

Summary
There are a multitude of resistance strategies that microbes can apply to avoid inhibition by antimicrobials. One of these strategies is the enzymatic modification of the antibiotic, in a process generally termed inactivation. Furthermore, some microorganisms may not be limited to the mere inactivation of the antimicrobial compounds. They can continue by further enzymatic degradation of the compounds' carbon backbone, taking nutritional and energetic advantage of the former antibiotic. This driving force to harness an additional food source in a complex environment adds another level of complexity to the reasonably well‐understood process of antibiotic resistance proliferation on a single cell level: It brings bioprotection into play at the level of microbial community. Despite the possible implications of a resistant community in a host and a lurking antibiotic failure, knowledge of degradation pathways of antibiotics and their connections is scarce. Currently, it is limited to only a few families of antibiotics (e.g. β‐lactams and sulfonamides). In this article, we discuss the fluctuating nature of the relationship between antibiotic resistance and the biodegradation of antibiotics. This distinction mainly depends on the genetic background of the microbe, as general resistance genes can be recruited to function in a biodegradation pathway.
Introduction
In the 21st century, antibiotics provide an ambivalent feeling of safety. On one hand, they constitute an effective weapon against many infectious diseases in human and veterinary medicine; on the other hand, there is the seemingly insurmountable problem of an onset of pathogenic microorganisms resistant to most antibiotics (Davies and Davies, 2010). This onset triggered increased efforts to investigate the determinants of antibiotic resistance development and propagation as well as the complex mechanisms involved in these processes (Vaz‐Moreira et al., 2014; Berendonk et al., 2015; Blair et al., 2015; Crofts et al., 2017). An antibiotic resistance gene (ARG) has been defined as a determinant, which gives a microbe the ability to resist certain antibiotics and which results in increased vulnerability when this specific gene is missing (Martínez et al., 2015). ARGs can provide resistance by a number of mechanisms. Antibiotics can be transported out of the cell via efflux pump systems, as with fluoroquinolone resistance mechanisms (Munita and Arias, 2016). Moreover, resistance genes can encode for isozymes, which are insensitive to inhibition, as in the case of sulfonamides (Wise Jr. and Abou‐Donia, 1975), while they are also able to overexpress targets that saturate the free antibiotic molecules, as it has been observed for cases resistant against the tandem use of sulfonamides‐trimethoprim. Yet another aspect is the modification of the antibiotic itself by enzymes, which change its structure to the extent that it is no longer active, for example chloramphenicol and β‐lactams (Munita and Arias, 2016). In addition to protection from antimicrobials via ARGs, decreased susceptibility may also be conferred by preventing the antimicrobial compound from entering the cell by changes in cell wall permeability as the result of a genetic mutation, which has been reported for β‐lactams (Munita and Arias, 2016).
There is an urgent need better to understand how new antibiotic resistances can be developed and disseminated. To this end, it is necessary to consider that there are two distinct milieus in which new antibiotic‐resistant bacteria can be generated: the environment and the various human microbiota (skin, gut, etc.). In fact, antibiotic treatments can lead to an alteration of the resistome with new prevalent ARGs becoming accessible to potentially pathogenic microorganisms (Sommer and Dantas, 2011). As the human microbiota are not hermetically closed systems, there is a possibility to exchange genetic information with other environments, making them potential reservoirs of ARGs (Pal et al., 2016). Nevertheless, distinctly different strategies have been found to resist the same antibiotic in different environments. Indeed, resistomes from different compartments (human gut, soil) can contain unrelated strategies to counteract the same antimicrobial. This has been shown in the case of tetracycline resistance, where soil bacteria mainly resist through efflux pumps while in the human gut, almost the only mechanisms found are the ribosomal protection (Gibson et al., 2015).
The mechanisms encompassed by the enzymatic inactivation of antimicrobials represent a crucial link between the mere ability to circumvent growth inhibition and the capacity to acquire a nutritional and energy advantage from the antibiotic. These mechanisms can further be distinguished based on the microorganism's genetic resources to chemically modify, cleave or mineralize the antibiotics. A mechanism would be categorized as biotransformation or primary biodegradation when the modification or the partial degradation of the antibiotic target finally result in the accumulation of biotransformation products by the resistant organism. Conversely, ultimate biodegradation pertains to the degradation process into simple inorganic molecules, with which the degrading organism can valorize the parent compound as a source of nutrition and energy. Thus, to settle the categorization of an antibiotic‐degrading gene, it is necessary to extend the definition of an ARG to include the capability of the gene product to proceed with the primary or ultimate degradation of the antibiotic. Unfortunately, the literature on the degradation of antimicrobials is scarce compared with that on antibiotic resistance and is limited to a few publications (Wright, 2005; Barra Caracciolo et al., 2015). Two recent and exhaustive reviews summarize the current knowledge regarding the degradation of antibiotics for the most important and most widely used classes of antibiotics (Reis et al., 2020a; Reis et al., 2020b). In summary, for almost all the antibiotic classes reported, primary or ultimate biodegradation processes have been identified at least on the level of the bacterial community. However, in few classes (sulfonamides, β‐lactams) the degradation pathways have been linked to corresponding genes and enzymes. These articles also emphasized how knowledge of the degradation mechanisms, as well as of the genes and enzymes engaged, can help elucidate the role of antibiotic biodegradation in the development of novel antibiotic resistance mechanisms and its possible contribution to the dissemination of known resistance mechanisms. Thoroughly understanding the interplay of antibiotic degradation and resistance could ultimately help to evaluate whether new strategies can help slow down the pace at which new antibiotic resistances are formed and propagated. An example for this would be the design of antibiotics with a shorter half‐life, in order to avoid long‐term exposition of microbes to this antimicrobial agent.
Antibiotic biodegradation: an opportunity for the degrading bacteria to access nutrients and a chance for sensitive microorganisms to survive
The evolution of antibiotic resistance started long before antimicrobials were applied in medicine to fight infections (D'Costa et al., 2011). Presumably, at an early stage in evolution, some microbes acquired means to produce antimicrobial compounds to inhibit the proliferation of others competing with them for resources. Additionally, the antibiotic‐producing microbe may have developed resistance mechanisms for their own protection and this feature may have been acquired later by other microbes. This could even be turned into an enormous advantage, if an antimicrobial compound synthetized by a competitor could actually be recruited as a source of nutrients and energy. After all, antibiotics are organic molecules principally composed of carbon, oxygen, nitrogen, hydrogen and sulfur. Hence, it is not difficult to imagine that microbes do not discriminate between antibiotics and other substrate molecules, provided they carry a genetic makeup encoding for enzymes able to digest these antimicrobial molecules. First indications for bacteria able to use antibiotics as nutritional resources were presented in the 1970s, where different strains of the genus Pseudomonas were found to be able to utilize benzylpenicillin and streptomycin as sources of carbon and nitrogen (Fenton et al., 1973; Johnsen, 1977). Another more recent study screened for the ability of bacteria isolated from different soils to grow on a set of antibiotics as the sole carbon source (Dantas et al., 2018). This work emphasized the phylogenetic distance between the different isolates subsisting on antibiotics, suggesting an intrinsic microbial capability to acquire degradative mechanisms. Genes coding for degradative enzymes that target antibiotics, such as β‐lactamases, have even been found in ancient permafrost sediment (D'Costa et al., 2011). Thus, antibiotic degradation can be seen as part of an intrinsic repertoire of microbes in their competition for nutrients. Heterotrophic microbes are involved in the turnover of various kinds of organic matter encountered in nature, including those with complex chemical structure. This may easily explain the ability of bacteria to degrade natural antibiotics. In addition, there are also numerous accounts of a similar ability to degrade antibiotics of synthetic origin, i.e. ‘new’ compounds in an evolutionary sense. Many microbes can metabolize a broad variety of compounds as carbon source and in some cases are also able to act on ‘natural’ moieties present in synthetic antibiotics and catabolize them (Dantas and Sommer, 2012). Hence, the presence and evolution of promiscuous enzymes, which are unspecific and capable of catalysing reactions on substrates structurally related to the native target, represent the keystone in the development of new antibiotic degradation mechanisms (Kolvenbach et al., 2014). Driving forces for the evolution of a novel machinery to degrade antibiotics to gain nutrients and energy may be bolstered in environments where the competition for nutrients is strong. As the natural environment is experiencing increasing levels of pollution with antibiotics due to anthropogenic activities, this could also exert a dual selective pressure in terms of selection for resistance and providing potential nutrients, warranting further discussion. Conversely, the pressure for evolution/recruitment of catabolic determinants might presumably be less intense in environments where carbon sources are abundant, as in the human body. Here, the mechanisms of mere resistance probably play a more relevant role (Dantas et al., 2018). The development of abilities in human microbiota to degrade antibiotics can be related to the phenomenon of bioprotection (Erb et al., 1997), which has also been referred to as indirect resistance (Nicoloff and Andersson, 2016). This phenomenon occurs when antibiotic‐degrading bacteria protect part of the population that is susceptible to antibiotics by removing/modifying the inhibiting compounds. Eventually, the degradation of the antibiotic will result in a decreased concentration. If this concentration is below the minimum inhibitory concentration for susceptible pathogens, it may result in antibiotic failure and subsequent microbial infection. However, bioprotection via primary biodegradation is still a poorly understood process and has yet only been linked to β‐lactamase producers (Yurtsev et al., 2013; Medaney et al., 2016; Murray et al., 2018). No similar examples for bioprotection from other antibiotic families have been reported so far (Nicoloff and Andersson, 2016). Taking this concept of indirect resistance into account increases the complexity of antibiotic resistance scenarios and adds a concept of ‘community‐based resistance’ to the aspects and forms of individual resistance. Even more so, following the Black Queen Hypothesis, the principle of bioprotection could even drive the loss of ARGs. This would be the case when enzymes rendering antimicrobials harmless would be excreted into the environment. Here, this ‘leaky function’ benefitting the whole community would relieve the pressure to keep individual copies of genes serving the same function and make them dispensable for other individuals (Morris et al., 2012).
The genes of the resistome might be either resistant or degradative and their role is influenced by the genetics of the host
The resistome is defined as the collection of all genes related to antimicrobial resistance within a given compartment (being either a single organism or even a community or ecosystem) (Wright, 2007). Genes related to antibiotic resistance and those related to antibiotic degradation are clustered heterogeneously inside the resistome. Likewise, one could define a sub‐category in the resistome (labelled ‘subsistome’), comprising genes that allow the degrading of antibiotic molecules and their use as a source of nutrients and energy. The subsistome classification would therefore exclude the other antibiotic resistance and degrading genes, which are not directly involved in the valorization of the antimicrobial pathways (Fig. 1). Here however, the possible role of a specific gene is strongly affected by its genomic context. β‐lactamase, for example, cleaves the lactam rings of penicillin derivatives and can be placed among the ARGs (Walsh et al., 2013). Yet, if it is present as a part of a larger degradation pathway to utilize the carbon backbone of the antibiotic, as in the case of the β‐lactamase bla with the concomitant presence of put operon and paa catabolon (Crofts et al., 2018), it will clearly fall into the subsistome category, as it can then serve to provide nutrients from its substrate. In fact, this genomic setup is capable of further processing and catabolizing the phenylacetamide moiety of the cleaved penicillin. In this way, a gene would behave as a resistance gene or antibiotic‐subsisting gene, depending on its wider genetic context. Furthermore, genes coding for the resistance and mineralization of the same antibiotic can be carried independently in a bacterial strain, as in the case of Microbacterium BR1 sp. On the one hand, this strain contains a modified dihydropteroate synthase sul1, a well‐characterized sulfonamide resistance gene (Wise Jr. and Abou‐Donia, 1975). On the other hand, it also bears the sad cluster, encoding for two monooxygenases (sadA and sadB) and a flavin reductase (sadC) respectively, which are responsible for the partial mineralization of sulfonamides (Ricken et al., 2017). This strain was actually the first bacterium for which the genes and enzymes involved in the use of an antibiotic as a carbon and energy source had been characterized. The sad cluster is flanked by sequences encoding for mobile genetic elements (Ricken et al., 2017), as was also the case for Candidatus Leucobacter sulfamidivorax, where multiple copies of sadA were found to be flanked by single IS1380 family transposases (Reis et al., 2019). Moreover, the sulfonamide‐resistant gene sul1 is commonly found in class 1 integrons and therefore known to be easily disseminated (Gillings, 2014). It is interesting to note the presence of distinct genomic islands distributed on separate regions of the genomes of different actinobacteria (Kim et al., 2019), previously identified as sulfonamide degraders (Tappe et al., 2013; Topp et al., 2013; Deng et al., 2016). These islands carry the sulfonamide resistance and the determinants for degradation. This strongly suggests the potential mobility of the degradative genes and the resistome determinants. It may also explain how the sulfonamide degrading machinery could have been acquired with the resistance determinants. In general, the resistance of a specific antibiotic could be a mandatory first step for bacteria to develop antimicrobial degrading mechanisms, in order to relieve stress during the evolving stage towards degradation (Kim et al., 2019). This is the case in Pseudomonas strain ABC07, which carries the resistance gene β‐lactamase, which is also an integral part of a wider framework (in collaboration with the put operon and the paa catabolon), enabling the strain to grow on penicillin, cephalosporins and carbapenems (Crofts et al., 2018).
Figure 1.
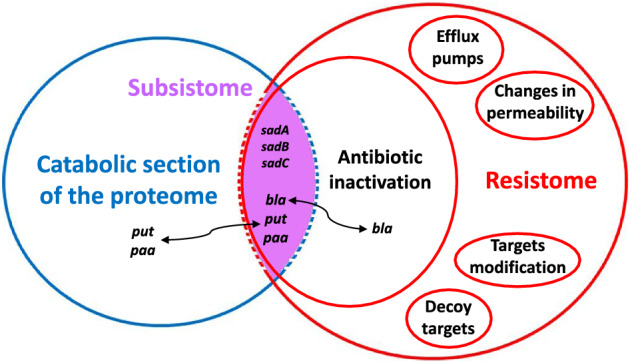
The subsistome as a junction between the resistome and the catabolic section of the proteome. Genes involved in the inactivation of antibiotics that belong to the resistome can cross the classification border and become part of the subsistome. This may occur if they are colocalized with other genes allowing the further valorization of the antibiotic as a source of nutrition and energy. This case demonstrates that the classifications are somewhat fuzzy and that, in certain cases, the antibiotic resistance and degradation functions are partially overlapping.
Conclusions and perspectives
In order to propose efficient solutions for the problem of antibiotic resistance, it may not be sufficient to compile and identify antibiotic resistance determinants and find out how these can be disseminated among microbes. As discussed in the previous section, there are wider implications if we expand the focus from the isolated genes to their context within a genome or even in a microbiome or any other ecosystem. Antibiotic resistances may misleadingly be classified as degradative pathways and resistance genes can even coincide with a part of the ‘subsistome’, resulting in bacterial machineries capable of more than mere resistance. Microbial degradation of antibiotics is strongly related to indirect resistance, encountered when antibiotic concentrations in a microbial environment fall below the inhibitory threshold due to the action of microbes capable of degrading antibiotics (compare Fig. 2). This development from resistance via degradation to indirect resistance, creating a need for novel, effective antibiotics, represents a vicious cycle.
Figure 2.
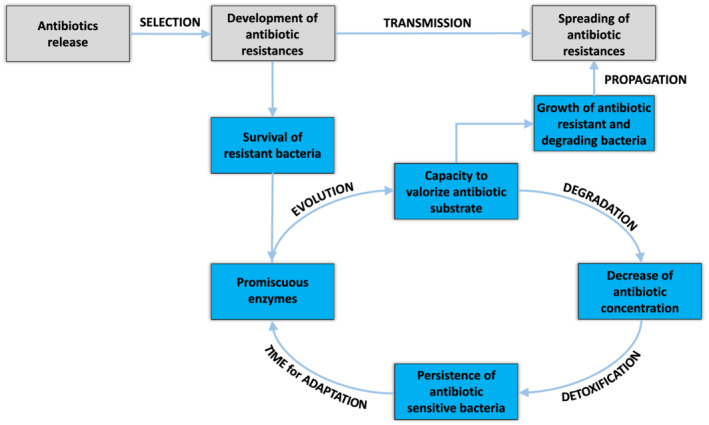
Schematic representation of the vicious cycle, which depicts the possible interplay between antibiotic resistance and degradation during the development of new degradation mechanisms and the dissemination of antibiotic resistances. The release of new antibiotics into the environment may apply selective pressure on the community, selecting for new antibiotic resistances in sensitive strains. After establishing this basal resistance, promiscuous enzymes can play a key role in the evolution of novel pathways to valorize the antibiotics as nutrition source. Valorization will result in the ability to grow on the antimicrobials and propagate the classic ARG. Moreover, the reduction of the antibiotic concentration will enable susceptible strains to survive and adapt under non‐lethal stress conditions. Finally, novel antibiotics have to be developed and prescribed.
Both from a medical and economic perspective, there is a desire to design antibiotics intended to be effective for longer periods without the risk of imminent onset of antibiotic‐resistant microbes and subsequent antibiotic failure. In order to provide improved antibiotics, we believe that a deeper knowledge of the (micro)biological fate of antibiotics is essential. This comprises information on possible biotransformation/biodegradation products, the genes and the enzymes that are involved, but also information on the environmental fate of the parent compound. For instance, the precise knowledge of sadA‐catalysed biodegradation of sulfonamides can be valorized to design sulfonamides, which cannot be degraded via the ipso‐substitution degradation mechanism. In that case, sulfonamides lacking chemical groups on the heteroatom‐bearing moiety that usually enable the delocalization of one free electron pair would be recalcitrant to bacterial degradation. Finally, in order to keep future drugs effective as long as possible, efforts to design new antibiotics should also include the study of potential resistance hot spots, such as the presence of silent‐ and proto‐resistance genes (Perry et al., 2014) in potential application environments, e.g. the patients' microbiomes.
Acknowledgements
We gratefully acknowledge funding from the Swiss National Science Foundation in the frame of Grant IZLCZ2_170272. We also acknowledge support from the European Union's Horizon 2020 research and innovation programme under grant agreement number 826244. We also thank Andrew Brown for proofreading the manuscript.
References
- Barra Caracciolo, A. , Topp, E. , and Grenni, P. (2015) Pharmaceuticals in the environment: biodegradation and effects on natural microbial communities. A review. J Pharm Biomed Anal 106: 25–36. [DOI] [PubMed] [Google Scholar]
- Berendonk, T.U. , Manaia, C.M. , Merlin, C. , Fatta‐Kassinos, D. , Cytryn, E. , Walsh, F. , et al (2015) Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol 13: 310–317. [DOI] [PubMed] [Google Scholar]
- Blair, J.M.A. , Webber, M.A. , Baylay, A.J. , Ogbolu, D.O. , and Piddock, L.J.V. (2015) Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 13: 42–51. [DOI] [PubMed] [Google Scholar]
- Crofts, T.S. , Gasparrini, A.J. , and Dantas, G. (2017) Next‐generation approaches to understand and combat the antibiotic resistome. Nat Rev Microbiol 15: 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofts, T.S. , Wang, B. , Spivak, A. , Gianoulis, T.A. , Forsberg, K.J. , Gibson, M.K. , et al (2018) Shared strategies for β‐lactam catabolism in the soil microbiome. Nat Chem Biol 14: 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas, G. , and Sommer, M.O.A. (2012) Ecological and Clinical Consequences of Antibiotic Subsistence by Environmental Microbes. Antimicrobial Resistance in the Environment, 1st ed. Hoboken, NJ: John Wiley & Sons. [Google Scholar]
- Dantas, G. , Sommer, M.O.A. , Oluwasegun, R.D. , and Church, G.M. (2018) Bacteria subsisting on antibiotics. Science 320: 100–103. [DOI] [PubMed] [Google Scholar]
- Davies, J. , and Davies, D. (2010) Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 74: 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Costa, V.M. , King, C.E. , Kalan, L. , Morar, M. , Sung, W.W.L. , Schwarz, C. , et al (2011) Antibiotic resistance is ancient. Nature 477: 457–461. [DOI] [PubMed] [Google Scholar]
- Deng, Y. , Mao, Y. , Li, B. , Yang, C. , and Zhang, T. (2016) Aerobic degradation of sulfadiazine by Arthrobacter spp.: kinetics, pathways, and genomic characterization. Environ Sci Technol 50: 9566–9575. [DOI] [PubMed] [Google Scholar]
- Erb, R.W. , Eichner, C.A. , Wagner‐Döbler, I. , and Timmis, K.N. (1997) Bioprotection of microbial communities from toxic phenol mixtures by a genetically designed pseudomonad. Nat Biotechnol 15: 378–382. [DOI] [PubMed] [Google Scholar]
- Fenton, J.J. , Harsch, H.H. , and Klein, D. (1973) Production of volatile nitrogenous compounds from the degradation of streptomycin by Pseudomonas maltophilia . J Bacteriol 116: 1267–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, M.K. , Forsberg, K.J. , and Dantas, G. (2015) Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J 9: 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillings, M.R. (2014) Integrons: past, present, and future. Microbiol Mol Biol Rev 78: 257–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen, J. (1977) Utilization of benzylpenicillin as carbon, nitrogen and energy source by a Pseudomonas fluorescens strain. Arch Microbiol 115: 271–275. [DOI] [PubMed] [Google Scholar]
- Kim, D.W. , Thawng, C.N. , Lee, K. , Wellington, E.M.H. , and Cha, C.J. (2019) A novel sulfonamide resistance mechanism by two‐component flavin‐dependent monooxygenase system in sulfonamide‐degrading actinobacteria. Environ Int 127: 206–215. [DOI] [PubMed] [Google Scholar]
- Kolvenbach, B.A. , Helbling, D.E. , Kohler, H.P.E. , and Corvini, P.X.F. (2014) Emerging chemicals and the evolution of biodegradation capacities and pathways in bacteria. Curr Opin Biotechnol 27: 8–14. [DOI] [PubMed] [Google Scholar]
- Martínez, J.L. , Coque, T.M. , and Baquero, F. (2015) What is a resistance gene? Ranking risk in resistomes. Nat Rev 13: 116–123. [DOI] [PubMed] [Google Scholar]
- Medaney, F. , Dimitriu, T. , Ellis, R.J. , and Raymond, B. (2016) Live to cheat another day: bacterial dormancy facilitates the social exploitation of β‐lactamases. ISME J 10: 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, J.J. , Lenski, R.E. , and Zinser, E.R. (2012) The black queen hypothesis: evolution of dependencies through adaptive gene loss. mBio 3: e00036‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munita, J.M. , and Arias, C.A. (2016) Mechanisms of antibiotic resistance. Microbiol Spectrum 4: 1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A.K. , Zhang, L. , Yin, X. , Zhang, T. , Buckling, A. , Snape, J. , and Gaze, W.H. (2018) Novel insights into selection for antibiotic resistance in complex microbial communities. mBio 9: e00969‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoloff, H. , and Andersson, D.I. (2016) Indirect resistance to several classes of antibiotics in cocultures with resistant bacteria expressing antibiotic‐modifying or ‐degrading enzymes. J Antimicrob Chemother 71: 100–110. [DOI] [PubMed] [Google Scholar]
- Pal, C. , Bengtsson‐Palme, J. , Kristiansson, E. , and Joakim Larsson, D.G. (2016) The structure and diversity of human, animal and environmental resistomes. Microbiome 4: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, J.A. , Westman, E.L. , and Wright, G.D. (2014) The antibiotic resistome: what's new? Curr Opin Microbiol 21: 45–50. [DOI] [PubMed] [Google Scholar]
- Reis, A.C. , Kolvenbach, B.A. , Chami, M. , Gales, L. , Egas, C. , Corvini, P.X.F. , and Nunes, O.C. (2019) Comparative genomics reveals a novel genetic organization of the sad cluster in the sulfonamide‐degrader ‘Candidatus Leucobacter sulfamidivorax’ strain GP. BMC Genomics 20: 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis, A.C. , Kolvenbach, B.A. , Nunes, O.C. , and Corvini, P.X.F. (2020a) Biodegradation of antibiotics: the new resistance determinants – part I. New Biotechnol 54: 34–51. [DOI] [PubMed] [Google Scholar]
- Reis, A.C. , Kolvenbach, B.A. , Nunes, O.C. , and Corvini, P.X.F. (2020b) Biodegradation of antibiotics: the new resistance determinants – part II. New Biotechnol 54: 13–27. [DOI] [PubMed] [Google Scholar]
- Ricken, B. , Kolvenbach, B.A. , Bergesch, C. , Benndorf, D. , Kroll, K. , Strnad, H. , et al (2017) FMNH2‐dependent monooxygenases initiate catabolism of sulfonamides in Microbacterium sp. strain BR1 subsisting on sulfonamide antibiotics. Sci Rep 7: 15783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer, M.O.A. , and Dantas, G. (2011) Antibiotics and the resistant microbiome. Curr Opin Microbiol 14: 556–563. [DOI] [PubMed] [Google Scholar]
- Tappe, W. , Herbst, M. , Hofmann, D. , Koeppchen, S. , Kummer, S. , Thiele, B. , and Groeneweg, J. (2013) Degradation of sulfadiazine by Microbacterium lacus strain SDZm4, isolated from Lysimeters previously manured with slurry from sulfadiazine‐medicated pigs. Appl Environ Microbiol 79: 2572–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp, E. , Chapman, R. , Devers‐Lamrani, M. , Hartmann, A. , Marti, R. , Martin‐Laurent, F. , et al (2013) Accelerated biodegradation of veterinary antibiotics in agricultural soil following long‐term exposure, and isolation of a sulfamethazine‐degrading Microbacterium sp. J Environ Qual 42: 173–178. [DOI] [PubMed] [Google Scholar]
- Vaz‐Moreira, I. , Nunes, O.C. , and Manaia, C.M. (2014) Bacterial diversity and antibiotic resistance in water habitats: searching the links with the human microbiome. FEMS Microbiol Rev 38: 761–778. [DOI] [PubMed] [Google Scholar]
- Walsh, F. , Amyes, S.G.B. , and Duffy, B. (2013) Challenging the concept of bacteria subsisting on antibiotics. Int J Antimicrob Agents 41: 558–563. [DOI] [PubMed] [Google Scholar]
- Wise, E.M., Jr. , and Abou‐Donia, M.M. (1975) Sulfonamide resistance mechanism in Escherichia coli: R plasmids can determine sulfonamide‐resistant dihydropteroate synthases. Proc Natl Acad Sci U S A 72: 2621–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, G.D. (2005) Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv Drug Deliv Rev 57: 1451–1470. [DOI] [PubMed] [Google Scholar]
- Wright, G.D. (2007) The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol 5: 175–186. [DOI] [PubMed] [Google Scholar]
- Yurtsev, E.A. , Chao, H.X. , Datta, M.S. , Artemova, T. , and Gore, J. (2013) Bacterial cheating drives the population dynamics of cooperative antibiotic resistance plasmids. Mol Syst Biol 9: 683. [DOI] [PMC free article] [PubMed] [Google Scholar]