

Abstract
Pseudomonas aeruginosa infection elicits the production of cytotoxic amyloids from lung endothelium, yet molecular mechanisms of host‐pathogen interaction that underlie the amyloid production are not well understood. We examined the importance of type III secretion system (T3SS) effectors in the production of cytotoxic amyloids. P aeruginosa possessing a functional T3SS and effectors induced the production and release of cytotoxic amyloids from lung endothelium, including beta amyloid, and tau. T3SS effector intoxication was sufficient to generate cytotoxic amyloid release, yet intoxication with exoenzyme Y (ExoY) alone or together with exoenzymes S and T (ExoS/T/Y) generated the most virulent amyloids. Infection with lab and clinical strains engendered cytotoxic amyloids that were capable of being propagated in endothelial cell culture and passed to naïve cells, indicative of a prion strain. Conversely, T3SS‐incompetent P aeruginosa infection produced non‐cytotoxic amyloids with antimicrobial properties. These findings provide evidence that (1) endothelial intoxication with ExoY is sufficient to elicit self‐propagating amyloid cytotoxins during infection, (2) pulmonary endothelium contributes to innate immunity by generating antimicrobial amyloids in response to bacterial infection, and (3) ExoY contributes to the virulence arsenal of P aeruginosa through the subversion of endothelial amyloid host‐defense to promote a lung endothelial‐derived cytotoxic proteinopathy.
Keywords: amyloid beta (Aβ), exoenzyme Y (ExoY), nosocomial pneumonia, prion, tau (τ)
Abbreviations
- AP‐1
activator protein 1
- Aβ
amyloid beta
- Aβ42
amyloid beta peptide variant that contains 42 of the possible 43 residues for Aβ
- Aβ40
amyloid beta peptide variant that contains 40 of the possible 43 residues for Aβ
- BALF
bronchoalveolar lavage fluid
- cAMP
cyclic adenosine monophosphate
- cCMP
cyclic cytosine monophosphate
- CFU
colony forming unit
- cGMP
cyclic guanosine monophosphate
- cNMP
cyclic nucleotide monophosphate
- cUMP
cyclic uridine monophosphate
- ExoS
exoenzyme S—a type III secretion system effector; an ADP‐ribosyltransferase
- ExoT
exoenzyme T—a type III secretion system effector; an ADP‐ribosyltransferase
- ExoU
exoenzyme U—a type III secretion system effector; a phospholipase A2
- ExoY
exoenzyme Y—a type III secretion system effector; a promiscuous nucleotidyl cyclase
- ExoYK81M
mutant of P aeruginosa strain PA103; exclusively expresses catalytically inactive ExoY
- ExoY+
mutant of P aeruginosa strain PA103; exclusively expresses ExoY
- LB
lysogeny broth
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B‐cells
- PA‐808
P aeruginosa isolated from BALF of nosocomial pneumonia patient (ExoY, ExoS, ExoT)
- PA103
Pseudomonas aeruginosa isolated from the sputum of patient 103 (ExoU, ExoT)
- PAO1
P aeruginosa strain 1 isolated from a wound (ExoY, ExoS, ExoT)
- PMVEC
pulmonary microvascular endothelial cell
- T3SS
type III Secretion System
- VAP
ventilator‐associated pneumonia
- YESCA
yeast extract casamino acids minimal media
- ZOI
zone of inhibition
- ΔPcrV
mutant of P aeruginosa strain PA103; expresses a T3SS with an incompetent needle
1. INTRODUCTION
Pseudomonas aeruginosa is a Gram‐negative opportunistic pathogen that poses a significant concern within the intensive care setting. 1 This bacterium is capable of upregulating virulence factors in response to environmental cues and quorum sensing. 2 , 3 P aeruginosa colonizes the endotracheal tubes of mechanically ventilated patients either alone or in conjunction with other nosocomial microbes, where they cooperatively form antibiotic‐resistant biofilms. 4 This colonization comprises a locus that can facilitate the seeding of bacteria into the lower airways as a source of ventilator‐associated pneumonia (VAP). 5 , 6 P aeruginosa is the predominant bacterial agent of VAP; it is responsible for the majority of nosocomial infections. 7 , 8 Within the distal airways the organism disrupts the alveolar (ie, epithelial)‐capillary (ie, capillary endothelial) membrane, leading to exudative edema that impairs oxygenation and promotes bacterial dissemination through the circulation. End‐organ damage, with increased rates of morbidity and mortality, 9 , 10 , 11 frequently plague the outcomes of patients recovering from VAP. Some studies report that fewer than 50% of these patients survive the first‐year post‐discharge, 12 , 13 and this number falls to 30% within the next 4 years. 14 Factors contributing to the poor health outcomes and compromised longevity of VAP survivors are currently unclear.
Virulent P aeruginosa bacteria extend a syringe‐and‐needle‐like type III secretion system (T3SS) from the bacterial surface within low calcium milieus or upon contact with the host cell. 15 Expression of a functional T3SS fundamentally modulates the severity of P aeruginosa‐mediated nosocomial pneumonia. 16 , 17 The T3SS is capable of injecting four primary exoenzyme effectors into target cells: ExoS, ExoT, ExoU, and ExoY. 18 Effectors ExoS and ExoT are ADP ribosyltransferases associated with cytoskeletal rearrangement, yet Shaver and Hauser reported that ExoS, but not ExoT, moderately enhanced virulence within the lung. 19 An acutely cytotoxic phospholipase A2, ExoU breaks down cell membranes leading to rapid cell death within both in vitro and in vivo acute lung infection models. 20 , 21 However, ExoU is less frequently encoded in the virulence arsenal of P aeruginosa than any of the other known T3SS exoenzymes. 22 , 23
ExoY is the most recently identified T3SS effector of P aeruginosa. It is a promiscuous nucleotidyl cyclase that preferentially generates canonical (cGMP and cAMP) and non‐canonical (cUMP and cCMP) cNMPs upon injection into host cells, including the lung microvasculature. 24 , 25 , 26 , 27 , 28 , 29 In contrast to ExoU, ExoY is abundantly found in hospital‐acquired strains of P aeruginosa, encoded by ~90%‐95% of clinical isolates. 22 , 23 However, the extent to which ExoY contributes to virulence is debated. ExoY intoxication induces a protracted pathogenesis resulting in cell rounding and the disruption of cell‐to‐cell junctions. Within the alveolar‐capillary membrane, ExoY induces marked interendothelial gap formation and consequent alveolar flooding, 30 , 31 although it does not cause frank cell lysis as does ExoU. 32 ExoY has been reported to suppress the innate immune response via inhibition of NF‐κB and AP‐1 with a consequent reduction in pro‐inflammatory cytokines and caspase‐1 activation within lung epithelia and monocytes. 33 , 34 The effect of ExoY intoxication upon the innate immune response of the pulmonary endothelium is currently unknown.
While the role of ExoY within the virulence arsenal of P aeruginosa remains incompletely understood, recent evidence suggests ExoY may significantly contribute, even in the aftermath of infection, by eliciting the production of transmissible and cytotoxic amyloids. 35 Ochoa and co‐workers established that ExoY intoxication of the pulmonary endothelium induces the hyperphosphorylation of a non‐neuronal form of microtubule‐stabilizing tau protein. ExoY‐induced hyperphosphorylated tau dissociates from microtubules leading to tubule catastrophe, cytoskeletal involution, endothelial cell rounding and gap formation, and the release of oligomeric tau into the extracellular space. 35 , 36 Later, Balczon and colleagues provided evidence that the transmissible cytotoxins arising secondary to T3SS effector intoxication are comprised of oligomeric beta amyloid (Aβ) as well as tau. 37 These cytotoxic amyloids are also capable of being propagated indefinitely in cell culture in a prion‐like manner in the absence of active bacterial infection. 37 The potential for ExoY to elicit Aβ from the endothelium as a component of this infection‐induced cytotoxic amyloid prionopathy has not been investigated.
Aβ is a product of the amyloid precursor protein (APP). Following translation and sorting, APP may be directed from the trans golgi network to either the surface of the cell or the endosomal pathway. APP inserted into the cell surface membrane may either be proteolytically processed through sequential cleavages of alpha and gamma secretases to produce non‐amyloidogenic products including, soluble APP alpha, or reinternalized via endocytosis. Endosomes are rich in the beta‐ (β‐secretase) and gamma secretases (γ‐secretase) required for the processing of APP into Aβ. Once sequestered within an endosome, APP may be broken down via endosomal‐lysosome fusion, transported back to the trans golgi, or proteolyzed to yield Aβ. Sequential cleavages by β‐secretase (also known as BACE1) and γ‐secretase produce Aβ and soluble APP beta, which are consequently exocytosed into the extracellular space. 38 , 39 The variant of Aβ that is generated depends upon the site(s) of cleavage by β‐secretase. The Aβ variants that have received the most attention in studies of neurodegenerative disease include Aβ40 (Aβ1‐40) and Aβ42 (Aβ1‐42). Aβ is aggregation prone and thermodynamics frequently favor oligomerization, polymerization of Aβ into fibrils, or potentially the formation of amorphous aggregates. Oligomeric Aβ is neurotoxic and intrinsic to the pathogenesis of various forms of dementia, including Alzheimer's Disease. 40 , 41
Recent studies have given credence to the long proposed notion that Aβ has antimicrobial properties, as recently reviewed. 42 , 43 These antimicrobial properties have been attributed to Aβ oligomers that form fibrils in the presence of bacterial surface epitopes. In particular, Moir and colleagues have reported that recombinant Aβ42 is a broad‐spectrum antimicrobial peptide exhibiting efficacy against both Gram‐negative and Gram‐positive bacteria, as well as viral and fungal pathogens. 44 , 45 The naturally produced antimicrobial amyloids released during infection are poorly understood, yet microorganisms are found in plaques, and in studies of Aβ lowering drugs, infection is a frequently reported side effect. 46 , 47 We questioned whether endothelium is capable of generating antimicrobial forms of Aβ and/or oligomeric tau, and further, whether Pseudomonas exoenzymes fulfill an inter‐kingdom function of converting the antimicrobial amyloids into cytotoxic forms. More specifically, we hypothesized that T3SS‐competent P aeruginosa elicit cytotoxic amyloids during infection and suppress an innate amyloid immune response from the pulmonary endothelium.
2. MATERIALS AND METHODS
2.1. Cell culture
The cell culture core at the University of South Alabama Center for Lung Biology provided PMVEC clones isolated from both male Sprague Dawley (four rats) and Fischer (one rat) rats. Clones from two female Fischer rats (five clones) and one male Fischer rat (one clone) were isolated and generously provided by Dr Sarah A. McMurtry. The isolation protocol has been previously described and is standard for our published studies. 25 , 30 , 31 , 35 , 36 , 37 , 48 Cells were cultivated in DMEM (4.5 g/L glucose) with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA, cat. no. S11550H) and 1% penicillin‐streptomycin and incubated at 37°C in room air with 5% CO2.
2.2. P aeruginosa isogenic strains
Isogenic strains of P aeruginosa have been previously described. 24 , 26 PA103 (ExoU/T) and PAO1 (ExoS/T/Y) each have a fully functional T3SS capable of injecting exoenzymes into the cytoplasmic compartment of the host cell. The ExoY+ (PA103ΔexoUexoT::Tc pUCPexoY) mutant is able to both express and secrete the functional ExoY cyclase into target cells. Conversely, the T3SS‐incompetent mutant ΔPcrV (PA103ΔPcrV; ExoU and ExoT) lacks the PcrV protein needed to facilitate the formation of a functional translocon channel. As such, the ΔPcrV mutant expresses ExoU and ExoT but is unable to inject them into the host cytosol. The ExoYK81M (PA103ΔexoUexoT::Tc pUCPexoYK81M) mutant has a patent T3SS translocation pore yet expresses a catalytically inactive ExoY that interacts with the cytoskeleton once injected into the host cell but does not generate a cyclic nucleotide signature.
2.3. Isolation and identification of clinical P aeruginosa
Bronchoalveolar lavage samples, submitted to the microbiology laboratory for bacterial culture, were vortexed for 30‐60 seconds followed by inoculation of agar (Trypticase Soy Agar II with 5% Sheep's Blood) plates, chocolate agar (CHOC) plates, and MacConkey Agar (MAC) plates. Using a disposable 1 µL calibrated loop, bacteria were streaked onto TSA II 5% Sheep's Blood (TSA II 5% SB) and CHOC agar by making a straight line down the center and a series of passes were made at 90° angles through the inoculum. MAC agar was streaked for isolation. Plates were placed in an incubator containing approximately 5% CO2 at 35°C for 24‐48 hours. Colonies from the quantitative primary media, TSA II 5% SB, were counted where each colony = 1000 colony‐forming units per milliliter. 49 Isolates were identified by Matrix Assisted Laser Desorption Ionization Time of Flight mass spectroscopy (MALDI‐TOF, Vitek©MS, Biomerieux).
2.4. Genotyping of clinical P aeruginosa strains
P aeruginosa isolates were screened by multiplex PCR for the presence of the genes listed in Table 1. Primers for DNA polymerase A (PolA) were used as the internal control. The thermocycler was programed for initial denaturation at 94°C for 5 minutes followed by 35 cycles consisting of 30 seconds of denaturation at 94°C, 20 seconds annealing at 55°C, and 3 minutes of extension at 72°C.
Table 1.
Primers for screening clinical isolates
| Primer name | Primer sequence | PCR fragment size |
|---|---|---|
| pcrV‐P.a.F | CCCTTCGAGAAGGACAACAA | 200 bp |
| pcrV‐P.a.R | CTAGATCGCGCTGAGAATGT | |
| exoY‐P.a.F | TGAGCGAGGACGGATTCTA | 309 bp |
| exoY‐P.a.R | GATAGCCGTTGCCCTTGAT | |
| exoU‐P.a.F | CTCAATGTACTCCCACGCATAG | 406 bp |
| exoU‐P.a.R | CATCCTGGAATTCTGTCCACTC | |
| exoT‐P.a.F | GCCGAGATCAAGCAGATGAT | 405 bp |
| exoT‐P.a.R | GACAGGCTCGCCCTTTAC | |
| exoT‐P.a.F | GTCTTTCGTGGCTGAGTTGA | 300 bp |
| exoT‐P.a.R | TGCAGCATCATCTGCTTGA | |
| exoS‐P.a.F | CATCAGGTAATGAGCGAGGTC | 410 bp |
| exoS‐P.a.R | TTCAGGGAGGTGGAGAGATAG | |
| PolA‐P.a.F | TTTCCTGCAGCCAGTTATCC | 707 bp |
| PolA‐P.a.R | CAAGCTCAAGAGCACCTACA | |
| Vfr‐P.a.F | GGAACTTCGCATAGCTGATCT | 307 bp |
| Vfr‐P.a.R | CACACACCCAAACTCAAACAC | |
| CyaB‐P.a.F | GGGAACCTACACCCAGTATTTC | 400 bp |
| CyaB‐P.a.R | GAAGGCGGTGATGCAGAT |
2.5. Generation of 1° bacterial supernatants
PMVECs from both male and female Fischer and male Sprague Dawley rats were grown to 24 hours post‐confluence. The monolayer from an identically seeded counting plate was trypsinized and cells were counted with a Countess II Automated Cell Counter (Invitrogen, Carlsbad, CA, cat. no. AMQAX1000) according to manufacturer's instructions. Bacteria from overnight Vogel‐Bonner plates were suspended in 1x Phosphate Buffered Saline (PBS) pH 7.4 (Invitrogen; Carlsbad, CA, cat. no. 10010‐049) to an OD540 of 0.25, previously determined to represent 2 × 108 CFUs/mL, and diluted to an MOI of 20:1 in Hank's Balanced Salt Solution (HBSS; Invitrogen; Carlsbad, CA, cat. no. 14025‐134). Infected monolayers were incubated (37°C and 5% CO2) for 4 hours (PA103 and ΔPcrV), 5 hours (PAO1, PA‐808), or 7 hours (ExoY+, ExoYK81M). Supernatants were then collected, centrifuged at 4000 g, and sterilized via passage through a PES 0.22 µm filter (EMD Millipore, Burlington, MA, cat. no. SLGP033RS). Vehicle control supernatant was generated as described for bacterial infection except bacteria were excluded from the PBS/HBSS solution used to treat the monolayers. “No Cell” supernatants were produced via the incubation of full DMEM media overnight in a cell culture plate. Following incubation, the media was aspirated and the plate was washed with HBSS and infected as detailed above. To calculate the bacterial MOI for these “No Cell” experiments, endothelial cell counts were estimated based on the prior infection experiments.
2.6. Protein analysis
Supernatants were concentrated 20‐fold with Amicon Ultra‐4 Centrifugal Filter Units with a 3 kDa MWCO (EMD Millipore, Burlington, MA, cat. no. UFC800324) to generate a concentrated stock solution of each sample. The protein concentration of each concentrated sample was determined via the microplate method of a Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, cat. no. 23227) according to manufacturer's instructions. HBSS was used to dilute the samples to the desired protein concentration.
2.7. Congo Red staining of supernatants
Supernatant protein concentration was standardized to 5 µg/10 µL. Samples were well‐suspended prior to applying 10 µL, carefully in a drop‐wise fashion, to a clean slide. Slides were heat‐fixed on a warm hot plate to provide consistency between results obtained among samples both with and without bacteria. Light sensitive Congo Red (Fisher Scientific; Hampton, NH, cat. no. C580‐25) was prepared in ultrapure water at a concentration of 3 mg/mL, vortexed thoroughly, and passed through a 0.22 µm syringe filter to disrupt any remaining clumps. Slides with fixed and cooled samples were flooded with Congo Red for 5 minutes, rinsed, destained with 70% ethanol for 3 minutes, rinsed again, and allowed to air dry while being protected from light. Slides were imaged on a Nikon Eclipse 80i upright microscope under (1) bright‐field (Crystal Violet control and Congo Red), and (2) with a Nikon rotatable polarizer and analyzer for the cross‐polarized light necessary to ascertain birefringence (Congo Red).
2.8. Thioflavin T binding assay
Thioflavin T (ThT) is a light‐sensitive stain that is often used to study amyloid fibrilization kinetics or as an end‐point read out to assess the quantity of fibrillar amyloid species in a given sample. 50 A 25 µM ThT (Acros Organics; Geel, Belgium, cat. no. AC211760250) stock solution was prepared in 100% ethanol, vortexed thoroughly, and passed through a PES 0.22 µm syringe filter to remove clumps. Black clear‐bottomed Corning Costar 96‐well polystyrene plates (Corning Inc; Corning, NY, cat. no. 3603) were used for assays. Supernatant samples were prepared as detailed for “Protein analysis” and supernatants were diluted into 1x PBS pH 7.4 (Invitrogen; Carlsbad, CA, cat. no. 10010‐049) to a protein concentration of 5 µg/mL. ThT from the 25 µM stock solution was added just prior to reading the plate at a final ThT concentration of 0.49 µM and a total volume of 200 µL/well. Fluorescence was measured on a Molecular Devices Spectramax iD5 Multi‐Mode Microplate reader (425 excitation; 490 emission), 1 minute orbital shaking prior to read, high gain, 400 ms integration time.
2.9. Antibody validation
The A11 antibody (Stressmarq; Victoria, British Columbia; cat. no.SPC‐506D) was validated against various conformations of recombinant rat Aβ42 (rPeptide; Athens, GA; cat. no. A‐1008‐1). Initially, Aβ42 was reconstituted with ice‐cold 1% ammonium hydroxide to a stock concentration of 1 mg/mL, or 226.4 µM, on ice. Stock was diluted with ice‐cold 1x pH 7.4 PBS to 100 µM on ice, aliquoted, and stored at −80°C. Monomeric Aβ quickly begins to aggregate into oligomers of various sizes once it is introduced into an aqueous solution. Increased ratios of oligomers were noted post‐freezing. Oligomer/monomer mixes were also generated by storing an aliquot of 100 µM Aβ on ice at 4°C for 48 hours. Largely pure samples of Aβ42 fibrils were produced through the rotation of 100 µM aliquots of Aβ42 for 7 days at 37°C. 200 pmol of recombinant Aβ42 was resolved on a 4%‐12% Bis‐Tris gel, transferred to PVDF, and probed with either A11, MOAB‐2, or OC. Primary antibodies: polyclonal A11 at 1:1200, monoclonal MOAB‐2 (Novus Biologicals; Centennial, CO; cat. no. NBP2‐13075) at 1:1000, or polyclonal OC (EMD Millipore; Burlington, MA; cat. no. AB2286) at 1:1000.
The T22 antibody (EMD Millipore; Billerica, MA; cat. no. ABN454) was validated against a recombinant tau ladder (rPeptide; Athens, GA, cat. no. T‐1007‐1) consisting of all six monomeric isoforms of neuronal tau. About 10 ng of the ladder was resolved on a 4%‐12% Bis‐Tris gel, transferred to PVDF, and probed with either the T22 or TAU5 antibodies. Primary antibodies: polyclonal anti‐oligomeric tau T22 (EMD Millipore; Burlington, MA; cat. no. ABN454) at 1:1000 or the monoclonal pan‐tau antibody TAU5 (MBL International; Woburn, MA, cat. no. AT‐5004) at 1:1000.
Alzheimer's brain hippocampal lysate (Genetex, Irvine, CA, cat no. GTX26445) was used as a positive control. All blocking and antibody incubation was conducted in 7% nonfat milk—TBST. A11, T22, and OC secondary antibody: Goat anti‐rabbit preadsorbed IgG (abcam; Cambridge, MA; AB7090) at 1:10 000. Secondary antibody for MOAB‐2 and TAU5: Goat anti‐mouse preadsorbed IgG (abcam; Cambridge, MA; AB97040) at 1:20 000.
The conformation of recombinant proteins was monitored by resolving either 200 pmol of Aβ42 or 10 ng of tau ladder on a 1 mm thick 4%‐12% Bis‐Tris gel and staining with SYPRO Ruby Protein Gel Stain (Invitrogen; Carlsbad, CA, cat. no. S12000) according to the manufacturer's instructions and imaging. All blots were stripped and reprobed with each of the respective alternate antibodies. When negative results were obtained using A11, blots were stripped and reprobed with either MOAB‐2 or OC to confirm the presence of Aβ. When negative results were obtained using T22, blots were stripped and reprobed with TAU5 to confirm the presence of tau.
2.10. Western blotting
Proteins were precipitated from supernatants with 100% trichloroacetic acid solution (TCA). Briefly, 100 µL of 100% TCA solution was added to a 1 mL sample of supernatant and kept on ice overnight at 4°C. The next morning, samples were centrifuged at 18 000 g (4°C) for 45 minutes. Supernatants were carefully discarded and the pellet washed with 100% ethanol. Samples were centrifuged again (18 000 g at 4°C) for 15 minutes. The ethanol wash was discarded, and the pellet was retained and dried in a heating block at 60°C for 10 minutes, cooled to room temperature, and then, resuspended in Laemmli sample buffer (Boston Bioproducts, Worcester, MA; cat. no. BP‐111R). Samples were heated for 5 minutes at 95°C and spun briefly in a microfuge prior to loading on a 4%‐12% Bis‐Tris gel for sample resolution. Proteins were transferred from the gel to PVDF and probed as indicated (A11 and T22: blocking and probing in 7% milk—1x TBST, MOAB‐2: blocking, and probing in 5% milk—1x TBST). Antibodies: A11 at 1:1000, T22 at 1:500, and MOAB‐2 at 1:500. All blocking and probing for A11 and T22 was done in 7% nonfat milk—1x TBST. Blocking and probing with MOAB‐2 was conducted in 5% nonfat milk—1x TBST whereas blocking and incubation with secondary antibody was done in 7% nonfat milk—1x TBST. Secondary antibody for A11 and T22: Goat anti‐rabbit preadsorbed IgG at 1:10 000. Secondary antibody for MOAB‐2: Goat anti‐mouse preadsorbed IgG at 1:20 000.
2.11. Immunoprecipitations
Amyloids were neutralized for cytotoxicity and antimicrobial studies through the addition of antibodies A11, T22, monoclonal antibody clone 11A50‐B10 (Biolegend; San Diego, CA; cat. no. 805401), and polyclonal anti‐Aβ1‐43 (Novus Biologicals; Centennial, CO; cat. no. NBP2‐25093) at a concentration of 2 µg/mL and rotated overnight at 4°C. Protein A agarose beads (Santa Cruz; Dallas, TX; cat. no. SC2001) for A11, T22, and polyclonal anti‐Aβ1‐43 or protein G agarose beads (Santa Cruz; Dallas, TX; cat. no. SC2002) for the11A50‐B10 anti‐Aβ40 antibody were added in accordance to the binding capacity of the beads/amount of antibody added to the sample and rotated for an additional 3 hours at 4°C. Samples were then centrifuged at 1000 g for 5 minutes and the supernatants collected. Recovered supernatants were filter‐sterilized, dialyzed against 4 changes of HBSS (1:200), filter‐sterilized again, and stored at −20°C until use.
2.12. Assessment of interendothelial gap formation
Wells of a 12‐well plate were seeded at identical densities from a single cell suspension and grown to confluence. Prior to treatment, media was removed and confluent monolayers were gently rinsed with HBSS. 300 µL of unconcentrated infection or vehicle supernatant (warmed to 37°C and thoroughly suspended with trituration) were gently applied to rinsed monolayers. Individual experiments were run across 1‐2 wells of the same plate with multiple images taken per well at 0, 6, 16, and 24 hours with a Nikon Eclipse TS100 microscope. All images used for analysis were taken at 10x magnification.
2.13. Resozurin reduction assay
PMVECs were seeded at identical densities from a single cell suspension and grown to confluence in a Corning CellBIND 48‐well microplate (Corning, NY, cat. no.3338) to prevent premature mechanical detachment post‐treatment that could otherwise falsely impact viability measurements. At cell confluence, media was removed, monolayers rinsed with HBSS, and 200 µL of vehicle or infection‐derived supernatant was added. Treated monolayers were incubated at 37°C for ~22.75 hours. Alamar Blue (Molecular Probes, Eugene, OR, cat. no. DAL1025) was added at 10% volume/well and the plates returned to 37°C for an additional 1.25 hours. Fluorescence was measured on a Spectramax iD5 Multi‐Mode Microplate reader (530 excitation; 590 emission).
2.14. Kirby‐Bauer disk diffusion assay
P aeruginosa strain PA103 was struck from frozen stocks onto Vogel‐Bonner minimal salts medium and incubated overnight at 37°C. 1 µL of the overnight culture was collected using a calibrated loop and inoculated into Mueller‐Hinton broth (Thermo Scientific, Waltham, MA, cat. no.CM0405B) and shaken at 240 rpm at 37°C to mid‐log phase. An aliquot of each broth culture was collected and standardized to an OD540 of 0.25 in 1x PBS pH 7.4 (previously determined to be 2 × 108 CFUs/mL). Using sterile cotton swabs, a uniform lawn was struck from each standardized bacterial suspension onto Mueller‐Hinton agar plates (plate depth: 4 mm) and allowed to rest for 15 minutes. Disks (Thermo Scientific, Waltham, MA, cat. no. R55054) were then inoculated with either gentamicin (10 µg/20 µL), HBSS (20 µL), or infection‐derived supernatants (10 µg/20 µL) and carefully applied with even pressure to lawns with disinfected forceps. Any irregularity in disk application resulted in the scratching of that disk from the experiment and all individual experiments were conducted in triplicate. Inoculated plates were incubated for 17 hours at 37°C. At the conclusion of the incubation period, the zones of inhibition (ZOI) were measured at the widest point to the nearest millimeter, including the disk. Clearing was confirmed via bright‐field microscopy. Assessment of progressive antimicrobial activity following the initial incubation period was conducted with plates held at room temperature and ZOIs re‐measured to the nearest millimeter and recorded at 24, 48, and 72 hours post‐inoculation. Assays were conducted in compliance with CLSI standards. 51
2.15. Bacterial aggregation on solid substrate
Yeast extract casamino acid (YESCA) minimal media agar was produced [Reagents all purchased from Fisher Scientific in Hampton, NH: 1% casamino acids (cat. no. BP1424‐100), 0.2% yeast extract (cat. no. BP1422‐100), and 4% agar (cat. no. BP1423‐500)] to screen for potential bacterial amyloid production. Following autoclaving, media was cooled to 50°C prior to the addition of 200 µg/mL of Congo Red and 200 µg/mL of Coomassie Brilliant Blue R‐250 while protecting the mixture from light. Plates were poured to a depth of 4 mm, allowed to set and dry while loosely covered with aluminum foil, and stored away from light at 4°C. Bacterial suspensions were prepared and streaked into lawns upon the YESCA‐Congo Red agar as described for the Kirby‐Bauer disk diffusion assays. Plates were incubated overnight at 37°C. Following initial incubation, gentamicin (10 µg/20 µL), HBSS (20 µL), or infection‐derived supernatants (10 µg/20 µL) were applied directly drop‐wise to established lawns. Of important note, the protein concentration of supernatants that had undergone antibody neutralization was not standardized as the protein concentration was so low the production of sufficient neutralized sample to meet the minimum concentration was cost and time prohibitive. Inoculated lawns were then held at 25°C for 72 hours and images of each treatment were obtained with a Nikon Eclipse TS100 inverted microscope. Images used for quantitation were taken at 10x.
2.16. Bacterial aggregation in suspension and plating experiments
Strain PA103 was struck from frozen stocks onto a Vogel‐Bonner agar plate and incubated overnight. A calibrated loop was used to obtain 1 µL of PA103 from the overnight plate to inoculate 10 mL of Mueller‐Hinton broth, and grown to an OD540 of 0.25 (2 × 108 CFUs/mL) at 37°C and 240 rpm. Bacteria were then diluted into Mueller‐Hinton broth to a density of 500 000 PA103 per 75 µL and dispensed into wells of a 96‐well plate. Wells containing bacteria were treated with 75 µL of either HBSS or supernatant (0.05 µg/100 µL) and incubated at 37°C for 24 hours with intermittent orbital shaking. Samples were collected, gently mixed, and 10 µL of each sample was applied drop‐wise onto glass slides and heat fixed. Slides were cooled and stained with either Crystal Violet or Congo Red (3 mg/mL) as described under “Congo Red staining of supernatants.” Slides were imaged on a Nikon Eclipse 80i upright microscope under (1) bright‐field (Crystal Violet and Congo Red), and (2) with a Nikon rotatable polarizer and analyzer for the cross‐polarization necessary to ascertain birefringent properties (Congo Red).
For plating experiments, 1 µL of PA103 from an overnight Vogel‐Bonner plate was inoculated into 10 mL of Mueller‐Hinton broth and grown to an OD540 of 0.25 (2 × 108 CFUs/mL) at 37°C and 240 rpm. Bacteria were diluted into 1x PBS pH 7.4 to a density of 500 000 bacteria/75 µL. 75 µL of the bacterial suspension was dispensed into the wells of a 96‐well plate and 75 µL of either HBSS or supernatant (0.05 µg/100 µL) were added to the wells. The plate was then incubated at 37°C for 3 hours and orbitally shaken for 30 seconds at 0, 1.5, and 3 hours. Samples were then serially diluted into 1x PBS pH 7.4, plated in triplicate on LB agar, and incubated overnight at 37°C. Colonies were counted the next day and CFUs/mL calculated.
2.17. Quantitation of endothelial gap formation and bacterial inhibition on Congo Red agar
Cell gap quantification was performed using a custom macro for ImageJ (National Institute of Health, Bethesda, Maryland) that automatically detected the fractional area of regions lacking cells to regions containing cells within a micrographic image, as previously described. 52
2.18. Statistical analysis
Data were analyzed using GraphPad Prism 5.0 software. Each n was comprised of an independent biological replicate with technical replicates as indicated. One‐way, two‐way, and repeated‐measures two‐way ANOVA were used to compare sample groups and Tukey's, Bonferroni, and Dunnett's, post hoc comparisons, as indicated and appropriate. In all studies, α = 0.05; significance was considered P < .05.
3. RESULTS
3.1. T3SS‐competent P aeruginosa instigates a transmissible cytotoxicity following endothelial infection
T3SS‐competency significantly contributes to the virulence of clinical isolates of P aeruginosa. 19 , 22 To test the cytotoxic potential of P aeruginosa on pulmonary endothelium, pulmonary microvascular endothelial cells (PMVECs) were infected with genetically engineered P aeruginosa strains. A strain that synthesizes the T3SS machinery but is unable to assemble a functional translocation pore (ΔPcrV) was used to determine whether the pore itself or delivery of T3SS effectors impacts gap formation in the monolayer. In addition, strains that express different combinations of injected effector proteins were tested and are simply referred to as the effectors that are delivered (ExoU/T or ExoY). Intoxication of pulmonary microvascular endothelium with strains that are able to deliver effectors (T3SS‐competent) induced marked cell rounding, interendothelial gap formation, and cell detachment. A strain that injects ExoU and ExoT induced acute damage that was detectable approximately 3 hours earlier than the damage that occurred by either T3SS pore formation or the injection of ExoY (Figure 1A).
Figure 1.
T3SS effector intoxication elicits transmissible injury. A, Infection of PMVECs with P aeruginosa ΔPcrV did not instigate interendothelial gap formation at 4 hours whereas strains capable of injecting ExoU/T induced marked endothelial barrier disruption within the same time frame. ExoY intoxication of PMVECs elicited pronounced interendothelial gap formation yet required roughly 3 hours longer to achieve damage equivalent to that observed in cells injected with ExoU/T. B, Cell supernatants collected from active bacterial infections with T3SS‐incompetent strains ΔPcrV and ExoYK81M, and strains injecting T3SS effectors ExoU/T (PA103), ExoY alone (ExoY+), and ExoS/T/Y (PAO1), as well as a non‐infected vehicle control, were collected and filter‐sterilized to render them bacteria‐free. C, Primary infection‐induced endothelial damage is qualitatively ranked, with injection of ExoU/T proving more acutely injurious than ExoY. D, Filter‐sterilized vehicle and infection supernatants were transferred to confluent, naïve PMVEC monolayers and imaged over a 24 hours time course. Vehicle, ΔPcrV, and ExoYK81M infection‐derived supernatants did not contain transmissible cytotoxins yet strains capable of injecting T3SS exoenzymes produced supernatants that profoundly compromised endothelial barrier integrity when transferred to naïve cells. E, Schematic illustrates the hierarchy of transmissible cytotoxicity relevant to the exoenzymes deployed during the original active infection. In contrast to the primary infection, endothelial intoxication with ExoS/T/Y as well as ExoY alone elicited a more virulent secondary cytotoxicity than endothelial intoxication with ExoU/T. Scale bar = 200 µm
To test for the potential toxicity of the T3SS translocation pore we used a strain that exclusively synthesizes and injects a derivative of ExoY that no longer expresses nucleotidyl cyclase activity, ExoYK81M. In addition, a strain was added that does not synthesize ExoU but synthesizes and can inject ExoS, ExoT, and ExoY. The toxicity of the primary infection is shown on the top tier of cellular images. Damage to the monolayer appeared most severe with the delivery of ExoU/T, followed by delivery of ExoS/T/Y and ExoY alone. Vehicle controls, a strain that cannot produce a functional translocation pore (ΔPcrV) and a strain that produces a functional pore but a non‐catalytically active ExoY (ExoYK81M) produced no damage to the monolayers (Figure 1B). These data suggest that the ability to deliver active effectors is important to endothelial cell toxicity. Severity of primary infections were qualitatively ranked with delivery of ExoU/ExoT being more acutely toxic than the injection of ExoY or the formation of the translocation pore (Figure 1C).
T3SS delivery of effectors requires bacterial contact with target cells. Toxins other than the T3SS effectors may contribute to the pathology detected in endothelial cells. To address this possibility, we collected and filter‐sterilized supernatants from infected cultures. Confluent monolayers of PMVECs were then treated with bacteria‐free supernatants and incubated for 24 hours prior to imaging. T3SS‐competent bacterial infections produced supernatants that were disruptive to the integrity of the endothelial barrier including interendothelial gap formation and cell detachment. The ΔPcrV T3SS‐incompetent infection, vehicle, and ExoYK81M infection supernatants did not induce endothelial injury. The production of another bacterial toxin as the cause of pathology was eliminated as all strains, except the ExoS/T/Y producing strain, are isogenic (Figure 1D). Moreover, the use of supernatants from infected cultures demonstrated a different hierarchy of toxicity than the results from direct contact (Figure 1A,B). Our results suggest that an endothelial cellular component, perhaps released from cells during infection, may be contributing to secondary pathology.
3.2. Infection‐derived supernatants contain amyloids
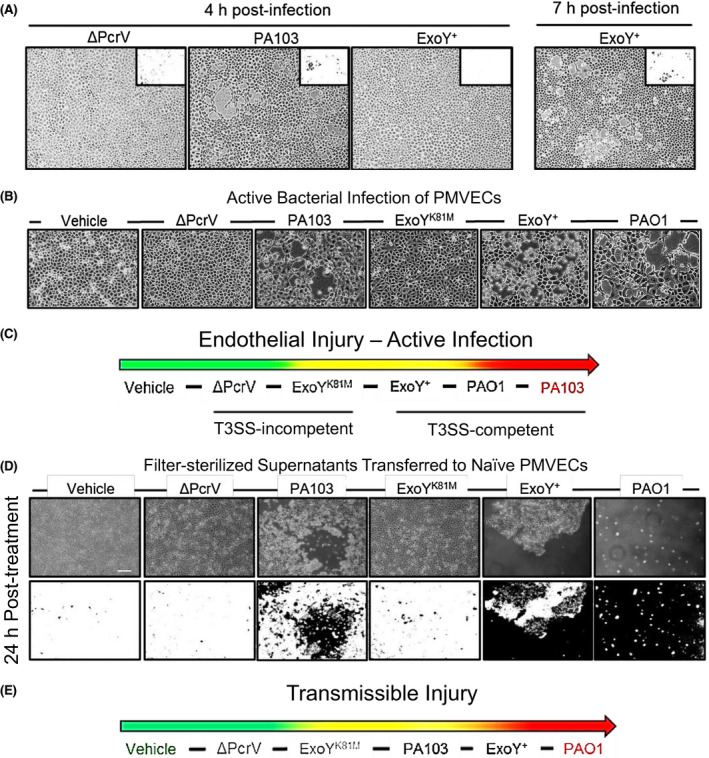
In previous studies, amyloid species were immunodetected in cell supernatants following ExoU/T and ExoY injection. 35 , 37 , 48 To determine whether cell supernatants contained amyloid constituents in our studies, we fixed and stained filter‐sterilized supernatants that were standardized to 0.5 µg protein/1 µL of supernatant with the amyloid differential Congo Red stain and imaged. Congo Red intercalates into the amyloid fold rendering amyloid species birefringent in response to polarized light. 53 , 54 The vehicle supernatant contained sparse, pinpoint species while the supernatant from ΔPcrV infected PMVECs produced small and punctate birefringent species. Larger puncta were associated with infection mediated by a strain able to inject ExoU/T. Strains capable of injecting ExoY into PMVECs produced large, birefringent aggregates (Figure 2A). These data suggest that amyloids are present and that physically distinct aggregates can be released based on the severity of the injury response.
Figure 2.
Endothelial supernatants contain amyloids. A, Congo Red stained vehicle supernatant contained sparse birefringent species suggesting constitutive amyloid release. Supernatant obtained from ΔPcrV infection exhibited numerous small, punctate amyloids. Birefringent puncta were distinctly larger in cell supernatants produced from infection with strains capable of injecting ExoU/T with the largest aggregates noted in supernatants from ExoY intoxicated cells. Scale bar = 200 µm. B, Thioflavin T (ThT) fluorescence (Ex: 425 nm; Em: 490 nm) was measured in supernatants obtained from uninfected cells and cells subjected to bacterial infection. ThT fluorescence was observed in supernatants from both vehicle control and ∆PcrV‐infections, but it was significantly higher in supernatants obtained from PA103 (ExoU/T)‐ and ExoY+ (only ExoY) infections. n ≥ 3 (three vehicle, 4‐6 infection supernatants) with six technical replicates per biological replicate; mean ± SEM; one‐way ANOVA with Tukey's post hoc; *P < .05, ***P < .001
Amyloids interact with ThT and elicit an increase in fluorescence, particularly with fibrillar amyloid species. 37 , 50 To determine whether supernatants contained ThT‐reactive amyloids, supernatant was standardized to protein (5 µg/mL) and probed with 0.49 µm ThT. Figure 2B demonstrates a positive ThT signal in the vehicle and T3SS‐incompetent ΔPcrV controls as well as supernatants collected from cells intoxicated with either ExoU/T or ExoY alone. Supernatants from ExoU/T and ExoY intoxicated cells exhibited a significantly higher ThT signal as compared to the vehicle control. We have previously shown that ∆PcrV‐infected cell supernatant is devoid of a ThT signal. 37 However, supernatant protein concentration was not standardized in our previous study design. Of note, non‐injurious ∆PcrV infection generates a much lower total protein concentration than infection with markedly damaging T3SS‐competent strains. The outcomes of our ThT experiments indicate that (1) amyloids are released both constitutively by PMVECs as well as in response to infection, (2) that these species are partially comprised of ThT‐reactive fibrillar species, and (3) T3SS effector intoxication of PMVECs elicits a significant increase in the extracellular concentration of these amyloids.
3.3. T3SS effector intoxication elicits virulent endothelial amyloids
Oligomers have been identified as the most cytotoxic conformation of amyloid species. 55 , 56 Intriguingly, intoxication of pulmonary endothelia with either T3SS effector ExoU or ExoY generates the release of transmissible, oligomeric tau during infection 35 and intoxication with ExoU/T generates oligomeric amyloids including Aβ. 37 However, whether clinical isolates of T3SS‐competent P aeruginosa are capable of (1) eliciting cytotoxic amyloids from the endothelium during infection; and (2) inducing transmissible injury in the absence of active bacterial infection has not been tested.
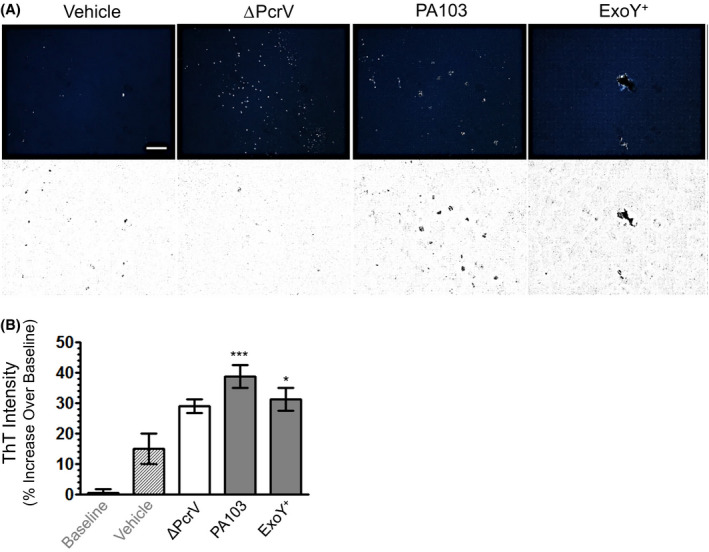
To address this point, supernatants from endothelial cultures infected with nosocomial pneumonia clinical isolate PA‐808 (ExoS/T/Y), reference strain PAO1 (ExoS/T/Y), PA103 (ExoU/T), ΔPcrV, ExoYK81M (catalytically inactive ExoY), and ExoY+ (ExoY alone) were collected, filter‐sterilized, and transferred to naïve endothelial monolayers. Treated cells were imaged over time to assess interendothelial gap formation. PMVECs infected with T3SS‐competent strains produced supernatants that were transmissibly injurious (Figure 3A). Again, supernatants generated through endothelial intoxication with ExoS/T/Y or ExoY alone were more acutely injurious than those produced through the cytosolic injection of ExoU/T (Figure 3A,C). Infection with ExoS/T/Y competent strains produced the most severe transmissible injury, marked by large interendothelial gaps at 16 hours and progressing to complete cell detachment by 24 hours (Figure 3C,D). Our findings suggest that PMVEC intoxication with ExoY alone, or in conjunction with ExoS/T, produces a more severe transmissible injury phenotype than ExoU/T intoxicated PMVECs. Moreover, these data indicate that infection with T3SS‐competent clinical isolates of P aeruginosa, in corroboration of observed outcomes with isogenic engineered strains, are capable of generating a transmissible injury that promotes endothelial barrier disruption.
Figure 3.
T3SS‐competent infection elicits transmissible and injurious amyloids from PMVECs. Naïve endothelial cell monolayers were treated with (A) vehicle or bacteria‐free infection supernatants generated from PMVEC intoxication with either ExoU/T (PA103), ExoY alone (ExoY+), and ExoS/T/Y (reference strain PAO1 and clinical isolate PA‐808). Neutralization of infection‐derived supernatants with anti‐pan oligomeric amyloid antibody (A11) or anti‐oligomeric tau antibody (T22) was used to determine the contribution of amyloids to the transmissible injury arising from the intoxication of PMVECs with T3SS effectors, as illustrated by representative images of the 24 hours time point in (B) with interendothelial gap quantitation presented in (G). The (‐) indicates infection‐derived supernatants were treated with antibodies targeting either oligomeric amyloid (A11) or oligomeric tau (T22), respectively, that were subsequently pulled‐down. Quantitative results from the imaging time course of untreated supernatants are plotted by (C) total gap area across all time points and (D) total gap area at the 24 hours time point. n ≥ 6 (6‐9) with ≥ three technical replicates for each individual experiment; mean ± SEM; two‐way ANOVA with Bonferroni post hoc. *P < .05, **P < .01, ***P < .001. The specificity of the A11 antibody for the oligomeric conformation of amyloids was validated against monomeric ((m) Aβ42), oligomeric ((o) Aβ42), and fibrillar ((f) Aβ42) conformations of recombinant rat Aβ42 in (E). The T22 oligomeric tau antibody was validated against a recombinant ladder of the six monomeric isoforms of neuronal tau in (F). Images of immunoblots are representative of ≥ three representative blots. G, Results of oligomeric amyloid neutralization studies are quantitatively expressed as percent gap area as normalized to corresponding untreated supernatants. n ≥ 6 (6‐9) with ≥ three technical replicates for each individual experiment. Kruskall‐Wallis with Dunn's post hoc; mean ± SD; *P < .05, **P < .01, ***P < .001. Scale bars = 200 µm
Next, to determine any potential contribution of oligomeric amyloids to the transmissible injury phenomenon we sought to utilize immunoprecipitation experiments using anti‐oligomeric amyloid and anti‐oligomeric tau antibodies, A11 and T22. 57 , 58 , 59 A11 (a polyclonal pan‐oligomeric amyloid antibody) and T22 (a monoclonal antibody specific for oligomeric tau) have been well‐characterized. Nonetheless, we validated these antibodies using recombinant rat Aβ42 and tau. Alzheimer's brain hippocampal lysate was used as a positive control for both A11 and T22.
To validate the specificity of A11, we generated a mix of monomeric and oligomeric conformations of recombinant Aβ42 as well as fibrillar Aβ42 alone. We resolved 200 pmol of each sample and probed each blot with either A11 or the MOAB‐2 antibody (recognizes unaggregated Aβ42 or Aβ40 oligomers). In corroboration of previous reports, A11 recognized mid to high molecular weight oligomers in both the Alzheimer's hippocampal lysate control and the sample of recombinant Aβ42 monomers and oligomers. However, A11 did not recognize either low molecular weight species consistent with monomeric Aβ (~4.5 kDa) in the lysate control or the monomeric recombinant rat Aβ42 (4.4 kDa) present in our mixed sample. Further, A11 did not recognize Aβ42 fibrils. Blots were then stripped and reprobed with the opposite antibody. In contrast to A11, MOAB‐2 identified the monomeric Aβ42 in our samples of mixed monomers and oligomers as well as high molecular weight oligomers. Further, whereas A11 did not recognize Aβ42 fibrils, an OC antibody specific for the fibril conformation of any amyloid readily recognized the fibrils still in the wells, as they were too large to pass into the gel (Figure 3E).
We investigated the specificity of T22 in a similar manner utilizing a recombinant tau ladder. The tau ladder is comprised of the 6 monomeric isotypes of neuronal tau. 60 We resolved 10 ng of the ladder and probed with either the TAU5 antibody, commonly used to detect total tau regardless of conformation, 60 or the T22 antibody. T22 showed robust immunoreactivity against the Alzheimer's hippocampal lysate control but it did not detect the monomeric isoforms of the tau ladder. TAU5, moreover, consistently recognized the monomeric tau ladder (Figure 3F).
Infection‐derived supernatants were treated with either an antibody recognizing oligomeric amyloids (A11) or an antibody specific for tau oligomers (T22) at a consistent antibody concentration. Supernatants were collected following the pull‐down of antibody reactive species, filter‐sterilized, and applied to naïve PMVEC monolayers along with untreated samples of each infection‐derived supernatant and imaged over a 24 hours time course to ascertain interendothelial disruption. Figure 3B is comprised of representative images of monolayers treated with A11 and T22 neutralized supernatants, respectively. Quantification of interendothelial gap area induced by exposure to neutralized samples vs untreated supernatants is presented in Figure 3G.
PMVEC exposure to either A11 or T22 neutralized vehicle supernatant was not injurious. A11 and T22 neutralization of infection supernatants collected from ExoU/T or ExoS/T/Y intoxicated cells was equally cytoprotective. Although A11 neutralization of supernatants produced exclusively from ExoY intoxication was cytoprotective, T22 neutralization completely ablated transmissible injury. Neutralization significantly attenuated transmissible injury secondary to ExoS/T/Y intoxication albeit we did not observe complete protection. The remaining cellular injury may be due to amyloid species that are not recognized by A11 or T22 antibodies, the production of oligomeric amyloids in excess of the binding capacity of the antibody or, potentially, other transmissible factors that have not yet been identified. (Figure 3E). Our results suggest that oligomeric amyloid species significantly contribute to the transmissible injury generated by endothelial infection with T3SS‐competent strains of P aeruginosa. Further, the efficacy of the oligomeric tau‐specific T22 antibody in eliminating transmissible injury secondary to intoxication with ExoY alone suggests that tau oligomers may fundamentally mediate this pathogenesis.
3.4. ExoY elicits amyloids that transmissibly ablate oxidative phosphorylation
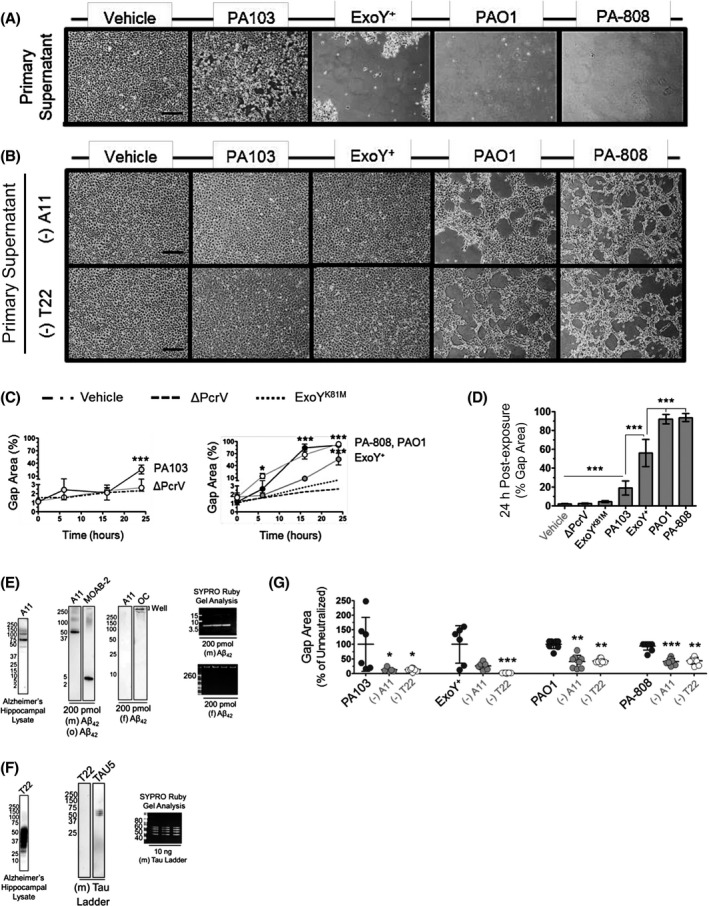
Previously, the injury of infection‐derived supernatants has been measured through the assessment of interendothelial gap formation and lactate dehydrogenase assay. 37 Here, we examined the potential of T3SS‐competent infection‐elicited amyloids to impact cell viability. Metabolically healthy cells capable of oxidative phosphorylation are able to reduce resozurin to resorufin to generate a commensurate increase in fluorescence with high fidelity. 61 , 62 Naïve endothelial monolayers were treated with either vehicle or bacteria‐free infection supernatants for 24 hours prior to assessing resozurin reduction induced fluorescence. Naïve cells exposed to the vehicle control and supernatants derived from infection with ΔPcrV, the ExoY analog lacking nucleotidyl cyclase activity (ExoYK81M), and ExoU/T intoxicated endothelium did not exhibit significantly attenuated oxidative phosphorylation. However, transfer of supernatants from ExoY intoxicated PMVECs to healthy, naïve cells abolished oxidative phosphorylation. Infection with strains that inject ExoS/T/Y into target cells engendered supernatants that either ablated or significantly attenuated oxidative phosphorylation in naïve cell populations exposed to it (Figure 4A). Our results here suggest that injection of either ExoS/T/Y or ExoY alone into PMVECs generates a transmissible toxicity that abrogates cell oxidative phosphorylation.
Figure 4.
ExoY intoxication generates transmissible amyloids that abolish oxidative phosphorylation. Naïve PMVECs were treated with either filter‐sterilized infection supernatants or a vehicle control and cellular oxidation‐reduction status was measured after 24 hours via resozurin. To determine whether amyloids generated secondary to T3SS effector intoxication contribute to the effect, oligomeric amyloids (A11 Ab) and oligomeric tau (T22 Ab) were neutralized in samples of each infection‐derived supernatant and then, transferred to naïve PMVECs. Fluorescence was plotted (A) as normalized to the vehicle control; and (B) as normalized to treatment with T3SS‐incompetent ΔPcrV‐supernatant. n ≥ 6 (6‐13); 3‐6 technical replicates each, One‐way ANOVA with Tukey's posthoc; mean ± SD; *P < .05, **P < .01, ***P < .001. C, Schematic qualitatively ranks the virulence of transmissible amyloid‐mediated injury arising secondary to P aeruginosa infection of lung endothelial cells
We then sought to clarify the potential contribution of oligomeric amyloid species to the suppression of metabolic efficiency following supernatant transfer to naïve cells. A11‐ or T22‐reactive oligomeric species were neutralized and pulled down from samples of each infection‐derived supernatant. Neutralized supernatants were then transferred to naïve endothelial cells. As compared to untreated supernatants, neutralized samples did not suppress oxidative phosphorylation in naïve cells. Notably, metabolic efficiency was protected via the neutralization of oligomeric amyloid and tau (Figure 4B). The virulence of each strain's infection‐induced transmissible injury is qualitatively ranked in Figure 4C. The outcomes of these studies indicate that transmissible oligomeric amyloids elicited from PMVECs secondary to T3SS effector intoxication negatively impact metabolic efficiency. Further, PMVEC intoxication with ExoY alone is sufficient to generate transmissible oligomeric amyloids that abolish oxidative phosphorylation in uninfected, previously healthy cells.
3.5. Infection‐induced amyloid cytotoxins are self‐propagating
Infection with P aeruginosa that express a patent T3SS with active effectors engenders a transmissible amyloid‐mediated cytotoxicity. Moreover, injection of ExoY alone or in combination with ExoS/T into PMVECs produces the most injurious and toxic transmissible phenotype. Previously, Balczon et al reported that ExoU/T intoxicated endothelium produced cytotoxic amyloids that were not only transmissible to naïve cells but could be further propagated from one cell population to another, across several passages, with little loss of virulence. 37 A self‐propagating protein, or prion, should be able to replicate in cell culture and pass from one naïve cell population to another while retaining its pathological characteristics. 63 , 64 , 65 However, the potential of ExoY, alone or in conjunction with other T3SS effectors, to generate self‐propagating amyloid cytotoxins has not been determined.
To address this gap, we serially passaged amyloids from filter‐sterilized infection supernatants across various populations of naïve PMVECs. In our passaging experiments, endothelial cells were briefly exposed to bacteria‐free infection supernatants prior to extensive rinsing and, finally, incubation in fresh media for 16‐20 hours prior to collecting, centrifuging, and filter‐sterilizing the resulting secondary (2º) supernatant. The process was sequentially repeated using the newly collected 2º supernatant to generate a tertiary (3º) supernatant from naïve cells (Figure 5A). Target naïve cell populations were comprised of PMVECs of different clone number, rat number, rat sex, and rat background.
Figure 5.
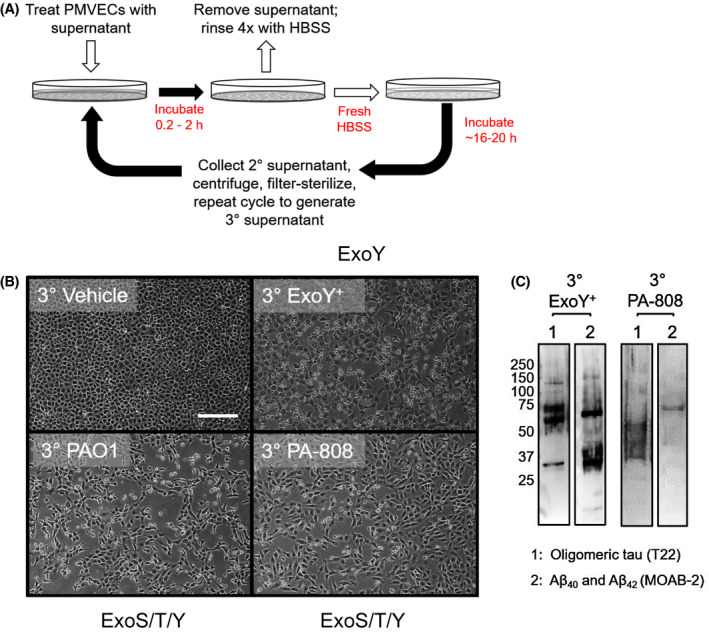
ExoY intoxication of endothelial cells elicits release of cytotoxic amyloid prions. A, Schematic of the passaging protocol to propagate cytotoxic amyloids from one naïve cell population to another. B, Images represent the second iteration (or production of a 3º cytotoxic supernatant) of amyloid passaging among various populations of PMVECs. All images represent at least three independent experiments. Naïve cells required variable exposure times to bacteria‐free supernatants to propagate cytotoxicity with supernatants obtained from ExoS/T/Y intoxicated cells requiring less time (45 minutes to 1.5 hours) than supernatants from cells injected with ExoY alone (2 hours). C, Immunoblotting reveals amyloid species in 3º cytotoxic supernatants. Blot was first probed with A11 (not shown), then T22, and finally MOAB‐2. Scale bars = 200 µm
Serially passaged vehicle supernatant was not injurious to naïve target cell populations. However, infection supernatants derived from ExoY or ExoS/T/Y intoxicated endothelium induced marked endothelial gap formation and cell lifting throughout the course of serial passaging (Figure 5B). Moreover, passaged ExoY or ExoS/T/Y generated amyloids were acutely cytotoxic. Naïve PMVECs exposed to passaged ExoY or ExoS/T/Y induced cytotoxic amyloids began to form interendothelial microgaps in as little as 45 minutes (ExoS/T/Y) up to 2 hours (ExoY alone) (data not shown). As such, naïve PMVECs required ~2 hours less exposure time to ExoY or ExoS/T/Y derived supernatants (data not shown) to initiate propagation than the 4 hours previously reported for ExoU/T. 37 We probed 3º supernatants resulting from ExoY and ExoS/T/Y induced self‐propagating cytotoxicities for unaggregated/oligomeric/fibrillar Aβ42 and Aβ40 (MOAB‐2 Ab) in addition to oligomeric tau (T22 Ab). Passaged supernatants contained both MOAB‐2 reactive Aβ species and oligomeric tau (Figure 5C). Our findings indicate that intoxication with ExoY alone, or in conjunction with ExoS/T, is sufficient to elicit self‐propagating, injurious amyloids from PMVECs.
3.6. T3SS‐incompetent infection elicits heat‐stable antimicrobial effector from PMVECs
We questioned why the endothelium would produce amyloids constitutively and in response to an infectious challenge (see Figure 2B). Accumulating evidence suggests that Aβ, specifically the oligomeric conformation of the Aβ42 variant, functions as a broad‐spectrum antimicrobial peptide. 42 , 44 , 45 , 66 In order to determine whether pulmonary endothelial amyloids might have a similar innate function, we began by testing infection‐derived supernatants via a standard Kirby‐Bauer disk diffusion assay. 51 However, many bacteria release antimicrobials such as bacteriocins to inhibit competing strains. 67 , 68 To control for this possibility, we used strain PA103 for the lawn, as it is isogenic to the majority of the strains we used to elicit endothelial amyloid release.
The zone of inhibition (ZOI) around disks treated with supernatant increased over time after the initial 24 hours measurement with no such increase observed for the HBSS or Gentamicin controls (Figure 6A). ZOI diameters around disks treated with supernatants obtained from infection with ΔPcrV or ExoYK81M, as well as supernatant from ExoU/T intoxicated PMVECs, notably increased over time. In accordance with CLSI standards, 61 ZOI diameter was measured across the area of inhibition at multiple points with the center of the disk serving as the point of rotation, or foci, and recording the greatest diameter measurement to the nearest mm. ZOIs ranged from circumferential/parabolic to undulate/jagged (Figure 6B). Non‐cytotoxic ΔPcrV infection‐derived supernatant induced a significantly greater ZOI than any other infection‐derived supernatant during the initial measurement, and over time, in the following rank order: ΔPcrV >> ExoU/T > ExoYK81M > ExoS/T/Y > ExoY alone. Further, supernatant derived from ExoU/T intoxicated endothelium displayed 2‐ to 3‐fold greater inhibition than supernatant from cells intoxicated with either ExoS/T/Y or ExoY alone at 72 hours. Supernatants generated from PMVEC intoxication with ExoY alone exhibited negligible antimicrobial activity with no increase in efficacy over time (Figure 6A). These data suggest that PMVEC infection with a strain incapable of injecting T3SS effectors into the cell elicits a compound with antimicrobial activity and, further, that T3SS effector intoxication, particularly with ExoY alone, significantly constrains the release of this antimicrobial.
Figure 6.
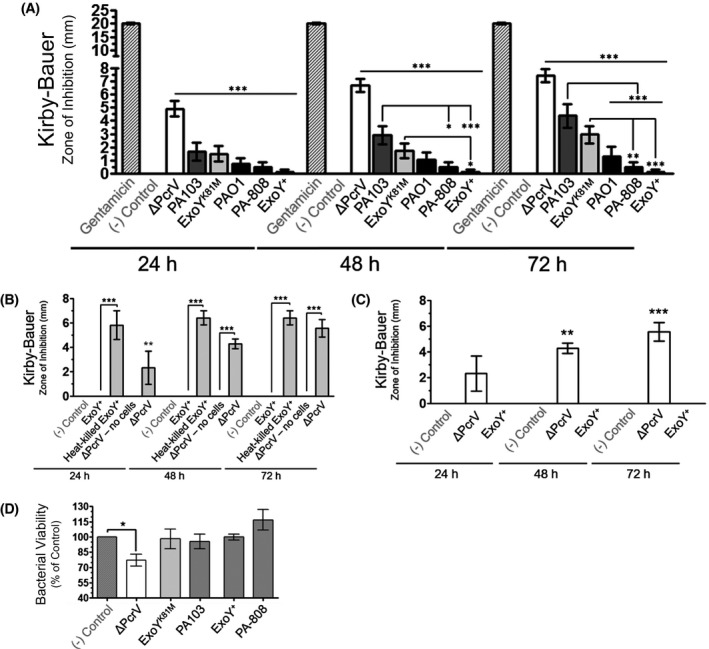
T3SS‐incompetent infection elicits an antimicrobial from PMVECs. A, Whereas infection with T3SS‐incompetent ΔPcrV produces amyloids that are not cytotoxic, they are distinctly and consistently antibacterial in a time‐dependent manner. n ≥ 7 (7‐34) with each independent experiment conducted in triplicate, repeated measures two‐way ANOVA with Bonferroni post hoc; mean ± SEM; *P < .05, **P < .01, ***P < .001. B, T3SS‐incompetent infection elicited endothelial antimicrobial production requires interaction between the bacterium and the host cell. Moreover, active injection of ExoY into host cells is required for the ablation of antimicrobial activity secondary to ExoY+ infection. n ≥ 3 with three technical replicates per each independent experiment, two‐way ANOVA with Bonferroni post hoc; mean ± SEM; *P < .05, **P < .01, ***P < .001. C, Boiling cell supernatants from ΔPcrV infection for 30 minutes followed by submersion in an ice bath does not diminish antimicrobial efficacy. n ≥ 4 with each independent experiment conducted in triplicate, repeated measures two‐way ANOVA with Bonferroni post hoc; mean ± SEM; *P < .05, **P < .01, ***P < .001. D, The potential of the endothelial antimicrobial to induce bacterial killing was ascertained by incubating supernatants with bacteria followed by serial dilution and plating experiments. n ≥ 4 with three technical replicates per biological replicate, one‐way ANOVA with Dunnet's multiple comparison post hoc; mean ± SEM; *P < .05
We questioned whether the ΔPcrV strain was secreting an antimicrobial that might contribute to the ZOI. Toward this point, we generated “no cell” supernatant by incubating full DMEM media overnight in a plate that did not contain any cells. The next day we removed media, rinsed the plate with HBSS, and “infected” these plates with ΔPcrV utilizing the same protocol that we use to conduct our usual cell infections. Supernatants obtained from “no cell” ΔPcrV infections did not exhibit antimicrobial activity (Figure 6B). Our results suggest that the antimicrobicity of supernatants obtained from infection with T3SS‐incompetent strain ΔPcrV is not a factor that is released by the bacteria independent of contact with PMVECs.
We also sought to determine whether the ablation of antimicrobial efficacy observed with supernatant obtained from cells intoxicated with ExoY alone might potentially be the result of another independent or confounding factor. To preclude this possibility, we performed infections of PMVECs with inactivated, or heat‐killed, P aeruginosa strain ExoY+ (injects ExoY only). Contrary to the results obtained from ExoY intoxicated PMVECs, infection supernatants produced from heat‐killed ExoY+ exhibited robust antimicrobial efficacy equivalent to that elicited with T3SS‐incompetent strain ΔPcrV infection (Figure 6B). The outcome of these experiments clarify that injection of ExoY into the host PMVEC is necessary to ablate the efficacy of the endothelial antimicrobial whereas endothelial contact with heat‐killed bacteria is sufficient to elicit antimicrobial release.
Amyloids are heat‐stable. To determine whether the antimicrobial compound elicited by T3SS‐incompetent infection is heat‐stable, we subjected samples of ΔPcrV infection‐derived cell supernatant to 30 minutes of boiling followed by immediate submersion in an ice bath to prevent potential refolding of heat denatured proteins. Boiled/iced samples of ΔPcrV infection‐derived supernatants were significantly antimicrobial over time, consistent to outcomes obtained with samples that did not undergo boiling (Figure 6C). Our data suggest that the antimicrobial elicited from PMVECs secondary to T3SS‐incompetent infection is highly heat‐stable.
The next question we addressed was whether the antimicrobial is capable of killing bacteria. We incubated either infection‐derived supernatants or HBSS negative control with bacteria for 3 hours prior to serial dilution and plating. Live bacteria generate colonies that can be counted to determine the number of viable bacteria, or colony forming units (CFUs), remaining after treatment with each supernatant. Supernatants generated from PMVECs intoxicated with ExoU/T, ExoY alone, or ExoS/T/Y did not negatively impact CFUs. Conversely, bacteria incubated with supernatant derived from infection of PMVECs with T3SS‐incompetent strain ΔPcrV significantly attenuated CFUs as compared to the negative control. (Figure 6D). These data suggest that endothelial infection in the absence of T3SS effector intoxication elicits a bactericidal antimicrobial.
3.7. Endothelial amyloids aggregate bacteria
Amyloids, particularly Aβ42, polymerizes into fibrils upon contact with bacterial surface epitopes and actively agglutinate bacteria prior to bactericidal activity. 45 , 69 Therefore, we questioned whether treating bacteria with infection‐derived supernatants would instigate the aggregation of bacteria. To test this idea, we grew lawns of bacteria on a solid media substrate and subjected the bacteria to infection‐elicited endothelial supernatant. Some bacteria, including Salmonella spp., Staphylococcus spp., Escherichia coli, and Pseudomonads, generate their own bacterial amyloids, 70 , 71 , 72 , 73 which could confound our experimental results. To control for this possibility, the amyloid differential Congo Red dye was incorporated into the media along with a counterstain at equal concentrations. If the target lawn expressed bacterial amyloid, then the differential Congo Red dye would be incorporated thereby producing a bright, scarlet red colony color. 74 Figure 7A illustrates a strain of S aureus plated on our YESCA Congo Red agar exhibiting the characteristic scarlet red color indicative of positive bacterial amyloid expression [Amyloid (+)] as well as a culture of our target strain, PA103, exhibiting the color of the agar and thereby indicating a lack of bacterial amyloid production [Amyloid (−)].
Figure 7.
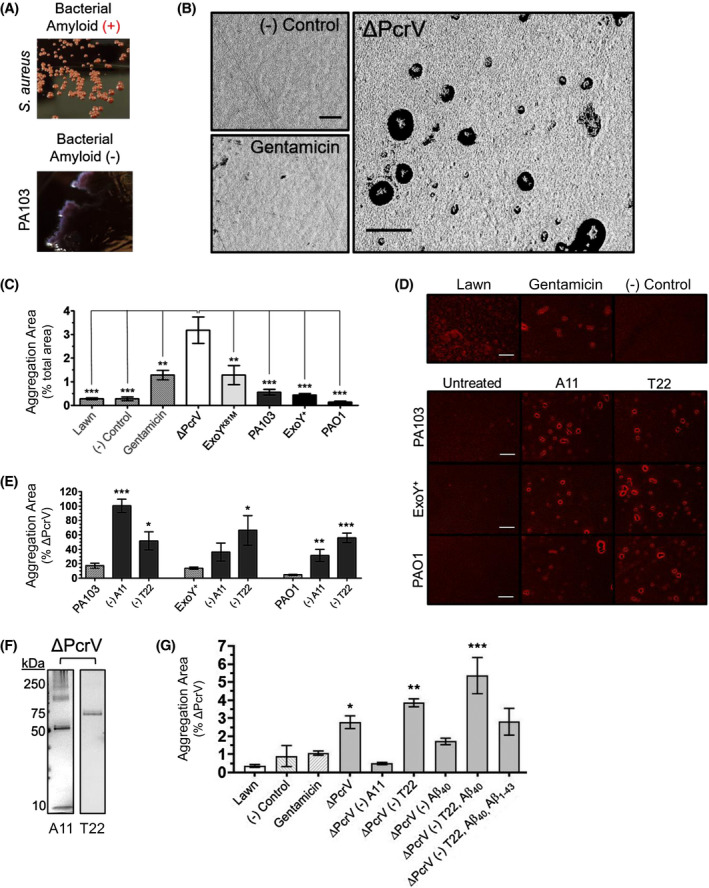
T3SS‐incompetent infection produces endothelial amyloids that aggregate bacteria on a solid substrate. Filter‐sterilized infection supernatants were applied to established lawns of P aeruginosa (strain PA103) to determine whether treatment would instigate the aggregation of target bacteria. A, Congo Red agar was used as the substrate to screen for potential confounding bacterial amyloid production, with example images of positive (S aureus; Amyloid (+)) and negative (PA103; Amyloid (‐)) bacterial amyloid production shown. High‐contrast binary masks of images of (B) negative HBSS control, positive gentamicin control, and ΔPcrV‐supernatant treatment. ΔPcrV infection supernatant initiated the formation of punctate aggregates. C, Quantification of lawn coalescence expressed as percent total aggregate area. n ≥ 4 (4‐7); one‐way ANOVA with Dunnet's post hoc; mean ± SEM; ***P < .001.; and (D) bright‐field images of lawns of P aeruginosa plated on Congo Red agar inoculated with controls (top row) and untreated infection supernatants vs A11 (pan‐amyloid oligomer) or T22 (oligomeric tau) neutralized samples of the same supernatant. E, Quantification of aggregation data from D was expressed as percent aggregation area induced by ΔPcrV‐supernatant. n ≥ 4 (4‐7); one‐way ANOVA with Bonferroni post hoc; mean ± SEM; *P < .05, **P < .01, ***P < .001. F, ΔPcrV infection supernatants probed for amyloids exhibit A11 and T22 immunoreactive bands. Images representative of ≥ four independent experiments. F, Anti‐amyloid antibodies were used to neutralize supernatants from ΔPcrV infected PMVECs either in standalone experiments (eg, A11, T22, and monoclonal Aβ40 antibody 11A50‐B10 independently) or sequentially (eg, (‐) T22, Aβ40: denotes antibody neutralization and pull‐down of captured antibody out of samples in series) to clarify potential contributions of different amyloid/Aβ/tau species to the observed antimicrobial effect. n ≥ 3 (3‐6); ≥ two technical replicates for each independent experiment; mean ± SEM; one‐way ANOVA with Dunnett's post hoc. *P < .05, **P < .01, ***P < .001. Scale bars = 200 µm
Supernatants were standardized by protein and applied to established lawns of P aeruginosa. High‐contrast binary masks (Figure 7B) and bright field images (Figure 7D) reveal the topology of both treated and untreated lawns of P aeruginosa on YESCA Congo Red agar. Treatment of bacterial lawns with endothelial supernatant from ΔPcrV infection induced the formation of compact, punctate bacterial aggregates, an effect that was not observed after treating lawns with endothelial supernatant from T3SS‐competent infections (Figure 7C). Thus, T3SS effectors elicit a change in endothelial amyloid function.
As neutralization using the A11 and T22 antibodies reduced/abolished T3SS effector induced transmissible cytotoxicity, we tested whether neutralization would rescue amyloid antimicrobial function. To do this, cytotoxic supernatant from ExoU/T, ExoS/T/Y, and ExoY intoxicated cells was neutralized with A11 and T22 antibodies and aggregation was tested. A11 and T22 neutralization significantly increased the aggregation area. Supernatant from ExoU/T intoxicated PMVECs exhibited a greater increase in aggregation area following A11 neutralization, whereas T22 neutralization appeared more effective in rescuing antimicrobial aggregation activity following ExoY and ExoS/T/Y intoxication (Figure 7D,E). These data suggest that T3SS intoxication elicits A11 and T22 reactive amyloids that suppress endothelial antimicrobial activity.
Outcomes of our studies to this point suggested that the endothelial antimicrobial released during T3SS‐incompetent infection could potentially be an antimicrobial amyloid. However, we had not previously detected amyloids in T3SS‐incompetent derived supernatants using volume controlled study designs. Here, we began to address this possibility using TCA precipitation to collect all of the protein from 1 mL of ΔPcrV infection‐derived supernatant and probing the sample with A11 and T22 antibodies. ΔPcrV infection‐derived supernatant was immunoreactive for both A11 and T22 (Figure 7F), suggesting that T3SS‐incompetent infection elicits endothelial amyloids partially comprised of oligomeric species.
To clarify the potential contribution of amyloid species to the antimicrobial aggregation activity of endothelial supernatants, we used antibody neutralization experiments and tested aggregation. A11 neutralization of supernatant from ΔPcrV infected cells ablated antimicrobial aggregation whereas T22 neutralization augmented aggregation efficacy (Figure 7G). As Aβ, particularly variant Aβ42, has been implicated as an antimicrobial peptide, we used a series of sequential neutralizations to assess the potential contribution of Aβ to the antimicrobial effect observed in our studies. In addition to T22, we also used the monoclonal antibody clone 11A50‐B10 to target Aβ40 75 and a polyclonal antibody to target the first 10 residues of the amino terminus of any variant of Aβ1‐43. 76 Neutralization of Aβ40 alone reduced the aggregation efficacy of ΔPcrV supernatant. However, neutralization and pull‐down of T22 immunoreactive species, followed by the same process with the 11A50‐B10 antibody in the same sample, markedly increased sample antimicrobial efficacy. Moreover, neutralizing the same ΔPcrV supernatant sample (having undergone sequential neutralization with the T22 and 11A50‐B10 antibodies, respectively) with the polyclonal antibody targeting any Aβ variant, reduced supernatant‐induced aggregation (Figure 7G). Results of our studies here indicate that infection elicited antimicrobial activity requires an A11 immunoreactive amyloid, Aβ contributes to this effect, and T22 immunoreactive tau inhibits antimicrobial efficacy. These data further suggest that the antimicrobial is comprised of multiple components and tau and Aβ40 may interact synergistically to inhibit efficacy.
3.8. Antimicrobial endothelial amyloids induce formation of annular aggregates
Although T3SS‐incompetent infection generates the release of endothelial amyloids that aggregate bacteria into punctate inclusions on solid substrate, we questioned whether these amyloids were capable of aggregating bacteria in suspension. To address this question, supernatants (0.5 µg/µL) were incubated with P aeruginosa at the mid‐log phase. Following incubation, aliquots of each treatment were stained with either Crystal Violet or the amyloid differential Congo Red stain and visualized through either bright field, to determine gross morphology (Figure 8A‐C), or cross‐polarized microscopy to assess potential amyloid birefringence (Figure 8D‐F). Bacteria incubated with HBSS negative control displayed little to no coalescence when stained with Crystal Violet (Figure 8A). In contrast, bacteria incubated with endothelial supernatants from ΔPcrV infection exhibited punctate areas of aggregation (Figure 8B). In addition to smaller straight and curved morphologies in Congo Red stained samples, we noted that Congophilic species frequently polymerized in circular, or annular, aggregate conformations (Figure 8C). Under polarized light, with the polarizer and analyzer in place, but not crossed, bacteria were faintly visualized in the background of a ΔPcrV‐supernatant treated sample (Figure 8D). However, as the polarizer is rotated 45°, crossing with the analyzer, the background darkens revealing only birefringent amyloids. Annular aggregates exhibit green‐yellow birefringence (Figure 8E,F). These results indicate that endothelial antimicrobial amyloids are capable of aggregating bacteria in suspension and corroborate their proclivity to form annular inclusions as also observed on solid agar substrate.
Figure 8.
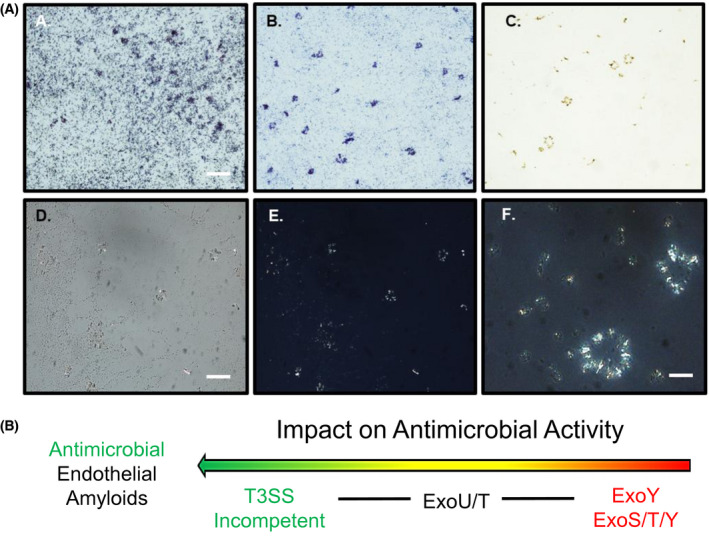
Endothelial amyloids aggregate bacteria in suspension. A, HBSS‐treated bacterial liquid culture stained with non‐differential Crystal Violet stain; B, ΔPcrV‐supernatant‐treated bacterial liquid culture stained with Crystal Violet exhibiting punctate bacterial aggregation; C, ΔPcrV‐supernatant‐treated bacterial culture stained with differential Congo Red stain displayed various morphologies of Congophilic species; D and E, Identical images of ΔPcrV‐supernatant‐treated bacterial liquid culture stained with Congo Red stain and assessed with (D) polarizer and analyzer in place but uncrossed; and (E) polarizer and analyzer crossed at 45°. F, Higher magnification image of annular aggregates. Scale bars = 100 µm (A and D) and 50 µm (F). Schematic illustrates the rank order of endothelial infection‐derived amyloids to exhibit antimicrobial activity. T3SS‐incompetent infection elicits endothelial amyloids with significant antimicrobial activity
4. DISCUSSION
Pathogens responsible for nosocomial pneumonia, including P aeruginosa, Klebsiella pneumonia, and Staphylococcus aureus, elicit lung endothelial production and release of amyloids including Aβ and tau. 37 , 48 These amyloids form oligomers and interact with one another in biological fluids during infection, and they can be retrieved from the bronchoalveolar lavage and cerebrospinal fluid of critically ill patients harboring infection. 37 , 48 Cytotoxic amyloids generated from endothelial infection with nosocomial pathogens are transmissible from cell‐to‐cell and may be propagated and enriched in cell culture without loss‐of‐function, thereby fulfilling the criteria of a prion or proteinopathy. 35 , 37 , 48 Furthermore, they have been hypothesized to contribute to morbidity and mortality during, and in the aftermath, of critical illness. 48 Despite the important implications to human health and disease, the molecular mechanisms underlying the generation of endothelial amyloids remain undetermined. Moreover, there is no current understanding as to why the pulmonary endothelium would produce amyloid species that could potentially give rise to a proteinopathy in the wake of bacterial infection. Here, we address both of these issues in studies examining P aeruginosa virulence. Novel outcomes of our studies indicate that: (1) Endothelial intoxication with P aeruginosa T3SS effectors instigates the release of self‐propagating cytotoxic amyloid prions; (2) these cytotoxic endothelial amyloids are comprised of A11‐ and T22‐immunoreactive species; (3) the composition of amyloids released from the endothelium secondary to PA103 and ExoY‐competent infection differ in function; and (4) infection with T3SS‐incompetent P aeruginosa induces the generation and release of lung endothelial amyloids that are both non‐cytotoxic and antimicrobial. This work implicates T3SS effectors, and ExoY in particular, as a molecular mechanism of inter‐kingdom communication, targeting amyloid function in innate immunity, and it reveals a previously unappreciated mechanism of immune surveillance (Figure 9).
Figure 9.
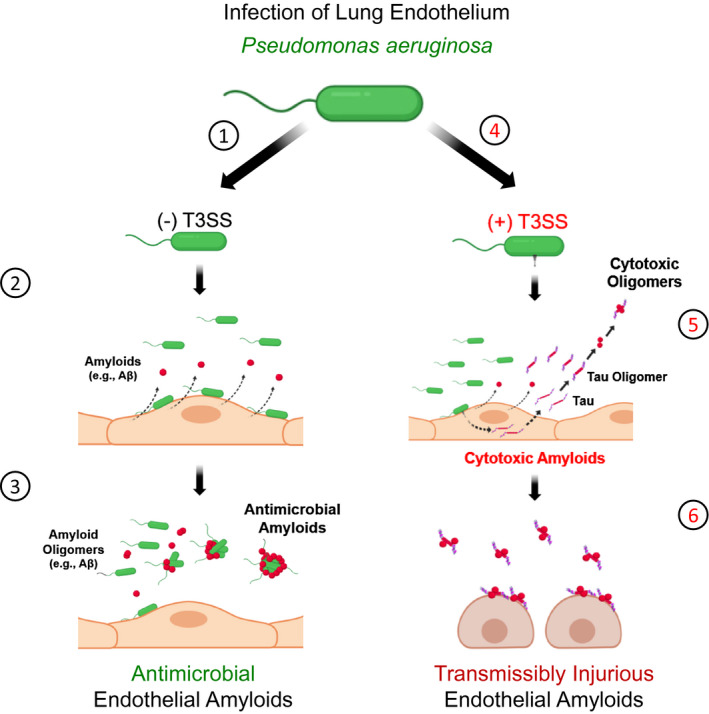
Infection elicits release of amyloids from endothelium that have antimicrobial or cytotoxic properties. Endothelial infection with T3SS incompetent P aeruginosa promotes the release of amyloids with antimicrobial properties, including Aβ. These antimicrobial properties include the aggregation of bacteria (see steps 1‐3). Endothelial infection with T3SS competent P aeruginosa, especially strains possessing ExoY, promote the release of transmissible amyloids with cytotoxic properties. These cytotoxic amyloids, partially comprised of oligomeric tau, are transmissible and propagate injury across multiple generations of naïve cells (see steps 4‐6). Oligomeric tau suppresses endothelial amyloid antimicrobial efficacy. Thus, T3SS effectors convert antimicrobial amyloids to cytotoxic species as part of the host‐pathogen interaction
ExoY was initially identified as an adenylate cyclase whose function required a mammalian cofactor. 24 Subsequent studies revealed ExoY to be a promiscuous nucleotidyl cyclase capable of generating both canonical and non‐canonical cyclic nucleotides. 17 , 25 , 27 , 36 In contrast to ExoU, an acutely cytolytic phospholipase, ExoY produces cell rounding and the ablation of cell viability without notable cell lysis. 31 Lung microvascular endothelial rounding corresponds with a breakdown of cellular adhesions leading to exudative edema in vivo; ExoY is an edema factor. 26 , 30 , 31 However, ExoY's effects are not transient in nature. We observed that ExoY+ instillation into the airways results in protracted vascular dysfunction, even a week after recovery from infection. 30 The mechanism for this effect was not immediately apparent, but in subsequent studies we found that ExoY intoxication induces the release of cytotoxic tau oligomers that promote edema in the alveolar‐capillary membrane. 35 In our present studies, we have identified that ExoY intoxication instigates the production and release of other oligomeric amyloids, including Aβ and tau, from endothelial cells. Importantly, both the ExoY+ mutant that possesses multiple copies of the ExoY gene and a clinical strain that possess a single copy of the ExoY gene along with ExoS and ExoT elicit cytotoxic amyloid production.
Oligomeric forms of amyloids can be cytotoxic, although there remains considerable controversy regarding how amyloid oligomers are generated and which of them exhibits cytotoxicity. 55 , 77 , 78 , 79 , 80 , 81 Aβ cytotoxicity depends upon the form of Aβ that is produced, that is, Aβ40 vs Aβ42 vs Aβ43, whether or not it is a soluble oligomer, and the extent to which it polymerizes into fibrillar species. 40 , 55 , 78 , 80 , 81 Soluble oligomers appear to be the most cytotoxic species, although how they are sensed by naïve cells and initiate cytotoxicity remain important areas of study. Post‐translational modification contributes significantly to the oligomerization process itself and to the relative amyloid cytotoxicity. 79 , 82 Both heparin sulfate and phosphorylation potentiate the cytotoxicity of Aβ. 83 , 84 Hyperphosphorylation is also a recognized mechanism of oligomeric tau formation. Hyperphosphorylated tau initially oligomerizes in a detergent soluble form that is cytotoxic, and then, develops into larger detergent insoluble aggregates that can progress to neurofibrillary tangles. 36 , 85 Oligomeric Aβ and tau have generally been considered to be discrete causes of cytotoxicity, yet recent studies indicate they interact to cause synergistic cytotoxicity. 86 , 87 How Aβ and tau interact and assemble is unknown. Here, we have identified that the neutralization of amyloid and tau oligomers from previously cytotoxic and non‐antimicrobial supernatants markedly attenuates cytotoxicity and promotes antimicrobial aggregation. Thus, our results are consistent with the idea that ExoY promotes oligomerization of a cytotoxic Aβ‐tau complex during the natural course of infection. The chemical nature of an Aβ‐tau interaction remains poorly understood as do the other species that contribute to the complex. Resolving the chemical nature of this Aβ‐tau interaction will be an important focus for future studies, including the identification of contributing amyloids and other potential molecular species. Better resolution of these issues will enable development of approaches to inactivate cytotoxic forms of the infection‐derived amyloid complex.
We questioned why endothelium would produce and release amyloids during P aeruginosa infection. Studies using a T3SS‐incompetent mutant provided insight into this question. Endothelium infected with P aeruginosa mutant ∆PcrV produced an amyloid signature that was not cytotoxic, but rather, was antimicrobial. It is notable that this amyloid pool was visible after either precipitation of all protein constituents in a given volume of supernatant or supernatant concentration (ie, protein‐controlled study designs) and it was not previously seen in volume‐controlled study designs. 37 The idea that amyloids have antimicrobial properties has been described for decades. 43 Amyloid species such as Aβ, prion protein, tau, alpha‐synuclein, and S100 have also been reported to be antimicrobial. 69 , 88 , 89 , 90 , 91 Recently, Moir and colleagues have shown assembled Aβ complexes have antimicrobial activity capable of killing bacteria (both Gram positive and Gram negative), viruses, and fungi. 45 , 66 , 69 Moreover, clinical trials using Aβ‐lowering drugs report infection as an important side effect. 43 There is currently no consensus regarding how antimicrobial forms of amyloids are generated and biodistributed, especially within the natural course of a disease. Aβ is constitutively found within the circulation although a function for it has not been ascribed. 92 Our present findings support the idea that Aβ has an important antimicrobial function at the alveolar‐capillary membrane and in the circulation, and suggests endothelium, by producing amyloids with antimicrobial properties, actively contributes to innate immunity. As we go forward, it will be important to learn whether endothelial antimicrobial amyloids are constitutively generated, and the extent to which they fulfill an essential immune surveillance function in the circulation.
The pulmonary endothelium releases amyloid species, including oligomeric forms of Aβ and tau, in response to P aeruginosa infection. Intriguingly, the function of these amyloids is modulated by T3SS effector intoxication of the endothelium. In the absence of T3SS competency, these endothelial amyloid species are non‐cytotoxic innate immune complexes. However, T3SS effector activity within the endothelium significantly impairs the innate antimicrobial function of endothelial amyloids while contributing to the conversion of these amyloid complexes into a cytotoxic form. The cytotoxic amyloid conformation is both self‐replicating and transmissible among naïve cells, that is, a prion. It is notable that endothelial amyloids such as Aβ and tau can either be cytotoxic to the mammalian host or antimicrobial. Therefore, P aeruginosa T3SS effectors contribute to the conversion of antimicrobial amyloids to cytotoxic forms. Although other T3SS effectors appear capable of promoting this functional conversion to some degree, ExoY alone and as a component of the T3SS arsenal exhibits the most profound effect. In this work, we did not address the extent to which ExoU, ExoS, or ExoT independently contribute to, or induce, this phenomenon. This will be a focus of our future studies along with clarifying the mechanism governing the antimicrobial‐to‐cytotoxic amyloid conversion. Albeit, ExoY intoxication alone, or in synergy with ExoS and/or ExoT, is sufficient to ablate innate amyloid antimicrobicity and elicit a markedly cytotoxic pool of self‐propagating amyloids. Here, we have identified a unique function for P aeruginosa T3SS effectors in the subversion of innate host defense to promote an endothelial‐derived proteinopathy—a form of nosocomial infection‐induced “friendly fire”—that may play an important role in the poor outcomes associated with survivors of VAP.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
S. Voth and T. Stevens designed research with input from D. W. Frank, N. Housley, and J. Audia; S. Voth., M. Gwin, R. Balczon, S. Piechocki, K. Madera, A. Simmons, and M. Crawford performed research; C. M. Francis contributed analytical tools and results of the analyses, D. W. Frank, J. P. Audia, J.‐F. Pittet, B. M. Wagener, and S. Moser contributed reagents and analytical results. S. Voth and T. Stevens analyzed data and wrote the paper; and all authors contributed to revising and editing the paper and approving the final manuscript.
ACKNOWLEDGMENTS
This work was funded by HL140182 (RB, TS, J‐FP, BMW), HL66299, HL135003 (to TS and RB), HL60024 (to TS), HL118334 (JPA), and HL136869 (CMF). SV and MG are predoctoral fellows (NIH 1F31HL147512‐01 and NIH T32 HL076125, SV) and (AHA 19PRE34380166, MG).
Voth S, Gwin M, Francis CM, et al. Virulent Pseudomonas aeruginosa infection converts antimicrobial amyloids into cytotoxic prions. The FASEB Journal. 2020;34:9156–9179. 10.1096/fj.202000051RRR
Kayla Madera and Autumn Simmons contributed equally to this study.
REFERENCES
- 1. Kollef MH, Chastre J, Fagon J‐Y, et al. Global prospective epidemiologic and surveillance study of ventilator‐associated pneumonia due to Pseudomonas aeruginosa . Crit Care Med. 2014;42:2178‐2187. [DOI] [PubMed] [Google Scholar]
- 2. Marden JN, Diaz MR, Walton WG, et al. An unusual CsrA family member operates in series with RsmA to amplify posttranscriptional responses in Pseudomonas aeruginosa . Proc Natl Acad Sci U S A. 2013;11:15055‐15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Intile PJ, Diaz MR, Urbanowski ML, Wolfgang MC, Yahr TL. The AlgZR two‐component system recalibrates the RsmAYZ posttranscriptional regulatory system to inhibit expression of the Pseudomonas aeruginosa type III secretion system. J Bacteriol. 2014;196:357‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Danin P‐E, Girou E, Legrand P, et al. Description and microbiology of endotracheal tube biofilm in mechanically ventilated subjects. Respir Care. 2015;60:21‐29. [DOI] [PubMed] [Google Scholar]
- 5. Gil‐Perotin S, Ramirez P, Marti V, et al. Implications of endotracheal tube biofilm in ventilator‐associated pneumonia response: a state of concept. Crit Care. 2012;16:R93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodrigues ME, Lopes SP, Pereira CR, et al. Polymicrobial ventilator‐associated pneumonia: fighting in vitro Candida albicans‐Pseudomonas aeruginosa biofilms with antifungal‐antibacterial combination therapy. PLoS ONE. 2017;12:e0170433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramírez‐Estrada S, Borgatta B, Rello J Pseudomonas aeruginosa ventilator‐associated pneumonia management. Infect Drug Resist. 2016;9:7‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsay T‐B, Jiang Y‐Z, Hsu C‐M, Chen L‐W Pseudomonas aeruginosa colonization enhances ventilator‐associated pneumonia‐induced lung injury. Respir Res. 2016;17:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melsen WG, Rovers MM, Groenwold RHH, et al. Attributable mortality of ventilator‐associated pneumonia: a meta‐analysis of individual patient data from randomised prevention studies. Lancet Infect Dis. 2013;13:665‐671. [DOI] [PubMed] [Google Scholar]
- 10. Tumbarello M, De Pascale G, Trecarichi EM, et al. Clinical outcomes of Pseudomonas aeruginosa pneumonia in intensive care unit patients. Intensive Care Med. 2013;39:682‐692. [DOI] [PubMed] [Google Scholar]
- 11. Mikkelsen ME, Christie JD, Lanken PN, et al. The adult respiratory distress syndrome cognitive outcomes study: long‐term neuropsychological function in survivors of acute lung injury. Am J Respir Crit Care Med. 2012;185:1307‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ibañez J, Riera M, Amezaga R, et al. Long‐term mortality after pneumonia in cardiac surgery patients: a propensity‐matched analysis. J Intensive Care Med. 2016;31:34‐40. [DOI] [PubMed] [Google Scholar]
- 13. Karhu J, Ala‐Kokko TI, Ylipalosaari P, Ohtonen P, Laurila JJ, Syrjälä H. Hospital and long‐term outcomes of ICU‐treated severe community‐ and hospital‐acquired, and ventilator‐associated pneumonia patients. Acta Anaesthesiol Scand. 2011;55:1254‐1260. [DOI] [PubMed] [Google Scholar]
- 14. Dick A, Liu H, Zwanziger J, et al. Long‐term survival and healthcare utilization outcomes attributable to sepsis and pneumonia. BMC Health Serv Res. 2012;12:432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vallis AJ, Finck‐Barbançon V, Yahr TL, Frank DW. Biological effects of Pseudomonas aeruginosa type III‐secreted proteins on CHO cells. Infect Immun. 1999;67:2040‐2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frank DW, Vallis A, Wiener‐Kronish JP, et al. Generation and characterization of a protective monoclonal antibody to Pseudomonas aeruginosa PcrV. J Infect Dis. 2002;186:64‐73. [DOI] [PubMed] [Google Scholar]
- 17. Kloth C, Schirmer B, Munder A, Stelzer T, Rothschuh J, Seifert R. The role of Pseudomonas aeruginosa ExoY in an acute mouse lung infection model. Toxins. 2018;10:E185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol. 2009;7:654‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shaver CM, Hauser AR. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect Immun. 2004;72:6969‐6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sato H, Frank DW. ExoU is a potent intracellular phospholipase. Mol Microbiol. 2004;53:1279‐1290. [DOI] [PubMed] [Google Scholar]
- 21. Finck‐Barbançon V, Goranson J, Zhu L, et al. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol. 1997;25:547‐557. [DOI] [PubMed] [Google Scholar]
- 22. Feltman H, Schulert G, Khan S, Jain M, Peterson L, Hauser AR. Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa . Microbiology. 2001;147:2659‐2669. [DOI] [PubMed] [Google Scholar]
- 23. Azimi S, Kafil HS, Baghi HB, et al. Presence of ExoY, ExoS, ExoU and ExoT genes, antibiotic resistance and biofilm production among Pseudomonas aeruginosa isolates in Northwest Iran. GMS Hyg Infect Control. 2016;11:Doc04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci U S A. 1998;95:13899‐13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morrow KA, Seifert R, Kaever V, et al. Heterogeneity of pulmonary endothelial cyclic nucleotide response to Pseudomonas aeruginosa ExoY infection. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1199‐L1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morrow KA, Frank DW, Balczon R, Stevens T. The Pseudomonas aeruginosa Exoenzyme Y: a promiscuous nucleotidyl cyclase edema factor and virulence determinant In: Seifert R, ed. Non‐canonical Cyclic Nucleotides. , Vol. 238, Handbook of Experimental Pharmacology Cham, Switzerland: Springer; 2017:67‐85. 10.1007/164_2016_5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beckert U, Wolter S, Hartwig C, et al. ExoY from Pseudomonas aeruginosa is a nucleotidyl cyclase with preference for cGMP and cUMP formation. Biochem Biophys Res Commun. 2014;450:870‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beckert U, Grundmann M, Wolter S, et al. cNMP‐AMs mimic and dissect bacterial nucleotidyl cyclase toxin effects. Biochem Biophys Res Commun. 2014;451:497‐502. [DOI] [PubMed] [Google Scholar]
- 29. Seifert R. cCMP and cUMP across the tree of life: from cCMP and cUMP generators to cCMP‐ and cUMP‐regulated cell functions In: Seifert R, ed. Non‐canonical Cyclic Nucleotides. Vol. 238, Handbook of Experimental Pharmacology Cham, Switzerland: Springer; 2016:3‐23. 10.1007/164_2016_5005. [DOI] [PubMed] [Google Scholar]
- 30. Stevens TC, Ochoa CD, Morrow KA, et al. The Pseudomonas aeruginosa Exoenzyme Y impairs endothelial cell proliferation and vascular repair following lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306:L915‐L924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sayner SL, Frank DW, King J, Chen H, VandeWaa J, Stevens T. Paradoxical cAMP‐induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ Res. 2004;95:196‐203. [DOI] [PubMed] [Google Scholar]
- 32. Benson MA, Schmalzer KM, Frank DW. A sensitive fluorescence‐based assay for the detection of ExoU‐mediated PLA(2) activity. Clin Chim Acta. 2010;411:190‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jeon J, Kim Y‐J, Shin H, Ha U‐H. T3SS effector ExoY reduces inflammasome‐related responses by suppressing bacterial motility and delaying activation of NF‐κB and caspase‐1. FEBS J. 2017;284:3392‐3403. [DOI] [PubMed] [Google Scholar]
- 34. He C, Zhou Y, Liu F, et al. Bacterial nucleotidyl cyclase inhibits the host innate immune response by suppressing TAK1 activation. Infect Immun. 2017;85:e00239‐17 10.1128/IAI.00239-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morrow KA, Ochoa CD, Balczon R, et al. Pseudomonas aeruginosa Exoenzymes U and Y induce a transmissible endothelial proteinopathy. Am J Physiol Lung Cell Mol Physiol. 2016;310:L337‐L353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ochoa CD, Alexeyev M, Pastukh V, Balczon R, Stevens T Pseudomonas aeruginosa Exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability. J Biol Chem. 2012;287:25407‐25418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balczon R, Morrow KA, Zhou C, et al. Pseudomonas aeruginosa infection liberates transmissible, cytotoxic prion amyloids. FASEB J. 2017;31:2785‐2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen G‐F, Xu T‐H, Yan Y, et al. Amyloid beta: structure, biology and structure‐based therapeutic development. Acta Pharmaco Sin. 2017;38:1205‐1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pauwels K, Williams TL, Morris KL, et al. Structural basis for increased toxicity of pathological Aβ42:Aβ40 ratios in Alzheimer disease. J Biol Chem. 2012;287:5650‐5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luo J, Wärmländer SKTS, Gräslund A, Abrahams JP. Cross‐interactions between the Alzheimer disease Amyloid‐β peptide and other amyloid proteins: a further aspect of the amyloid cascade hypothesis. J Biol Chem. 2016;291:16485‐16493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spitzer P, Condic M, Herrmann M, et al. Amyloidogenic amyloid‐β‐peptide variants induce microbial agglutination and exert antimicrobial activity. Sci Rep. 2016;6:32228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gosztyla ML, Brothers HM, Robinson SR. Alzheimer's Amyloid‐β is an antimicrobial peptide: a review of the evidence. J Alzheimers Dis. 2018;62:1495‐1506. [DOI] [PubMed] [Google Scholar]
- 44. Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer's disease‐associated amyloid beta‐protein is an antimicrobial peptide. PLoS ONE. 2010;5:e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar DKV, Choi SH, Washicosky KJ, et al. Amyloid‐β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. 2016;8:340ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miklossy J. Bacterial amyloid and DNA are important constituents of senile plaques: further evidence of the spirochetal and biofilm nature of senile plaques. J Alzheimers Dis. 2016;53:1459‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shima K, Kuhlenbäumer G, Rupp J Chlamydia pneumoniae infection and Alzheimer's disease: a connection to remember? Med Microbiol Immunol. 2010;199:283‐289. [DOI] [PubMed] [Google Scholar]
- 48. Lin MT, Balczon R, Pittet J‐F, et al. Nosocomial pneumonia elicits an endothelial proteinopathy: evidence for a source of neurotoxic amyloids in critically ill patients. Am J Respir Crit Care Med. 2018;198(12):1575‐1578. 10.1164/rccm.201801-0060LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leber AL, ed., Clinical Microbiology Procedures Handbook. 4th ed Washington, DC: American Society of Microbiology; 2016. 10.1128/9781555818814. [DOI] [Google Scholar]
- 50. Xue C, Lin TY, Chang D, Guo Z. Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R Soc Open Sci. 2017;4:160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Clinical Laboratory and Standards Institute . CLSI supplement M100 In: Jenkins CEM, Leirer KL, Martin L, eds. Performance Standards for Antimicrobial Susceptibility Testing, 29th ed Wayne, PA: Clinical and Laboratory Standards Institute; 2019. [Google Scholar]
- 52. Balczon R, Francis M, Leavesley S, Stevens T. Methods for detecting cytotoxic amyloids following infection of pulmonary endothelial cells by Pseudomonas aeruginosa . J Vis Exp. 2018;137:57447 10.3791/57447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Howie AJ, Brewer DB, Howell D, Jones AP. Physical basis of colors seen in Congo Red‐stained amyloid in polarized light. Lab Invest. 2008;88:232‐242. [DOI] [PubMed] [Google Scholar]
- 54. Prusiner SB, McKinley MP, Bowman KA, et al. Scrapie prions aggregate to form amyloid‐like birefringent rods. Cell. 1983;35:349‐358. [DOI] [PubMed] [Google Scholar]
- 55. Sengupta U, Nilson AN, Kayed R. The role of Amyloid‐β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. 2016;6:42‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ow S‐Y, Dunstan DE. A brief overview of amyloids and Alzheimer's disease. Protein Sci. 2014;23:1315‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486‐489. [DOI] [PubMed] [Google Scholar]
- 58. Glabe CG. Conformation‐dependent antibodies target diseases of protein misfolding. Trends Biochem Sci. 2004;29:542‐547. [DOI] [PubMed] [Google Scholar]
- 59. Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 2012;26:1946‐1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ercan E, Eid S, Weber C, et al. A validated antibody panel for the characterization of tau post‐translational modifications. Mol Neurodegener. 2017;12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rampersad SN. Multiple applications of Alamar Blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors. 2012;12:12347‐12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hamid R, Rotshteyn Y, Rabadi L, Parikh R, Bullock P. Comparison of Alamar Blue and MTT assays for high through‐put screening. Toxicol In Vitro. 2004;18:703‐710. [DOI] [PubMed] [Google Scholar]
- 63. Giles K, Woerman AL, Berry DB, Prusiner SB. Bioassays and inactivation of prions. Cold Spring Harb Perspect Biol. 2017;9:a023499 10.1101/cshperspect.a023499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aoyagi A, Condello C, Stöhr J, et al. Aβ and tau prion‐like activities decline with longevity in the Alzheimer's disease human brain. Sci Transl Med. 2019;11:eaat8462 10.1126/scitranslmed.aat8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, et al. Alzheimer's disease‐associated β‐amyloid is rapidly seeded by Herpesviridae to protect against brain infection. Neuron. 2018;99:56‐63.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hetz C, Bono MR, Barros LF, Lagos R. Microcin E492, a channel‐forming bacteriocin from Klebsiella pneumoniae, induces apoptosis in some human cell lines. Proc Natl Acad Sci U S A. 2002;99:2696‐2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ming L, Zhang Q, Yang L, Huang J‐A. Comparison of antibacterial effects between antimicrobial peptide and bacteriocins isolated from Lactobacillus plantarum on three common pathogenic bacteria. Int J Clin Exp Med. 2015;8:5806‐5811. [PMC free article] [PubMed] [Google Scholar]
- 69. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement. 2018;14:1602‐1614. [DOI] [PubMed] [Google Scholar]
- 70. Romero D, Kolter R. Functional amyloids in bacteria. Int Microbiol. 2004;17:65‐73. [DOI] [PubMed] [Google Scholar]
- 71. Zhou Y, Smith DR, Hufnagel DA, Chapman MR. Experimental manipulation of the microbial functional amyloid called curli. Methods Mol Biol. 2013;966:53‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012;8:e1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dueholm MS, Søndergaard MT, Nilsson M, et al. Expression of Fap amyloids in Pseudomonas aeruginosa, P fluorescens, and P putida results in aggregation and increased biofilm formation. Microbiologyopen. 2013;2:365‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Reichhardt C, Jacobson AN, Maher MC, et al. Congo Red interactions with curli‐producing E coli and native curli amyloid fibers. PLoS ONE. 2015;10:e0140388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Portelius E, Westman‐Brinkmalm A, Zetterberg H, Blennow K. Determination of beta‐amyloid peptide signatures in cerebrospinal fluid using immunoprecipitation‐mass spectrometry. J Proteome Res. 2006;5:1010‐1016. [DOI] [PubMed] [Google Scholar]
- 76. He W, Barrow CJ. The A beta 3‐pyroglutamyl and 11‐pyroglutamyl peptides found in senile plaque have greater beta‐sheet forming and aggregation propensities in vitro than full‐length A beta. Biochemistry. 1999;38:10871‐10877. [DOI] [PubMed] [Google Scholar]
- 77. Castillo‐Carranza DL, Nilson AN, Van Skike CE, et al. Cerebral microvascular accumulation of tau oligomers in Alzheimer's disease and related tauopathies. Aging Dis. 2017;8:257‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mroczko B, Groblewska M, Litman‐Zawadzka A, Kornhuber J, Lewczuk P. Amyloid β oligomers (AβOs) in Alzheimer's disease. J Neural Transm. 2018;125:177‐191. [DOI] [PubMed] [Google Scholar]
- 79. Mietelska‐Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G. Tau protein modifications and interactions: their role in function and dysfunction. Int J Mol Sci. 2014;15:4671‐4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Forloni G, Balducci C. Alzheimer's disease, oligomers, and inflammation. J Alzheimers Dis. 2018;62:1261‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cowan CM, Quraishe S, Mudher A. What is the pathological significance of tau oligomers? Biochem Soc Trans. 2012;40(4):693‐697. [DOI] [PubMed] [Google Scholar]
- 82. Grochowska KM, Yuanxiang P, Bär J, et al. Posttranslational modification impact on the mechanism by which amyloid‐β induces synaptic dysfunction. EMBO Rep. 2017;18:962‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nguyen K, Rabenstein DL. Interaction of the heparin‐binding consensus sequence of β‐amyloid peptides with heparin and heparin‐derived oligosaccharides. J Phys Chem B. 2016;120:2187‐2197. [DOI] [PubMed] [Google Scholar]
- 84. Kumar S, Walter J. Phosphorylation of amyloid beta (Aβ) peptides—a trigger for formation of toxic aggregates in Alzheimer's disease. Aging. 2011;3:803‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Alavi Naini SM, Soussi‐Yanicostas N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev. 2015:151979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pascoal TA, Mathotaarachchi S, Shin M, et al. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimers Dement. 2017;13:644‐653. [DOI] [PubMed] [Google Scholar]
- 87. Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to Beta ‐Amyloid‐induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364‐6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kobayashi N, Masuda J, Kudoh J, Shimizu N, Yoshida T. Binding sites on tau proteins as components for antimicrobial peptides. Biocontrol Sci. 2008;13:49‐56. [DOI] [PubMed] [Google Scholar]
- 89. Park S‐C, Moon JC, Shin SY, et al. Functional characterization of alpha‐synuclein protein with antimicrobial activity. Biochem Biophys Res Commun. 2016;47:924‐928. [DOI] [PubMed] [Google Scholar]
- 90. Lathe R, Darlix J‐L. Prion protein PRNP: a new player in innate immunity? The aβ connection. J Alzheimers Dis Rep. 2017;1:263‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Realegeno S, Kelly‐Scumpia KM, Dang AT, et al. S100A12 is part of the antimicrobial network against Mycobacterium leprae in human macrophages. PLoS Pathog. 2016;12:e1005705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid‐beta peptide across the blood‐brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2009;8:16‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]