report results of a pooled analysis of published TKI response rates in ctDNA‐identified driver mutation‐positive cases, supplemented by a patient cohort newly reported herein.
Materials and Methods
Patients
Clinical history and molecular test results from all individuals with a diagnosis of advanced (defined on the test request form as stage IIIB‐IV) lung adenocarcinoma (LUAD) or NSCLC not otherwise specified (NSCLC‐NOS) who underwent ctDNA analysis using clinical Guardant360 testing between June 2014 and October 2016 were reviewed for inclusion (see Supporting Methods). The generation of de‐identified data sets by Guardant Health for research purposes was approved by the Quorum Institutional Review Board.
Clinical outcomes data were collected by chart review and analyzed for a subset of patients who consented to the Clinical Outcomes of Cancer Patients with Cell Free DNA Tumor Sequencing study (Science37 Registry) (see Supporting Methods). Response rates were assessed using modified Response Evaluation Criteria in Solid Tumors (RECIST) criteria (version 1.1).
ctDNA Analysis
ctDNA for the Guardant360 assay, a New York State Department of Health‐approved test, was isolated from plasma, and NGS was performed as previously described at Guardant Health Inc, a Clinical Laboratory Improvement Amendments‐certified, College of American Pathologists‐accredited laboratory.13, 15 These data span 3 versions of Guardant360, which included additions to the genes and/or variant types detected, without changing the underlying test methodology. Point mutations were analyzed in 54 to 70 genes, CNG was analyzed in up to 18 genes, fusions were analyzed in up to 6 genes, and small insertions and/or deletions (indels) were analyzed in up to 3 genes, depending on the panel version performed (see Supporting Table 1). Detailed descriptions of the assay and its validation were published previously.13, 15
Data Analysis
Demographic data, mutation frequencies, and variant allele fractions (VAFs) were analyzed and reported for the full cohort. VAFs were defined as the proportions of variant alleles relative to wild‐type alleles. Detailed analyses regarding oncogene mutation prevalence and resistance landscape were conducted on the subset of patients tested using the 70‐gene panel (N = 6087; 70% of total; Table 1). An oncoprint was constructed using data from the 2844 patients who had an alteration in ≥1 oncogenes of interest (n = 3956 alterations), including only ≥3 CNGs.16, 17 A 2‐tailed chi‐square test was used to assess the mutual exclusivity of EGFR and KRAS mutations, and a Fisher exact test was used to compare frequencies of classic and nonclassic EGFR and KRAS mutations.
Table 1.
Demographics
| Variable | No. (%) |
|---|---|
| Patient characteristics, N = 8388 | |
| Sex | |
| Women | 4799 (57) |
| Men | 3589 (43) |
| Pathology | |
| Adenocarcinoma | 4142 (49) |
| NSCLC‐NOS | 4246 (51) |
| ctDNA alterations detected | |
| Yes | 7301 (87) |
| No | 1087 (13) |
| Panel version a | |
| 54‐Gene panel | 358 (4) |
| 68‐Gene panel | 2230 (26) |
| 70‐Gene panel | 6087 (70) |
| Sample characteristics, N = 9202 | |
| Panel version | |
| 54‐Gene panel | 361 (4) |
| 68‐Gene panel | 2345 (25) |
| 70‐Gene panel | 6496 (71) |
| ctDNA alterations detected | |
| Yes | 7921 (86) |
| No | 1281 (14) |
Abbreviations: ctDNA, circulating tumor DNA; NSCLC‐NOS, non–small cell lung cancer, not otherwise specified.
Some patients had ctDNA analysis at more than 1 time point, some with different panel versions.
A literature review was conducted for MORs occurring in NSCLC after treatment with an EGFR TKI, and the frequencies of these MORs were calculated among patients tested with the 70‐gene panel whose blood was known to have been drawn at progression on an EGFR TKI (N = 447).
Another literature review was conducted for NSCLC studies that used the Guardant360 assay and reported response rates for ≥3 patients. EGFR activating mutations; ALK, ROS1, and RET fusions; BRAF V600E; ERBB2 exon 20 insertions; MET E14skip mutations; and MET high‐level CNGs were included in counts of targetable driver mutations, and patients were subsequently treated in the first or second line and beyond. Case‐specific data were abstracted, and pooled objective response rates (ORRs) and disease control rates (DCRs) were calculated. Treatment data and line of treatment status were only available for a subset of patients with driver mutations. In some cases, necessary patient‐specific data were not published, and the authors were contacted and shared the necessary data. Patients without line of therapy data were excluded from analyses regarding line of therapy but were included in gene‐specific and therapy‐specific calculations. Response was assessed by RECIST version 1.1 unless otherwise specified. The z ratio was used to assess differences between the ORRs of patients treated based on Guardant360 results and those treated based on tissue results, with a 2‐tailed P value <.05 indicating significance.
Results
From June 2014 through October 2016, 9202 consecutive plasma samples from 8388 patients with advanced NSCLC underwent Guardant360 testing (for demographics, see Table 1). The mean number of days between initial diagnosis and ctDNA collection was 467 days (median, 177 days). The submitting sites indicated that all patients had NSCLC, with an additional 49% further described as LUAD. Most samples (70%) were assessed using the 70‐gene panel (Table 1), and all gene‐specific and mutation‐specific analyses were conducted with this largest subcohort unless otherwise noted.
Somatic alterations were detected in 86% of all samples and increased with each assay iteration, with alterations detected in 78%, 84%, and 87% of samples for the 54‐gene, 68‐gene, and 70‐gene panels, respectively. The median number of alterations per sample for all panels was 3 (range, 0‐93 alterations), and the median VAF was 0.43% (range, 0.03%‐97.62%) (see Supporting Fig. 1 and Supporting Methods).
Oncogene Mutation Detection
Driver oncogene mutations were identified in 2948 of 6087 patients (48.4%), or 57.2% of those who had ≥1 alteration detected (2948 of 5151 patients) (see Supporting Table 2). In this cohort, EGFR driver mutations were most frequent (26.4%), followed by KRAS mutations (17.2%), MET CNG (5.7%), BRAF mutations (2.8%), ERBB2 (HER2) mutations (2.3%), ALK fusions (1.3%), RET fusions (0.9%), MET E14skip mutations (0.4%), and ROS1 fusions (0.2%) (see Fig. 1).
Figure 1.

Non–small cell lung cancer tumorigenesis pathways and mutation frequencies are illustrated. Pathway representations show the genes assessed and the number of patients who had mutations identified. Mutation counts are of pathogenic single nucleotide variants unless otherwise specified.
EGFR driver mutations included exon 19 deletions (E19dels) (51.8%), L858R (34.4%), exon 20 insertions (E20ins) (4.5%), other commonly accepted activators (G719A, G719C, G719D, G719R, and G719S, L861Q and L861R; S768I; 9.0%), and other previously reported abnormalities (4.5%) (see Supporting Table 3). Sixty‐four percent (n = 454) of E19dels were the canonical E746_A750del; however, the remaining 36% were composed of 50 distinct sequence variants (see Supporting Table 4). Similarly, 22 unique E20ins were observed, led by S768_N770 duplications (S768_N770dup) (23.0%) and A767_V769dup (16.4%) (see Supporting Table 5). In 65 cases, rare mutations occurred in tandem with other activating EGFR mutations (see Supporting Table 6). Notably, 35 of 66 G719 mutations, 16 of 19 S768I mutations, and 19 of 19 E709 mutations were accompanied by a second known EGFR activating mutation (see Supporting Table 6).
Among KRAS‐mutant samples, codon 12 substitutions were most common (80.6%), followed by G13 and Q61 (8.1% each). Within codon 12, the smoking‐associated G12C (35%) and G12V (17%) represented the most common substitutions, followed by G12D (14%) (see Supporting Table 7). KRAS exon 4 activating mutations (K117, D119, and A146) were observed in 1.3% of the cohort, along with other weakly activating alterations comprising nearly 3%.18 In 22 samples, multiple KRAS mutations were detected simultaneously (see Supporting Table 8), of which 9 consisted of 2 commonly mutated loci (ie, codon 12, 13, or 61).
Among BRAF mutations, V600E was the most frequent (40.3%) followed by G469A, G469V, G469R, and G469E (22.3%) substitutions (Table 2; see Supporting Table 9). NRAS mutations (n = 56) were observed in 55 samples (1.1%) and were largely comprised of substitutions at codons 12, 13, and 61 (91%) (see Supporting Table 10), whereas HRAS mutations were seen in 15 samples (0.3%). Activating mutations in MEK1 (MAP2K1) were observed in 17 samples, dominated by K57E, K57N, K57T substitutions (n = 8) and Y130C substitutions (n = 4). In all but 1 sample, MAP2K1 mutations were mutually exclusive with each other and with other drivers (Fig. 2A).
Table 2.
Key Oncogene Mutation Spectrum
| Oncogene Mutation | No. a | % b | Oncogene Mutation | No. a | % b |
|---|---|---|---|---|---|
| EGFR driver mutations, n = 1418 mutations in 1361 patients c | ERBB2 mutations, n = 126 mutations in 124 patients | ||||
| Exon 19 deletions | 705 | 51.8 | Exon 20 insertion | 76 | 61.3 |
| L858R | 468 | 34.4 | S310F, S310Y | 16 | 12.9 |
| G719A, G719C, G719D, G719R, G719S | 66 | 4.8 | V659E | 7 | 5.6 |
| Exon 20 insertion | 61 | 4.5 | L755P, L755S, L755A | 6 | 4.8 |
| L861Q, L861R | 38 | 2.8 | V777L | 4 | 3.2 |
| S768I | 19 | 1.4 | V842I | 3 | 2.4 |
| L833V | 9 | 0.7 | D769Y, D769D | 3 | 2.4 |
| Others | 52 | 3.8 | Others | 11 | 8.9 |
| KRAS mutations, n = 910 mutations in 888 patients c | ALK fusions, n = 65 | ||||
| G12C, G12V, G12D, G12A, G12S, G12F, G12R, G12E | 716 | 80.6 | EML4‐ALK fusion | 58 | 89.2 |
| G13C, G13D, G13E, G13F, G13P, G13R, G13V | 72 | 8.1 | STRN‐ALK fusion | 3 | 4.6 |
| Q61H, Q61L, Q61R, Q61K | 72 | 8.1 | KLC1‐ALK fusion | 2 | 3.1 |
| V14I | 10 | 1.1 | KIF5B‐ALK fusion | 1 | 1.5 |
| A146V, A146T, A146P | 10 | 1.1 | TFG‐ALK fusion | 1 | 1.5 |
| Others | 30 | 3.4 | |||
| BRAF mutations, n = 142 mutations in 139 patients | ROS1 fusions, n = 9 mutations | ||||
| V600E, V600K | 56 | 40.3 | CD74‐ROS1 fusion | 5 | 55.6 |
| G469A, G469V, G469R, G469E | 31 | 22.3 | EZR‐ROS1 fusion | 2 | 22.2 |
| G466V, G466R, G466A | 12 | 8.6 | SDC4‐ROS1 fusion | 1 | 11.1 |
| N581S | 11 | 7.9 | TPM3‐ROS1 fusion | 1 | 11.1 |
| K601E | 7 | 5.0 | RET fusions, n = 45 | ||
| D594G, D594A | 5 | 3.6 | KIF5B‐RET fusion | 28 | 62.2 |
| G596R, G596 | 4 | 2.9 | CCDC6‐RET fusion | 13 | 28.9 |
| Others | 16 | 11.5 | NCOA4‐RET fusion | 4 | 8.9 |
No. indicates the number of mutations identified.
The percentage (%) indicates the proportion of patients with a mutation in that gene who had the respective mutation.
Double mutation profiles are detailed in Supporting Tables 6 and 8.
Figure 2.
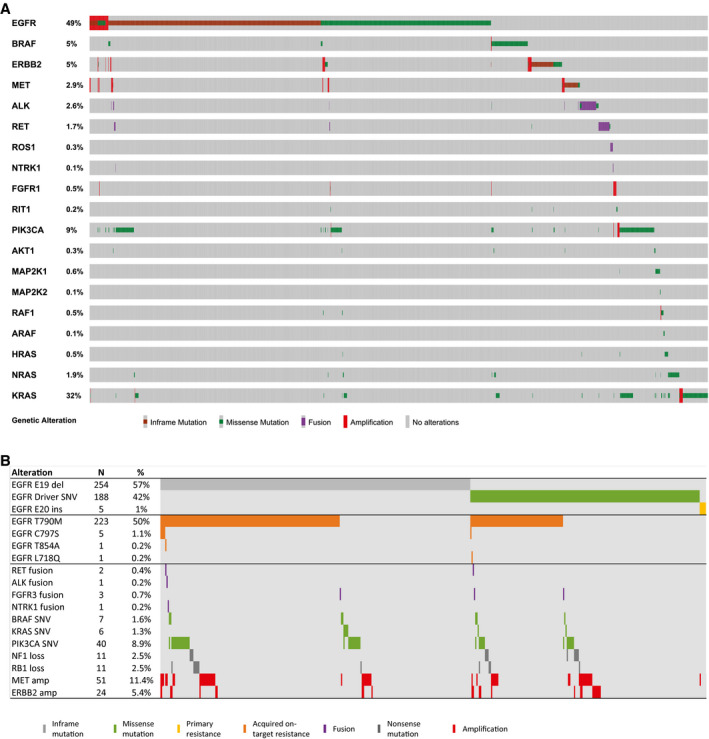
Driver and resistance mutation distribution and co‐occurrences are illustrated. (A) An oncoprint illustrates the mutual exclusivity of the oncogenes studied (n = 3956 alterations, n = 2844 patients). Percentages shown are of the 2844 patients who had ≥1 mutation in ≥1 gene(s) of interest. (B) The epidermal growth factor receptor (EGFR) resistance landscape is illustrated. This analysis was limited to patients tested with the 70‐gene panel whose blood was known to have been drawn at progression when receiving an EGFR tyrosine kinase inhibitor (TKI) according to test request form notes (N = 447). Treatment at the time of blood draw was erlotinib (n = 310 patients; 69%), afatinib (n = 87 patients; 19%), gefitinib (n = 2 patients; 0.4%), osimertinib (n = 32 patients; 7%), rociletinib (n = 10 patients; 2%), or other not specified, third‐generation EGFR TKI on a trial (n = 6 patients; 1%). The 5 C797S events all occurred among patients who were receiving a third‐generation TKI (3 osimertinib, 1 rociletinib, 1 trial drug not specified) for a prevalence of 10% among the 48 patients who had blood drawn at the time of progression on a third‐generation TKI.
Activating alterations in PIK3CA were observed in samples from 253 patients. In total, 37 unique substitutions known to constitutively activate PI3K were observed, dominated by E545 (34.2%), E542 (22.5%), and H1047 (13.5%). In addition, 9 AKT1 activating mutations were seen at E17K or E40K. As observed previously in NSCLC, PI3K‐AKT alterations were mutually exclusive with each other but were not mutually exclusive with the driver oncogenes listed in Table 2 (see Fig. 2A).
Activating alterations in ERBB2 (HER2) and MET were observed in 2.3% and 6.1% of samples, respectively. Small, in‐frame insertions in the tyrosine kinase domain accounted for 61.3% of ERBB2 mutant cases (see Supporting Table 2). MET E14skip mutations and gene CNGs were seen in 19 and 295 samples, respectively, and 2 samples were positive for both.
Fusion events were underrepresented in this cohort (collectively, 2%). The most frequent fusion partners were EML4 (89.2% of ALK fusions), KIF5B (62.2% of RET fusions), and CD74 (55.6% of ROS1 fusions), respectively (Table 2). In addition, FGFR3‐TACC3 fusions were seen in 6 samples, and NTRK1 fusions in 2 samples (LMNA‐NTRK1, SQSTM1‐NTRK1). The spectrum of additional mutation frequencies (excluding synonymous alterations and variants of uncertain significance) are shown in Supporting Table 11.
Mutual Exclusivity of Oncogenic Drivers
NSCLC driver mutations in 18 known lung‐associated oncogenes showed a pattern of mutual exclusivity (Fig. 2A), consistent with prior analysis on a smaller subset.5 Of the 5151 patients, 1361 harbored EGFR mutations, and 888 harbored KRAS mutations; however, only 25 were positive for both (P < .0001). Of these, 14 patients (56%) harbored nonstandard EGFR or KRAS abnormalities, including KRAS A146T, V14I, Q22R, L19F, and T50I mutations and/or EGFR R451C, T725M, D761N, and E330K mutations. Thus the few samples that were positive for both EGFR and KRAS mutations had significantly more nonstandard mutations than the single‐mutant samples (P < .0001). Seven of these also harbored EGFR T790M mutations, suggesting that, in many cases, the presence of a KRAS mutation may be associated with emergent resistance. To further assess this, we evaluated the clonality of these mutations by calculating the relative VAF and correcting for copy number, as previously described.5 The results showed that KRAS was subclonal in 5 of 7 patients, suggesting acquired resistance, and was not subclonal in the other 2 patients, suggesting possible innate resistance or possibly the presence of 2 different primary cancers.
Acquired Resistance in EGFR‐Mutant and ALK‐Mutant Cases
Among patients whose blood was known to have been drawn at the time of progression while they were receiving an EGFR TKI (N = 447), 310 (69%) had received prior erlotinib, 87 (19%) had received prior afatinib, 2 (0.4%) had received prior gefitinib, 32 (7%) had received prior osimertinib, 10 (2%) had received prior rociletinib, and 6 (1%) had received another unspecified, third‐generation EGFR TKI on a trial.
A typical distribution of EGFR intragene resistance mutations was identified (Fig. 2B). EGFR L747S (n = 2) and T854S (n = 1) mutations were also observed but in patients who had unknown treatment status and thus are excluded from Figure 2B. All 5 C797S‐positive cases also harbored T790M and occurred in patients who were receiving a third‐generation TKI (3 osimertinib, 1 rociletinib, and 1 trial drug not specified), accounting for 10% of the 48 patients who had samples drawn at the time of progression on a third‐generation TKI. Among previously recognized bypass MORs, the largest subgroups were MET CNG, PIK3CA activating mutations, and ERBB2 CNG. Activation of BRAF (3 V600E, 3 G469A/G469R, and 1 L485F) and KRAS secondary activating fusions and loss of RB1 also were detected.
Among the 65 patients who had ALK fusions, 32 (49%) were naive to ALK inhibitor therapy, and 11 had unknown prior treatment status. Five occurred in patients who progressed on an EGFR TKI at VAFs that were subclonal to a primary EGFR mutation, likely as an acquired MOR, as described previously.19 Of the remaining 17 patients, 7 (41%) had ≥1 ALK single‐nucleotide variant (SNV) known to confer acquired resistance, including G1202R (N = 4), I1171T (N = 2), L1196M (N = 2), and 1 each of F1174C and F1174V. Two patients had alternative drivers suggestive of ALK‐independent resistance. An additional 3 patients had ≥1 ALK resistance SNV without a detectable ALK fusion at progression (detected on tissue testing at diagnosis). An expanded description of ALK fusions and MORs was previously published.19
Expanded Mutation Detection at Diagnosis
Chart review of consecutive patients identified a subset (n = 1288) for whom tissue biomarker analysis results were available or were reported as incomplete because of insufficient tissue. Among these, 32% had a National Comprehensive Cancer Network‐listed or KRAS driver mutation (Fig. 3A) reported or had a completely negative finding on a comprehensive test. The remaining 68% were considered undergenotyped, defined as a lack of results for at least 1 of the 7 guideline‐recommended genes in tissue analysis. ctDNA analysis identified 252 additional patients who had an actionable biomarker missed or not evaluated by tissue analysis (Fig. 3B). This represents a 65% increase in biomarker detection beyond what was initially achieved in tissue. One‐half of these patients became eligible for targeted therapy. The other one‐half excluded targeted therapy based on KRAS mutation identification.
Figure 3.

The clinical utility of circulating tumor DNA (ctDNA) genotyping is illustrated in patients who had undergenotyped non–small cell lung cancer. The analysis was limited to patients for whom tissue biomarker analysis results were available or reported as incomplete because of insufficient tissue (n = 1288). (A) Tissue genotyping status is illustrated (biomarker positive, 30%; undergenotyped, 68%). QNS indicates quantity/quality not sufficient; UG, undergenotyped (not evaluated for all guideline‐recommended genes because of insufficient tissue or test not ordered). (B) Genotyping of ctDNA increases biomarker identification by 65%. This analysis identified 252 biomarkers not previously detected in tissue QNS and undergenotyped cases. Neg indicates negative; Pos, positive.
Fifty patients who had targetable driver mutations consented to participate in the Science37 Registry. Data regarding outcomes of targeted therapy were available for 12 of these patients (see Supporting Table 12). Among 8 patients who were identified by ctDNA but not tissue, 2 had stable disease, and 6 had a partial response (see Supporting Fig. 2).
ctDNA‐Directed Therapy and Response Rates
To expand our analysis of the relation between plasma‐based biomarker identification and treatment outcomes after appropriate, targeted therapy, a pooled analysis of published literature was conducted to supplement the patients identified in this study (see Supporting Fig. 2). Ten publications met criteria for inclusion.19, 20, 21, 22, 23, 24, 25, 26, 27, 28 Line of therapy was available for 130 patients, with an ORR of 68.8% (95% CI, 53.6%‐80.9%) and a DCR of 93.8% (95% CI, 81.8%‐98.4%) in the first‐line setting and ORR of 58.5% (95% CI, 47.1%‐69.2%) and a DCR of 86.6% (95% CI, 76.9%‐92.8%) in the second‐line and beyond (see Supporting Table 12). Pooled response rates also were stratified by gene and targeted therapy (n = 186). For ALK and ROS1 fusions, EGFR activating mutations, and EGFR T790M‐positive patients, ORRs were not significantly different from those in the FDA registrational study for each drug (Fig. 4).
Figure 4.
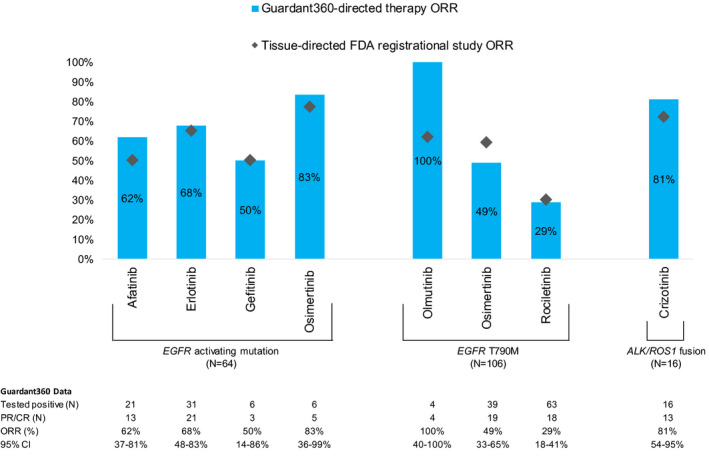
Pooled response rates of patients identified by circulating tumor DNA (ctDNA) compared with the US Food and Drug Administration (FDA) label. No statistically significance differences in the overall response rate (ORR) were identified between Guardant360‐directed therapy and the FDA registrational study (P > .12 for each therapy). Note that, for drugs without an FDA label, the ORR in a pivotal trial was used as a reference comparison. CR indicates complete response; PR, partial response.
Discussion
Here, we present the largest investigation of liquid biopsies in advanced NSCLC conducted to date, drawn from a consecutive series of over 8000 real‐world patients from typical clinical settings across the United States. Larger NGS landscape studies were pancancer studies, relied on tissue biopsies rather than blood samples, and only a portion included LUAD or NSCLC.29, 30
Previous tissue sequencing studies of patients with metastatic NSCLC reported similar mutation spectra, providing corroboration for this assay's ability to detect mutations.12, 31 Higher rates of EGFR mutations and concomitantly lower rates of other driver mutations were anticipated because of the frequent clinical use of this test in patients progressing on EGFR TKIs.32, 33 Another recent study prospectively assessed the detection of targetable driver mutations in first‐line advanced NSCLC with the same liquid biopsy assay and confirmed its ability to detect driver mutations at frequencies equivalent to those detected in tissue.14 The significant structural diversity among E19dels and E20ins and the identification of rare KRAS mutations highlight the benefits of comprehensive NGS because rare variants often are not detected by hotspot mutation panels. In addition, variants were detected at allele frequencies as low as 0.03% (median, 0.43%), emphasizing the need for a highly sensitive test in this arena, as previous studies have demonstrated that patients who have driver mutations with a low VAF have response rates to targeted therapy similar to the rates among patients who have driver mutations with a higher VAF.25, 34
The clinical utility of liquid biopsies is enhanced by the identification of actionable biomarkers that are not detected in the diagnostic tissue specimen. This may result from insufficient tissue availability, failed quality control, or the use of a biomarker panel with inadequate depth or breadth of analysis (undergenotyping). In a subset of 1288 patients from the current series, comprehensive liquid biopsies provided a 65% increase in driver oncogene detection, giving 252 additional patients the opportunity to consider targeted therapy and/or clinical trials enrollment. This finding is consistent with a recent study in which the addition of comprehensive liquid biopsy nearly doubled the number of targetable alterations identified in an NSCLC cohort.28
The optimal indicator of biomarker clinical utility is its ability to predict response to appropriately targeted therapies. Because our data set was largely divorced from patient outcomes, we supplemented the available, fully annotated patients with a pooled analysis of the literature using this assay in which outcome associations were reported. The analysis demonstrated response rates statistically equivalent to the expected outcomes for each targeted agent, as observed in their respective FDA registrational studies. This provides additional support for the use of comprehensive liquid biopsy to identify patients for targeted therapy in the first‐line and progression settings. Similarly, a recent prospective study reported a disease control rate of 85.7% among patients receiving targeted therapy for mutations identified using ctDNA NGS.28 CNG events detected in plasma are a function of the number of gene copies in the tumor, how much ctDNA is shed into the peripheral circulation, and the basal levels of normal cell‐free DNA in circulation. Recent studies have developed and validated a method for calculating an adjusted plasma copy number that statistically correlates with patient outcomes on targeted therapy.35, 36 The predictive utility of this assay in identifying effective therapies for patients who progress on targeted agents strongly suggests that the positive identification of an actionable biomarker by ctDNA is sufficient evidence for treatment decision making without the need for further confirmation in tissue. Because not all tumors shed adequate quantities of ctDNA for detection, lack of detection of oncogenic driver mutations in plasma would require follow‐up testing by tissue biopsy in patients with newly diagnosed, advanced NSCLC. Thus clinicians may consider a plasma‐first option, defaulting to a new tissue biopsy only in the absence of a clinically meaningful finding in plasma.
By its nature, this resource does not represent a random cross‐section of NSCLC in the United States; rather, it is composed largely of patients with a significant pretreatment history who were analyzed at the time of progression on a targeted therapy, which influenced the mutation patterns, as discussed above. Another key limitation is the availability of patient treatment outcomes and matched tissue genotyping for only a limited subset, an observation that led us to conduct the pooled outcome analysis (Fig. 4; see Supporting Table 12).3, 16 Some alterations identified by NGS may be caused by clonal hematopoiesis of indeterminant potential (CHIP), not arising from the tumor. Mutations caused by CHIP have been identified both in ctDNA and in tissue sequencing37 and are most frequently identified in the DNMT3A, TET2, ASXL1, SF3B1, and PPM1D genes, which are not sequenced in the assay studied here, as well as JAK2 and TP53, which are included in this assay but are not clinically actionable in solid tumors (see Supporting Table 1).38, 39 KRAS and IDH2 mutations are clinically relevant and may be CHIP‐derived, but this is very rare.38, 39 Of the 15,801 patients tested by Jaiswal et al, only 3 each had CHIP‐based KRAS and IDH2 mutations.38
In summary, we conducted a large‐scale assessment of the Guardant360 digital NGS platform in more than 8000 consecutively tested patients with NSCLC and identified actionable driver mutations and emergent resistance mutations in patient plasma at frequencies and distributions matching expectations for this clinical setting. Subset analyses showed that liquid biopsies markedly increased the number of actionable/informative biomarkers detectable at baseline and that abnormalities identified by plasma, both in the first line and at progression, respond to matched targeted therapy at rates similar to published results based on tissue assessment. Thus, at diagnosis, plasma NGS may be useful to obtain complete genotyping when tissue is insufficient or incomplete (undergenotyped) for all recommended biomarkers and can identify targetable driver and resistance mutations in NSCLC, supporting the concept of a plasma‐first algorithm at progression on targeted therapy.
Funding Support
No specific funding was disclosed.
Conflict of Interest Disclosures
Philip C. Mack reports personal fees from Guardant Health Inc, AstraZeneca, Pfizer, and Novartis outside the submitted work. Kimberly C. Banks is a current employee of and shareholder in Guardant Health Inc. Carin R. Espenschied is a former employee of Ambry Genetics and a current employee of and shareholder in Guardant Health Inc. Oliver A. Zill is a former employee of and shareholder in Guardant Health Inc and is currently an employee of Genentech. Christine E. Lee is a current employee of and shareholder in Guardant Health Inc and reports an issued patent (general early tech; Guardant Health license PCT/US2017/027809). Jonathan W. Riess reports institutional research grants from Merck & Company, AstraZeneca, Spectrum, and Novartis outside the submitted work and personal fees from Takeda, Celgene, Spectrum, Boehringer Ingelheim, and Loxo Oncology outside the submitted work. Stefanie A. Mortimer is a current employee of and shareholder in Guardant Health Inc; she reports a patent pending (20190316185) a patent issued (9850523). AmirAli Talasaz is cofounder and president of Guardant Health Inc, owns shares in the company, and reports personal fees from the company outside the submitted work. Richard B. Lanman serves on the board of directors of Biolase Inc and owns shares in the company; serves as an advisor to Forward Medical Inc and owns shares in the company; and he is a current employee of Guardant Health Inc, owns shares in the company, and has a leadership position. David R. Gandara reports grants from Bristol‐Myers Squibb and Novartis outside the submitted work; grants and personal fees from Roche‐Genentech and Merck & Company outside the submitted work; and personal fees from AstraZeneca, Celgene, CellMax Life, FujiFilm, Guardant Health Inc, Inivata, IO Biotech, Lilly, Liquid Genomics, Samsung Bioepis, and Pfizer all outside the submitted work. Rebekah A. Burich made no disclosures.
Author Contributions
Philip C. Mack: Conceptualization, formal analysis, writing–original draft, and writing–review and editing. Kimberly C. Banks: Conceptualization, data curation, formal analysis, writing–original draft, and writing–review and editing. Carin R. Espenschied: Data curation, formal analysis, writing–original draft, and writing–review and editing. Rebekah A. Burich: Formal analysis and writing–review and editing. Oliver A. Zill: Conceptualization, formal analysis, and writing–review and editing. Christine E. Lee: Data curation and writing–review and editing. Jonathan W. Riess: Conceptualization and writing–review and editing. Stefanie A. Mortimer: Formal analysis and writing–review and editing. AmirAli Talasaz: Conceptualization and writing–review and editing. Richard B. Lanman: Conceptualization and writing–review and editing. David R. Gandara: Conceptualization and writing–review and editing.
Supporting information
Figures1‐2
Tables1‐12
Supplementary Material
See editorial on pages 3176‐80, this issue.
References
- 1. Ettinger DS, Wood DE, Aisner DL, et al. Non‐Small Cell Lung Cancer, Version 1.2019. National Comprehensive Cancer Network (NCCN) Guidelines. NCCN; 2018. Accessed January 2019. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf [Google Scholar]
- 2. Lindeman NI, Cagle PT, Aisner DL, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J Mol Diagn. 2018;20:129‐159. [DOI] [PubMed] [Google Scholar]
- 3. Rolfo C, Mack PC, Scagliotti GV, et al. Liquid biopsy for advanced non‐small cell lung cancer (NSCLC): a statement paper from the IASLC. J Thorac Oncol. 2018;13:1248‐1268. [DOI] [PubMed] [Google Scholar]
- 4. Collisson EA, Campbell JD, Brooks AN, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zill OA, Banks KC, Fairclough SR, et al. The landscape of actionable genomic alterations in cell‐free circulating tumor DNA from 21,807 advanced cancer patients. Clin Cancer Res. 2018;24:3528‐3538. [DOI] [PubMed] [Google Scholar]
- 6. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR‐mutated advanced non–small‐cell lung cancer. N Engl J Med. 2018;378:113‐125. [DOI] [PubMed] [Google Scholar]
- 7. Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first‐ and second‐generation ALK inhibitors in ALK‐rearranged lung cancer. Cancer Discov. 2016;6:1118‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dagogo‐Jack I, Brannon AR, Ferris LA, et al. Tracking the evolution of resistance to ALK tyrosine kinase inhibitors through longitudinal analysis of circulating tumor DNA. JCO Precis Oncol. 2018;2018:00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Facchinetti F, Loriot Y, Kuo MS, et al. Crizotinib‐resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1‐ and ALK‐rearranged lung cancers. Clin Cancer Res. 2016;22:5983‐5991. [DOI] [PubMed] [Google Scholar]
- 10. Engstrom L, Aranda R, Lee M, et al. Glesatinib exhibits antitumor activity in lung cancer models and patients harboring MET exon 14 mutations and overcomes mutation‐mediated resistance to type I MET inhibitors in nonclinical models. Clin Cancer Res. 2017;23:6661‐6672. [DOI] [PubMed] [Google Scholar]
- 11. Subbiah V, Gainor JF, Rahal R, et al. Precision targeted therapy with BLU‐667 for RET‐driven cancers. Cancer Discov. 2018;8:836‐849. [DOI] [PubMed] [Google Scholar]
- 12. Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a plasma‐based comprehensive cancer genotyping assay utilizing orthogonal tissue‐ and plasma‐based methodologies. Clin Cancer Res. 2018;24:3539‐3549. [DOI] [PubMed] [Google Scholar]
- 14. Leighl NB, Page RD, Raymond VM, et al. Clinical utility of comprehensive cell‐free DNA analysis to identify genomic biomarkers in patients with newly diagnosed metastatic non‐small cell lung cancer. Clin Cancer Res. 2019;25:4691‐4700. [DOI] [PubMed] [Google Scholar]
- 15. Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell‐free circulating tumor DNA. PLoS One. 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCoach CE, Blakely CM, Banks KC, et al. Clinical utility of cell‐free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non‐small cell lung cancer. Clin Cancer Res. 2018;24:2758‐2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thompson JC, Yee SS, Troxel AB, et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next‐generation sequencing of cell‐free circulating tumor DNA. Clin Cancer Res. 2016;22:5772‐5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Santos ES, Raez LE, Castillero L, Marana C, Hunis B. Genomic tissue analysis and liquid biopsy profiles from patients diagnosed with advanced adenocarcinoma of the lung. Clin Oncol. 2016;1:1‐4. [Google Scholar]
- 22. Rozenblum AB, Ilouze M, Dudnik E, et al. Clinical impact of hybrid capture‐based next‐generation sequencing on changes in treatment decisions in lung cancer. J Thorac Oncol. 2017;12:258‐268. [DOI] [PubMed] [Google Scholar]
- 23. Kim ST, Banks KC, Lee SH, et al. Prospective feasibility study for using cell‐free circulating tumor DNA‐guided therapy in refractory metastatic solid cancers: an interim analysis. JCO Precis Oncol. 2017;1:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwaederle MC, Patel SP, Husain H, et al. Utility of genomic assessment of blood‐derived circulating tumor DNA (ctDNA) in Patients with advanced lung adenocarcinoma. Clin Cancer Res. 2017;23:5101‐5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacobs MT, Mohindra NA, Shantzer L, et al. Use of low‐frequency driver mutations detected by cell‐free circulating tumor DNA to guide targeted therapy in non–small‐cell lung cancer: a multicenter case series. JCO Precis Oncol. 2018;2:1‐10. [DOI] [PubMed] [Google Scholar]
- 26. Laufer‐Geva S, Rozenblum AB, Twito T, et al. The clinical impact of comprehensive genomic testing of circulating cell‐free DNA in advanced lung cancer. J Thorac Oncol. 2018;13:1705‐1716. [DOI] [PubMed] [Google Scholar]
- 27. Blakely CM, Watkins TBK, Wu W, et al. Evolution and clinical impact of co‐occurring genetic alterations in advanced‐stage EGFR‐mutant lung cancers. Nat Genet. 2017;49:1693‐1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aggarwal C, Thompson JC, Black TA, et al. Clinical implications of plasma‐based genotyping with the delivery of personalized therapy in metastatic non‐small cell lung cancer. JAMA Oncol. 2019;5:173‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hartmaier RJ, Albacker LA, Chmielecki J, et al. High‐throughput genomic profiling of adult solid tumors reveals novel insights into cancer pathogenesis. Cancer Res. 2017;77:2464‐2475. [DOI] [PubMed] [Google Scholar]
- 30. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sholl LM, Aisner DL, Varella‐Garcia M, et al. Multi‐institutional oncogenic driver mutation analysis in lung adenocarcinoma: the Lung Cancer Mutation Consortium experience. J Thorac Oncol. 2015;10:768‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473‐481. [DOI] [PubMed] [Google Scholar]
- 33. Stewart EL, Tan SZ, Liu G, Tsao MS. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations—a review. Transl Lung Cancer Res. 2015;4:67‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helman E, Nguyen M, Karlovich CA, et al. Cell‐free DNA next‐generation sequencing prediction of response and resistance to third‐generation EGFR inhibitor. Clin Lung Cancer. 2018;19:518‐530.e7. [DOI] [PubMed] [Google Scholar]
- 35. Lee J, Kim ST, Kim K, et al. Tumor genomic profiling guides metastatic gastric cancer patients to targeted treatment: the VIKTORY Umbrella Trial. Cancer Discov. 2019;9:1388‐1405. [DOI] [PubMed] [Google Scholar]
- 36. Siravegna G, Sartore‐Bianchi A, Nagy RJ, et al. Plasma HER2 (ERBB2) copy number predicts response to HER2‐targeted therapy in metastatic colorectal cancer. Clin Cancer Res. 2019;25:3046‐3053. [DOI] [PubMed] [Google Scholar]
- 37. Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small‐cell lung cancer. J Clin Oncol. 2016;34:3375‐3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jaiswal S, Fontanillas P, Flannick J, et al. Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488‐2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures1‐2
Tables1‐12
Supplementary Material