

Abstract
In the rapidly growing group of rare genetic disorders, data scarcity demands an intelligible use of available data, in order to improve understanding of underlying pathophysiology. We hypothesize, based on the principle that clinical similarities may be indicative of shared pathophysiology, that determining phenotypic specificity could provide unsuspected insights in pathophysiology of rare genetic disorders. We explored our hypothesis by studying subunit deficiencies of the conserved oligomeric Golgi (COG) complex, a subgroup of congenital disorders of glycosylation (CDG). In this systematic data assessment, all 45 reported patients with COG‐CDG were included. The vocabulary of the Human Phenotype Ontology was used to annotate all phenotypic features and to assess occurrence in other genetic disorders. Gene occurrence ratios were calculated by dividing the frequency in the patient cohort over the number of associated genes, according to the Human Phenotype Ontology. Prioritisation based on phenotypic specificity was highly informative and captured phenotypic features commonly associated with glycosylation disorders. Moreover, it captured features not seen in any other glycosylation disorder, among which episodic fever, likely reflecting underappreciated other cellular functions of the COG complex. Interestingly, the COG complex was recently implicated in the autophagy pathway, as are more than half of the genes underlying disorders that present with episodic fever. This suggests that whereas many phenotypic features in these patients are caused by disrupted glycosylation, episodic fever might be caused by disrupted autophagy. Thus, we here demonstrate support for our hypothesis that determining phenotypic specificity could facilitate understanding of pathophysiology in rare genetic disorders.
Keywords: COG complex, conserved oligomeric Golgi complex, episodic fever, HPO, human phenotype ontology, pathophysiology, phenotypic specificity
Abbreviations
- CDG
congenital disorder of glycosylation
- COG
conserved oligomeric Golgi
- HPO
human phenotype ontology
1. INTRODUCTION
Whole exome sequencing has induced an explosion of newly discovered rare genetic disorders. Understanding the underlying pathophysiology of each of these disorders would improve disease recognition, would allow insight in a patient's disease course and would create the opportunity for conceiving treatment strategies. However, the small number of patients and the inherent scarcity of data pose a new and significant challenge which demands for an intelligible use of available data.
Attempting to meet this challenge, “deep phenotyping” is being advocated to increase insight into pathophysiology of rare disorders, by describing the physical state of a single patient as comprehensively and thoroughly as possible.1, 2 To enable interpretation, these in‐depth phenotypic descriptions of single patients should be pooled. To facilitate this process, a standard vocabulary of phenotypic abnormalities is provided by the Human Phenotype Ontology (HPO).3 Its openly accessible unambiguous terminology allows aligning in‐depth heterogeneous phenotypic descriptions. HPO thereby effectively enhances specificity of phenotypic description, so that slightly different symptoms can be better distinguished. Moreover, it allows trustworthy delineation of the frequency of phenotypic features.4
Deep phenotyping using HPO results in a large list of disease phenotypic features and their frequencies. To gain insight in pathophysiology, this list requires prioritisation. However, as exemplified by developmental delay and failure to thrive, prioritisation based on frequency alone is insufficient, as it may merely reflect the commonness of a feature across a range of genetic disorders. In contrast, features that are pathognomonic for a disease tend to reflect underlying pathophysiology. This is exemplified by the “Kayser‐Fleischer ring” in Wilson disease, indicating the copper depositions in the cornea that arise as a result of excessive copper accumulation, caused by defective copper transport. Interestingly, as illustrated by mucopolysaccharidoses, a group of disorders involving one degradation pathway, striking clinical similarities between different diseases may be indicative of shared pathophysiology.
By analogy, we here hypothesize that prioritisation based on highly specific phenotypic features in poorly understood rare genetic disorders combined with a search for better understood disorders sharing these features, would allow us to extrapolate pathophysiological insights from known diseases to less known diseases. We here test our hypothesis, by exploring a strategy focusing on systematically assessing phenotypic specificity and shared pathophysiology to improve understanding of underlying pathophysiology.
We use COG‐CDG (deficiencies in the conserved oligomeric Golgi complex (COG), causing a congenital disorder of glycosylation (CDG)) to explore this strategy, as COG‐CDG patients illustrate the problem of data scarcity and vocabulary heterogeneity in rare genetic disorders. Along with the increase in the number of identified COG‐CDG patients in the past 15 years, some insights in the pathophysiology of COG‐CDG have been gained,5 but it still remains largely unknown how the clinical picture of COG‐CDG patients evolves. In the most recent case series about COG‐CDG, all available clinical data was assembled.6 However, distilling a relevant phenotypic description from different case reports of many different authors was seriously impeded by an ambiguous nomenclature. For example, facial dysmorphism was noted in 70% of patients,6 implying phenotypic overlap, but do all patients look the same?
2. MATERIALS AND METHODS
2.1. Study population
In this systematic data assessment, all reported patients with a deficiency of one of the eight subunits of the COG complex were included. The COG complex is a tethering protein complex in the Golgi apparatus and is crucial for proper glycosylation. Since the first reported patient in 2004,7 45 patients from 32 families have been reported5, 8, 9, 10, 11 in 22 case series and case reports, with mutations in all but one (COG3) of the eight subunits (MIM: #611209, #606974, #613489, #613612, #614576, #608779, and #611182 respectively for COG1‐CDG to COG8‐CDG). All procedures followed were in accordance with the ethical standards of the Helsinki Declaration of 1975, as revised in 2000. This study does not contain any studies with human or animal subjects performed by any of the authors.
2.2. Phenotypic frequency
Phenotypic traits were aligned using the openly accessible terminology of HPO. HPO has placed all phenotypic features in a tree‐like hierarchical structure. For each reported phenotypic feature in the case series and case reports included in this study, the most specifically described HPO code was retrieved. Signs and symptoms not specifically addressed in phenotypic descriptions of COG‐CDG patients were considered absent. All first and corresponding authors of the case‐series and case‐reports were contacted to confirm the presence and absence of phenotypic features. For each of the HPO codes and all their associated super classes, the number of patients that presented with that feature was counted. Next, phenotypic frequency was calculated, dividing the number of patients that present with a phenotypic feature over the total number of patients (n = 45).
2.3. Phenotypic specificity
Phenotypic specificity was assessed by determining occurrence of the phenotypic feature in other genetic diseases. In HPO it is noted for each phenotypic feature with which genetic diseases and corresponding genes it is associated. For each phenotypic feature this number of associated genes and the list of associated genes were retrieved. HPO was last consulted on 1 November 2018.
Subsequently, for each phenotypic feature a “gene occurrence ratio” was calculated, dividing the frequency in the COG‐CDG patient cohort over the number of associated genes. To meet a minimum sensitivity, a cut‐off value of how many patients in the cohort should at least present with a phenotypic feature was needed. To explore the best cut‐off value, for each percentage of patients presenting with a phenotypic feature, the number of phenotypic features was noted, and the differences between these counts were studied. In addition, a “specificity ratio” was calculated for each percentage of patients presenting with a phenotypic feature, dividing the median gene occurrence ratio of the top 10 phenotypic features over the median gene occurrence ratio of all other phenotypic features. Data analysis was performed in R programming language. R code is supplied in Supporting Information.
2.4. Studying pathophysiology
Phenotypic features with the 10 highest occurrence ratios were studied in detail. For all genes that were associated with any of the 10 selected features, gene encoded protein functions were derived from GeneCards. Protein functions were classified into data‐driven categories. Since glycosylation is expected to play an important role in pathophysiology of COG‐CDG, these categories included the category “glycosylation”. The number of genes per category was noted to assess differences in distribution over categories for the different phenotypic features. Hereby, a special interest was in the category “glycosylation”, to assess if specific phenotypic features also occur in other glycosylation disorders.
3. RESULTS
3.1. Aligning phenotypic descriptions using HPO
All reported signs and symptoms in COG‐CDG patients were aligned and pooled by using the HPO vocabulary, allowing a multi‐faceted but unambiguous phenotypic description (Tables S1 and S2). Accuracy of the pooled data was confirmed by the corresponding author of 1 of 45 cases. From the aligned and pooled phenotypic data, listing 666 phenotypic features, recurrent features in COG‐CDG were delineated. Largely in line with the report of Rymen et al6 the nervous system is the most frequently affected organ system, affected in all COG‐CDG patients. This comprises among others of microcephaly (76%), global developmental delay (73%), and intellectual disability (53%) (Table S1). The second most frequently affected organ is the abdomen (67%), mainly due to abnormalities of the liver (60%) (Table S1).
The enhanced specificity of phenotypic description provided by HPO allowed us to differentiate whether or not a common phenotype at a higher aggregation level reflected true clinical overlap. Facial dysmorphism (present in 71%) turned out to include abnormalities of the mouth (29%), nose (27%), orbital region (27%), and skull (27%) (Table S1). This can be further distinguished in a list of many different characteristic features (Table S2). Thus, the use of an unambiguous nomenclature confirmed that COG‐CDG patients with facial dysmorphisms do not look the same at all.
Despite the harmonisation of all phenotypic descriptions using HPO, the most frequently observed features (microcephaly and global developmental delay) fail to be discriminatory, nor do they contribute to an increased pathophysiological understanding of COG‐CDG.
3.2. Determining phenotypic specificity to prioritise phenotypic features
In contrast to ascertaining phenotypic frequency, ascertaining phenotypic specificity provided more relevant prioritisation. A sliding scale was noted, ranging from one phenotypic feature that is described in all COG‐CDG patients (eg, abnormality of the nervous system, Table S1), towards >250 phenotypic features that are described only in a single COG‐CDG patient (eg, gingival bleeding, Table S2 and Figure S1). The best trade‐off between frequency and specificity was explored. We noted that for features described in at least six COG‐CDG patients the number of phenotypic features added to the total list per added patient frequency is very constant (Figure S1), suggesting that zooming in on features described in at least six COG‐CDG patients is optimal. The calculated specificity ratio (the ratio of the median gene occurrence ratio of the top 10 phenotypic features over the median gene occurrence ratio of all other phenotypic features) supported this cut‐off value of at least six patients presenting with a certain phenotype (Figure S2). A wider zoom (at least four patients) resulted in a longer list of less relevant features (Figure S3) while a narrower zoom (at least eight patients) resulted in missing relevant features (Figure S4).
Plotting all occurrence ratios of phenotypic features present in at least six COG‐CDG patients illustrated the diversity in phenotypic specificity and clearly highlighted the top 10 of very specific phenotypes (Figure 1A). Features with the highest occurrence ratios were severe hearing impairment (present in 7 patients, associated with only 5 genetic diseases) and episodic fever (present in 20 patients, associated with only 22 genetic diseases), contrasting with for example global developmental delay (present in 33 patients, but associated with 1123 other genetic diseases). Strikingly, for episodic fever even its two super classes (fever (HP:0001945) and abnormality of temperature regulation (HP:0004370)) stood out as highly specific phenotypic features (Figure 1A).
Figure 1.
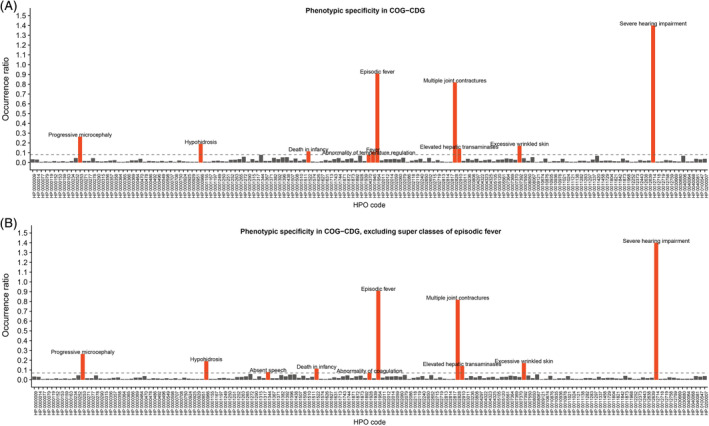
Phenotypic specificity in COG‐CDG. The ratio of the number of COG‐CDG patients over the number of HPO associated genes is depicted on the y‐axis, while all the different phenotypic features, indicated by their HPO code, are depicted on the x‐axis. A, In red, the 10 most specific COG‐CDG phenotypic features. B, In red, the 10 most specific COG‐CDG phenotypic features after excluding “fever” and “abnormality of temperature regulation” as super classes of “episodic fever”. Now “absent speech” and “abnormality of coagulation” reach top 10. CDG, congenital disorder of glycosylation; COG, conserved oligomeric Golgi; HPO, human phenotype ontology
3.3. Studying pathophysiology in diseases sharing highly specific phenotypic features
Next, we assessed potential shared pathophysiology among diseases sharing highly specific phenotypic features. Hereto, the super classes of episodic fever were excluded (Figure 1B). Gene encoded protein functions of all 677 genes associated with the 10 most specific phenotypic features (Figure 1B) were classified into 26 data‐driven categories, including the category “glycosylation” (Table 1). Demonstrating the power of this approach, seven out of the 10 extracted highly specific phenotypic features were clearly associated with other CDG (Table 1; bottom lines). This included the features elevated hepatic transaminases and abnormality of coagulation, features that are commonly associated with classical CDG and are mainly seen in CDG affecting N‐glycosylation (Figure S5). It also included absent speech, a feature described in a subset of non‐classical CDG affecting O‐mannosylation and glycosylphosphatidyl‐inositol anchoring (Figure S5). These findings corroborate the known importance of the tethering function of the COG complex for proper glycosylation and provide support for its suspected involvement in solid glycosylphosphatidylinositol anchoring. Altogether, these findings demonstrate the relevance of prioritising phenotypic features based on phenotypic specificity.
Table 1.
Ten highly specific phenotypic features in conserved oligomeric Golgi (COG)‐congenital disorder of glycosylation (CDG) associated with other genetic diseases according to human phenotype ontology (HPO)
| Severe hearing impairment | Episodic fever | Multiple joint contractures | Progressive microcephaly | Hypo‐hidrosis | Excessive wrinkled skin | Elevated hepatic transam. | Death in infancy | Absent speech | Abnormality of coagulation | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of COG‐CDG patients | 7 | 20 | 9 | 14 | 11 | 6 | 22 | 15 | 6 | 9 | |
| Number of associated genes | 5 | 22 | 11 | 53 | 58 | 35 | 157 | 132 | 77 | 127 | 677 |
| Ratio COG‐CDG patients/genes | 1.400 | 0.909 | 0.818 | 0.264 | 0.190 | 0.171 | 0.140 | 0.114 | 0.078 | 0.071 | |
| Purine and pyrimidine metabolism | 1 | 1 | 1 | 2 | 3 | 2 | 1 | 12 | |||
| DNA | 1 | 4 | 1 | 1 | 12 | 14 | 4 | 1 | 38 | ||
| Transcription | 1 | 2 | 14 | 3 | 14 | 10 | 10 | 7 | 62 | ||
| RNA | 12 | 1 | 8 | 4 | 4 | 7 | 36 | ||||
| Amino acid metabolism | 3 | 3 | 2 | 2 | 5 | 2 | 1 | 19 | |||
| Protein metabolism | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 9 | |||
| Mitosis | 2 | 1 | 3 | 1 | 5 | 1 | 1 | 5 | 8 | 27 | |
| Cell proliferation | 1 | 4 | 2 | 3 | 4 | 10 | 12 | 4 | 6 | 46 | |
| Cell adhesion | 1 | 1 | 4 | 6 | |||||||
| Cell cilia | 1 | 6 | 2 | 1 | 10 | ||||||
| Cytoskeleton | 1 | 2 | 1 | 7 | 6 | 9 | 2 | 3 | 32 | ||
| Cell signalling | 1 | 1 | 2 | 7 | 3 | 5 | 5 | 5 | 8 | 337 | |
| Cargo trafficking | 1 | 3 | 1 | 6 | 3 | 6 | 5 | 25 | |||
| Cell homeostasis | 4 | 5 | 4 | 3 | 12 | 6 | 7 | 7 | 49 | ||
| Mitochondrial homeostasis and energy metabolism | 5 | 27 | 14 | 6 | 6 | 58 | |||||
| Peroxisome biogenesis | 2 | 13 | 13 | 28 | |||||||
| Sterol and bile acid metabolism | 2 | 1 | 9 | 6 | 18 | ||||||
| Glycosaminoglycan metabolism | 1 | 3 | 1 | 5 | |||||||
| Ectodermal development | 2 | 1 | 10 | 1 | 2 | 3 | 1 | 23 | |||
| Mesodermal development | 3 | 8 | 10 | 2 | 1 | 27 | |||||
| Neurotransmitters and synapses | 3 | 2 | 3 | 1 | 3 | 12 | |||||
| Muscle cell homeostasis | 1 | 2 | 4 | 1 | 1 | 9 | |||||
| Immune system | 3 | 1 | 6 | 1 | 3 | 14 | |||||
| Coagulation | 1 | 23 | 24 | ||||||||
| Other | 1 | 1 | 7 | 2 | 1 | 12 | |||||
| Glycosylation | 1 | 3 | 2 | 15 | 5 | 8 | 17 | 51 | |||
| Glycosylation (%) | 0 | 0 | 9.1 | 5.7 | 3.4 | 0 | 9.6 | 3.8 | 10.4 | 13.3 | 7.4 |
Two out of the 10 extracted highly specific phenotypic features, severe hearing impairment and excessive wrinkled skin, are not linked to any other CDG according to HPO. However, their super classes (hearing impairment (HP:0000365) and abnormally lax or hyperextensible skin (HP:0008067)) have poor specificity (969 and 109 associated genes, ratios 0.007 and 0.055, respectively) and are associated with other CDG. According to HPO, hearing impairment has been described in seven CDG (MIM #212066; #610768; #616721; #617082; #606056; #612015, and #613661) and abnormally lax or hyperextensible skin has been described in four (MIM #612379; #219200; #617403, and #617402; Table S3).
In contrast, one of the 10 extracted highly specific phenotypic features, episodic fever, is not linked to any other CDG. According to HPO, its highly specific super classes (fever (HP:0001945) and abnormality of temperature regulation (HP:0004370) are linked to only one other CDG, TMEM165‐CDG (MIM #614727; Table S3). TMEM165 encodes a protein which is considered an ion transporter and is presumed to play a key role in manganese homeostasis, and to influence calcium and pH homeostasis of the Golgi apparatus.12 Manganese is considered a cofactor for several glycosyltransferases in the glycosylation process.12 To date, five TMEM165‐CDG patients have been described, of which two presented with recurrent episodes of unexplained fever.13, 14
The scarcity of clinical overlap with other CDG suggests a function of the COG‐complex other than glycosylation. To explore this hypothesis, we focused on other genetic diseases that present with episodic fever, to extrapolate pathophysiological knowledge from other, better understood diseases that present with episodic fever to COG‐CDG.
3.4. Studying pathophysiology in diseases presenting with episodic fever
All but two (CYP11B2 and LPIN2) of the proteins encoded by the 22 genes associated with episodic fever have an established function in temperature regulation, implying involvement of the COG complex in temperature regulation. Aspects ranged from inflammation induction and control to cytokine response, second messenger function, neuronal pathways, neurotransmitter synthesis and calcium signalling (Table 2), which at first glance appear unrelated to known functions of the COG complex. However, we noted that autophagy is thought to play a considerable role in pathophysiology in more than half of the diseases presenting with episodic fever15, 16, 17, 18, 19, 20, 21, 22, 23, 24 (Table 2). This is of interest, since the COG complex has recently been shown to be involved in the non‐selective autophagy pathway.25, 26, 27 Specifically, in the cytoplasm‐to‐vesicle pathway in yeast, the COG complex is expected to promote vesicle fusion to form double‐membrane autophagosomes and/or to deliver Atg9 to the post‐Golgi compartment.25, 26, 27
Table 2.
Genes associated to one of the most specific phenotypic features in COG‐CDG, episodic fever (HP:0001954)
| Gene | Gene name | Disease name | OMIMa | Protein function | Autophagy associatedb | Protein effect on autophagy |
|---|---|---|---|---|---|---|
| ELP1 | Elongator complex protein 1 | Neuropathy, hereditary sensory and autonomic, type III | #223900 | Inflammation induction | No | |
| PSMB8 | Proteasome subunit beta 8 | Autoinflammation, lipodystrophy and dermatosis syndrome | #256040 | No | ||
| STXBP2 | Syntaxin binding protein 2 | Familial hemophagocytic lymphohistiocytosis V | #613101 | No | ||
| MVK | Mevalonate kinase | Mevalonic aciduria | #610377 | Inflammation control | Yes15 | Impaired RhoA transloc. |
| MEFV | MEFV, pyrin innate immunity regulator | Familial Mediterranean fever | #249100 | Yes15, 16 | No NLRP3 degradation | |
| NLRP3 | NLR family pyrin domain containing 3 | Familial cold autoinflammatory syndrome I | #120100 | No | ||
| NLRP12 | NLR family pyrin domain containing 12 | Familial cold autoinflammatory syndrome II | #611762 | No | ||
| CRLF1 | Cytokine receptor like factor 1 | Cold‐induced sweating syndrome I | #272430 | Cytokine response | No | |
| LIFR | LIF receptor alpha | Stuve‐Wiedemann syndrome | #601559 | No | ||
| SLC29A3 | Solute carrier family 29 member 3 | Histiocytosis‐lymphadenopathy plus syndrome | #602782 | No | ||
| TNFAIP3 | TNF alpha induced protein 3 | Familial autoinflammatory syndrome, Behcet‐like | #616744 | Yes17 | mTORC1 inactivation | |
| TNFRSF1A | TNF receptor superfamily member 1A | Familial periodic fever, autosomal dominant | #142680 | Yes17 | No NF‐κB pathway act. | |
| GALC | Galactosylceramidase | Krabbe disease | #245200 | Second messenger | Yes18, 19 | LC3‐II overexpression |
| NGF | Nerve growth factor | Neuropathy, hereditary sensory and autonomic, type V | #608654 | Neuronal cell growth | Yes20 | PI3K signalling |
| NTRK1 | Neurotrophic receptor tyrosine kinase 1 | Congenital insensitivity to pain, with anhidrosis | #256800 | Yes21 | PI3K signalling | |
| PTS | 6‐Pyruvoyltetrahydropterin synthase | BH4‐deficient hyperphenyl. type A | #261640 | Tetrahydrobiopterin synthesis | Yes22 | mTORC1 inactivation |
| GCH1 | GTP cyclohydrolase 1 | BH4‐deficient hyperphenyl. type B | #233910 | Yes23 | mTORC1 inactivation | |
| QDPR | Quinoid dihydropteridine reductase | BH4‐deficient hyperphenyl. type C | #261630 | Yes23 | mTORC1 inactivation | |
| ORAI1 | ORAI calcium release‐activated calcium modulator 1 | Immunodeficiency 9 | #612782 | Calcium signalling | Yes24 | mTORC1 inactivation |
| STIM1 | Stromal interaction molecule 1 | Immunodeficiency 10 | #612783 | Yes24 | mTORC1 inactivation | |
| LPIN2 | Lipin 2 | Majeed syndrome | #609628 | Adipose tissue metabolism | No | |
| CYP11B2 | Cytochrome P450 family 11 subfamily b member 2 | Corticosterone methyloxidase type I deficiency | #203400 | Aldosterone synthesis | No |
OMIM, online inheritance in man phenotype number.
Ref. 12.
The links between episodic fever and autophagy, and autophagy and the COG complex suggest that episodic fever in COG‐CDG patients, unlike other COG‐CDG phenotypic features caused by the disrupted glycosylation process, might be caused by disruption of its function in the autophagy pathway. The veracity of this hypothesis remains to be further elucidated.
4. DISCUSSION
Since improving understanding of underlying pathophysiology in the rapidly growing group of rare genetic disorders demands for an intelligible use of available data, we here explored a strategy focusing on systematically assessing phenotypic specificity and shared pathophysiology. Our results confirm the power of HPO in reliably aligning heterogeneous phenotypic information from multiple case series and case reports. We showed that calculating phenotypic frequency, while revealing the most prevalent phenotypic features, did not aid pathophysiological understanding. In contrast, determining phenotypic specificity did lead the way towards features easily linked to the underlying pathophysiology. Seven features could be linked to the major known pathophysiological process glycosylation, whereas episodic fever could be linked to disturbances of cellular processes (eg, autophagy) of which the importance was previously uncertain. Thus, we here provide support for our hypothesis that phenotypic specificity could facilitate progress in understanding underlying pathophysiology of rare genetic disorders.
We note four important limitations of our approach, all regarding the risk of reporting bias. Due to importance of reporting bias for the validity of the conclusions, we here urge awareness of these limitations when our approach will be used in daily practice. Firstly, there is a risk that phenotypic features noted in case series and case reports are incorrectly attributed to a certain disease. This could be the case in genetically isolated populations, or in patients from consanguineous parents, who may harbour more than one genetic disease. As the risk for incorrectly attributed phenotypic features decreases when the number of patients increases, this limitation should be considered especially in case series with a limited sample size. Secondly, reporting bias might be introduced by the potential inclination of authors of case series and case reports to include phenotypic features that are in line with related genetic disorders, and to reduce emphasis on phenotypic features that are not known to overlap with related genetic disorders. This might cause overrepresentation of phenotypic features commonly associated with a certain disease, and underrepresentation of less usual phenotypic features. For example, when reporting on COG‐CDG patients, authors might be more inclined to report on features such as elevated hepatic transaminases and abnormality of coagulation, because these features are commonly associated with CDG. This might cause overrepresentation of CDG among genes associated with these features. Thirdly, although HPO provides a standard vocabulary, enhancing reliable alignment and pooling of phenotypic data, ascertaining phenotypic frequency and specificity heavily relies on the extent, specificity and completeness of the initial phenotypic description. We illustrate this limitation using two examples. According to HPO, the phenotypic feature abnormally lax or hyperextensible skin (HP:0008067) is defined as “abnormally loose or stretchable skin” or “abnormally loose or hyperelastic skin”. This feature is associated to 109 genes (Table S3) and when mentioned in case series and case reports, it can be readily recognised. However, it might be hard to distinguish the three underlying subclasses, excessive wrinkled skin (HP:0007392, 35 genes), cutis laxa (HP:0000973, 73 genes, defined as “inelastic skin, cutaneous laxity, generalised elastolysis, hanging skin, lax skin, dermatochalasia, skin laxity, hypoelastic skin, loose skin, chalazoderma, or dermatomegaly) and increased number of skin folds (HP:0007522, 3 genes), based on phenotypic descriptions in case series and case reports. For COG‐CDG, three patients were described as having “loose, wrinkled skin”.7, 28 These patients were noted to have both cutis laxa and excessive wrinkled skin. Three other patients were described to have “wrinkled skin”.29 For these patients, only excessive wrinkled skin was noted (Table S2). Similarly, while episodes of recurrent fever described in COG‐CDG patients were noted as episodic fever in HPO, episodes of recurrent, unexplained fever, described in TMEM165‐CDG patients,13, 14 were noted as unexplained fever in HPO. Altogether, these examples illustrate the need for an unambiguous terminology in case series and case reports, which would allow for a more trustworthy delineation of phenotypic features, especially when features are described in much detail. In addition, it illustrates the need for further improvements of the residual unambiguity of the HPO vocabulary. We advocate that when classification is still ambiguous, both HPO classifications should be noted, rather than making an arbitrary decision. Fourth and lastly, the strategy presented here depends on the completeness of HPO. Unfortunately, the HPO database is incomplete, especially in subclasses most distant from the root of the HPO‐tree. This is illustrated by the phenotypic feature hearing impairment (HP:0000365). This phenotypic feature is associated to 969 genes, according to HPO (Table S3). However, while hearing impairment is well‐specified based on the type in either conductive or sensorineural hearing loss (810/969 genes), only a few genes are further classified based on other characteristics of hearing impairment: only 15 of 969 genes on severity (mild/moderate/severe/profound hearing impairment), 9 of 969 genes on frequency (low/mid/high frequency hearing impairment), and 50 of 969 genes on the course (transient/progressive hearing impairment), clearly demonstrating the incompleteness of HPO. This is important, as this incompleteness can potentially lead to underestimation of the denominator of the gene occurrence ratio, and thus an overestimation of the degree of phenotypic specificity. Likely as a consequence of the underestimation of the number of associated genes, in COG‐CDG patients hearing impairment has poor specificity (ratio 0.0007), while severe hearing impairment has very high specificity (1.400). To overcome this limitation, we assessed whether the superclasses of highly specific phenotypic features were also highly specific, and whether these superclasses were associated to other CDG. This revealed that hearing impairment was associated to seven CDG, whereas the two superclasses of episodic fever were associated to only one CDG, underlining a potential function of the COG complex other than glycosylation.
Taking into account these limitations, phenotypic specificity can be determined as reliably as possible if (a) an unambiguous vocabulary is used, (b) both HPO classifications are noted when classification is debatable, (c) a cut‐off value of how many patients in the cohort should at least present with a phenotypic feature to meet a minimum sensitivity is calculated, (d) superclasses of potentially highly specific phenotypic features are also assessed, and (e) if potentially relevant findings are confirmed using the available literature. To further improve the validity of our approach, we advocate the use of an unambiguous vocabulary in case series and case reports, as it enhances the reliability of the type of systematic data assessment as we present here. Perhaps even more importantly, we encourage to include detailed phenotypic descriptions of all patients included in case series and case reports, for example as Supporting Information. Moreover, we urge curation and completion of HPO, to better serve deep phenotyping approaches.
Interestingly, we had the opportunity to assess the effect of the addition of newly described patients on the results of the approach, when during the development of our strategy four additional case reports were published and HPO was updated. This necessitated us to retake our steps, now including the 12 newly described COG‐CDG patients.8, 9, 10, 11 Both analyses, including first 33 and now 45 patients, were strikingly similar and both put forward episodic fever as highly specific feature not associated with any other CDG, supporting the soundness of our approach.
The approach that we present here can be performed relatively easy and rapidly by anyone with access to medical literature, HPO, and GeneCards. For this reason, we anticipate that our approach could readily find its way to daily research and clinical practice, when the problem of data scarcity hampering pathophysiological understanding of a rare genetic disease is faced. We invite anyone interested to test our hypothesis that determining phenotypic specificity could facilitate understanding of pathophysiology for their own rare genetic disorder of interest. To facilitate this, we provide a standard operating procedure, explaining the here described approach step by step (Supporting Information).
In summary, we here demonstrate support for our hypothesis that despite data scarcity, determining phenotypic specificity allows relevant prioritisation of phenotypic features in rare genetic disorders, thereby facilitating progress in understanding underlying pathophysiology in a hypothesis‐free manner.
CONFLICT OF INTEREST
H.A.H., J.J., and P.M.H. state that they have no conflict of interests to declare. None of the authors accepted any reimbursements, fees or funds from any organisation that may in any way gain or lose financially from the results of this study. The authors have not been employed by such an organisation. The authors do not have any stock ownership, or other equity interests or patent licensing arrangements.
AUTHOR CONTRIBUTIONS
H.A.H. and P.M.H. conceived the research and P.M.H. and J.J. supervised the research. H.A.H. performed the systematic data assessment and wrote the manuscript. All authors edited and reviewed the manuscript and approved the final version. P.M.H. declares that he will accept full responsibility for the work and the conduct of the study. He had access to all the data and controlled the decision to publish.
WEB RESOURCES
GeneCards (The Human Gene Database). Weizmann Institute of Science; 1996. http://genecards.org/. Accessed November 1, 2018
HPO (Human Phenotype Ontology Browser). http://compbio.charite.de/hpoweb/showterm?id=HP:0000118#id=HP_0000118. Accessed November 1, 2018
Supporting information
Data S1 Supporting Information
ACKNOWLEDGMENT
This work was supported by the personal Alexandre Suerman Stipend of the University Medical Centre Utrecht (H.A.H.).
Haijes HA, Jaeken J, van Hasselt PM. Hypothesis: determining phenotypic specificity facilitates understanding of pathophysiology in rare genetic disorders. J Inherit Metab Dis. 2020;43:701–711. 10.1002/jimd.12201
Communicating Editor: Eva Morava
Funding information Alexandre Suerman Stipend
Contributor Information
Hanneke A. Haijes, Email: h.a.siepel-3@umcutrecht.nl.
Peter M. van Hasselt, Email: p.vanhasselt@umcutrecht.nl.
REFERENCES
- 1. Haring R, Wallaschofski H. Diving through the “‐omics”: the case for deep phenotyping and systems epidemiology. Omics. 2012;16:231‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tracy RP. ‘Deep phenotyping’: characterizing populations in the era of genomics and systems biology. Curr Opin Lipidol. 2008;19:151‐157. [DOI] [PubMed] [Google Scholar]
- 3. Köhler S, Vasilevsky N, Engelstad M, et al. The human phenotype ontology in 2017. Nucl Acids Res. 2017;45:865‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Westbury SK, Turro E, Greene D, et al. Human phenotype ontology annotation and cluster analysis to unravel genetic defects in 707 cases with unexplained bleeding and platelet disorders. Genome Med. 2015;7(36):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haijes HA, Jaeken J, Foulquier F, van Hasselt PM. Hypothesis: lobe a (COG1‐4)‐CDG causes a more severe phenotype than lobe B (COG5‐8)‐CDG. J Med Genet. 2018;55:137‐142. [DOI] [PubMed] [Google Scholar]
- 6. Rymen D, Winter J, van Hasselt PM, et al. Key features and clinical variability of COG6‐CDG. Mol Genet Metab. 2015;116:163‐170. [DOI] [PubMed] [Google Scholar]
- 7. Wu X, Steet RA, Bohorov O, et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004;10:518‐523. [DOI] [PubMed] [Google Scholar]
- 8. Kim YO, Yun M, Jeong JH, et al. A mild form of COG5 defect showing early‐childhood‐onset Friedreich's‐ataxia‐like phenotypes with isolated cerebellar atrophy. J Korean Med Sci. 2017;32:1885‐1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alsubhi S, Alhashen A, Faqeih E, et al. Congenital disorders of glycosylation: the Saudi experience. Am J Med Genet A. 2017;173:2614‐2621. [DOI] [PubMed] [Google Scholar]
- 10. Althonaian N, Alsultan A, Morava E, Alfadhel M. Secondary hemophagocytic syndrome associated with COG6 gene defect: report and review. JIMD Reports. 2018;42:105‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang A, Cho SY, Jang JH, et al. Further delineation of COG8‐CDG: a case with novel compound heterozygous mutations diagnosed by targeted exome sequencing. Clin Chim Acta. 2017;471:191‐195. [DOI] [PubMed] [Google Scholar]
- 12. Dulary E, Potelle S, Legrand D, Foulquier F. TMEM165 deficiencies in congenital disorders of glycosylation type II (CDG‐II): clues and evidences for roles of the protein in Golgi functions and ion homeostasis. Tissue Cell. 2017;49:150‐156. [DOI] [PubMed] [Google Scholar]
- 13. Foulquier F, Amyere M, Jaeken J, et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am J Hum Genet. 2012;91:15‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zeevaert R, de Zegher F, Sturiale L, et al. Bone dysplasia as a key feature in three patients with a novel congenital disorder of glycosylation (CDG) type II due to a deep intronic splice mutation in TMEM165. JIMD Rep. 2013;8:145‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kwak SS, Suk J, Choi JH, et al. Autophagy induction by tetrahydrobiopterin deficiency. Autophagy. 2011;7:1323‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Si Q, Sun S, Gu Y. A278C mutation of dihydropteridine reductase decreases autophagy via mTOR signaling. Acta Biochim Biophys Sin. 2017;49:706‐712. [DOI] [PubMed] [Google Scholar]
- 17. Tanwar J, Motiani RK. Role of SOCE architects STIM and Orai proteins in cell death. Cell Calcium. 2018;69:19‐27. [DOI] [PubMed] [Google Scholar]
- 18. Ohashi Y, Munro S. Membrane delivery to the yeast autophagosome from the Golgi‐endosomal system. Mol Biol Cell. 2010;21:3998‐4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yen WL, Shintani T, Nair U, et al. The conserved oligomeric Golgi complex is involved in double‐membrane vesicle formation during autophagy. J Cell Biol. 2010;188:101‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang IH, Chen YJ, Hsu JW, Lee FJS. The Arl3 and Arl1 GTPases co‐operate with Cog8 to regulate selective autophagy via Atg9 trafficking. Traffic. 2017;18:580‐589. [DOI] [PubMed] [Google Scholar]
- 21. Hua Y, Shen M, McDonald C, Yao Q. Autophagy dysfunction in autoinflammatory diseases. J Autoimmun. 2018;88:11‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aslan D. Leukopenia in familial Mediterranean fever: case series and literature review with special emphasis on pathogenesis. Pediatr Hematol Oncol. 2014;31:120‐128. [DOI] [PubMed] [Google Scholar]
- 23. Matsuzawa Y, Oshima S, Takahara M, et al. TNFAIP3 promotes survival of CD4 T cells by restricting MTOR and promoting autophagy. Autophagy. 2015;11:1052‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ribbens JJ, Moser AB, Hubbard WC, Bongarzone ER, Maegawa GH. Characterization and application of a disease‐cell model for a neurodegenerative lysosomal disease. Mol Genet Metab. 2014;111:172‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Del Grosso A, Antoninin S, Angella L, Tonazzini I, Signore G, Cecchini M. Lithium improves cell viability in psychosine‐treated MO3.13 human oligodendrocyte cell line via autophagy activation. J Neurosci Res. 2016;94:1246‐1260. [DOI] [PubMed] [Google Scholar]
- 26. Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathic neurons: an alternative mechanism of death execution. Mol Cell Neurosci. 1999;14:180‐198. [DOI] [PubMed] [Google Scholar]
- 27. Franco ML, Melero C, Sarasola E, et al. Mutations in TrkA causing congenital insensitivity to pain with anhidrosis (CIPA) induce misfolding, aggregation, and mutation‐dependent neurodegeneration by dysfunction of the autophagic flux. J Biol Chem. 2016;291:21363‐21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rymen D, Keldermans L, Race V, et al. COG5‐CDG: expanding the clinical spectrum. Orphanet J Rare Dis. 2012;7:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morava E, Zeevaert R, Korsch E, et al. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur J Hum Genet. 2017;15(6):638‐645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting Information