

Abstract
Drug repurposing is potentially the fastest available option in the race to identify safe and efficacious drugs that can be used to prevent and/or treat COVID‐19. By describing the life cycle of the newly emergent coronavirus, SARS‐CoV‐2, in light of emerging data on the therapeutic efficacy of various repurposed antimicrobials undergoing testing against the virus, we highlight in this review a possible mechanistic convergence between some of these tested compounds. Specifically, we propose that the lysosomotropic effects of hydroxychloroquine and several other drugs undergoing testing may be responsible for their demonstrated in vitro antiviral activities against COVID‐19. Moreover, we propose that Niemann‐Pick disease type C (NPC), a lysosomal storage disorder, may provide new insights into potential future therapeutic targets for SARS‐CoV‐2, by highlighting key established features of the disorder that together result in an “unfavorable” host cellular environment that may interfere with viral propagation. Our reasoning evolves from previous biochemical and cell biology findings related to NPC, coupled with the rapidly evolving data on COVID‐19. Our overall aim is to suggest that pharmacological interventions targeting lysosomal function in general, and those particularly capable of reversibly inducing transient NPC‐like cellular and biochemical phenotypes, constitute plausible mechanisms that could be used to therapeutically target COVID‐19.
Keywords: angiotensin‐converting enzyme‐2 (ACE2), cathepsins, cholesterol, COVID‐19, lipid rafts, lysosomal storage diseases, pandemic
Abbreviations
- 25‐HC
25‐hydroxycholesterol
- 7‐KC
7‐ketocholesterol
- ACE2
angiotensin‐converting enzyme 2
- ADAM17
disintegrin and metallopeptidase domain‐containing protein 17, also known as TACE for Tumor necrosis factor (TNF)‐alpha‐converting enzyme
- COVID‐19
coronavirus disease, previously known as nCoV19 (“2019 novel coronavirus”)
- FCoV‐I
feline infectious peritonitis‐related coronavirus 1
- HCoV‐229E
human coronavirus‐229E
- LSD
lysosomal storage disease
- MERS‐CoV
middle east respiratory syndrome‐related coronavirus
- NPC
Niemann‐Pick disease type C
- ROS
reactive oxygen species
- SARS‐CoV
severe acute respiratory syndrome‐related coronavirus
- SARS‐CoV‐2
severe acute respiratory syndrome‐related coronavirus 2
- SP‐A
surfactant protein‐A
- TMPRSS2
transmembrane serine protease 2
1. THE SARS‐COV‐2 INFECTION CYCLE REQUIRES INTACT LYSOSOMAL FUNCTION
SARS‐CoV‐2 is the newly emergent coronavirus implicated in the current COVID‐19 pandemic that appeared first in Wuhan, Hubei province, China, in late December 2019. 1 Shortly after its discovery, SARS‐CoV‐2 was shown to carry nearly 80% sequence homology to SARS‐CoV, another member of the coronaviridae family that was responsible for the SARS epidemic in 2002‐2003. 2 , 3 Since then, SARS‐CoV‐2 has increasingly been viewed as molecularly “similar” to SARS‐CoV, especially in regards to carrying a similar spike (S) protein in its envelope, which mediates viral binding to host cells. 1 , 3 Specifically, the S protein docks the viral particle onto angiotensin‐converting enzyme 2 (ACE2), 4 a membrane protein particularly abundant in the plasma membrane of type II pneumocytes. 5 , 6 Upon binding ACE2 via its S protein, SARS‐CoV‐2 gets internalized and undergoes intracellular trafficking within endosomes, which eventually fuse with mature lysosomes, a requirement for viral uncoating and fusion (Figure 1A). 7 , 8 , 9
FIGURE 1.
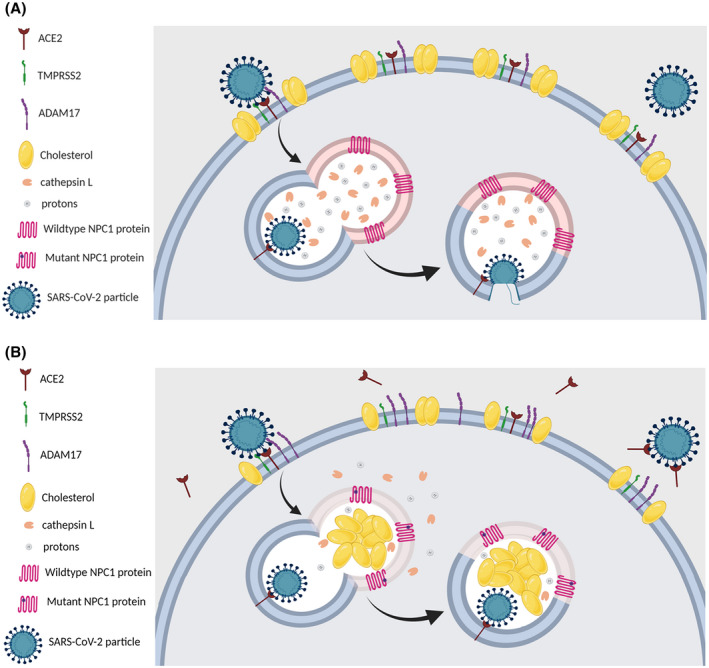
- The reduced number and cholesterol‐depleted nature of lipid rafts in the plasma membrane of NPC cells influence the stability of ACE2 and TMPRSS2 which reside within these membrane domains.
- The NPC‐related increase in plasma membrane levels of ADAM17 induces increased the shedding of ACE2, which hinders viral attachment/docking to host cells.
- The NPC‐related abnormalities in the localization and activities of cathepsin L would blunt the chances of a successful viral fusion, after the endosome carrying the viral particle fuses with the NPC1‐deficient lysosome.
- The elevated levels of the antiviral oxysterols 25‐HC and 7‐KC in NPC cells, also impede viral fusion and subsequent replication.
Altogether, these NPC‐related intracellular aberrations may reduce the likelihood of successful entry, trafficking, and fusion of SARS‐CoV‐2 in NPC cells (Figure 1B)
However, after binding ACE2, but prior to undergoing internalization, the S protein in the viral envelope must undergo enzymatic activation, formally referred to as “priming”, by the transmembrane serine protease 2, TMPRSS2, a membrane‐bound enzyme that resides within the vicinity of ACE2 in lipid rafts. 10 , 11 , 12 , 13 This TMPRSS2‐mediated cleavage of the S protein has been shown to constitute an important prerequisite for the endocytic entry of SARS‐CoV‐2 into the host cell, a process that is abrogated when cells are treated with selective inhibitors of TMPRSS2. 10 , 12 , 14 , 15
Within the lysosome, the S protein undergoes another series of enzymatic cleavages and modifications, mediated primarily by cathepsin L, and to a lesser degree, by cathepsin B, which are crucial for viral membrane fusion and subsequent release of the viral RNA genome into the host cytoplasm. 9 , 16 , 17 It is important to note that the aforementioned modifications are highly pH‐dependent, such that the fusion of the viral envelope can only occur after the endocytosed viral particle has reached a highly acidic intracellular compartment, namely the lysosome, which possesses the highest intracellular concentration and activity of cathepsin L. 7 In support of this notion, several lysosomotropic agents, that is, compounds that interfere with normal lysosomal fusion or function, have been shown to interfere with the intracellular processing of key proteins within the viral envelope that mediate fusion, thereby halting viral infectivity. 8 In fact, SARS‐CoV entry into host cells has been shown to be prevented by pre‐treating target cells with lysosomotropic agents. 15
Thus, it is reasonable to propose that, shedding ACE2 so as to reduce its presentation at the host cell membrane, disrupting its internalization, and/or interfering with its trafficking upon binding SARS‐CoV‐2 and subsequent cleavage within the lysosomes, 18 constitute three possible points of therapeutic intervention against COVID‐19. However, testing either of these therapeutic strategies may be time‐consuming and complicated, especially in the absence of an “appropriate” or representative physiological model. But, what if, there is already an existing cellular model that possesses these characteristics, that is, reduced ACE2 presentation and internalization, and impaired trafficking and fusion of ACE2‐bound SARS‐CoV‐2 particles in the lysosomes, added to several other intracellular biochemical characteristics that create an “unfavorable” host cell environment to SARS‐CoV‐2?
Here, we hypothesize that the inherent cellular and biochemical abnormalities of lysosomal storage diseases (LSDs) in general, and Niemann‐Pick disease type C (NPC) in particular, create possibly “unfavorable” environments for SARS‐CoV‐2 infectivity in the corresponding host cells (Figure 1B). Moreover, the fact that lysosomal proteases are the key mediators of coronavirus tropism and infection rates in bats, the natural reservoir of these viruses, 19 supports the importance of investigating the lysosome as a potential therapeutic target of intervention against COVID‐19.
2. NPC‐RELATED PERTURBATIONS IN ACE2 AND ADAM17 INTERFERE WITH VIRAL INTERNALIZATION
The ACE2 protein has been previously shown to reside mainly within the lipid raft domains of the plasma membrane, which are tightly packed and cholesterol‐enriched microdomains within the lipid bilayer. 20 Because they mainly consist of cholesterol and sphingomyelin, along with several other sphingolipid species, these plasma membrane microdomains (ie, lipid rafts) are easily perturbed by intrinsic or extrinsic processes that affect the synthesis and/or intracellular trafficking of either lipid species. 21 As a result, lipid rafts are disrupted in NPC, a condition notable for having impaired intracellular trafficking of both, cholesterol and sphingomyelin, 22 due to autosomal recessive mutations in NPC1 or NPC2 (90% and 4%, respectively) that result in the entrapment and accumulation of cholesterol and various sphingolipids within late endosomes and lysosomes. 23 Consequently, the partitioning of various proteins to lipid rafts, which is often a requirement for their normal functionality, is disrupted in NPC. 24 Specifically, Lusa et al, followed by Garver et al, have each independently shown that NPC cells have reduced numbers of lipid rafts in their plasma membrane, with the available ones being relatively cholesterol‐depleted, compared with those of wild‐type (ie, healthy) cells. 25 , 26 Moreover, membrane proteins that normally reside within these domains have also been shown to exhibit higher affinity for cholesterol within the membranes of NPC cells, which is believed to at least partially account for their altered functions/activities in NPC. 24 These NPC‐related alterations in lipid raft numbers and reduced cholesterol composition would, therefore, be expected to adversely impact both, the presentation and subsequent internalization and trafficking of the raft‐resident protein ACE2, which in turn, would impede SARS‐CoV‐2 entry and subsequent infection of host cells. Moreover, it has also been shown that membrane cholesterol levels play an important role in the S protein‐mediated binding of SARS‐CoV to ACE2 in the membrane of host cells, a model that finds its support in the previous work of Glende et al, who showed that cyclodextrin‐induced cholesterol‐depletion of lipid rafts in the plasma membrane abolishes the S protein‐mediated binding of SARS‐CoV particles to ACE2, reducing viral infectivity in a dose‐dependent manner. 27 Moreover, while it is not known whether SARS‐CoV requires a functional NPC1 protein to proceed through its infectious cycle as the Ebola virus does for instance, 28 it has been shown that the intracellular trafficking of the SARS‐CoV particle to NPC1‐positive compartments of the endo‐lysosomal system is important for establishing successful infection. 7 Therefore, SARS‐CoV‐2, which has a similar infectious life cycle as SARS‐CoV would also be expected to require entry into NPC1‐positive subcellular compartments, that is, late endosomes and lysosomes, in order for it to successfully establish infection. 7
Importantly, the same cells notable for their high expression of ACE2 and therefore highly susceptible to infection by SARS‐CoV‐2, that is, the type II pneumocytes, 4 , 6 also rely heavily on the functionality of the NPC1 and NPC2 proteins. The latter play key roles in modulating the lipid composition of the predominant secretory product of these cells, that is, pulmonary surfactant. 29 , 30 In fact, NPC1‐deficient type II alveolar cells contain enlarged lipid‐rich lamellar bodies within their cytoplasm, such that the surfactant these cells secrete is particularly abundant in cholesterol and surfactant protein A (SP‐A), a notable endogenous antimicrobial peptide with potent antiviral activities. 30 , 31 , 32 , 33 , 34 Thus, type II pneumocytes deficient for the NPC1 protein would be expected to not only be “altered” and possibly less recognizable to SARS‐CoV‐2 particles, but the SP‐A‐enriched pulmonary surfactant that they secrete may pose another barrier that precludes SARS‐CoV‐2 attachment/binding to these pneumocytes.
In contrast to TMPRSS2, which plays an important role in inducing key conformational changes in the S protein that promote the endocytic entry of the viral particle, 35 ADAM metallopeptidase domain 17 (ADAM17), also known as TACE (tumor necrosis factor‐α‐converting enzyme), competes with TMPRSS2 to counteract its viral entry‐promoting role. Specifically, by inducing the shedding of ACE2, ADAM17 prevents TMPRSS2 from modifying ACE2 in the manner required for viral entry. In addition, the shed ACE2 avidly binds to the S protein in the envelope of SARS‐CoV‐2 particles, thereby preventing their attachment to host cells. ADAM17 activity may, therefore, reduce the likelihood for entry of SARS‐CoV‐2 particles into host cells. 36 Intriguingly, both membrane proteases, TMPRSS2 and ADAM17, partition primarily to the detergent‐resistant membrane domains, that is, lipid rafts of the plasma membrane. 13 , 37 While no formal evaluation for the expression and/or activity level of TMPRSS2 has been reported in NPC, it is suggested that the plasma membrane levels of ADAM17 are elevated in NPC, 38 , 39 , 40 presumably due to the previously mentioned NPC‐related alterations in lipid rafts. As a result, it is possible to speculate that this elevated level of ADAM17 in the plasma membrane of NPC cells increases ACE2 shedding and counteracts TMPRSS2‐mediated entry‐favoring modifications, which grants these cells an increased “protection” against the viral binding.
3. NPC‐INDUCED ABERRATIONS IN CATHEPSIN LOCALIZATION AND ACTIVITY INTERFERE WITH VIRAL FUSION
In addition to their defective egress of intra‐lysosomal cholesterol, NPC cells are also known to have impaired localization and activities of various lysosomal enzymes, including cathepsins L and B, a finding also reported in other LSDs. 41 , 42 Specifically, the chronic accumulation of various substances within the lysosomes of NPC cells, with sphingomyelin and sphingosine being notable examples of these substances, has been found to disrupt the integrity of the lysosomal membrane, leading to the leakage of several lysosomal enzymes, such as cathepsins, into the cytosol. 41 , 43 , 44 , 45 Other studies have also shown direct inhibitory effects of the lipids accumulating in NPC toward lysosomal cathepsins. 46 Moreover, the same lipid substrates that accumulate within the lysosomes of LSD cells, have also been shown to disrupt normal acidification of the lysosomes, thereby increasing the intra‐lysosomal pH and adversely affecting the activities of enzymes within those lysosomes. 47 , 48 , 49 , 50 In fact, these substrates have also been found to simultaneously impede normal fusion of vesicles transporting lysosomal cargo, including enzymes, such as cathepsins, into the lysosome. This further depletes the intra‐lysosomal stores of key hydrolytic enzymes. 41 , 47 , 51 Thus, it is also reasonable to speculate that these NPC‐related aberrations in lysosomal enzyme localization, transport into the lysosomes, and intracellular activity levels, especially those of cathepsins B and L, pose additional “barriers” in NPC that could prevent the trafficking and intracellular processing of viral membrane proteins, a step that is required for successful viral fusion. Therefore, it may also be worth testing various inhibitors of cathepsins B and L, such as recombinant cystatins or stefins, for their therapeutic potential against SARS‐CoV‐2 infection, given their ability to induce an NPC‐resembling lysosomal dysfunctional state. 17 , 18 , 52
4. THE OXYSTEROLS THAT ACCUMULATE IN NPC POSSESS POTENT ANTIVIRAL ACTIVITIES
Among the several oxysterols that accumulate in NPC, two are particularly worth highlighting, given their potential relevance to COVID‐19, namely 7‐ketocholesterol (7‐KC) and 25‐hydroxycholesterol (25‐HC). 53 Both 7‐KC and 25‐HC have been previously reported to possess potent antiviral activities, against various viral families. 54 , 55 , 56 , 57 Specifically, elevated intracellular levels of 25‐HC, have been previously shown to reduce infectivity by several members of the coronaviridae, filoviridae (eg, Ebola virus), and flaviviridae (eg, Zika and hepatitis C viruses). 54 , 55 , 56 , 57 Similarly, increased intracellular levels of 7‐KC have been shown to interfere with viral maturation, and subsequent budding and release from host cells. 58 While the precise mechanism(s) underlying their antiviral activities remain a subject of ongoing debate, these oxysterols have been shown to perturb the normal trafficking of cholesterol, such that they replace the latter within the lipid rafts, altering their properties, which impairs subsequent viral entry, endocytic transport of viral particles, and their eventual fusion intracellularly. 56 , 59
5. THE LYSOSOMOTROPIC ACTIVITIES OF DIFFERENT DRUGS UNDERGOING TESTING AGAINST COVID‐19
Multiple available drugs are undergoing repurposed testing to evaluate their safety and efficacy against COVID‐19, in an attempt to expedite drug discovery and approval for use in treating patients with COVID‐19. 60 In what follows, we discuss a subset of these drugs currently being tested for COVID‐19, highlighting some of their lysosomotropic effects.
5.1. Chloroquine ± Azithromycin
Among the various repurposed drugs currently under investigation for the treatment of COVID‐19, chloroquine and its derivatives, such as hydroxychloroquine, emerged as the first potentially efficacious existing drug for COVID‐19, based on documented pre‐clinical efficacy and expert consensus by several Chinese scientific authorities. 61 , 62 , 63 As a result, there has been cumulative interest in testing the efficacy of chloroquine and its derivatives, in the treatment of COVID‐19, such that there are now, over 20 different related clinical trials in trial registries. 64 Additionally, two different phase III clinical trials are currently investigating the use of hydroxychloroquine for pre‐ and post‐exposure prophylaxis against COVID‐19 in healthcare workers (NCT04303507 and NCT04328285). However, it remains unclear how an anti‐malarial drug‐like chloroquine could also exert antimicrobial activity against a viral pathogen‐like SARS‐CoV‐2, raising the question of whether the same mechanism underlying chloroquine's antimalarial activity may also be responsible for its antiviral effects.
Importantly, chloroquine has been used for many years in lysosomal storage disease (LSD) research, given its ability to inhibit lysosomal fusion with endosomes, as well as inhibit the activity of various lysosomal enzymes. These properties of chloroquine allowed it to be used in vitro to pharmacologically induce transient LSD‐like cellular pathology. 65 , 66 , 67 In fact, this lysosomotropic activity of chloroquine is what is thought to be responsible for its anti‐malarial mode of action. Specifically, chloroquine is believed to undergo trafficking into the lysosomes of Plasmodium trophozoites, where it gets protonated and entrapped, thereby disrupting the fusion of these lysosomes with the “food vacuoles” (ie, phagosomes) of the trophozoites, hampering the latter's ability to feed on engulfed red blood cells. 68 , 69 However, this same propensity of chloroquine to traffic into, and concentrate within intracellular acidic organelles, also cross‐reacts with mammalian cells, inducing similar disruptions in the functions of their lysosomes as the ones it induces for protozoal food vacuoles, that is, interfering with endo‐lysosomal fusion, elevating intra‐lysosomal pH, and inducing partial permeabilization of lysosomal membranes, which altogether mirror the lysosomal pathology intrinsic to several LSDs. 65 , 69 , 70
Such lysosomal “disruptions” are actually intrinsic to NPC in particular, as previously discussed, which further supports the possibility of a lysosome‐mediated antiviral activity for chloroquine against SARS‐CoV‐2, since chloroquine is capable of inducing transient NPC‐like lysosomal abnormalities that may interfere with intracellular viral trafficking and fusion.
In support of this hypothesis, previous studies have successfully shown that the antiviral activity of chloroquine against several caliciviridae, another family of RNA viruses, occurs through chloroquine's ability to inhibit cathepsin L. 71 Furthermore, chloroquine also inhibits the transport of cholesterol out of the lysosomes, including to the plasma membrane, which would be expected to reduce the abundance, and alter the composition of membrane rafts, 72 thereby mimicking the raft alterations seen in NPC. Additionally, chloroquine has also been shown to interfere with the trimming of the N‐glycosylated side chain of ACE2, which may affect the internalization of ACE2, and subsequently, viral entry. 73 Interestingly, N‐glycosylation has actually been shown to be altered in NPC, 74 further suggesting that NPC cells likely possess inherent chloroquine‐like effects of altered N‐glycosylation modification of ACE2, which potentially further offers these cells with “protection” against SARS‐CoV‐2 infection. In that regard, a small open‐label non‐randomized clinical trial conducted in France (EU CTR 2020‐000890‐25) has recently gained considerable interest, after it showed a statistically significant difference in the rates of SARS‐CoV‐2 viral clearance from the nasal swabs of COVID‐19‐positive patients receiving a combination of hydroxychloroquine and azithromycin, the latter being a macrolide antibiotic, compared with those receiving hydroxychloroquine only (P = .002 at Day 3 post‐inclusion), or no antimicrobial therapy whatsoever (P = .005 at Day 3 post‐inclusion). 75 In this context, it is also important to highlight the lysosomotropic activity of azithromycin itself, the drug combined with hydroxychloroquine in that trial, as an add‐on therapy. 75 Similar to chloroquine or its derivatives, azithromycin also undergoes trafficking to, and accumulation within the lysosomes, where it alkalinizes the luminal pH of these organelles, thereby inhibiting the activity of resident enzymes. 76 In fact, the combination of both drugs, chloroquine and azithromycin, has been previously shown to exhibit synergistic lysosomotropic effects, especially with regards to increasing lysosomal pH. 76 Moreover, chronic azithromycin treatment in patients with cystic fibrosis has been shown to increase susceptibility to mycobacterial infections, which usually rely heavily on adequate phagocytosis and bacterial containment within phagosomes. 77 Azithromycin has been particularly shown to block the lysosome‐mediated acidification of phagosomes containing the mycobacteria, allowing the latter to escape the phagosomes and multiply uncontrollably. 77 However, in contrast to mycobacteria where the intact lysosomal function is required to contain/control the infection, 78 , 79 in SARS‐CoV‐2 infections, the intact lysosomal function is actually needed for successful viral fusion and establishment of infection, as discussed earlier. Thus, it is possible that the observed synergistic efficacy of azithromycin combination with hydroxychloroquine, in the treatment of SARS‐CoV‐2, is the result of their similar lysosome‐mediated antiviral activities, that is, their independent inhibition of endosomal‐lysosomal fusion and lysosomal proteases, which are key for successful viral fusion. In addition, azithromycin's tropism toward the lysosomes has also been shown to induce an accumulation of neutral lipids, namely free cholesterol and phospholipids, within these organelles, 80 , 81 which phenocopies the “natural” cellular phenotype of NPC cells. 23
5.2. Remdesivir
Another promising drug with documented pre‐clinical efficacy against COVID‐19, and undergoing testing in multiple different phase III clinical trials, is remdesivir (NCT04292899, NCT04292730, etc), an adenosine analog originally developed to treat Ebola. 61 , 82 However, besides its efficacy against the Ebola virus, remdesivir has been shown to possess remarkable in vivo antiviral activity against several members of the coronaviridae family, including the feline coronavirus that is implicated in feline infectious peritonitis type I (FCoV‐I), 83 as well as the middle east respiratory syndrome coronavirus (MERS‐CoV) and SARS‐CoV, 84 , 85 two human coronaviruses that belong to the same genus as SARS‐CoV‐2, that is, the beta coronaviruses. 86
Interestingly, infection by FCoV‐I, the coronavirus species showing the greatest response to remdesivir treatment, was inhibited in host cells pre‐treated with U18666A, a well‐known direct pharmacological inhibitor of the NPC1 protein that is used to induce NPC‐like lysosomal dysfunction, with a resultant lysosomal accumulation of cholesterol and sphingolipids. 87 , 88 This raises the possibility of a mechanistic convergence or synergy, albeit being a partial one, between the U18666A‐mediated direct inhibition of NPC1, and remdesivir's demonstrated activity against SARS‐CoV‐2, besides its primary mechanism of action being the inhibition of the viral RNA‐dependent RNA polymerase. 89 We, therefore, propose evaluating the antiviral potential of U18666A against SARS‐CoV‐2, given its capacity to induce an NPC cellular phenocopy, as an easy starting point to support our hypothesis. Although the safety of U18666A has not been established yet in humans, it has been shown to be without major toxicity in at least two animal models, that is, rats and cats. 88 , 90
In addition, various adenosine analogs, especially the ribose‐modified subtypes such as those used as antivirals or antineoplastic agents, have been shown to possess cross‐binding and activation or inhibition capacities for adenosine receptors, 91 with different affinities toward the A1 and A2a adenosine receptor types. 92 , 93 The reason for mentioning this is to highlight that stimulating A2a receptors has been previously shown to rescue the cholesterol entrapment seen in NPC, with that effect being abolished by A2a antagonism. 94 Thus, despite a current lack of data on remdesivir's ability to bind and subsequently, activate or inhibit any of the adenosine receptors, it is worth entertaining the possibility that, given its structural similarity to adenosine, remdesivir may potentially act as an A2a receptor antagonist, causing it to induce a transient NPC‐like cellular environment as part of its antiviral activity against SARS‐CoV‐2. In fact, several adenosine analogs have been previously shown to directly inhibit lysosomal activity in neutrophils, 95 as well as compete with intracellular adenosine on binding adenosine deaminase, an intra‐lysosomal adenosine‐metabolizing enzyme, 96 leading to the accumulation of adenosine within the lysosomal lumen 97 and inducing an LSD‐like cellular pathology with impaired regulation of lysosomal calcium stores. 98 Such features have all been previously shown to be part of the cytopathological findings in NPC. 99
5.3. Triazoles
Another class of drugs that is being considered for testing against COVID‐19, is the triazole antifungals, 100 namely posaconazole, itraconazole, and their modified derivatives. This is based on previous studies that have demonstrated potent antiviral activities for these compounds, against a wide array of coronaviridae members, including FCoV‐I, human coronavirus‐229E (HCoV‐229E), MERS‐CoV, and SARS‐CoV. 101 , 102 , 103 , 104 , 105 While triazoles are widely used as antifungal agents owing to their inhibition of ergosterol synthesis, which is an essential component of fungal cell membranes, they have also been shown to possess antiviral activities that were attributed to their differential inhibitory actions on viral helicases, including coronavirus helicase, thereby interfering with viral replication. 101 , 104 Interestingly, however, triazole antifungals are also notable for their cross‐interference with mammalian cholesterol homeostasis, including both cholesterol synthesis and trafficking, due the molecular resemblance between cholesterol and ergosterol. 106 In fact, triazoles have been actually shown to inhibit lysosomal cholesterol efflux, through directly binding to, and inhibiting the activity of NPC1 within the lysosomal membrane. 107 , 108 Thus, besides their inhibition of viral helicase, which would interfere with SARS‐CoV‐2 replication, the predicted antiviral activities of triazole antifungals against SARS‐CoV‐2 could also be mediated by their direct interference with lysosomal cholesterol egress and its eventual trafficking to the plasma membrane and membrane rafts, via NPC1‐targeted inhibition.
5.4. Glycopeptides
An additional class of drugs undergoing testing against COVID‐19 is the glycopeptide antibiotics, primarily vancomycin, teicoplanin, and their modified derivatives. 100 , 109 In vitro studies evaluating the antiviral potentials of these compounds against SARS‐CoV‐2 have successfully demonstrated their efficacy in blocking viral entry into host cells. 110 Speculations of a possible antiviral efficacy exhibited by these compounds against SARS‐CoV‐2 were based on their previously demonstrated activities against several members of the coronaviridae family, including FCoV‐I, SARS‐CoV, and MERS‐CoV. 111 , 112 Unlike their established anti‐bacterial activity that is mediated by their inhibition of cell wall synthesis, the antiviral activity of glycopeptide antibiotics is attributed to their various intracellular effects on host cells; these mainly include their direct inhibition of cathepsin L 110 , 112 and their trafficking to, and accumulation within the lysosomes, such that they “overload” the latter organelles. 113 , 114 Additionally, glycopeptide antibiotics have also been shown to induce increased ROS production within eukaryotic cells, which arguably may not be necessarily relevant for viral killing as they normally are for bacterial killing. 115 However, such findings further support our hypothesis, because NPC cells not only have an already reduced cathepsin L activity and overloaded lysosomes, but they also have elevated baseline levels of ROS intracellularly. 116 These combined intracellular effects of glycopeptides, therefore, create intracellular characteristics that mimic those intrinsic to NPC, which possibly accounts for their demonstrated antiviral activity against SARS‐CoV‐2. It is, however, worth mentioning that within the NPC patient community, several patients receiving glycopeptide antibiotics to treat hospital‐acquired pneumonia were subsequently found to develop focal pyogenic skin abscesses, whose underlying mechanism remains unclear (unpublished data). This anecdotal yet consistent finding could be due to the lysosomal accumulation of glycopeptide antibiotics, and their disruption of lysosomal function, 113 , 114 which causes these drugs to further burden the already overloaded lysosomes in NPC cells, including phagocytes, thereby hindering the latter's ability to destroy engulfed debris, resulting in abscess formation. 78 , 117
5.5. Cepharanthine
Finally, another repurposed drug being tested against COVID‐19 is cepharanthine, a plant‐derived alkaloid with prominent anti‐inflammatory effects. 118 In fact, cepharanthine demonstrated the highest potency among 2406 different clinically approved drugs that were screened against COVID‐19, with preclinical data suggesting it targets the entry of SARS‐CoV‐2. 118 Interestingly, cepharanthine has also been shown to undergo intracellular trafficking to the lysosomes, where it physically interacts with and inhibits the NPC1 protein, resulting in lysosomal cholesterol accumulation and elevated intra‐lysosomal pH. 119 It is, therefore, possible that cepharanthine's exhibited activity against SARS‐CoV‐2 is mediated, at least partially, by its lysosomotropic effects of directly inhibiting the NPC1 protein and inducing a cellular phenocopy of NPC.
6. CONCLUSION
To summarize, this report raises the hypothesis that the intracellular biochemical abnormalities inherent to LSDs in general and NPC in particular, may pose an “unfavorable” host cell environment for the entry, trafficking, and fusion of SARS‐CoV‐2. Specifically, we postulate that the altered composition of the plasma membrane and lipid rafts in NPC may affect the trafficking of ACE2, the primary host cell membrane receptor responsible for viral docking, thereby interfering with viral infection. Moreover, the increased levels of ADAM17 in the plasma membrane of NPC cells promote ACE2 shedding, thereby inhibiting viral docking at the plasma membrane of host cells. Additionally, the NPC‐related lysosomal membrane permeabilization, which leads to cathepsin L leakage, and the increased intra‐lysosomal pH seen in NPC, impair the activity of cathepsin L, a key protease required for the successful fusion of SARS‐CoV‐2. Furthermore, we highlight how two key oxysterols whose levels are notably elevated in NPC, 25‐HC, and 7‐KC, possess potent antiviral activities, which further grants NPC cells the characteristic of being an unfavorable host cell environment for successful SARS‐CoV‐2 infectivity. We also discuss how the different repurposed drugs demonstrating preliminary efficacy in the treatment of COVID‐19 (chloroquine, azithromycin, remdesivir, triazoles, glycopeptide antibiotics, and cepharanthine) possess lysosomotropic activities, which we propose as being the unifying mechanism underlying their demonstrated and shared antiviral activity against SARS‐CoV‐2. Overall, we propose that pharmacologically targeting one or more of the metabolic facets that comprise the NPC cellular phenotype, may prove beneficial in identifying and rapidly developing treatments for COVID‐19 (Figure 1).
Finally, it is important to note that most of the evidence we present here in support of a role of the lysosomes in SARS‐CoV‐2 infectivity is mainly circumstantial and inferred from studies originally designed to test other hypotheses. Thus, going forward, it will be important to test some of our proposed connections between LSD‐related lysosomal dysfunction, especially those pertaining to NPC, and reduced SARS‐CoV‐2 infectivity, for example, by comparing the rates of successful infection of human NPC vs wild‐type cells, following incubation with labeled SARS‐CoV‐2 pseudovirions. Alternatively, it would also be interesting to compare infection rates in wild‐type pneumocytes before and after the transient induction of an NPC cellular phenotype, for example, via U18666A treatment or treatment with other lysosomotropic agents. It would also be important to establish the safe yet efficacious dosage ranges for the different lysosomotropic compounds discussed here, that would be capable of sufficiently preventing SARS‐CoV‐2 infection. Such information will be key for determining the precise mechanisms of action underlying the demonstrated antiviral activities of the different agents undergoing testing against COVID‐19, which may expedite drug design and development for proper therapeutic targeting of this pandemic. Ultimately, however, large‐sized, randomized, and double‐blinded controlled clinical trials are needed to determine the safety and efficacy of any drug that may be used in the future for COVID‐19, including for the various agents/compounds we described here.
CONFLICT OF INTEREST
All authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
AUTHOR CONTRIBUTIONS
R.A. Ballout conceived and researched the hypothesis proposed in this manuscript and drafted the entire preliminary version of it. A.T. Remaley revised and edited the first draft, and with R.A. Ballout, added a conclusion section. A.T. Remaley then consulted a virologist (M. Bukrinsky), and an expert in intracellular lipid metabolism and lipid rafts (D. Sviridov), for them to revise, update and edit the sections of the draft falling within their areas of expertise. R.A. Ballout consulted with two outside NPC experts, who revised and edited the draft, shared some valuable reference with R.A. Ballout, and helped in updating the figure. However, they opted to remain unknown at their own discretion, after we offered them co‐authorship status, or at least, to acknowledge them for all their efforts.
Ballout RA, Sviridov D, Bukrinsky MI, Remaley AT. The lysosome: A potential juncture between SARS‐CoV‐2 infectivity and Niemann‐Pick disease type C, with therapeutic implications. The FASEB Journal. 2020;34:7253–7264. 10.1096/fj.202000654R
Funding information
RB and AR are supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute (NHLBI) [HL006092] at the National Institutes of Health. MB and D.S are supported by a research grant from the National Heart, Lung, and Blood Institute (NHLBI) under award number [R01‐HL131473]. MB is also supported by the District of Columbia Center for AIDS Research (DC‐CFAR), an NIH‐funded program [P30‐AI117970]
REFERENCES
- 1. Zhu NA, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilder‐Smith A, Chiew CJ, Lee VJ. Can we contain the COVID‐19 outbreak with the same measures as for SARS? Lancet Infect Dis. 2020;20:E102‐E107. 10.1016/S1473-3099(20)30129-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID‐19 coronavirus (SARS‐CoV‐2) based on SARS‐CoV immunological studies. Viruses. 2020;12(3):254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li W, Moore MJ, Vasilieva N, et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. To KF, Lo AW. Exploring the pathogenesis of severe acute respiratory syndrome (SARS): the tissue distribution of the coronavirus (SARS‐CoV) and its putative receptor, angiotensin‐converting enzyme 2 (ACE2). J Pathol. 2004;203(3):740‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mingo RM, Simmons JA, Shoemaker CJ, et al. Ebola virus and severe acute respiratory syndrome coronavirus display late cell entry kinetics: evidence that transport to NPC1+ endolysosomes is a rate‐defining step. J Virol. 2015;89(5):2931‐2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang H, Yang P, Liu K, et al. SARS coronavirus entry into host cells through a novel clathrin‐ and caveolae‐independent endocytic pathway. Cell Res. 2008;18(2):290‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bosch BJ, Bartelink W, Rottier PJ. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J Virol. 2008;82(17):8887‐8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reinke LM, Spiegel M, Plegge T, et al. Different residues in the SARS‐CoV spike protein determine cleavage and activation by the host cell protease TMPRSS2. PLoS ONE. 2017;12(6):e0179177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bertram S, Dijkman R, Habjan M, et al. TMPRSS2 activates the human coronavirus 229E for cathepsin‐independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol. 2013;87(11):6150‐6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shulla A, Heald‐Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85(2):873‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heald‐Sargent T, Gallagher T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses. 2012;4(4):557‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nat Commun. 2020;11(1):1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang IC, Bosch BJ, Li F, et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2‐expressing cells. J Biol Chem. 2006;281(6):3198‐3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond Sl, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A. 2005;102(33):11876‐11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shah PP, Wang T, Kaletsky RL, et al. A small‐molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol Pharmacol. 2010;78(2):319‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng Y, Shang J, Yang Y, et al. Lysosomal proteases are a determinant of coronavirus tropism. J Virol. 2018;92(24):e01504‐18 10.1128/JVI.01504-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lingwood D, Simons K. Lipid rafts as a membrane‐organizing principle. Science. 2010;327(5961):46‐50. [DOI] [PubMed] [Google Scholar]
- 21. Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol. 2010;11(10):688‐699. [DOI] [PubMed] [Google Scholar]
- 22. Devlin C, Pipalia NH, Liao X, Schuchman EH, Maxfield FR, Tabas I. Improvement in lipid and protein trafficking in Niemann‐Pick C1 cells by correction of a secondary enzyme defect. Traffic. 2010;11(5):601‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patterson M. Niemann‐Pick disease type C In: Adam MP, Ardinger HH, Pagon RA, et al., GeneReviews®. Seattle, WA: University of Washington; 2000:1993‐2020. [PubMed] [Google Scholar]
- 24. Vainio S, Bykov I, Hermansson M, Jokitalo E, Somerharju P, Ikonen E. Defective insulin receptor activation and altered lipid rafts in Niemann‐Pick type C disease hepatocytes. Biochem J. 2005;391(Pt 3):465‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garver WS, Krishnan K, Gallagos JR, Michikawa M, Francis GA, Heidenreich RA. Niemann‐Pick C1 protein regulates cholesterol transport to the trans‐Golgi network and plasma membrane caveolae. J Lipid Res. 2002;43(4):579‐589. [PubMed] [Google Scholar]
- 26. Lusa S, Blom TS, Eskelinen EL, et al. Depletion of rafts in late endocytic membranes is controlled by NPC1‐dependent recycling of cholesterol to the plasma membrane. J Cell Sci. 2001;114(Pt 10):1893‐1900. [DOI] [PubMed] [Google Scholar]
- 27. Glende J, Schwegmann‐Wessels C, Al‐Falah M, et al. Importance of cholesterol‐rich membrane microdomains in the interaction of the S protein of SARS‐coronavirus with the cellular receptor angiotensin‐converting enzyme 2. Virology. 2008;381(2):215‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carette JE, Raaben M, Wong AC, et al. Ebola virus entry requires the cholesterol transporter Niemann‐Pick C1. Nature. 2011;477(7364):340‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roszell BR, Tao J‐Q, Yu KJ, Huang S, Bates SR. Characterization of the Niemann‐Pick C pathway in alveolar type II cells and lamellar bodies of the lung. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L919‐L932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodriguez‐Gil JL, Watkins‐Chow DE, Baxter LL, et al. NPC1 deficiency in mice is associated with fetal growth restriction, neonatal lethality and abnormal lung pathology. J Clin Med. 2019;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lawson PR, Reid KB. The roles of surfactant proteins A and D in innate immunity. Immunol Rev. 2000;173:66‐78. [DOI] [PubMed] [Google Scholar]
- 32. Roszell BR, Tao J‐Q, Yu KJ, et al. Pulmonary abnormalities in animal models due to Niemann‐Pick type C1 (NPC1) or C2 (NPC2) disease. PLoS ONE. 2013;8(7):e67084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hartshorn KL. Role of surfactant protein A and D (SP‐A and SP‐D) in human antiviral host defense. Front Biosci (Schol Ed). 2010;2:527‐546. [DOI] [PubMed] [Google Scholar]
- 34. Li L, Zheng Q, Zhang Y, et al. Antiviral activity of recombinant porcine surfactant protein A against porcine reproductive and respiratory syndrome virus in vitro. Arch Virol. 2016;161(7):1883‐1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shirato K, Kawase M, Matsuyama S. Wild‐type human coronaviruses prefer cell‐surface TMPRSS2 to endosomal cathepsins for cell entry. Virology. 2018;517:9‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heurich A, Hofmann‐Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tellier E, Canault M, Rebsomen L, et al. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp Cell Res. 2006;312(20):3969‐3980. [DOI] [PubMed] [Google Scholar]
- 38. Jelinek D, Heidenreich RA, Orlando RA, Garver WS. The Niemann‐Pick C1 and caveolin‐1 proteins interact to modulate efflux of low density lipoprotein‐derived cholesterol from late endocytic compartments. J Mol Biochem. 2014;3(1):14‐26. [PMC free article] [PubMed] [Google Scholar]
- 39. Moreno‐Caceres J, Caja L, Mainez J, et al. Caveolin‐1 is required for TGF‐beta‐induced transactivation of the EGF receptor pathway in hepatocytes through the activation of the metalloprotease TACE/ADAM17. Cell Death Dis. 2014;5:e1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang H, Wang Y, Kar S. Effects of cholesterol transport inhibitor U18666A on APP metabolism in rat primary astrocytes. Glia. 2017;65(11):1728‐1743. [DOI] [PubMed] [Google Scholar]
- 41. Chung C, Puthanveetil P, Ory DS, Lieberman AP. Genetic and pharmacological evidence implicates cathepsins in Niemann‐Pick C cerebellar degeneration. Hum Mol Genet. 2016;25(7):1434‐1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vitner EB, Dekel H, Zigdon H, et al. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum Mol Genet. 2010;19(18):3583‐3590. [DOI] [PubMed] [Google Scholar]
- 43. Amritraj A, Peake K, Kodam A, et al. Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann‐Pick type C1‐deficient mice. Am J Pathol. 2009;175(6):2540‐2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gabandé‐Rodríguez E, Boya P, Labrador V, Dotti CG, Ledesma MD. High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell Death Differ. 2014;21(6):864‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kågedal K, Zhao M, Svensson I, Brunk UT. Sphingosine‐induced apoptosis is dependent on lysosomal proteases. Biochem J. 2001;359(Pt 2):335‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Elrick MJ, Yu T, Chung C, Lieberman AP. Impaired proteolysis underlies autophagic dysfunction in Niemann‐Pick type C disease. Hum Mol Genet. 2012;21(22):4876‐4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Folts CJ, Scott‐Hewitt N, Pröschel C, Mayer‐Pröschel M, Noble M. Lysosomal re‐acidification prevents lysosphingolipid‐induced lysosomal impairment and cellular toxicity. PLoS Biol. 2016;14(12):e1002583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bach G, Chen CS, Pagano RE. Elevated lysosomal pH in Mucolipidosis type IV cells. Clin Chim Acta. 1999;280(1–2):173‐179. [DOI] [PubMed] [Google Scholar]
- 49. Holopainen JM, Saarikoski J, Kinnunen PKJ, Järvelä I. Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs). Eur J Biochem. 2001;268(22):5851‐5856. [DOI] [PubMed] [Google Scholar]
- 50. Sillence DJ. Glucosylceramide modulates endolysosomal pH in Gaucher disease. Mol Genet Metab. 2013;109(2):194‐200. [DOI] [PubMed] [Google Scholar]
- 51. Fraldi A, Annunziata F, Lombardi A, et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010;29(21):3607‐3620. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52. Cermak S, Kosicek M, Mladenovic‐Djordjevic A, Smiljanic K, Kanazir S, Hecimovic S. Loss of cathepsin B and L leads to lysosomal dysfunction, NPC‐like cholesterol sequestration and accumulation of the key Alzheimer's proteins. PLoS ONE. 2016;11(11):e0167428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Porter FD, Scherrer DE, Lanier MH, et al. Cholesterol oxidation products are sensitive and specific blood‐based biomarkers for Niemann‐Pick C1 disease. Sci Transl Med. 2010;2(56):p. 56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Y, Wang S, Yi Z, et al. Interferon‐inducible cholesterol‐25‐hydroxylase inhibits hepatitis C virus replication via distinct mechanisms. Sci Rep. 2014;4:7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li C, Deng Y‐Q, Wang S, et al. 25‐Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity. 2017;46(3):446‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu S‐Y, Aliyari R, Chikere K, et al. Interferon‐inducible cholesterol‐25‐hydroxylase broadly inhibits viral entry by production of 25‐hydroxycholesterol. Immunity. 2013;38(1):92‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang Y, Song Z, Wang MI, et al. Cholesterol 25‐hydroxylase negatively regulates porcine intestinal coronavirus replication by the production of 25‐hydroxycholesterol. Vet Microbiol. 2019;231:129‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Willard K, Elling C, Stice S, Brindley M. The oxysterol 7‐ketocholesterol reduces Zika virus titers in vero cells and human neurons. Viruses. 2019;11(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Civra A, Cagno V, Donalisio M, et al. Inhibition of pathogenic non‐enveloped viruses by 25‐hydroxycholesterol and 27‐hydroxycholesterol. Sci Rep. 2014;4:7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harrison C. Coronavirus puts drug repurposing on the fast track. Nat Biotechnol. 2020;38(4):379‐381. [DOI] [PubMed] [Google Scholar]
- 61. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30(3):269‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gao J, Tian Z, Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID‐19 associated pneumonia in clinical studies. Biosci Trends. 2020;14(1):72‐73. [DOI] [PubMed] [Google Scholar]
- 63. Multicenter Collaboration Group of Department of Science and Technology of Guangdong Province and Health Commission of Guangdong Province for Chloroquine in the Treatment of Novel Coronavirus Pneumonia . [Expert consensus on chloroquine phosphate for the treatment of novel coronavirus pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43(3):185‐188. [DOI] [PubMed] [Google Scholar]
- 64. Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID‐19. J Crit Care. 2020:5. 10.1016/j.jcrc.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen PM, Gombart ZJ, Chen JW. Chloroquine treatment of ARPE‐19 cells leads to lysosome dilation and intracellular lipid accumulation: possible implications of lysosomal dysfunction in macular degeneration. Cell Biosci. 2011;1(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gonzalez‐Noriega A, Grubb JH, Talkad V, Sly WS. Chloroquine inhibits lysosomal enzyme pinocytosis and enhances lysosomal enzyme secretion by impairing receptor recycling. J Cell Biol. 1980;85(3):839‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome‐lysosome fusion. Autophagy. 2018;14(8):1435‐1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Slater AF. Chloroquine: mechanism of drug action and resistance in Plasmodium falciparum. Pharmacol Ther. 1993;57(2–3):203‐235. [DOI] [PubMed] [Google Scholar]
- 69. Homewood CA, Warhurst DC, Peters W, Baggaley VC. Lysosomes, pH and the anti‐malarial action of chloroquine. Nature. 1972;235(5332):50‐52. [DOI] [PubMed] [Google Scholar]
- 70. Ashoor R, Yafawi R, Jessen B, Lu S. The contribution of lysosomotropism to autophagy perturbation. PLoS ONE. 2013;8(11):e82481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shivanna V, Kim Y, Chang KO. Endosomal acidification and cathepsin L activity is required for calicivirus replication. Virology. 2014;465:287‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pagler TA, Neuhofer A, Laggner H, Strobl W, Stangl H. Cholesterol efflux via HDL resecretion occurs when cholesterol transport out of the lysosome is impaired. J Lipid Res. 2007;48(10):2141‐2150. [DOI] [PubMed] [Google Scholar]
- 73. Vincent MJ, Bergeron E, Benjannet S, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005;2:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kosicek M, Gudelj I, Horvatic A, et al. N‐glycome of the lysosomal glycocalyx is altered in Niemann‐Pick type C disease (NPC) model cells. Mol Cell Proteomics. 2018;17(4):631‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gautret P, Lagier JC, Parola P, et al. Hydroxychloroquine and azithromycin as a treatment of COVID‐ 19: results of an open‐label non‐randomized clinical trial. Int J Antimicrob Agents. 2020;105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76. Nujić K, Banjanac M, Munić V, Polančec D, Eraković Haber V. Impairment of lysosomal functions by azithromycin and chloroquine contributes to anti‐inflammatory phenotype. Cell Immunol. 2012;279(1):78‐86. [DOI] [PubMed] [Google Scholar]
- 77. Renna M, Schaffner C, Brown K, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest. 2011;121(9):3554‐3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fineran P, Lloyd‐Evans E, Lack NA, et al. Pathogenic mycobacteria achieve cellular persistence by inhibiting the Niemann‐Pick Type C disease cellular pathway. Wellcome Open Res. 2016;1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Koo IC, Ohol YM, Wu P, Morisaki JH, Cox JS, Brown EJ. Role for lysosomal enzyme beta‐hexosaminidase in the control of mycobacteria infection. Proc Natl Acad Sci U S A. 2008;105(2):710‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu Y, Kam WR, Ding J, Sullivan DA. One man's poison is another man's meat: using azithromycin‐induced phospholipidosis to promote ocular surface health. Toxicology. 2014;320:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu Y, Kam WR, Ding J, Sullivan DA. Effect of azithromycin on lipid accumulation in immortalized human meibomian gland epithelial cells. JAMA Ophthalmol. 2014;132(2):226‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS‐5734 against Ebola virus in rhesus monkeys. Nature. 2016;531(7594):381‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pedersen NC, Perron M, Bannasch M, et al. Efficacy and safety of the nucleoside analog GS‐441524 for treatment of cats with naturally occurring feline infectious peritonitis. J Feline Med Surg. 2019;21(4):271‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. de Wit E, Feldmann F, Cronin J, et al. Prophylactic and therapeutic remdesivir (GS‐5734) treatment in the rhesus macaque model of MERS‐CoV infection. Proc Natl Acad Sci U S A. 2020;117(12):6771‐6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sheahan TP, Sims AC, Graham RL, et al. Broad‐spectrum antiviral GS‐5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017;9(396):eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS‐CoV‐2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Takano T, Endoh M, Fukatsu H, Sakurada H, Doki T, Hohdatsu T. The cholesterol transport inhibitor U18666A inhibits type I feline coronavirus infection. Antiviral Res. 2017;145:96‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Doki T, Tarusawa T, Hohdatsu T, Takano T. Vivo antiviral effects of u18666a against type i feline infectious peritonitis virus. Pathogens. 2020;9(1):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tchesnokov E, Feng J, Porter D, Götte M. Mechanism of inhibition of ebola virus RNA‐dependent RNA polymerase by remdesivir. Viruses. 2019;11(4):326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cenedella RJ, Jacob R, Borchman D, et al. Direct perturbation of lens membrane structure may contribute to cataracts caused by U18666A, an oxidosqualene cyclase inhibitor. J Lipid Res. 2004;45(7):1232‐1241. [DOI] [PubMed] [Google Scholar]
- 91. Taylor MD, Moos WH, Hamilton HW, et al. Ribose‐modified adenosine analogues as adenosine receptor agonists. J Med Chem. 1986;29(3):346‐353. [DOI] [PubMed] [Google Scholar]
- 92. Jensen K, Johnson LA, Jacobson PA, et al. Cytotoxic purine nucleoside analogues bind to A1, A2A, and A3 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 2012;385(5):519‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Janać B, Pešić V, Peković S, Rakić L, Stojiljković M. Different effects of adenosine A1 agonist ribavirin on amphetamine‐induced total locomotor and stereotypic activities in rats. Ann N Y Acad Sci. 2005;1048:396‐399. [DOI] [PubMed] [Google Scholar]
- 94. Visentin S, De Nuccio C , Bernardo A, et al. The stimulation of adenosine A2A receptors ameliorates the pathological phenotype of fibroblasts from Niemann‐Pick type C patients. J Neurosci. 2013;33(39):15388‐15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Schrier DJ, Imre KM. The effects of adenosine agonists on human neutrophil function. J Immunol. 1986;137(10):3284‐3289. [PubMed] [Google Scholar]
- 96. Lindley ER, Pisoni RL. Demonstration of adenosine deaminase activity in human fibroblast lysosomes. Biochem J. 1993;290(Pt 2):457‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wu JZ, Lin CC, Hong Z. Ribavirin, viramidine and adenosine‐deaminase‐catalysed drug activation: implication for nucleoside prodrug design. J Antimicrob Chemother. 2003;52(4):543‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhong XZ, Zou Y, Sun X, et al. Inhibition of transient receptor potential channel mucolipin‐1 (TRPML1) by lysosomal adenosine involved in severe combined immunodeficiency diseases. J Biol Chem. 2017;292(8):3445‐3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lloyd‐Evans E, Morgan AJ, He X, et al. Niemann‐Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14(11):1247‐1255. [DOI] [PubMed] [Google Scholar]
- 100. Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019‐nCoV). Nat Rev Drug Discov. 2020;19(3):149–150. [DOI] [PubMed] [Google Scholar]
- 101. Adedeji AO, Singh K, Kassim A, et al. Evaluation of SSYA10‐001 as a replication inhibitor of severe acute respiratory syndrome, mouse hepatitis, and Middle East respiratory syndrome coronaviruses. Antimicrob Agents Chemother. 2014;58(8):4894‐4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Karypidou K, Ribone SR, Quevedo MA, et al. Synthesis, biological evaluation and molecular modeling of a novel series of fused 1,2,3‐triazoles as potential anti‐coronavirus agents. Bioorg Med Chem Lett. 2018;28(21):3472‐3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Takano T, Akiyama M, Doki T, Hohdatsu T. Antiviral activity of itraconazole against type I feline coronavirus infection. Vet Res. 2019;50(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zaher NH, Mostafa MI, Altaher AY. Design, synthesis and molecular docking of novel triazole derivatives as potential CoV helicase inhibitors. Acta Pharm. 2020;70(2):145‐159. [DOI] [PubMed] [Google Scholar]
- 105. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS‐CoV‐2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020:23. 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zarn JA, Bruschweiler BJ, Schlatter JR. Azole fungicides affect mammalian steroidogenesis by inhibiting sterol 14 alpha‐demethylase and aromatase. Environ Health Perspect. 2003;111(3):255‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Trinh MN, Lu F, Li X, et al. Triazoles inhibit cholesterol export from lysosomes by binding to NPC1. Proc Natl Acad Sci U S A. 2017;114(1):89‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Long T, Qi X, Hassan A, Liang Q, De Brabander JK, Li X. Structural basis for itraconazole‐mediated NPC1 inhibition. Nat Commun. 2020;11(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Baron SA, Devaux C, Colson P, Raoult D, Rolain JM. Teicoplanin: an alternative drug for the treatment of coronavirus COVID‐19? Int J Antimicrob Agents. 2020;55(4):105944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang J, Ma X, Yu F, et al. Teicoplanin potently blocks the cell entry of 2019‐nCoV. BioRxiv, 2020. 10.1101/2020.02.05.935387. [DOI] [Google Scholar]
- 111. Balzarini J, Keyaerts E, Vijgen L, et al. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antiviral Res. 2006;72(1):20‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zhou N, Pan T, Zhang J, et al. Glycopeptide antibiotics potently inhibit cathepsin L in the late endosome/lysosome and block the entry of ebola virus, middle east respiratory syndrome coronavirus (MERS‐CoV), and severe acute respiratory syndrome coronavirus (SARS‐CoV). J Biol Chem. 2016;291(17):9218‐9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Barcia‐Macay M, Mouaden F, Mingeot‐Leclercq M‐P, Tulkens PM, Van Bambeke F. Cellular pharmacokinetics of telavancin, a novel lipoglycopeptide antibiotic, and analysis of lysosomal changes in cultured eukaryotic cells (J774 mouse macrophages and rat embryonic fibroblasts). J Antimicrob Chemother. 2008;61(6):1288‐1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Beauchamp D, Gourde P, Simard M, Bergeron MG. Subcellular localization of tobramycin and vancomycin given alone and in combination in proximal tubular cells, determined by immunogold labeling. Antimicrob Agents Chemother. 1992;36(10):2204‐2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yano T, Sakamoto Y, Hanada Y, Takeshita A, Inagaki F, Masuda S. Vancomycin induced renal tubular cell injury by reactive oxygen species generation and mitochondrial dysfunction. FASEB J. 2015;29(1_supplement): 777.1. [Google Scholar]
- 116. Fu R, Yanjanin NM, Bianconi S, Pavan WJ, Porter FD. Oxidative stress in Niemann‐Pick disease, type C. Mol Genet Metab. 2010;101(2–3):214‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Berg RD, Levitte S, O'Sullivan MP, et al. Lysosomal disorders drive susceptibility to tuberculosis by compromising macrophage migration. Cell. 2016;165(1):139‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Fan H‐H, Wang L‐Q, Liu W‐L, et al. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019‐novel coronavirus (2019‐nCoV) related coronavirus model. Chin Med J (Engl). 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lyu J, Yang EJ, Head SA, et al. Pharmacological blockade of cholesterol trafficking by cepharanthine in endothelial cells suppresses angiogenesis and tumor growth. Cancer Lett. 2017;409:91‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]