

Abstract
LPA1 is one of six known receptors (LPA1‐6) for lysophosphatidic acid (LPA). Constitutive Lpar1 null mutant mice have been instrumental in identifying roles for LPA‐LPA1 signaling in neurobiological processes, brain development, and behavior, as well as modeling human neurological diseases like neuropathic pain. Constitutive Lpar1 null mutant mice are protected from partial sciatic nerve ligation (PSNL)‐induced neuropathic pain, however, the cell types that are functionally responsible for mediating this protective effect are unknown. Here, we report the generation of an Lpar1flox/flox conditional null mutant mouse that allows for cre‐mediated conditional deletion, combined with a PSNL pain model. Lpar1flox/flox mice were crossed with cre transgenic lines driven by neural gene promoters for nestin (all neural cells), synapsin (neurons), or P0 (Schwann cells). CD11b‐cre transgenic mice were also used to delete Lpar1 in microglia. PSNL‐initiated pain responses were reduced following cre‐mediated Lpar1 deletion with all three neural promoters as well as the CD11b promoter, supporting involvement of Schwann cells, central and/or peripheral neurons, and microglia in mediating pain. Interestingly, rescue responses were nonidentical, implicating distinct roles for Lpar1‐expressing cell types. Our results with a new Lpar1 conditional mouse mutant expand an understanding of LPA1 signaling in the PSNL model of neuropathic pain.
Keywords: conditional knockout, LPA, PSNL, Schwann cells, neurons
Abbreviations
- CNS
central nervous system
- DRG
dorsal root ganglia
- LPA
lysophosphatidic acid
- Lpar1
(LPA receptor 1)
- PNS
peripheral nervous system
- PNSL
partial sciatic nerve ligation
1. INTRODUCTION
Neuropathic pain is produced by nerve lesions or neurological conditions such as multiple sclerosis, diabetes, and cancer and affects an estimated 10% of the general population. 1 Treatment options for individuals affected by neuropathic pain are limited and ineffective, often leading to a worsened condition and disability. Initiation and propagation of pain signaling occurs through afferent nerve fibers that relay peripheral signals through dorsal root ganglia (DRG) to signal via the central nervous system (CNS) spinal cord dorsal horn and brain (reviewed in 2, 3). Neuropathic pain involves central sensitization, a process that results in allodynia (painful response to normally innocuous stimuli) and hyperalgesia (increased pain sensation to noxious stimuli). 5
One identified modulator of neuropathic pain is the bioactive lipid lysophosphatidic acid (LPA). LPA normally signals through six known G protein‐coupled receptors, LPA1‐6, 6 which are involved in myriad biological and pathological processes affecting most of the physiological systems in the body, including the nervous system. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 LPA1 is expressed in the peripheral nervous system (PNS) and CNS. Schwann cells represent one of the LPA1 expressing cell types that may be involved in the induction of neuropathic pain. LPA signaling through this receptor influences Schwann cell morphology, migration, and survival. 14 , 15 In vivo, sciatic nerves of Lpar1 deficient mice show abnormalities including an increased number of apoptotic Schwann cells, reduced myelin thickness, and a proportionately lower number of small nerve fiber interacting Schwann cells. 14 , 16 Neurons can also be affected through LPA1‐mediated changes to cell morphology, motility, growth cone collapse, calcium signaling, and proliferation. 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 Mice deficient for this receptor display alterations in cortical development and neurogenesis as well as behavioral abnormalities. 22 , 23 , 24
A role for LPA in pain sensation was first identified through intrathecal (i.t.) injection of LPA, where mice that received a single i.t. injection of LPA developed thermal hyperalgesia and mechanical allodynia. 25 LPA‐induced neuropathic pain was accompanied by other sequelae including demyelination in the dorsal root and increased expression of pain associated markers including, protein kinase Cγ(PKCγ) in the spinal cord dorsal horn, and voltage‐gated calcium channel Caα2δ1 in the DRG. 25 Interestingly, i.t. injection of LPA also induced de novo production of LPA in the dorsal horn and dorsal root, implicating a feed‐forward role in pain generation. 26 , 27 De novo LPA production was also observed in the dorsal horn and dorsal root following PSNL. 28 , 29 , 30 Wildtype (Wt) mice subject to PSNL displayed pain behaviors similar to those of mice that received i.t. LPA. and showed similar demyelination as well as upregulation of PKCγ and Caα2δ1. 25
LPA’s effects in PSNL were shown to be receptor‐dependent through the use of constitutive null receptor mutants. Lpar1 null mutant mice were protected from both PSNL and i.t. LPA injection induced mechanical allodynia, and did not show accompanying increases of PKCγ and Caα2δ1. 25 Lpar5 null mutant mice were also protected from PSNL‐induced neuropathic pain, albeit through CNS mechanisms distinct from those of Lpar1 null mutants. 31
While Lpar1 null mutant mice are protected from PSNL‐induced neuropathic pain, the cell types responsible for mediating this protection remain unclear. To address this issue, we generated a Lpar1 conditional null mutant mouse and targeted deletion of Lpar1 in all neural lineages, peripheral and CNS neurons, Schwann cells, and microglia/myeloid cells to identify the cell types responsible for mediating Lpar1's protective effect in the PSNL neuropathic pain model.
2. MATERIALS AND METHODS
2.1. Mice
All procedures performed on animals were IACUC approved and performed in accordance with the regulations of The Scripps Research Institute (TSRI) Department of Animal Resources and the Sanford Burnham Prebys Medical Discovery Institute animal care and use committee. Mice used in this study were nestin‐cre (Jackson Laboratory Stock Number 003 771), P0‐cre (Jackson Laboratory Stock Number 017 927), synapsin‐cre (Jackson Laboratory Stock Number 003 966), and CD11b‐cre (obtained from Don Cleveland) transgenic lines.
2.2. Synthesis of the Lpar1 conditional gene targeting vector
Creation of the Lpar1 conditional gene targeting vector was accomplished by PCR amplification of mouse Lpar1 genomic fragments using a bacterial artificial chromosome (BAC RP23‐149020 Children's Hospital Oakland Research Institute (CHORI)) containing the Lpar1 genomic locus as a template. PCR amplification was performed using Pfx50 DNA polymerase (Invitrogen) and amplified genomic fragments were assembled into pBluescript II. During the process of assembly, a loxP site was inserted into a HindIII site 5′ of Lpar1 exon 3 and a neomycin cassette under the control of the phosphoglycerate kinase promoter (PGK‐neo) flanked by loxP sites was inserted directionally (all loxP sites in the same orientation) into an XbaI site 3′ of Lpar1 exon 3 (Figure 1A). The construct was engineered so that 3.4 and 6.7 kb of Lpar1 genomic DNA flanked the PGK‐neo insertion site. To aid in cloning, BamHI and AatII restriction enzyme sites were added to the distal 5′ and 3′ ends of the Lpar1 genomic segment chosen for targeting vector design. An EcoRI restriction enzyme site was included in the loxP flanked PGK‐neo cassette to identify ES cell clones containing an allele that recombined homologously with the targeting vector.
Figure 1.
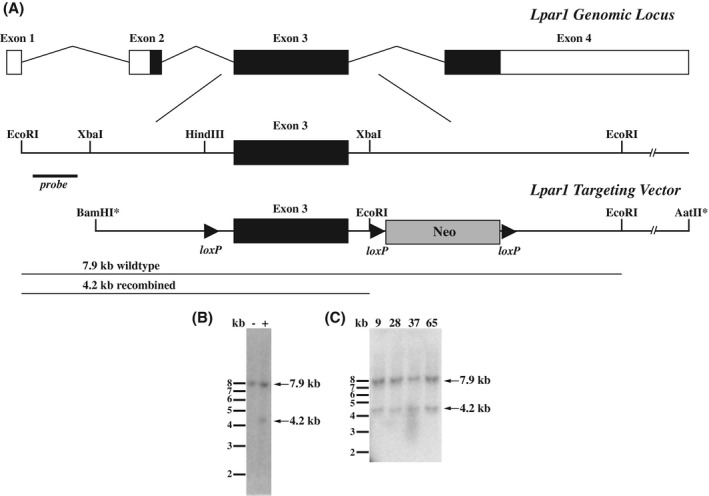
Conditional gene targeting of the Lpar1 gene locus and identification of ES cells positive for homologous recombination. A, Schematic of the Lpar1 genomic locus, the region used for gene targeting, and the Lpar1 targeting vector. In the targeting vector, loxP sites flank Lpar1 exon 3 and the neomycin cassette used for ES cell drug resistance selection screening, an introduced EcoRI site allows for identification of homologous recombination events with the indicated external probe. Asterisks represent artificial restriction enzyme sites used in the construction of the targeting vector. B, Southern blot of EcoRI digested ES cell DNA hybridized with the radiolabeled probe shown in (A) identified an ES cell clone positive (+) for homologous recombination, as indicated by the presence of a 4.2 kb band. An ES cell clone with an incorrect recombination event (−) is shown for comparison and shows only the Wt 7.9 kb Lpar1 band. C, Four identified ES cell clones (9, 28, 37, and 65) were grown and homologous recombination was reconfirmed by Southern blotting. These clones were chosen for loxP site retention screening, clones 37 and 65 were used for used for blastocyst injections.
2.3. Production of Lpar1flox/flox and Lpar1flox/flox‐cell type‐specific null mutant mice
To create the Lpar1flox/flox mice, 1 × 107 R1 ES cells were mixed with 50 µg of linearized Lpar1 targeting vector in a 0.4 cm electroporation cuvette and the cells were pulsed with a Bio‐Rad Gene Pulser II (200 mVolts × 800 µF capacitance). The electroporated ES cells were plated on mitotically inactive mouse feeder cells and allowed to recover for 24 hours at 37°C; 24 hours after electroporation and plating, 150 µg/mL Geneticin (Invitrogen) was added to the ES cell medium and the cells were placed under selection for 7 days. ES cell clones were then isolated and grown individually for subsequent DNA isolation and screening for homologous recombination events by Southern blotting and hybridization with a Lpar1 DNA probe containing sequence external to that of the 5′ end of the Lpar1 targeting vector. Clones with homologous recombination events were then screened for retention of the loxP site 5′ to Lpar1 exon 3 with the following primers: 5′ loxP Forward 5′‐gttgggacatggatgctattc‐3′ and 5′ loxP Reverse 5′‐aatctgttctcatcccacacg‐3′. Correctly targeted ES cell clones were then injected into C57BL/6J blastocysts at the TSRI Murine Genetics Core.
To delete the loxP flanked PGK‐neo cassette in vivo, gene targeted mice were crossed to nestin‐cre transgenic mice and resultant males were then bred to C57BL/6J female mice. Male mice were chosen because cre is expressed in the germline of nestin‐cre male mice. Offspring were then screened by PCR for the presence of the 5′ loxP site with the primers listed above, for the presence or absence of the PGK‐neo cassette with primers A1 Exon 3 Forward 5′‐agactgtggtcattgtgcttg‐3′ and Neo Reverse 5′‐tggatgtggaatgtgtgcgag‐3′, and for retention of the loxP site 3′ to Lpar1 exon 3 with primers 3′ loxP Forward 5′‐tgcagaattatgagtggacagg‐3′ and 3′ loxP Reverse 5′‐ggtttagtggtgtgggatcg‐3′. Mice that retained the loxP sites 5′ and 3′ to Lpar1 exon 3 but deleted the PGK‐neo cassette were selected for propagation and crossing with nestin‐cre, P0‐cre, synapsin‐cre, and CD11b‐cre transgenic mice. 32 , 33 , 34 , 35
PCR genotyping of the Lpar1 conditional mutant mice was done with the following primers: 5′ loxP Forward 5′‐gttgggacatggatgctattc‐3′, 3′ loxP Reverse 5′‐ggtttagtggtgtgggatcg‐3′, and A1 Exon 3 Forward 5′‐agactgtggtcattgtgcttg‐3′. PCR amplification of genomic DNA with these primers identified Wt, Lpar1flox, and Lpar1 deleted products of 316, 354, and 242 bp, respectively. Synapsin‐cre, CD11b, P0‐cre, and nestin‐cre transgenes were identified by PCR amplification of genomic DNA with a common reverse PCR primer, (Cre Reverse 5′‐CAG CAT TGC TGT CAC TTG GTC‐3′), and forward primers specific for synapsin (SynCreForward 5′‐CCCAAGAAGAAGAGGAAGGTG‐3′), CD11b (CD11b Forward 5′‐ACACCTCAGCCTGTCCAGTAG‐3′), P0 (MPZ Forward (P0 Cre) 5′‐ATT GGT CAC TGG CTC AAG AC‐3′), and nestin (Nestin Prom: 5′‐ACT CCC TTC TCT AGT GCT CCA‐3′) yielding products of 350 bp, 1 kb, 525 bp, and 550 bp, respectively.
2.4. Southern blotting and DNA hybridization
ES cell clones were screened for homologous recombination by digesting 10 µg of ES cell DNA with EcoRI, running the DNA on a 0.8% 1 x TAE agarose gel, and transferring the digested DNA to Nytran SuPerCharge membrane (GE Healthcare Life Sciences) in 20 × SSPE. Transferred DNA was UV crosslinked to the membrane and hybridized with a 32P‐labeled (Prime‐It II Random Primer Labeling Kit, Agilent) Lpar1 probe with sequence external to the 5′ end of the targeting vector. The 800 bp probe was produced by PCR from a BAC containing Lpar1 with the following primers: A1 Ext Forward 5′‐actgaggtcacttactcagag‐3′ and A1 Ext Reverse 5′‐gtctatggctgtggaattcaag‐3′. Probe hybridization was carried out overnight at 42°C in a .05 M pH 7.4 phosphate buffer containing 50% formamide, 5 × SSPE, 1 × Denhart's, 1% SDS, containing .1% denatured 10 mg/mL salmon sperm DNA following a 1 hour pre‐hybridization. Blots were washed and visualized using a phosphorimager. The presence of a 4.2 kb recombined band and a 7.9 kb Wt band was indicative of ES cells with homologous recombination events.
2.5. Partial sciatic nerve ligation and behavioral testing
The partial sciatic nerve ligation (PSNL) procedure was performed as described. 31 Adult Lpar1flox/flox and Lpar1flox/flox‐cre transgenic mice in a C57BL/6J background were anesthetized via nosecone delivery of isoflurane and the right limb sciatic nerve exposed and tightly ligated with 10‐0 fine sutures. The wound and skin were closed and stitched, and the animals allowed to recover. For behavioral testing, animals were acclimated in cages with wire mesh bottoms for 1 hour prior to testing in an environmentally controlled testing room. Paw withdrawal threshold (gram (g)) against increasing mechanical stimuli (0‐50 g in 20 seconds) were measured before and following PSNL surgery with tests conducted four separate times with at least a 1 minute interval between tests. The average response was normalized to presurgery controls ± SEM.
2.6. Immunohistochemistry
DRG were isolated from the lumbar region of Lpar1flox/flox control and Lpar1flox/flox‐conditional null‐mutant mice. Tissues were embedded in OCT compound and 5 µM sections were cut and immunolabeled with antibodies to mouse LPA1 (PA1 10401, Thermo Fisher Scientific) and MBP (ab134018, Abcam). Secondary antibodies were used against the listed primary antibodies and 60x images were acquired on a Zeiss Axio Imager.D2 microscope.
2.7. Reverse transcription PCR
DRG were isolated from the lumbar region of Lpar1flox/flox and Lpar1 flox/flox‐nestin‐cre conditional null‐mutant mice. DRG were placed in 1 mL of TRIzol Reagent (Thermo Fisher Scientific) and total RNA was isolated according to the manufacturer's directions. cDNA was synthesized from total RNA using a Bio‐Rad iScript cDNA synthesis kit and β‐actin and Lpar1‐specific oligonucleotide primer pairs were used to amplify target gene transcripts. Primers used to amplify a 350 bp product from β actin cDNA were M β Actin Forward 5′‐tggaatcctgtggcatccatg‐3′ and M β Actin Reverse 5′‐aaacgcagctcagtaacagtc‐3′; primers used to amplify a 194 bp product from Lpar1 cDNA were M LPA1 Forward RT 5′‐gacaccatgatgagccttctg‐3′ and M LPA1 Reverse RT 5′‐tcgcggtaggagtagatgatg‐3′. An equivalent amount of cDNA from each sample was calibrated to produce equal amounts of β‐actin PCR product to amplify the Lpar1 cDNA.
3. RESULTS
3.1. Generation of Lpar1 conditional null mutant mice
We selected a portion of the Lpar1 genomic locus for conditional gene targeting in embryonic stem (ES) cells to create a mutant mouse (Lpar1flox/flox) where Lpar1 exon 3 is selectively deleted in the presence of the cre recombinase (Figure 1A). The targeting vector contained a loxP site that was introduced into a restriction enzyme site 5′ of exon 3, and a neomycin drug selection cassette flanked by loxP sites in a restriction enzyme site 3′ of exon 3. Following electroporation of the linearized Lpar1 targeting construct, drug selection, and screening of DNA isolated from selected ES cell clones by Southern blotting and hybridization, several clones with a homologously recombined Lpar1 allele were identified (Figure 1B,C). PCR with primers flanking the 5' loxP site was used to select ES cell clones for blastocyst injection. Mice positive for germline transmission of the recombined allele were then crossed with nestin‐cre transgenic mice to produce Lpar1flox/flox‐nestin‐cre mice. 34 Because nestin is expressed in the testis, male Lpar1flox/flox‐nestin‐cre mice were bred to C57BL/6J female mice to produce offspring with germline cre‐mediated loxP site recombination. Selective deletion of the floxed neomycin cassette and retention of the 5′ loxP site in offspring were identified by PCR (Figure 2A,B). Heterozygous Lpar1flox/+ mice with the correct recombination events were then crossed together to produce Wt, Lpar1flox/+, and Lpar1flox/flox mice (Figure 2C). A high level of embryonic lethality was observed for Lpar1 constitutive null mutant mice in a C57BL/6J background, whereas Lpar1flox/flox mice in this background strain were healthy and indistinguishable from Wt littermates. Wt, Lpar1flox/+, and Lpar1flox/flox mice were differentiated by PCR (Figure 2C) and are behaviorally the same.
Figure 2.
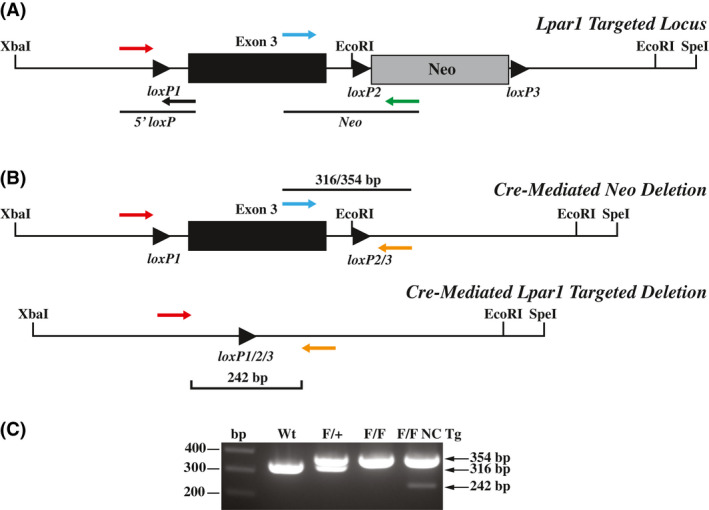
Cre‐mediated deletion in mice with a recombined Lpar1 allele. A, Schematic showing PCR primer pairs used to screen for cre‐mediated deletion of the floxed neomycin cassette. The primers shown assay for the presence of the 5′ loxP site and the presence or absence of the neomycin cassette. B, Diagrams showing the finished Lpar1 targeted allele produced through in vivo cre‐mediated deletion of the neomycin cassette (top) and cre‐mediated targeted deletion of floxed Lpar1 exon 3 (bottom). The three‐primer combination used for PCR genotyping is indicated. C, PCR genotyping of tail DNA from wildtype (Wt), Lpar1flox/+(F/+), Lpar1flox/flox(F/F), and Lpar1flox/flox‐nestin‐cre transgenic (F/F NC Tg) mice. Primers shown in (B) were used for PCR. Wt Lpar1 produced bands of 316 bp, while floxed alleles produced bands 354 bp. The presence of a 242 bp band in the Lpar1flox/flox‐nestin‐cre sample is indicative of cre‐mediated deletion in neural tissue present in the mouse tail.
3.2. Cre‐mediated Lpar1 targeted deletion
To delete Lpar1 in all neural cell types, neurons, Schwann cells, and myeloid lineage cells, Lpar1floxflox mice were crossed to nestin, synapsin, P0, and CD11b‐cre transgenic mice, respectively. 32 , 33 , 34 , 35 To confirm that Lpar1 was deleted in the presence of cre, genomic DNA was isolated from DRG of Lpar1flox/flox and Lpar1flox/flox‐nestin‐cre mice and PCR was used to verify genomic recombination of the Lpar1 genomic locus to produce a null allele (Figure 3A). DRG contain both neural and nonneural cells, with conditional deletion limited to neural cells, thus producing a recombined (neural) and unrecombined (nonneural) signal in conditional mutants. As expected, PCR products indicative of both an unrecombined and recombined Lpar1flox/flox allele can be amplified from genomic DNA isolated from Lpar1flox/flox‐nestin‐cre DRG, while only an unrecombined product can be produced from the DRG of control Lpar1flox/flox mice (Figure 3A). In agreement with genomic deletion of Lpar1, RT‐PCR showed Lpar1 mRNA transcripts are absent in Lpar1flox/flox‐nestin‐cre DRG (Figure 3B). Following Schwann cell‐specific P0‐cre crossing, PCR analyses of sciatic nerve showed deletion of Lpar1 (Figure 3C) compared to Wt. Neuronal deletion was confirmed in the cerebral cortex (Ctx) of Lpar1flox/flox‐synapsin‐cre mice (Figure 3D). Immunofluorescent labeling of peripheral myelinated axons for myelin basic protein (MBP, red) and satellite glia expressing LPA1 (green) in Wt DRG (Figure 3E) was not observed in Lpar1flox/flox‐nestin‐cre mice (Figure 3F). These data demonstrate conditional deletion of Lpar1 in the presence of targeted cre recombinase expression.
Figure 3.
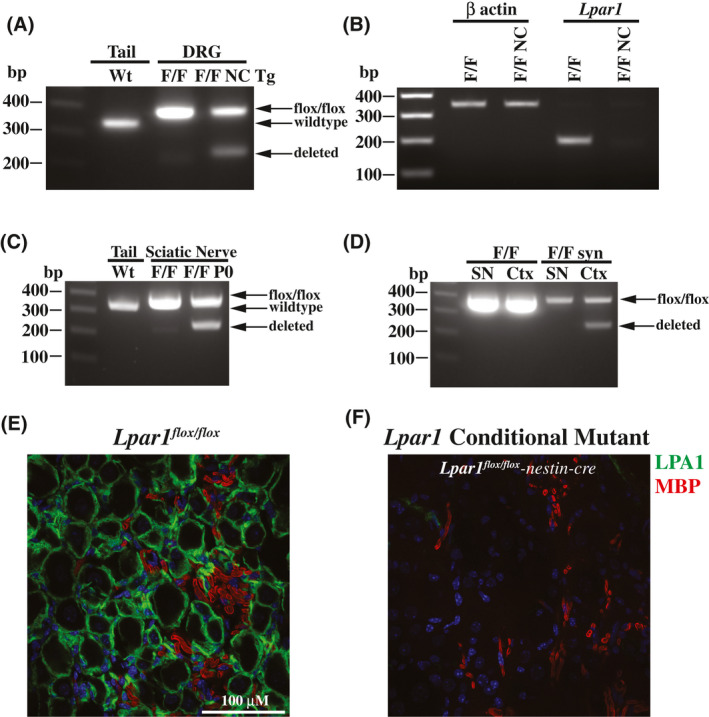
Functional deletion of Lpar1 is cre‐dependent. A, PCR products of DNA isolated from Lpar1flox/flox (F/F) and Lpar1flox/flox‐nestin‐cre transgenic (F/F NC Tg) DRG shows genomic deletion of Lpar1 exon 3 DNA from the tail of a Wt mouse is shown for comparison. B, qPCR products of cDNA prepared from Lpar1flox/flox (F/F) and Lpar1flox/flox‐nestin‐cre transgenic (F/F NC Tg) DRG shows that Lpar1 transcripts are lost in neural tissues. C, PCR of genomic DNA isolated from the tail of a Wt mouse (Wt) and the sciatic nerve of Lpar1flox/flox (F/F) and Lpar1flox/flox‐P0‐cre (F/F P0) mice. D, PCR amplification of genomic DNA isolated from the sciatic nerve (SN) and cortex (Ctx) of Lpar1flox/flox and Lpar1flox/flox‐synapsin‐cre mice. The PCR primers used for amplification of Wt (316 bp), Lpar1 floxed alleles (354 bp), and Lpar1 deleted products (242 bp) are identical to those used in Figure 2B. The 100, 200, 300, and 400 bp bands of the 1kb plus DNA ladder are indicated for reference. E and F, Immunofluorescent labeling of peripheral myelinated axons identify Wt LPA1 immunolabeling in Lpar1flox/flox mice (E) and its absence (F) in Lpar1flox/flox‐nestin‐cre transgenic mice. LPA1 labeling is in green and MBP (myelin) in red for individual samples. Scale bar = 100 µM.
3.3. Lpar1 expressing neural cell types contribute to PSNL‐induced neuropathic pain phenotypes
To determine which Lpar1 expressing neural cell types mediate PSNL‐induced neuropathic pain protection, paw withdrawal threshold responses following cre recombination for Lpar1flox / flox‐nestin, Lpar1 flox/flox‐synapsin, Lpar1 flox/flox‐P0, and Lpar1 flox/flox‐CD11b‐cre were assessed. A rescued pain phenotype was observed for all genotypes compared to controls (Figure 4A‐D). Lpar1flox/flox‐nestin‐cre conditional mutant mice challenged with PSNL had similar paw withdrawal threshold responses compared to previously defined Lpar1 constitutive null mutant mice 25 (Figure 4A). By contrast, Lpar1flox/flox‐P0‐cre mice initially responded like control mice at early time points (Figure 4B; days 3 and 6), but then showed sustained protection at later time points (Figure 4E; day 9 through day 21). Lpar1flox/flox‐synapsin‐cre mice were initially refractory to PSNL‐induced neuropathic pain (Figure 4C) but lost protection over time (Figure 4E; day 12 through day 21). Pain rescue was observed in Lpar1 flox/flox‐CD11b‐cre mice compared to Lpar1flox / flox controls, with statistically significant protection observed at day 9 post‐PSNL (Figure 4D). It is notable that the combined protection of P0 and synapsin‐cre recombination approximated the protection produced by nestin‐cre recombination (Figure 4A), implicating an additive rescue effect produced by both Schwann cells and neuronal LPA1 activation in PSNL‐initiated pain.
Figure 4.
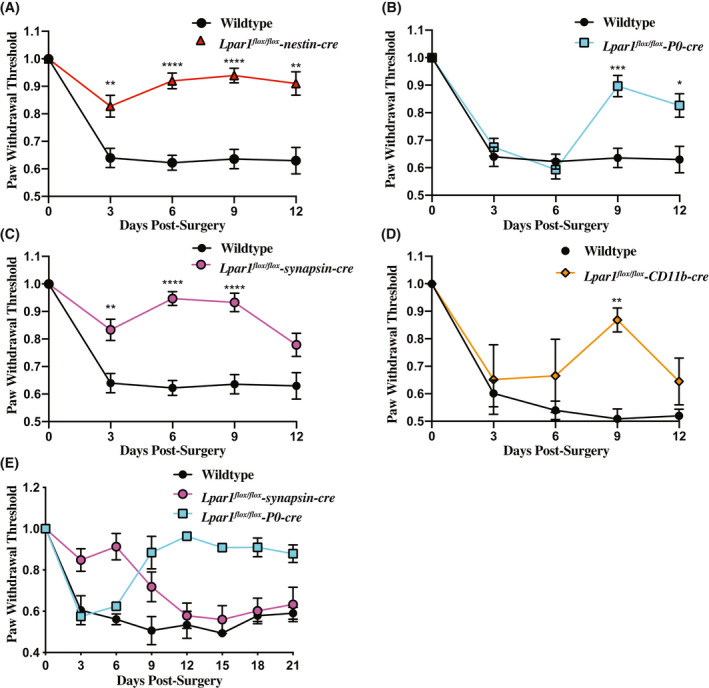
Deletion of Lpar1 in neuronal lineages protects against PSNL induced neuropathic pain. A, Targeted nestin‐cre‐mediated deletion of Lpar1 in all neural lineages protects against neuropathic pain in the PSNL mouse model. B, Schwann cell‐specific deletion of Lpar1 through a P0‐cre transgene protects mice from PSNL at later, but not earlier, time points. C, Specific deletion of Lpar1 in neurons protects mice from PSNL induced neuropathic pain only at early time points. D, Deletion of Lpar1 in CD11b expressing cell types provides protection from neuropathic pain at day 9 post‐PSNL. E, Schwann cell‐specific deletion of Lpar1 occurs at later time points and is long‐lasting. The plotted data are the average paw withdrawal threshold time observed for Lpar1 conditional null mutants normalized to Lpar1flox/flox control animal responses ± SEM. For (A, B, and C), N = 10 Lpar1flox/flox, N = 10 Lpar1flox/flox‐nestin‐cre, N = 9 Lpar1flox/flox‐P0 cre, and N = 8 Lpar1flox/flox‐synapsin‐cre animals. For (D), N = 4 Lpar1flox/flox, and N = 4 Lpar1flox/flox‐CD11b‐cre. For (E), N = 2 for all genotypes used. Statistical analysis was performed using a two‐way ANOVA followed by a Sidak's multiple comparisons test and differences were considered significant when P ≤ .05 (*=P ≤ .05, **≤.001, ***≤.001, ****P ≤ .0001)
4. DISCUSSION
Lpar1 conditional null mutant mice were generated and shown to undergo cre‐mediated recombination, enabling identification of Lpar1‐expressing neurons and Schwann cells as functionally important for the PSNL phenotype. In the absence of cre, Lpar1flox/flox mice developed a pain phenotype comparable to Wt control mice, 25 demonstrating that this new floxed mutant gene functions normally in mice subject to PSNL. Lpar1flox/flox‐nestin‐cre mice with a pan‐neural lineage deletion of Lpar1 were protected from PSNL‐induced neuropathic pain which supports neural LPA1 signaling as important despite Lpar1’s ubiquitous tissue expression. By comparison, P0 and synapsin‐cre recombination produced only partial rescue with complementary temporal phases of protection that appeared additive to account for the degree of rescue by nestin‐cre recombination. Rescue from neuropathic pain was only observed at day 9 in Lpar1flox/flox‐CD11b‐cre mice. It was recently reported that microglial depletion protects mice from PSNL‐induced thermal hyperalgesia, therefore, we cannot exclude the possibility that CD11b expressing myeloid lineage cells also play a role in PSNL‐induced neuropathic pain. 36
The actions of LPA1 in Schwann cells affecting PSNL phenotypes have not, to our knowledge, been previously reported, and the observed phenotype was unexpected with regard to the clear and differential time‐dependence of the effect. Explanations for these temporal changes in pain protection may be due to differences in de novo synthesis of LPA and the varied activation states documented for LPA1 8 , 11 , 37 , 38 , 39 , 40 that may occur in neurons and Schwann cells. Such LPA signaling effects could be altered by receptor removal to produce the time‐course differences observed for PSNL‐initiated pain rescue. Long‐lasting protection from neuropathic pain at later time points may also reflect changes in nerve myelination that may interfere with the transmission of pain stimuli as previously suggested. 14 , 25 Nerve fibers in Lpar1flox/flox‐P0‐cre mice may be abnormally myelinated, and nerve injury‐induced demyelination may alter normal pain signal transmission. However, we note that the nerve fibers that respond to noxious stimuli are lightly myelinated Aδ fibers and unmyelinated C‐fibers, 2 , 3 requiring a more complex scenario that might involve central pain consolidation through myelinated fibers.
Effects of Lpar1 deletion from neurons in Lpar1flox/flox‐synapsin‐cre mice showed early protection in PSNL, contrasting with later protection of Schwann cell receptor deletion, while supporting the involvement of neurons in LPA1‐mediated PSNL‐induced pain. Synapsin‐cre deletion is effective in CNS neurons 41 but can be less effective in peripheral (DRG) neurons, 33 , 42 implicating central neuronal mechanisms. A possible explanation for rescue at early timepoints could involve a lack of de novo LPA synthesis from Lpar1 deficient neurons. LPA can be released by neurons following nerve transection and neurons can synthesize LPA de novo through an LPA receptor dependent feed‐forward mechanism (as evidenced by LPA3). 15 , 26 , 43 De novo LPA synthesis from other cell types following PSNL may result in LPA accumulation to drive neuropathic pain at later time points, particularly through activation of Schwann cell receptors in the neuron‐specific mutants. Alternatively, PSNL may cause damage and vascular leakage that exposes peripheral nerves to LPA by activating cognate receptors to produce aberrant pain signaling. 9 , 44 , 45 , 46
Other LPA receptor subtypes can contribute in distinct ways to neuropathic pain based on analyses of different LPA receptor‐null mutants. 13 , 16 , 25 , 31 , 39 , 47 , 48 Lpar5 null mutant mice are also protected from PSNL‐induced neuropathic pain and show decreased sensitivity to acute pain stimuli and faster recovery responses when challenged in an inflammatory pain model. 31 , 49 Additionally, deletion of Lpar3 in mice prevents i.t. LPA‐induced de novo production of LPA in the dorsal horn and dorsal root and also prevents LPA‐induced allodynia and hyperalgesia, 26 suggesting an LPA3 mediated feed‐forward mechanism for LPA in neuropathic pain initiation. 27 Prevention of LPA de novo synthesis and neuropathic pain in the i.t. LPA and PSNL neuropathic pain models using minocycline combined with Lpar3 expression in microglia indicate that this feed‐forward mechanism is likely mediated by microglia. 50 , 51 In the present study, we observed a rescue effect of Lpar1 loss from microglia only at day 9 post‐PSNL, suggesting that maintained LPA3 could sustain PSNL‐initiated pain, and the possible involvement of Lpar1 expressing CD11b expressing myeloid cell‐types. Cx3cr1CreER transgenic mice that express the cre recombinase fused to a mutant estrogen ligand‐binding domain, would be useful in delineating the contribution of microglia vs other CD11b expressing cell types in PSNL, 52 which could be pursued in the future.
The generated Lpar1 conditional mutant mice will be useful in identifying other cell types involved in LPA1 signaling in neuropathic pain models, as recently described for astrocytes, 36 as well as many other conditions and disease models. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 39 , 47 , 53 , 54 The tractability of LPA1 as a member of the lysophospholipid receptor family supports its potential as a druggable GPCR 8 , 10 , 37 , 38 and the development of novel therapies that target LPA1.
CONFLICT OF INTEREST
The authors declare no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
R. Rivera, M. Lin, and J. Chun designed research; R. Rivera, M. Lin, and E. Bornhop performed research; R. Rivera created new conditional knockout mice; R. Rivera and M. Lin analyzed data; R. Rivera and J. Chun wrote the paper.
ACKNOWLEDGMENTS
We thank Dr Andras Nagy for the R1 ES cells used for gene targeting, Grace Kennedy for histology expertise, Dr Gwendolyn Kaeser for statistical analysis, and Dr Gwendolyn Kaeser and Danielle Jones for editorial assistance. Funding was provided by the NIMH of the National Institutes of Health under award number R01MH051699 to JC and non‐Federal funds from a predoctoral fellowship from Amira Pharmaceuticals to ML. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Rivera RR, Lin M‐E, Bornhop EC, Chun J. Conditional Lpar1 gene targeting identifies cell types mediating neuropathic pain. The FASEB Journal. 2020;34:8833–8842. 10.1096/fj.202000317R
Richard R. Rivera and Mu‐En Lin contributed equally to this work.
REFERENCES
- 1. van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain. 2014;155:654‐662. [DOI] [PubMed] [Google Scholar]
- 2. D'Mello R, Dickenson AH. Spinal cord mechanisms of pain. Br J Anaesth. 2008;101:8‐16. [DOI] [PubMed] [Google Scholar]
- 3. Dubin AE, Patapoutian A. Nociceptors: the sensors of the pain pathway. J Clin Invest. 2010;120:3760‐3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ueda H. Molecular mechanisms of neuropathic pain‐phenotypic switch and initiation mechanisms. Pharmacol Ther. 2006;109:57‐77. [DOI] [PubMed] [Google Scholar]
- 5. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171:3575‐3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yung YC, Stoddard NC, Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res. 2014;55:1192‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blaho VA, Chun J. “Crystal” clear? Lysophospholipid receptor structure insights and controversies. Trends Pharmacol Sci. 2018;39:953‐966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choi JW, Herr DR, Noguchi K, et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010;50:157‐186. [DOI] [PubMed] [Google Scholar]
- 10. Kihara Y, Mizuno H, Chun J. Lysophospholipid receptors in drug discovery. Exp Cell Res. 2015;333:171‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yung YC, Stoddard NC, Mirendil H, Chun J. Lysophosphatidic acid signaling in the nervous system. Neuron. 2015;85:669‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chun J, Hla T, Moolenaar W, Spiegel S, eds. Lysophospholipid Receptors : Signaling and Biochemistry. Hoboken, NJ: Wiley; 2014. [Google Scholar]
- 13. Yang AH, Ishii I, Chun J. In vivo roles of lysophospholipid receptors revealed by gene targeting studies in mice. Biochim Biophys Acta. 2002;1582:197‐203. [DOI] [PubMed] [Google Scholar]
- 14. Anliker B, Choi JW, Lin ME, et al. Lysophosphatidic acid (LPA) and its receptor, LPA1, influence embryonic schwann cell migration, myelination, and cell‐to‐axon segregation. Glia. 2013;61:2009‐2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weiner JA, Fukushima N, Contos JJ, Scherer SS, Chun J. Regulation of Schwann cell morphology and adhesion by receptor‐mediated lysophosphatidic acid signaling. J Neurosci. 2001;21:7069‐7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci U S A. 2000;97:13384‐13389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dubin AE, Herr DR, Chun J. Diversity of lysophosphatidic acid receptor‐mediated intracellular calcium signaling in early cortical neurogenesis. J Neurosci. 2010;30:7300‐7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fincher J, Whiteneck C, Birgbauer E. G‐protein‐coupled receptor cell signaling pathways mediating embryonic chick retinal growth cone collapse induced by lysophosphatidic acid and sphingosine‐1‐phosphate. Dev Neurosci. 2014;36:443‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukushima N, Weiner JA, Kaushal D, et al. Lysophosphatidic acid influences the morphology and motility of young, postmitotic cortical neurons. Mol Cell Neurosci. 2002;20:271‐282. [DOI] [PubMed] [Google Scholar]
- 20. Hecht JH, Weiner JA, Post SR, Chun J. Ventricular zone gene‐1 (vzg‐1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J Cell Biol. 1996;135:1071‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suckau O, Gross I, Schrotter S, et al. LPA1, LPA2, LPA4, and LPA6 receptor expression during mouse brain development. Dev Dyn. 2019;248:375‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Estivill‐Torrus G, Llebrez‐Zayas P, Matas‐Rico E, et al. Absence of LPA1 signaling results in defective cortical development. Cereb Cortex. 2008;18:938‐950. [DOI] [PubMed] [Google Scholar]
- 23. Matas‐Rico E, Garcia‐Diaz B, Llebrez‐Zayas P, et al. Deletion of lysophosphatidic acid receptor LPA1 reduces neurogenesis in the mouse dentate gyrus. Mol Cell Neurosci. 2008;39:342‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harrison SM, Reavill C, Brown G, et al. LPA1 receptor‐deficient mice have phenotypic changes observed in psychiatric disease. Mol Cell Neurosci. 2003;24:1170‐1179. [DOI] [PubMed] [Google Scholar]
- 25. Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712‐718. [DOI] [PubMed] [Google Scholar]
- 26. Ma L, Uchida H, Nagai J, et al. Lysophosphatidic acid‐3 receptor‐mediated feed‐forward production of lysophosphatidic acid: an initiator of nerve injury‐induced neuropathic pain. Mol Pain. 2009;5:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ueda H. Lysophosphatidic acid signaling is the definitive mechanism underlying neuropathic pain. Pain. 2017;158(Suppl 1):S55‐S65. [DOI] [PubMed] [Google Scholar]
- 28. Ma L, Nagai J, Chun J, Ueda H. An LPA species (18:1 LPA) plays key roles in the self‐amplification of spinal LPA production in the peripheral neuropathic pain model. Mol Pain. 2013;9:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ma L, Uchida H, Nagai J, Inoue M, Aoki J, Ueda H. Evidence for de novo synthesis of lysophosphatidic acid in the spinal cord through phospholipase A2 and autotaxin in nerve injury‐induced neuropathic pain. J Pharmacol Exp Ther. 2010;333:540‐546. [DOI] [PubMed] [Google Scholar]
- 30. Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205‐218. [DOI] [PubMed] [Google Scholar]
- 31. Lin ME, Rivera RR, Chun J. Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. J Biol Chem. 2012;287:17608‐17617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boillee S, Yamanaka K, Lobsiger CS, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389‐1392. [DOI] [PubMed] [Google Scholar]
- 33. Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0‐Cre transgenic mice for inactivation of adhesion molecules in schwann cells. Ann N Y Acad Sci. 1999;883:116‐123. [PubMed] [Google Scholar]
- 34. Tronche F, Kellendonk C, Kretz O, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99‐103. [DOI] [PubMed] [Google Scholar]
- 35. Zhu Y, Romero MI, Ghosh P, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ueda H, Neyama H, Nagai J, Matsushita Y, Tsukahara T, Tsukahara R. Involvement of lysophosphatidic acid‐induced astrocyte activation underlying the maintenance of partial sciatic nerve injury‐induced neuropathic pain. Pain. 2018;159:2170‐2178. [DOI] [PubMed] [Google Scholar]
- 37. Chrencik JE, Roth CB, Terakado M, et al. Crystal structure of antagonist bound human lysophosphatidic acid receptor 1. Cell. 2015;161:1633‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chun J, Kihara Y, Jonnalagadda D, Blaho VA. Fingolimod: lessons learned and new opportunities for treating multiple sclerosis and other disorders. Annu Rev Pharmacol Toxicol. 2019;59:149‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lummis NC, Sanchez‐Pavon P, Kennedy G, et al. LPA1/3 overactivation induces neonatal posthemorrhagic hydrocephalus through ependymal loss and ciliary dysfunction. Sci Adv. 2019;5:eaax2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mirendil H, Thomas EA, De Loera C, Okada K, Inomata Y, Chun J. LPA signaling initiates schizophrenia‐like brain and behavioral changes in a mouse model of prenatal brain hemorrhage. Transl Psychiatry. 2015;5:e541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choi JW, Gardell SE, Herr DR, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1‐phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A. 2011;108:751‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feltri ML, Graus Porta D, Previtali SC, et al. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fukushima N, Weiner JA, Chun J. Lysophosphatidic acid (LPA) is a novel extracellular regulator of cortical neuroblast morphology. Dev Biol. 2000;228:6‐18. [DOI] [PubMed] [Google Scholar]
- 44. Eichholtz T, Jalink K, Fahrenfort I, Moolenaar WH. The bioactive phospholipid lysophosphatidic acid is released from activated platelets. Biochem J. 1993;291(Pt 3):677‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schumacher KA, Classen HG, Spath M. Platelet aggregation evoked in vitro and in vivo by phosphatidic acids and lysoderivatives: identity with substances in aged serum (DAS). Thromb Haemost. 1979;42:631‐640. [PubMed] [Google Scholar]
- 46. Lim TK, Shi XQ, Johnson JM, et al. Peripheral nerve injury induces persistent vascular dysfunction and endoneurial hypoxia, contributing to the genesis of neuropathic pain. J Neurosci. 2015;35:3346‐3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yung YC, Mutoh T, Lin ME, et al. Lysophosphatidic acid signaling may initiate fetal hydrocephalus. Sci Transl Med. 2011;3:99ra87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ye X, Hama K, Contos JJ, et al. LPA3‐mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature. 2005;435:104‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Callaerts‐Vegh Z, Leo S, Vermaercke B, Meert T, D'Hooge R. LPA5 receptor plays a role in pain sensitivity, emotional exploration and reversal learning. Genes Brain Behav. 2012;11:1009‐1019. [DOI] [PubMed] [Google Scholar]
- 50. Ma L, Nagai J, Ueda H. Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. J Neurochem. 2010;115:643‐653. [DOI] [PubMed] [Google Scholar]
- 51. Moller T, Contos JJ, Musante DB, Chun J, Ransom BR. Expression and function of lysophosphatidic acid receptors in cultured rodent microglial cells. J Biol Chem. 2001;276:25946‐25952. [DOI] [PubMed] [Google Scholar]
- 52. Goldmann T, Wieghofer P, Muller PF, et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci. 2013;16:1618‐1626. [DOI] [PubMed] [Google Scholar]
- 53. Gennero I, Laurencin‐Dalicieux S, Conte‐Auriol F, et al. Absence of the lysophosphatidic acid receptor LPA1 results in abnormal bone development and decreased bone mass. Bone. 2011;49:395‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14:45‐54. [DOI] [PubMed] [Google Scholar]