

Abstract
Several drugs targeting members of the TNF superfamily or TNF receptor superfamily (TNFRSF) are widely used in medicine or are currently being tested in therapeutic trials. However, their mechanism of action remains poorly understood. Here, we explored the effects of TNFRSF co‐stimulation on murine Foxp3+ regulatory T cell (Treg) biology, as they are pivotal modulators of immune responses. We show that engagement of TNFR2, 4‐1BB, GITR, and DR3, but not OX40, increases Treg proliferation and survival. Triggering these TNFRSF in Tregs induces similar changes in gene expression patterns, suggesting that they engage common signal transduction pathways. Among them, we identified a major role of canonical NF‐κB. Importantly, TNFRSF co‐stimulation improves the ability of Tregs to suppress colitis. Our data demonstrate that stimulation of discrete TNFRSF members enhances Treg activation and function through a shared mechanism. Consequently, therapeutic effects of drugs targeting TNFRSF or their ligands may be mediated by their effect on Tregs.
Keywords: Autoimmunity, Costimulation, NF‐κB, Regulatory T cells, TNF receptor family
Triggering some TNF receptor superfamily (TNFRSF) members, such as TNFR2, 4‐1BB, GITR or DR3, on purified Foxp3+ regulatory T cells (Treg) induces activation of common signaling pathways, including canonical NF‐κB. This leads to increased proliferation, survival, and suppressive function of Treg, enhancing their capacity to suppress colitis.
Introduction
The TNF superfamily (TNFSF) is composed of 19 structurally related cytokines that play major roles in the immune system. There is a considerable interest in targeting the TNFSF ligands and receptors of the TNF receptor superfamily (TNFRSF) in medicine. Beyond PD‐1 and CTLA4, several TNFRSF members (4‐1BB, GITR, CD27, OX40, CD40, CD30, HVEM, BAFFR, or TACI) are emerging targets in cancer immunotherapy and are currently tested in clinical trials [1, 2, 3]. TNFSF and TNFRSF members are also important targets for the treatment of autoimmune or inflammatory bowel diseases to dampen inflammation. Some drugs targeting the TNFRSF/TNFSF are clinically approved, such as anti‐TNF drugs, and many others are under investigation in clinical trials.
Experiments carried out in mice have started to elucidate the mode of action of some TNFRSF agonists in cancer. 4‐1BB, GITR, and OX40 agonistic monoclonal antibodies (mAbs) have been proposed to achieve tumor rejection by depleting or inhibiting tumor infiltrating regulatory T cells (Tregs) and by stimulating conventional T cells (Tconvs) [4, 5, 6, 7, 8, 9]. Intriguingly, these same agonists of 4‐1BB and OX40 are also able to suppress autoimmune diseases [6, 7, 9, 10]. Some mechanisms have been proposed to explain these paradoxical findings. 4‐1BB agonists would increase IFN‐γ production by Tconvs, leading to protective IDO production in an autoimmune context and tumor rejection in cancer [11, 12]. OX40 agonistic mAb can promote protective Treg expansion in an autoimmune context while depleting tumor‐infiltrating Tregs [6, 13]. Thus, although the therapeutic potential of drugs targeting TNFSF ligands or TNFRSF receptors is major, their mode of action is still poorly defined and challenging to predict.
Transcriptome analyses have clearly demonstrated that several members of the TNFRSF are preferentially expressed on human and mouse Tregs when compared to other immune cell subsets. For instance, Tnfrsf1b (TNFR2), Tnfrsf9 (4‐1BB), Tnfrsf18 (GITR), and Tnfrsf4 (OX40) belong to the very restrictive core Treg signature expressed by all Treg subsets and composed of the top 10 genes that are the most highly expressed by Tregs compared to Tconvs [14, 15]. Also, Tnfrsf9, Tnfrsf18, and Tnfrsf4 are among the top genes that are the most highly expressed by tumor infiltrating Tregs compared to healthy tissue Tregs and tumor infiltrating Tconvs in mice and humans [16, 17]. Moreover, GITR protein expression is often considered as a prototypic Treg marker and early upregulation of 4‐1BB is used to discriminate activated Tregs from activated Tconvs in humans [18]. Together, this suggests that in vivo triggering of these TNFRSF proteins is likely to preferentially target Tregs.
However, the cell‐intrinsic functions of TNFRSF receptors in Treg biology are poorly defined. Experiments done in mice with germline ablation of TNFRSFs are difficult to interpret, since it is unclear whether effects on Tregs are direct or indirect. To date, conditional KO models of TNFRSF receptors in Tregs are lacking. Injection of agonist molecules to TNFR2, 4‐1BB, GITR, DR3, or OX40 led to Treg expansion in mice, and for some of them, to therapeutic effects in autoimmunity [13, 19‐22], but it was unclear whether this relied on a direct effect on Tregs. In vitro experiments showed that agonists of TNFR2, 4‐1BB, GITR, and DR3 increased Treg expansion [23, 24, 25, 26, 27]. Importantly, the effect of several TNFRSF agonists on Treg suppressive function led to some apparently contradictory findings. It was suggested that stimulation of TNFR2, GITR, and OX40 inhibited Treg suppressive function. However, in the meantime these agonists rendered Tconvs refractory to Treg‐mediated suppression. When using experimental design to restrict the effects of the agonists on Tregs, it was usually found that most TNFRSF agonists did not inhibit their suppressive function in vitro [9, 28‐31].
Most of what we know about signal transduction induced by triggering of TNFRSF receptors comes from studies performed in cell lines, showing activation of the NF‐κB, PI3K, and MAPK pathways [32, 33]. Some studies performed with primary Tconvs and very few on primary Tregs suggested activation of these same pathways [34, 35, 36, 37, 38]. Moreover, only two studies have investigated the effect of TNFRSF agonists in vitro on the Treg transcriptome, one for TNF and one for OX40 [39, 40]. Because a thorough comparison of the effects of different TNFRSF agonists on Tregs has to date not been performed, we know very little on what is common and what is specific to each TNFRSF. Here, we performed a global in vitro and in vivo analysis to explore the effects of TNFRSF engagement on Treg biology, function, and identity. We found that different TNFRSF agonists modulated the expression of a common core set of genes and activate the canonical NF‐κB pathway. These stimulations resulted in Tregs with enhanced proliferation, survival, and suppression of inflammation, opening new perspectives for therapeutic interventions using TNFRSF co‐stimulated Tregs.
Results
Costimulation with TNFR2, 4‐1BB, GITR, and DR3 agonists increases Treg proliferation and survival
We first analyzed expression kinetics of TNFRSF members in Tregs after T‐cell activation in vitro. Except for GITR that was constitutively expressed at high levels, TNFR2, 4‐1BB, or OX40 was expressed at low levels on resting Tregs. The expression of all these receptors was upregulated as early as 24 h after TCR/CD28 activation (Fig. 1A). DR3 was reported to be highly expressed on freshly purified Treg [22]. To address the effect of the direct stimulation of these TNFRSF on Treg biology, we stimulated highly purified Tregs with anti‐CD3 and anti‐CD28 mAbs for 3 days in the presence of variable concentrations of TNFRSF agonists. Addition of TNFR2‐, 4‐1BB‐, GITR‐, and DR3‐specific agonists increased the numbers of living Tregs by three‐ to fourfold at their optimal concentration compared to control Tregs. In contrast, addition of agonists of OX40 (OX40L and mAb), DR5 (TRAIL and mAb), HVEM (LIGHT), CD27 (CD70), or RANK (RANKL) had minimal or no effects, whereas Fas engagement induced Treg death (Supporting Information Fig. S1). For the rest of the study, we therefore analyzed the effects of agonists of TNFR2, 4‐1BB, GITR, and DR3 at their optimal concentration, along with OX40 agonist because of its well‐characterized capacity to stimulate Tregs in vivo. When we compared these five agonists, TNFR2 and 4‐1BB agonists displayed superior ability to increase Treg proliferation and survival, while GITR and DR3 agonists had a weaker effect and OX40 agonist had no effect (Fig. 1B–D). Cotreatments combining some of these agonists showed an additive effect on Treg survival and proliferation (Supporting Information Fig. S2). In conclusion, stimulation of TNFR2, 4‐1BB, GITR, and DR3 by agonistic molecules had a direct effect on Tregs to increase their survival and proliferation with some additive effects when they were combined.
Figure 1.
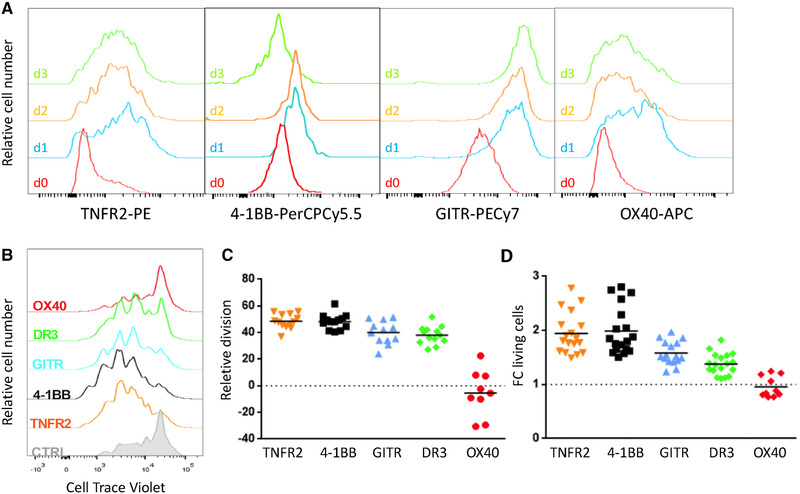
Costimulation with agonists of TNFR2, 4‐1BB, GITR, and DR3 increased Treg proliferation and survival in vitro. (A) Expression of TNFR2, 4‐1BB, GITR, and OX40 was assessed by flow cytometry on freshly purified Tregs (d0) and after 24, 48, and 72 h of culture with agonists and beads coated with anti‐CD3/CD28 mAbs. Representative experiment of three independent experiments with biological duplicates. (B–D) Purified Tregs were stimulated by APCs, anti‐CD3 mAb, and agonists of TNFRSF receptors for 3 days to quantify proliferation and survival. (B) Representative cell proliferation profile of at least nine independent experiments with biological duplicates. Increased proliferation (C) and fold change (FC) of living cells (D) relative to the control culture (without agonist, dotted lines), with each symbol representing the mean of biological duplicates of independent experiments (at least nine). For each agonist, the horizontal bar represents the mean. In (A) and (B), around 20 000 cells were analyzed per gated Tregs.
TNFRSF agonists induce a shared transcriptome signature in Tregs
To further investigate the modulation of Treg biology by TNFRSF agonists, we performed high‐throughput RNA‐sequencing experiments. Tregs were stimulated with anti‐CD3/CD28 mAbs in the presence or absence of agonists of TNFR2, 4‐1BB, DR3, or OX40 for 18 and 36 h before transcriptome analysis. Of note, GITR agonist was omitted in this analysis because of its relative weak capacity to costimulate Treg proliferation, whereas OX40 agonist was included as a “negative control.” Principal component analysis showed that Tregs costimulated by each of the four agonists clearly separated from control Tregs without TNFRSF costimulation (Fig. 2A and Supporting Information Fig. S3A). To analyze similarities between the effects of the different TNFRSF agonists, we analyzed fold change (FC) of differentially expressed genes (DEG) compared to control. We represented a FC/FC plot to compare the effects of TNFR2 and 4‐1BB agonists. Most of the values gathered on the diagonal with a high positive correlation (Pearson correlation factors of r = 0.85 at 18 h and r = 0.87 at 36 h), suggesting that these two agonists induced a very similar response (Fig. 2B and Supporting Information Fig. S3B). Similar analysis comparing the effects of TNFR2 and DR3 agonists also showed strong positive correlation (r = 0.83 at 18 h and r = 0.87 at 36 h). Interestingly, the off‐diagonal placement of the gene cloud indicates that the global impact of the DR3 agonist was weaker than the impact of the TNFR2 agonist. Finally, a lower correlation was observed between TNFR2 and OX40 agonists (r = 0.81 at 18 and 36 h) (Fig. 2B and Supporting Information Fig. S3B). The heatmap representation of a two‐group comparison (control vs. the four agonists together, using false discovery rate (FDR) < 0.05 and log2FC > 0.5) confirmed similarities of the effects of the four agonists. This analysis also highlighted that 4‐1BB and TNFR2 agonists induced the strongest gene expression changes compared to the control, both at 18 h and 36 h (Fig. 2C and Supporting Information Fig. S3C). Interestingly, a number of the top DEG induced by the TNFRSF agonists at 36 h belonged to the core Treg transcriptomic signature (Supporting Information Fig. S3D), suggesting that TNFRSF stimulation modified the identity of Treg.
Figure 2.
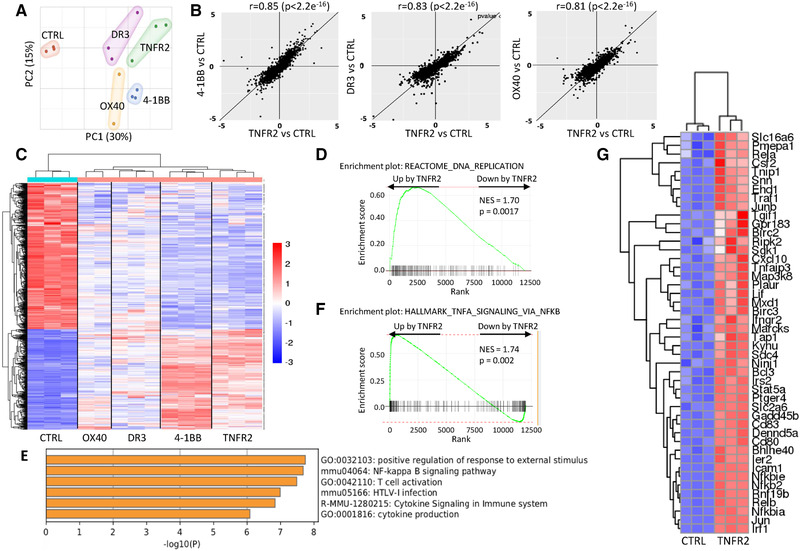
Different TNFRSF agonists induced a similar transcriptomic signature. RNA sequencing was performed on Tregs stimulated with anti‐CD3/CD28 mAbs and agonists of TNFRSF for 18 h. Biological triplicates of one experiment is shown. (A) Principal component analysis. (B) FC/FC plots (expressed in log2) of differentially expressed genes (DEG) compared to controls (FDR < 0.05) to compare the effects of TNFR2 agonist with the effects of the three other agonists (4‐1BB, DR3, and OX40) on Tregs using the Pearson correlation coefficient. (C) Heatmap of DEG (FDR < 0.05 and log2FC > 0.5) in a two‐group comparison analysis between control Tregs and Tregs costimulated by TNFRSF agonists. (D) Gene set enrichment analysis (GSEA) plot showing significant enrichment of genes regulated by TNFR2 costimulation among the indicated DNA replication gene set. (E) Most significantly represented pathways analyzed by gene ontology of DEG upregulated by TNFR2 costimulation (FDR < 0.05, FC > 0.5). (F) GSEA plot showing significant enrichment of genes upregulated by TNFR2 costimulation among the indicated “TNF signaling via NF‐κB” pathway. (G) Heatmap representation of the leading edge subset genes extracted from the GSEA in (F).
Because of the strong similarities in gene expression profiles induced by the different TNFRSF agonists, we had to exclude that TNFR2, 4‐1BB, GITR and DR3 co‐stimulation triggered the same TNFRSF receptor, directly or indirectly, to promote Treg proliferation and survival. None of the agonists was able to increase significantly the mRNA expression of the other TNFRSF receptors (Supporting Information Fig. S4A). Also, since Tregs are able to produce TNF, we performed experiments using Tregs isolated from Tnf and Tnfrsf1b (coding for TNFR2) KO mice to exclude a possible role of autocrine TNF. Agonists of TNFR2, 4‐1BB, GITR and DR3 increased proliferation and survival of Tregs to a similar extent in WTand Tnf KO Tregs, showing that their effects were not TNF‐mediated (Supporting Information Fig. S4B). When using Tnfrsf1b KO Tregs, the increased Treg proliferation and survival was abolished only with the TNFR2 agonist, showing that agonists of 4‐1BB, GITR or DR3 did not co‐stimulate Tregs via TNFR2 (Supporting Information Fig. S4C). In conclusion, triggering of TNFR2, 4‐1BB, DR3 and OX40 induced a shared program of gene expression with the strongest response observed with agonists of TNFR2 and 4‐1BB. Thus, this comprehensive analysis highlighted TNFR2 and 4‐1BB agonist agents as superior candidates for modulating Treg activity.
TNFRSF costimulation of Tregs involves canonical NF‐κB activation
We next focused our transcriptomic analysis at 18 h on the effect of TNFR2 agonist because of its capacity to induce a strong response as attested when studying the proliferation, survival and transcriptome. Gene set enrichment analysis highlighted that genes involved in DNA replication were up‐regulated by TNFR2 agonist, which was consistent with the increased Treg proliferation induced by this agonist (Fig. 2D). Enrichment analysis of genes up‐regulated by TNFR2 agonist (FDR < 0.05, Log2FC > 0.5) showed that the “NF‐κB signaling pathway” and “T‐cell activation” pathway were among the most significantly represented (Fig. 2E). Also, genes of the “TNF signaling via NF‐κB” pathway were upregulated by TNFR2 agonist in Tregs (Fig. 2F). Among them, we could find Rela, Relb, Nfkb2, Traf1, Birc2, Birc3, or Ripk2 (Fig. 2G and Supporting Information Fig. S5). Similar findings were obtained when we analyzed the effect of the 4‐1BB agonist on Tregs at 18 h (Supporting Information Fig. S6).
To assess whether NF‐κB activation was involved in TNFRSF Treg costimulation, we measured NF‐κB activation by electrophoretic mobility shift assay (EMSA), combined with supershift analysis to assess the involvement of individual NF‐κB subunits. TNFR2, 4‐1BB, GITR, and DR3 agonists induced strong NF‐κB activation 30 min after initial costimulation with sustained activation for at least 4 h (Fig. 3A). Compared to these four agonists, OX40 agonist induced very minor NF‐κB activation, suggesting once more that OX‐40 had a separate mechanism of action on Tregs. When looking at the identity of the different NF‐κB activated subunits by supershift, RelA and cRel were detected in cells co‐stimulated by TNFR2, 4‐1BB, GITR, and DR3 (Fig. 3B). This biochemical analysis shows that these TNFRSF agonists strongly activate the canonical NF‐κB pathway in Tregs. To assess the mechanistic relevance of this observation, we analyzed whether the effect of these agonists would be lost using Tregs purified from mice with conditional ablation of Rela in Tregs. Compared to control Tregs, Rela‐deficient Tregs had significant reduction of proliferation and major reduction of survival induced by the TNFRSF agonists (Fig. 3C–E). Altogether, these experiments demonstrated that the costimulatory effects of agonists of TNFR2, 4‐1BB, GITR, and DR3 on Tregs required activation of the canonical NF‐κB pathway.
Figure 3.
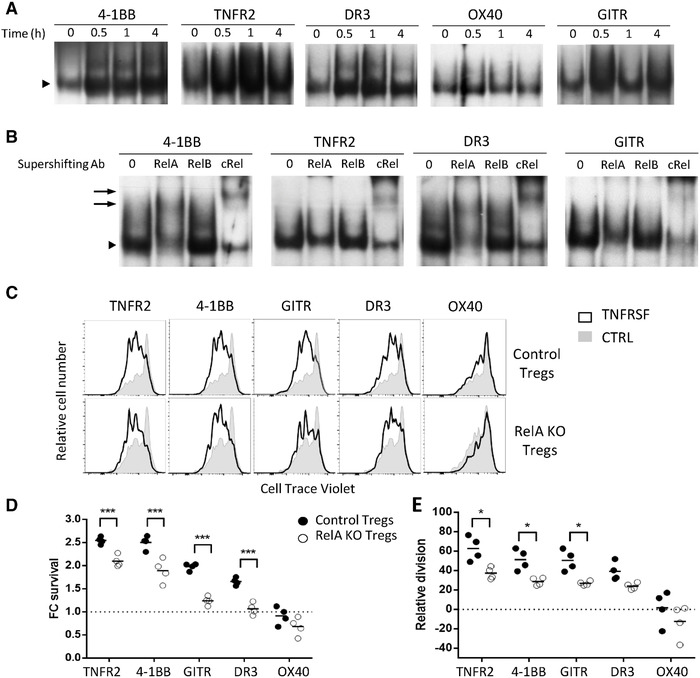
TNFRSF receptor costimulation activated the canonical NF‐κB pathway in Tregs. (A, B) Tregs, pre‐activated with anti‐CD3/CD28 mAbs for 24 h, were restimulated with TNFRSF agonists alone. The EMSA (A) was performed at 0, 0.5, 1, and 4 h of restimulation. The supershift (B) was performed at 0.5 h of restimulation to investigate the subset composition of the NF‐κB containing complex. Arrowheads indicate the position of the NF‐κB containing complex and arrows indicate the positions of the supershifting complexes bound to mAb specific to RelA and c‐Rel. A representative of three experiments is shown. Proliferation (C, D) and survival (E) of Rela KO and control Tregs, stimulated and analyzed as in Fig. 1. One representative proliferation profile (C) and increased proliferation and FC living cell numbers relative to the control culture (D, E) from the pool of two independent experiments with two mice per experiment. Each circle represents a mouse (two‐way ANOVA, *p < 0.05, ***p < 0.001.) and the horizontal bar represents the mean. In (C), around 20 000 cells were analyzed per gated Tregs.
Triggering of TNFRSF in Tregs favors expression of molecules of type 2 and type 17 immunity
It is well described that the Treg population can be further subdivided into Tregs with different properties that differentially express cytokines, chemokine receptors, and transcription factors [41]. To explore whether TNFRSF costimulation would change expression of these molecules, we first analyzed cytokine production by Tregs after 3 days of culture. As expected, control Tregs mainly produced IL‐10, whereas other cytokines (IL‐4, IL‐5, IL‐13, IL‐17a, IL‐23p19, IFN‐γ, and TNF) were detected at lower levels (Fig. 4A). Interestingly, agonists of TNFR2, 4‐1BB, GITR, and DR3 increased type 2 (IL‐4, IL‐5, and IL‐13) and type 17 (IL23p19) cytokines, whereas TNF was increased only by the TNFR2 agonist. The expression of IL‐10, IL‐17a, and IFN‐γ, the other studied cytokines, was not reproducibly modified by TNFRSF agonists (Fig. 4A). Consistent with these findings, we detected increased expression of Il5, Ccr4, Ccr6, Jun, Stat3, Stat6, and Fosl2, all known markers of type 2 and 17 immune responses, and decreased expression of Cxcr3 (Fig. 4B), a marker of type 1 immunity, upon TNFR2 or 4‐1BB costimulation. The increased expression of CCR6 and GATA3 and decreased expression of CXCR3 were also observed at the protein level in Tregs triggered by TNFRSF agonists (Fig. 4C). Thus, TNFRSF costimulation in Tregs favors expression of molecules of type 2 and 17 immunity.
Figure 4.
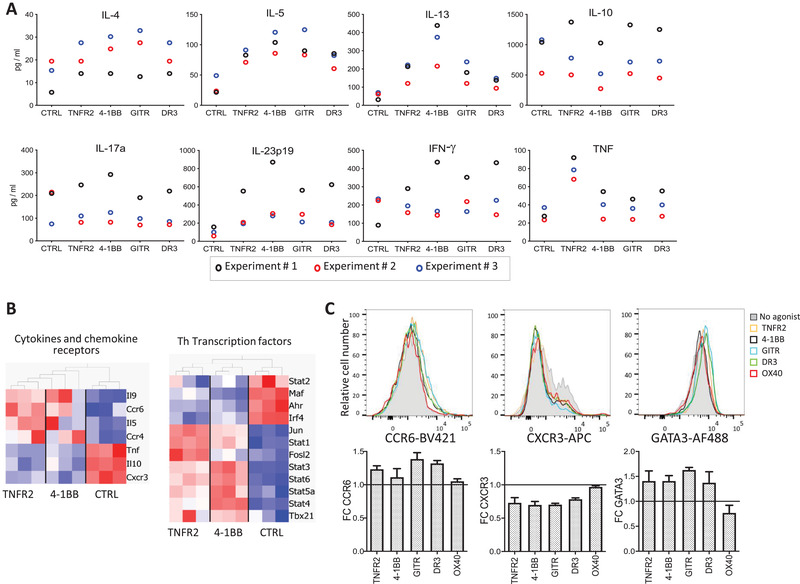
TNFRSF agonists favored polarization toward type 2 and type 17 Tregs. (A) Cytokine production (measured in the supernatant) by Tregs activated by anti‐CD3/CD28 mAb and TNFRSF agonists for 3 days. Each symbol represents mean values of biological triplicates from an independent experiment. (B) DEG (FDR < 0.05) between control and Tregs costimulated by TNFR2 or 4‐1BB for 36 h among gene sets of “cytokine and chemokine receptors” and “T helper” (Th) transcription factors, listed in the Materials and Methods section. Biological triplicates of one experiment. (C) Representative profiles (upper panels) and FC compared to control (lower panels) of CXCR3, CCR6, and GATA3 expression, analyzed by flow cytometry, on Tregs stimulated by anti‐CD3/CD28 mAbs and TNFRSF agonists for 3 days. Mean (±SEM) values of three independent experiments with biological duplicates. Around 20 000 cells were analyzed per gated Tregs.
Triggering of TNFRSF increases Treg expansion in vivo
We then studied the in vivo fate of Tregs costimulated by TNFRSF in vitro. To assess the effect of TNFR2, 4‐1BB, GITR, and DR3 engagement on Treg homeostasis, we used an adoptive cell transfer approach. Tregs, first stimulated in vitro with TNRSF agonists, were cotransferred intravenously with the same number of control Tregs into WT recipients to compare the homeostatic properties of the two Treg subsets, identified using congenic markers (Fig. 5A). Early migration was assessed 12 h after cell transfer, before later homeostatic reorganization that would complicate data interpretations. Proportions of TNFRSF costimulated and control Tregs were comparable in all the analyzed lymphoid and nonlymphoid tissues, except the blood, suggesting that TNFRSF treatments did not affect early migration in lymphoid or nonlymphoid tissues (Supporting Information Fig. S7A).
Figure 5.
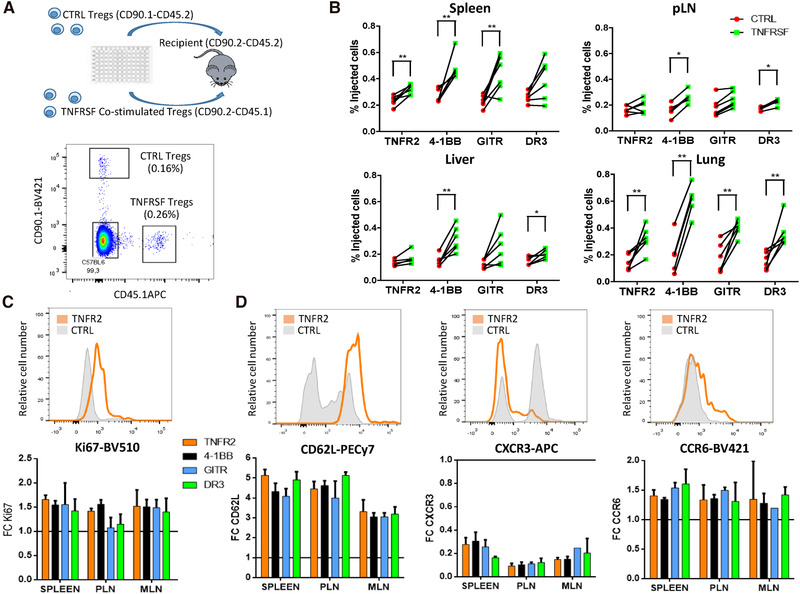
TNFRSF costimulation increased Treg in vivo expansion and modified Treg tissue recirculation. Tregs, preactivated for 3 days with anti‐CD3/CD28 mAbs alone (control Tregs) or combined with TNFRSF costimulation (co‐stimulated Tregs), were cotransferred in equal numbers to assess their in vivo homeostasis 7 days later. (A) Experimental design (upper panel) and representative plot showing cotransferred Tregs identified with congenic markers (lower panel). (B) Proportions of coinjected Tregs in spleen, peripheral lymph nodes (pLN), liver, and lung. Each dot is a mouse and lines connect cells from the same mouse. Unpaired Mann–Whitney test was used. (C, D) Ki67, CD62L, CXCR3, and CCR6 expression among injected cells. Upper panels show representative histograms for control and TNFR2 costimulated Tregs in the spleen. Lower panels show the mean (±SD) of FC MFI expression of costimulated compared to control Tregs in spleen, pLN, and mesenteric (MLN) LN. Data were obtained from two independent experiments with six mice per group in total. From 100 to 500 cells were analyzed per gated Tregs.
Having established that TNFRSF‐treated Tregs have the same early tissue distribution than control Tregs, we then analyzed donor cells 7 days after co‐transfer to assess their recirculation, survival, and proliferation. Overall, costimulated Tregs were present in higher numbers than control Tregs in the spleen, lymph nodes, lung, and liver, with variations depending on the tissue and the TNFRSF agonist used (Fig. 5B). 4‐1BB costimulation induced the strongest effect, followed by GITR, TNFR2, and DR3 costimulation. Importantly, costimulated Tregs expressed higher levels of the Ki67 proliferation marker in all tissues compared to control (Fig. 5C). Since Ki67 differential expression was not yet observed 12 h after transfer (Supporting Information Fig. S7B), costimulated Tregs proliferated more than control Tregs or proliferating costimulated Tregs had better survival after transfer. The increased mRNA expression of Bcl2l1 (Bcl‐XL) and decreased expression of Bcl2l11 (Bim) in costimulated Tregs, two molecules critical in cell survival, may indicate an effect on cell survival (Supporting Information Fig. S8). Compared to control cells, costimulated Tregs expressed higher levels of CD62L and CCR6 and lower levels of CXCR3 7 days after transfer (Fig. 5D), a phenomenon that was already observed 12 h after transfer (Supporting Information Fig. S7C). In conclusion, Tregs that were costimulated with TNFRSF agonists modified their phenotype and acquired an increased capacity to expand in lymphoid and nonlymphoid tissues.
Tregs costimulated by TNFRSF have increased capacity to suppress colitis
We finally studied the effect of TNFRSF agonists on Treg function. Tregs that were costimulated for 3 days with agonists of TNFR2, 4‐1BB, GITR, or DR3 were tested for their in vitro capacity to suppress proliferation of conventional T cells. None of the agonists had an impact (Fig. 6A). However, it is well known that only a fraction of the numerous in vivo Treg suppressive mechanisms is tested in this classical in vitro assay. To further explore putative changes induced by TNFRS agonists in the Treg suppressive profile, we analyzed RNA expression of a gene set involved in immune suppression. We observed that some of them were downregulated and others were upregulated upon TNFR2 and 4‐1BB engagements (e.g., Il10/Ctla4 and Ebi3, respectively) (Fig. 6B). This suggests that, beyond their similar suppressive profile in vitro, TNFRSF‐stimulated Tregs may use different means to achieve target inhibition. We then assessed the impact of short‐term incubation (2 h) of Tregs with TNFRSF agonists on their in vivo function in a colitis model induced by Tconv injection in Rag2−/− mice. We only studied the effect of TNFR2 and GITR agonists because these receptors were already expressed by resting Tregs. Remarkably, Tregs that were preincubated with agonists of TNFR2 or GITR displayed a significantly increased capacity to control the disease compared to control Tregs. Indeed, mice injected with these costimulated Tregs gained weight over time (Fig. 6C). Histological examination of the colon further supported these findings. Mice injected with Tconvs alone had marked inflammatory cell infiltrates associated with major epithelial hyperplasia and loss of intestinal glands (Fig. 6D). Mice coinjected with control Tregs displayed reduced but clear colitis with moderate inflammatory cell infiltrates and mild epithelial hyperplasia. Mice coinjected with Tregs costimulated with agonists of TNFR2 or GITR had no or minor colitis with no or minimal scattered mucosal inflammatory cell infiltrates without epithelial hyperplasia. Thus, triggering of TNFR2 or GITR increased the capacity of Tregs to suppress colitis.
Figure 6.
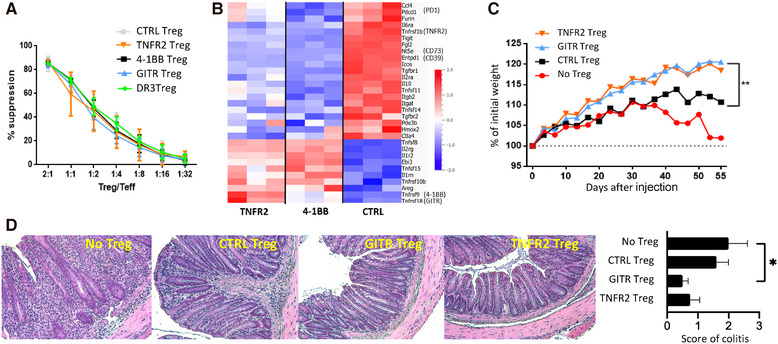
Costimulation of Tregs by TNFRSF receptors modified their expression of suppressive molecules and increased their capacity to suppress colitis. (A) Tregs, preactivated with anti‐CD3/CD28 mAb and TNFRSF agonists, were tested for their in vitro suppressive function. Mean (±SD) values from one representative of three independent experiments is shown. (B) DEG (FDR < 0.05) between control and Tregs co‐stimulated by TNFR2 or 4‐1BB for 36 h among a gene sets of Treg effector molecules, listed in the Materials and Methods section. (C, D) Tregs preincubated with agonists of GITR or TNFR2 or control (CTRL) Tregs were injected in Rag2−/− mice to assess their ability to control colitis induced by Tconvs. Pool of two independent experiments with a total of six mice per group. Colitis was assessed by measuring mouse weight (C) and by histological examination of the colon at sacrifice (10× magnification) (D). (D) Representative (left panels) and mean (±SEM) colitis score (right panel) of tissue sections are shown. Statistical significance was analyzed using a one‐way ANOVA test, *p < 0.05.
Discussion
There are multiple drugs targeting TNFSF ligands and TNFRSF receptors. Some are widely used to treat autoimmune diseases (e.g. anti‐TNF) and many others are in clinical trials for cancers, and autoimmune and inflammatory diseases. Although their mechanisms of action are likely diverse, there is strong evidence that part of them are Treg mediated for different reasons: (i) Tregs specifically express high levels of several TNFRSF members (i.e., TNFR2, 4‐1BB, GITR, and OX40), (ii) different agonists of TNFRSF members increase Treg expansion in vitro, and (iii) injecting agonists of several TNFRSF induced Treg expansion, although it could be via indirect mechanisms. Besides these findings, we know surprisingly very little on the direct biologic effect of TNFRSF agonists on Tregs.
Our study shows for the first time the following new findings: (i) Triggering of 4‐1BB, GITR, and DR3 increases Treg survival in vitro; (ii) there is an additive effect of combining agonists of some TNFRSF for Treg proliferation and survival; (iii) at the whole transcriptome level, agonists of TNFRSF induce a surprisingly similar signature, although the global effect is stronger with agonists of TNFR2 and 4‐1BB; (iv) agonists of 4‐1BB, GITR, and DR3 do not costimulate Tregs simply by increasing TNF/TNFR2 signaling; (v) triggering TNFRSF activates the canonical NF‐κB pathway in Tregs by increasing RelA and cRel DNA binding activity; (vi) part of the increased Treg proliferation and survival induced by agonists of TNFRSF is RelA dependent; (vii) triggering TNFRSF induces Tregs that express higher levels of molecules of type 2 and 17 immunity, (viii) agonists of TNFRSF directly increase Treg expansion in vivo; and (viii) agonists of TNFR2 and GITR increase the capacity of Tregs to control colitis. The long‐term impact on colitis of the short‐term preincubation (only 2 h) with TNFR2 or GITR agonists was quite impressive. It might be due partly to the fact that the agonists would remain bound to the injected Tregs, prolonging a signal transduction in vivo. Overall, we show here that triggering several members of the TNFRSF in Tregs modifies their biology, increases their capacity to proliferate and survive through NF‐κB activation and improves their capacity to suppress in vivo.
One of the major surprises of our data is the strong similarity of the effects of agonists of TNFR2, 4‐1BB, GITR, and OX40 when looking at the whole transcriptome level. Since we excluded the possibility that this was due to triggering the same receptor (TNFR2) via endogenous TNF, this indicates that their downstream signaling pathways are likely related or even identical. Illustrating this point, the absence of co‐stimulatory activity of OX40 is best explained by its inability to induce canonical NF‐κB activation.
It was shown previously that triggering of TNFR2, 4‐1BB, GITR, or DR3 increases Treg proliferation in vitro [23, 24, 25, 26, 27] and we confirmed these findings. However, there has been no report showing that this would be the case in vivo as well. It was shown that injection of agonists of several TNFRSF molecules did induce Treg expansion but this could be due to an indirect effect [13, 19‐22]. For instance, triggering of some TNFRSF receptors also activate Tconvs, which produce IL‐2 that in turn may boost Treg proliferation [42, 43]. Interestingly, it was proposed that injection of an OX40 agonist increased Treg expansion by increasing their sensitivity to IL‐2 due to decreased SOCS1 and increased miR155 expression [44]. Thus, the absence of an effect of OX40 agonist in vitro could be due to the high IL‐2 level in our cultures. Here, we show that triggering of TNFR2, 4‐1BB, GITR, and DR3 on Tregs increases their expansion in vivo by a direct effect on the cells.
We show here that agonists of TNFR2, 4‐1BB, GITR, and DR3 increased Treg survival. This was shown previously for Tconvs but not for Tregs [45]. For Tconvs, increased survival was associated with increased expression of pro‐survival molecules, such as survivin, Bcl‐2, Bcl‐XL, cFLIP‐short, or Bfl‐1 and decreased expression of pro‐apoptotic molecules, such as Bim and Bad [45, 46, 47]. The mechanisms of Treg survival induced by TNFRSF agonists may be only partly similar to those of Tconvs. Indeed, on the one hand, we also observed increased expression of Bcl2l1 (coding for Bcl‐XL) and decreased expression of Bcl2l11 (coding for Bim) but, on the other hand, we had reduced expression of Bcl2 and no significant difference for the other described molecules (Supporting Information Fig. S8). Other molecules may play a role since we found changes in the expression of caspases (Casp2 and 7), Ripk1 and Ripk3 and TRAF molecules (Traf1, 3, 6 and 7), all involved in T‐cell survival or apoptosis.
The signal transduction triggered by TNFRSF agonists in vitro has hardly been studied in Tregs. To date and to our knowledge, transcriptomic analyses have been reported in only two studies. In the one study, the authors studied the effect of TNF on human Tregs but there was no TCR/CD28 stimulation [40]. In the other study, the effect of an agonist of OX40 was studied in combination with TCR activation on purified mouse Tregs in vitro [39], a condition that resembles our experimental setting. However, only 21 DEG were detected versus over 180 DEG (FDR < 0.05, FC > 2) in our study, suggesting that their culture condition may not be optimal. Also, we observed that OX40 is the one, among the five studied TNFRSF, that had no effect on Treg proliferation and survival. Thus, using our transcriptomic analysis, comparing the effect of agonist of TNFR2, 4‐1BB, DR3, and OX40 on Tregs, opens a new field of investigation. As for the transcriptome, very few biochemical data exploring the signals transduced by TNFRSF on Tregs have been published. These reports, measuring amounts of IκBα, and NF‐κB members (NFκB1, NFκB2, RelA, c‐Rel, and RelB) by Western blot suggested that triggering of TNFR2, GITR, and DR3 activates both the canonical and noncanonical NF‐κB pathways [34, 35, 36, 37, 38]. However, these assays are less informative than the EMSA and supershift assays that we performed since the amount of a transcription factor does not necessarily reflects its DNA binding activity. Our study challenges previous findings and indicates that TNFRSF agonists mainly activate the canonical NF‐κB pathway with activation of cRel and RelA subunits. This was confirmed using Rela‐deficient Tregs in which part of the biological activity induced by TNFRSF costimulation was lost. We did not find any evidence of the activation of the noncanonical pathway, which may be even inhibitory for Tregs since GITR‐costimulation induced an increase in proliferation when cells were deficient for the NF‐κB‐inducing kinase, an up‐stream activator of this pathway [36].
We also demonstrated that triggering of TNFRSF did not modify Treg suppressive function in vitro, as previously shown for TNFR2, GITR, and OX40 [9, 28‐31]. However, these in vitro assays only reflect the “tip of the iceberg” of the suppressive potential of Tregs that use very diverse mechanisms of suppression in vivo. We found that agonists of TNFR2 or GITR increased the capacity of Tregs to suppress colitis. This may be important with respect to the use of Tregs in cell therapy, which is intensively investigated to control graft rejection, graft versus host disease or autoimmune and inflammatory diseases [48]. The major challenge is to obtain high amounts of pure, stable, and functional Tregs after in vitro cell expansion. Adding TNFRSF agonists to the culture might help to reach this goal given their capacity to increase Treg proliferation, expansion, and probably in vivo suppressive function. The type 2/17 immune profile of Tregs co‐stimulated by the TNFRSF agonists might be due to the increased expression of Gata3, Stat3, Stat5, Stat6, and AP‐1 factors (Jun and Fosl2), although decreased expression of Maf, Irf4, or Ahr may limit this differentiation. Our findings may suggest that Tregs costimulated with TNFRSF agonists would be more adapted to control Th2 and Th17 type diseases, rather than Th1 type diseases, notably because the increased expression of CCR4 and CCR6 and decreased expression of CXCR3 would favor their migration toward Th2‐ and Th17‐type inflammation sites. This is also compatible with the increased capacity of Treg costimulated by the GITR or TNFR2 agonists to control colitis because of the known pathogenic IL‐23/Th17 axis in this disease [49]. Overall, we show here that triggering several members of the TNFRSF in Tregs increased their proliferation and survival by activating NF‐κB and improve their capacity to suppress colitis. Thus, the TNFRSF may play a major role in Treg biology and part of the beneficial effects of drugs targeting TNFSF or TNFRSF molecules is likely Treg mediated.
Materials and Methods
Mice
Foxp3‐CRE‐IRES‐YFP (Foxp3Cre) [50] and Foxp3‐IRES‐GFP [51] (Foxp3GFP) knock‐in mice were kindly given by Prs. Alexander Rudensky and Bernard Malissen, respectively, and Relaflox knock‐in mice were previously described [52]. Foxp3‐DTR (Foxp3tm3(DTR/GFP)Ayr/J), Tnf−/− (Tnftm1Gk1/J), and Tnfrsf1b−/− (Tnfrsf1btm1Mwm/J) were obtained from the Jackson laboratory. Cd3−/− (CD3etm1Mal) and Rag2−/− mice were obtained from the cryopreservation distribution typing and animal archiving department (Orléans, France). All mice were on a C57Bl/6J background. Mice were housed under specific pathogen‐free conditions. All experimental protocols were approved by the local ethics committee and are in compliance with European Union guidelines.
Cell preparation from tissues
For lymphoid tissues, cells were isolated by mechanical dilacerations. For nonlymphoid tissues, anesthetized mice were perfused intracardially with cold PBS. Small pieces of livers and lungs were digested in type IV collagenase (0.3 mg/mL) and DNase I (100 μg/mL) for 30 min at 37°C, followed by Percoll gradient (30–70%) separation.
Antibodies and flow cytometry analysis
The following mAbs from BD Biosciences were used: anti‐CD45 (30‐F11), anti‐CD4 (RM4‐5), anti‐CD62L (MEL‐14), anti‐CD90.1 (OX‐7), anti‐CD90.2 (30‐H12), anti‐CD45.1 (A20), anti‐CD45.2 (104), anti‐CD25 (PC61 or 7D4), anti‐GITR (DTA‐1), anti‐GATA3 (L50‐823), anti‐CXCR3 (CXCR3‐173), anti‐CCR6 (140706), and anti‐TNFR2 (TR75‐89). Anti‐GFP antibody was purchased from Life Technologies. Anti‐CD3 (145‐2C11), anti‐Foxp3 (FJK‐16s), anti‐Ki‐67 (SOLA15), anti‐4‐1BB (17B5), and anti‐OX40 (OX86) were purchased from eBioscience, and Foxp3 staining was performed using the eBioscience kit and protocol. Cell survival was assessed with fixable viability dyes (e780 and e506). Flow cytometry analyses and sorting were performed according to previously described guidelines [53]. Cells were acquired on a BD LSRII or a BD Fortessa X20 cytometers and analyzed using FlowJo software.
Treg and Tconv purification
Tconvs were purified after enrichment of CD25− cells using biotinylated anti‐CD25 mAb (7D4) or of CD8− CD19− CD11b− cells using biotinylated anti‐CD8 (53‐6.7), CD19 (1D3), and CD11b (M1/70) mAbs and anti‐biotin microbeads (Miltenyi Biotec), followed by CD4 staining (RM4.5) and cell sorting of CD4+ Foxp3/YFP− cells or CD4+ Foxp3/GFP− using the BD FACSAria II. Control Tregs were purified from spleen and lymph nodes after enrichment of CD25+ cells using biotinylated anti‐CD25 mAb (7D4) and anti‐biotin microbeads (Miltenyi Biotec), followed by CD4 staining (RM4.5) and cell sorting of CD4+ Foxp3/YFP+ cells or CD4+ Foxp3/GFP+ using the BD FACSAria II. Treg purification was over 99%. TNF‐ and TNFR2‐deficient Tregs were similarly purified from Tnf−/− x Foxp3GFP and Tnfrsf1b−/− x Foxp3GFP mice, respectively. RelA‐deficient Tregs and their control Tregs were similarly purified from Foxp3Cre/+ x Relaflox and Foxp3Cre/+ mice, respectively. We used Foxp3Cre/+ x Relaflox mice because they did not develop autoimmunity, contrary to Foxp3Cre x Relaflox mice as we recently reported [52].
Cell cultures
Purified Tregs were cultivated in a complete medium composed of RPMI 1640, glutamax, and 10% fetal bovine serum, which was supplemented with IL‐2 (10 ng/mL, Peprotech) in all the cultures. Tregs were stimulated either with coated anti‐CD3 (coating at 1 or 2 μg/mL, 2C11; BioXcell) and anti‐CD28 (coating at 2 μg/mL) mAbs in 96‐well flat plates (15 x 104 cells/well) or with irradiated splenocytes from Cd3−/− mice (7.5 x 104 cells/well) used as APCs and soluble anti‐CD3 (0.05 μg/mL) mAb in 96‐well round plate (2.5 x 104 cells/well). The following soluble TNFRSF agonists were used to costimulate Tregs: anti‐4‐1BB mAb (10 μg/mL, 3H3; BioXcell), anti‐GITR mAb (3 μg/mL, DTA‐1; BioXcell), OX40L (100 ng/mL; AdipoGen), TNC‐sc(mu)TNF80 (STAR2) (12 ng/mL [19]), TL1A (12 ng/mL), LIGHT (AdipoGen), CD70, RANKL (Adipogen), FASL (Adipogen), TRAIL (Adipogen), anti‐DR5 mAb (MD5‐1; BioXCell), and anti‐OX40 mAb (OX86; BioXcell). Proliferation was assessed by labeling Tregs with CellTrace Violet (Life Technologies).
The “Relative Division” value was calculated using the following formula: [(CellTrace mean fluorescence intensity (CT MFI) of CTRL Tregs minus CT MFI of TNFRSF co‐stimulated Tregs) divided by CT MFI of CTRL Tregs] x 100, among living cells. The “FC living cells” value was calculated using the following formula: percentage of TNFRSF co‐stimulated living Tregs divided by percentage of control living Tregs. Living cells were assessed using a fixable viability dye (live/dead assay). For the additive effect of two TNFRSF agonists, the relative proliferation was calculated using the following formula: CT MFI of Tregs cultivated with one agonist divided by CT MFI of Tregs cultivated with two agonists and the FC living cells was calculated using the following formula: percentage of living Tregs cultivated with two agonists divided by percentage of living Tregs cultivated with one agonist.
For the suppression assay
Tconvs (2.5 x 104 cells/well), labeled with CellTrace Violet, were incubated with different concentrations of Tregs and splenocytes from Cd3−/− mice (7.5 x 104 cells/well) with an anti‐CD3 mAb (0.05 μg/mL, 2C11) in a round‐bottom plate in complete medium. The percentage suppression was calculated using the following formula: (% of divided cells in Tconvs alone minus % of divided cells in Tconvs + Tregs) divided by % of divided cells in Tconvs alone.
Cytokine quantification
After 3 days of Treg stimulation with coated antibodies and agonists, cytokine production in the supernatant was assessed using a Luminex kit from R&D systems.
Colitis
Tregs were first preincubated for 2 h at 37°C in complete medium and IL‐2 and anti‐GITR antibody (12 μg/mL) or STAR2 (48 ng/mL). Then, Tconvs (CD4+GFP−, 1 x 105 cells) and Tregs (CD4+YFP+, 2 x 104 cells) were injected intravenously into sex‐matched Rag2−/− mice. The clinical evaluation was performed three times a week by measuring body weight. For histological examination, colons were fixed in 10% formaldehyde. Five micrometer paraffin‐embedded sections were stained with hematoxylin and eosin and blindly scored. Colitis was scored on tissue sections as described previously [54].
T‐cell adoptive transfer
Tregs purified from Foxp3GFP mice expressing the CD90.1 CD45.2 congenic markers and from Foxp3‐DTR mice expressing the CD90.2 CD45.1 congenic markers were cultivated for 3 days as explained above, rested for 24 h in complete medium and IL‐2, and then coinjected intravenously (5 x 105 of each group) in C57BL/6J mice (CD90.2/CD45.2). Donor cells were analyzed 12 h and 7 days postinjection.
RNA sequencing and bioinformatics analyses
Tregs were stimulated by coated anti‐CD3 and CD28 mAbs as above for 18 and 36 h to generate biological triplicates. RNA was extracted using the NucleoSpin RNA XS kit from Macherey‐Nagel, quantified using a ND‐1000 NanoDrop spectrophotometer (NanoDrop Technologies), and purity/integrity was assessed using disposable RNA chips (Agilent High Sensitivity RNA ScreenTape) and an Agilent 2200 Tapestation (Agilent Technologies, Waldbrunn, Germany). mRNA library preparation was performed following manufacturer's recommendations (SMART‐Seq v4 Ultra Low Input RNA Kit TAKARA). Final‐17 samples pooled library prep was sequenced on Nextseq 500 ILLUMINA with HighOutPut cartridge (2x400Millions of 75 bases reads), corresponding to 2 times 23 x 106 reads per sample after demultiplexing. Poor quality sequences have been trimmed or removed with Trimmomatic software to retain only good quality paired reads. Star v2.5.3a [55] has been used to align reads on reference genome mm10 using standard options. Quantification of gene and isoform abundances has been done with rsem 1.2.28 [56] prior to normalization on library size with DESEq2 bioconductor package. Finally, differential analysis has been conducted with edgeR bioconductor package. Multiple hypothesis adjusted p‐values were calculated with the Benjamini–Hochberg procedure to control FDR. Principal component analysis was performed using Partek‐flow® (Partek). Heatmaps, gene ontology, and gene set enrichment analysis were performed using Qlucore® omics explorer 3.5 (Qlucore), Metascape (Metascape.org), and the shiny interface of the Institut du Cerveau et de la Moelle épinière (Paris, France).
Gene sets of supervised RNA‐Seq analyses
The “cytokines and chemokine receptors” and “T helper transcription factors” lists of genes used in Fig. 4B are the following: Ccr10, ccr4, Ccr6, Ccr8, Cxcr3, Ifng, Il10, Il13, Il17, Il2, Il21, Il22, Il23r, Il4, Il5, Il9, Klrb1c, Ptgdr2, Tnf and Ahr , Fosl2, Foxo4, Gata3, Irf4, Jun, Maf, Rorc, Spi1, Stat*, and Tbx21. The list of genes involved in T‐cell survival and apoptosis used in Supporting Information Fig. S8 is the following: Akt1 (Akt), Bad, Bak1 (Bak), Bax, Bbc3 (Puma), Bcl10, Bcl2, Bcl2a* (Bfl‐1), Bcl2l* (Bcl‐XL, Bim, etc.), Birc5 (Survivin), Casp*, Cflar (c‐Flip), Cybb (Nox2), Dffa, Fadd, Fas, Fasl, Foxp3, Gpam, Il7r, Il2ra, Il2rb, Il2rg, Jun, Lgals* (Galectin‐*), Max, Mcl1, Mnt, Myc, Nfkb1, Nfkb2, Nfkbi* (Ikb*), Nos2 (iNOS), Pdcd1 (Pd1), Pip, Pmaip1 (Noxa), Prelid1, Prkcq (PKC‐theta), Ripk1 (Rip1), Ripk3 (Rip3), Sirt1, Siva1, Stat3, Stat5a, Stat5b, Tgfb*, Traf*, and Trp53 (P53). The “Treg effector” list of genes used in Fig. 6B is the following: Areg, Ccl3, Ccl4, Cd274 (PDL1), Ctla4, Ebi3, Entpd1, Ezh2, Fgl2, Foxp3, Furin, Gzm*, Hmox1, Hmox2, Icos, Il10, Il1r*, Il2r*, Il6ra, Itgal, Itgb2, Itgb8, Lag3, Lgals1, Lrrc32, Nrp1, Nt5e, Pdcd1, Pde3b, Tgfb*, Tigit, Tnfsf*, and Tnfrsf*. Heatmaps represented DEG (FDR < 0.05) using Qlucore® omics explorer 3.5 (Qlucore).
Electrophoretic mobility shift assays (EMSA) combined with supershift assays
Tregs from Foxp3GFP mice were stimulated for 24 h with coated anti‐CD3/CD28 mAbs, rested for 1.5 or 4 h in complete medium and IL‐2, and restimulated with TNFRSF agonists and IL‐2 for 0.5, 1, and 4 h. Total protein extracts were prepared and analyzed for DNA‐binding activity using the HIV‐LTR tandem κB oligonucleotide as κB probe as previously described [57]. For supershifts, protein extracts were incubated with specific antibodies for 30 min on ice before incubation with the labeled probe.
Statistics
Statistical analyses were performed using GraphPad Prism Software. Statistical tests used were indicated in the figure legends: *p < 0.05, **p < 0.01, ***p < 0.001.
Author contributions
M.L.d.R., E.R., D.C., J.D., and S.G. performed experiments and analyzed data. H.W. provided essential reagents and edited the manuscript. Y.G.‐B., T.G., and G.M. performed bioinformatic analyses and edited the manuscript. V.B. contributed to experimental design and edited the manuscript. B.L.S. designed and analyzed experiments and wrote the manuscript.
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Abbreviations
- DEG
differentially expressed genes
- FC
fold change
- FDR
false discovery rate
- Tconvs
conventional T cells
- TNFRSF
TNF receptor superfamily
- TNFSF
TNF superfamily
Supporting information
supplemental information
Acknowledgements
We thank the great expertise of Yannick Marie, Delphine Bouteiller, Beata Gyorgy, and Justine Guegan from the bio‐informatic platform of the Institut du Cerveau et de la Moelle Epinière (Paris) and of Vincent Alcacer from the Centre de Recherche en Cancérologie de Lyon. We thank Flora Issert and Doriane Foret from the animal facility for taking care of the mice. We thank Prs. Alexander Rudensky and Bernard Malissen who kindly provided us with the Foxp3Cre and Foxp3GFP mice, respectively. We thank Odile Devergne for critical reading of the manuscript.
MLdR and TG were funded by the European Union's H2020 research and innovation program “ENLIGHT‐TEN” under the Marie Sklodowska‐Curie grant agreement 675395. This work was supported by the ENLIGHT‐TEN program, the Fondation pour la Recherche Médicale (Equipes FRM 2015), and the Agence Nationale de la Recherche (ANR‐15‐CE15‐0015‐01 and ANR‐17‐CE15‐0030‐01) to B.L.S. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummer 324392634—TRR 221 to H.W.
See accompanying commentary by Ayroldi and Grohmann
See accompanying commentary: https://onlinelibrary.wiley.com/doi/10.1002/eji.202048711
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.201948393
References
- 1. Assal, A. , Kaner, J. , Pendurti, G. and Zang, X. , Emerging targets in cancer immunotherapy: beyond CTLA‐4 and PD‐1. Immunotherapy 2015. 7: 1169–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burugu, S. , Dancsok, A. R. and Nielsen, T. O. , Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2018. 52: 39–52. [DOI] [PubMed] [Google Scholar]
- 3. Croft, M. , Benedict, C. A. and Ware, C. F. , Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug. Discov. 2013. 12: 147–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buchan, S. L. , Dou, L. , Remer, M. , Booth, S. G. , Dunn, S. N. , Lai, C. , Semmrich, M. et al., Antibodies to costimulatory receptor 4‐1BB enhance anti‐tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity 2018. 49: 958‐970, e957. [DOI] [PubMed] [Google Scholar]
- 5. Bulliard, Y. , Jolicoeur, R. , Windman, M. , Rue, S. M. , Ettenberg, S. , Knee, D. A. , Wilson, N. S. et al., Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor‐targeting antibodies. J. Exp. Med. 2013. 210: 1685–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bulliard, Y. , Jolicoeur, R. , Zhang, J. , Dranoff, G. , Wilson, N. S. and Brogdon, J. L. , OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol. Cell. Biol. 2014. 92: 475–480. [DOI] [PubMed] [Google Scholar]
- 7. Kocak, E. , Lute, K. , Chang, X. , May, K. F., Jr. , Exten, K. R. , Zhang, H. , Abdessalam, S. F. et al., Combination therapy with anti‐CTL antigen‐4 and anti‐4‐1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res. 2006. 66: 7276–7284. [DOI] [PubMed] [Google Scholar]
- 8. Menk, A. V. , Scharping, N. E. , Rivadeneira, D. B. , Calderon, M. J. , Watson, M. J. , Dunstane, D. , Watkins, S. C. et al., 4‐1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018. 215: 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Piconese, S. , Valzasina, B. and Colombo, M. P. , OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med. 2008. 205: 825–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun, Y. , Lin, X. , Chen, H. M. , Wu, Q. , Subudhi, S. K. , Chen, L. and Fu, Y. X. , Administration of agonistic anti‐4‐1BB monoclonal antibody leads to the amelioration of experimental autoimmune encephalomyelitis. J. Immunol. 2002. 168: 1457–1465. [DOI] [PubMed] [Google Scholar]
- 11. Chen, S. , Lee, L. F. , Fisher, T. S. , Jessen, B. , Elliott, M. , Evering, W. , Logronio, K. et al., Combination of 4‐1BB agonist and PD‐1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol. Res. 2015. 3: 149–160. [DOI] [PubMed] [Google Scholar]
- 12. Choi, B. K. , Asai, T. , Vinay, D. S. , Kim, Y. H. and Kwon, B. S. , 4‐1BB‐mediated amelioration of experimental autoimmune uveoretinitis is caused by indoleamine 2,3‐dioxygenase‐dependent mechanisms. Cytokine 2006. 34: 233–242. [DOI] [PubMed] [Google Scholar]
- 13. Ruby, C. E. , Yates, M. A. , Hirschhorn‐Cymerman, D. , Chlebeck, P. , Wolchok, J. D. , Houghton, A. N. , Offner, H. et al., Cutting Edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J. Immunol. 2009. 183: 4853–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arvey, A. , van der Veeken, J. , Plitas, G. , Rich, S. S. , Concannon, P. and Rudensky, A. Y. , Genetic and epigenetic variation in the lineage specification of regulatory T cells. Elife 2015. 4: e07571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zemmour, D. , Zilionis, R. , Kiner, E. , Klein, A. M. , Mathis, D. and Benoist, C. , Single‐cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 2018. 19: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Magnuson, A. M. , Kiner, E. , Ergun, A. , Park, J. S. , Asinovski, N. , Ortiz‐Lopez, A. , Kilcoyne, A. et al., Identification and validation of a tumor‐infiltrating Treg transcriptional signature conserved across species and tumor types. Proc. Natl. Acad. Sci. USA 2018. 115: E10672–E10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Plitas, G. , Konopacki, C. , Wu, K. , Bos, P. D. , Morrow, M. , Putintseva, E. V. , Chudakov, D. M. et al., Regulatory T cells exhibit distinct features in human breast cancer. Immunity 2016. 45: 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bacher, P. , Heinrich, F. , Stervbo, U. , Nienen, M. , Vahldieck, M. , Iwert, C. , Vogt, K. et al., Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell 2016. 167: 1067‐1078, e1016. [DOI] [PubMed] [Google Scholar]
- 19. Chopra, M. , Biehl, M. , Steinfatt, T. , Brandl, A. , Kums, J. , Amich, J. , Vaeth, M. et al., Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med. 2016. 213: 1881–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ephrem, A. , Epstein, A. L. , Stephens, G. L. , Thornton, A. M. , Glass, D. and Shevach, E. M. , Modulation of Treg cells/T effector function by GITR signaling is context‐dependent. Eur. J. Immunol. 2013. 43: 2421–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Irie, J. , Wu, Y. , Kachapati, K. , Mittler, R. S. and Ridgway, W. M. , Modulating protective and pathogenic CD4+ subsets via CD137 in type 1 diabetes. Diabetes 2007. 56: 186–196. [DOI] [PubMed] [Google Scholar]
- 22. Schreiber, T. H. , Wolf, D. , Tsai, M. S. , Chirinos, J. , Deyev, V. V. , Gonzalez, L. , Malek, T. R. et al., Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J. Clin. Invest. 2010. 120: 3629–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen, X. , Baumel, M. , Mannel, D. N. , Howard, O. M. and Oppenheim, J. J. , Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. 2007. 179: 154–161. [DOI] [PubMed] [Google Scholar]
- 24. Nguyen, D. X. and Ehrenstein, M. R. , Anti‐TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF‐TNF‐RII binding in rheumatoid arthritis. J. Exp. Med. 2016. 213: 1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taraban, V. Y. , Slebioda, T. J. , Willoughby, J. E. , Buchan, S. L. , James, S. , Sheth, B. , Smyth, N. R. et al., Sustained TL1A expression modulates effector and regulatory T‐cell responses and drives intestinal goblet cell hyperplasia. Mucosal Immunol. 2011. 4: 186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Valzasina, B. , Guiducci, C. , Dislich, H. , Killeen, N. , Weinberg, A. D. and Colombo, M. P. , Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood 2005. 105: 2845–2851. [DOI] [PubMed] [Google Scholar]
- 27. Zheng, G. , Wang, B. and Chen, A. , The 4‐1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. J. Immunol. 2004. 173: 2428–2434. [DOI] [PubMed] [Google Scholar]
- 28. Chen, X. , Hamano, R. , Subleski, J. J. , Hurwitz, A. A. , Howard, O. M. and Oppenheim, J. J. , Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3‐ conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J. Immunol. 2010. 185: 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stephens, G. L. , McHugh, R. S. , Whitters, M. J. , Young, D. A. , Luxenberg, D. , Carreno, B. M. , Collins, M. et al., Engagement of glucocorticoid‐induced TNFR family‐related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J. Immunol. 2004. 173: 5008–5020. [DOI] [PubMed] [Google Scholar]
- 30. Takeda, I. , Ine, S. , Killeen, N. , Ndhlovu, L. C. , Murata, K. , Satomi, S. , Sugamura, K. et al., Distinct roles for the OX40‐OX40 ligand interaction in regulatory and nonregulatory T cells. J. Immunol. 2004. 172: 3580–3589. [DOI] [PubMed] [Google Scholar]
- 31. Zaragoza, B. , Chen, X. , Oppenheim, J. J. , Baeyens, A. , Gregoire, S. , Chader, D. , Gorochov, G. et al., Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat. Med. 2016. 22: 16–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Croft, M. and Siegel, R. M. , Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017. 13: 217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. So, T. and Croft, M. , Cutting edge: OX40 inhibits TGF‐beta‐ and antigen‐driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J. Immunol. 2007. 179: 1427–1430. [DOI] [PubMed] [Google Scholar]
- 34. Bittner, S. and Ehrenschwender, M. , Multifaceted death receptor 3 signaling‐promoting survival and triggering death. FEBS Lett. 2017. 591: 2543–2555. [DOI] [PubMed] [Google Scholar]
- 35. Joetham, A. , Ohnishi, H. , Okamoto, M. , Takeda, K. , Schedel, M. , Domenico, J. , Dakhama, A. et al., Loss of T regulatory cell suppression following signaling through glucocorticoid‐induced tumor necrosis receptor (GITR) is dependent on c‐Jun N‐terminal kinase activation. J. Biol. Chem. 2012. 287: 17100–17108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu, L. F. , Gondek, D. C. , Scott, Z. A. and Noelle, R. J. , NF kappa B‐inducing kinase deficiency results in the development of a subset of regulatory T cells, which shows a hyperproliferative activity upon glucocorticoid‐induced TNF receptor family‐related gene stimulation. J. Immunol. 2005. 175: 1651–1657. [DOI] [PubMed] [Google Scholar]
- 37. Vasanthakumar, A. , Liao, Y. , Teh, P. , Pascutti, M. F. , Oja, A. E. , Garnham, A. L. , Gloury, R. et al., The TNF receptor superfamily‐NF‐kappaB axis is critical to maintain effector regulatory T cells in lymphoid and non‐lymphoid tissues. Cell. Rep. 2017. 20: 2906–2920. [DOI] [PubMed] [Google Scholar]
- 38. Wang, J. , Ferreira, R. , Lu, W. , Farrow, S. , Downes, K. , Jermutus, L. , Minter, R. et al., TNFR2 ligation in human T regulatory cells enhances IL2‐induced cell proliferation through the non‐canonical NF‐kappaB pathway. Sci. Rep. 2018. 8: 12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burocchi, A. , Pittoni, P. , Gorzanelli, A. , Colombo, M. P. and Piconese, S. , Intratumor OX40 stimulation inhibits IRF1 expression and IL‐10 production by Treg cells while enhancing CD40L expression by effector memory T cells. Eur. J. Immunol. 2011. 41: 3615–3626. [DOI] [PubMed] [Google Scholar]
- 40. Nagar, M. , Jacob‐Hirsch, J. , Vernitsky, H. , Berkun, Y. , Ben‐Horin, S. , Amariglio, N. , Bank, I. et al., TNF activates a NF‐kappaB‐regulated cellular program in human CD45RA‐ regulatory T cells that modulates their suppressive function. J. Immunol. 2010. 184: 3570–3581. [DOI] [PubMed] [Google Scholar]
- 41. Josefowicz, S. Z. , Lu, L. F. and Rudensky, A. Y. , Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 2012. 30: 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grinberg‐Bleyer, Y. , Baeyens, A. , You, S. , Elhage, R. , Fourcade, G. , Gregoire, S. , Cagnard, N. et al., IL‐2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J. Exp. Med. 2010. 207: 1871–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grinberg‐Bleyer, Y. , Saadoun, D. , Baeyens, A. , Billiard, F. , Goldstein, J. D. , Gregoire, S. , Martin, G. H. et al., Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J. Clin. Invest. 2010. 120: 4558–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piconese, S. , Pittoni, P. , Burocchi, A. , Gorzanelli, A. , Care, A. , Tripodo, C. and Colombo, M. P. , A non‐redundant role for OX40 in the competitive fitness of Treg in response to IL‐2. Eur. J. Immunol. 2010. 40: 2902–2913. [DOI] [PubMed] [Google Scholar]
- 45. So, T. and Croft, M. , Regulation of PI‐3‐kinase and Akt signaling in T lymphocytes and other cells by TNFR family molecules. Front. Immunol. 2013. 4: 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Song, J. , So, T. , Cheng, M. , Tang, X. and Croft, M. , Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity 2005. 22: 621–631. [DOI] [PubMed] [Google Scholar]
- 47. Starck, L. , Scholz, C. , Dorken, B. and Daniel, P. T. , Costimulation by CD137/4‐1BB inhibits T cell apoptosis and induces Bcl‐xL and c‐FLIP(short) via phosphatidylinositol 3‐kinase and AKT/protein kinase B. Eur. J. Immunol. 2005. 35: 1257–1266. [DOI] [PubMed] [Google Scholar]
- 48. Brusko, T. M. , Putnam, A. L. and Bluestone, J. A. , Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol. Rev. 2008. 223: 371–390. [DOI] [PubMed] [Google Scholar]
- 49. Uhlig, H. H. and Powrie, F. , Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu. Rev. Immunol. 2018. 36: 755–781. [DOI] [PubMed] [Google Scholar]
- 50. Rubtsov, Y. P. , Rasmussen, J. P. , Chi, E. Y. , Fontenot, J. , Castelli, L. , Ye, X. , Treuting, P. et al., Regulatory T cell‐derived interleukin‐10 limits inflammation at environmental interfaces. Immunity 2008. 28: 546–558. [DOI] [PubMed] [Google Scholar]
- 51. Wang, Y. , Kissenpfennig, A. , Mingueneau, M. , Richelme, S. , Perrin, P. , Chevrier, S. , Genton, C. et al., Th2 lymphoproliferative disorder of LatY136F mutant mice unfolds independently of TCR‐MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J. Immunol. 2008. 180: 1565–1575. [DOI] [PubMed] [Google Scholar]
- 52. Ronin, E. , Lubrano di Ricco, M. , Vallion, R. , Divoux, J. , Kwon, H. K. , Gregoire, S. , Collares, D. et al., The NF‐kappaB RelA transcription factor is critical for regulatory T cell activation and stability. Front. Immunol. 2019. 10: 2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cossarizza, A. , Chang, H. D. , Radbruch, A. , Acs, A. , Adam, D. , Adam‐Klages, S. , Agace, W. W. et al., Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 2019. 49: 1457–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martin, B. , Auffray, C. , Delpoux, A. , Pommier, A. , Durand, A. , Charvet, C. , Yakonowsky, P. et al., Highly self‐reactive naive CD4 T cells are prone to differentiate into regulatory T cells. Nat. Commun. 2013. 4: 2209. [DOI] [PubMed] [Google Scholar]
- 55. Dobin, A. , Davis, C. A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. et al., STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013. 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li, B. and Dewey, C. N. , RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 2011. 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Authier, H. , Billot, K. , Derudder, E. , Bordereaux, D. , Riviere, P. , Rodrigues‐Ferreira, S. , Nahmias, C. et al., IKK phosphorylates RelB to modulate its promoter specificity and promote fibroblast migration downstream of TNF receptors. Proc. Natl. Acad. Sci. USA 2014. 111: 14794–14799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplemental information