

Abstract
A straightforward methodology for the synthesis of anti‐Markovnikov‐type alcohols is presented. By using a specific cobalt triphos complex in the presence of Zn(OTf)2 as an additive, the hydrogenation of epoxides proceeds with high yields and selectivities. The described protocol shows a broad substrate scope, including multi‐substituted internal and terminal epoxides, as well as a good functional‐group tolerance. Various natural‐product derivatives, including steroids, terpenoids, and sesquiterpenoids, gave access to the corresponding alcohols in moderate‐to‐excellent yields.
Keywords: anti-Markovnikov, cobalt, epoxides, homogeneous catalysis, hydrogenation
Non‐noble‐metal catalysis: The cobalt‐catalyzed hydrogenation of epoxides for the synthesis of anti‐Markovnikov alcohols is reported. This method is suitable for internal, as well as terminal, epoxides and works smoothly even with multi‐substituted derivatives under mild conditions.

Alcohols are a class of important organic compounds that are ubiquitous in bulk and fine chemicals, as well as in natural products.1 Among the numerous established procedures for the synthesis of alcohols, the classic hydroboration/oxidation protocol still prevails on a laboratory scale (Figure 1 A). Advantageously, this methodology allows for a formal anti‐Markovnikov functionalization of linear alcohols from olefins, however, stoichiometric amounts of borane agents have to be employed.2 To overcome this problem, various catalytic approaches to provide similar products have been developed. For example, a formal anti‐Markovnikov hydration of monosubstituted styrenes by triple‐relay catalysis was demonstrated by Grubbs and co‐workers.3 More recently, Lei and co‐workers established a visible‐light‐mediated anti‐Markovnikov hydration of water to olefins, by using a photoredox catalyst in combination with a redox‐active hydrogen‐atom donor.4 Additionally, the Arnold group realized a regioselective redox hydration of styrenes catalyzed by a metal‐oxo enzyme.5
Figure 1.
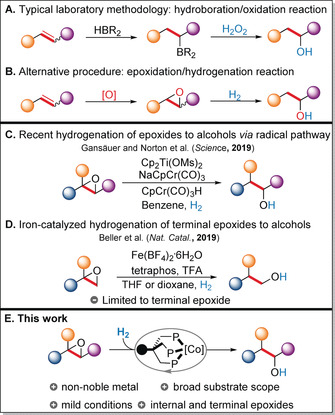
Synthesis of alcohols from olefins and hydrogenation of epoxides to alcohols.
Conceptually, the selective hydrogenation of epoxides, which are readily available from alkenes by a one‐step oxidation using peroxyacids or hydrogen peroxide,6 offers an attractive alternative (Figure 1 B).7 Heterogeneous catalysts such as Pd/C generally facilitate this transformation, but are limited to aryl epoxides, whereas Markovnikov‐type alcohols are formed as the major products in the case of alkyl epoxides.8 On the contrary, homogeneous catalysts have been scarcely investigated for this task. Until very recently, the only known examples featured rhodium‐ and ruthenium‐based systems and suffered from poor product selectivities.9
In 2019, Gansäuer, Norton, and co‐workers disclosed an elegant strategy that used cooperative catalysis to give linear alcohols by combining titanocene‐mediated10 epoxide opening with chromium‐catalyzed hydrogen activation and radical reduction (Figure 1 C).11 Independently, our group developed the first non‐noble‐metal‐catalyzed hydrogenation of terminal epoxides to give primary alcohols. Using a combination of Fe(BF4)2⋅6 H2O and tris(2‐(diphenylphosphanyl)phenyl)phosphane (tetraphos) the desired products are obtained in high yields and selectivities.12 However, a drawback of this procedure was that only terminal epoxides were suitable substrates and internal epoxides did not undergo hydrogenation to provide secondary alcohols (Figure 1 D).
Herein, we describe a general and efficient non‐noble‐metal catalyst system13 to enable selective hydrogenation14 of both internal and terminal epoxides to the corresponding alcohols under mild conditions (Figure 1 E).
Based on our former studies, we explored the hydrogenation of 2‐methyl‐3‐phenyloxirane (1 a) as a benchmark substrate. As expected, the previous Fe(BF4)2/tetraphos system gave no significant amount of alcohols. Similarly, in the presence of related multidentate phosphines, for example, 1,1,1‐tris(diphenyl‐phosphinomethyl)ethane (triphos), no desired product formation occurred. Moreover, well‐known molecularly defined noble‐metal complexes [Ru(acac)3/triphos]15 and [Rh(PPh3)3Cl] also failed to furnish the desired product under otherwise identical reaction conditions (for experimental details, see the Supporting Information). Interestingly, applying the combination of Co(BF4)2⋅6 H2O and tetraphos in the presence of HNTf2 resulted in formation of the desired anti‐Markovnikov‐type product 1‐phenylpropan‐2‐ol (2 a), albeit in a low yield (17 %), with 1‐phenylpropan‐2‐one (2 a′) produced as a side product (for experimental details, see the Supporting Information). When triphos was tested as the ligand, a slight increase in activity was observed (Table 1, entry 1). Changing the catalyst precursor to Co(NTf2)2 further improved the observed yield (Table 1, entry 2); however, when the reaction temperature was decreased (to 100 °C), the hydrogenation process almost completely stopped and only minor amounts of 2 a could be detected (Table 1, entry 3). Notably, the addition of catalytic amounts of Zn(OTf)2 (3.0 mol %) significantly improved catalyst activity, even at a lower temperature (80 °C) (Table 1, entries 4 and 5).
Table 1.
Optimization of Cobalt‐catalyzed hydrogenation of epoxide (1 a).[a]

|
Entry |
Catalyst |
Additive |
T [°C] |
2 a [%][b] |
|---|---|---|---|---|
|
1[c] |
Co(BF4)2 ⋅6 H2O |
HNTf2 |
120 |
23 |
|
2[c] |
Co(NTf2)2 |
– |
120 |
43 |
|
3 |
Co(NTf2)2 |
– |
100 |
<10 |
|
4 |
Co(NTf2)2 |
Zn(OTf)2 |
100 |
74 |
|
5 |
Co(NTf2)2 |
Zn(OTf)2 |
80 |
80 |
|
6 |
Co(NTf2)2 |
In(OTf)3 |
80 |
74 |
|
7 |
Co(NTf2)2 |
Al(OTf)3 |
80 |
<10 |
|
8 |
Co(NTf2)2 |
Fe(OTf)2 |
80 |
18 |
|
9 |
– |
Zn(OTf)2 |
80 |
– |
|
10[d] |
Co(NTf2)2 |
Zn(OTf)2 |
80 |
– |
|
11[e] |
Co(NTf2)2 |
Zn(OTf)2 |
80 |
85 |
|
12[e] |
Co(BF4)2 ⋅6 H2O |
Zn(OTf)2 |
80 |
80 |
|
13[e] |
Co(ClO4)2 ⋅6 H2O |
Zn(OTf)2 |
80 |
73 |
[a] Reaction conditions: 1 a (0.5 mmol), [Co] (3.0 mol %), triphos (6.0 mol %), additive (3.0 mol %), THF (4 mL), 16 h, yields were determined by GC analysis with n‐hexadecane as an internal standard. [b] 1‐Phenylpropan‐2‐one 2 a′ is the main side product. [c] triphos (3.0 mol %). [d] Without triphos. [e] Zn(OTf)2 (7.0 mol %).
Other Lewis acids, such as In(OTf)3, Al(OTf)3, and Fe(OTf)2, provided inferior results (Table 1, entries 6–8). Control experiments indicated that the synergistic combination of triphos and cobalt precursor is crucial for the epoxide hydrogenation process (Table 1, entries 9 and 10). Increasing the amount of additive further improved the obtained yield of 2 a to 85 % (Table 1, entry 11). In general, other cobalt precursors could be applied in this benchmark process, but resulted in slightly lower catalyst activities (Table 1, entries 12 and 13). It should be noted that standard heterogeneous catalysts, such as PtO2, Pd/C, and Raney‐Ni, exhibited significantly lower or even no activity, even in the presence of Zn(OTf)2 (for experimental details, see the Supporting Information).
With the optimized reaction conditions in hand, we tested the suitability of our methodology towards various internal epoxides. As shown in Scheme 1, various di‐ and tri‐substituted internal epoxides were successfully applied and yielded the desired secondary alcohols in good yields and high regioselectivities. All the reactions occurred under relatively mild conditions and importantly, tolerate a variety of valuable substituents and functional groups irrespective of their location at the ortho‐, meta‐, or para‐position. Notably, ester 2 q, which is typically reduced by cobalt/triphos catalysts, remained unaffected under the applied conditions and led to the corresponding alcohol in 74 % yield (Scheme 1).13a In addition, asymmetric dialkyl‐substituted internal epoxides 2 k, 2 m, and 2 r were successfully transformed to diastereomeric secondary alcohols.
Scheme 1.

Cobalt‐catalyzed hydrogenation of internal epoxides. Reaction condition: 1 (0.5 mmol), Co(NTf2)2 (3.0 mol %), triphos (6.0 mol %), Zn(OTf)2 (7.0 mol %), THF, 80 °C, 16 h. [a] The diastereoisomer ratio is 1:1.1. [b] Co(BF4)2⋅6 H2O (3.0 mol %), 1,4‐dioxane, 60 °C, 20 h, the diastereoisomer ratio (2.8:1) and yield were determined by GC analysis.
However, when the tetra‐substituted epoxide 2,2,3,3‐tetramethyloxirane 1 s (see the Supporting Information) was applied to our reaction conditions, only the formation of a complex product mixture was observed.
The applied cobalt‐based catalyst system is not restricted to the hydrogenation of internal oxiranes. In fact, numerous terminal epoxides, including several natural‐product derivatives (steroids, terpenoids, and sesquiterpenoids), were effectively hydrogenated to the linear alcohols with high regioselectivities. Compared to our previously disclosed iron/tetraphos catalyst system, the cobalt/triphos catalyst demonstrated a wider applicability for such substrates (Scheme 2).12 More specifically, both mono‐ and di‐substituted terminal epoxides were suitable substrates and provided the desired anti‐Markovnikov‐type alcohols in good yields, tolerating amide, silyloxy, alkene, and ester substituents. Using renewable terpenes, such as (±)‐camphene (3 g), (−)‐β‐pinene (3 h), and (+)‐aromadendrene (3 i), which are the main constituents of essential oil, the respective primary alcohols were isolated in high yields and selectivities.
Scheme 2.

Cobalt‐catalyzed hydrogenation of terminal epoxides. Reaction condition: 3 (0.5 mmol), Co(BF4)2⋅6 H2O (3.0 mol %), triphos (6.0 mol %), 1,4‐dioxane (6 mL), 80 °C, 16 h. [a] THF (4 mL) as solvent. [b] Yields were determined by GC analysis with n‐hexadecane as an internal standard. [c] Co(NTf2)2 (3.0 mol %), Zn(OTf)2 (7.0 mol %). [d] The major isomers are shown. TBS=tert‐Butyldimethylsilyl.
Additionally, the bioactive pentacyclic triterpenoid betulin (3 k), which is abundant in the bark of birch trees and, moreover, plays an active role in antiviral, analgesic, and antineoplastic agents, also furnished the hydrogenated products in good yields. Similarly, pregnenolone (3 j), an important steroid, underwent the hydrogenation process smoothly, providing the isolated diastereomeric alcohols in 77 % yield.
To better understand the strong performance of our cobalt/triphos catalyst, a set of mechanistic experiments was performed. Firstly, kinetic studies using the model substrate 2‐methyl‐3‐phenyloxirane (1 a) were performed. As shown in Scheme 3 a, 1 a is quickly isomerized16 to 1‐phenylpropan‐2‐one (2 a′), followed by subsequent hydrogenation to the desired alcohol 2 a.17 To further prove that 2 a′ is indeed a reaction intermediate, a control experiment employing 1 a as the starting material under an argon atmosphere without hydrogen present was conducted. Accordingly, 1‐phenylpropan‐2‐one (2 a′) was isolated in almost quantitative yield. Next, 2 a′ was used as the substrate under the standard reaction conditions, yielding the corresponding hydrogenation product 2 a in 92 % yield (Scheme 3 b). In agreement with these observations, [D]2‐2 a was obtained as the final product in a deuterium‐labelling experiment that applied D2 instead of H2 (Scheme 3 c). Finally, to understand whether the cobalt or zinc salts catalyze the isomerization of epoxides to the corresponding ketones, several control experiments were conducted (Supporting Information). Interestingly, the Meinwald rearrangement of 1 a to 2 a′ also proceeded without a co‐catalyst; however, addition of Zn(OTf)2 improved this reaction step. Based on the obtained results, we propose that the reaction takes place via Meinwald rearrangement of the epoxide to the corresponding carbonyl compound, followed by subsequent cobalt/triphos‐catalyzed hydrogenation to the desired anti‐Markovnikov alcohols.
Scheme 3.

Selected mechanistic studies.
In summary, the first cobalt‐catalyzed hydrogenation of epoxides for the synthesis of anti‐Markovnikov alcohols is reported. The presented methodology is suitable for internal, as well as terminal, epoxides and works smoothly even with multi‐substituted derivatives under mild conditions. This novel cascade transformation is well‐suited for the reduction of natural‐product‐derived epoxides, including steroids, terpenoids, and sesquiterpenoids. Mechanistic studies indicate that initially a Meinwald rearrangement of the epoxides to the corresponding ketones/aldehydes takes place followed by cobalt/triphos‐catalyzed hydrogenation. In general, this transformation offers an attractive alternative compared to the traditional hydroboration/oxidation protocol of olefins.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the analytic department (LIKAT) for their support, the State of Mecklenburg‐Western Pomerania, the Federal State of Germany (BMBF), and the EU (Grant 670986) for financial support.
W. Liu, T. Leischner, W. Li, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2020, 59, 11321.
References
- 1. Weissermel K., Arpe H.-J., Industrial organic chemistry, Wiley, Hoboken, 2008. [Google Scholar]
- 2.
- 2a. Brown H. C., Zweifel G., J. Am. Chem. Soc. 1959, 81, 247–247; [Google Scholar]
- 2b. Burgess K., Ohlmeyer M. J., Chem. Rev. 1991, 91, 1179–1191; [Google Scholar]
- 2c. Obligacion J. V., Chirik P. J., Nat. Rev. Chem. 2018, 2, 15–34; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Wen H., Liu G., Huang Z., Coord. Chem. Rev. 2019, 386, 138–153. [Google Scholar]
- 3. Dong G., Teo P., Wickens Z. K., Grubbs R. H., Science 2011, 333, 1609–1612. [DOI] [PubMed] [Google Scholar]
- 4. Hu X., Zhang G., Bu F., Lei A., ACS Catal. 2017, 7, 1432–1437. [Google Scholar]
- 5. Hammer S. C., Kubik G., Watkins E., Huang S., Minges H., Arnold F. H., Science 2017, 358, 215. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Katsuki T., Sharpless K. B., J. Am. Chem. Soc. 1980, 102, 5974–5976; [Google Scholar]
- 6b. Zhang W., Loebach J. L., Wilson S. R., Jacobsen E. N., J. Am. Chem. Soc. 1990, 112, 2801–2803; [Google Scholar]
- 6c. Denard C. A., Bartlett M. J., Wang Y., Lu L., Hartwig J. F., Zhao H., ACS Catal. 2015, 5, 3817–3822. [Google Scholar]
- 7. Huang C.-Y., Doyle A. G., Chem. Rev. 2014, 114, 8153–8198. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Sajiki H., Hattori K., Hirota K., Chem. Commun. 1999, 1041–1042; [Google Scholar]
- 8b. Ley S. V., Mitchell C., Pears D., Ramarao C., Yu J.-Q., Zhou W., Org. Lett. 2003, 5, 4665–4668; [DOI] [PubMed] [Google Scholar]
- 8c. Kwon M. S., Park I. S., Jang J. S., Lee J. S., Park J., Org. Lett. 2007, 9, 3417–3419; [DOI] [PubMed] [Google Scholar]
- 8d. Thiery E., Le Bras J., Muzart J., Eur. J. Org. Chem. 2009, 961–985. [Google Scholar]
- 9.
- 9a. Mochida I., Shirahama S.-i., Fujitsu H., Takeshita K., Chem. Lett. 1977, 6, 421–422; [Google Scholar]
- 9b. Fujitsu H., Shirahama S., Matsumura E., Takeshita K., Mochida I., J. Org. Chem. 1981, 46, 2287–2290; [Google Scholar]
- 9c. Fujitsu H., Matsumura E., Shirahama S., Takeshita K., Mochida I., J. Chem. Soc. Perkin Trans. 1 1982, 855–859; [Google Scholar]
- 9d. Ito M., Hirakawa M., Osaku A., Ikariya T., Organometallics 2003, 22, 4190–4192; [Google Scholar]
- 9e. O W. W. N., Lough A. J., Morris R. H., Chem. Commun. 2010, 46, 8240–8242; [DOI] [PubMed] [Google Scholar]
- 9f. Murru S., Nicholas K. M., Srivastava R. S., J. Mol. Catal. A 2012, 363–364, 460–464; [Google Scholar]
- 9g. Thiyagarajan S., Gunanathan C., Org. Lett. 2019, 21, 9774–9778. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Gansäuer A., Fan C.-A., Piestert F., J. Am. Chem. Soc. 2008, 130, 6916–6917; [DOI] [PubMed] [Google Scholar]
- 10b. Gansäuer A., Klatte M., Brändle G. M., Friedrich J., Angew. Chem. Int. Ed. 2012, 51, 8891–8894; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9021–9024; [Google Scholar]
- 10c. Ye K.-Y., McCallum T., Lin S., J. Am. Chem. Soc. 2019, 141, 9548–9554. [DOI] [PubMed] [Google Scholar]
- 11. Yao C., Dahmen T., Gansäuer A., Norton J., Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]
- 12. Liu W., Li W., Spannenberg A., Junge K., Beller M., Nat. Catal. 2019, 2, 523–528. [Google Scholar]
- 13.
- 13a. Korstanje T. J., van der Vlugt J. I., Elsevier C. J., de Bruin B., Science 2015, 350, 298–302; [DOI] [PubMed] [Google Scholar]
- 13b. Adam R., Bheeter C. B., Cabrero-Antonino J. R., Junge K., Jackstell R., Beller M., ChemSusChem 2017, 10, 842–846; [DOI] [PubMed] [Google Scholar]
- 13c. Adam R., Cabrero-Antonino J. R., Spannenberg A., Junge K., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2017, 56, 3216–3220; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3264–3268; [Google Scholar]
- 13d. Cabrero-Antonino J. R., Adam R., Junge K., Beller M., Chem. Sci. 2017, 8, 6439–6450; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13e. Cabrero-Antonino J. R., Adam R., Papa V., Holsten M., Junge K., Beller M., Chem. Sci. 2017, 8, 5536–5546; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13f. Schieweck B. G., Klankermayer J., Angew. Chem. Int. Ed. 2017, 56, 10854–10857; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10994–10997; [Google Scholar]
- 13g. Schneidewind J., Adam R., Baumann W., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2017, 56, 1890–1893; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1916–1919; [Google Scholar]
- 13h. Liu W., Sahoo B., Spannenberg A., Junge K., Beller M., Angew. Chem. Int. Ed. 2018, 57, 11673–11677; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11847–11851; [Google Scholar]
- 13i. Emayavaramban B., Chakraborty P., Sundararaju B., ChemSusChem 2019, 12, 3089–3093. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Bullock R. M., Science 2013, 342, 1054–1055; [DOI] [PubMed] [Google Scholar]
- 14b. Chirik P. J., Acc. Chem. Res. 2015, 48, 1687–1695; [DOI] [PubMed] [Google Scholar]
- 14c. Murugesan S., Kirchner K., Dalton Trans. 2016, 45, 416–439; [DOI] [PubMed] [Google Scholar]
- 14d. Du X., Huang Z., ACS Catal. 2017, 7, 1227–1243; [Google Scholar]
- 14e. Kallmeier F., Kempe R., Angew. Chem. Int. Ed. 2018, 57, 46–60; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 48–63; [Google Scholar]
- 14f. Filonenko G. A., van Putten R., Hensen E. J. M., Pidko E. A., Chem. Soc. Rev. 2018, 47, 1459–1483; [DOI] [PubMed] [Google Scholar]
- 14g. Liu W., Sahoo B., Junge K., Beller M., Acc. Chem. Res. 2018, 51, 1858–1869; [DOI] [PubMed] [Google Scholar]
- 14h. Mukherjee A., Milstein D., ACS Catal. 2018, 8, 11435–11469; [Google Scholar]
- 14i. Alig L., Fritz M., Schneider S., Chem. Rev. 2019, 119, 2681–2751; [DOI] [PubMed] [Google Scholar]
- 14j. Ai W., Zhong R., Liu X., Liu Q., Chem. Rev. 2019, 119, 2876–2953; [DOI] [PubMed] [Google Scholar]
- 14k. Junge K., Papa V., Beller M., Chem. Eur. J. 2019, 25, 122–143. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Núñez Magro A. A., Eastham G. R., Cole-Hamilton D. J., Chem. Commun. 2007, 3154–3156; [DOI] [PubMed] [Google Scholar]
- 15b. Geilen F. M. A., Engendahl B., Hölscher M., Klankermayer J., Leitner W., J. Am. Chem. Soc. 2011, 133, 14349–14358; [DOI] [PubMed] [Google Scholar]
- 15c. vom Stein T., Meuresch M., Limper D., Schmitz M., Hölscher M., Coetzee J., Cole-Hamilton D. J., Klankermayer J., Leitner W., J. Am. Chem. Soc. 2014, 136, 13217–13225; [DOI] [PubMed] [Google Scholar]
- 15d. Savourey S., Lefèvre G., Berthet J.-C., Thuéry P., Genre C., Cantat T., Angew. Chem. Int. Ed. 2014, 53, 10466–10470; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10634–10638; [Google Scholar]
- 15e. Cui X., Li Y., Topf C., Junge K., Beller M., Angew. Chem. Int. Ed. 2015, 54, 10596–10599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10742–10745; [Google Scholar]
- 15f. Li Y., Topf C., Cui X., Junge K., Beller M., Angew. Chem. Int. Ed. 2015, 54, 5196–5200; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5285–5289; [Google Scholar]
- 15g. Zhang L., Han Z., Zhang L., Li M., Ding K., Chin. J. Org. Chem. 2016, 36, 1824–1838; [Google Scholar]
- 15h. Adam R., Cabrero-Antonino J., Junge K., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2016, 55, 11049–11053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11215–11219. [Google Scholar]
- 16.
- 16a. Jiang G., Chen J., Thu H.-Y., Huang J.-S., Zhu N., Che C.-M., Angew. Chem. Int. Ed. 2008, 47, 6638–6642; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6740–6744; [Google Scholar]
- 16b. Hrdina R., Müller C. E., Wende R. C., Lippert K. M., Benassi M., Spengler B., Schreiner P. R., J. Am. Chem. Soc. 2011, 133, 7624–7627; [DOI] [PubMed] [Google Scholar]
- 16c. Vyas D. J., Larionov E., Besnard C., Guénée L., Mazet C., J. Am. Chem. Soc. 2013, 135, 6177–6183; [DOI] [PubMed] [Google Scholar]
- 16d. Lamb J. R., Mulzer M., LaPointe A. M., Coates G. W., J. Am. Chem. Soc. 2015, 137, 15049–15054; [DOI] [PubMed] [Google Scholar]
- 16e. Tian Y., Jürgens E., Kunz D., Chem. Commun. 2018, 54, 11340–11343. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Zhang G., Scott B. L., Hanson S. K., Angew. Chem. Int. Ed. 2012, 51, 12102–12106; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12268–12272; [Google Scholar]
- 17b. Zhang G., Vasudevan K. V., Scott B. L., Hanson S. K., J. Am. Chem. Soc. 2013, 135, 8668–8681; [DOI] [PubMed] [Google Scholar]
- 17c. Gärtner D., Welther A., Rad B. R., Wolf R., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2014, 53, 3722–3726; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3796–3800; [Google Scholar]
- 17d. Rösler S., Obenauf J., Kempe R., J. Am. Chem. Soc. 2015, 137, 7998–8001; [DOI] [PubMed] [Google Scholar]
- 17e. Zhong R., Wei Z., Zhang W., Liu S., Liu Q., Chem 2019, 5, 1552–1566. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary