

To the Editor,
The airway epithelial barrier in asthma may be more susceptible to damage and/or less capable of repair in response to aeroallergens like house dust mite (HDM), resulting in loss of barrier function, as evidenced by reduced expression of cell‐cell adhesion protein E‐cadherin.1 This may not only lead to increased permeability to allergens and impaired epithelial differentiation, but also result in increased pro‐inflammatory responses, including the release of cytokines such as CCL20, GM‐CSF, CCL17, IL‐33 and TSLP to attract and activate immune cells.2 Epithelial cells from asthma patients display a more basal phenotype than healthy epithelium, along with an inability to re‐differentiate into a functionally intact epithelium and reconstitute normal barrier function upon damage by allergens.3 The loss of E‐cadherin releases β‐catenin, which translocates to the nucleus, inducing divergent gene expression profiles depending on recruitment of different transcriptional co‐activators. Recruitment of CREB‐binding protein (CBP) results in expression of genes associated with epithelial de‐differentiation, migration and proliferation, while p300 induces gene transcription associated with cell differentiation.4 We previously observed that epithelial exposure to HDM results in E‐cadherin and β‐catenin loss from adherens junctions, accompanied by increased CCL20 release, and that asthma‐derived airway epithelial cells are more susceptible to these HDM‐induced effects. It is currently unknown whether dysregulated β‐catenin signalling contributes to this abnormal epithelial phenotype in asthma.
We hypothesized that binding of β‐catenin to p300 suppresses pro‐inflammatory responses and improves epithelial barrier function. We tested this by pharmacological inhibition of β‐catenin/CBP activity using ICG‐001, a highly specific inhibitor of β‐catenin/CBP binding promoting β‐catenin/p300 binding,5 in human primary airway epithelial cells (PAECs). We assessed effects on HDM‐induced CCL20 release and barrier function at baseline and after injury.
We first investigated whether β‐catenin/CBP signalling regulates epithelial pro‐inflammatory responses by quantifying the effect of ICG‐001 on HDM‐induced CCL20 release. HDM significantly increased CCL20 production in PAECs obtained by bronchial brushings from both asthma and healthy donors (Figure 1A,C). As there was no significant difference in HDM‐induced CCL20 increase between PAECs from asthma (4.4 ± 2.0‐fold) and healthy donors (2.9 ± 1.1‐fold), we combined the groups to assess the effect of ICG‐001. Pretreatment with ICG‐001 significantly decreased the HDM‐induced CCL20 release in the combined group of asthma and heathy donors (0.48 ± 0.35‐fold, Figure 1B,D). Similar effects were observed for GM‐CSF (Figure S1A,B), where ICG‐001 significantly reduced basal and HDM‐induced GM‐CSF levels, while CCL17, TSLP and IL‐33 were not detectable. Furthermore, a similar effect of HDM on CCL20 was observed in PAECs derived from human tracheobronchial tissue of normal lung transplant donors, with a trend towards inhibition of HDM‐induced CCL20 levels by ICG‐001 (Figure S1C,D).
Figure 1.
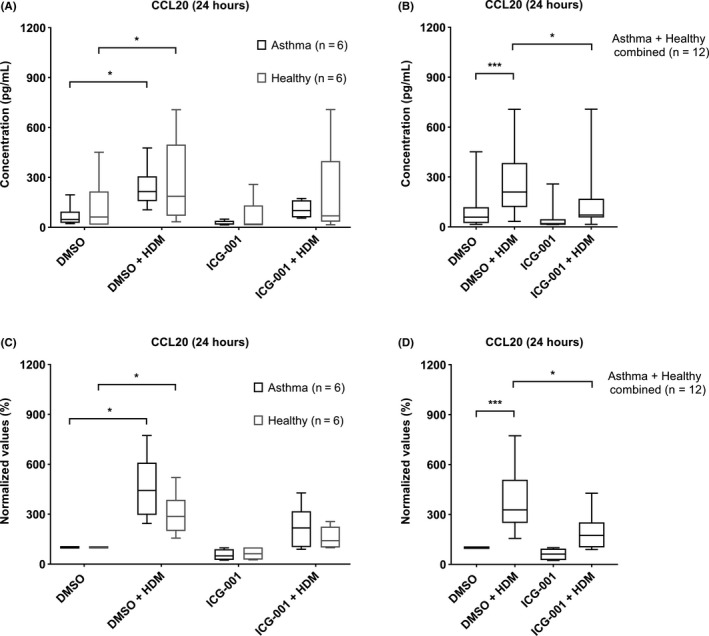
ICG‐001 inhibits HDM‐induced CCL20 production in PAECs. PAECs were obtained by bronchial brushings from healthy (n = 6) and asthmatic (n = 6) donors. Cells were seeded in duplicates at a density of 5 × 104 cells/well in a 24‐well plate, grown to confluence, growth factor/hormone deprived overnight, pretreated with 10 µM of ICG‐001/vehicle control for 3 h, followed by 50 µg/mL HDM. Cell‐free supernatants were collected after 24 hours, and protein levels of CCL20 were measured. CCL20 levels are presented as absolute values (pg/mL) (A, B) or as percentage of the unstimulated levels (C, D). CCL20 levels in separate (A, C), and in combined group (B, D) of asthma and healthy‐derived PAECs. Data are presented as median ± IQR, and Friedman test was used to determine the statistical significance; *P < .05, ***P < .001
We next investigated if ICG‐001 treatment improves epithelial barrier function, a hypothesized prerequisite for epithelial differentiation, in PAECs derived from human bronchial tissue of normal lung transplant donors because of the limited availability of PAECs from the bronchial brushings from asthma and healthy donors. Electric resistance was measured as read‐out for epithelial barrier function using Electric Cell‐substrate Impedance Sensing (ECIS), a highly accurate technique for real‐time monitoring of cell adhesion/spreading, barrier function and wound healing.6 While low‐frequency resistance is most sensitive for changes in cell‐cell contacts, high‐frequency capacitance is more sensitive for changes in cell‐substrate contacts. Low‐frequency resistance, stabilizing at 2572.4 ± 294.9 Ω upon hormone/growth factor deprivation, significantly increased after addition of ICG‐001 (~1.5‐fold), which was evident within 6 hours and lasted up to 30 hours (Figure 2A). The high‐frequency capacitance altered to a lesser extent (Figure 2B), indicating that this effect is primarily due to increased cell‐cell adhesion. We next investigated if ICG‐001 promotes epithelial cell repair after injury induced by electroporation, using 5 V pulses at 40 kHz for 30 seconds, resulting in almost complete detachment of the cells from the electrode.6 This was reflected by an immediate decrease in resistance (Figure 2C) and comparable increase in capacitance (Figure 2D), followed by migration of cells over the wounded area, as evident from the increase in resistance and decrease in capacitance, restoring the integrity of the monolayer within ~3 hours. While 3‐hour pretreatment with ICG‐001 did not affect this initial repair response, ICG‐001 further enhanced low‐frequency resistance once capacitance stabilized, indicating increased recovery of cell‐cell contacts (Figure 2C,D).
Figure 2.

ICG‐001 increases the epithelial barrier function in PAECs. PAECs were obtained from human bronchial tissue of normal lung transplant donors (n = 6). Cells were seeded in duplicates at a density of 5 × 104 cells/well in 8‐well electrode arrays connected to ECIS system, grown to confluence for 3 days, growth factor/hormone deprived overnight, and pretreated with 10 µM of ICG‐001 or vehicle control for 3 h. (A) Resistance and (B) capacitance values were normalized to the time point of addition of ICG‐001. After 3 h, the cells were wounding by electroporation. (C) Resistance and (D) capacitance values were normalized to the lowest and highest points respectively after wounding. Data are presented as mean ± SEM, and two‐way ANOVA was used to determine the statistical significance; *P < .05
Because of the limited PAEC numbers, we used human bronchial epithelial 16HBE cells for further mechanistic studies. We observed that in 16HBE both HDM and thapsigargin increased the release of CCL20, which was inhibited by ICG‐001 pretreatment (Figure S2A,B). ICG‐001 did not reduce CCL20 mRNA expression (Figure S2C‐F), indicating that ICG‐001 inhibits HDM‐induced CCL20 release through post‐transcriptional regulation. Indeed, ICG‐001 still inhibited CCL20 release upon inhibition of de novo synthesis using cycloheximide (Figure S3A,B). Of note, disrupting cortical actin filaments, and thus localization of E‐cadherin at cell‐cell contacts, by cytochalasin D significantly increased CCL20 release, with a trend towards inhibition by ICG‐001 (Figure S3C).
As for the mechanisms of barrier function, we previously observed that 16HBE barrier function is comparable to that of differentiated PAECs.6 We used thapsigargin to induce barrier damage, a sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor that mimics the Ca2+‐mediated effects of HDM,2 yet causing a more robust effect. Thapsigargin induced a similar decrease in 16HBE barrier function (Figure S4C) as observed for electroporation in PAECs. ICG‐001 increased barrier function at baseline and improved recovery from thapsigargin‐induced damage (Figure S4). This was accompanied by a significant increase in the localization of E‐cadherin to cell‐cell contacts, which may be regulated by the organization of the actin cytoskeleton and cortical expression of F‐actin (Figure S5A‐C). Accordingly, blocking actin polymerization using cytochalasin D caused a transient decrease in barrier function, with a disorganized actin network and junctional disruption of E‐cadherin, which could at least in part be mitigated by ICG‐001 (Figure S5D,E). Collectively, these data suggest that ICG‐001 may improve epithelial barrier function by increasing β‐catenin recycling to adherens junctions to form a complex with E‐cadherin and the actin cytoskeleton. We previously showed that junctional E‐cadherin loss leads to increased EGFR activity,7 which can induce Ca2+ signalling and subsequently activate ADAM10, a well‐known sheddase of E‐cadherin.8 Our current data suggest that inhibition of CCL20 release by ICG‐001 may be mediated by stabilizing E‐cadherin at the membrane, inhibiting EGFR activity,7 Ca+2 signalling and subsequent ADAM10 activation (Figure S6), which we have previously shown to be involved in CCL20 release.9 Further studies are currently being directed towards investigating the effect of inhibition of the β‐catenin/CBP pathway on barrier function and mucociliary differentiation of PAECs from healthy and asthma donors cultured at air‐liquid interface.
In conclusion, our data show that inhibition of β‐catenin/CBP signalling promotes cell‐cell contacts and recovery of epithelial barrier function upon damage, while attenuating CCL20 release after HDM exposure. Interference of β‐catenin/CBP signalling may constitute a novel treatment strategy aimed at the restoration of the mucosal barrier in asthma and protection against pro‐inflammatory activity in asthma.
CONFLICTS OF INTEREST
Mr Kuchibhotla, Dr Nawijn and Dr Heijink report grant from Stichting Astma Bestrijding (SAB) during the conduct of the study. Dr Nawijn also reports a grant from Lung Foundation (Netherlands) during the conduct of the study. Mr Jonker, Ms Noordhoek, Mr de Bruin and Dr Knight have nothing to disclose.
Supporting information
{kind=link}
REFERENCES
- 1. Heijink IH, Nawijn MC, Hackett T‐L. Airway epithelial barrier function regulates the pathogenesis of allergic asthma. Clin Exp Allergy. 2014;44(5):620‐630. [DOI] [PubMed] [Google Scholar]
- 2. Post S, Nawijn MC, Jonker MR, et al. House dust mite‐induced calcium signaling instigates epithelial barrier dysfunction and CCL20 production. 2013;68(9):1117‐1125. [DOI] [PubMed] [Google Scholar]
- 3. Hackett T‐L, Warner SM, Stefanowicz D, et al. Induction of epithelial‐mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor‐β1. Am J Respir Crit Care Med. 2009;180(2):122‐133. [DOI] [PubMed] [Google Scholar]
- 4. Ma H, Nguyen C, Lee K‐S, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/b‐catenin‐mediated survivin gene expression. Oncogene 2005;24:3619‐3631. [DOI] [PubMed] [Google Scholar]
- 5. Emami KH, Nguyen C, Ma H, et al. A small molecule inhibitor of beta‐catenin/CREB‐binding protein transcription [corrected]. Proc Natl Acad Sci USA. 2004;101(34):12682‐12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heijink IH, Brandenburg SM, Noordhoek JA, Postma DS, Slebos DJ, Van Oosterhout AJM. Characterisation of cell adhesion in airway epithelial cell types using electric cell‐substrate impedance sensing. Eur Respir J. 2010;35(4):894‐903. [DOI] [PubMed] [Google Scholar]
- 7. Heijink IH, Kies PM, Kauffman HF, Postma DS, van Oosterhout AJM, Vellenga E. Down‐regulation of E‐cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor‐dependent Th2 cell‐promoting activity. J Immunol. 2007;178(12):7678‐7685. [DOI] [PubMed] [Google Scholar]
- 8. Maretzky T, Reiss K, Ludwig A, et al. ADAM10 mediates E‐cadherin shedding and regulates epithelial cell‐cell adhesion, migration, and ‐catenin translocation. Proc Natl Acad Sci USA. 2005;102(26):9182‐9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Post S, Rozeveld D, Jonker MR, Bischoff R, Van Oosterhout AJ, Heijink IH. ADAM10 mediates the house dust mite‐induced release of chemokine ligand CCL20 by airway epithelium. 2015;70(12):1545‐1552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials